Modeling of supramolecular biopolymers: Leading the in silico revolution of tissue engineering and nanomedicine
-
Federico Fontana


Abstract
The field of tissue engineering is poised to be positively influenced by the advent of supramolecular biopolymers, because of their promising tailorability coming from the bottom-up approach used for their development, absence of toxic byproducts from their gelation reaction and intrinsic better mimicry of extracellular matrix nanotopography and mechanical properties. However, a deep understanding of the phenomena ruling their properties at the meso- and macroscales is still missing. In silico approaches are increasingly helping to shine a light on questions still of out of reach for almost all empirical methods. In this review, we will present the most significant and updated efforts on molecular modeling of SBP properties, and their interactions with the living counterparts, at all scales. In detail, the currently available molecular mechanic approaches will be discussed, paying attention to the pros and cons related to their representability and transferability. We will also give detailed insights for choosing different biomolecular modeling strategies at various scales. This is a systematic overview of tools and approaches yielding to advances at atomistic, molecular, and supramolecular levels, with a holistic perspective demonstrating the urgent need for theories and models connecting biomaterial design and their biological effect in vivo.
Nomenclature
- AA-MD
-
all-atom molecular dynamics
- AFM
-
atomic force microscopy
- AI
-
artificial intelligence
- CAPs
-
complementary co-assembling peptides
- CG
-
coarse grained
- CG-MD
-
coarse-grained molecular dynamics
- DFT
-
density functional theory
- DPD
-
dissipative particle dynamics
- dsDNA
-
double-stranded DNA
- dsRNA
-
double-stranded RNA
- ECM
-
extracellular matrix
- FE
-
finite element
- FS
-
fully saturated
- GQDs
-
graphene quantum dots
- HA
-
hyaluronic acid
- MD
-
molecular dynamics
- PAs
-
peptide amphiphiles
- PU
-
polyunsaturated
- QM
-
quantum mechanics
- RMD
-
reactive molecular dynamics
- SAPs
-
self-assembling peptides
- SBPs
-
supramolecular biopolymers
- SEM
-
scanning electron microscopy
- SLPs
-
surfactant-like peptides
- SMD
-
steered molecular dynamics
- TE
-
tissue engineering
- UCG
-
ultra-coarse grained
- µFE
-
micro-finite element
1 Introduction
The future of medical treatments relies on the investigation and understanding of the mechanisms underlying the complexity of life [1,2,3,4,5,6,7]. In the last two decades, particular attention has been paid to tissue engineering (TE) and regenerative medicine applications. TE, like regenerative medicine, is an interdisciplinary research field aiming to heal injured or degenerated tissues to regain their own physiological states [8,9,10,11]. Healthy tissues are characterized by precise interactions among cells and the extracellular environment, that are finely regulated, among others, by the architecture of extracellular matrix (ECM). ECM mainly consists of differently glycosylated intertwined fibrillar proteins.
The advances in bioorganic chemistry enabled the design of supramolecular biopolymers (SBPs), which can spontaneously organize into fibrillar structures resembling the main structural and mechanical features of the ECM with no (or minimal) chemical reactions involved. As shown in Figure 1, SBPs found several applications in TE due to their promising biocompatibility and biodegradability. Among SBPs, collagen was used as injectable hydrogel for central nervous system repair of traumatic injuries, correction of peripheral nerve defects, remediation to cardiovascular defects, regeneration of renal tissue, and a component for scaffolds designed for bone and skin regeneration [12,13,14,15,16,17,18,19,20,21,22].
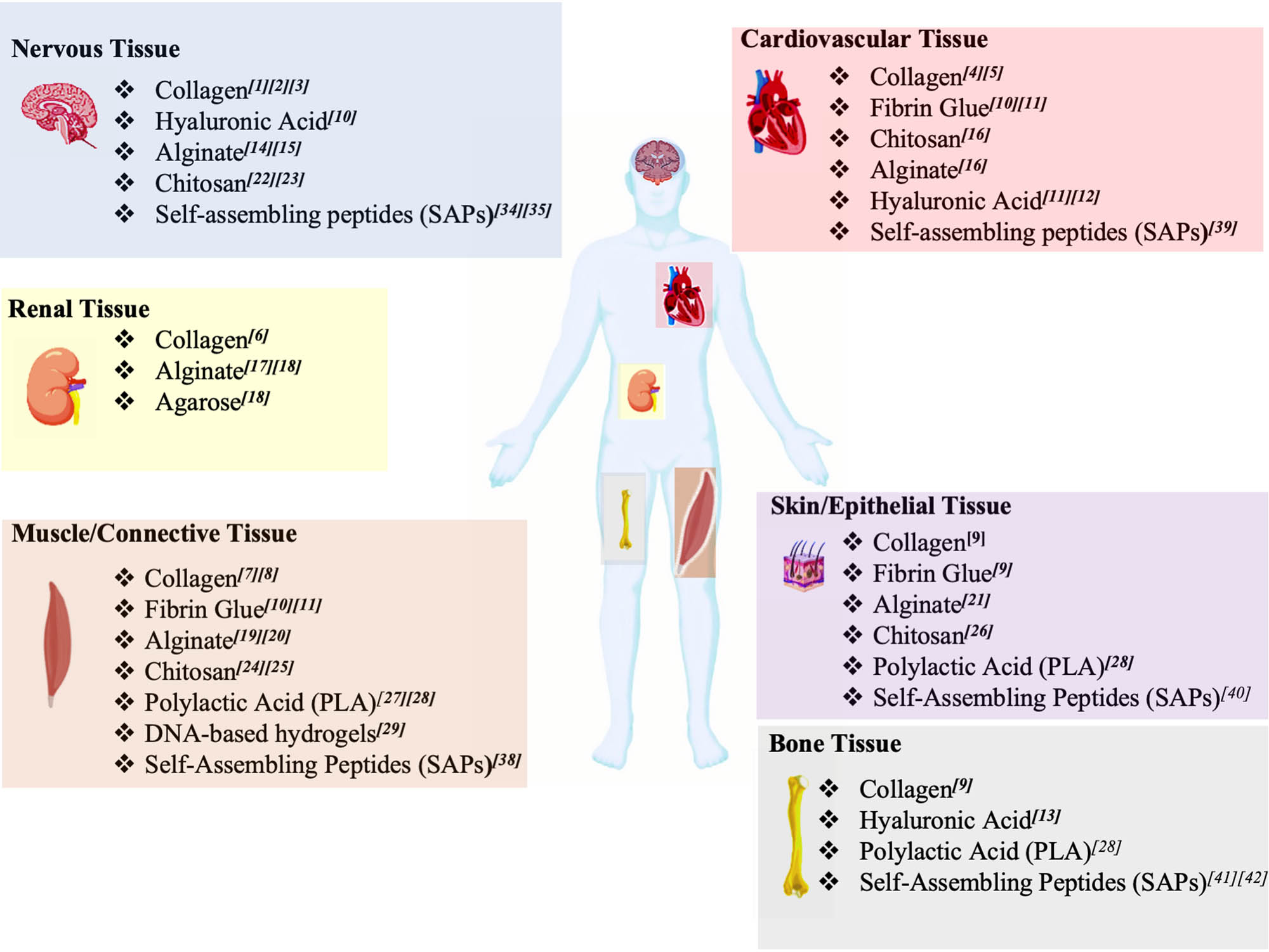
Schematic overview of SBPs used in TE. TE applications of SBPs according to the target tissue. Different SBPs are suitable for neural TE applications, such as collagen, HA, alginate, chitosan, and SAPs. These SBPs are versatile enough to be used also for the regenerative approaches of cardiovascular, renal, skin, muscle, and bone tissues. Lastly, SAPs and DNA origami, also known as programmable SBPs, were found to be suitable for other various TE applications.
Another promising SBP is hyaluronic acid (HA) used as a component for hydrogels in TE of nervous and cardiovascular systems; when mixed with chitosan and fibrin glue hydrogels, HA was adopted to reestablish skin, bone, cardiovascular and connective tissue properties [23,24,25,26]. 3D bio-printed chitosan-based hydrogels, thanks to their good biocompatibility and mechanical properties, were used for the TE of hearth and cardiovascular system, skin, bone, and connective tissues [19,26,27,28,29,30]. Some applications in TE also employed alginate, amenable of ready processing properties by using physical and chemical methods. Alginate and its derivative materials have largely been used for the healing of bone, connective, muscle, renal, cardiovascular, and nervous tissues [31,32,33,34,35,36,37,38,39].
Polylactic acid was also combined with other SBPs in diverse TE applications [40,41,42]. Furthermore, other synthetic SBPs were used for the development of electrospun self-standing implants as reported in other review articles [43,44,45,46]. Among the realm of synthetic animal-free SBPs, programmable SBP hydrogels show intriguing properties, such as reversible and tunable self-assembly, high biocompatibility, and good mechanical properties. Programmable SBPs can encompass almost all the SBPs’ molecular classes: however, for the sake of conciseness, such classification, in this review, will be used for the most advanced classes of SBP biomaterials, such as DNA-based bioconjugates and self-assembling peptides (SAPs) [47,48,49].
Both DNA-based bioconjugates and SAPs are widely adopted in TE applications as hydrogel-forming molecules. DNA–peptide hybrid hydrogels, with assembling properties of informational molecules, feature an easy control of the assembly via both pH shifts and the addition of oligonucleotides [50,51,52].
1.1 DNA–peptide based biomaterials
Nucleic acids represent one of the most used molecular building blocks for the design of SBPs. Nucleic acid nanotechnology encompasses DNA origami, DNA hydrogel, DNA nanorobot, molecular tweezers, and DNA walkers [53,54] and found applications in drug delivery, biosensing, and bioimaging [55,56]. Just recently, the progress in click-chemistry allowed the design of DNA conjugates: among them, DNA–peptide bioconjugates found several applications because they combine the properties of both informational molecules.
For example, DNA–peptide bioconjugates could find application in systems biology. A DNA–peptide tethering system was demonstrated to exert control on the adhesion of the cells on surfaces. It was made of three components: (1) an oligonucleotide sequence immobilized on a solid surface; (2) a DNA moiety complementary to the immobilized oligonucleotide; and (3) a competitive oligonucleotide sequence capable of replacing the DNA–peptide oligonucleotide, thus modulating the anchoring of the cells on the surface. Indeed, the cells adhered and spread on the surface by establishing receptor–ligand interactions with the functional motif of the DNA–peptide conjugates when bound to the immobilized oligonucleotide sequences [57,58]. Thus, DNA–peptide bioconjugates, which exhibit bioactive peptide sequences, such as RGD, IKVAV or growth factors, were used for promoting the proliferation of the neuronal or osteoclast cells [57].
Such tailorability encouraged the use of DNA–peptide hybrids in TE. Indeed, the DNA–peptide hybrids have been demonstrated to be suitable hydrogel-forming molecules. DNA–peptide hydrogels showed intriguing properties, such as the reversibility of the aggregation state. Insights from coarse-grained molecular dynamics (CG-MD) and experimental characterization unveiled that the DNA–peptide hybrids spontaneously assemble into nanofibrils. The growth of such fibril bundles has been manipulated by adding “invaders” oligonucleotides, thus modulating the resulting mechanical properties of the hydrogels [50]. DNA bioconjugates were demonstrated to be suitable also for nanomedicine applications, such as drug delivery or gene therapy. DNA aptamers can be anisotropically functionalized onto branched DNA nanostructures to control cell adhesion and for the fine-tuning of 3D cell cultures [52].
1.2 SAPs
As shown in Figure 1, SAPs found large applications in TE applications for skin regeneration, muscle repair, heart failure, and trauma at the level of central nervous systems [59,60,61,62,63,64,65,66]. For example, three main classes of SAPs were used for neural TE applications, i.e., RADA-like SAPs, complementary co- assembling peptides (CAPs), and peptide amphiphiles (PAs) [2,67,68]. The serendipitous discovery of RADA-like SAPs comes from studies of Shuguang Zhang on Zuotin, a Z-DNA-binding protein [69]. Since then, different variants of RADA-like SAPs have been designed and studied, such as Ac-(RADA)3-CONH2, Ac-(KLDL)3-CONH2, and Ac-(RADADADA)2-CONH2 [70,71,72].
As demonstrated through CG-MD investigations, peptides belonging to this class of SAPs self-assemble into cross-ß structures. CG-MD simulations showed that interactions among hydrophobic residues lead to peptide assembly into nanofibers with charged amino acid side chains exposed to the aqueous environment [2,68,73–75]. In addition to TE applications, RADA-like SAPs have been used as hemostat solutions. CAPs are made of positive Ac-(LKLK)3-CONH2 and negative Ac-(LDLD)3-CONH2 modules, eventually functionalized, i.e., Ac-KLPGWSG-(LDLD)3-CONH2 [60,68]. The electrostatic interactions between positively and negatively charged modules drive the co-assembly, while the strong self-repulsion of each module allows for mixing of one module of CAPs and cells at neutral pH without gelation taking place, thus preserving cell viability in a less harmful environment than in classic SAPs. This finely tuned process leads to the formation of nanofibrillar networks. At the mesoscale, both SAP and CAP nanofibers yield to 3D hydrogels that retain water and form porous scaffolds [68].
PAs’ molecular structures resemble those of phospholipids in cell membranes, comprising hydrophobic alkyl tails and hydrophilic heads. In functionalized PAs, the peptide moiety is usually formed by three sections: (1) a hydrophobic sequence capable of forming ß-sheet structures; (2) a hydrophilic, eventually charged, section; and (3) a bioactive epitope, also named functional motif [5,50]. In PAs, the hydrophobic peptide sequence consists of non-polar amino acid residues (G, A, V, L, I, P, F), while the hydrophilic head consists of positively charged (H, K, R) or negatively charged (D, E) residues. PA’s self-assembly is ruled by the hydrophobic interactions of alkyl tails, hydrogen bond formation, and electrostatic repulsion among charged amino acids [50,65]. Other SAPs can assemble into tubular structures because of π–π interactions among aromatic side chains of amino acid residues, such as diphenylalanine SAPs. Instead, cyclic peptides self-assemble into hollow ß-sheet-rich cylinders by stacking on top of each other through stable hydrogen bond formation [76–78].
Functional motifs or other bioactive moieties can be incorporated into the sequence of SAPs to provide them with biomimetic properties or to alter their self-organization. Functional motifs are short peptides linked to N- and C-terminus of the self-assembling backbone and can be spaced with flexible linkers, usually a few glycine, to ensure flexibility and proper exposure to biological targets. Notably, functional motifs may alter the self-assembling propensity of the original SAP backbone [79]: therefore, a proper in silico design should be always performed to optimize functional motif, self-assembling backbone, and spacer sequences.
According to the evidence from proteomics and glycomics of ECM, glycosylation is a fundamental post-translation modification, which affects protein stability, folding, and cellular localization. Glycoproteins are also involved in cellular communication, extracellular vesicles’ recognition, and modulation of immune response [80]. The modification of SAPs with carbohydrates represents an intriguing strategy to facilitate the control over supramolecular arrangements and confer additional biomimetic features [81–84]. Chemical cross-linking represents another promising strategy for improving the control over supramolecular architectures of SBPs’ nanostructures and in particular SAP hydrogels. Such strategy was reported for the development of self-standing bioprothesis for neural TE [85]. Furthermore, physical and chemical cross-linking is a method suitable to provide a safe release of therapeutic compounds [86–88].
1.3 The role of computational modeling in supramolecular bio-polymers design
The advent of SBPs fostered the engineering of molecular structures with programmable shapes and properties. Indeed, molecular self-assembly and subsequent hierarchical organization arise from the interplay of non-covalent interactions (i.e., hydrogen bonding, hydrophobic forces, Van der Waals forces, π–π interactions, and electrostatic interactions) among molecular subunits. Flexible approaches have been developed to design a variety of programmable SBPs, such as DNA origami, DNA brick, DNA wireframe, SAPs, CAPs, PAs, DNA–peptide hybrid, and glycosylated SAPs [89–91]. A large variety of these SBPs have been realized by combining design software, dedicated computational workflows, and experimental validation [10,59,68,91–93].
As a result, efficient computational methods for accelerating the design process of SBPs became a priority. Modeling and simulation approaches were introduced but balancing computational demands and prediction accuracy was the winning strategy. For example, quantum mechanic (QM) simulations provide results at atomic detail, accurately describing covalent and non-covalent interactions, but are prohibitively expensive in terms of computational cost [94,95].
Molecular dynamic (MD) simulations too, due to their high level of cost/accuracy, can feasibly elucidate non-covalent interactions and structural features of limited-size systems only [78,96–98].
CG-MD simulations played a pivotal role to further reduce the computational cost associated with the simulations of large SBP systems [96,99–104]. For example, several coarse-grained (CG) models were developed for reproducing the fundamental properties of different SBPs (see Table 1): among them, OxDNA and MARTINI force fields deserve a short digression. OxDNA captures nucleic acid structural information more accurately than MARTINI [105,106]: indeed just recently, MARTINI applications have been extended to the simulations of carbohydrates and nucleic acids [107–112]. On the other hand, main applications of MARTINI are traditionally located in the realm of protein and lipid system simulations. OxDNA represents each nucleotide as a single bead that interacts with other ones through potentials implicitly reproducing the effect of the ionic solution [106]. Instead, MARTINI condenses the atomic models of nucleotides and solvents to four grains or heavy atoms (N, C, O, S) [107–112].
Biomolecules vs force fields
| Biomolecules | Atomistic (AA) | CG |
|---|---|---|
| Nucleic acids | AMBER | AWSEM |
| AMOEBA | MARTINI | |
| CHARMM | OPEP | |
| Drude | OxDNA | |
| Gromos | TIS | |
| OPLS | 3SPN | |
| Protein/peptides | AMBER | AWSEM |
| AMOEBA | Bereau and Deresno | |
| CHARMM | CABS | |
| GROMOS | MARTINI | |
| OPLS | OPEP | |
| PaLaCe | ||
| PRIMO | ||
| Rosetta | ||
| Scorpion | ||
| UNRES | ||
| Lipids | AMBER | M3B |
| CHARMM | MARTINI | |
| GLYCAM | ||
| GROMOS | ||
| OPLS | ||
| Carbohydrates | AMBER | MARTINI |
| CHARMM | ||
| GROMOS | ||
| OPLS | ||
| SLIPIDS |
Although these models have the main advantages of describing detailed characteristics, such as the hybridization process, thermal dissociation, and partition coefficient, they implement iterative numerical methods like the classical MDs, simulating large supramolecular structures, which are characterized by wide motion ranges, and requiring a time scale of days/weeks for their production. Combining supramolecular and multi-resolution models was reported as the most promising approach to predict structural features while achieving computational efficiency, i.e., lowering production times to a few hours. For example, DNA nanostructures were approximated as continuous medium or beam-like structures, achieving higher computational efficiency [114]. Nonetheless, such models still rely on the validation through experimental characterization because electrostatic interactions and sequence-dependent mechanical properties are usually ignored [115]. To overcome this problem, Lee and coworkers developed an innovative multiscale analysis framework for fast analysis of engineered DNA assemblies at quasi-atomic resolution [115]. Their framework sequentially integrates the atomistic properties of DNA with a continuum description [116]. All-atom MDs (AA-MDs) simulate the structural motifs constituting the structured DNA and determine the sequence-dependent geometric/mechanical properties of DNA assemblies. Subsequently, these properties and electrostatic interactions between the DNA helices are embedded into a finite element (FE) structural model. As shown in Figure 2a and b, this method predicted the angle and curvature of different DNA origami nanostructures, providing results in good agreement with experimental characterization [116].
![Figure 2
Computational analysis workflow for SBP development. (a) Analysis of bundle DNA nanostructures through AA-MD simulations. Angle control by hinge stiffness or adjuster module length. The designed DNA origami include angles ranging from 15 to 150 and controlled by the stiffness of the hinge module and of the adjuster module length. (b) Analysis of bundles of DNA nanostructures through AA-MD simulation. Curvature control of DNA origami by BP insertion or deletion. For the quarter (Q) and the half (H) circle designs, the radius of curvature and the angle of curvature are quantified by fit to positions in the predicted structures. Figures (a) and (b) have been adapted from Lee et al. [116], with the permission of ACS publications. (c) Workflow (from left to right columns) for the development of hydrogels from branched SAPs mixed with linear (LDLK)3. Branched SAPs have been developed starting from (LDLK)3 backbone moieties, connected with one or multiple “lysine knots.” Branched SAPs were created by increasing the number of (LDLK)3-branches and/or by adding the BMHP1 neuro-regenerative functional motif as a single branch. CG-MD simulations unveiled the conformations of branched SAPs within (LDLK)3 self-assembled nanostructures. Then, SAPs were classified according to their contribution to the stability of fibril nanostructures by calculating their degree of integration. Lastly (right column), SAP hydrogels were synthesized and characterized via AFM imaging.](/document/doi/10.1515/ntrev-2022-0455/asset/graphic/j_ntrev-2022-0455_fig_002.jpg)
Computational analysis workflow for SBP development. (a) Analysis of bundle DNA nanostructures through AA-MD simulations. Angle control by hinge stiffness or adjuster module length. The designed DNA origami include angles ranging from 15 to 150 and controlled by the stiffness of the hinge module and of the adjuster module length. (b) Analysis of bundles of DNA nanostructures through AA-MD simulation. Curvature control of DNA origami by BP insertion or deletion. For the quarter (Q) and the half (H) circle designs, the radius of curvature and the angle of curvature are quantified by fit to positions in the predicted structures. Figures (a) and (b) have been adapted from Lee et al. [116], with the permission of ACS publications. (c) Workflow (from left to right columns) for the development of hydrogels from branched SAPs mixed with linear (LDLK)3. Branched SAPs have been developed starting from (LDLK)3 backbone moieties, connected with one or multiple “lysine knots.” Branched SAPs were created by increasing the number of (LDLK)3-branches and/or by adding the BMHP1 neuro-regenerative functional motif as a single branch. CG-MD simulations unveiled the conformations of branched SAPs within (LDLK)3 self-assembled nanostructures. Then, SAPs were classified according to their contribution to the stability of fibril nanostructures by calculating their degree of integration. Lastly (right column), SAP hydrogels were synthesized and characterized via AFM imaging.
