Comparing the differences of prokaryotic microbial community between pit walls and bottom from Chinese liquor revealed by 16S rRNA gene sequencing
-
Shu Fang
 ,
Chuanxiang Wang
,
Chuanxiang Wang

Abstract
This study aims to explore the prokaryotic microbial community structures and diversity in pit mud from different depths, and provide a theoretical basis for the liquor production and further study of pit mud. The fermented pit muds of strong-flavor liquor from Yun distillery were taken as samples. The high-throughput sequencing approach, followed by bioinformatics analyses, was used to compare the differences in the prokaryotic microbial community between pit walls and bottom represented by samples. A total of 31 bacteria phyla and 2 archaea phyla were detected. The dominant phyla in YJ-S, YJ-Z, and YJ-X (sample name) were Proteobacteria and Firmicutes, while the dominant genera in them were Acinetobacter, Aminobacterium, and Lactobacillus. YJ-Z and YJ-X were the closest in species diversity. In species richness analysis, YJ-X was the highest, followed by YJ-Z, and YJ-S was the lowest; in species uniformity analysis, YJ-S was the highest, followed by YJ-Z, and YJ-X was the lowest. The function predicted by 16S rRNA genome showed that prokaryotic microbial function in pit mud was mainly concentrated in “Carbohydrate transport and metabolism” and “Amino acid transport and metabolism.” Significant differences in prokaryotic microbial community and gene function prediction between pit walls and bottom were found in YJ-S, YJ-Z, and YJ-X (p < 0.05).
1 Introduction
China is a country with a long history of traditional liquor (baijiu) production. Baijiu is one of the world’s six major distilled liquors [1]. Liquor is mainly produced in the way of solid-state fermentation of certain crops, such as sorghum, corn, wheat, rice, and glutinous rice [2]. During the fermentation process of liquor, liquor has gradually developed with 12 kinds of different styles in the land of China, due to the differences in brewing raw materials, brewing technology, ecological environment, saccharification, distillation, and other factors in different regions of China, such as sauce-flavored, strong-flavored, light-flavored, rice-flavored, and medicinal flavors [3,4].
Among these liquors, strong-flavor liquor, which is produced by distilling fermented grains (mainly sorghum), has the strong fragrance, sweetness, and lasting aftertaste [1,2]. Strong-flavor liquor is the most popular type of liquor in China, accounting for more than half of the liquor consumption market [5,6]. During the production process of solid-state fermentation of traditional Chinese liquor, a bucket-shaped pit must be excavated underground in advance. The inner wall and bottom of the pit are coated with special fermented clay, which is called pit mud [7]. Pit mud consists of cooked sticky loess, humus soil, cooked bean cake powder, bran koji powder, yellow water, caproic acid bacteria solution, fragrant mud, yeast solution, ammonium phosphate, urea, and wine tail. The above materials are mixed and stirred in a certain proportion, then it is formed after a complex fermentation process in mother-pit. Pit mud is essential for the fermentation and production of strong-flavor liquor, due to the presence of many microorganisms, such as bacteria, archaea, and fungi [6,7]. These microorganisms, which can produce aromatic compounds for the fermentation of strong-flavor liquor, rely strongly on the suitable microecological environment provided by the pit mud [8]. Many studies have shown that the flavor of liquor is closely related to the microorganisms used for fermenting liquor raw materials in the pit mud [9]. The microbial community structure of pit mud is quite complex and closely related to the age and geographical location of the pit [4]. Different microorganisms ferment the raw materials and produce various enzymes and aromatic compounds in the liquor, forming various types of liquors with distinctive flavors [9,10].
In order to increase the output of liquor, liquor-production enterprises generally build larger pits. The pit provides a closed environment for microbial fermentation in pit mud and Daqu. The temperature in the environment is 25–32°C, the humidity is 40–45%, and the pH is 3.0–5.0 [5]. Due to the differences in the microecological environment at different locations of the pit, such as pit walls and bottom, there are differences in the microbial community abundance at different locations in the pit, which leads to differences in the quality of the fermented liquor, and affects the quality of liquor production; such studies have rarely been reported [4].
In this study, Anhui Yun distillery Group Co., Ltd, located in Ma’anshan city, east of Anhui province, China, established some pits with 2.85 m in length, 2.65 m in width, and 2.2 m in depth for the production of strong-flavor liquor in 2003, the pit muds of the pits were cultivated in the same microorganism enriched mother-pit. Since the quality of liquor produced in these pits was not exactly the same, the pit producing the best quality liquor was selected as our research subject by consulting the distillery technician. High-throughput sequencing technologies have developed rapidly and emerged as an effective tool, both for the microbial diversity analysis of complex samples and studying metabolic pathways in recent years [11–14]. The technique of high-throughput sequencing has also been recently used for microbial community structure analysis of Chinese liquor [2,4–6,10,15]. In prokaryotic cells, the 16S rRNA genes are composed of variable regions and conserved regions, the former of which can be sequenced to identify between microbes for bacterial phylogenetic and taxonomic identification [11]. In this study, the variables V3 and V4 regions of the 16S rRNA genes were used to determine the prokaryotic microbial diversity in pit walls and bottom. Our study provides a theoretical basis for the daily maintenance of pit mud, the specifications of new pits construction, and the quality of liquor production.
2 Materials and methods
2.1 Sample collection
After the selected pit was opened and the distiller’s grains were taken out, the pit muds in 24 positions (1 g/each sample position) were taken vertically and equidistant from the fermentation pit in upper (0.2 m in depth), middle (1.2 m in depth), and bottom layer (2.2 m in depth), respectively (Figure 1). The pit muds in the same layer (eight positions/each sample layer) were transferred into a sterile bag, sealed, named as YJ-S, YJ-Z, YJ-X, and then stored at −80°C until further analysis.
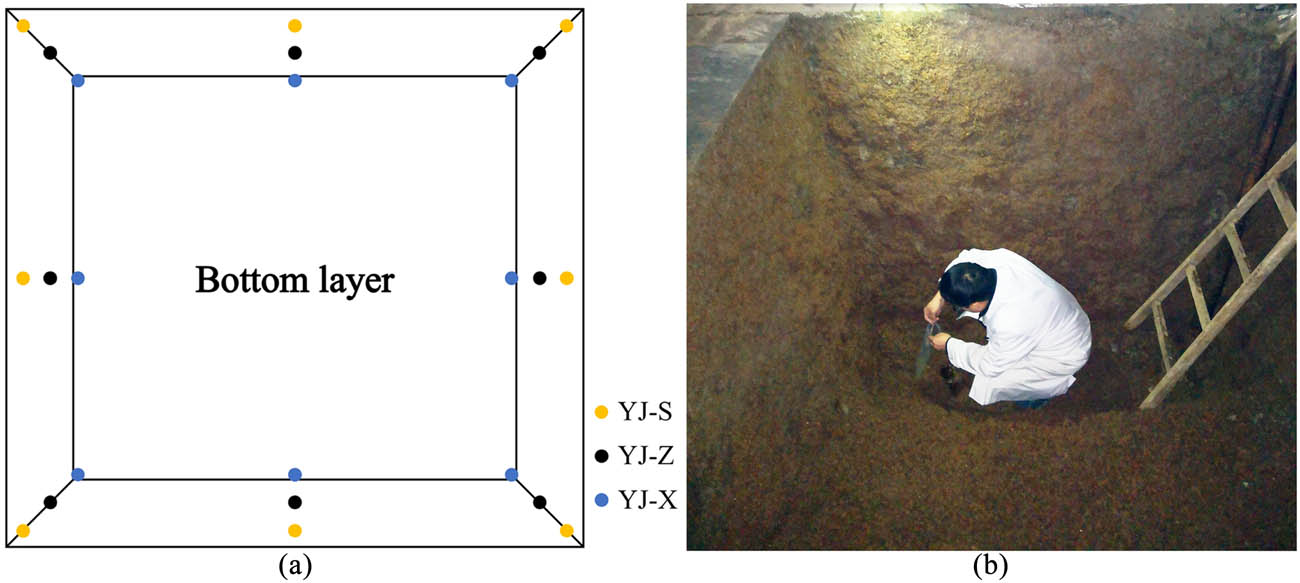
Sampling points of mud in the pit: (a) sampling diagram and (b) sampling in the pit.
2.2 DNA extraction
Pit mud samples needed to be transformed into pellets through pretreatment, then the resulted pellets from the three samples were performed using centrifuge following the method described by Ding et al. [16]. The pellets were subjected to DNA extraction using the commercial Mag-Bind Soil DNA Kit (Omega Bio-tek Corporation, USA) according to the manufacturer’s instruction. The concentration and purity of extracted DNAs were measured using 0.8% (w/v) agarose gel electrophoresis and a micro-spectrophotometer, respectively. The resulted DNA was diluted to a DNA concentration of 1 ng/μL and subsequently used as a template for PCR to amplify 16S rRNA genes.
2.3 PCR amplification
The V3 and V4 regions of 16S rRNA genes were amplified using the universal forward primer 341F(5′-CCCTACACGACGCTCTTCCGATCTG(barcode)CCTACGGGNGGCWGCAG-3′) and the reverse primer 805R (5′-GACTGGAGTTCCTTGGCACCCGAGAATTCCAGACTACHVGGGTATCTAATCC-3′) with a set of six-nucleotide barcodes, the primers were modified and synthesized by Sangon Biotech Company (Shanghai, China) [17,18]. The detailed PCR procedures were modified according to a method described by Tang et al. [19]. The PCR amplification conditions were as follows: 94°C for 3 min; 94°C for 30 s, 45°C for 20 s, 65°C for 30 s, five cycles; 94°C for 20 s, 55°C for 20 s, 72°C for 30 s, 20 cycles and 72°C for 5 min. The amplified PCR products were detected by 2% agarose gel electrophoresis and purified using the commercial QIAquick Gel Extraction Kit (QIAGEN Corporation, Germany). After gel purification, the PCR product was quantified using the commercial Qubit ssDNA Assay Kit (Life Technologies, USA). Finally, the purified 16S rRNA gene amplicons were to construct DNA library using TruSeq DNA PCR-Free Sample Preparation Kit (Illumina Inc, USA) and pair-end sequenced on an Illumina HiSeq2500 PE250 platform at Sangon Biotech Company (Shanghai, China).
2.4 Genome data processing
The original image data file obtained by high-throughput sequencing was transformed into the original sequence by CASAVA base recognition analysis, which was called raw reads. Cutadapt software (V1.2.1) [20] was used to remove the primer splice sequence of raw reads, pear software (V0.9.6) [21] was used to splice sequences, Prinseq software (V0.20.4) [22] was used to filter sequences, Usearch software (V5.2.236) [23] was used to remove chimeras and nonspecific amplification sequences, and finally, the qualified sequences were obtained.
2.5 Bioinformatics analysis of the 16S rRNA genome sequences
Usearch software contains a suite of software tools that were used to cluster all effective tags to the operational taxonomic units (OTU) based on 97% identity of the sequences. The taxonomy of each OTU representative sequence was assigned using the Ribosomal Database Project (http://rdp.cme.msu.edu/misc/resources.jsp) classifier with a minimum bootstrap threshold of 80%. Subsequently, taxonomic information at the six-level classification level domain, phylum, class, order, family, and genus was obtained [24]. Based on the results of the OTU, a Venn diagram was generated using R software (V3.2) [25] to show the number of OTU of microorganisms that were shared in the three samples and the variance among them. The Bray–Curtis algorithm was used to construct a sample clustering tree and species abundance histogram based on the abundance of different sample species. Mothur software (V1.2.7) [26] was used to calculate the five kinds of alpha diversity index, such as Shannon, ACE, Chao1, Coverage, and Simpson; R software was used to draw dilution curve and rank abundance curve. Heat maps were drawn based on unifrac distance matrix between different samples; PICRUSt software (V1.1.4) [27] was used to predict the metagenomic function of the three samples, and abundance heat maps based on the COG function and the KEGG metabolic pathway were both drawn. STAMP software (V2.1.3) [28] was used to compare with the differences of species abundance and gene functional abundance among the three samples, the 25 ranks with the lowest p value were listed in the figures, when p was less than 0.05, the species classification was marked in red. In statistical analysis, significance was determined at 95% confidence interval (p = 0.05).
2.6 Nucleic acid sequence accession number
The 16S rRNA genome sequences obtained by high-throughput sequencing in this study had been submitted to NCBI database under the accession number: SRR15900602, SRR15900603, and SRR15900777.
3 Results
3.1 OTU clustering of microorganisms in pit mud
After high-throughput sequencing of PCR amplification products, a total of 208,366 raw reads were obtained from the three samples, and 175,150 qualified reads were obtained after processing (Table 1). These 16S rRNA gene sequences were clustered into 11,806 OTUs for species analysis with 97% similarity. Among them, YJ-S had 58,282 qualified reads and a cluster of 5,071 OTUs, YJ-Z had 49,669 qualified reads and a cluster of 3,563 OTUs, YJ-X had 67,199 qualified reads and a cluster of 3,172 OTUs (Table 1). The Venn diagram can be used to count the number of shared and unique OTUs in samples. There were 464 OTUs in both YJ-S and YJ-Z, accounting for 3.93% of the total OTUs; 391 OTUs in both YJ-S and YJ-X, accounting for 3.31% of the total OTUs; and 413 OTUs in both YJ-Z and YJ-X, accounting for 3.50% of the total OTUs. There were 277 OTUs in YJ-S, YJ-Z, and YJ-X, accounting for 2.35% of the total OTUs (Figure 2). To sum up, YJ-S and YJ-Z had the highest similarity in OTU number composition, followed by YJ-Z and YJ-X, finally YJ-S and YJ-X.