Differently from DNA assemblies, a complete computational analysis workflow for SAP hydrogels is still missing. CG MARTINI models found large applications in determining structural and mechanical features of peptide assemblies and eventually validated through experimental data [108,113,117–120]. For example, MARTINI CG-MD simulations, combined with customized software analysis tools, have been used to high-light structural features of high-performance SAP hydrogels composed of a mixture of branched and linear SAPs (see Figure 2c) [91]. In another work, atomistic steered MD (SMD) and GoMARTINI CG-SMD were used to derive the elastic and shear moduli of SAP fibrils [120,121]. The comparison between the abovementioned SMD approaches suggested that GoMARTINI CG-SMD simulations provide comparable results to atomistic SMD simulations.
Although these results highlight a clear connection between atomistic features of SAPs and their nanomechanical properties, a demonstration of how they reverberate at the macroscale on SAPs’ mechanical properties is still missing.
Other research groups used a multiscale model to elucidate SAPs’ hierarchical organization [122–124]. Indeed, Liu et al. employed a bottom-up multiscale theoretical model to analyze co-assembling complementary di-peptides systems. Their model combined density functional theory (DFT), AA-MD, CG-MD, and dissipative particle dynamic (DPD) simulations. The morphologies obtained from DPD simulations were in good agreement with scanning electron microscopy (SEM) and transmission electron microscopy measurements. Their approach represents a promising strategy to study the mechanism of spontaneous hierarchical self-assembly, fostering a better rational design of peptide nanostructures [124].
In a recent work, Zhao and coworkers introduced another intriguing strategy for elucidating structural features of peptide nanostructures [125]: MARTINI CG simulations unveiled the early stages of self-assembling while AA-MD and mixed resolution models simulated the assembly and reorganization of peptide fibrils [125]. Nonetheless, it must be kept in mind the limited reliability of MARTINI CG-MD simulations in predicting the hydrogen bond dynamics that play a pivotal role in the first stage of peptide self-assembly [120,121,126].
1.4 The multiscale paradigm in material science and TE
SBPs’ high versatility makes them suited for different purposes. This is mainly due the tailorability of their properties capable of matching various requirements in terms of (1) molecular architectures and nanostructuring propensities; (2) mesoscale topological arrangements; and (3) mechanical properties. Such features can be investigated in silico and potentially validated via dedicated experimental characterization tests.
The multiscale approach (see Figure 3) has been widely used for elucidating the hierarchical organization and mechanical properties of animal-derived materials, such as collagen or spider silk [118,125,126]. This approach relies on the combination of multidisciplinary theories, multiscale simulation methods, and multiscale experiments. The molecular interactions across many hierarchical scales heavily affect the mechanical behavior of biological materials. At the microscale and nanoscale, SBPs feature molecular unfolding or sliding when subjected to mechanical stresses, while at larger length scales, where the interactions with biological structures become more evident, more complex mechanisms contribute to SBPs’ biomechanics. As an example, the multiscale analysis framework of SAP hydrogels is depicted in Figure 3. At the nanoscale, recent progress in solid-state nuclear magnetic resonance (ssNMR) technique allows retrieving information about molecular movements at natural isotope abundance, such as MD restraints needed to run atomistic MD simulations [104,121].
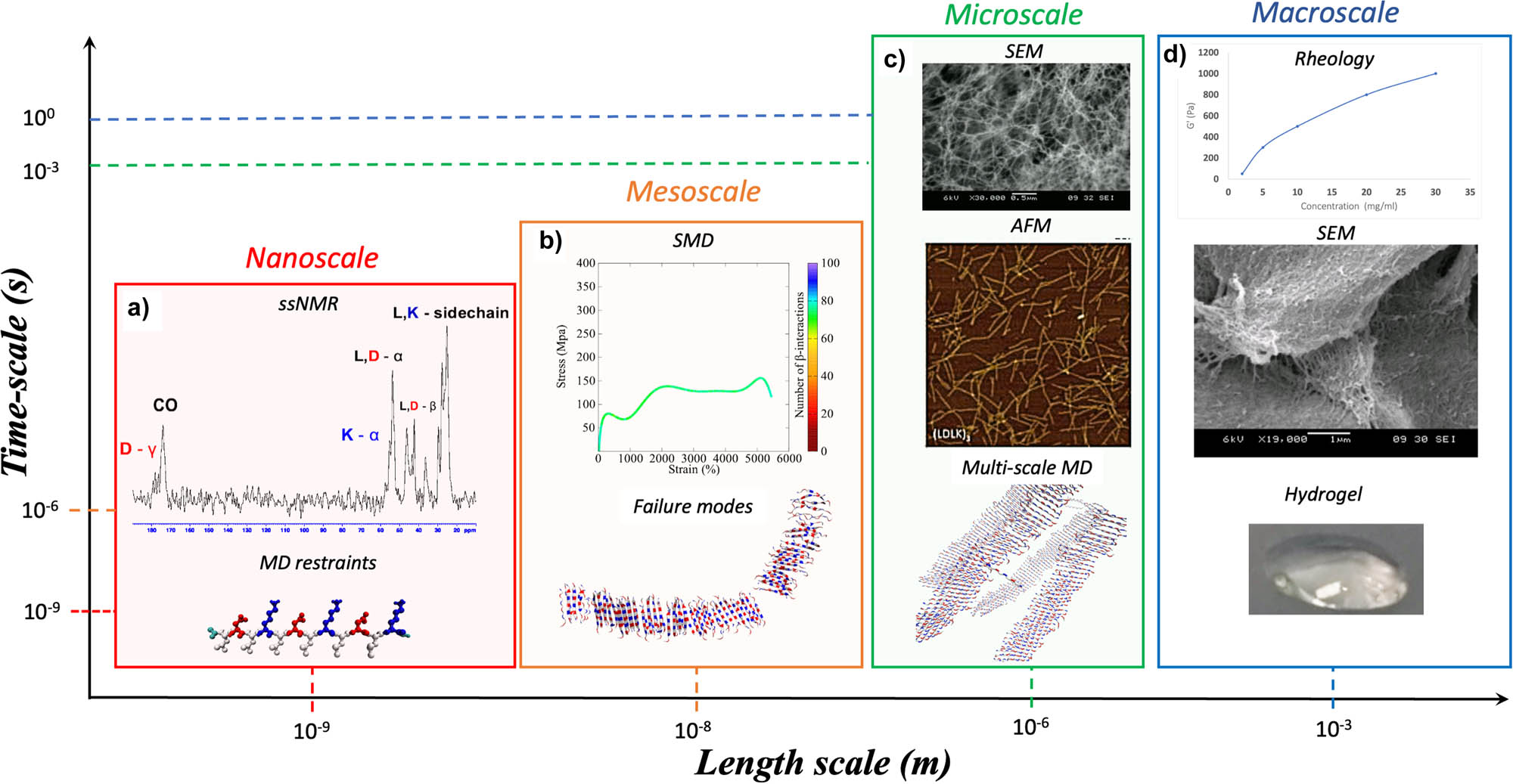
SBP multiscale modeling. A full understanding of the hierarchical organization properties of biomaterials requires an ensemble of computational and experimental techniques. Computational approaches, supported and validated by experimental evidence, can be used to transverse through a wide range of length and time scales. As an example, here is described the multiscale analysis framework for SAP hydrogels. (a) ssNMR characterization can be used to derive the MD restraints for running atomistic MD simulations. The results from MD simulations can be used for validating atomistic or CG model of SAP fibril and fibril seeds. (b) SMD simulations of atomistic and CG models are useful to derive mechanical properties (e.g., Young and Bending moduli) of fibril and fibril seeds. (c) Then, multiscale MDs can be used to predict the supramolecular arrangement of SAP fibrils at the mesoscale. Such results can be validated and directly compared with mesoscale experimental analysis, such as AFM and SEM. (d) Lastly, these data integrated in dedicated workflows, also encompassing FEM analysis, could be used for corroborating macroscale analyses, such as rheology measurements.
The structures of SAP fibril seeds obtained from restrained MD simulations can be used to run MARTINI CG-MD simulations at the mesoscale level [105,121], such as SMD simulations to extract mechanical properties. Such approach allows to derive the Young and Bending moduli mechanical properties of fibril seeds (nucleation seeds) [121,127]. CG-MD simulation has been used for the characterization of SAPs at meso- and microscale levels.
Indeed, the supramolecular arrangement of SAP fibrils can be predicted by combining atomistic and validated and directly compared with nano- and microscale characterization techniques, such as atomic force microscope (AFM) and SEM [104]. On the other hand, DPD simulations and FE analyses are likely the most promising candidates enabling quantitative validations of macroscale simulations via mechanical tests [128].
Despite significant advances in biomaterial informatics, thanks to the application of machine learning and artificial intelligence (AI) algorithms, a multiscale description of SBPs is still missing [129,130]. Just recently, voxel-based FE and micro-finite element (μFE) methods were employed for the simulation of non-linear deformations of bone tissues [131,132] that are crucial phenomena to be considered for the prediction of biocompatibility and stability of implanted prosthetic devices [132].
To this purpose, non-linear FE analysis will likely play a pivotal role in the design and development of innovative SBPs for bone TE applications. In a recent critical review of Roy and Chatterjee, the deformations of bacteria nanostructures are described as key players for the biocompatibility of mechanobactericidal biomaterials [133].
It is author’s opinion that the combination of CG-MD simulations and FE analysis of cell membrane structures and biomaterials will be a promising strategy to tackle the challenge of predicting biomaterials’ biocompatibility and pro-regenerative potential.
1.5 Summary of the review
The remaining chapters of this review are structured as follows and dedicated to the main methods shown in Table 2. In Sections 2.1.1 and 2.1.2, the molecular mechanics’ (MM) methods for simulating biopolymer’s building blocks are introduced. In Section 2.1.3, the authors point out the potential applications of such methods in drug discovery. In Section 3, the attention moves toward density functional theory for predicting SBPs’ electronic structures. Section 4 introduces the classification of different CG (Section 4.1) strategies and their potential impact in multiscale modeling (Section 4.2). Section 5 addresses the duality of the problem of thermodynamic-mechanical stability of SBPs. Section 5.1 introduces the role of SMD for calculating the mechanical properties of SBPs. Section 5.2 describes MD for studying the thermodynamic stability of SBPs. In Section 6, the authors showcase the potential beneficial impact of combining the different modeling methods. Section 6.1 highlights the potential applications of parallel and serial multiscale simulations for studying the properties of SBPs. Section 6.2 addresses the ultimate challenge of reactive MD simulations for predicting cross-linking reactions products. Section 6.3 points out the lack of a connection between the microscale and macroscale simulation methods. Lastly, Section 6.4 reports a few examples of multiscale MD used for the simulation of organelles and cells.
Main MD simulation methods described in this review
| Methods | Description | Advantages | Limitations |
|---|---|---|---|
| AA-MD | MD approach in which each atom is explicitly considered | Temporal and spatial resolution of position of all atoms | High computational cost required for the simulation of systems of biological interest |
| CG-MD | MD approach in which each interaction sites correspond to multiple atoms | Simulation of larger molecular systems compared to those simulated in AA-MD approaches | Limited monitoring of non-covalent interactions interplay, such as H-bond formation and breakage |
| DFT | Quantum mechanical method used to investigate the electronic structure of atoms, molecules and condensed phase | High accuracy in prediction of electronic configuration of molecular systems | Many approximations and limits for strongly correlated systems. Low computational efficiency for liquids |
| DPD | Stochastic simulation method for simulating the properties of complex fluids. Applied in rheology and fluid dynamics | Simulation of the behavior of molecular systems on longer time and length scales | Simulation of mesoscale systems, not appliable to small size systems |
| RMD | MD simulations of the formation of covalent bonds, enabling the description of chemical reactions | Simulation of molecular systems involving formation or breakage of covalent bonds | High computational cost |
2 MM for biopolymers development
2.1 Modeling self-assembling biopolymers building blocks: proteins, carbohydrates, lipids, and nucleic acids
2.1.1 Atomistic MD simulations of polymeric building blocks
MD approaches are largely used for the investigation of supramolecular systems, due to the high accuracy in the prediction of exact conformation of molecular building blocks. Indeed, AA-MD allow to monitor H-bonding as well as aromatic and charge–charge interactions by representing them using Coulomb and Lennard-Jones potential [134–136].
Historically, proteins were the first simulated biomolecular class. The route for the simulations of proteins lays its foundation in 1977 with the study of bovine pancreatic trypsin inhibitor [137,138]. Such work demonstrated that AA-MD simulations are suitable tools for the description and understanding of the protein folding mechanisms. Nowadays, AA-MD simulations allow the investigation of complex phenomena inherent to protein and peptide folding, for example, allosteric regulation of biochemical reactions [139,140]. AA-MD applications also encompass the study of protein/DNA interactions shedding new light on genomic regulation mechanisms [141].
As shown in Table 1, different types of force fields have been developed for the simulation of proteins. The AMBER force field family, commonly associated with the AMBER software package, was extensively used in the study of SAPs and intrinsically disordered proteins (IDPs) [142]. Indeed, amber AA-MD simulations allowed to elucidate the mechanism underlying the self-assembly of ß-sheet forming SAPs, which mimic ß-barrel proteins [143]. Besides, thanks to amber AA-MD simulations, a system for the controlled drug release of glucagon-like peptides was developed, setting up a milestone in the treatment of type 2 diabetes [144]. Amber AA-MD simulations were also employed for the investigation of halogenated amyloidogenic peptides to expand the applications of halogen bond chemistry [145–147].
Further applications of amber AA-MD simulations, relying on QM and machine learning approaches, consisted in the molecular design and elucidation of π-conjugated oligopeptides structural properties [148,149]. Subsequently, AMBER was upgraded with the introduction of novel parameters for the simulation of carbohydrates, nucleic acid, and lipids. In particular, the new AMBER parameters for carbohydrates laid the foundation of GLYCAM [136], opening new opportunities for the investigation of protein–carbohydrate interactions [150].
AMBER was exploited for the simulation of glycosylated protein systems in various fields, such as cancer treatment as well as in computational virology [151]. In other works, AMBER simulations were used for nucleic acid structural characterization: for example, they allowed to elucidate structural transitions among A-DNA, B-DNA, and Z-DNA [152–154].
AMBER parameters have also been optimized for the simulation of protein-lipids systems [155–157] and even for larger lipid membrane systems [156,157]: hence, AMBER simulations were used to elucidate the molecular mechanisms of membrane proteins, a prominent target of many pharmacological therapies.
On the other hand, OPLS force field, initially developed for the simulations of small organic molecules and peptides, features a strong influence from AMBER. Indeed, some parameters of OPLS force field have been retrieved from AMBER simulations and were used to reproduce the gas-phase structures and thermodynamic properties of organic liquids [158,159], as well as for the simulations of DNA base pairs [160]. Nonetheless, published works comparing AMBER, CHARMM, OPLS, and GROMOS MD simulations unveiled that OPLS underestimates the H-bond strengths between the complementary bases of B-DNA and poorly reproduces properties heavily dependent on torsional energetics [161,162]. After additional optimizations of OPLS-AA parameters, consisting of better fitted QM torsional energetics of dipeptides [168], OPLS has been successfully used for AA-MD simulations of SAPs, even crosslinked, in explicit solvent [158,159]. Recently, the parameters for MD simulations of RNA molecules have been introduced in OPLS: AA-MD simulations of dinucleotides/tetranucleotides revealed good accuracy in reproducing 3J couplings found in ssNMR studies without the onset of several unphysical states observed with CHARMM and AMBER simulations. OPLS AA-MD correctly quantified the interactions among hydrogen bonds belonging to different atomic groups [158,163,164].
CHARMM is a force field suitable for supramolecular biochemistry studies: it has been largely used for simulating systems comprising SAPs, lipids, and nucleic acids. SAP fibrils and the influence of functional motif on SAPs’ self-assembling propensity was investigated with CHARMM MD simulations [165,166]. Thanks to the last CHARMM update, it became possible to simulate small drug-like molecules and other organic moieties (e.g., Fmoc moiety) currently unrepresented in AA-MD simulations [167].
These advances enabled the applications of the CHARMM force field for comprehensive characterizations of diverse supramolecular systems [165,167]. GROMOS denotes another family of force fields initially developed for performing MD simulations through its bundled software package. The first version of GROMOS, released in 1987, was developed as a united-atom force field to cope with the limited computational resources. In this way, the heavy atoms (carbon) and the attached hydrogen were represented as single-interaction sites.
Since then, GROMOS force field has been repeatedly improved and refined. In 2004, the GROMOS force field has been reparametrized with the 53A5 and 53A6 parameter sets and optimized by fitting the solvation-free energies of small polar molecules [168–170].
In the validation of the 53A6 parameter set, the simulated structures of protein (egg-white lysozyme), peptides (ß3-dodecapeptide), and DNA dodecamer were in good agreement with ssNMR data [171]. Still, helical folding of different proteins did not comply with experimental characterization. The new 54A7 parameter set was introduced for protein structures and further extended to peptides [169,172]. Lastly, the introduction of the 53A6GLY C parameter set overcame the main limitations of the 53A6 and 54A7 and laid the foundation of realistic simulations of carbohydrates and nucleic acids [168]. Additional parameters were introduced for the simulations of halogen bonds and furanose-based carbohydrates [147,173] to allow the investigation of glycosylated SBPs and halogenated SBPs, such as SAPs [145,146].
Large efforts were devoted to improving atomistic force fields based on fixed-charged models, such as AMBER, CHARMM, and GROMOS; however, QM calculations have demonstrated that fixed-charged models are not appropriate to represent ionic interactions, hydrogen bonding, and base stacking [162]. To overcome these drawbacks, polarizable force field models, such as CHARMM Drude and AMOEBA (atomic multiple optimized energetics for biomolecular applications), have been developed. Both CHARMM Drude and AMOEBA exhibit significant advances over fixed-charged models in the simulations of nucleic acid, protein, and organic molecules [174–176]. The parameters of AMOEBA force field have been recently validated for nucleic acid simulation by investigating the stability of the native structures and comparing their properties with experimental results (3J coupling, nuclear Overhauser effect) [174,175].
3 CG-MD simulations of biopolymers building blocks
CG force field has proven to be useful for the investigation of systems of biological relevance, such as soft matter systems. The main advantage of CG modeling relies on the reduction of computational cost associated with the MD simulations iterative workflow. CG force fields map diverse atom groups and monomers (nucleotides/amino acids) as single interaction sites. However, the main drawback of CG force fields is represented by the limited compatibility with different biomolecular classes: most CG models were developed to reproduce specific molecular features, such as partition coefficient or enthalpy of solvation.
In details, the parameter sets of CG force fields can be derived to reproduce microscopic properties from fine-grained simulations (bottom-up approach) or macroscopic thermodynamic quantities (top-down approach). The validation of a new CG model requires the assessment of the ability of the model to predict of the system properties at the thermodynamic state point used during its development (representability) and at different state points (transferability).
A multitude of CG force fields has been developed to simulate different biomolecular systems (see Table 1). Each CG force field relies on different mapping strategies and works accordingly to its limited assumptions: thus, each CG force field is suitable for limited applications.
The AWSEM (associative memory, water mediated, structure, and energy model) is a CG force field suitable for the investigation of protein folding mechanisms [190]. Indeed, the AWSEM parameter sets were derived through bioinformatics approaches, which consider the many-body effects modulated by the primary sequences of proteins. Besides, the AWSEM parameter set encompasses physically derived terms, like hydrogen bonds and electrostatic interactions [177]. AWSEM force field has been successfully adopted to demonstrate that the mechanisms underlying the protein dimerization process is driven by heterogeneity and flexibility of monomers [178]. AWSEM CG-MD simulations were also employed for elucidating genetic switches found in both transcription factors and DNA: de facto obsoleting the classic interpretation of the “binding and release process” involved in genetic regulation and introducing the so-called “molecular stripping” concept [177]. Furthermore, the server tool, dubbed AWSEM-Suite, was adopted for predicting monomer structures when a suitable structure template is not available [177]. It was also possible to simulate protein–DNA systems, within a reasonable amount of clock time, by combining AWSEM and 3SPN, a supramolecular force field for DNA molecules [143].
Similarly, to the AWSEM model, OPEP (optimized potential for efficient protein structure prediction) was developed through bioinformatics approaches and validated through AMBER MD simulations [179,180]. In the OPEP model, each amino acid is represented by six beads: five for the backbone and one for the sidechain [179,180]: OPEP model was used to study protein folding and amyloid fibrils formation and to model protein, DNA, and RNA complexes [179].
The CG Rosetta model is one of the leading approaches used for ab initio (from first principles, i.e., the pursue of Schrodinger equations solution from the positions of nuclei and electronic densities) structure predictions.
The Rosetta model represents each protein structure with the distribution of the center of masses of its own backbone. Recently, the Rosetta model was upgraded to enable protein–protein docking, protein–ligand docking, and modeling of protein–DNA interactions [131,132].