Sample sequencing information and alpha diversity of microorganism in pit mud
| Sample | Raw reads | Qualified reads | OTU | Alpha diversity index | ||||
|---|---|---|---|---|---|---|---|---|
| Shannon | ACE | Chao1 | Coverage | Simpson | ||||
| YJ-S | 73,608 | 58,282 | 5,071 | 3.65 | 90410.65 | 34646.12 | 0.93 | 0.17 |
| YJ-Z | 59,100 | 49,669 | 3,563 | 3.66 | 33985.33 | 23344.33 | 0.94 | 0.12 |
| YJ-X | 75,658 | 67,199 | 3,172 | 4.38 | 60690.04 | 22079.02 | 0.97 | 0.08 |

Venn diagram of number of OTUs of microorganism in pit mud.
3.2 Taxonomic annotation of microorganisms in pit mud
Both bacteria and archaea were detected in YJ-S, YJ-Z, and YJ-X. The abundance of bacteria and that of archaea in YJ-S were 99 and 1%, respectively, in YJ-Z 98% and 2%, respectively, and in YJ-X 99% and 0.7% ,respectively. Among the three samples, bacterial abundance was absolutely dominant compared with archaea. At the phylum level, a total of 33 phyla including Firmicutes, Proteobacteria, Bacteroidetes, Synergistetes, and Actinobacteria were identified in the 16S rRNA gene sequences of the three samples, and the abundances were significantly different (Figure 3; Figures S1–S3 in Supplementary 1, p < 0.05). In addition, Euryarchaeota and Crenarchaeota were both identified in all three samples, belonging to the Archaea phylum, but only the abundance of Euryarchaeota was significantly different (Figure 3; Figures S1–S3 in Supplementary 1, p < 0.05). Firmicutes was the prominent phylum in YJ-Z and YJ-X with the most abundances, while it was Proteobacteria in YJ-S (p < 0.05). At the genus level, the distribution of microbial communities in YJ-S, YJ-Z, and YJ-X was also different, and the abundance of different genera was also different. Having abundance levels of more than 1%, the main microorganisms in YJ-S were Acinetobacter (43.53%), Lysinibacillus (14.96%), Caryophanon (7.05%), Petrimonas (4.29%), Proteiniphilum (4.13%), Clostridium IV (1.79%), Solibacillus (1.5%), and Aminobacterium (1.03%), and 8.18% of the microorganisms were unclassified genus (Table 2). The main microorganisms in YJ-Z were Aminobacterium (25.04%), Clostridium IV (21.39%), Caloramator (17.01%), Lactobacillus (2.49%), Semimentibacter (1.93%), Olsenella (1.91%), Tissierella (1.6%), Syntrophimonas (1.48%), Garciella (1.25%), Syntrophaceticus (1.09%), and unclassified genus (12.75%) (Table 2). The main microorganisms in YJ-X were Lactobacillus (6.49%), Bifidobacterium (4.71%), Bacteroides (4.16%), Faecalibacterium (3.88%), Clostridium sensu stricto (3.83%), Proteiniphilum (3.55%), Syntrophomonas (3.41%), Petrimonas (2.83%), Clostridium IV (2.63%), Blautia (2.06%), Aminobacterium (1.75%), Alistipes (1.32%), Roseburia (1.14%), Burkholderia (1.07%), and unclassified genus (35.54%) (Table 2). To sum up, the numbers of genera that have an abundance of 1% or higher in YJ-S, YJ-Z, and YJ-X were 9, 10, and 14, respectively, each making up 78.28, 75.19, and 42.83% of the total abundance in the respective sample (Figure 4, Table 2). The abundances of genera mentioned above were significantly different in YJ-S, YJ-Z, and YJ-X (p < 0.05), and the p value is shown in Supplementary 2.
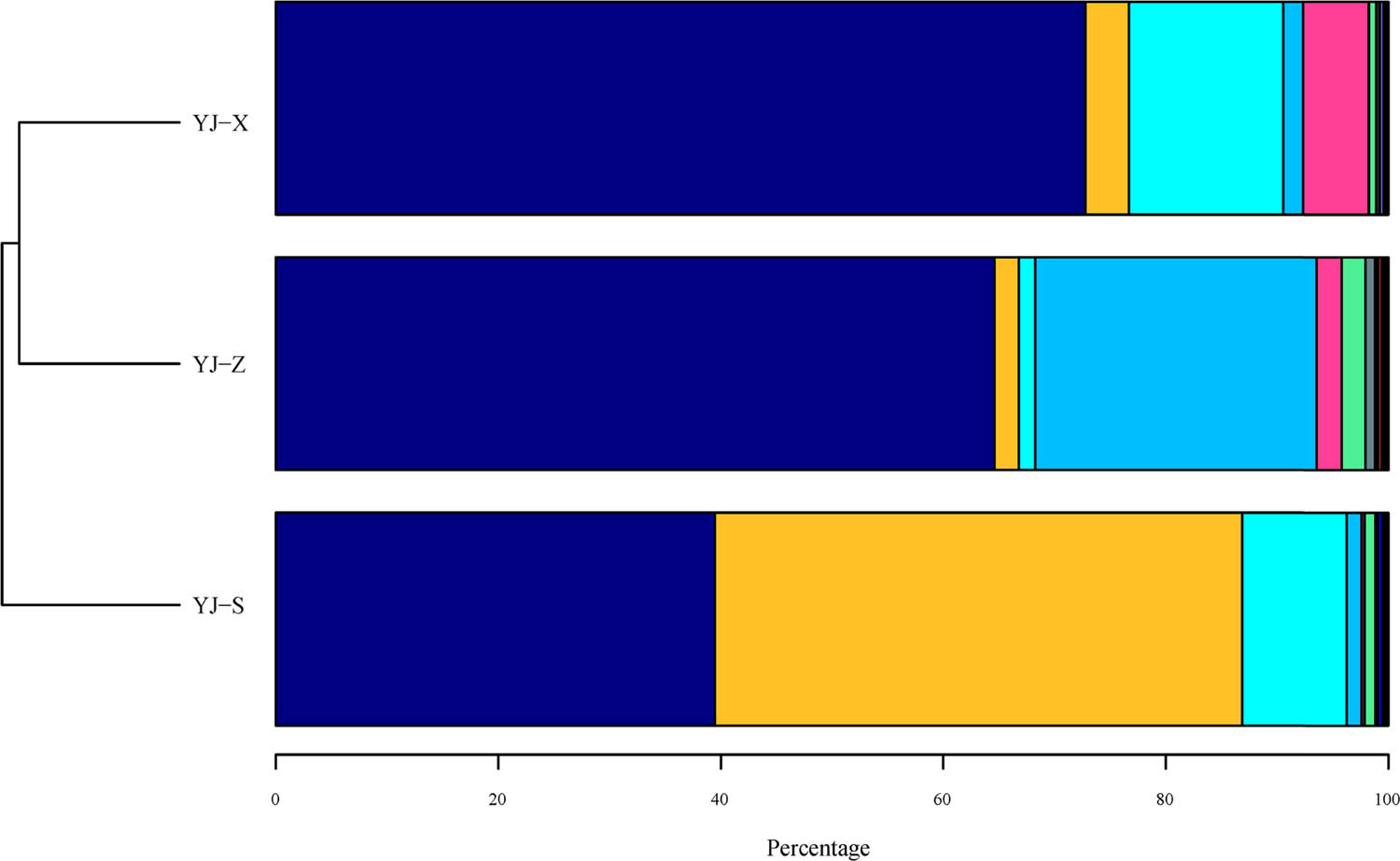
Phylum level distribution of microorganism population present in pit mud. Note: ( ) Firmicutes; (
) Firmicutes; ( ) Proteobacteria; (
) Proteobacteria; ( ) Bacteroidetes; (
) Bacteroidetes; ( ) Synergistetes; (
) Synergistetes; ( ) Actinobacteria; (
) Actinobacteria; ( ) Euryarchaeota; (
) Euryarchaeota; ( ) unclassified; (
) unclassified; ( ) Chloroflexi; (
) Chloroflexi; ( ) Verrucomicrobia; (
) Verrucomicrobia; ( ) Acidobacteria; (
) Acidobacteria; ( ) Tenericutes; (
) Tenericutes; ( ) Cloacimonetes; (
) Cloacimonetes; ( ) Planctomycetes; (
) Planctomycetes; ( ) Armatimonadetes; (
) Armatimonadetes; ( ) Spirochaetes; (
) Spirochaetes; ( ) Gemmatimonadetes; (
) Gemmatimonadetes; ( ) Candidatus Saccharibacteria; (
) Candidatus Saccharibacteria; ( ) Deinococcus-Thermus; (
) Deinococcus-Thermus; ( ) Atribacteria; (
) Atribacteria; ( ) Chlamydiae; (
) Chlamydiae; ( ) Parcubacteria; (
) Parcubacteria; ( ) Nitrospirae; (
) Nitrospirae; ( ) Ignavibacteriae; (
) Ignavibacteriae; ( ) Lentisphaerae; (
) Lentisphaerae; ( ) candidate division WPS-1; (
) candidate division WPS-1; ( ) Aminicenantes; (
) Aminicenantes; ( ) BRC1; (
) BRC1; ( ) candidate division WPS-2; (
) candidate division WPS-2; ( ) Latescibacteria; (
) Latescibacteria; ( ) Thermotogae; (
) Thermotogae; ( ) Elusimicrobia; (
) Elusimicrobia; ( ) Microgenomates; (
) Microgenomates; ( ) Hydrogenedentes; (
) Hydrogenedentes; ( ) Crenarchaeota.
) Crenarchaeota.
Abundance (%) of major genera (≥1%) from each pit mud sample
| Genus | YJ-S (%) | Genus | YJ-Z (%) | Genus | YJ-X (%) |
|---|---|---|---|---|---|
| Acinetobacter | 43.53 | Aminobacterium | 25.04 | Lactobacillus | 6.49 |
| Lysinibacillus | 14.96 | Clostridium IV | 21.39 | Bifidobacterium | 4.71 |
| Caryophanon | 7.05 | Caloramator | 17.01 | Bacteroides | 4.16 |
| Petrimonas | 4.29 | Lactobacillus | 2.49 | Faecalibacterium | 3.88 |
| Proteiniphilum | 4.13 | Sedimentibacter | 1.93 | Clostridium sensu stricto | 3.83 |
| Clostridium IV | 1.79 | Olsenella | 1.91 | Proteiniphilum | 3.55 |
| Solibacillus | 1.50 | Tissierella | 1.60 | Syntrophomonas | 3.41 |
| Aminobacterium | 1.03 | Syntrophomonas | 1.48 | Petrimonas | 2.83 |
| Garciella | 1.25 | Clostridium IV | 2.63 | ||
| Syntrophaceticus | 1.09 | Blautia | 2.06 | ||
| Aminobacterium | 1.75 | ||||
| Alistipes | 1.32 | ||||
| Roseburia | 1.14 | ||||
| Burkholderia | 1.07 |

Genus level distribution of microorganism population present in pit mud. Note: ( ) unclassified; (
) unclassified; ( ) Acinetobacter; (
) Acinetobacter; ( ) Aminobacterium; (
) Aminobacterium; ( ) Clostridium IV; (
) Clostridium IV; ( ) Caloramator; (
) Caloramator; ( ) Lysinibacillus; (
) Lysinibacillus; ( ) Lactobacillus; (
) Lactobacillus; ( ) Proteiniphilum; (
) Proteiniphilum; ( ) Petrimonas; (
) Petrimonas; ( ) Caryophanon; (
) Caryophanon; ( ) Syntrophomonas; (
) Syntrophomonas; ( ) Clostridium sensu stricto; (
) Clostridium sensu stricto; ( ) Bifidobacterium; (
) Bifidobacterium; ( ) Bacteroides; (
) Bacteroides; ( ) Faecalibacterium; (
) Faecalibacterium; ( ) Sedimentibacter; (
) Sedimentibacter; ( ) Tissierella; (
) Tissierella; ( ) Blautia; (
) Blautia; ( ) Olsenella; (
) Olsenella; ( ) Garciella; (
) Garciella; ( ) Alistipes; (
) Alistipes; ( ) Syntrophaceticus; (
) Syntrophaceticus; ( ) Clostridium III; (
) Clostridium III; ( ) Solibacillus; (
) Solibacillus; ( ) Roseburia; (
) Roseburia; ( ) Burkholderia; (
) Burkholderia; ( ) Clostridium XlVa; (
) Clostridium XlVa; ( ) Lutispora; (
) Lutispora; ( ) Methanoculleus; (
) Methanoculleus; ( ) Gemmiger; (
) Gemmiger; ( ) Methanomassiliicoccus; (
) Methanomassiliicoccus; ( ) Rummeliibacillus; (
) Rummeliibacillus; ( ) Bacillus; (
) Bacillus; ( ) Sporanaerobacter; (
) Sporanaerobacter; ( ) Collinsella; (
) Collinsella; ( ) Anaerostipes; (
) Anaerostipes; ( ) Methanobacterium; (
) Methanobacterium; ( ) Oscillibacter; (
) Oscillibacter; ( ) Lachnospiracea_incertae_sedis; (
) Lachnospiracea_incertae_sedis; ( ) Tepidimicrobium; (
) Tepidimicrobium; ( ) Intestinimonas; (
) Intestinimonas; ( ) Barnesiella; (
) Barnesiella; ( ) Parabacteroides; (
) Parabacteroides; ( ) Klebsiella; (
) Klebsiella; ( ) Fusicatenibacter; (
) Fusicatenibacter; ( ) Ralstonia; (
) Ralstonia; ( ) Pelotomaculum; (
) Pelotomaculum; ( ) Tepidanaerobacter; (
) Tepidanaerobacter; ( ) Anaerovorax; (
) Anaerovorax; ( ) other.
) other.
3.3 Microbial diversity in the pit mud
In the analysis of alpha diversity, diversity index, dilution curve, and rank abundance curve are important tools for the analysis of species diversity in different samples. The results from the Shannon index showed YJ-X (4.38) > YJ-Z (3.66) > YJ-S (3.65), and Simpson index showed YJ-X (0.08) < YJ-Z (0.12) < YJ-S (0.17) (Table 1). In microbial diversity between pit walls and bottom, the sample with the richest abundance was YJ-X, followed by YJ-Z and finally YJ-S (Figure 5). The rank abundance curve showed the species evenness: the highest being YJ-S, followed by YJ-Z and YJ-X (Figure 6). In the analysis of beta diversity, the weighted unifrac distance matrix between the two samples was used to draw a heat map for measuring the difference of the two samples. YJ-Z and YJ-X were the most similar in microbial diversity, while YJ-S and YJ-Z were the most different (Figure 7).

Dilution cure analysis of samples.
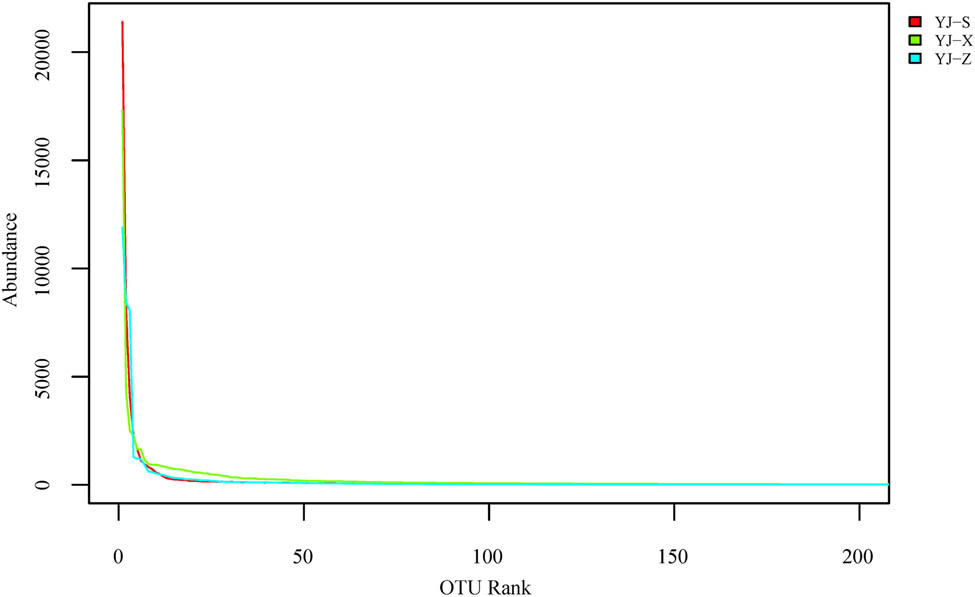
Rank abundance cure analysis of samples.

Distance heatmap of samples.
3.4 Gene functional diversity of microorganisms in pit mud
In COG analysis, the gene functional abundance ranked the top five in the three samples: “General function prediction only” followed by “Amino acid transport and metabolism,” “Function unknown,” “Transcription” and “Translation, ribosomal structure and biogenesis” in YJ-S; “General function prediction only” followed by “Carbohydrate transport and metabolism,” “Amino acid transport and metabolism,” “Transcription” and “function unknown” in YJ-Z; “General function prediction only” followed by “Carbohydrate transport and metabolism,” “Transcription,” “Amino acid transport and metabolism” and “Function unknown” in YJ-X (Figure 8, Table 3). And the abundances of gene functional were significantly different (Figures S4–S6 in Supplementary 1, p < 0.05).
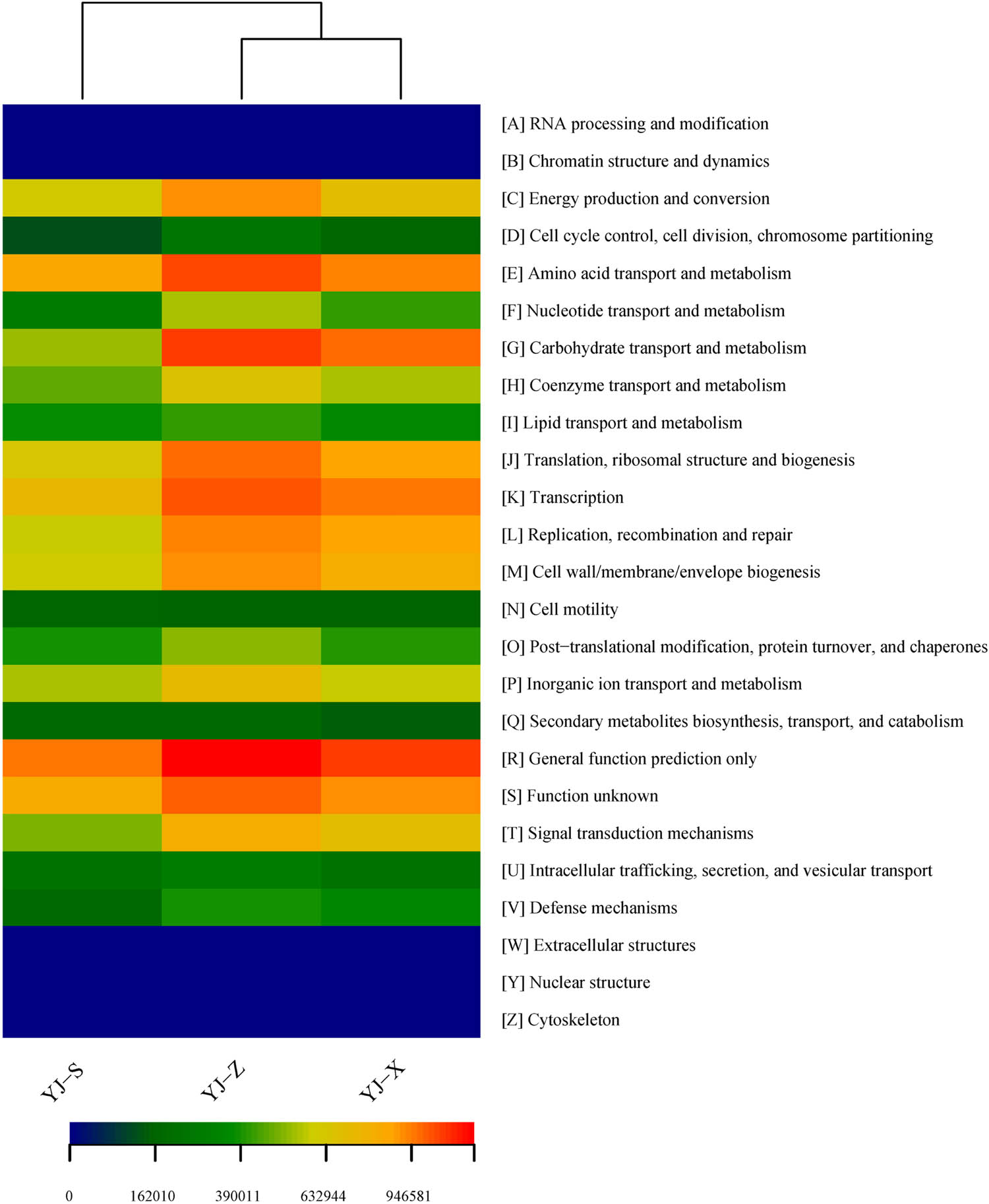
Heatmap of gene functional predictions of samples based on COG.
Abundance of top five COG classification of microorganism in pit mud
| YJ-S | Abundance | YJ-Z | Abundance | YJ-X | Abundance |
|---|---|---|---|---|---|
| General function prediction only | 1265413 | General function prediction only | 2064464 | General function prediction only | 1620397 |
| Amino acid transport and metabolism | 922062 | Carbohydrate transport and metabolism | 1615582 | Carbohydrate transport and metabolism | 1271587 |
| Function unknown | 881872 | Amino acid transport and metabolism | 1518171 | Transcription | 1194181 |
| Transcription | 790853 | Transcription | 1426763 | Amino acid transport and metabolism | 1158436 |
| Translation, ribosomal structure and biogenesis | 698184 | Function unknown | 1420056 | Function unknown | 1095976 |
In the analysis of KEGG metabolic pathway, the gene functional abundance ranks in the top five: in YJ-S it was “Amino acid Metabolism” followed by “Membrane Transport,” “Carbohydrate Metabolism,” “Replication and Repair” and “Energy Metabolism.” In YJ-Z it was “Membrane Transport” followed by “Carbohydrate Metabolism,” “Amino Acid Metabolism,” “Replication and Repair” and “Translation.” In YJ-X it was “Membrane Transport” followed by “Carbohydrate Metabolism,” “Amino Acid Metabolism,” “Replication and Repair” and “Translation” (Figure 9, Table 4). And the abundances of gene function were significantly different (Figures S7–S9 in Supplementary 1, p < 0.05).

Heatmap of gene functional predictions of samples based on KEGG.
Abundance of top five KEGG metabolic pathways of microorganism in pit mud
| YJ-S | Abundance | YJ-Z | Abundance | YJ-X | Abundance |
|---|---|---|---|---|---|
| Amino acid metabolism | 1245752 | Membrane transport | 2156445 | Membrane transport | 1765974 |
| Membrane transport | 1234878 | Carbohydrate metabolism | 1584091 | Carbohydrate metabolism | 2080906 |
| Carbohydrate metabolism | 1077914 | Amino acid metabolism | 1871697 | Amino acid metabolism | 1360333 |
| Replication and repair | 883471 | Replication and repair | 1677025 | Replication and repair | 1211644 |
| Energy metabolism | 654153 | Translation | 1142174 | Translation | 803222 |
4 Discussion
In recent years, microbial diversity in different types of fermented foods, such as fermented seafood, kimchi, traditional fermented mustard, traditional Myanmar fermented tea leaves, and traditional fermented vegetables, were all investigated using rRNA gene sequencing technology [13,14,29–31]. Based on the analysis of coverage index (≥0.93, Table 1) and dilution curve (Figure 5), all microorganisms in the three samples were covered relatively comprehensive, which could truly reflect the prokaryotic microbial community differences between pit walls and bottom. The results showed that YJ-Z and YJ-X were the most similar in terms of microbial diversity. However, due to the differences in vertical height and the microecological environment, the prokaryotic microbial community structure and abundance in YJ-Z and YJ-X were different. At the phylum level, the dominant phylum of YJ-Z and YJ-X was Firmicutes, while in YJ-S it was Proteobacteria. Previous studies also showed that Firmicutes and Bacteroidetes were the dominant phyla in pit muds [2,6,32–34]. Firmicutes can produce endophytic spores and resist extreme environments, degrade volatile fatty acids such as butyrate and its analogs, and is represented mainly by the Clostridia and Bacilli [10]. Although the relative abundance of Firmicutes, Proteobacteria, Bacteroidetes, and Synergistetes differed among YJ-S, YJ-Z, and YJ-X, they were still the key components of the microbiota in the pit mud of Yun liquor. And the similar results were also reported in the pit mud of Yibin liquor [2]. Bacteroidetes was the major contributor to the synthesis of terpenes [32]. In general, at increased depth within the pit mud, the relative abundance of Firmicutes and Actinobacteria shows an upward trend. At the genus level, the dominant genus of the three samples were also different. For example, Aminobacterium was the dominant genus in YJ-Z, with an abundance of 25.04%; while in YJ-S and YJ-X, Aminobacterium’s abundance was much lower, being 1.03% and 1.75%, respectively. Aminobacterium is a gram-negative, single arrangement, slightly curved rod, no spore formation, non-motile, and strictly anaerobic mesophilic bacteria. It can increase the content of ammonium nitrogen in pit mud by amino acid fermentation, which was first found in the sludge of the wastewater treatment tank of the dairy factory, and the model species of this bacterium was Aminobacterium colombense [35,36]. In this study, the abundance of Aminobacterium was the highest in YJ-Z, which may be related to the anaerobic environment in the middle of the pit and the fermentation temperature suitable for the growth of Aminobacterium. Aminobacterium and Sedimentibacter can ferment amino acids to generate ammonium nitrogen through metabolism, which could provide nitrogen sources for the growth of other microbes [10]. In YJ-X, the abundance of unclassified genus was as high as 35.54%, the dominant genus was Lactobacillus, and its abundance was only 6.49%; the abundance of Lactobacillus in YJ-Z was also as low as 2.49%; the abundance is the lowest in YJ-S, being only 0.03%. Lactobacillus is a gram-positive, rod-shaped, non-spore producing, facultative anaerobic, and fermenting with glucose to produce lactic acid. Lactic acid is further converted to ethyl acetate through enzymatic reaction, which increases the mellow smell of liquor. The repulsion relationship Lactobacillus has with other bacteria contributes to the production of caproic acid, which inhibits the growth of bacteriocin by pathogenic microorganisms and mold rot microorganisms, maintaining the quality of liquor fermentation [37,38]. Lactobacillus was accounted for the most in YJ-X, which was consistent with the reported viewpoints of Gao et al. [6]. Lactobacillus was critical for liquor brewing, also as a dominant genus among liquor pit muds of several flavor types [2]. Acinetobacter was the dominant genus in YJ-S, and the abundance was as high as 43.53%. Acinetobacter is a gram-negative, oxidase negative, and non-motile aerobic bacterium, which is widely distributed in soil and water, and even found in hot spring with an optimum growth temperature between 33 and 35°C [39,40]. Acinetobacter is the main microorganism for brewing maotai flavor and light flavor liquor by balancing the taste of alcohol and coordinating the effect of alcohol through the fermentation of glucose and other carbohydrates. In this study, Acinetobacter accounted for the main proportion in the upper part of the pit, indicating that YJ-S was partially similar to maotai flavor and light flavor in pit mud. The abundance of Acinetobacter in YJ-Z and YJ-X was very low, 0.14 and 0.51%, respectively. This may be caused by the fact that YJ-Z and YJ-X are further from the ground and have less contaction with oxygen, causing them to have different microecological environments than YJ-S. Aminobacterium, Syntrophomonas, and Petrimonas play a positive role in the maturation of pit mud, which indicate that the tested pit mud samples are in the mature state [10]. Based on the abundance of major genera in pit muds, the mature state of YJ-Z was more than YJ-S and YJ-X.