The elucidation of the mechanisms underlying protein–protein interactions represents the last frontier in systems biology. To this purpose, Bereau and Deresno developed a model where each amino acid is mapped using three-to-four beads: (1) one bead for the amide group (N), (2) another one for the central carbon (Cα), (3) a third one for the carbonyl group (C′), and (4) eventually, a side chain bead (Cß) [181,182]. Analogously to AWSEM model, the Bereau and Deresno CG-MD simulations demonstrated that peptide folding is competitive with peptide aggregation [182].
Similarly, to the Bereau and Deresno model, in the CABS (C-Alpha, C-Beta, Sidechain) model, each amino acid is mapped using three-to-four beads [183,184]. CABS CG-MD simulations were used for the modeling of loop formation [185,186], protein folding [185,187], and the binding of IDPs [184]. In particular, the multiscale framework, obtained from the combination of AMBER and CABS MD simulations, has been used for elucidating the folding pathway of the B1-binding domain of G protein [187].
In the Palace (Pasi–Levery–Ceres) CG model, amino acids are mapped using three-to-five beads [128]. The Palace CG model found applications in analyses of the mechanical properties and conformational flexibility of different proteins, giving better results than traditional elastic network models [109,128].
The PRIMO model makes use of more accurate protein representations than those of previously mentioned force fields: each amino acid is represented with five-to-seven interaction sites. Nonetheless, due to its higher computational costs, the PRIMO model was only employed for the prediction of small protein structures [129,130]. Lastly, SCORPION (solvated CG protein interactions) and UNRES (united residue) models represent each amino acid with three beads: one for the backbone and two for the sidechains: they have been used for the investigation of protein–protein interactions, considering their solvation conditions [133,134].
Nucleic acid structuring attracted great interest for a better understanding the genomic regulations, but nowadays CG-MD simulations are also employed for elucidating structural and mechanical properties of nucleic acid-based hydrogels. For example, OxDNA force field has been widely used for their mechanical characterization. The original OxDNA parameters sets were derived through a “bottom-up” approach: indeed, each DNA base pair was represented by two beads of the same type with potentials derived from AA-MD simulations [106,107]. In 2015, an improved version of the OxDNA force field, dubbed OxDNA2 [106,107,139], showed a clear signature of twist-bend coupling, still absent in OxDNA, in good agreement with experimental characterization [107,139].
Recently, the OxDNA2 force field was used for the simulation of DNA nanodevices, such as DNA origami [116]. OxRNA model was also adopted to elucidate the complex phenomenon involved in toehold RNA displacement [188] and enabled the development of autonomous DNA motors [189]. The TIS (three interaction sites) model, where nucleotides are mapped by TIS (a phosphate, sugar, and base), has also been used to investigate the mechanical unfolding of single-stranded RNA hairpins and ribozyme folding [141,190], giving results in agreement with empirical data.
While all CG models introduced so far have been designed to simulate a limited number of biomolecules, the MARTINI force field represents the most flexible and used CG model nowadays (see Table 1).
Indeed, MARTINI features good properties of representability and transferability [100,108], enabling the simulations of several biomolecular classes, such as DNA, proteins, lipids, and carbohydrates [110,112,113,191]. MARTINI MD simulations demonstrated the self-assembly pathways and structuring propensities of several SAPs at both nano- and mesoscale. In the last years, MARTINI CG-MD simulations were also used for the elucidation of emerging fibrillar networks of bioconjugated SAPs currently used in TE applications [68,91]. Lastly, being carbohydrates crucially involved in protein folding, stability, cellular localization, and modulation of immune reactions, the M3B CG model was specifically designed for the simulation of malto-oligosaccharides and their aqueous mixtures [192]. Its mapping strategy was used for the extension of MARTINI to the modeling of carbohydrates and glycans [112,193].
3.1 Modeling of cellular membranes and their interactions with biomaterials/pharmaceutical compounds
Cellular membranes are complex assemblies of lipids, proteins that separate intracellular from the extracellular environment and are also involved in signal transduction, cellular homeostasis, and recognition. Improved experimental techniques (e.g., nuclear magnetic resonance, single-particle tracking, mass spectroscopy) give more and more insights into the organization of the cell membranes [232,233]. Also, synthetic biology methods, making use of synthetic membrane-like structures, have been used for the investigation of cell membranes [194–197]. However, detailed membranes organization is still far from being fully described. To this purpose, MD simulation techniques became precious tools for describing the interactions among all the components of the membrane systems. Thanks to the steadfast improvements in force field parametrization and computer performances, membrane systems became a target at hand of MD simulations. While different force fields were developed to allow for reliable simulations of lipids, just a limited number was successfully adopted for the simulation of lipid membrane systems: in case of atomistic force fields, CHARMM played a major role (see Table 1).
CHARMM represents the most elaborated force field for lipids, encompassing the main families of lipids found in higher organisms and bacteria [155–157]. The repository of lipid parameters is CHARMM-GUI, an online web server suitable for building realistic cell membranes. Slipids is another promising force field parametrized to be consistent with AMBER for simulating lipidic systems [166,198–200]. Despite these efforts, atomistic force fields allow the investigation of limited size systems and show limited reliability in the simulation of complex membranes featuring embedded proteins. To overcome these limitations, several CG models have been developed and tested.
As shown in Figure 4a, the MARTINI CG-MD simulations were reported to be suitable for the simulations of the cell membranes. The proposed model consists of 63 different lipid species asymmetrically distributed across two bilayers. MARTINI CG-MD simulations elucidated the “lipids domain” dynamics [201] and unveiled an enrichment of the cholesterol in the outer layer contemporaneous with its depletion in the inner one [202]. These simulations unveiled a complex interplay among fully saturated (FS) and polyunsaturated (PU) lipids that was detected in simulations of neural plasma membrane as well [201,203]: again, the concentration of FS lipids increased in the outer leaflet and decreased in the inner leaflet while PU lipids showed an opposite tendency [202,203]. These CG-MD simulations enabled the investigation of complex cell membranes embedding different protein structures [201,203].
![Figure 4
Realistic cellular membrane modeling. (a) Modeling of an idealized plasma membrane. The model of plasma membrane comprises 63 different lipid types, including cholesterol, phosphatidylcholines (PC), sphingomyelins (SM), phosphatidylethanolamines (PE), gangliosides (GM), phosphatidylserines (PS), and phosphatidylinositol phosphates (PIPs). The overall headgroups composition and number of unsaturated bonds in the lipid tails are shown for the outer and inner leaflet (pie charts), together with snapshots of both leaflets (full leaflets with a zoom-in underneath) after 80 μs of simulation. The lipid headgroups and tails are depicted with the same colors in the pie charts. This figure has been adapted from Figure 2 of Ingólfsson et al. [203], under the license number 5380391174086 provided by Elsevier. (b) Cross-sectional view of local lipid environment around AQP1. The simulation setup consists of a plasma membrane with four embedded membrane proteins. The two-dimensional lateral density maps show local fluctuations around AQP1 in upper leaflet (top row) and lower leaflet (bottom row) of PU lipids, FS lipids and cholesterol (CHOL). Major variations are pointed by arrows: I, nonspecific binding; II, nonuniform distribution; III, leaflet asymmetry; IV, specific binding; V, membrane fluctuations. This figure has been adapted from Figures 1a and b and 2a of Corradi et al. [201]. (c) Unbiased MD simulations can be employed for the investigation of interactions among amphiphiles anticancer drugs and phospholipid membranes, resembling the chemical physical features of cellular membrane. This figure has been adapted from Figure 1c of Tang et al. [207].](/document/doi/10.1515/ntrev-2022-0455/asset/graphic/j_ntrev-2022-0455_fig_004.jpg)
Realistic cellular membrane modeling. (a) Modeling of an idealized plasma membrane. The model of plasma membrane comprises 63 different lipid types, including cholesterol, phosphatidylcholines (PC), sphingomyelins (SM), phosphatidylethanolamines (PE), gangliosides (GM), phosphatidylserines (PS), and phosphatidylinositol phosphates (PIPs). The overall headgroups composition and number of unsaturated bonds in the lipid tails are shown for the outer and inner leaflet (pie charts), together with snapshots of both leaflets (full leaflets with a zoom-in underneath) after 80 μs of simulation. The lipid headgroups and tails are depicted with the same colors in the pie charts. This figure has been adapted from Figure 2 of Ingólfsson et al. [203], under the license number 5380391174086 provided by Elsevier. (b) Cross-sectional view of local lipid environment around AQP1. The simulation setup consists of a plasma membrane with four embedded membrane proteins. The two-dimensional lateral density maps show local fluctuations around AQP1 in upper leaflet (top row) and lower leaflet (bottom row) of PU lipids, FS lipids and cholesterol (CHOL). Major variations are pointed by arrows: I, nonspecific binding; II, nonuniform distribution; III, leaflet asymmetry; IV, specific binding; V, membrane fluctuations. This figure has been adapted from Figures 1a and b and 2a of Corradi et al. [201]. (c) Unbiased MD simulations can be employed for the investigation of interactions among amphiphiles anticancer drugs and phospholipid membranes, resembling the chemical physical features of cellular membrane. This figure has been adapted from Figure 1c of Tang et al. [207].
Corradi and coworkers used MARTINI CG-MD simulations for the characterization of the lipid environment of 10 different membrane proteins [201]. Figure 4b shows the simulation setup consisting of a plasma membrane embedding four aquaporins (AQP1): a two-dimensional lateral density analysis revealed domain fluctuations of the lipids surrounding the proteins [201]. The FS enrichment was observed in the outer leaflet near the AQP1, while PU concentration increased in the inner leaflet close to AQP1. Besides, a series of specific interactions has been detected in both leaflets, between CHOL and AQP1 [201]. These tendencies have been also observed for other proteins, such as prostaglandin H2 synthase (COX1), dopamine transporter (DAT), epidermal growth factor receptor (EGFR), AMPA-sensitive glutamate receptor (GluA2), glucose transporter (GLUT1), voltage-dependent Shaker potassium channel 1.2 (Kv1.2), sodium, potassium pump (Na, K-ATPase), δ-opioid receptor (δ-OPR), and P-glycoprotein (P-gp) [201]. This inspiring work gave new insights on lipid-mediated interactions by showing how different sets of lipids and bilayers can mediate cellular recognition via specific interactions with proteins [201,203].
Intriguingly, non-uniform lipid mixing and lipid segregation can also modulate membrane properties and heavily influence membrane permeability, cell aggregation, and trafficking [204–206]. As shown in Figure 3c, in MARTINI CG-MD simulations, we detected potential interactions among self-assembling amphiphilic drugs and phospholipid membranes. Results suggest that the shape of drug delivery vehicles can have a tremendous impact over bioavailability of the carried drugs, then affecting the overall therapeutical treatment efficacy [207].
4 QM approaches for biopolymers: density functional theory
Non-covalent interactions, such as hydrogen bonding, metal coordination, hydrophobic and Van der Waals forces, π–π, electrostatic interactions, and rule molecular self-assembly. Therefore, an accurate modeling of such interactions is crucial for the development of reliable tools predicting the stability of supramolecular complexes. To this purpose, QM approaches were largely adopted for the accurate descriptions of both covalent than non-covalent interactions in molecular systems [94,96,208,209].
Among ab initio QM methods, DFT, making use of electronic functional of electronic densities instead of the exact form of the electronic distributions, attracted increasing attention in supramolecular biomaterials modeling. Indeed, DFT, used for the prediction of the electronic structure of DNA base pairs and amino acids [210], reduces the computational costs related to the calculation of empirical potentials. Being difficult to determine the empirical potentials of the various energy contributions of H-bonding and/or their electronic distributions, DFT is a good compromise, balancing computational costs and accuracy, to investigate small atom clusters resembling the main features of biopolymers [210].
Further applications of DFT consisted in the elucidation of the adsorption mechanism of biomolecules on different material surfaces, such as graphene, metal oxides, or hydroxyapatite [96,208,209,211,212].
DFT molecular simulations demonstrated that interactions among amino acids and graphene nanostructures are mainly driven by Van der Waals interactions [208], while their aggregation state on graphene nanostructures is mainly stabilized by hydrogen bonding [96,208]. In addition, DFT simulations demonstrated that the electric properties of graphene are affected by the interactions with amino acids. These results enabled the development of different nanotechnological applications, exploiting the peculiar properties of graphene in biosensors, storage devices, nanoporous membranes, and biomolecule delivery devices [208,213].
DFT approaches have also been used for the investigation of binding mechanisms of several ligands in ß-amyloid structures involved in Alzheimer’s disease [95,214]. DFT simulations demonstrated that interactions among metal ions and amyloid-like peptides are fluxional, with most of the cationic groups mainly interacting with metal ions. This prodromic work laid the foundation for MD simulation analyses of conformational flexibility of Aß peptides [95].
Interestingly, the similarity of SAPs’ supra-molecular structures resembling the organization of amyloid fibrils encouraged the application of DFT approaches to the prediction of structural and vibrational properties of SAP aggregates [78,215]. DFT methods allowed to predict IR spectra of SAPs and demonstrated that such spectra can be altered by the presence of functional groups, such as Fmoc [215].
Ab initio DFT methods unveiled the effects of chirality on the self-assembly of l-diphenylalanine and d-diphenylalanine peptides. While both types of diphenylalanine peptides self-assemble into nanotube-like structures, the effect of chirality plays a pivotal role in the self-assembling process. D-FF peptides form thicker and shorter peptide nanotubes than those detected on L-FF. Also, in terms of nanotubes’ topology, L-FF form helical turns, while D-FF form rings [79]. These results demonstrated that DFT calculations, combined with infrared spectroscopy, can be used for the characterization of SAPs largely used in TE [216–218].
Other applications of DFT approaches comprise simulations of carbohydrates and nucleic acid structures to develop different nanotechnological applications, such as nanodots and quantum dots [111,219–224].
Applications of DFT methods in lipid simulations are limited as they have been used mainly for the improvement of CG-MD simulations [225,226].
Nonetheless, the plethora of applications of DFT for SBP development is expanding rapidly: e.g., DFT was used to calculate the binding energy of cross-linked supramolecular binders for the improvement of lithium batteries capacity [227]. DFT simulations also unveiled that glycine could affect the ß-structuring propensity of SAP nanofibrils [228]. Lastly, DFT allowed to elucidate the role of aryl amino acids (such as phenylalanine or tyrosine) in the self-aggregation of different structures, ranging from crystalline to organogels [229].
The development of conductive SBPs represents the last frontier in TE. Conductive SBPs have been developed to comply with electrical properties of muscle, bone, and nervous tissues. A limited number of SBPs have been used for the development of conductive 3D-scaffolds (e.g., silk fibroin, chitosan, peptides, nanofibers, and DNA nanowires) [230–233], while other biopolymers (e.g., alginate, agarose, lipid-based, and carbohydrate-based gels), usually characterized by low conductivity, need supplements of metals or conductive polymers to match the good conductivity properties of the target tissues [231,232]. DFT simulations played a pivotal role in calculating the conductivity properties of bioinorganic SBP hybrids. DFT simulations were used for building a graphene-based biosensor for tyrosine: DFT simulations demonstrated the high reactivity of graphene toward tyrosine and how it influences its electronic properties. Authors demonstrated that graphene, shaped as nanoribbons, can be a promising candidate for the detection of amino acids and a novel biosensor in different biomedical applications [208,213]. In another work, DFT simulations quantified graphene quantum dots’ (GQDs) adsorption on DNA fragments, demonstrating the low genotoxicity of GQDs, potentially suited for biomedical applications [224].
5 CG modeling of supra-molecular biopolymers
5.1 The phylogeny of CG approaches
CG strategies have been largely used for the simulations of biomolecular building blocks: they were proven to be suitable for the prediction of mesoscopic phenomena and to provide precious insights for empirical experimental tests [234].
CG approaches for SBPs can be categorized in three classes: structure-based, knowledge-based, and dynamic-based approaches.
Structure-based approaches make use of atomic-scale data of molecular structures to derive the respective CG models [235–237]. MARTINI CG force field represents the most used structure-based approach where each CG interaction site represents a different residue or functional group [106,108,113].
Two approaches are currently used to describe the CG interactions in structure-based models: (1) a model predicting minimal energy configurations (i.e., the ground state) of the reference structures, to be used for the parametrization, and (2) a model where native contacts are represented using attractive non-bonded interactions, while other interactions are assumed to be purely repulsive. This method, known as Go-Model, has been recently combined with network models to investigate large amplitude protein conformational transitions [238].
Knowledge-based approaches leverage on the growing collection of solved experimental structures (or measured macroscopic properties) of different macromolecules and their conformers. These approaches aim to design CG models with high degree of transferability and chemical specificity. A major example of knowledge-based models is represented by the critical assessment of structure prediction experiments for protein structure prediction and homology modeling [239], where training sets are composed by related proteins. Other knowledge-based approaches deploy Bayesian inference techniques for the calibration of the optimal force field for CG-MD simulations [240,241].
Dynamic-based approaches use systematic algorithms to derive CG parameters from atomistic simulations. Despite the clear connection between atomistic and CG scales, these methods do not recap in CG models’ some phenomena strictly linked to the atomistic scale, such as hydrophobicity. Nonetheless, these approaches were used to parametrize molecular interactions as elastic networks (REACH) and ultra-CG (UCG) models [242–244].
Lyman and coworkers employed the dynamic-based approach to parametrize a heterogeneous elastic network model for proteins, laying the foundation for its potential applications at any level of coarse graining. Such approach was used to extrapolate the CG potentials for the REACH force field, designed for investigating the dynamics of dimeric protein interactions [244].
Dama and coworkers recently defined the theoretical bases of UCG modeling: they introduced the UCG-fitting procedures from atomistic-scale data [245] and they introduced the UCG rapid local equilibrium simulations, an UCG force field method suitable for the simulations of several related molecules [245]. UCG approaches have also been adopted and improved by Zhang’s group: authors proposed an intriguing advance in UCG simulations by using the fluctuation matching method for building a stable UCG model with a Go-like potential particularly suited for the simulations of SBPs. The same group introduced another advance: the double-well UCG model, which was used to describe the conformational transitions of different proteins, such as adenylated kinase, glutamine-binding protein, and lactoferrin [246].
5.2 Challenging the multiscale paradigm: limits and assets
The development of multi-resolution modeling approaches marks another important step toward the multiscale simulation framework for SBPs. As discussed in Section 1.3, multiscale frameworks take advantage of computational techniques, ranging from simulations of QM to continuous modeling. These approaches have already been used to study the main SBPs found in the human tissues, such as keratin, elastin, collagen, and actin. In particular, the multiscale modeling of keratin elucidated the molecular origin of keratin-related diseases, such as epidermolysis bullosa simplex, epidermolytic palmoplantar keratoderma, and epidermolysis hyperkeratosis (EHK) [247]. The combination of AA-MD and CG-MD simulations provided better understandings of different highly conserved regions of keratin: authors showed that supramolecular arrangement of keratin depends on punctual mutations tremendously influencing the assembling of keratin into filament structures. Their study allowed personalized medical treatments for EHK, caused by point mutations along one of the keratin subunits [247].
Multiscale simulations of collagen led to the “continuum modeling” of collagen-based biomaterials: such studies constituted the first attempt of multiscale investigation of muscle and bone tissues to unravel the molecular roots of various pathological conditions [248].
The multiscale framework has been extended to different SBPs used in TE, such as spider silk or SAPs. Atomistic MD simulations demonstrated that the promising mechanical properties of spider-silk were closely related to the presence of β-sheet nanocrystals and a well-balanced ratio of hydrophilic and hydrophobic domains in the silk fibroin protein [249,250]. CG-MD simulations identified a combination of crystalline phase and semi-amorphous protein matrix as a key factor endowing silk with superior mechanical properties [249,250]. Such developments enabled to process silk into differently shaped biomaterials, such as membranes, composites, or hydrogels.
Similarly, multiscale modeling simulations are currently exploited for understanding the supramolecular organization and the mechanical properties of SAPs’ nanofibers or amyloid-like structures [91,167,230,251], demonstrating how mechanical properties of SAPs are closely related to the presence of specific structural domains, such as cross-β structures [79,85,167,215,230], thus complementing the experimental characterization of SAP-based hydrogels [79,85,167,215,230]. At larger length scales, CG-MD simulations demonstrated that the mechanical properties of SAP nanofibrils are also dependent on the mutual SAPs’ alignment within cross-β domains [127,252]. It is authors’ opinion that next promising steps in this area should be (1) finding innovative methods to integrate CG-MD information into a continuum model and (2) developing algorithms based on machine learning techniques to accelerate the design and investigation of SBPs [148,253,254]. Currently, machine learning techniques play a pivotal role in simulations of biomolecules by reducing the computational costs associated with MD simulations approaches. For example, AlphaFold is by far the most successful application of AI approaches for the prediction of protein tertiary structures from their amino-acid sequences. Recently, two machine-learning systems, which outperformed AlphaFold, have been developed and published: AlphaFold 2 and RoseTTaFold [255,256].
Although AI approaches for predicting the structure of SBPs are still lacking, such advances are going to open new opportunities in biomaterials design and drug discovery as well. Therefore, machine learning techniques will likely be employed for multiscale investigations of complex supramolecular systems soon.
6 Assessing biomaterial biomechanics via SMD
6.1 SMD simulations vs experimental characterization
The success/failure of TE applications of SBPs is also closely dependent on their mechanical properties: adequate mechanical features promote ingrowth of host regenerating tissues into the implanted scaffolds and foster appropriate integration of the implants within the surrounding host tissue. Such evidence lays the foundation of mechanobiology, which is a rapidly emerging holistic approach for the development of innovative TE scaffolds [307–309].
Since mechanical properties are related to the structural properties of the scaffolds at the nano- and microscales, SMD simulations of fibrils/seeds are a valid approach for the investigation of macromolecular structure biomechanics. Indeed, SMD simulations were proven to be suitable for the investigation of different SBPs, from nucleic acid to carbohydrate macromolecular chains (see Table 3).
SMD approaches provide results comparable to those obtained through experimental techniques, such as AFM or optical tweezers. More specifically, via SMD simulations researcher can extrapolate biopolymers properties, such as bending or elastic stiffness, as well as persistence length, which defines the rigid behavior of the polymer chains. More specifically, persistence length is intimately connected to other mechanical properties, such as twisting, in long biopolymer chains [308,309].
Genomic regulation relies on both nucleic acid sequences and their mechanic features: as such, it is also influenced by twisting and torsional rigidities of nucleic acids [310–315]. Single-molecule experiments of AFM and optical tweezers unveiled that double-stranded DNA (dsDNA) and RNA (dsRNA) show similar values of persistence length, ranging from 51 to 66 nm (see Table 3) [310,314]. Hence, dsDNA and dsRNA show similar folding patterns, as confirmed by investigation through MD and SMD simulations (see Table 3) [312,313]. However, further investigations unveiled that dsDNA molecules are about 3-fold stiffer than dsRNA molecules (see Table 1) [310,313,314]. Such results, combined with analysis of torsional rigidity, depicted an unexpected behavior of dsRNA molecules [310,315]. Indeed, it was demonstrated that the twist-stretch coupling of dsRNA could affect the typical size of dsRNA molecules when injected into cells in RNA interference experiments.
The investigation of the mechanical properties of amyloid fibers is attracting increasing attention due to various factors: (1) amyloid fibers are associated with a range of human diseases; (2) they serve biological functions, such as biofilm formation in bacteria; and (3) their chemical–physical features inspired several researchers to develop ß-structuring biomimetic SBPs for TE applications. The mechanical properties of amyloid fibers have been elucidated by fluorescent-force or atomic force microscopy [257,258]. Additional investigations with SMD simulations unveiled how their high ß-sheet content well correlates with strong mechanical features [259,260].
As highlighted in Table 3, the difference among computational and experimental characterizations is negligible. Hence, the combination of different MD simulations techniques can be used for the development of amyloid-inspired SAPs [68,72,105,127,258–261]. Indeed, atomistic SMD approaches elucidated the role of hydrogen bonding and charge interactions on the bending stiffness of ER/K alpha-helix peptides [262]. Other SMD simulations provided clear links between mechanical properties of peptide aggregates and structural motifs of Aß1-42 amyloid fibrils or coiled coil structures [258,261,263].
A combination of experimental and computational approaches was adopted for the investigation of other biomacromolecules like collagen [264–267], cellulose [268–272], actin [273–275], silk [252,269,276,277], and HA [278,279]. Historically, studies on the mechanical properties of collagen represented the first attempts to approach a multiscale model [119,265,267,280]: CG-MD simulations were used to investigate the structural and mechanical features of collagen fibrils, but results were in poor agreement with experimental data (see Table 3) [264–267,280].
Conversely, SMD simulations of silk nanostructures provided values comparable to experimental data and elucidated the phenomena underlying brittle failure of silk domains [252]. A similar approach was also employed for investigating mechanical failures of nanotubes of cyclic peptides [263]. Atomistic SMD simulations unraveled the connection between mechanical behavior of actin and its binding propensity to ATP/ADP molecules, thus giving new insights on the functioning of muscle cells [275]. Such evidence encouraged the development of CG models for understanding the molecular mechanisms of the mechanic failure of SAP fibrils. CG-SMD simulations were also used to investigate the mechanical properties of large SAP aggregates, but MARTINI SMD approach has been modestly used for this purpose since it does not allow conformational transitions typical of biological systems [108,113]. On the other hand, GoMARTINI-SMD [121] was feasibly adopted to elucidate mechanical properties of SAP fibrils and provided comparable results to atomistic SMD simulations [121,127,281]: UA-SMD and GoMARTINI simulations’ maximum bending stresses were comparable (163 MPa for UA-SMD, 150 MPa for GoMARTINI SMD) (Figure 5b). Instead, the axial UA-SMD stretching simulations of fibrils showed lower mechanical stress value than those observed in GoMARTINI SMD simulations (UA-SMD axial stretching failure stress was 146 MPa while in GoMARTINI SMD the maximum stress was equal to 305 MPa) (Figure 5c). The same tendency is observed in bending simulations of fibrils (UA-SMD axial bending failure stress was equal to 195 MPa while in GoMARTINI SMD it was equal to 280 MPa) (Figure 5d). The figure has been adapted from Figures 1a and b and 2a, c, d, and f from Fontana et al. (https://doi.org/10.1039/C9NA00621D).
![Figure 5
Computational nanomechanic characterization of (LDLK)3 fibril seeds and fibrils. Mechanical failure of SAP fibril seeds and fibrils was studied via SMD simulations. In each plot, the number of ß-interactions refers to the number of ß-contacts among (LDLK)3 self-assembling backbone identified through a customized software dubbed Morphoscanner [72]. (a) Axial UA-SMD stretching simulations of fibril seeds showed higher mechanical stress values than those observed in GoMARTINI SMD simulations. (UA-SMD maximum stress of axial stretching was equal to 323 MPa, while in GoMARTINI it was equal to 236). (b) UA-SMD and GoMARTINI simulations’ maximum bending stresses were comparable (163 MPa for UA-SMD, 150 MPa for GoMARTINI SMD). Instead, (c) the axial UA-SMD stretching simulations of fibrils showed lower mechanical stress value than those observed in GoMARTINI SMD simulations (UA-SMD axial stretching failure stress was 146 MPa while in GoMARTINI SMD the maximum stress was equal to 305 MPa). (d) The same tendency is observed in bending simulations of fibrils. (UA-SMD axial bending failure stress was equal to 195 MPa while in GoMARTINI SMD it was equal to 280 MPa). The figure has been adapted from Figures 1a and b and 2a, c, d, and f from Fontana and Gelain [127].](/document/doi/10.1515/ntrev-2022-0455/asset/graphic/j_ntrev-2022-0455_fig_005.jpg)
Computational nanomechanic characterization of (LDLK)3 fibril seeds and fibrils. Mechanical failure of SAP fibril seeds and fibrils was studied via SMD simulations. In each plot, the number of ß-interactions refers to the number of ß-contacts among (LDLK)3 self-assembling backbone identified through a customized software dubbed Morphoscanner [72]. (a) Axial UA-SMD stretching simulations of fibril seeds showed higher mechanical stress values than those observed in GoMARTINI SMD simulations. (UA-SMD maximum stress of axial stretching was equal to 323 MPa, while in GoMARTINI it was equal to 236). (b) UA-SMD and GoMARTINI simulations’ maximum bending stresses were comparable (163 MPa for UA-SMD, 150 MPa for GoMARTINI SMD). Instead, (c) the axial UA-SMD stretching simulations of fibrils showed lower mechanical stress value than those observed in GoMARTINI SMD simulations (UA-SMD axial stretching failure stress was 146 MPa while in GoMARTINI SMD the maximum stress was equal to 305 MPa). (d) The same tendency is observed in bending simulations of fibrils. (UA-SMD axial bending failure stress was equal to 195 MPa while in GoMARTINI SMD it was equal to 280 MPa). The figure has been adapted from Figures 1a and b and 2a, c, d, and f from Fontana and Gelain [127].
6.2 Thermodynamic stability and mechanical properties of biopolymers: two sides of the same coin
SMD simulations were also used for the estimation of the interaction strength in peptide monolayers or aggregates [282–285], allowing to quantify the thermodynamic stability in aqueous solution of surfactant-like peptide (SLP) fibrils and oligomers (see Figure 6). In these simulations, an external mechanical force is applied to one SLP, which is dragged out from the core of an aggregate: the potential of mean force (PMF) profile, representing the dissociation energy from the fibril (or the oligomer), is then calculated [285]. Results showed that the required energy to pull one SLP out of the fibril structure is 3-fold of one for the oligomeric structure. Therefore, fibril structures are more stable than the oligomer configurations. Since the magnitude of the dissociation energy is an indicator for the thermodynamic stability of the supramolecular assemblies, these results suggest that an increasing number of charges in the head-group, when subjected to a mechanical stress, causes more perturbations in the fibril structures than in the less-ordered oligomers.
![Figure 6
Control over the fibrillization yield by varying the oligomeric nucleation propensities of SLPs. (a) Primary sequence of SLPs, isoleucine and leucine, are colored in green, while anionic groups are colored in red. (b) Schematic representation of the two-step nucleation of peptide self-assembly, in which peptides first assemble into oligomeric particles through condensation; nucleates are then formed within the oligomeric particles. (c) PMF profiles of fibril (left) and oligomer (right) models along the reaction coordinate derived from SMD simulations provides information on the thermodynamic stability of fibrils and oligomer structures. Increasing the number of charges of the head-groups reduced the structural stability of the fibrils, but, not in the oligomers. A representative snapshot of SMD trajectory is shown in each graph. The figure has been adapted from Figures 1a, b and 6a from Lau et al. [285].](/document/doi/10.1515/ntrev-2022-0455/asset/graphic/j_ntrev-2022-0455_fig_006.jpg)
Control over the fibrillization yield by varying the oligomeric nucleation propensities of SLPs. (a) Primary sequence of SLPs, isoleucine and leucine, are colored in green, while anionic groups are colored in red. (b) Schematic representation of the two-step nucleation of peptide self-assembly, in which peptides first assemble into oligomeric particles through condensation; nucleates are then formed within the oligomeric particles. (c) PMF profiles of fibril (left) and oligomer (right) models along the reaction coordinate derived from SMD simulations provides information on the thermodynamic stability of fibrils and oligomer structures. Increasing the number of charges of the head-groups reduced the structural stability of the fibrils, but, not in the oligomers. A representative snapshot of SMD trajectory is shown in each graph. The figure has been adapted from Figures 1a, b and 6a from Lau et al. [285].
Such results extended the conclusions of the work of Yu et al. [283], which used SMD to force the transition of 90 PAs in aqueous solution from the bound state (corresponding to a cylinder nanofiber) to a free state. They found that Pas’ assembly pathway is mainly dominated by conformational disorder-to-order transition, encompassing secondary structures’ formation along with tail–head core–shell alignments and condensation, leading to the total exclusion of water from their cores [283,284]. These fragments of evidence lead to a new understanding of the thermodynamic characteristics, which underly the mechanical properties of SAPs and other SBPs.
7 Outlook
The complexity and heterogeneity of biological systems will require complex in silico approaches capable of recapitulating larger systems for longer timeframes: biomaterials will be investigated but the biological counterpart will soon become the priority. Different tools should also be used depending on the accuracy needed for the phenomena investigated and the computational cost required. Also, being biomacromolecules and biosystems intrinsically evolving over time, it will be essential to capture chemical modifications to better simulate and predict the biological effect of existing and novel biomaterials.
7.1 Mixed approaches: serial and parallel multiscale MG simulations (QM/MM/CG)
Understanding physicochemical reactions at different scales in biological systems represents the ultimate challenge in the field of biomateriomics, the science behind the development of innovative SBPs for TE applications. The issue must be addressed across different spatial–temporal scales, ranging from the nanoscale to the macroscale. As shown in the previous chapters, several research groups developed an arsenal of computational and theoretical approaches for investigating SBP systems. Intriguingly, two opposed philosophies emerged for modeling multiscale phenomena: (1) serial multiscale approaches and (2) parallel multiscale approaches.
Rad-Malekshai and coworkers used a solid serial multiscale approach comprising CG and AA-MD simulations for elucidating the structural details of a peptide-based nanocarrier [104]. Even though computational results agreed with the experimental characterization data, their work suffers from using the backmapping approach of that time for peptide structures from CG to the atomistic model [285,286]. Indeed, since then, backmapping has been ameliorated and validated for membrane and protein–ligand systems [109,287].
The QM/MM methods represent a relevant example of parallel multiscale approach that has become the method of choice for modeling reactions in biomolecular systems [287–291]. Since one of the main drawbacks of this approach was the limited electrostatic coupling among different models [279,292], Wassenaar and coworkers introduced an efficient electrostatic coupling in hybrid AA/CG MARTINI systems [285].
Such advance opened new opportunities in the parallel multiscale approaches: for example, QM/MM/CG modeling was used for investigating the factors that influence the potential energy profiles of the enzymatic reaction catalyzed by chorismate mutase [292,293].
Likely, future applications of QM/MM/CG approaches will also rely on elastic networks like GoMARTINI to elucidate the connections between mechanical properties and molecular features of SBPs at respectively the macro- and nanoscales [121,127,293].
7.2 Reactive coarse-grained (RCG) MD for covalent cross-linking
The development of reactive force fields represents one major challenge in molecular modeling. Atomistic reactive force fields are applied for the simulation of diverse polymeric systems [294,295] and represent the key factor for implementing reactive MD (RMD).
In RMD simulations, the topology of the molecular system changes at each integration step: hence, RMD simulations come at the price of high computational costs if compared to serial multiscale MD simulations [117,295].
Indeed, implementing CG-MD simulations and subsequent AA-MD simulations require less computational power than RMD simulations [104].
Few applications, mainly related to organic polymers, have been reported in literature due to the high computational costs associated with this approach [294,296]. Voth and Lafage developed a promising RCG model using AA data to create a CG model able to represent chemical reactions with an evolving bonding topology [297]. However, RCG applications for SBPs are still lacking and not yet suitable for macroscale studies [234,297].
It is authors’ opinion that in the near future RCG simulations could find applications in the investigation of cross-linked SBPs [85,87,91,298,299].
Chemical cross-linking represents a promising strategy for tailoring architectural and mechanical features of SBPs [85,87,88,91,298,299]: it was used for enhancing structural and mechanical properties of peptide-based systems [300]. SAP hydrogels crosslinked with genipin, a natural extract from gardenia jasminoides, were more suitable for TE applications due to their improved mechanical stability enabling their electro-spinning into well-defined solid flexible channels or mats [85,91]. Also, chemical cross-linking led to the development of DNA-based systems [87] or chitosan microspheres [88] for the controlled release of active biomolecules. Presumably, RCG models will be frontrunners in the improvement of cross-linking, by bringing additional understandings to enable the control of both cross-linked scaffold topologies and reaction yield.
7.3 Linking molecular scale to macroscale: moving toward the continuum models
Most of TE approaches rely on the development of scaffolds providing specific mechanical cues to the cells through their microarchitectures [302]. Indeed, cells can “react” to physical and chemical signals generated by other cells and/or the extracellular environment [303]: a deep understanding of these complex signaling interplay will allow the development of novel high-performing scaffolds.
To this purpose, several FE approaches have been adopted to describe the propagation of mechanical deformations through scaffold architectures [302,304] but were mainly limited to the mechanical properties of scaffolds, disregarding the interplay of biochemical signaling [302,304].
The combined use of FE methods and MD simulations features the potential to elucidate the complex relationships between scaffold biomechanics and molecular properties [305].
To tackle this issue Solernou’s lab developed the fluctuating finite element analysis (FFEA) software package [306]. FFEA is suited for simulations of large proteins and protein complexes at the mesoscale (from 5 nm to 1 μm), where modeling tools are lacking the most. As input FFEA requires volumetric information like cryo-electron tomography (cryo-ET) maps or high-resolution atomistic coordinates [306].
Still, efficient mesoscale MD simulation methods will be highly needed to be accessible to mesoscale characterization data. In this way, a mesoscale model could be easily validated using easily accessible experimental characterization methods. Thus, if the experimental validation confirms the results of mesoscale MD simulations, this evidence could guide the understanding of how individual changes at the molecular level affect the macroscale properties of SBPs.
7.4 In silico models of “whole” viruses, organelles, and unicellular organisms
Recent progresses in molecular modeling enabled the simulation of complex biomolecular structures, like cell membranes (see Section 2.2). In a recent work, Pezeshkian and coworkers introduced a multiscale algorithm that backmaps a continuum membrane model, represented as a dynamically triangulated surface (DTS), to its corresponding CG molecular model based on the Martini force field [288].
DTS simulations enabled the investigation of slow conformational changes of large-scale membranes, demonstrating how MARTINI CG simulations may be applied to explore the local properties of complex membrane systems [288]. This approach was used for investigating the local deformations of a vesicular bud induced by the binding of Shiga Toxin. Remarkably, the same multiscale model was adopted to elucidate the organization of an entire mitochondrion to near-atomic resolution, as shown in Figure 7a, where the mitochondrion system (without solvent molecules) contains more than 83 million particles and represents over six million lipids. To note, their system did not show significant changings in the membrane shape throughout 2 ns of MD simulation [287,288].
![Figure 7
MD of large organelles. (a) Modeling scheme of mitochondrial membrane at near-atomic resolution. (Ai) Composition of mitochondrial membrane. POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; SAPE, 1-stearoyl-2-arachidonoyl-sn-glycero-3-phospho-ethanolamine; SAPI, 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoinositol; CHOL, cholesterol; CRDL, cardiolipin. (Aii) Cross-section of the generated surface points for the outer monolayer of outer membrane (blue), inner monolayer of outer membrane (green), outer monolayer of inner membrane (red), and inner monolayer of inner membrane (yellow). Figures (Ai) and (Aii) have been adapted from Pezeshkian et al. [288]. (b) MD simulation of transfection using large lipoplexes. (Bi) Schematic structure of large lipoplex, showing connecting channels. (Bii) Snapshot of MD trajectory, showing the fusion of lipoplex on top of a large endosomal model bilayer patch. (Biii) Last stage of dsDNA transfection: release of dsDNA from large lipoplex. Figures (Bi), (Bii), and (Biii) have been adapted from Bruininks et al. [314], with the permission of eLife Sciences Publications Ltd.](/document/doi/10.1515/ntrev-2022-0455/asset/graphic/j_ntrev-2022-0455_fig_007.jpg)
MD of large organelles. (a) Modeling scheme of mitochondrial membrane at near-atomic resolution. (Ai) Composition of mitochondrial membrane. POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; SAPE, 1-stearoyl-2-arachidonoyl-sn-glycero-3-phospho-ethanolamine; SAPI, 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoinositol; CHOL, cholesterol; CRDL, cardiolipin. (Aii) Cross-section of the generated surface points for the outer monolayer of outer membrane (blue), inner monolayer of outer membrane (green), outer monolayer of inner membrane (red), and inner monolayer of inner membrane (yellow). Figures (Ai) and (Aii) have been adapted from Pezeshkian et al. [288]. (b) MD simulation of transfection using large lipoplexes. (Bi) Schematic structure of large lipoplex, showing connecting channels. (Bii) Snapshot of MD trajectory, showing the fusion of lipoplex on top of a large endosomal model bilayer patch. (Biii) Last stage of dsDNA transfection: release of dsDNA from large lipoplex. Figures (Bi), (Bii), and (Biii) have been adapted from Bruininks et al. [314], with the permission of eLife Sciences Publications Ltd.
Recently, Bruininks’s lab used CG MARTINI simulations to investigate the molecular mechanisms underlying the DNA transfer from lipoplexes (see Figure 7B): they unveiled the fusion mechanisms of large lipoplexes with lipid membranes in 10-μs-long CG-MD simulations. Their findings opened the door to new applications of non-viral vectors for in vivo gene therapies [307].
Lastly, simulations of large organelles may also allow to elucidate mechanisms underlying different metabolic cell states.
The simulations of complete viruses represent the last host topic in multiscale modeling: the current coronavirus pandemic and the annual surge of diverse influenza strains represent a crucial challenge for modern societies and healthcare systems. Indeed, multiscale simulations are deeply used to discover new medical treatments targeting specific virus structures [151,308].
As shown in Figure 8a, Durrant and coworkers developed an all-atom model of an intact H1N1 virus. MD atomistic simulations, coupled with Markov state model theory, have provided new insights on the potential role of the neuraminidase secondary site, where terminal sialic acid residues can bind before transferring to the primary site (responsible for the enzymatic cleavage). Their work broke new ground in terms of molecular simulation size, complexity, and methodological analyses. It also provided fundamental insights into the understanding of substrate recognition processes, enabling new strategies for influenza treatments [151].
![Figure 8
Computational virology applications: mesoscale simulations enhance conformational sampling of viral glycoproteins. AA-MD simulation of a fully intact all-atom model of the influenza A H1N1 2009 (pH1N1) viral envelope (a). Hemagglutinin (HA) glycoproteins are shown in dark blue; neuraminidase (NA) glycoproteins are shown in light blue. In (b), a single NA monomer is shown (top view) with its catalytic site (white), secondary site (yellow), 150-loop (green), and 430-loop (red). In (c–e), principal component analysis (PCA) is performed by considering the motions of C atoms of 19 1° pocket residues. In (c), the PCA of the four monomers is sampled during a single-NA-tetramer simulation. In (d), the PCA of the 120 monomer trajectories is extracted during the last 8.33 ns of the viral envelope simulation. In (e), the PCA of all 120 monomer simulations is extracted from the full simulation of the viral envelope. The figure has been adapted from Figure 1a–e from Durrant et al. [151]. In (f) and (g), a multiscale model of the SARS-Cov-2 virion is shown. An exterior view and an interior view of the SARS-CoV-2 virion are given in (f) and (g), respectively. The S-protein trimers are depicted in teal, with the glycosylation sites represented as black spheres. M-protein dimers are in blue, with pentameric E ion channels in orange. The experimental characterization addressed the appropriate density of S, M, and E proteins. The diameters of the membrane envelope span from 100 to 140 nm, including the S proteins on the virion surface. The figure has been adapted from Figure 3a and b of Yu et al. (2021) [316].](/document/doi/10.1515/ntrev-2022-0455/asset/graphic/j_ntrev-2022-0455_fig_008.jpg)
Computational virology applications: mesoscale simulations enhance conformational sampling of viral glycoproteins. AA-MD simulation of a fully intact all-atom model of the influenza A H1N1 2009 (pH1N1) viral envelope (a). Hemagglutinin (HA) glycoproteins are shown in dark blue; neuraminidase (NA) glycoproteins are shown in light blue. In (b), a single NA monomer is shown (top view) with its catalytic site (white), secondary site (yellow), 150-loop (green), and 430-loop (red). In (c–e), principal component analysis (PCA) is performed by considering the motions of C atoms of 19 1° pocket residues. In (c), the PCA of the four monomers is sampled during a single-NA-tetramer simulation. In (d), the PCA of the 120 monomer trajectories is extracted during the last 8.33 ns of the viral envelope simulation. In (e), the PCA of all 120 monomer simulations is extracted from the full simulation of the viral envelope. The figure has been adapted from Figure 1a–e from Durrant et al. [151]. In (f) and (g), a multiscale model of the SARS-Cov-2 virion is shown. An exterior view and an interior view of the SARS-CoV-2 virion are given in (f) and (g), respectively. The S-protein trimers are depicted in teal, with the glycosylation sites represented as black spheres. M-protein dimers are in blue, with pentameric E ion channels in orange. The experimental characterization addressed the appropriate density of S, M, and E proteins. The diameters of the membrane envelope span from 100 to 140 nm, including the S proteins on the virion surface. The figure has been adapted from Figure 3a and b of Yu et al. (2021) [316].