In this study, PICRUSt software was used to predict macrogenomic information from 16S rRNA gene of the three samples, and COG database and KEGG database were used to analyze microorganism gene COG classification and KEGG metabolic pathway in YJ-S, YJ-Z, and YJ-X, respectively. Since YJ-S, YJ-Z, and YJ-X were located in different microecological environments, their microorganism gene COG classification and KEGG metabolic pathway abundance were different from each other. In COG analysis, “General function prediction only” was in the first position in the three samples. The abundance of “Amino acid transport and metabolism” was decreased with the decrease of the vertical distance of the sample in the pit, which were in the second, third, and fourth positions in YJ-S, YJ-Z, and YJ-X, respectively. The function of “Amino acid transport and metabolism” may be related to the contact amount of oxygen. Further down the pit, the oxygen contact amount decreases, so the abundance of “Amino acid transport and metabolism” decreases. The abundance of “Function unknown” was the highest in YJ-S, and the abundance ranking was the same in YJ-Z and YJ-X. This may be due to the fact that YJ-S was closer to the ground and had more exposure to oxygen. “Carbohydrate transport and metabolism,” “Amino acid transport and metabolism,” and “Transcription” were the main genes function in YJ-S, YJ-Z, and YJ-X, which were consistent with the conclusion of Wang et al. [41] in studying the microbial community structure and gene function of high-temperature Daqu liquor in a factory in Xiangyang, Hubei Province. In KEGG analysis, “Amino Acid Metabolism” was in the first position in YJ-S, and in the third position in both YJ-Z and YJ-X. This may be due to YJ-S having a closer vertical distance to the ground, more contact with oxygen, more aerobic bacteria in pit mud, and more bacteria using amino acid fermentation. “Membrane Transport” and “Carbohydrate Transport and Metabolism” were in the second and third positions in YJ-Z and YJ-X, indicating that there may be a large number of cellulose degrading enzyme genes in the macrogenomes of the two samples, and a large number of cellulose degrading enzymes synthesized by bacteria were transported outside the cells to degrade cellulose in fermented grains. “Translation” was present at the fifth position in both YJ-Z and YJ-X, after the fifth position in YJ-S. It showed that the enzyme synthesis and metabolism of microorganism, and the characteristics that are beneficial to grain fermentation, in YJ-Z and YJ-X were stronger than those in YJ-S. “Carbohydrate Metabolism” and “Amino Acid Metabolism” were arranged at the second and third positions in YJ-Z and YJ-X, respectively, and the first and third positions in YJ-S, indicating that the gene functions of pit mud microorganism were mainly concentrated in carbohydrate metabolism and amino acid metabolism, which was consistent with the predicted function of “Amino acid transport and metabolism” and that of “Carbohydrate transport and metabolism.” The prediction was based on COG database in this study.
5 Conclusion
The study above showed that prokaryotic microbial community structures and diversity, and gene function prediction is significantly different in pit walls from those in the bottom. The dominant phyla in YJ-S, YJ-Z, and YJ-X were Proteobacteria and Firmicutes, and the dominant genera in them were Acinetobacter, Aminobacterium, and Lactobacillus. In species diversity analysis, YJ-Z and YJ-X were the closest; in species richness analysis, the order was YJ-X > YJ-Z > YJ-S; in species uniformity analysis, the order was YJ-S > YJ-Z > YJ-X. The prokaryotic microbial function in pit mud was mainly concentrated in “Carbohydrate transport and metabolism” and “Amino acid transport and metabolism.” These scientific data provide a theoretical basis for the future maintenance of pit mud, the specifications of building new pits, the input of fermentation material, and the quality of liquor production. In future, the study can be used as a groundwork for revealing the relationship among physicochemical properties, microbiome, and volatiles of pit mud.
-
Funding information: This study was financially supported by the school level key construction discipline of food science and engineering in Chaohu University (grant no. kj16zdjsxk02) and the project of Chaohu University (grant no. kj21kctd03).
-
Author contributions: Shu Fang participated in the experiments design, data analysis, and drafted the manuscript. Chuanxiang Wang provided these pit mud samples. Juan Yan participated in the experiment operation. All authors have read and approved the final manuscript.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
[1] Tu W, Cao X, Cheng J, Li L, Zhang T, Wu Q, et al. Chinese Baijiu: the perfect works of microorganisms. Front Microbiol. 2022;13:919044.10.3389/fmicb.2022.919044Search in Google Scholar PubMed PubMed Central
[2] Chen L, Li Y, Jin L, He L, Ao X, Liu S, et al. Analyzing bacterial community in pit mud of Yibin Baijiu in China using high throughput sequencing. PeerJ. 2020;8:e9122.10.7717/peerj.9122Search in Google Scholar PubMed PubMed Central
[3] Ling Y, Li W, Tong T, Li Z, Li Q, Bai Z, et al. Assessing the microbial communities in four different daqus by using PCR-DGGE, PLFA, and biolog analyses. Pol J Microbiol. 2020;69(1):1–11.10.33073/pjm-2020-004Search in Google Scholar PubMed PubMed Central
[4] Cai W, Xue Y, Tang F, Wang Y, Yang S, Liu W, et al. The depth-depended fungal diversity and non-depth-depended aroma profiles of pit mud for strong-flavor Baijiu. Front Microbiol. 2022;12:789845.10.3389/fmicb.2021.789845Search in Google Scholar PubMed PubMed Central
[5] Tan G, Zhou R, Zhang W, Hu Y, Ruan Z, Li J, et al. Detection of viable and total bacterial community in the pit mud of Chinese strong-flavor liquor using propidium monoazide combined with quantitative PCR and 16S rRNA gene sequencing. Front Microbiol. 2020;11:896.10.3389/fmicb.2020.00896Search in Google Scholar PubMed PubMed Central
[6] Gao Z, Wu Z, Zhang W. Effect of pit mud on bacterial community and aroma components in yellow water and their changes during the fermentation of Chinese strong-flavor liquor. Foods. 2020;9(3):372.10.3390/foods9030372Search in Google Scholar PubMed PubMed Central
[7] Pu S, Yan S. Fungal diversity profiles in pit mud samples from Chinese strong-flavour liquor pit. Foods. 2022;11(22):3544.10.3390/foods11223544Search in Google Scholar PubMed PubMed Central
[8] Sakandar HA, Hussain R, Farid Khan Q, Zhang H. Functional microbiota in Chinese traditional Baijiu and Mijiu Qu (starters): a review. Food Res Int. 2020;138(Pt B):109830.10.1016/j.foodres.2020.109830Search in Google Scholar PubMed
[9] Guan T, Lin Y, Chen K, Ou M, Zhang J. Physicochemical factors affecting microbiota dynamics during traditional solid-state fermentation of Chinese strong-flavor Baijiu. Front Microbiol. 2020;11:2090.10.3389/fmicb.2020.02090Search in Google Scholar PubMed PubMed Central
[10] Liu Y, Sun M, Hou P, Wang W, Shen X, Zhang L, et al. Analysis of microbial community structure and volatile compounds in pit mud used for manufacturing Taorong-type Baijiu based on high-throughput sequencing. Sci Rep. 2022;12(1):7347.10.1038/s41598-022-10412-8Search in Google Scholar PubMed PubMed Central
[11] Fang S, Yan J. Analysis of prokaryotic microbial diversity in hot spring water from Bantang (China) using the targeted amplicon analysis. All Life. 2022;15(1):325–39.10.1080/26895293.2022.2049899Search in Google Scholar
[12] Liu Z, Li J, Wei B, Huang T, Xiao Y, Peng Z, et al. Bacterial community and composition in Jiang-shui and Suan-cai revealed by high-throughput sequencing of 16S rRNA. Int J Food Microbiol. 2019;306:108271.10.1016/j.ijfoodmicro.2019.108271Search in Google Scholar PubMed
[13] Cai W, Tang F, Wang Y, Zhang Z, Xue Y, Zhao X, et al. Bacterial diversity and flavor profile of Zha-Chili, a traditional fermented food in China. Food Res Int. 2021;141:110112.10.1016/j.foodres.2021.110112Search in Google Scholar PubMed
[14] Lee M, Song JH, Park JM, Chang JY. Bacterial diversity in Korean temple kimchi fermentation. Food Res Int. 2019;126:108592.10.1016/j.foodres.2019.108592Search in Google Scholar PubMed
[15] Tan G, Hu Y, Huang Y, Liu H, Dong W, Li J, et al. Analysis of bacterial communities in pit mud from Zhijiang Baijiu distillery using denaturing gradient gel electrophoresis and high-throughput sequencing. J Inst Brew. 2020;126(1):90–7.10.1002/jib.595Search in Google Scholar
[16] Ding XF, Wu CD, Zhang LQ, Zheng J, Zhou RQ. Characterization of eubacterial and archaeal community diversity in the pit mud of Chinese Luzhou-flavor liquor by nested PCR-DGGE. World J Microbiol Biotechnol. 2014;30(2):605–12.10.1007/s11274-013-1472-4Search in Google Scholar PubMed
[17] Yang C, Liu W, He Z, Thangavel S, Wang L, Zhou A, et al. Freezing/thawing pretreatment coupled with biological process of thermophilic Geobacillus sp. G1: acceleration on waste activated sludge hydrolysis and acidification. Bioresour Technol. 2015;175:509–16.10.1016/j.biortech.2014.10.154Search in Google Scholar PubMed
[18] Sinclair L, Osman OA, Bertilsson S, Eiler A. Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the illumina platform. PLoS One. 2015;10(2):e0116955.10.1371/journal.pone.0116955Search in Google Scholar PubMed PubMed Central
[19] Tang Q, He G, Huang J, Wu C, Jin Y, Zhou R. Characterizing relationship of microbial diversity and metabolite in Sichuan Xiaoqu. Front Microbiol. 2019;10:696.10.3389/fmicb.2019.00696Search in Google Scholar PubMed PubMed Central
[20] He B, Zhu R, Yang H, Lu Q, Wang W, Song L, et al. Assessing the impact of data preprocessing on analyzing next generation sequencing data. Front Bioeng Biotechnol. 2020;8:817.10.3389/fbioe.2020.00817Search in Google Scholar PubMed PubMed Central
[21] Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30(5):614–20.10.1093/bioinformatics/btt593Search in Google Scholar PubMed PubMed Central
[22] Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–4.10.1093/bioinformatics/btr026Search in Google Scholar PubMed PubMed Central
[23] Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1.10.1093/bioinformatics/btq461Search in Google Scholar PubMed
[24] Balvočiūtė M, Huson DH. SILVA, RDP, Greengenes, NCBI and OTT – how do these taxonomies compare? BMC Genomics. 2017;18(Suppl 2):114.10.1186/s12864-017-3501-4Search in Google Scholar PubMed PubMed Central
[25] Mair P, Hofmann E, Gruber K, Hatzinger R, Zeileis A, Hornik K. Motivation, values, and work design as drivers of participation in the R open source project for statistical computing. Proc Natl Acad Sci U S A. 2015;112(48):14788–92.10.1073/pnas.1506047112Search in Google Scholar PubMed PubMed Central
[26] Schloss PD. Reintroducing mothur: 10 years later. Appl Environ Microbiol. 2020;86(2):e02343–19.10.1128/AEM.02343-19Search in Google Scholar PubMed PubMed Central
[27] Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–21.10.1038/nbt.2676Search in Google Scholar PubMed PubMed Central
[28] Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30(21):3123–4.10.1093/bioinformatics/btu494Search in Google Scholar PubMed PubMed Central
[29] Jiang S, Ma C, Peng Q, Huo D, Li W, Zhang J. Microbial profile and genetic polymorphism of predominant species in some traditional fermented seafoods of the Hainan Area in China. Front Microbiol. 2019;10:564.10.3389/fmicb.2019.00564Search in Google Scholar PubMed PubMed Central
[30] Yu Y, Li L, Xu Y, An K, Shi Q, Yu Y, et al. Evaluation of the relationship among biogenic amines, nitrite and microbial diversity in fermented mustard. Molecules. 2021;26(20):6173.10.3390/molecules26206173Search in Google Scholar PubMed PubMed Central
[31] Bo B, Kim SA, Han NS. Bacterial and fungal diversity in Laphet, traditional fermented tea leaves in Myanmar, analyzed by culturing, DNA amplicon-based sequencing, and PCR-DGGE methods. Int J Food Microbiol. 2020;320:108508.10.1016/j.ijfoodmicro.2020.108508Search in Google Scholar PubMed
[32] Fu J, Chen L, Yang S, Li Y, Jin L, He X, et al. Metagenome and analysis of metabolic potential of the microbial community in pit mud used for Chinese strong-flavor liquor production. Food Res Int. 2021;143:110294.10.1016/j.foodres.2021.110294Search in Google Scholar PubMed
[33] Liu MK, Tang YM, Zhao K, Liu Y, Guo XJ, Tian XH, et al. Contrasting bacterial community structure in artificial pit mud-starter cultures of different qualities: a complex biological mixture for Chinese strong-flavor baijiu production. 3 Biotech. 2019;9(3):89.10.1007/s13205-019-1622-ySearch in Google Scholar PubMed PubMed Central
[34] Qian W, Lu ZM, Chai LJ, Zhang XJ, Li Q, Wang ST, et al. Cooperation within the microbial consortia of fermented grains and pit mud drives organic acid synthesis in strong-flavor Baijiu production. Food Res Int. 2021;147:110449.10.1016/j.foodres.2021.110449Search in Google Scholar PubMed
[35] Baena S, Fardeau ML, Labat M, Ollivier B, Thomas P, Garcia JL, et al. Aminobacterium colombiensegen. nov. sp. nov., an amino acid-degrading anaerobe isolated from anaerobic sludge. Anaerobe. 1998;4(5):241–50.10.1006/anae.1998.0170Search in Google Scholar PubMed
[36] Baena S, Fardeau ML, Labat M, Ollivier B, Garcia JL, Patel BK. Aminobacterium mobile sp. nov., a new anaerobic amino-acid-degrading bacterium. Int J Syst Evol Microbiol. 2000;50(Pt 1):259–64.10.1099/00207713-50-1-259Search in Google Scholar PubMed
[37] He G, Huang J, Wu C, Jin Y, Zhou R. Bioturbation effect of fortified Daqu on microbial community and flavor metabolite in Chinese strong-flavor liquor brewing microecosystem. Food Res Int. 2020;129:108851.10.1016/j.foodres.2019.108851Search in Google Scholar PubMed
[38] He G, Huang J, Zhou R, Wu C, Jin Y. Effect of fortified Daqu on the microbial community and flavor in Chinese strong-flavor liquor brewing process. Front Microbiol. 2019;10:56.10.3389/fmicb.2019.00056Search in Google Scholar PubMed PubMed Central
[39] Rawat N, Joshi GK. Bacterial community structure analysis of a hot spring soil by next generation sequencing of ribosomal RNA. Genomics. 2019;111(5):1053–8.10.1016/j.ygeno.2018.06.008Search in Google Scholar PubMed