Recently, Yu and coworkers reported an ongoing development of a largely “bottom-up” CG model of the SARS-Cov-2 virion, shown in Figure 8b [309]. Their multiscale model of the SARS-CoV-2 virion, built by retrieving CG interactions from AA-MD simulations, it is deemed to reveal new routes to target the virus and enable new antiviral treatments [308,309].
8 Conclusions
The remarkable progresses in HPC have nurtured different modeling approaches for SBPs. We foresee that diverse TE applications of SBPs will likely need dedicated different computational workflows. Multiscale methods will soon connect the atomistic changes and the macroscale morpho-mechanical characteristics of SBPs. Besides, recent advances in the modeling of membrane structures will enable accurate investigations of biomaterial–cell interactions. Further progresses in modeling of cell compartments will drive the study of complex cellular systems, thus leading the way to the computational characterization of tissues. Lastly, SBP–tissue interactions will also be elucidated by novel AI-based approaches so far applied to proteins only. Hence, the development of safer and more precise biomedical treatments will be soon made possible by a multi-facet arsenal of computational approaches targeting most of the different phenomena to be considered in biomaterial design and characterization.
Experimental and computational characterization of the most important SBPs used in TE and nanomedicine
| Biopolymers | Young modulus experimental (GPa) | Persistence length experimental (nm) | Young modulus computational (GPa) | Persistence length computational (nm) |
|---|---|---|---|---|
| dsDNA | 0.3–1 [301] | 51 [302] | 0.3 [303] | 63 [304] |
| dsRNA | 0.09 [305]* | 63–65 [306] | 0.205–0.259 [305]* | 66.3 [305] |
| 57–60 [302,305] | ||||
| Prion | 0.35–0.80 [257,258] | 1500–3300 [257,261] | 13–18 [259,307] | 1000–4000 [259,307] |
| Collagen | 3 [280] | 10–20 [264] | 4.5–4.75 [265,266] | 51.7 [267] |
| Actin | 2.6 [274] | 15690 [274] | 1.80[ATP] [275] 1.90[ADP] [275] | 15410 [275] |
| Silk | 14–36 [276] | 3 [269] | 6.2–22.6 [252,277] | 2.5 [252] |
| Cellulose | 78 [270] | 5–20 [271] | 161 [268] | 14.5 [272] |
| CPNs | 18.5–40 [263] | 600 [263] | 7.8 [263] | 460 [263] |
| HA | 5.9 [279] | 7.1 [278] | ||
| SAP fibril seed | 0.5–1.626 [127] | 593–1928 [127]* | ||
| SAP fibril | 2–6 [127] | 2110–7116 [127]* |
Note: Values marked with * were calculated using the following length (P) formula: P = B s/(TKP) indicates the length of the persistence and B s Young’s modulus.
-
Funding information: The authors’ funding was granted by INAIL (BRIC2019-ID25) and by the “Ministero della Salute Ricerca Corrente 2018-2021” from the Italian Ministry of Health and by the “5 × 1,000” voluntary contributions. Financial support also came from Revert Onlus.
-
Author contributions: All authors have accepted responsibility for the entire content of this article and approved its submission.
-
Conflict of interest: The authors state no conflict of interest.
References
[1] Whitesides GM. Self-assembly at all scales. Science. 2002;295:2418.10.1126/science.1070821Search in Google Scholar PubMed
[2] Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171.10.1038/nbt874Search in Google Scholar PubMed
[3] Woodson SA. RNA folding pathways and the self-assembly of ribosomes. Acc Chem Res. 2011;44:1312.10.1021/ar2000474Search in Google Scholar PubMed PubMed Central
[4] Kim JH, Jin HM, Yang GG, Han KH, Yun T, Shin JY, et al. Smart nanostructured materials based on self-assembly of block co-polymers. Adv Funct Mater. 2020;30:1902049.10.1002/adfm.201902049Search in Google Scholar
[5] Kokkoli E, Mardilovich A, Wedekind A, Rexeisen EL, Garg A, Craig JA. Self-assembly and applications of biomimetic and bioactive peptide-amphiphiles. Soft Matter. 2006;2:1015.10.1039/b608929aSearch in Google Scholar PubMed
[6] Serag MF, Aikeremu A, Tsukamoto R, Piwon´ski H, Abadi M, Kaji N, et al. Geometry-based self-assembly of histone–DNA nanostructures at single-nucleotide resolution. ACS Nano. 2019;13:8155.10.1021/acsnano.9b03259Search in Google Scholar PubMed
[7] Subramani K, Ahmed W. Self-assembly of proteins and peptides and their applications in bionanotechnology and dentistry. Emerg Nanotechnol Dent. 2018;231:209–24.10.1016/B978-0-12-812291-4.00012-1Search in Google Scholar
[8] Lee EJ, Kasper FK, Mikos AG. Biomaterials for tissue engineering. Ann Biomed Eng. 2014;42:323.10.1007/978-981-10-2723-9_2Search in Google Scholar
[9] Pérez-Pedroza R, Ávila-Ramírez A, Khan Z, Moretti M, Hauser CAE. Supramolecular biopolymers for tissue engineering. Adv Polym Technol. 2021;2021:8815006.10.1155/2021/8815006Search in Google Scholar
[10] Saracino GAA, Cigognini D, Silva D, Caprini A, Gelain F. Nano-materials design and tests for neural tissue engineering. Chem Soc Rev. 2013;42:225.10.1039/C2CS35065CSearch in Google Scholar
[11] Kyle S, Aggeli A, Ingham E, McPherson MJ. Production of self-assembling biomaterials for tissue engineering. Trends Biotechnol. 2009;27:423.10.1016/j.tibtech.2009.04.002Search in Google Scholar PubMed PubMed Central
[12] Klein S, Vykoukal J, Felthaus O, Dienstknecht T, Prantl L. Collagen type I conduits for the regeneration of nerve defects. Materials. 2016;9:219.10.3390/ma9040219Search in Google Scholar PubMed PubMed Central
[13] Yeh J-Z, Wang D-H, Cherng J-H, Wang Y-W, Fan G-Y, Liou N-H, et al. A collagen-based scaffold for promoting neural plasticity in a rat model of spinal cord injury. Polymers. 2020;12:2245.10.3390/polym12102245Search in Google Scholar PubMed PubMed Central
[14] Copes F, Pien N, van Vlierberghe S, Boccafoschi F, Mantovani D. Collagen-based tissue engineering strategies for vascular medicine. Front Bio-Eng Biotechnol. 2019;7:00166.10.3389/fbioe.2019.00166Search in Google Scholar PubMed PubMed Central
[15] Majid QA, Fricker ATR, Gregory DA, Davidenko N, Hernandez Cruz O, Jabbour RJ, et al. Natural biomaterials for cardiac tissue engineering: A highly biocompatible solution. Front Cardiovascular Med. 2020;7:55459.10.3389/fcvm.2020.554597Search in Google Scholar PubMed PubMed Central
[16] Lee SJ, Wang H-J, Kim T-H, Choi JS, Kulkarni G, Jackson JD, et al. In situ tissue regeneration of renal tissue induced by collagen hydrogel injection. Stem Cell Transl Med. 2018;7:241–50.10.1002/sctm.16-0361Search in Google Scholar PubMed PubMed Central
[17] Chen S, Nakamoto T, Kawazoe N, Chen G. Engineering multi-layered skeletal muscle tissue by using 3D microgrooved collagen scaffolds. Biomaterials. 2015;73:23–31.10.1016/j.biomaterials.2015.09.010Search in Google Scholar PubMed
[18] Jha BS, Ayres CE, Bowman JR, Telemeco TA, Sell SA, Bowlin GL, et al. Electrospun collagen: A tissue engineering scaffold with unique functional properties in a wide variety of applications. J Nanomater. 2011;2011:348268.10.1155/2011/348268Search in Google Scholar
[19] Han CM, Zhang LP, Sun JZ, Shi HF, Zhou J, Gao CY. Application of collagen-chitosan/fibrin glue asymmetric scaffolds in skin tissue engineering. J Zhejiang Univ Sci B. 2010;11(7):524–30.10.1631/jzus.B0900400Search in Google Scholar PubMed PubMed Central
[20] Ucar B, Humpel, C. Collagen for brain repair: Therapeutic perspectives. Neural Regenerat Res. 2018;13:595–8.10.4103/1673-5374.230273Search in Google Scholar PubMed PubMed Central
[21] Russo L, Sgambato A, Lecchi M, Pastori V, Raspanti M, Natalello A, et al. Neoglucosylated collagen matrices drive neuronal cells to differentiate. ACS Chem Neurosci. 2014;5:261.10.1021/cn400222sSearch in Google Scholar PubMed PubMed Central
[22] Figuereido I, Paiotta A, Dal Magro R, Tinelli F, Corti R, Re F, et al. A new approach for glyco-functionalization of collagen-based biomaterials. Int J Mol Sci. 2019;20:1747.10.3390/ijms20071747Search in Google Scholar PubMed PubMed Central
[23] Wang X, He J, Wang Y, Cui F-Z. Hyaluronic acid-based scaffold for central neural tissue engineering. Interface Focus. 2012;2:278–91.10.1098/rsfs.2012.0016Search in Google Scholar PubMed PubMed Central
[24] Jang Y, Park Y, Kim J. Engineering biomaterials to guide heart cells for matured cardiac tissue. Coatings. 2020;10:925.10.3390/coatings10100925Search in Google Scholar
[25] Bonafè F, Govoni M, Giordano E, Caldarera CM, Guarnieri C, Muscari C. Hyaluronan and cardiac regeneration. J Biomed Sci. 2014;21:100.10.1186/s12929-014-0100-4Search in Google Scholar PubMed PubMed Central
[26] Hemshekhar M, Thushara RM, Chandranayaka S, Sherman LS, Kem-paraju K, Girish KS. Emerging roles of hyaluronic acid bioscaffolds in tissue engineering and regenerative medicine. Int J Biol Macromol. 2016;86:917–28.10.1016/j.ijbiomac.2016.02.032Search in Google Scholar PubMed
[27] Zakhem E, Raghavan S, Gilmont RR, Bitar KN. Chitosan-based scaffolds for the support of smooth muscle constructs in intestinal tissue engineering. Biomaterials. 2012;33:4810–7.10.1016/j.biomaterials.2012.03.051Search in Google Scholar PubMed PubMed Central
[28] Hajiabbas M, Mashayekhan S, Nazaripouya A, Naji M, Hunkeler D, Rajabi Zeleti S, et al. Chitosan-gelatin sheets as scaffolds for muscle tissue engineering, artificial cells. Nanomed Biotechnol. 2015;43(2):124–32, 10.3109/21691401.2013.852101.Search in Google Scholar PubMed
[29] Gnavi S, Barwig C, Freier T, Haastert-Talini K, Grothe C, Geuna S. The use of chitosan-based scaffolds to enhance regeneration in the nervous system. Int Rev Neurobiol. 2013;109:1–62.10.1016/B978-0-12-420045-6.00001-8Search in Google Scholar PubMed
[30] Ojeda-Hernández DD, Canales-Aguirre AA, Matias-Guiu J, Gomez-Pinedo U, Mateos-Díaz JC. Potential of chitosan and its derivatives for biomedical applications in the central nervous system. Front Bioeng Biotechnol. 2020;8:389.10.3389/fbioe.2020.00389Search in Google Scholar PubMed PubMed Central
[31] Han Y, Li Y, Zeng Q, Li H, Peng J, Xu Y, et al. Injectable bioactive akermanite/alginate composite hydrogels for in situ skin tissue engineering. J Mater Chem B. 2017;5:3315–26.10.1039/C7TB00571GSearch in Google Scholar
[32] Liu J, Zhou H, Weir MD, Xu HHK, Chen Q, Trotman CA. Fast-degradable microbeads encapsulating human umbilical cord stem cells in alginate for muscle tissue engineering. Tissue Eng Part A. 2012;18:2303–14.10.1089/ten.tea.2011.0658Search in Google Scholar PubMed PubMed Central
[33] Baniasadi H, Mashayekhan S, Fadaoddini S, Haghirsharifzamini Y. Design, fabrication and characterization of oxidized alginate–gelatin hydrogels for muscle tissue engineering applications. J Biomater Appl. 2016;31:152–61.10.1177/0885328216634057Search in Google Scholar PubMed
[34] Jansen K, Schuurmans CCL, Jansen J, Masereeuw R, Vermonden T. Hydrogel-based cell therapies for kidney regeneration: current trends in biofabrication and in vivo repair. Curr Pharm Des. 2017;23:3845–57.10.2174/1381612823666170710155726Search in Google Scholar PubMed PubMed Central
[35] Amirian J, Van TTT, Bae S-H, Jung H-I, Choi H-J, Cho H-D, et al. Examination of in vitro and in vivo biocompatibility of alginate-hyaluronic acid microbeads as a promising method in cell delivery for kidney regeneration. Int J Biol Macromol. 2017;105:143–53.10.1016/j.ijbiomac.2017.07.019Search in Google Scholar PubMed
[36] Tamimi M, Rajabi S, Pezeshki-Modaress M. Cardiac ECM/chitosan/alginate ternary scaffolds for cardiac tissue engineering application. Int J Biol Macromol. 2020;164:389–402.10.1016/j.ijbiomac.2020.07.134Search in Google Scholar PubMed
[37] Pawar K, Prang P, Müller R, Caioni M, Bogdahn U, Kunz W, et al. Intrinsic and extrinsic determinants of central nervous system axon outgrowth into alginate-based anisotropic hydrogels. Acta Biomater. 2015;27:131–9.10.1016/j.actbio.2015.08.032Search in Google Scholar PubMed
[38] Grijalvo S, Nieto-Díaz M, Maza RM, Eritja R, Díaz DD. Alginate hydrogels as scaffolds and delivery systems to repair the damaged spinal cord. Biotechnol J. 2019;14:e190027.10.1002/biot.201900275Search in Google Scholar PubMed
[39] Moran JM, Pazzano D, Bonassar, LJ. Characterization of polylactic acid–polyglycolic acid composites for cartilage tissue engineering. Tissue Eng. 2003;9:63–70.10.1089/107632703762687546Search in Google Scholar PubMed
[40] Santoro M, Shah SR, Walker JL, Mikos AG. Poly (Lactic Acid) nanofibrous scaffolds for tissue engineering. Adv Drug Delivery Rev. 2016;107:206–12.10.1016/j.addr.2016.04.019Search in Google Scholar PubMed PubMed Central
[41] Sotoudeh A, Darbemamieh G, Goodarzi V, Shojaei S, Asefnejad A. Tissue engineering needs new biomaterials: Poly (Xylitol-Dodecanedioic Acid)–Co-Polylactic Acid (PXDDA-Co-PLA) and its nanocomposites. Eur Polym J. 2021;152:110469.10.1016/j.eurpolymj.2021.110469Search in Google Scholar
[42] Liu R, Zhang S, Zhao C, Yang D, Cui T, Liu Y, et al. Regulated surface morphology of polyaniline/polylactic acid composite nanofibers via various inorganic acids doping for enhancing biocompatibility in tissue engineering. Nanoscale Res Lett. 2021;16:4.10.1186/s11671-020-03457-zSearch in Google Scholar PubMed PubMed Central
[43] Baolin G, Ma PX. Synthetic biodegradable functional polymers for tissue engineering: A brief review. Sci China Chem. 2014;57(4):490–500.10.1007/s11426-014-5086-ySearch in Google Scholar PubMed PubMed Central
[44] Bolívar-Monsalve EJ, Alvarez MM, Hosseini S, Espinosa-Hernandez MA, Ceballos-González CF, Sanchez-Dominguez M, et al. Engineering bioactive synthetic polymers for biomedical applications: A review with emphasis on tissue engineering and controlled release. Mater Adv. 2021;2:4447.10.1039/D1MA00092FSearch in Google Scholar
[45] Campa-Siqueiros PI, Madera-Santana TJ, Castillo-Ortega MM, López-Cervantes J, Ayala-Zavala JF, Ortiz-Vazquez EL. Electrospun and co-electrospun biopolymer nanofibers for skin wounds on diabetic patients: An overview. RSC Adv. 2021;11(25):15340–50.10.1039/D1RA02986JSearch in Google Scholar
[46] Ciarfaglia N, Laezza A, Lods L, Lonjon A, Dandurand J, Pepe A, et al. Thermal and dynamic mechanical behavior of poly (Lactic Acid) (PLA)-based electrospun scaffolds for tissue engineering. J Appl Polym Sci. 2021;138:51313.10.1002/app.51313Search in Google Scholar
[47] Ng DYW, Wu Y, Kuan SL, Weil T. Programming supramolecular biohybrids as precision therapeutics. Acc Chem Res. 2014;47:3471–80.10.1021/ar5002445Search in Google Scholar PubMed
[48] Lai J, Jiang P, Gaddes ER, Zhao N, Abune L, Wang Y. Aptamer-functionalized hydrogel for self-programmed protein release via sequential photo-reaction and hybridization. Chem Mater. 2017;29:5850–7.10.1021/acs.chemmater.7b00875Search in Google Scholar PubMed PubMed Central
[49] Sur S, Newcomb CJ, Webber MJ, Stupp SI. Tuning supramolecular mechanics to guide neuron development. Biomaterials. 2013;34:4749.10.1016/j.biomaterials.2013.03.025Search in Google Scholar PubMed PubMed Central
[50] Freeman R, Han M, Álvarez Z, Lewis JA, Wester JR, Stephanopou-los N, et al. , Reversible self-assembly of superstructured networks. Science. 2018;362:808.10.1126/science.aat6141Search in Google Scholar PubMed PubMed Central
[51] Sato Y, Sakamoto T, Takinoue M. Sequence-based engineering of dynamic functions of micrometer-sized DNA droplets. Appl Sci Eng. 2020;6(23):eaba3471.10.1126/sciadv.aba3471Search in Google Scholar PubMed PubMed Central
[52] Nam K, Im BI, Kim T, Kim YM, Roh YH. Anisotropically functionalized aptamer-DNA nanostructures for enhanced cell proliferation and target-specific adhesion in 3D cell cultures. Biomacromolecules. 2021;22(7):3138–47.10.1021/acs.biomac.1c00619Search in Google Scholar PubMed
[53] He J-Y, Shang X, Yang C-L, Zuo S-Y, Yuan R, Xu W-J. Antibody-responsive ratiometric fluorescence biosensing of biemissive silver nanoclusters wrapped in switchable DNA tweezers. Anal Chem. 2021;93:11634.10.1021/acs.analchem.1c02444Search in Google Scholar PubMed
[54] Li S, Jiang Q, Liu S, Zhang Y, Tian Y, Song C, et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat Biotechnol. 2018;36:258.10.1038/nbt.4071Search in Google Scholar PubMed
[55] Hong F, Zhang F, Liu Y, Yan H. DNA origami: Scaffolds for creating higher order structures. Chem Rev. 2017;117:12584.10.1021/acs.chemrev.6b00825Search in Google Scholar PubMed
[56] Morya V, Walia S, Mandal BB, Ghoroi C, Bhatia D. Functional DNA based hydrogels: Development, properties and biological applications. ACS Biomater Sci Eng. 2020;6(11):6021–35.10.1021/acsbiomaterials.0c01125Search in Google Scholar PubMed
[57] Freeman R, Stephanopoulos N, Álvarez Z, Lewis JA, Sur S, Serrano CM, et al. Instructing cells with programmable peptide DNA hybrids. Nat Commun. 2017;8:15982.10.1038/ncomms15982Search in Google Scholar PubMed PubMed Central
[58] Freeman R, Boekhoven J, Dickerson MB, Naik RR, Stupp SI. Biopolymers and supramolecular polymers as biomaterials for biomedical applications. MRS Bull. 2015;40:1089–101.10.1557/mrs.2015.270Search in Google Scholar PubMed PubMed Central
[59] Silva D, Natalello A, Sanii B, Vasita R, Saracino G, Zuckermann RN, et al. Synthesis and characterization of designed BMHP1-derived self-assembling peptides for tissue engineering applications. Nanoscale. 2013;5:704–18.10.1039/C2NR32656FSearch in Google Scholar PubMed
[60] Marchini A, Favoino C, Gelain F. Multi-functionalized self-assembling peptides as reproducible 3D cell culture systems enabling differentiation and survival of various human neural stem cell lines. Front Neurosci. 2020;14:00413.10.3389/fnins.2020.00413Search in Google Scholar PubMed PubMed Central
[61] Gelain F, Cigognini D, Caprini A, Silva D, Colleoni B, Donegá M, et al. New bioactive motifs and their use in functionalized self-assembling peptides for NSC differentiation and neural tissue engineering. Nanoscale. 2012;4:2946.10.1039/c2nr30220aSearch in Google Scholar PubMed
[62] Karavasili C, Fatouros DG. Self-assembling peptides as vectors for local drug delivery and tissue engineering applications. Adv Drug Delivery Rev. 2021;174:387–405.10.1016/j.addr.2021.04.024Search in Google Scholar PubMed
[63] Guo H, Cui G, Yang J, Wang C, Zhu J, Zhang L, et al. Sustained delivery of VEGF from designer self-assembling peptides improves cardiac function after myocardial infarction. Biochem Biophys Res Commun. 2012;424:105–11.10.1016/j.bbrc.2012.06.080Search in Google Scholar PubMed
[64] Hsu BB, Conway W, Tschabrunn CM, Mehta M, Perez-Cuevas MB, Zhang S, et al. Clotting mimicry from robust hemostatic bandages based on self-assembling peptides. ACS Nano. 2015;9:9394–406.10.1021/acsnano.5b02374Search in Google Scholar PubMed PubMed Central
[65] Alshehri S, Susapto HH, Hauser CAE. Scaffolds from self-assembling tetrapeptides support 3D spreading, osteogenic differentiation, and angiogenesis of mesenchymal stem cells. Biomacromolecules. 2021;22:2094–106.10.1021/acs.biomac.1c00205Search in Google Scholar PubMed PubMed Central
[66] Xu H, Wang C, Liu C, Li J, Peng Z, Guo J, et al. Stem cell-seeded 3D-printed scaffolds combined with self-assembling peptides for bone defect repair. Tissue Eng Part A. 2021;111:111–24.10.1089/ten.tea.2021.0055Search in Google Scholar PubMed
[67] Cui H, Webber MJ, Stupp SI. Self-Assembly of Peptide Am-phiphiles: From molecules to nanostructures to biomaterials. Biopolymers. 2010;94:1.10.1002/bip.21328Search in Google Scholar PubMed PubMed Central
[68] Raspa A, Saracino GAA, Pugliese R, Silva D, Cigognini D, Vescovi A, et al. Complementary co-assembling peptides: from in silico studies to in vivo application. Adv Funct Mater. 2014;24:6317.10.1002/adfm.201400956Search in Google Scholar
[69] Zhang S, Lockshin C, Herbert A, Winter E, Rich, A. Zuotin, a putative Z-DNA binding protein in saccharomyces cerevisiae. EMBO J. 1992;11:3787.10.1002/j.1460-2075.1992.tb05464.xSearch in Google Scholar PubMed PubMed Central
[70] Gelain F, Bottai D, Vescovi A, Zhang S. Designer self-assembling peptide nanofiber scaffolds for adult mouse neural stem cell 3-dimensional cultures. PLoS One. 2006;1:e119.10.1371/journal.pone.0000119Search in Google Scholar PubMed PubMed Central
[71] Horii A, Wang X, Gelain F, Zhang S. Biological designer self-assembling peptide nanofiber scaffolds significantly enhance osteoblast proliferation, differentiation and 3-D migration. PLoS ONE. 2007;2:e190.10.1371/journal.pone.0000190Search in Google Scholar PubMed PubMed Central
[72] Saracino GAA, Fontana F, Jekhmane S, Silva JM, Weingarth M, Gelain F. Elucidating self-assembling peptide aggregation via morphoscanner: A new tool for protein-peptide structural characterization. Adv Sci. 2018;5:1800471.10.1002/advs.201800471Search in Google Scholar PubMed PubMed Central
[73] Zhao X, Zhang S. Molecular designer self-assembling peptides. Chem Soc Rev. 2006;35:1105.10.1039/b511336aSearch in Google Scholar PubMed
[74] Caprini A, Silva D, Zanoni I, Cunha C, Volontè C, Vescovi A, et al. A novel bioactive peptide: Assessing its activity over murine neural stem cells and its potential for neural tissue engineering. N Biotechnol. 2013;30:552.10.1016/j.nbt.2013.03.005Search in Google Scholar PubMed
[75] Cigognini D, Satta A, Colleoni B, Silva D, Donegà M, Antonini S, et al. Evaluation of early and late effects into the acute spinal cord injury of an injectable functionalized self-assembling scaffold. PLoS ONE. 2011;6:e19782.10.1371/journal.pone.0019782Search in Google Scholar PubMed PubMed Central
[76] Clark TD, Buehler LK, Ghadiri MR. Self-assembling cyclic β3-peptide nanotubes as artificial transmembrane ion channels. J Am Chem Soc. 1998;120:651.10.1021/ja972786fSearch in Google Scholar
[77] Rodriguez-Vazquez N, Ozores H, Guerra A, Gonzalez-Freire E, Fuertes A, Panciera M, et al. Membrane-targeted self-assembling cyclic peptide nanotubes. Curr Top Medicinal Chem. 2015;14:2647.10.2174/1568026614666141215143431Search in Google Scholar PubMed
[78] Bystrov VS, Zelenovskiy PS, Nuraeva AS, Kopyl S, Zhulyabina OA, Tverdislov VA. Molecular modeling and computational study of the chiral-dependent structures and properties of self-assembling diphenylalanine peptide nan-otubes. J Mol Modeling. 2019;25:199.10.1007/s00894-019-4080-xSearch in Google Scholar PubMed
[79] Taraballi F. Glycine-spacers influence functional motifs exposure and self-assembling propensity of functionalized substrates tailored for neural stem cell cultures. Front Neuroeng. 2010;3:1–9.10.3389/neuro.16.001.2010Search in Google Scholar PubMed PubMed Central
[80] Macedo-da-Silva J, Santiago VF, Rosa-Fernandes L, Marinho CRF, Palmisano G. Protein glycosylation in extracellular vesicles: Structural characterization and biological functions. Mol Immunology. 2021;135:226–46.10.1016/j.molimm.2021.04.017Search in Google Scholar PubMed
[81] Moradi SV, Hussein WM, Varamini P, Simerska P, Toth I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem Sci. 2016;7:2492.10.1039/C5SC04392ASearch in Google Scholar PubMed PubMed Central
[82] Restuccia A, Seroski DT, Kelley KL, O’Bryan CS, Kurian JJ, Knox KR, et al. Hierarchical self-assembly and emergent function of densely glycosylated peptide nanofibers. Commun Chem. 2019;2:53.10.1038/s42004-019-0154-zSearch in Google Scholar
[83] Hendrikse SIS, Su L, Hogervorst TP, Lafleur RPM, Lou X, van der Marel GA, et al. Elucidating the ordering in self-assembled glycocalyx mimicking supramolecular copolymers in water. J Am Chem Soc. 2019;141:13877–86.10.1021/jacs.9b06607Search in Google Scholar PubMed PubMed Central
[84] Yuan D, Shi J, Du X, Zhou N, Xu B. Supramolecular glycosylation accelerates proteolytic degradation of peptide nanofibrils. J Am Chem Soc. 2015;137:10092–5.10.1021/jacs.5b05888Search in Google Scholar PubMed PubMed Central
[85] Pugliese R, Maleki M, Zuckermann RN, Gelain F. Self-assembling peptides cross-linked with genipin: resilient hydrogels and self-standing electrospun scaffolds for tissue engineering applications. Biomater Sci. 2019;7:76.10.1039/C8BM00825FSearch in Google Scholar PubMed
[86] Kitayama Y, Harada A. Interfacial photo-cross-linking: simple but powerful approach for fabricating capsule polymer particles with tunable PH-responsive controlled release capability. ACS Appl Mater & Interfaces. 2021;13.10.1021/acsami.0c20152Search in Google Scholar PubMed
[87] Epstein-Barash H, Stefanescu CF, Kohane DS. An in situ cross-linking hybrid hydrogel for controlled release of proteins. Acta Biomaterialia. 2012;8:1703–9.10.1016/j.actbio.2012.01.028Search in Google Scholar PubMed PubMed Central
[88] Hussain Md R, Devi RR, Maji TK. Controlled release of urea from chitosan microspheres prepared by emulsification and cross-linking method. Iran Polym J. 2012;21:473–9.10.1007/s13726-012-0051-0Search in Google Scholar
[89] Radvar E, Azevedo HS. Supramolecular peptide/polymer hybrid hydrogels for biomedical applications. Macromol Biosci. 2019;19(1):1800221.10.1002/mabi.201800221Search in Google Scholar PubMed
[90] Chhabra R, Sharma J, Liu Y, Rinker S, Yan H. DNA Self-assembly for nanomedicine. Adv Drug Delivery Rev. 2010;62(6):617–25.10.1016/j.addr.2010.03.005Search in Google Scholar PubMed
[91] Pugliese R, Fontana F, Marchini A, Gelain F. Branched peptides integrate into self-assembled nanostructures and enhance biomechanics of peptidic hydrogels. Acta Biomaterialia. 2018;66:258.10.1016/j.actbio.2017.11.026Search in Google Scholar
[92] Saracino GAA, Gelain F. Modelling and analysis of early aggregation events of BMHP1-derived self-assembling peptides. J Biomolecular Structure Dyn. 2014;32:759.10.1080/07391102.2013.790848Search in Google Scholar
[93] Pugliese R, Marchini A, Saracino GAA, Zuckermann RN, Gelain F. Cross-linked self-assembling peptide scaffolds. Nano Res. 2018;11:586.10.1007/s12274-017-1834-6Search in Google Scholar
[94] Gustafsson C, Linares M, Norman P. Quantum mechanics/molecular mechanics density functional theory simulations of the optical properties finger-printing the ligand-binding of pentameric formyl thiophene acetic acid in amyloid-β (1–42). J Phys Chem A. 2020;124:875.10.1021/acs.jpca.9b09779Search in Google Scholar
[95] Platts JA. Quantum chemical molecular dynamics and metadynamics simulation of aluminium binding to amyloid-β and related peptides. R Soc Open Sci. 2020;7:191562.10.1098/rsos.191562Search in Google Scholar
[96] Kamel M, Raissi H, Hashemzadeh H, Mohammadifard, K. Theoretical elucidation of the amino acid interaction with graphene and functionalized graphene nanosheets: Insights from DFT calculation and MD simulation. Amino Acids. 2020;52:1465–78.10.1007/s00726-020-02905-5Search in Google Scholar
[97] Georgoulia PS, Glykos NM. On the foldability of tryptophan-containing tetra- and pentapeptides: An exhaustive molecular dynamics study. J Phys Chem B. 2013;117:5522.10.1021/jp401239vSearch in Google Scholar
[98] Wymore T, Wong TC. Molecular dynamics study of substance P peptides in a biphasic membrane mimic. Biophys J. 1999;76:1199.10.1016/S0006-3495(99)77284-XSearch in Google Scholar
[99] He X, Lin M, Lu T, Qu Z, Xu F. Molecular analysis of interactions between a PAMAM dendrimer–paclitaxel conjugate and a biomembrane. Phys Chem Chem Phys. 2015;17:29507–17.10.1039/C5CP02242HSearch in Google Scholar PubMed
[100] Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: Coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812.10.1021/jp071097fSearch in Google Scholar PubMed
[101] Kroenke CD, Ziemnicka-Kotula D, Xu J, Kotula L, Palmer AG. Solution conformations of a peptide containing the cytoplasmic domain sequence of the β amyloid precursor protein. Biochemistry. 1997;36:8145.10.1021/bi9705669Search in Google Scholar PubMed
[102] Rosal R, Pincus MR, Brandt-Rauf PW, Fine RL, Michl J, Wang H. NMR solution structure of a peptide from the Mdm-2 binding domain of the P53 protein that is selectively cytotoxic to cancer cells. Biochemistry. 2004;43:1854.10.1021/bi035718gSearch in Google Scholar PubMed
[103] Zhao L, Cao Z, Bian Y, Hu G, Wang J, Zhou Y. Molecular dynamics simulations of human antimicrobial peptide LL-37 in model POPC and POPG lipid bilayers. Int J Mol Sci. 2018;19:1186.10.3390/ijms19041186Search in Google Scholar PubMed PubMed Central
[104] Rad-Malekshahi M, Visscher KM, Rodrigues JPGLM, de Vries R, Hennink WE, Baldus M, et al. The supramolecular organization of a peptide-based nanocarrier at high molecular detail. J Am Chem Soc. 2015;137:7775.10.1021/jacs.5b02919Search in Google Scholar PubMed
[105] Jekhmane S, Prachar M, Pugliese R, Fontana F, Medeiros-Silva J, Gelain F, et al. Design parameters of tissue-engineering scaffolds at the atomic scale. Angew Chem Int Ed. 2019;58:16943.10.1002/anie.201907880Search in Google Scholar PubMed PubMed Central
[106] Naskar S, Maiti PK. Mechanical properties of DNA and DNA nanostructures: Comparison of atomistic, martini and OxDNA models. J Mater Chem B. 2021;9:5102–13.10.1039/D0TB02970JSearch in Google Scholar
[107] Poppleton E, Romero R, Mallya A, Rovigatti L, Šulc P. OxDNA.Org: A public webserver for coarse-grained simulations of DNA and RNA nanostructures. Nucleic Acids Res. 2021;49:W491–8.10.1093/nar/gkab324Search in Google Scholar PubMed PubMed Central
[108] Marrink SJ, Tieleman DP. Perspective on the martini model. Chem Soc Rev. 2013;42:6801.10.1039/c3cs60093aSearch in Google Scholar PubMed
[109] Souza PCT, Thallmair S, Conflitti P, Ramírez-Palacios C, Alessandri R, Raniolo S, et al. Protein–ligand binding with the coarse-grained martini model. Nat Commun. 2020;11:3714.10.1038/s41467-020-17437-5Search in Google Scholar PubMed PubMed Central
[110] Uusitalo JJ, Ingólfsson HI, Akhshi P, Tieleman DP, Marrink SJ. Martini coarse-grained force field: Extension to DNA. J Chem Theory Comput. 2015;11:3932.10.1021/acs.jctc.5b00286Search in Google Scholar PubMed
[111] Zhou C, Liu K. Molecular dynamics simulation of reversible electroporation with martini force field. Biomed Eng Online. 2019;18:123.10.1186/s12938-019-0743-1Search in Google Scholar PubMed PubMed Central
[112] López CA, Rzepiela AJ, de Vries AH, Dijkhuizen L, Hünen-berger PH, Marrink SJ. Martini coarse-grained force field: Extension to carbohy-drates. J Chem Theory Comput. 2009;5:3195.10.1021/ct900313wSearch in Google Scholar PubMed
[113] Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink S-J. The MARTINI coarse-grained force field: Extension to proteins. J Chem Theory Comput. 2008;4:819.10.1021/ct700324xSearch in Google Scholar PubMed
[114] Kalliauer J, Kahl G, Scheiner S, Hellmich C. A new approach to the mechanics of DNA: Atoms-to-beam homogenization. J Mech Phys Solids. 2020;143:104040.10.1016/j.jmps.2020.104040Search in Google Scholar
[115] Kim Y-J, Kim D-N. Structural basis for elastic mechanical properties of the DNA double helix. PLOS ONE. 2016;11:e015322.10.1371/journal.pone.0153228Search in Google Scholar PubMed PubMed Central
[116] Lee JY, Lee JG, Yun G, Lee C, Kim Y-J, Kim KS, et al. Rapid computational analysis of DNA origami assemblies at near-atomic resolution. ACS Nano. 2021;15:1002–15.10.1021/acsnano.0c07717Search in Google Scholar PubMed
[117] Ayton GS, Noid WG, Voth GA. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr OpStruct Biol. 2007;17:192–8.10.1016/j.sbi.2007.03.004Search in Google Scholar PubMed
[118] Grünewald F, Souza PCT, Abdizadeh H, Barnoud J, de Vries AH, Marrink SJ. Titratable martini model for constant PH simulations. J Chem Phys. 2020;153:024118.10.1063/5.0014258Search in Google Scholar PubMed
[119] Gautieri A, Russo A, Vesentini S, Redaelli A, Buehler MJ. Coarse-grained model of collagen molecules using an extended MARTINI force field. J Chem Theory Comput. 2010;6:1210.10.1021/ct100015vSearch in Google Scholar
[120] Seo M, Rauscher S, Pomès R, Tieleman DP. Improving internal peptide dynamics in the coarse-grained MARTINI Model: Toward large-scale simulations of amyloid-and elastin-like peptides. J Chem Theory Comput. 2012;8:1774.10.1021/ct200876vSearch in Google Scholar PubMed PubMed Central
[121] Poma AB, Cieplak M, Theodorakis PE. Combining the MARTINI and structure-based coarse-grained approaches for the molecular dynamics studies of conformational transitions in proteins. J Chem Theory Comput. 2017;13:1366.10.1021/acs.jctc.6b00986Search in Google Scholar PubMed
[122] Carmichael SP, Shell MS. A new multiscale algorithm and its application to coarse-grained peptide models for self-assembly. J Phys Chem B. 2012;116:8383.10.1021/jp2114994Search in Google Scholar PubMed
[123] Narayanan T, Rüter A, Olsson U. Multiscale structural elucidation of peptide nanotubes by X-Ray scattering methods. Front Bioeng Biotechnol. 2021;9:654339.10.3389/fbioe.2021.654339Search in Google Scholar PubMed PubMed Central
[124] Yuan C, Li S, Zou Q, Ren Y, Yan X. Multiscale simulations for understanding the evolution and mechanism of hierarchical peptide self-assembly. Phys Chem Chem Phys. 2017;19(35):23614–31.10.1039/C7CP01923HSearch in Google Scholar
[125] Zhao X, Liao C, Ma YT, Ferrell JB, Schneebeli ST, Li J. Top-down multiscale approach to simulate peptide self-assembly from monomers. J Chem Theory Comput. 2019;15:1514.10.1021/acs.jctc.8b01025Search in Google Scholar PubMed
[126] Schmalhorst PS, Deluweit F, Scherrers R, Heisenberg CP, Sikora M. Overcoming the limitations of the MARTINI force field in simulations of polysaccharides. J Chem Theory Comput. 2017;13:5039.10.1021/acs.jctc.7b00374Search in Google Scholar PubMed
[127] Fontana F, Gelain F. Probing mechanical properties and failure mechanisms of fibrils of self-assembling peptides. Nanoscale Adv. 2020;2(1):190–8.10.1039/C9NA00621DSearch in Google Scholar
[128] Pasi M, Lavery R, Ceres N. PaLaCe: A coarse-grain protein model for studying mechanical properties. J Chem Theory Comput. 2013;9:785–93.10.1021/ct3007925Search in Google Scholar PubMed
[129] Kar P, Gopal SM, Cheng Y-M, Predeus A, Feig M. PRIMO: A transferable coarse-grained force field for proteins. J Chem Theory Comput. 2013;9:3769–88.10.1021/ct400230ySearch in Google Scholar PubMed PubMed Central
[130] Gopal SM, Mukherjee S, Cheng Y-M, Feig M. PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins: Structure, Function, Bioinforma. 2010;78:1266–81.10.1002/prot.22645Search in Google Scholar PubMed PubMed Central
[131] Kappel K, Das R. Sampling native-like structures of RNA-protein complexes through rosetta folding and docking. Structure. 2019;27:140–51.10.1016/j.str.2018.10.001Search in Google Scholar
[132] Fleishman SJ, Leaver-Fay A, Corn JE, Strauch E-M, Khare SD, Koga N, et al. Rosetta scripts: A scripting language interface to the rosetta macromolecular modeling suite. PLoS ONE. 2011;6:e20161.10.1371/journal.pone.0020161Search in Google Scholar
[133] Basdevant N, Borgis D, Ha-Duong T. Modeling protein–protein recognition in solution using the coarse-grained force field SCORPION. J Chem Theory Comput. 2013;9.10.1021/ct300943wSearch in Google Scholar
[134] Liwo A, Baranowski M, Czaplewski C, Gołas E, He Y, Jagieła D, et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole–multipole interactions. J Mol Modeling. 2014;20:2306.10.1007/s00894-014-2306-5Search in Google Scholar
[135] Senderowitz H, Parish C, Still WC. Carbohydrates: united atom AMBER* parameterization of pyranoses and simulations yielding anomeric free energies. J Am Chem Soc. 1996;118(8):2078–86.10.1021/ja9529652Search in Google Scholar
[136] Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, et al. GLYCAM06: A generalizable biomolecular force field. Carbohyd J Comput Chem. 2008;29:622.10.1002/jcc.20820Search in Google Scholar
[137] Wlodawer A, Nachman J, Gilliland GL, Gallagher W, Woodward C. Structure of form III crystals of bovine pancreatic trypsin inhibitor. J Mol Biol. 1987;198(3):469–80.10.1016/0022-2836(87)90294-4Search in Google Scholar
[138] Brunne RM, van Gunsteren WF. Dynamical properties of bovine pancreatic trypsin inhibitor from a molecular dynamics simulation at 5000 atm. FEBS Lett. 1993;323(3):215–7.10.1016/0014-5793(93)81342-WSearch in Google Scholar
[139] Sengar A, Ouldridge TE, Henrich O, Rovigatti L, Šulc P. A primer on the OxDNA model of DNA: When to use it, how to simulate it and how to interpret the results. Front Mol Biosci. 2021;8:693710.10.3389/fmolb.2021.693710Search in Google Scholar PubMed PubMed Central
[140] Denesyuk NA, Thirumalai, D. Coarse-grained model for predicting RNA folding thermodynamics. J Phys Chem B. 2013;117:4901–11.10.1021/jp401087xSearch in Google Scholar PubMed
[141] Denesyuk NA, Thirumalai D. How do metal ions direct ribozyme folding? Nat Chem. 2015;7:793–801.10.1038/nchem.2330Search in Google Scholar PubMed
[142] Denesyuk NA, Hori N, Thirumalai D. Molecular simulations of ion effects on the thermodynamics of RNA folding. J Phys Chem B. 2018;122:11860–7.10.1021/acs.jpcb.8b08142Search in Google Scholar PubMed
[143] Lu W, Bueno C, Schafer NP, Moller J, Jin S, Chen X, et al. OpenAWSEM with Open3SPN2: A fast, flexible, and accessible framework for large-scale coarse-grained biomolecular simulations. 2020;17(2):e1008308.10.1371/journal.pcbi.1008308Search in Google Scholar PubMed PubMed Central