[40] Castelán-Sánchez HG, Meza-Rodríguez PM, Carrillo E, Ríos-Vázquez DI, Liñan-Torres A, Batista-García RA, et al. The microbial composition in circumneutral thermal springs from Chignahuapan, Puebla, Mexico reveals the presence of particular sulfur-oxidizing bacterial and viral communities. Microorganisms. 2020;8(11):1677.10.3390/microorganisms8111677Search in Google Scholar PubMed PubMed Central
[41] Wang Y, Cai W, Wang W, Shu N, Zhang Z, Hou Q, et al. Analysis of microbial diversity and functional differences in different types of high-temperature Daqu. Food Sci Nutr. 2021;9(2):1003–16.10.1002/fsn3.2068Search in Google Scholar PubMed PubMed Central
© 2023 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Biomedical Sciences
- Systemic investigation of inetetamab in combination with small molecules to treat HER2-overexpressing breast and gastric cancers
- Immunosuppressive treatment for idiopathic membranous nephropathy: An updated network meta-analysis
- Identifying two pathogenic variants in a patient with pigmented paravenous retinochoroidal atrophy
- Effects of phytoestrogens combined with cold stress on sperm parameters and testicular proteomics in rats
- A case of pulmonary embolism with bad warfarin anticoagulant effects caused by E. coli infection
- Neutrophilia with subclinical Cushing’s disease: A case report and literature review
- Isoimperatorin alleviates lipopolysaccharide-induced periodontitis by downregulating ERK1/2 and NF-κB pathways
- Immunoregulation of synovial macrophages for the treatment of osteoarthritis
- Novel CPLANE1 c.8948dupT (p.P2984Tfs*7) variant in a child patient with Joubert syndrome
- Antiphospholipid antibodies and the risk of thrombosis in myeloproliferative neoplasms
- Immunological responses of septic rats to combination therapy with thymosin α1 and vitamin C
- High glucose and high lipid induced mitochondrial dysfunction in JEG-3 cells through oxidative stress
- Pharmacological inhibition of the ubiquitin-specific protease 8 effectively suppresses glioblastoma cell growth
- Levocarnitine regulates the growth of angiotensin II-induced myocardial fibrosis cells via TIMP-1
- Age-related changes in peripheral T-cell subpopulations in elderly individuals: An observational study
- Single-cell transcription analysis reveals the tumor origin and heterogeneity of human bilateral renal clear cell carcinoma
- Identification of iron metabolism-related genes as diagnostic signatures in sepsis by blood transcriptomic analysis
- Long noncoding RNA ACART knockdown decreases 3T3-L1 preadipocyte proliferation and differentiation
- Surgery, adjuvant immunotherapy plus chemotherapy and radiotherapy for primary malignant melanoma of the parotid gland (PGMM): A case report
- Dosimetry comparison with helical tomotherapy, volumetric modulated arc therapy, and intensity-modulated radiotherapy for grade II gliomas: A single‑institution case series
- Soy isoflavone reduces LPS-induced acute lung injury via increasing aquaporin 1 and aquaporin 5 in rats
- Refractory hypokalemia with sexual dysplasia and infertility caused by 17α-hydroxylase deficiency and triple X syndrome: A case report
- Meta-analysis of cancer risk among end stage renal disease undergoing maintenance dialysis
- 6-Phosphogluconate dehydrogenase inhibition arrests growth and induces apoptosis in gastric cancer via AMPK activation and oxidative stress
- Experimental study on the optimization of ANM33 release in foam cells
- Primary retroperitoneal angiosarcoma: A case report
- Metabolomic analysis-identified 2-hydroxybutyric acid might be a key metabolite of severe preeclampsia
- Malignant pleural effusion diagnosis and therapy
- Effect of spaceflight on the phenotype and proteome of Escherichia coli
- Comparison of immunotherapy combined with stereotactic radiotherapy and targeted therapy for patients with brain metastases: A systemic review and meta-analysis
- Activation of hypermethylated P2RY1 mitigates gastric cancer by promoting apoptosis and inhibiting proliferation
- Association between the VEGFR-2 -604T/C polymorphism (rs2071559) and type 2 diabetic retinopathy
- The role of IL-31 and IL-34 in the diagnosis and treatment of chronic periodontitis
- Triple-negative mouse breast cancer initiating cells show high expression of beta1 integrin and increased malignant features
- mNGS facilitates the accurate diagnosis and antibiotic treatment of suspicious critical CNS infection in real practice: A retrospective study
- The apatinib and pemetrexed combination has antitumor and antiangiogenic effects against NSCLC
- Radiotherapy for primary thyroid adenoid cystic carcinoma
- Design and functional preliminary investigation of recombinant antigen EgG1Y162–EgG1Y162 against Echinococcus granulosus
- Effects of losartan in patients with NAFLD: A meta-analysis of randomized controlled trial
- Bibliometric analysis of METTL3: Current perspectives, highlights, and trending topics
- Performance comparison of three scaling algorithms in NMR-based metabolomics analysis
- PI3K/AKT/mTOR pathway and its related molecules participate in PROK1 silence-induced anti-tumor effects on pancreatic cancer
- The altered expression of cytoskeletal and synaptic remodeling proteins during epilepsy
- Effects of pegylated recombinant human granulocyte colony-stimulating factor on lymphocytes and white blood cells of patients with malignant tumor
- Prostatitis as initial manifestation of Chlamydia psittaci pneumonia diagnosed by metagenome next-generation sequencing: A case report
- NUDT21 relieves sevoflurane-induced neurological damage in rats by down-regulating LIMK2
- Association of interleukin-10 rs1800896, rs1800872, and interleukin-6 rs1800795 polymorphisms with squamous cell carcinoma risk: A meta-analysis
- Exosomal HBV-DNA for diagnosis and treatment monitoring of chronic hepatitis B
- Shear stress leads to the dysfunction of endothelial cells through the Cav-1-mediated KLF2/eNOS/ERK signaling pathway under physiological conditions
- Interaction between the PI3K/AKT pathway and mitochondrial autophagy in macrophages and the leukocyte count in rats with LPS-induced pulmonary infection
- Meta-analysis of the rs231775 locus polymorphism in the CTLA-4 gene and the susceptibility to Graves’ disease in children
- Cloning, subcellular localization and expression of phosphate transporter gene HvPT6 of hulless barley
- Coptisine mitigates diabetic nephropathy via repressing the NRLP3 inflammasome
- Significant elevated CXCL14 and decreased IL-39 levels in patients with tuberculosis
- Whole-exome sequencing applications in prenatal diagnosis of fetal bowel dilatation
- Gemella morbillorum infective endocarditis: A case report and literature review
- An unusual ectopic thymoma clonal evolution analysis: A case report
- Severe cumulative skin toxicity during toripalimab combined with vemurafenib following toripalimab alone
- Detection of V. vulnificus septic shock with ARDS using mNGS
- Novel rare genetic variants of familial and sporadic pulmonary atresia identified by whole-exome sequencing
- The influence and mechanistic action of sperm DNA fragmentation index on the outcomes of assisted reproduction technology
- Novel compound heterozygous mutations in TELO2 in an infant with You-Hoover-Fong syndrome: A case report and literature review
- ctDNA as a prognostic biomarker in resectable CLM: Systematic review and meta-analysis
- Diagnosis of primary amoebic meningoencephalitis by metagenomic next-generation sequencing: A case report
- Phylogenetic analysis of promoter regions of human Dolichol kinase (DOLK) and orthologous genes using bioinformatics tools
- Collagen changes in rabbit conjunctiva after conjunctival crosslinking
- Effects of NM23 transfection of human gastric carcinoma cells in mice
- Oral nifedipine and phytosterol, intravenous nicardipine, and oral nifedipine only: Three-arm, retrospective, cohort study for management of severe preeclampsia
- Case report of hepatic retiform hemangioendothelioma: A rare tumor treated with ultrasound-guided microwave ablation
- Curcumin induces apoptosis in human hepatocellular carcinoma cells by decreasing the expression of STAT3/VEGF/HIF-1α signaling
- Rare presentation of double-clonal Waldenström macroglobulinemia with pulmonary embolism: A case report
- Giant duplication of the transverse colon in an adult: A case report and literature review
- Ectopic thyroid tissue in the breast: A case report
- SDR16C5 promotes proliferation and migration and inhibits apoptosis in pancreatic cancer
- Vaginal metastasis from breast cancer: A case report
- Screening of the best time window for MSC transplantation to treat acute myocardial infarction with SDF-1α antibody-loaded targeted ultrasonic microbubbles: An in vivo study in miniswine
- Inhibition of TAZ impairs the migration ability of melanoma cells
- Molecular complexity analysis of the diagnosis of Gitelman syndrome in China
- Effects of maternal calcium and protein intake on the development and bone metabolism of offspring mice
- Identification of winter wheat pests and diseases based on improved convolutional neural network
- Ultra-multiplex PCR technique to guide treatment of Aspergillus-infected aortic valve prostheses
- Virtual high-throughput screening: Potential inhibitors targeting aminopeptidase N (CD13) and PIKfyve for SARS-CoV-2
- Immune checkpoint inhibitors in cancer patients with COVID-19
- Utility of methylene blue mixed with autologous blood in preoperative localization of pulmonary nodules and masses
- Integrated analysis of the microbiome and transcriptome in stomach adenocarcinoma
- Berberine suppressed sarcopenia insulin resistance through SIRT1-mediated mitophagy
- DUSP2 inhibits the progression of lupus nephritis in mice by regulating the STAT3 pathway
- Lung abscess by Fusobacterium nucleatum and Streptococcus spp. co-infection by mNGS: A case series
- Genetic alterations of KRAS and TP53 in intrahepatic cholangiocarcinoma associated with poor prognosis
- Granulomatous polyangiitis involving the fourth ventricle: Report of a rare case and a literature review
- Studying infant mortality: A demographic analysis based on data mining models
- Metaplastic breast carcinoma with osseous differentiation: A report of a rare case and literature review
- Protein Z modulates the metastasis of lung adenocarcinoma cells
- Inhibition of pyroptosis and apoptosis by capsaicin protects against LPS-induced acute kidney injury through TRPV1/UCP2 axis in vitro
- TAK-242, a toll-like receptor 4 antagonist, against brain injury by alleviates autophagy and inflammation in rats
- Primary mediastinum Ewing’s sarcoma with pleural effusion: A case report and literature review
- Association of ADRB2 gene polymorphisms and intestinal microbiota in Chinese Han adolescents
- Tanshinone IIA alleviates chondrocyte apoptosis and extracellular matrix degeneration by inhibiting ferroptosis
- Study on the cytokines related to SARS-Cov-2 in testicular cells and the interaction network between cells based on scRNA-seq data
- Effect of periostin on bone metabolic and autophagy factors during tooth eruption in mice
- HP1 induces ferroptosis of renal tubular epithelial cells through NRF2 pathway in diabetic nephropathy
- Intravaginal estrogen management in postmenopausal patients with vaginal squamous intraepithelial lesions along with CO2 laser ablation: A retrospective study
- Hepatocellular carcinoma cell differentiation trajectory predicts immunotherapy, potential therapeutic drugs, and prognosis of patients
- Effects of physical exercise on biomarkers of oxidative stress in healthy subjects: A meta-analysis of randomized controlled trials
- Identification of lysosome-related genes in connection with prognosis and immune cell infiltration for drug candidates in head and neck cancer
- Development of an instrument-free and low-cost ELISA dot-blot test to detect antibodies against SARS-CoV-2
- Research progress on gas signal molecular therapy for Parkinson’s disease
- Adiponectin inhibits TGF-β1-induced skin fibroblast proliferation and phenotype transformation via the p38 MAPK signaling pathway
- The G protein-coupled receptor-related gene signatures for predicting prognosis and immunotherapy response in bladder urothelial carcinoma
- α-Fetoprotein contributes to the malignant biological properties of AFP-producing gastric cancer
- CXCL12/CXCR4/CXCR7 axis in placenta tissues of patients with placenta previa
- Association between thyroid stimulating hormone levels and papillary thyroid cancer risk: A meta-analysis
- Significance of sTREM-1 and sST2 combined diagnosis for sepsis detection and prognosis prediction
- Diagnostic value of serum neuroactive substances in the acute exacerbation of chronic obstructive pulmonary disease complicated with depression
- Research progress of AMP-activated protein kinase and cardiac aging
- TRIM29 knockdown prevented the colon cancer progression through decreasing the ubiquitination levels of KRT5
- Cross-talk between gut microbiota and liver steatosis: Complications and therapeutic target
- Metastasis from small cell lung cancer to ovary: A case report
- The early diagnosis and pathogenic mechanisms of sepsis-related acute kidney injury
- The effect of NK cell therapy on sepsis secondary to lung cancer: A case report
- Erianin alleviates collagen-induced arthritis in mice by inhibiting Th17 cell differentiation
- Loss of ACOX1 in clear cell renal cell carcinoma and its correlation with clinical features
- Signalling pathways in the osteogenic differentiation of periodontal ligament stem cells
- Crosstalk between lactic acid and immune regulation and its value in the diagnosis and treatment of liver failure
- Clinicopathological features and differential diagnosis of gastric pleomorphic giant cell carcinoma
- Traumatic brain injury and rTMS-ERPs: Case report and literature review
- Extracellular fibrin promotes non-small cell lung cancer progression through integrin β1/PTEN/AKT signaling
- Knockdown of DLK4 inhibits non-small cell lung cancer tumor growth by downregulating CKS2
- The co-expression pattern of VEGFR-2 with indicators related to proliferation, apoptosis, and differentiation of anagen hair follicles
- Inflammation-related signaling pathways in tendinopathy
- CD4+ T cell count in HIV/TB co-infection and co-occurrence with HL: Case report and literature review
- Clinical analysis of severe Chlamydia psittaci pneumonia: Case series study
- Bioinformatics analysis to identify potential biomarkers for the pulmonary artery hypertension associated with the basement membrane
- Influence of MTHFR polymorphism, alone or in combination with smoking and alcohol consumption, on cancer susceptibility
- Catharanthus roseus (L.) G. Don counteracts the ampicillin resistance in multiple antibiotic-resistant Staphylococcus aureus by downregulation of PBP2a synthesis
- Combination of a bronchogenic cyst in the thoracic spinal canal with chronic myelocytic leukemia
- Bacterial lipoprotein plays an important role in the macrophage autophagy and apoptosis induced by Salmonella typhimurium and Staphylococcus aureus
- TCL1A+ B cells predict prognosis in triple-negative breast cancer through integrative analysis of single-cell and bulk transcriptomic data
- Ezrin promotes esophageal squamous cell carcinoma progression via the Hippo signaling pathway
- Ferroptosis: A potential target of macrophages in plaque vulnerability
- Predicting pediatric Crohn's disease based on six mRNA-constructed risk signature using comprehensive bioinformatic approaches
- Applications of genetic code expansion and photosensitive UAAs in studying membrane proteins
- HK2 contributes to the proliferation, migration, and invasion of diffuse large B-cell lymphoma cells by enhancing the ERK1/2 signaling pathway
- IL-17 in osteoarthritis: A narrative review
- Circadian cycle and neuroinflammation
- Probiotic management and inflammatory factors as a novel treatment in cirrhosis: A systematic review and meta-analysis
- Hemorrhagic meningioma with pulmonary metastasis: Case report and literature review
- SPOP regulates the expression profiles and alternative splicing events in human hepatocytes
- Knockdown of SETD5 inhibited glycolysis and tumor growth in gastric cancer cells by down-regulating Akt signaling pathway
- PTX3 promotes IVIG resistance-induced endothelial injury in Kawasaki disease by regulating the NF-κB pathway
- Pancreatic ectopic thyroid tissue: A case report and analysis of literature
- The prognostic impact of body mass index on female breast cancer patients in underdeveloped regions of northern China differs by menopause status and tumor molecular subtype
- Report on a case of liver-originating malignant melanoma of unknown primary
- Case report: Herbal treatment of neutropenic enterocolitis after chemotherapy for breast cancer
- The fibroblast growth factor–Klotho axis at molecular level
- Characterization of amiodarone action on currents in hERG-T618 gain-of-function mutations
- A case report of diagnosis and dynamic monitoring of Listeria monocytogenes meningitis with NGS
- Effect of autologous platelet-rich plasma on new bone formation and viability of a Marburg bone graft
- Small breast epithelial mucin as a useful prognostic marker for breast cancer patients
- Continuous non-adherent culture promotes transdifferentiation of human adipose-derived stem cells into retinal lineage
- Nrf3 alleviates oxidative stress and promotes the survival of colon cancer cells by activating AKT/BCL-2 signal pathway
- Favorable response to surufatinib in a patient with necrolytic migratory erythema: A case report
- Case report of atypical undernutrition of hypoproteinemia type
- Down-regulation of COL1A1 inhibits tumor-associated fibroblast activation and mediates matrix remodeling in the tumor microenvironment of breast cancer
- Sarcoma protein kinase inhibition alleviates liver fibrosis by promoting hepatic stellate cells ferroptosis
- Research progress of serum eosinophil in chronic obstructive pulmonary disease and asthma
- Clinicopathological characteristics of co-existing or mixed colorectal cancer and neuroendocrine tumor: Report of five cases
- Role of menopausal hormone therapy in the prevention of postmenopausal osteoporosis
- Precisional detection of lymph node metastasis using tFCM in colorectal cancer
- Advances in diagnosis and treatment of perimenopausal syndrome
- A study of forensic genetics: ITO index distribution and kinship judgment between two individuals
- Acute lupus pneumonitis resembling miliary tuberculosis: A case-based review
- Plasma levels of CD36 and glutathione as biomarkers for ruptured intracranial aneurysm
- Fractalkine modulates pulmonary angiogenesis and tube formation by modulating CX3CR1 and growth factors in PVECs
- Novel risk prediction models for deep vein thrombosis after thoracotomy and thoracoscopic lung cancer resections, involving coagulation and immune function
- Exploring the diagnostic markers of essential tremor: A study based on machine learning algorithms
- Evaluation of effects of small-incision approach treatment on proximal tibia fracture by deep learning algorithm-based magnetic resonance imaging
- An online diagnosis method for cancer lesions based on intelligent imaging analysis
- Medical imaging in rheumatoid arthritis: A review on deep learning approach
- Predictive analytics in smart healthcare for child mortality prediction using a machine learning approach
- Utility of neutrophil–lymphocyte ratio and platelet–lymphocyte ratio in predicting acute-on-chronic liver failure survival
- A biomedical decision support system for meta-analysis of bilateral upper-limb training in stroke patients with hemiplegia
- TNF-α and IL-8 levels are positively correlated with hypobaric hypoxic pulmonary hypertension and pulmonary vascular remodeling in rats
- Stochastic gradient descent optimisation for convolutional neural network for medical image segmentation
- Comparison of the prognostic value of four different critical illness scores in patients with sepsis-induced coagulopathy
- Application and teaching of computer molecular simulation embedded technology and artificial intelligence in drug research and development
- Hepatobiliary surgery based on intelligent image segmentation technology
- Value of brain injury-related indicators based on neural network in the diagnosis of neonatal hypoxic-ischemic encephalopathy
- Analysis of early diagnosis methods for asymmetric dementia in brain MR images based on genetic medical technology
- Early diagnosis for the onset of peri-implantitis based on artificial neural network
- Clinical significance of the detection of serum IgG4 and IgG4/IgG ratio in patients with thyroid-associated ophthalmopathy
- Forecast of pain degree of lumbar disc herniation based on back propagation neural network
- SPA-UNet: A liver tumor segmentation network based on fused multi-scale features
- Systematic evaluation of clinical efficacy of CYP1B1 gene polymorphism in EGFR mutant non-small cell lung cancer observed by medical image
- Rehabilitation effect of intelligent rehabilitation training system on hemiplegic limb spasms after stroke
- A novel approach for minimising anti-aliasing effects in EEG data acquisition
- ErbB4 promotes M2 activation of macrophages in idiopathic pulmonary fibrosis
- Clinical role of CYP1B1 gene polymorphism in prediction of postoperative chemotherapy efficacy in NSCLC based on individualized health model
- Lung nodule segmentation via semi-residual multi-resolution neural networks
- Evaluation of brain nerve function in ICU patients with Delirium by deep learning algorithm-based resting state MRI
- A data mining technique for detecting malignant mesothelioma cancer using multiple regression analysis
- Markov model combined with MR diffusion tensor imaging for predicting the onset of Alzheimer’s disease
- Effectiveness of the treatment of depression associated with cancer and neuroimaging changes in depression-related brain regions in patients treated with the mediator-deuterium acupuncture method
- Molecular mechanism of colorectal cancer and screening of molecular markers based on bioinformatics analysis
- Monitoring and evaluation of anesthesia depth status data based on neuroscience
- Exploring the conformational dynamics and thermodynamics of EGFR S768I and G719X + S768I mutations in non-small cell lung cancer: An in silico approaches
- Optimised feature selection-driven convolutional neural network using gray level co-occurrence matrix for detection of cervical cancer
- Incidence of different pressure patterns of spinal cerebellar ataxia and analysis of imaging and genetic diagnosis
- Pathogenic bacteria and treatment resistance in older cardiovascular disease patients with lung infection and risk prediction model
- Adoption value of support vector machine algorithm-based computed tomography imaging in the diagnosis of secondary pulmonary fungal infections in patients with malignant hematological disorders
- From slides to insights: Harnessing deep learning for prognostic survival prediction in human colorectal cancer histology
- Ecology and Environmental Science
- Monitoring of hourly carbon dioxide concentration under different land use types in arid ecosystem
- Comparing the differences of prokaryotic microbial community between pit walls and bottom from Chinese liquor revealed by 16S rRNA gene sequencing
- Effects of cadmium stress on fruits germination and growth of two herbage species
- Bamboo charcoal affects soil properties and bacterial community in tea plantations
- Optimization of biogas potential using kinetic models, response surface methodology, and instrumental evidence for biodegradation of tannery fleshings during anaerobic digestion
- Understory vegetation diversity patterns of Platycladus orientalis and Pinus elliottii communities in Central and Southern China
- Studies on macrofungi diversity and discovery of new species of Abortiporus from Baotianman World Biosphere Reserve
- Food Science
- Effect of berrycactus fruit (Myrtillocactus geometrizans) on glutamate, glutamine, and GABA levels in the frontal cortex of rats fed with a high-fat diet
- Guesstimate of thymoquinone diversity in Nigella sativa L. genotypes and elite varieties collected from Indian states using HPTLC technique
- Analysis of bacterial community structure of Fuzhuan tea with different processing techniques
- Untargeted metabolomics reveals sour jujube kernel benefiting the nutritional value and flavor of Morchella esculenta
- Mycobiota in Slovak wine grapes: A case study from the small Carpathians wine region
- Elemental analysis of Fadogia ancylantha leaves used as a nutraceutical in Mashonaland West Province, Zimbabwe
- Microbiological transglutaminase: Biotechnological application in the food industry
- Influence of solvent-free extraction of fish oil from catfish (Clarias magur) heads using a Taguchi orthogonal array design: A qualitative and quantitative approach
- Chromatographic analysis of the chemical composition and anticancer activities of Curcuma longa extract cultivated in Palestine
- The potential for the use of leghemoglobin and plant ferritin as sources of iron
- Investigating the association between dietary patterns and glycemic control among children and adolescents with T1DM
- Bioengineering and Biotechnology
- Biocompatibility and osteointegration capability of β-TCP manufactured by stereolithography 3D printing: In vitro study
- Clinical characteristics and the prognosis of diabetic foot in Tibet: A single center, retrospective study
- Agriculture
- Biofertilizer and NPSB fertilizer application effects on nodulation and productivity of common bean (Phaseolus vulgaris L.) at Sodo Zuria, Southern Ethiopia
- On correlation between canopy vegetation and growth indexes of maize varieties with different nitrogen efficiencies
- Exopolysaccharides from Pseudomonas tolaasii inhibit the growth of Pleurotus ostreatus mycelia
- A transcriptomic evaluation of the mechanism of programmed cell death of the replaceable bud in Chinese chestnut
- Melatonin enhances salt tolerance in sorghum by modulating photosynthetic performance, osmoregulation, antioxidant defense, and ion homeostasis
- Effects of plant density on alfalfa (Medicago sativa L.) seed yield in western Heilongjiang areas
- Identification of rice leaf diseases and deficiency disorders using a novel DeepBatch technique
- Artificial intelligence and internet of things oriented sustainable precision farming: Towards modern agriculture
- Animal Sciences
- Effect of ketogenic diet on exercise tolerance and transcriptome of gastrocnemius in mice
- Combined analysis of mRNA–miRNA from testis tissue in Tibetan sheep with different FecB genotypes
- Isolation, identification, and drug resistance of a partially isolated bacterium from the gill of Siniperca chuatsi
- Tracking behavioral changes of confined sows from the first mating to the third parity
- The sequencing of the key genes and end products in the TLR4 signaling pathway from the kidney of Rana dybowskii exposed to Aeromonas hydrophila
- Development of a new candidate vaccine against piglet diarrhea caused by Escherichia coli
- Plant Sciences
- Crown and diameter structure of pure Pinus massoniana Lamb. forest in Hunan province, China
- Genetic evaluation and germplasm identification analysis on ITS2, trnL-F, and psbA-trnH of alfalfa varieties germplasm resources
- Tissue culture and rapid propagation technology for Gentiana rhodantha
- Effects of cadmium on the synthesis of active ingredients in Salvia miltiorrhiza
- Cloning and expression analysis of VrNAC13 gene in mung bean
- Chlorate-induced molecular floral transition revealed by transcriptomes
- Effects of warming and drought on growth and development of soybean in Hailun region
- Effects of different light conditions on transient expression and biomass in Nicotiana benthamiana leaves
- Comparative analysis of the rhizosphere microbiome and medicinally active ingredients of Atractylodes lancea from different geographical origins
- Distinguish Dianthus species or varieties based on chloroplast genomes
- Comparative transcriptomes reveal molecular mechanisms of apple blossoms of different tolerance genotypes to chilling injury
- Study on fresh processing key technology and quality influence of Cut Ophiopogonis Radix based on multi-index evaluation
- An advanced approach for fig leaf disease detection and classification: Leveraging image processing and enhanced support vector machine methodology
- Erratum
- Erratum to “Protein Z modulates the metastasis of lung adenocarcinoma cells”