[144] Li Y, Shao M, Zheng X, Kong W, Zhang J, Gong M. Self-assembling peptides improve the stability of glucagon-like peptide-1 by forming a stable and sustained complex. Mol Pharmaceutics. 2013;10:3356.10.1021/mp4001734Search in Google Scholar PubMed
[145] Gautieri A, Milani A, Pizzi A, Rigoldi F, Redaelli A, Metrangolo P. Molecular dynamics investigation of halogenated amyloidogenic peptides. J Mol Modeling. 2019;25:124.10.1007/s00894-019-4012-9Search in Google Scholar PubMed
[146] Nunes RS, Vila-Viçosa D, Costa PJ. Halogen bonding: An underestimated player in membrane–ligand interactions. J Am Chem Soc. 2021;143:4253–67.10.1021/jacs.0c12470Search in Google Scholar PubMed
[147] Nunes R, Vila-Viçosa D, Machuqueiro M, Costa PJ. Biomolecular simulations of halogen bonds with a GROMOS force field. J Chem Theory Comput. 2018;14:5383.10.1021/acs.jctc.8b00278Search in Google Scholar PubMed
[148] Thurston BA, Ferguson AL. Machine learning and molecular design of self-assembling π-conjugated oligopeptides. Mol Simul. 2018;44(11):930–45.10.1080/08927022.2018.1469754Search in Google Scholar
[149] Thurston BA, Shapera EP, Tovar JD, Schleife A, Ferguson AL. Revealing the sequence-structure-electronic property relation of self-assembling π-conjugated oligopeptides by molecular and quantum mechanical modeling. Lang-Muir. 2019;35:15221.10.1021/acs.langmuir.9b02593Search in Google Scholar PubMed
[150] Fadda E, Woods RJ. Molecular simulations of carbohydrates and protein-carbohydrate interactions: Motivation, issues and prospects. Drug Discovery Today. 2010;15(15–16):596–609.10.1016/j.drudis.2010.06.001Search in Google Scholar PubMed PubMed Central
[151] Durrant JD, Kochanek SE, Casalino L, Ieong PU, Dommer AC, Amaro RE. Mesoscale all-atom influenza virus simulations suggest new substrate binding mechanism. ACS Cent Sci. 2020;6:189–96.10.1021/acscentsci.9b01071Search in Google Scholar PubMed PubMed Central
[152] Zgarbová M, Jurečka P, Šponer J, Otyepka M. A to B-DNA transition in AMBER force fields and its coupling to sugar pucker. J Chem Theory Comput. 2018;14:319.10.1021/acs.jctc.7b00926Search in Google Scholar PubMed
[153] Arora N, Jayaram B. Energetics of base pairs in B-DNA in solution: An appraisal of potential functions and dielectric treatments. J Phys Chem B. 1998;102(31):6139–44.10.1021/jp9813692Search in Google Scholar
[154] Zgarbová M, Šponer J, Otyepka M, Cheatham TE, Galindo-Murillo R, Jurecˇka P. Refinement of the sugar-phosphate backbone torsion beta for amber force fields improves the description of Z- and B-DNA. J Chem Theory Comput. 2015;11:5723.10.1021/acs.jctc.5b00716Search in Google Scholar PubMed
[155] Cordomí A, Caltabiano G, Pardo L. Membrane protein simulations using AMBER force field and berger lipid parameters. J Chem Theory Comput. 2012;8:948.10.1021/ct200491cSearch in Google Scholar PubMed
[156] Dickson CJ, Madej BD, Skjevik ÅA, Betz RM, Teigen K, Gould IR, et al. Lipid14: The amber lipid force field. J Chem Theory Comput. 2014;10:865.10.1021/ct4010307Search in Google Scholar PubMed PubMed Central
[157] Skjevik ÅA, Madej BD, Walker RC, Teigen K. LIPID11: A modular framework for lipid simulations using amber. J Phys Chem B. 2012;116:11124.10.1021/jp3059992Search in Google Scholar PubMed PubMed Central
[158] Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved peptide and protein torsional energetics with the OPLS-AA force field. J Chem Theory Comput. 2015;11:3499.10.1021/acs.jctc.5b00356Search in Google Scholar PubMed PubMed Central
[159] Maciejewski A, Pasenkiewicz-Gierula M, Cramariuc O, Vattulainen I, Rog T. Refined OPLS all-atom force field for saturated phosphatidylcholine bilayers at full hydration. J Phys Chem B. 2014;118:4571–81.10.1021/jp5016627Search in Google Scholar PubMed
[160] Robertson MJ, Qian Y, Robinson MC, Tirado-Rives J, Jorgensen WL. Development and testing of the OPLS-AA/M force field for RNA. J Chem Theory Comput. 2019;15:2734.10.1021/acs.jctc.9b00054Search in Google Scholar PubMed PubMed Central
[161] Krepl M, Zgarbová M, Stadlbauer P, Otyepka M, Banáš P, Kocˇa J, et al. Reference simulations of noncanonical nucleic acids with different X variants of the AMBER force field: Quadruplex DNA, quadruplex RNA, and Z-DNA. J Chem Theory Comput. 2012;8:2506.10.1021/ct300275sSearch in Google Scholar PubMed PubMed Central
[162] Smith MD, Rao JS, Segelken E, Cruz L. Force-field induced bias in the structure of Aβ21-30: A comparison of OPLS, AMBER, CHARMM, and GRO-MOS force fields. J Chem Inf Modeling. 2015;55:2587.10.1021/acs.jcim.5b00308Search in Google Scholar PubMed
[163] Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the opls all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–36.10.1021/ja9621760Search in Google Scholar
[164] Kony D, Damm W, Stoll S, van Gunsteren WF. An improved OPLS-AA force field for carbohydrates. J Comput Chem. 2002;23:1416–29.10.1002/jcc.10139Search in Google Scholar PubMed
[165] Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671.10.1002/jcc.21367Search in Google Scholar PubMed PubMed Central
[166] Venable RM, Sodt AJ, Rogaski B, Rui H, Hatcher E, MacKerell AD, et al. CHARMM all-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive curvature. Biophys J. 2014;107:134–45.10.1016/j.bpj.2014.05.034Search in Google Scholar PubMed PubMed Central
[167] Ramos Sasselli I, Ulijn RV, Tuttle T. CHARMM force field parameterization protocol for self-assembling peptide amphiphiles: The fmoc moiety. Phys Chem Chem Phys. 2016;18:4659.10.1039/C5CP06770GSearch in Google Scholar PubMed
[168] Pol-Fachin L, Rusu VH, Verli H, Lins RD. GROMOS 53A6 GLYC, an improved GROMOS force field for hexopyranose-based carbohydrates. J Chem Theory Comput. 2012;8:4681.10.1021/ct300479hSearch in Google Scholar PubMed
[169] Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, et al. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophysics J. 2011;40:843.10.1007/s00249-011-0700-9Search in Google Scholar PubMed
[170] Oostenbrink C, Villa A, Mark AE, van Gunsteren, WF. A Biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656.10.1002/jcc.20090Search in Google Scholar PubMed
[171] Oostenbrink C, Soares TA, van der Vegt NFA, Van Gun-Steren WF. Validation of the 53A6 GROMOS force field. Eur Biophysics J. 2005;34:273.10.1007/s00249-004-0448-6Search in Google Scholar PubMed
[172] Huang W, Lin Z, Van Gunsteren WF. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J Chem Theory Comput. 2011;7:1237.10.1021/ct100747ySearch in Google Scholar PubMed
[173] Nester K, Gaweda K, Plazinski W. A GROMOS force field for furanose-based carbohydrates. J Chem Theory Comput. 2019;15:1168.10.1021/acs.jctc.8b00838Search in Google Scholar PubMed
[174] Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, et al. Current status of the AMOEBA polarizable force field. J Phys Chem B. 2010;114:2549.10.1021/jp910674dSearch in Google Scholar PubMed PubMed Central
[175] Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, et al. Polarizable atomic multipole-based AMOEBA force field for proteins. J Chem Theory Comput. 2013;9:4046.10.1021/ct4003702Search in Google Scholar PubMed PubMed Central
[176] Zhang C, Lu C, Jing Z, Wu C, Piquemal JP, Ponder JW, et al. AMOEBA polarizable atomic multipole force field for nucleic acids. J Chem Theory Comput. 2018;14:2084.10.1021/acs.jctc.7b01169Search in Google Scholar PubMed PubMed Central
[177] Jin S, Contessoto VG, Chen M, Schafer NP, Lu W, Chen X, et al. AWSEM-Suite: A protein structure prediction server based on template-guided, coevolutionary-enhanced optimized folding landscapes. Nucleic Acids Res. 2020;48:W25.10.1093/nar/gkaa356Search in Google Scholar PubMed PubMed Central
[178] Davtyan A, Schafer NP, Zheng W, Clementi C, Wolynes PG, Papoian GA. AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012;116:8494–503.10.1021/jp212541ySearch in Google Scholar PubMed PubMed Central
[179] Sterpone F, Melchionna S, Tuffery P, Pasquali S, Mousseau N, Crag-nolini T, et al. The OPEP protein model: From single molecules, amyloid formation, crowding and hydrodynamics to DNA/RNA systems. Chem Soc Rev. 2014;43:4871–93.10.1039/C4CS00048JSearch in Google Scholar
[180] Sterpone F, Derreumaux P, Melchionna S. Protein simulations in fluids: Coupling the OPEP coarse-grained force field with hydrodynamics. J Chem Theory Comput. 2015;11:1843–53.10.1021/ct501015hSearch in Google Scholar PubMed PubMed Central
[181] Bereau T, Deserno M. Generic coarse-grained model for protein folding and aggregation. J Chem Phys. 2009;130(23):235106.10.1063/1.3152842Search in Google Scholar PubMed PubMed Central
[182] Bereau T, Bachmann M, Deserno M. Interplay between secondary and tertiary structure formation in protein folding cooperativity. J Am Chem Soc. 2010;132:13129.10.1021/ja105206wSearch in Google Scholar PubMed PubMed Central
[183] Kolinski A. Protein modeling and structure prediction with a reduced representation. Acta Biochimica Polonica. 2004;51(2):349–71.10.18388/abp.2004_3575Search in Google Scholar
[184] Kurcinski M, Kolinski A, Kmiecik S. Mechanism of folding and binding of an intrinsically disordered protein as revealed by ab initio simulations. J Chem Theory Comput. 2014;10:2224–31.10.1021/ct500287cSearch in Google Scholar PubMed
[185] Kmiecik S, Kolinski A. Simulation of chaperonin effect on protein folding: A shift from nucleation – condensation to framework mechanism. J Am Chem Soc. 2011;133:10283.10.1021/ja203275fSearch in Google Scholar PubMed PubMed Central
[186] Jamroz M, Kolinski A. Modeling of loops in proteins: A multi-method approach. BMC Struct Biol. 2010;10(1):1–9.10.1186/1472-6807-10-5Search in Google Scholar PubMed PubMed Central
[187] Kmiecik S, Kolinski A. Folding pathway of the B1 domain of protein G explored by multiscale modeling. Biophys J. 2008;94:726.10.1529/biophysj.107.116095Search in Google Scholar PubMed PubMed Central
[188] Šulc P, Ouldridge TE, Romano F, Doye JPK, Louis AA. Modelling toehold-mediated RNA strand displacement. Biophys J. 2015;108:1238.10.1016/j.bpj.2015.01.023Search in Google Scholar PubMed PubMed Central
[189] Shi Z, Arya G. Free energy landscape of salt-actuated reconfigurable DNA nanodevices. Nucleic Acids Res. 2020;48:548.10.1093/nar/gkz1137Search in Google Scholar PubMed PubMed Central
[190] Hyeon C, Thirumalai D. Mechanical unfolding of RNA hairpins. Proc Natl Acad Sci. 2005;102(19):6789–94.10.1073/pnas.0408314102Search in Google Scholar PubMed PubMed Central
[191] Faustino I, Marrink SJ. CgHeliParm: Analysis of DsDNA helical parameters for coarse-grained MARTINI molecular dynamics simulations. Bioinformatics. 2017;33:3813.10.1093/bioinformatics/btx444Search in Google Scholar PubMed
[192] Molinero V, Goddard WA. M3B: A coarse grain force field for molecular simulations of malto-oligosaccharides and their water mixtures. J Phys Chem B. 2004;108:1414–27.10.1021/jp0354752Search in Google Scholar
[193] Shivgan AT, Marzinek JK, Huber RG, Krah A, Henchman RH, Matsudaira P, et al. Extending the martini coarse-grained force field to N-glycans. J Chem Inf Modeling. 2020;60:3864–83.10.1021/acs.jcim.0c00495Search in Google Scholar PubMed
[194] Zhang D, Howarth GS, Parkin LA, McDermott AE. NMR studies of lipid regulation of the K+ channel KcsA. Biochim Biophys Acta (BBA) – Biomembranes. 2021;1863:183491.10.1016/j.bbamem.2020.183491Search in Google Scholar PubMed PubMed Central
[195] Phyo P, Zhao X, Templeton AC, Xu W, Cheung JK, Su Y. Understanding molecular mechanisms of biologics drug delivery and stability from NMR spectroscopy. Adv Drug Delivery Rev. 2021;174:1–29.10.1016/j.addr.2021.02.007Search in Google Scholar PubMed
[196] Maeda YT, Nakadai T, Shin J, Uryu K, Noireaux V, Libchaber A. Assembly of MreB filaments on liposome membranes: A synthetic biology approach. ACS Synth Biol. 2012;1:53–9.10.1021/sb200003vSearch in Google Scholar PubMed
[197] Schwille P, Diez S. Synthetic biology of minimal systems. Crit Rev Biochem Mol Biol. 2009;44:223–42.10.1080/10409230903074549Search in Google Scholar PubMed
[198] Venable RM, Krämer A, Pastor RW. Molecular dynamics simulations of membrane permeability. Chem Rev. 2019;119(9):5954–97.10.1021/acs.chemrev.8b00486Search in Google Scholar PubMed PubMed Central
[199] Ermilova I, Lyubartsev AP. Extension of the slipids force field to polyunsaturated lipids. J Phys Chem B. 2016;120:12826–42.10.1021/acs.jpcb.6b05422Search in Google Scholar PubMed
[200] Grote F, Lyubartsev AP. Optimization of slipids force field parameters describing headgroups of phospholipids. J Phys Chem B. 2020;124:8784–93.10.1021/acs.jpcb.0c06386Search in Google Scholar PubMed PubMed Central
[201] Corradi V, Mendez-Villuendas E, Ingolfsson HI, Gu R, Siuda I, Melo MN, et al. Lipid−protein interactions are unique fingerprints for membrane proteins. ACS Cent Sci. 2018;4:709–17.10.1021/acscentsci.8b00143Search in Google Scholar PubMed PubMed Central
[202] Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, Lopez CA, Wassenaar TA, et al. Lipid organization of the plasma membrane. J Am Chem Soc. 2014;136:14554.10.1021/ja507832eSearch in Google Scholar PubMed
[203] Ingólfsson HI, Carpenter TS, Bhatia H, Bremer P-T, Marrink SJ, Lightstone FC. Computational lipidomics of the neuronal plasma membrane. Biophys J. 2017;113:2271.10.1016/j.bpj.2017.10.017Search in Google Scholar PubMed PubMed Central
[204] Lim L, Wenk MR. Neuronal membrane lipids – their role in the synaptic vesicle cycle. In: Lajtha, A., Tettamanti, G., Goracci, G. (eds) Handbook of neurochemistry and molecular neurobiology. Boston, MA: Springer; 2009. p. 223–38.10.1007/978-0-387-30378-9_9Search in Google Scholar
[205] Levitan I, Fang Y, Rosenhouse-Dantsker A, Romanenko V. Cholesterol and ion channels. Subcell Biochem. 2010;51:509–49. 10.1007/978-90-481-8622-8_19. PMID: 20213557; PMCID: PMC2895485.Search in Google Scholar PubMed PubMed Central
[206] Dart C. SYMPOSIUM REVIEW: Lipid microdomains and the regulation of ion channel function. J Physiol. 2010;588:3169.10.1113/jphysiol.2010.191585Search in Google Scholar PubMed PubMed Central
[207] Tang PK, Manandhar A, Hu W, Kang M, Loverde SM. The interaction of supramolecular anticancer drug amphiphiles with phospholipid membranes. Nanoscale Adv. 2021;3:370.10.1039/D0NA00697ASearch in Google Scholar PubMed PubMed Central
[208] Qin W, Li X, Bian WW, Fan XJ, Qi JY. Density functional theory calculations and molecular dynamics simulations of the adsorption of biomolecules on graphene surfaces. Biomaterials. 2010;31:1007.10.1016/j.biomaterials.2009.10.013Search in Google Scholar PubMed
[209] Garrain P-A, Costa D, Marcus P. Biomaterial−biomolecule interaction: DFT-D study of glycine adsorption on Cr2O3. J Phys Chem C. 2011;115:719.10.1021/jp109704bSearch in Google Scholar
[210] Jurecˇka P, Šponer J, JCˇP, Hobza. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys Chem Chem Phys. 2006;8:1985.10.1039/B600027DSearch in Google Scholar PubMed
[211] Lou Z, Zeng Q, Chu X, Yang F, He D, Yang M, et al. First-principles study of the adsorption of lysine on hydroxyapatite (100) surface. Appl Surf Sci. 2012;258:4911–6.10.1016/j.apsusc.2012.01.116Search in Google Scholar
[212] Corno M, Rimola A, Bolis V, Ugliengo P. Hydroxyapatite as a key biomaterial: Quantum-mechanical simulation of its surfaces in interaction with biomolecules. Phys Chem Chem Phys. 2010;12:6309.10.1039/c002146fSearch in Google Scholar PubMed
[213] Ajeel FN, Khudhair AM, Mohammed MH, Mahdi KM. DFT investigation of graphene nanoribbon as a potential nanobiosensor for tyrosine amino acid. Russian J Phys Chem A. 2019;93:778.10.1134/S0036024419040022Search in Google Scholar
[214] Espargaró A, Llabrés S, Saupe SJ, Curutchet C, Luque FJ, Sabaté R. On the binding of congo red to amyloid fibrils. Angew Chem. 2020;132:8104–7.10.1002/anie.201916630Search in Google Scholar PubMed
[215] Fleming S, Frederix PWJM, Ramos Sasselli I, Hunt NT, Ulijn RV, Tuttle T. Assessing the utility of infrared spectroscopy as a structural diagnostic tool for β-sheets in self-assembling aromatic peptide amphiphiles. Langmuir. 2013;29:9510.10.1021/la400994vSearch in Google Scholar PubMed
[216] Silva CB, da Silva Filho JG, Pinheiro GS, Teixeira AMR, Freire PTC. Vibrational and structural properties of L-alanyl-L-phenylalanine dipeptide by raman spectroscopy, infrared and DFT calculations. Vibrational Spectrosc. 2018;98:128.10.1016/j.vibspec.2018.08.001Search in Google Scholar
[217] Li L, Zhan H, Duan P, Liao J, Quan J, Hu Y, et al. Self-assembling nanotubes consisting of rigid cyclic γ-peptides. Adv Funct Mater. 2012;22:3051.10.1002/adfm.201200488Search in Google Scholar
[218] Raskatov JA, Foley AR, Louis JM, Yau W-M, Tycko R. Constraints on the structure of fibrils formed by a racemic mixture of amyloid-β peptides from solid-state NMR, electron microscopy, and theory. J Am Chem Soc. 2021;143(33):13299–313.10.1021/jacs.1c06339Search in Google Scholar PubMed PubMed Central
[219] Momany F, Schnupf U. DFT optimization and DFT-MD studies of glucose, ten explicit water molecules enclosed by an implicit solvent. COSMO, Comput Theor Chem. 2014;1029:57.10.1016/j.comptc.2013.12.007Search in Google Scholar
[220] Momany FA, Willett JL, Schnupf U. DFT molecular dynamics (DFTMD) simulations of carbohydrates: COSMO solvated α-Maltose. J Mol Structure THEOCHEM. 2010;953:61.10.1016/j.theochem.2010.05.004Search in Google Scholar
[221] Xue J, Guo X, Wang X, Xiao Y. Density functional theory studies on cytosine analogues for inducing double-proton transfer with guanine. Sci Rep. 2020;10:9671.10.1038/s41598-020-66530-8Search in Google Scholar PubMed PubMed Central
[222] Deng A, Li H, Bo M, Huang ZK, Li L, Yao C, et al. Understanding atomic bonding and electronic distributions of a DNA molecule using DFT calculation and BOLS-BC model. Biochem Biophysics Rep. 2020;24:100804.10.1016/j.bbrep.2020.100804Search in Google Scholar PubMed PubMed Central
[223] Faramarzi V, Ahmadi V, Fotouhi B, Abasifard M. A potential sensing mechanism for DNA nucleobases by optical properties of GO and MoS2 nanopores. Sci Rep. 2019;9:6230.10.1038/s41598-019-41165-6Search in Google Scholar PubMed PubMed Central
[224] Kong Z, Hu W, Jiao F, Zhang P, Shen J, Cui B, et al. Theoretical evaluation of DNA genotoxicity of graphene quantum dots: A combination of density functional theory and molecular dynamics simulations. J Phys Chem B. 2020;124:9335.10.1021/acs.jpcb.0c05882Search in Google Scholar PubMed