- Erratum to “BRCA1 subcellular localization regulated by PI3K signaling pathway in triple-negative breast cancer MDA-MB-231 cells and hormone-sensitive T47D cells”
- Retraction
- Retraction to “Protocatechuic acid attenuates cerebral aneurysm formation and progression by inhibiting TNF-alpha/Nrf-2/NF-kB-mediated inflammatory mechanisms in experimental rats”
Articles in the same Issue
- Biomedical Sciences
- Systemic investigation of inetetamab in combination with small molecules to treat HER2-overexpressing breast and gastric cancers
- Immunosuppressive treatment for idiopathic membranous nephropathy: An updated network meta-analysis
- Identifying two pathogenic variants in a patient with pigmented paravenous retinochoroidal atrophy
- Effects of phytoestrogens combined with cold stress on sperm parameters and testicular proteomics in rats
- A case of pulmonary embolism with bad warfarin anticoagulant effects caused by E. coli infection
- Neutrophilia with subclinical Cushing’s disease: A case report and literature review
- Isoimperatorin alleviates lipopolysaccharide-induced periodontitis by downregulating ERK1/2 and NF-κB pathways
- Immunoregulation of synovial macrophages for the treatment of osteoarthritis
- Novel CPLANE1 c.8948dupT (p.P2984Tfs*7) variant in a child patient with Joubert syndrome
- Antiphospholipid antibodies and the risk of thrombosis in myeloproliferative neoplasms
- Immunological responses of septic rats to combination therapy with thymosin α1 and vitamin C
- High glucose and high lipid induced mitochondrial dysfunction in JEG-3 cells through oxidative stress
- Pharmacological inhibition of the ubiquitin-specific protease 8 effectively suppresses glioblastoma cell growth
- Levocarnitine regulates the growth of angiotensin II-induced myocardial fibrosis cells via TIMP-1
- Age-related changes in peripheral T-cell subpopulations in elderly individuals: An observational study
- Single-cell transcription analysis reveals the tumor origin and heterogeneity of human bilateral renal clear cell carcinoma
- Identification of iron metabolism-related genes as diagnostic signatures in sepsis by blood transcriptomic analysis
- Long noncoding RNA ACART knockdown decreases 3T3-L1 preadipocyte proliferation and differentiation
- Surgery, adjuvant immunotherapy plus chemotherapy and radiotherapy for primary malignant melanoma of the parotid gland (PGMM): A case report
- Dosimetry comparison with helical tomotherapy, volumetric modulated arc therapy, and intensity-modulated radiotherapy for grade II gliomas: A single‑institution case series
- Soy isoflavone reduces LPS-induced acute lung injury via increasing aquaporin 1 and aquaporin 5 in rats
- Refractory hypokalemia with sexual dysplasia and infertility caused by 17α-hydroxylase deficiency and triple X syndrome: A case report
- Meta-analysis of cancer risk among end stage renal disease undergoing maintenance dialysis
- 6-Phosphogluconate dehydrogenase inhibition arrests growth and induces apoptosis in gastric cancer via AMPK activation and oxidative stress
- Experimental study on the optimization of ANM33 release in foam cells
- Primary retroperitoneal angiosarcoma: A case report
- Metabolomic analysis-identified 2-hydroxybutyric acid might be a key metabolite of severe preeclampsia
- Malignant pleural effusion diagnosis and therapy
- Effect of spaceflight on the phenotype and proteome of Escherichia coli
- Comparison of immunotherapy combined with stereotactic radiotherapy and targeted therapy for patients with brain metastases: A systemic review and meta-analysis
- Activation of hypermethylated P2RY1 mitigates gastric cancer by promoting apoptosis and inhibiting proliferation
- Association between the VEGFR-2 -604T/C polymorphism (rs2071559) and type 2 diabetic retinopathy
- The role of IL-31 and IL-34 in the diagnosis and treatment of chronic periodontitis
- Triple-negative mouse breast cancer initiating cells show high expression of beta1 integrin and increased malignant features
- mNGS facilitates the accurate diagnosis and antibiotic treatment of suspicious critical CNS infection in real practice: A retrospective study
- The apatinib and pemetrexed combination has antitumor and antiangiogenic effects against NSCLC
- Radiotherapy for primary thyroid adenoid cystic carcinoma
- Design and functional preliminary investigation of recombinant antigen EgG1Y162–EgG1Y162 against Echinococcus granulosus
- Effects of losartan in patients with NAFLD: A meta-analysis of randomized controlled trial
- Bibliometric analysis of METTL3: Current perspectives, highlights, and trending topics
- Performance comparison of three scaling algorithms in NMR-based metabolomics analysis
- PI3K/AKT/mTOR pathway and its related molecules participate in PROK1 silence-induced anti-tumor effects on pancreatic cancer
- The altered expression of cytoskeletal and synaptic remodeling proteins during epilepsy
- Effects of pegylated recombinant human granulocyte colony-stimulating factor on lymphocytes and white blood cells of patients with malignant tumor
- Prostatitis as initial manifestation of Chlamydia psittaci pneumonia diagnosed by metagenome next-generation sequencing: A case report
- NUDT21 relieves sevoflurane-induced neurological damage in rats by down-regulating LIMK2
- Association of interleukin-10 rs1800896, rs1800872, and interleukin-6 rs1800795 polymorphisms with squamous cell carcinoma risk: A meta-analysis
- Exosomal HBV-DNA for diagnosis and treatment monitoring of chronic hepatitis B
- Shear stress leads to the dysfunction of endothelial cells through the Cav-1-mediated KLF2/eNOS/ERK signaling pathway under physiological conditions
- Interaction between the PI3K/AKT pathway and mitochondrial autophagy in macrophages and the leukocyte count in rats with LPS-induced pulmonary infection
- Meta-analysis of the rs231775 locus polymorphism in the CTLA-4 gene and the susceptibility to Graves’ disease in children
- Cloning, subcellular localization and expression of phosphate transporter gene HvPT6 of hulless barley
- Coptisine mitigates diabetic nephropathy via repressing the NRLP3 inflammasome
- Significant elevated CXCL14 and decreased IL-39 levels in patients with tuberculosis
- Whole-exome sequencing applications in prenatal diagnosis of fetal bowel dilatation
- Gemella morbillorum infective endocarditis: A case report and literature review
- An unusual ectopic thymoma clonal evolution analysis: A case report
- Severe cumulative skin toxicity during toripalimab combined with vemurafenib following toripalimab alone
- Detection of V. vulnificus septic shock with ARDS using mNGS
- Novel rare genetic variants of familial and sporadic pulmonary atresia identified by whole-exome sequencing
- The influence and mechanistic action of sperm DNA fragmentation index on the outcomes of assisted reproduction technology
- Novel compound heterozygous mutations in TELO2 in an infant with You-Hoover-Fong syndrome: A case report and literature review
- ctDNA as a prognostic biomarker in resectable CLM: Systematic review and meta-analysis
- Diagnosis of primary amoebic meningoencephalitis by metagenomic next-generation sequencing: A case report
- Phylogenetic analysis of promoter regions of human Dolichol kinase (DOLK) and orthologous genes using bioinformatics tools
- Collagen changes in rabbit conjunctiva after conjunctival crosslinking
- Effects of NM23 transfection of human gastric carcinoma cells in mice
- Oral nifedipine and phytosterol, intravenous nicardipine, and oral nifedipine only: Three-arm, retrospective, cohort study for management of severe preeclampsia
- Case report of hepatic retiform hemangioendothelioma: A rare tumor treated with ultrasound-guided microwave ablation
- Curcumin induces apoptosis in human hepatocellular carcinoma cells by decreasing the expression of STAT3/VEGF/HIF-1α signaling
- Rare presentation of double-clonal Waldenström macroglobulinemia with pulmonary embolism: A case report
- Giant duplication of the transverse colon in an adult: A case report and literature review
- Ectopic thyroid tissue in the breast: A case report
- SDR16C5 promotes proliferation and migration and inhibits apoptosis in pancreatic cancer
- Vaginal metastasis from breast cancer: A case report
- Screening of the best time window for MSC transplantation to treat acute myocardial infarction with SDF-1α antibody-loaded targeted ultrasonic microbubbles: An in vivo study in miniswine
- Inhibition of TAZ impairs the migration ability of melanoma cells
- Molecular complexity analysis of the diagnosis of Gitelman syndrome in China
- Effects of maternal calcium and protein intake on the development and bone metabolism of offspring mice
- Identification of winter wheat pests and diseases based on improved convolutional neural network
- Ultra-multiplex PCR technique to guide treatment of Aspergillus-infected aortic valve prostheses
- Virtual high-throughput screening: Potential inhibitors targeting aminopeptidase N (CD13) and PIKfyve for SARS-CoV-2
- Immune checkpoint inhibitors in cancer patients with COVID-19
- Utility of methylene blue mixed with autologous blood in preoperative localization of pulmonary nodules and masses
- Integrated analysis of the microbiome and transcriptome in stomach adenocarcinoma
- Berberine suppressed sarcopenia insulin resistance through SIRT1-mediated mitophagy
- DUSP2 inhibits the progression of lupus nephritis in mice by regulating the STAT3 pathway
- Lung abscess by Fusobacterium nucleatum and Streptococcus spp. co-infection by mNGS: A case series
- Genetic alterations of KRAS and TP53 in intrahepatic cholangiocarcinoma associated with poor prognosis
- Granulomatous polyangiitis involving the fourth ventricle: Report of a rare case and a literature review
- Studying infant mortality: A demographic analysis based on data mining models
- Metaplastic breast carcinoma with osseous differentiation: A report of a rare case and literature review
- Protein Z modulates the metastasis of lung adenocarcinoma cells
- Inhibition of pyroptosis and apoptosis by capsaicin protects against LPS-induced acute kidney injury through TRPV1/UCP2 axis in vitro
- TAK-242, a toll-like receptor 4 antagonist, against brain injury by alleviates autophagy and inflammation in rats
- Primary mediastinum Ewing’s sarcoma with pleural effusion: A case report and literature review
- Association of ADRB2 gene polymorphisms and intestinal microbiota in Chinese Han adolescents
- Tanshinone IIA alleviates chondrocyte apoptosis and extracellular matrix degeneration by inhibiting ferroptosis
- Study on the cytokines related to SARS-Cov-2 in testicular cells and the interaction network between cells based on scRNA-seq data
- Effect of periostin on bone metabolic and autophagy factors during tooth eruption in mice
- HP1 induces ferroptosis of renal tubular epithelial cells through NRF2 pathway in diabetic nephropathy
- Intravaginal estrogen management in postmenopausal patients with vaginal squamous intraepithelial lesions along with CO2 laser ablation: A retrospective study
- Hepatocellular carcinoma cell differentiation trajectory predicts immunotherapy, potential therapeutic drugs, and prognosis of patients
- Effects of physical exercise on biomarkers of oxidative stress in healthy subjects: A meta-analysis of randomized controlled trials
- Identification of lysosome-related genes in connection with prognosis and immune cell infiltration for drug candidates in head and neck cancer
- Development of an instrument-free and low-cost ELISA dot-blot test to detect antibodies against SARS-CoV-2
- Research progress on gas signal molecular therapy for Parkinson’s disease
- Adiponectin inhibits TGF-β1-induced skin fibroblast proliferation and phenotype transformation via the p38 MAPK signaling pathway
- The G protein-coupled receptor-related gene signatures for predicting prognosis and immunotherapy response in bladder urothelial carcinoma
- α-Fetoprotein contributes to the malignant biological properties of AFP-producing gastric cancer
- CXCL12/CXCR4/CXCR7 axis in placenta tissues of patients with placenta previa
- Association between thyroid stimulating hormone levels and papillary thyroid cancer risk: A meta-analysis
- Significance of sTREM-1 and sST2 combined diagnosis for sepsis detection and prognosis prediction
- Diagnostic value of serum neuroactive substances in the acute exacerbation of chronic obstructive pulmonary disease complicated with depression
- Research progress of AMP-activated protein kinase and cardiac aging
- TRIM29 knockdown prevented the colon cancer progression through decreasing the ubiquitination levels of KRT5
- Cross-talk between gut microbiota and liver steatosis: Complications and therapeutic target
- Metastasis from small cell lung cancer to ovary: A case report
- The early diagnosis and pathogenic mechanisms of sepsis-related acute kidney injury
- The effect of NK cell therapy on sepsis secondary to lung cancer: A case report
- Erianin alleviates collagen-induced arthritis in mice by inhibiting Th17 cell differentiation
- Loss of ACOX1 in clear cell renal cell carcinoma and its correlation with clinical features
- Signalling pathways in the osteogenic differentiation of periodontal ligament stem cells
- Crosstalk between lactic acid and immune regulation and its value in the diagnosis and treatment of liver failure
- Clinicopathological features and differential diagnosis of gastric pleomorphic giant cell carcinoma
- Traumatic brain injury and rTMS-ERPs: Case report and literature review
- Extracellular fibrin promotes non-small cell lung cancer progression through integrin β1/PTEN/AKT signaling
- Knockdown of DLK4 inhibits non-small cell lung cancer tumor growth by downregulating CKS2
- The co-expression pattern of VEGFR-2 with indicators related to proliferation, apoptosis, and differentiation of anagen hair follicles
- Inflammation-related signaling pathways in tendinopathy
- CD4+ T cell count in HIV/TB co-infection and co-occurrence with HL: Case report and literature review
- Clinical analysis of severe Chlamydia psittaci pneumonia: Case series study
- Bioinformatics analysis to identify potential biomarkers for the pulmonary artery hypertension associated with the basement membrane
- Influence of MTHFR polymorphism, alone or in combination with smoking and alcohol consumption, on cancer susceptibility
- Catharanthus roseus (L.) G. Don counteracts the ampicillin resistance in multiple antibiotic-resistant Staphylococcus aureus by downregulation of PBP2a synthesis
- Combination of a bronchogenic cyst in the thoracic spinal canal with chronic myelocytic leukemia
- Bacterial lipoprotein plays an important role in the macrophage autophagy and apoptosis induced by Salmonella typhimurium and Staphylococcus aureus
- TCL1A+ B cells predict prognosis in triple-negative breast cancer through integrative analysis of single-cell and bulk transcriptomic data
- Ezrin promotes esophageal squamous cell carcinoma progression via the Hippo signaling pathway
- Ferroptosis: A potential target of macrophages in plaque vulnerability
- Predicting pediatric Crohn's disease based on six mRNA-constructed risk signature using comprehensive bioinformatic approaches
- Applications of genetic code expansion and photosensitive UAAs in studying membrane proteins
- HK2 contributes to the proliferation, migration, and invasion of diffuse large B-cell lymphoma cells by enhancing the ERK1/2 signaling pathway
- IL-17 in osteoarthritis: A narrative review
- Circadian cycle and neuroinflammation
- Probiotic management and inflammatory factors as a novel treatment in cirrhosis: A systematic review and meta-analysis
- Hemorrhagic meningioma with pulmonary metastasis: Case report and literature review
- SPOP regulates the expression profiles and alternative splicing events in human hepatocytes
- Knockdown of SETD5 inhibited glycolysis and tumor growth in gastric cancer cells by down-regulating Akt signaling pathway
- PTX3 promotes IVIG resistance-induced endothelial injury in Kawasaki disease by regulating the NF-κB pathway
- Pancreatic ectopic thyroid tissue: A case report and analysis of literature
- The prognostic impact of body mass index on female breast cancer patients in underdeveloped regions of northern China differs by menopause status and tumor molecular subtype
- Report on a case of liver-originating malignant melanoma of unknown primary
- Case report: Herbal treatment of neutropenic enterocolitis after chemotherapy for breast cancer
- The fibroblast growth factor–Klotho axis at molecular level
- Characterization of amiodarone action on currents in hERG-T618 gain-of-function mutations
- A case report of diagnosis and dynamic monitoring of Listeria monocytogenes meningitis with NGS
- Effect of autologous platelet-rich plasma on new bone formation and viability of a Marburg bone graft
- Small breast epithelial mucin as a useful prognostic marker for breast cancer patients
- Continuous non-adherent culture promotes transdifferentiation of human adipose-derived stem cells into retinal lineage
- Nrf3 alleviates oxidative stress and promotes the survival of colon cancer cells by activating AKT/BCL-2 signal pathway
- Favorable response to surufatinib in a patient with necrolytic migratory erythema: A case report
- Case report of atypical undernutrition of hypoproteinemia type
- Down-regulation of COL1A1 inhibits tumor-associated fibroblast activation and mediates matrix remodeling in the tumor microenvironment of breast cancer
- Sarcoma protein kinase inhibition alleviates liver fibrosis by promoting hepatic stellate cells ferroptosis
- Research progress of serum eosinophil in chronic obstructive pulmonary disease and asthma
- Clinicopathological characteristics of co-existing or mixed colorectal cancer and neuroendocrine tumor: Report of five cases
- Role of menopausal hormone therapy in the prevention of postmenopausal osteoporosis
- Precisional detection of lymph node metastasis using tFCM in colorectal cancer
- Advances in diagnosis and treatment of perimenopausal syndrome
- A study of forensic genetics: ITO index distribution and kinship judgment between two individuals
- Acute lupus pneumonitis resembling miliary tuberculosis: A case-based review
- Plasma levels of CD36 and glutathione as biomarkers for ruptured intracranial aneurysm
- Fractalkine modulates pulmonary angiogenesis and tube formation by modulating CX3CR1 and growth factors in PVECs
- Novel risk prediction models for deep vein thrombosis after thoracotomy and thoracoscopic lung cancer resections, involving coagulation and immune function
- Exploring the diagnostic markers of essential tremor: A study based on machine learning algorithms
- Evaluation of effects of small-incision approach treatment on proximal tibia fracture by deep learning algorithm-based magnetic resonance imaging
- An online diagnosis method for cancer lesions based on intelligent imaging analysis
- Medical imaging in rheumatoid arthritis: A review on deep learning approach
- Predictive analytics in smart healthcare for child mortality prediction using a machine learning approach
- Utility of neutrophil–lymphocyte ratio and platelet–lymphocyte ratio in predicting acute-on-chronic liver failure survival
- A biomedical decision support system for meta-analysis of bilateral upper-limb training in stroke patients with hemiplegia
- TNF-α and IL-8 levels are positively correlated with hypobaric hypoxic pulmonary hypertension and pulmonary vascular remodeling in rats
- Stochastic gradient descent optimisation for convolutional neural network for medical image segmentation
- Comparison of the prognostic value of four different critical illness scores in patients with sepsis-induced coagulopathy
- Application and teaching of computer molecular simulation embedded technology and artificial intelligence in drug research and development
- Hepatobiliary surgery based on intelligent image segmentation technology
- Value of brain injury-related indicators based on neural network in the diagnosis of neonatal hypoxic-ischemic encephalopathy
- Analysis of early diagnosis methods for asymmetric dementia in brain MR images based on genetic medical technology
- Early diagnosis for the onset of peri-implantitis based on artificial neural network
- Clinical significance of the detection of serum IgG4 and IgG4/IgG ratio in patients with thyroid-associated ophthalmopathy
- Forecast of pain degree of lumbar disc herniation based on back propagation neural network
- SPA-UNet: A liver tumor segmentation network based on fused multi-scale features
- Systematic evaluation of clinical efficacy of CYP1B1 gene polymorphism in EGFR mutant non-small cell lung cancer observed by medical image
- Rehabilitation effect of intelligent rehabilitation training system on hemiplegic limb spasms after stroke
- A novel approach for minimising anti-aliasing effects in EEG data acquisition
- ErbB4 promotes M2 activation of macrophages in idiopathic pulmonary fibrosis
- Clinical role of CYP1B1 gene polymorphism in prediction of postoperative chemotherapy efficacy in NSCLC based on individualized health model
- Lung nodule segmentation via semi-residual multi-resolution neural networks
- Evaluation of brain nerve function in ICU patients with Delirium by deep learning algorithm-based resting state MRI
- A data mining technique for detecting malignant mesothelioma cancer using multiple regression analysis
- Markov model combined with MR diffusion tensor imaging for predicting the onset of Alzheimer’s disease
- Effectiveness of the treatment of depression associated with cancer and neuroimaging changes in depression-related brain regions in patients treated with the mediator-deuterium acupuncture method
- Molecular mechanism of colorectal cancer and screening of molecular markers based on bioinformatics analysis
- Monitoring and evaluation of anesthesia depth status data based on neuroscience
- Exploring the conformational dynamics and thermodynamics of EGFR S768I and G719X + S768I mutations in non-small cell lung cancer: An in silico approaches
- Optimised feature selection-driven convolutional neural network using gray level co-occurrence matrix for detection of cervical cancer
- Incidence of different pressure patterns of spinal cerebellar ataxia and analysis of imaging and genetic diagnosis
- Pathogenic bacteria and treatment resistance in older cardiovascular disease patients with lung infection and risk prediction model
- Adoption value of support vector machine algorithm-based computed tomography imaging in the diagnosis of secondary pulmonary fungal infections in patients with malignant hematological disorders
- From slides to insights: Harnessing deep learning for prognostic survival prediction in human colorectal cancer histology
- Ecology and Environmental Science
- Monitoring of hourly carbon dioxide concentration under different land use types in arid ecosystem
- Comparing the differences of prokaryotic microbial community between pit walls and bottom from Chinese liquor revealed by 16S rRNA gene sequencing
- Effects of cadmium stress on fruits germination and growth of two herbage species
- Bamboo charcoal affects soil properties and bacterial community in tea plantations
- Optimization of biogas potential using kinetic models, response surface methodology, and instrumental evidence for biodegradation of tannery fleshings during anaerobic digestion
- Understory vegetation diversity patterns of Platycladus orientalis and Pinus elliottii communities in Central and Southern China
- Studies on macrofungi diversity and discovery of new species of Abortiporus from Baotianman World Biosphere Reserve
- Food Science
- Effect of berrycactus fruit (Myrtillocactus geometrizans) on glutamate, glutamine, and GABA levels in the frontal cortex of rats fed with a high-fat diet
- Guesstimate of thymoquinone diversity in Nigella sativa L. genotypes and elite varieties collected from Indian states using HPTLC technique
- Analysis of bacterial community structure of Fuzhuan tea with different processing techniques
- Untargeted metabolomics reveals sour jujube kernel benefiting the nutritional value and flavor of Morchella esculenta
- Mycobiota in Slovak wine grapes: A case study from the small Carpathians wine region
- Elemental analysis of Fadogia ancylantha leaves used as a nutraceutical in Mashonaland West Province, Zimbabwe
- Microbiological transglutaminase: Biotechnological application in the food industry
- Influence of solvent-free extraction of fish oil from catfish (Clarias magur) heads using a Taguchi orthogonal array design: A qualitative and quantitative approach
- Chromatographic analysis of the chemical composition and anticancer activities of Curcuma longa extract cultivated in Palestine
- The potential for the use of leghemoglobin and plant ferritin as sources of iron
- Investigating the association between dietary patterns and glycemic control among children and adolescents with T1DM
- Bioengineering and Biotechnology
- Biocompatibility and osteointegration capability of β-TCP manufactured by stereolithography 3D printing: In vitro study
- Clinical characteristics and the prognosis of diabetic foot in Tibet: A single center, retrospective study
- Agriculture
- Biofertilizer and NPSB fertilizer application effects on nodulation and productivity of common bean (Phaseolus vulgaris L.) at Sodo Zuria, Southern Ethiopia
- On correlation between canopy vegetation and growth indexes of maize varieties with different nitrogen efficiencies
- Exopolysaccharides from Pseudomonas tolaasii inhibit the growth of Pleurotus ostreatus mycelia
- A transcriptomic evaluation of the mechanism of programmed cell death of the replaceable bud in Chinese chestnut
- Melatonin enhances salt tolerance in sorghum by modulating photosynthetic performance, osmoregulation, antioxidant defense, and ion homeostasis
- Effects of plant density on alfalfa (Medicago sativa L.) seed yield in western Heilongjiang areas
- Identification of rice leaf diseases and deficiency disorders using a novel DeepBatch technique
- Artificial intelligence and internet of things oriented sustainable precision farming: Towards modern agriculture
- Animal Sciences
- Effect of ketogenic diet on exercise tolerance and transcriptome of gastrocnemius in mice
- Combined analysis of mRNA–miRNA from testis tissue in Tibetan sheep with different FecB genotypes
- Isolation, identification, and drug resistance of a partially isolated bacterium from the gill of Siniperca chuatsi
- Tracking behavioral changes of confined sows from the first mating to the third parity
- The sequencing of the key genes and end products in the TLR4 signaling pathway from the kidney of Rana dybowskii exposed to Aeromonas hydrophila
- Development of a new candidate vaccine against piglet diarrhea caused by Escherichia coli
- Plant Sciences
- Crown and diameter structure of pure Pinus massoniana Lamb. forest in Hunan province, China
- Genetic evaluation and germplasm identification analysis on ITS2, trnL-F, and psbA-trnH of alfalfa varieties germplasm resources
- Tissue culture and rapid propagation technology for Gentiana rhodantha
- Effects of cadmium on the synthesis of active ingredients in Salvia miltiorrhiza
- Cloning and expression analysis of VrNAC13 gene in mung bean
- Chlorate-induced molecular floral transition revealed by transcriptomes
- Effects of warming and drought on growth and development of soybean in Hailun region
- Effects of different light conditions on transient expression and biomass in Nicotiana benthamiana leaves
- Comparative analysis of the rhizosphere microbiome and medicinally active ingredients of Atractylodes lancea from different geographical origins
- Distinguish Dianthus species or varieties based on chloroplast genomes
- Comparative transcriptomes reveal molecular mechanisms of apple blossoms of different tolerance genotypes to chilling injury
- Study on fresh processing key technology and quality influence of Cut Ophiopogonis Radix based on multi-index evaluation
- An advanced approach for fig leaf disease detection and classification: Leveraging image processing and enhanced support vector machine methodology
- Erratum
- Erratum to “Protein Z modulates the metastasis of lung adenocarcinoma cells”
- Erratum to “BRCA1 subcellular localization regulated by PI3K signaling pathway in triple-negative breast cancer MDA-MB-231 cells and hormone-sensitive T47D cells”
- Retraction
- Retraction to “Protocatechuic acid attenuates cerebral aneurysm formation and progression by inhibiting TNF-alpha/Nrf-2/NF-kB-mediated inflammatory mechanisms in experimental rats”