[225] Frink LJD, Frischknecht AL, Heroux MA, Parks ML, Salinger AG. Toward quantitative coarse-grained models of lipids with fluids density functional theory. J Chem Theory Comput. 2012;8:1393.10.1021/ct200707bSearch in Google Scholar PubMed
[226] Schahl A, Réat V, Jolibois F. Structures and NMR spectra of short amylose-lipid complexes. insight using molecular dynamics and DFT quantum chemical calculations. Carbohydr Polym. 2020;235:115846.10.1016/j.carbpol.2020.115846Search in Google Scholar PubMed
[227] Man L, Yang Y, Wang H, Wang Y, An Y, Bao J, et al. In situ-cross-linked supramolecular eco-binders for improved capacity and stability of lithium-sulfur batteries. ACS Appl Energy Mater. 2021;4:3803.10.1021/acsaem.1c00207Search in Google Scholar
[228] Wang M, Zhao Y, Zhang L, Deng J, Qi K, Zhou P, et al. Unexpected role of achiral glycine in determining the suprastructural handedness of peptide nanofibrils. ACS Nano. 2021;15:10328.10.1021/acsnano.1c02547Search in Google Scholar PubMed
[229] Movilla F, Rey JM, Huck-Iriart C, di Salvo F. Amine-derivatized l-phenylalanine and l-tyrosine as versatile self-assembled platforms of diverse supramolecular architectures: From mesocrystals to organogels. Cryst Growth Des. 2021;21:3487.10.1021/acs.cgd.1c00274Search in Google Scholar
[230] Tao K, Makam P, Aizen R, Gazit E. Self-assembling peptide Semiconductors. Science. 2017;358(6365):eaam9756.10.1126/science.aam9756Search in Google Scholar PubMed PubMed Central
[231] Khan MA, Cantù E, Tonello S, Serpelloni M, Lopomo NF, Sardini E. A review on biomaterials for 3D conductive scaffolds for stimulating and monitoring cellular activities. Appl Sci. 2019;9(5):961.10.3390/app9050961Search in Google Scholar
[232] Mostafavi E, Medina-Cruz D, Kalantari K, Taymoori A, Soltantabar P, Webster TJ. Electroconductive nanobiomaterials for tissue engineering and regenerative medicine. Bioelectricity. 2020;2:120.10.1089/bioe.2020.0021Search in Google Scholar
[233] Nazari ZE, Herrero JG, Fojan P, Gurevich L. Formation of conductive DNA-based nanowires via conjugation of DsDNA with cationic peptide. Nano-Materials. 2017;7:128.10.3390/nano7060128Search in Google Scholar
[234] Pak AJ, Voth GA. Advances in coarse-grained modeling of macro-molecular complexes. Curr OpStruct Biol. 2018;52:119–26.10.1016/j.sbi.2018.11.005Search in Google Scholar
[235] Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505.10.1016/S0006-3495(01)76033-XSearch in Google Scholar
[236] Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys Rev Lett. 1997;79(16):3090.10.1103/PhysRevLett.79.3090Search in Google Scholar
[237] Tirion MM. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett. 1996;77(9):1905.10.1103/PhysRevLett.77.1905Search in Google Scholar PubMed
[238] Zhang Y, Cao Z, Xia F. Construction of ultra-coarse-grained model of protein with a Gō-like potential. Chem Phys Lett. 2017;681:1.10.1016/j.cplett.2017.05.039Search in Google Scholar
[239] Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP)–Round XII. Proteins: Structure, Funct Bioinforma. 2018;86:7.10.1002/prot.25415Search in Google Scholar PubMed PubMed Central
[240] Schöberl M, Zabaras N, Koutsourelakis P-S. Predictive coarse-graining. J Comput Phys. 2017;333:49–77.10.1016/j.jcp.2016.10.073Search in Google Scholar
[241] Farrell K, Oden JT, Faghihi, D. A bayesian framework for adaptive selection, calibration, and validation of coarse-grained models of atomistic systems. J Comput Phys. 2015;295:189–208.10.1016/j.jcp.2015.03.071Search in Google Scholar
[242] Moritsugu K, Kurkal-Siebert V, Smith JC. REACH coarse-grained normal mode analysis of protein dimer interaction dynamics. Biophys J. 2009;97:1158.10.1016/j.bpj.2009.05.015Search in Google Scholar PubMed PubMed Central
[243] Orellana L, Yoluk O, Carrillo O, Orozco M, Lindahl E. Prediction and validation of protein intermediate states from structurally rich ensembles and coarse-grained simulations. Nat Commun. 2016;7:12575.10.1038/ncomms12575Search in Google Scholar PubMed PubMed Central
[244] Lyman E, Pfaendtner J, Voth GA. Systematic multiscale parameterization of heterogeneous elastic network models of proteins. Biophys J. 2008;95:4183.10.1529/biophysj.108.139733Search in Google Scholar PubMed PubMed Central
[245] Dama JF, Jin J, Voth GA. The theory of ultra-coarse-graining. 3. coarse-grained sites with rapid local equilibrium of internal states. J Chem Theory Comput. 2017;13:1010.10.1021/acs.jctc.6b01081Search in Google Scholar PubMed
[246] Zhang Y, Cao Z, Zhang JZ, Xia F. Double-well ultra-coarse-grained model to describe protein conformational transitions. J Chem Theory Comput. 2020;16:6678.10.1021/acs.jctc.0c00551Search in Google Scholar PubMed
[247] Yeo J, Jung GS, Tarakanova A, Martín-Martínez FJ, Qin Z, Cheng Y, et al. Multiscale modeling of keratin, collagen, elastin and related human diseases: Perspectives from atomistic to coarse-grained molecular dynamics simulations. Extreme Mech Lett. 2018;20:112.10.1016/j.eml.2018.01.009Search in Google Scholar PubMed PubMed Central
[248] Seob Jung G, Buehler MJ. Downloaded from Www.Annualreviews.Org Access Provided by 172.58.187.196 on 08/13/21. For personal use only. Annu Rev Biomed Eng. 2017;19:435.Search in Google Scholar
[249] López Barreiro D, Yeo J, Tarakanova A, Martin-Martinez FJ, Buehler MJ. Multiscale modeling of silk and silk-based biomaterials–A review. Macro-Molecular Biosci. 2019;19:1800253.10.1002/mabi.201800253Search in Google Scholar PubMed
[250] Golas EI, Czaplewski C. Rapid communication: computational simulation and analysis of a candidate for the design of a novel silk-based biopolymer. Biopolymers. 2014;101:915–23.10.1002/bip.22494Search in Google Scholar PubMed
[251] Crowet J, Nasir M, Dony N, Deschamps A, Stroobant V, Morsomme P, et al. Insight into the self-assembling properties of peptergents: A molecular dynamics simulation study. Int J Mol Sci. 2018;19:2772.10.3390/ijms19092772Search in Google Scholar PubMed PubMed Central
[252] Keten S, Xu Z, Ihle B, Buehler MJ. Nanoconfinement controls stiffness, strength and mechanical toughness of β-sheet crystals in silk. Nat Mater. 2010;9:359–67.10.1038/nmat2704Search in Google Scholar PubMed
[253] Cheng J, Baldi P. Improved residue contact prediction using support vector machines and a large feature set. BMC Bioinforma. 2007;8:113.10.1186/1471-2105-8-113Search in Google Scholar PubMed PubMed Central
[254] Kerner J, Dogan A, von Recum H. Machine learning and big data provide crucial insight for future biomaterials discovery and research. Acta Biomater. 2021;130:54–65.10.1016/j.actbio.2021.05.053Search in Google Scholar PubMed
[255] Baek M, Dimaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373(6557):871–6.10.1126/science.abj8754Search in Google Scholar PubMed PubMed Central
[256] Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with alphafold. Nature. 2021;596:583.10.1038/s41586-021-03819-2Search in Google Scholar PubMed PubMed Central
[257] Castro CE, Dong J, Boyce MC, Lindquist S, Lang MJ. Physical properties of polymorphic yeast prion amyloid fibers. Biophys J. 2011;101:439–48.10.1016/j.bpj.2011.06.016Search in Google Scholar PubMed PubMed Central
[258] Lamour G, Yip CK, Li H, Gsponer J. High intrinsic mechanical flexibility of mouse prion nanofibrils revealed by measurements of axial and radial Young’s moduli. ACS Nano. 2014;8:3851–61.10.1021/nn5007013Search in Google Scholar PubMed
[259] Vandenakker CC, Engel MFM, Velikov KP, Bonn M, Koenderink GH. Morphology and persistence length of amyloid fibrils are correlated to peptide molecular structure. J Am Chem Soc. 2011;133:18030.10.1021/ja206513rSearch in Google Scholar PubMed
[260] Choi B, Kim T, Ahn ES, Lee SW, Eom K. Mechanical Deformation mechanisms and properties of prion fibrils probed by atomistic simulations. Nanoscale Res Lett. 2017;12:228.10.1186/s11671-017-1966-3Search in Google Scholar PubMed PubMed Central
[261] Lamour G, Nassar R, Chan PHW, Bozkurt G, Li J, Bui JM, et al. Mapping the broad structural and mechanical properties of amyloid fibrils. Biophys J. 2017;112:584.10.1016/j.bpj.2016.12.036Search in Google Scholar PubMed PubMed Central
[262] Sivaramakrishnan S, Spink BJ, Sim AYL, Doniach S, Spudich JA. Dynamic charge interactions create surprising rigidity in the ER/K-helical protein motif. Proc Nat Acad Sci. 2008;105:1335610.1073/pnas.0806256105Search in Google Scholar
[263] Ruiz L, VonAchen P, Lazzara TD, Xu T, Keten S. Persistence length and stochastic fragmentation of supramolecular nanotubes under mechanical force. Nanotechnology. 2013;24:195103.10.1088/0957-4484/24/19/195103Search in Google Scholar
[264] Sun Y-L, Luo Z-P, Fertala A, An K-N. Direct quantification of the flexibility of type I collagen monomer. Biochem Biophys Res Commun. 2002;295:382–6.10.1016/S0006-291X(02)00685-XSearch in Google Scholar
[265] Pradhan SM, Katti DR, Katti KS. Steered molecular dynamics study of mechanical response of full length and short collagen molecules. J Nanomech Micromech. 2011;1:104–10.10.1061/(ASCE)NM.2153-5477.0000035Search in Google Scholar
[266] Ghodsi H, Darvish K. Investigation of mechanisms of viscoelastic behavior of collagen molecule. J Mech Behav Biomed Mater. 2015;51:194–204.10.1016/j.jmbbm.2015.07.015Search in Google Scholar PubMed PubMed Central
[267] Gautieri A, Vesentini S, Montevecchi FM, Redaelli A. Mechanical properties of physiological and pathological models of collagen peptides investigated via steered molecular dynamics simulations. J Biomech. 2008;41:3073–7.10.1016/j.jbiomech.2008.06.028Search in Google Scholar PubMed
[268] Muthoka RM, Kim HC, Kim JW, Zhai L, Panicker PS, Kim J. Steered pull simulation to determine nanomechanical properties of cellulose nanofiber. Materials. 2020;13:710.10.3390/ma13030710Search in Google Scholar PubMed PubMed Central
[269] Trulove PC, Reichert WM, de Long HC, Kline S, Rahatekar S, Gilman J, et al. The structure and dynamics of silk and cellulose dissolved in ionic liquids. ECS Trans. 2019;16.10.1149/1.3159315Search in Google Scholar
[270] Hoogendam CW, de Keizer A, Cohen Stuart MA, Bijsterbosch BH, Smit JAM, van Dijk JAPP, et al. Persistence length of carboxymethyl cellulose as evaluated from size exclusion chromatography and potentiometric titrations. Macromolecules. 1998;31:6297–309.10.1021/ma971032iSearch in Google Scholar
[271] Guhados G, Wan W, Hutter JL. Measurement of the elastic modulus of single bacterial cellulose fibers using atomic force microscopy. Langmuir. 2005;21:6642–6.10.1021/la0504311Search in Google Scholar PubMed
[272] Kroon-Batenburg LMJ, Kruiskamp PH, Vliegenthart JFG, Kroon J. Estimation of the persistence length of polymers by MD simulations on small fragments in solution. Application to cellulose. J Phys Chem B. 1997;101:8454–9.10.1021/jp971717kSearch in Google Scholar
[273] Isambert H, Venier P, Maggs AC, Fattoum A, Kassab R, Pantaloni D, et al. Flexibility of actin filaments derived from thermal fluctuations. J Biol Chem. 1995;270:11437–44.10.1074/jbc.270.19.11437Search in Google Scholar
[274] Yanagida T, Nakase M, Nishiyama K, Oosawa F. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature. 1984;307:58–60.10.1038/307058a0Search in Google Scholar
[275] Mehrafrooz B, Shamloo A. Mechanical differences between ATP and ADP actin states: A molecular dynamics study. J Theor Biol. 2018;448:94–103.10.1016/j.jtbi.2018.04.010Search in Google Scholar
[276] Lepore E, Isaia M, Mammola S, Pugno N. The effect of ageing on the mechanical properties of the silk of the bridge spider larinioides cornutus (Clerck, 1757). Sci Rep. 2016;6:24699.10.1038/srep24699Search in Google Scholar
[277] Lee M, Kwon J, Na S. Mechanical behavior comparison of spider and silkworm silks using molecular dynamics at atomic scale. Phys Chem Chem Phys. 2016;18:4814–21.10.1039/C5CP06809FSearch in Google Scholar
[278] Bathe M, Rutledge GC, Grodzinsky AJ, Tidor B. A coarse-grained molecular model for glycosaminoglycans: Application to chondroitin, chondroitin sulfate, and hyaluronic acid. Biophys J. 2005;88:3870–87.10.1529/biophysj.104.058800Search in Google Scholar
[279] Berezney JP, Saleh OA. Electrostatic effects on the conformation and elasticity of hyaluronic acid, a moderately flexible polyelectrolyte. Macromolecules. 2017;50:1085–9.10.1021/acs.macromol.6b02166Search in Google Scholar
[280] Sasaki N, Odajima S. Stress-strain curve and Young’s modulus of a collagen molecule as determined by the X-ray diffraction technique. J Biomech. 1996;29:655–8.10.1016/0021-9290(95)00110-7Search in Google Scholar
[281] Serapian SA, Marchetti F, Triveri A, Morra G, Meli M, Moroni E, et al. The answer lies in the energy: How simple atomistic molecular dynamics simulations may hold the key to epitope prediction on the fully glycosylated SARS-CoV-2 spike protein. J Phys Chem Lett. 2020;11:8084.10.1021/acs.jpclett.0c02341Search in Google Scholar PubMed PubMed Central
[282] Iscen A, Schatz GC. Peptide amphiphile self-assembly. EPL (Euro-physics Lett). 2017;119:38002.10.1209/0295-5075/119/38002Search in Google Scholar
[283] Yu T, Lee O-S, Schatz GC. Steered molecular dynamics studies of the potential of mean force for peptide amphiphile self-assembly into cylindrical nanofibers. J Phys Chem A. 2013;117:7453.10.1021/jp401508wSearch in Google Scholar PubMed
[284] Lee O-S, Cho V, Schatz GC. Modeling the self-assembly of peptide amphiphiles into fibers using coarse-grained molecular dynamics. Nano Lett. 2012;12:4907.10.1021/nl302487mSearch in Google Scholar PubMed
[285] Lau C, Fontana F, Mandemaker LDB, Wezendonk D, Vermeer B, Bonvin AMJJ, et al. Control over the fibrillization yield by varying the oligomeric nucleation propensities of selfassembling peptides. Commun Chem. 2020;3:164.10.1038/s42004-020-00417-7Search in Google Scholar
[286] Wassenaar TA, Ingólfsson HI, Prieß M, Marrink SJ, Schäfer LV. Mixing MARTINI: Electrostatic coupling in hybrid atomistic-coarse-grained biomolecular simulations. J Phys Chem B. 2013;117:3516.10.1021/jp311533pSearch in Google Scholar PubMed
[287] Wassenaar TA, Pluhackova K, Böckmann RA, Marrink SJ, Tieleman DP. Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J Chem Theory Comput. 2014;10:676.10.1021/ct400617gSearch in Google Scholar PubMed
[288] Pezeshkian W, König M, Wassenaar TA, Marrink SJ. Backmapping triangulated surfaces to coarse-grained membrane models. Nat Commun. 2020;11:2296.10.1038/s41467-020-16094-ySearch in Google Scholar PubMed PubMed Central
[289] Pezeshkian W, Marrink SJ. Simulating realistic membrane shapes. Curr OpCell Biol. 2021;71:103–11.10.1016/j.ceb.2021.02.009Search in Google Scholar PubMed
[290] Maity S, Daskalakis V, Elstner M, Kleinekathöfer U. Multiscale QM/MM molecular dynamics simulations of the trimeric major light-harvesting complex II. Phys Chem Chem Phys. 2021;23:7407.10.1039/D1CP01011ESearch in Google Scholar PubMed
[291] Senn HM, Thiel W. QM/MM methods for biomolecular systems. Angew Chem – Int Ed. 2009;48(7):1198–229.10.1002/anie.200802019Search in Google Scholar PubMed
[292] Böselt L, Thürlemann M, Riniker S. Machine learning in QM/MM molecular dynamics simulations of condensed-phase systems. J Chem Theory Comput. 2021;17:2641.10.1021/acs.jctc.0c01112Search in Google Scholar
[293] Dohn AO. Multiscale electrostatic embedding simulations for modeling structure and dynamics of molecules in solution: A tutorial review. Int J Quantum Chem. 2020;120(21):e26343.10.1002/qua.26343Search in Google Scholar
[294] Sokkar P, Boulanger E, Thiel W, Sanchez-Garcia E. Hybrid quantum mechanics/molecular mechanics/coarse grained modeling: A triple-resolution approach for biomolecular systems. J Chem Theory Comput. 2015;11:1809.10.1021/ct500956uSearch in Google Scholar
[295] Stoliarov SI, Westmoreland PR, Nyden MR, Forney GP. A reactive molecular dynamics model of thermal decomposition in polymers: I. Poly(methyl methacrylate). Polymer. 2003;44(3):883–94.10.1016/S0032-3861(02)00761-9Search in Google Scholar
[296] Metin Aktulga H, Pandit SA, Van Duin ACT, Grama AY. Reactive molecular dynamics: Numerical methods and algorithmic techniques. SIAM J Sci Comput. 2012;34(1):C1–23.10.1137/100808599Search in Google Scholar
[297] Hahn SH, van Duin ACT. Surface reactivity and leaching of a sodium silicate glass under an aqueous environment: A ReaxFF molecular dynamics study. J Phys Chem C. 2019;123(25):15606–17.10.1021/acs.jpcc.9b02940Search in Google Scholar
[298] Dannenhoffer-Lafage T, Voth GA. Reactive coarse-grained molecular dynamics. J Chem Theory Comput. 2020;16:2541.10.1021/acs.jctc.9b01140Search in Google Scholar PubMed
[299] Bourne JW, Torzilli PA. Molecular simulations predict novel collagen conformations during cross-link loading. Matrix Biol. 2011;30:356.10.1016/j.matbio.2011.03.010Search in Google Scholar PubMed PubMed Central
[300] Garrec J, Patel C, Rothlisberger U, Dumont E. Insights into intra-strand cross-link lesions of DNA from QM/MM molecular dynamics simulations. J Am Chem Soc. 2012;134:2111.10.1021/ja2084042Search in Google Scholar PubMed
[301] Aulisa L, Dong H, Hartgerink JD. Self-assembly of multidomain peptides: Sequence variation allows control over cross-linking and viscoelasticity. Biomacromolecules. 2009;10:2694.10.1021/bm900634xSearch in Google Scholar PubMed
[302] Bloom KS. Beyond the code: The mechanical properties of DNA as they relate to mitosis. Chromosoma. 2008;117:103–10.10.1007/s00412-007-0138-0Search in Google Scholar PubMed PubMed Central
[303] Herrero-Galán E, Fuentes-Perez ME, Carrasco C, Valpuesta JM, Carrascosa JL, Moreno-Herrero F, et al. Mechanical identities of RNA and DNA double helices unveiled at the single-molecule level. J Am Chem Soc. 2013;135(1):122–31.10.1021/ja3054755Search in Google Scholar PubMed
[304] Naserian-Nik AM, Tahani M, Karttunen M. Molecular dynamics study of DNA oligomers under angled pulling. RSC Adv. 2014;4:10751.10.1039/c3ra45604hSearch in Google Scholar
[305] Mitchell JS, Glowacki J, Grandchamp AE, Manning RS, Maddocks JH. Sequence-dependent persistence lengths of DNA. J Chem Theory Comput. 2017;13:1539.10.1021/acs.jctc.6b00904Search in Google Scholar PubMed
[306] Lipfert J, Skinner GM, Keegstra JM, Hensgens T, Jager T, Dulin D, et al. Double-stranded RNA under force and torque: Similarities to and striking differences from double-stranded DNA. Proc Natl Acad Sci. 2014;111:15408–13.10.1073/pnas.1407197111Search in Google Scholar PubMed PubMed Central
[307] Abels JA, Moreno-Herrero F, van der Heijden T, Dekker C, Dekker NH. Single-molecule measurements of the persistence length of double-stranded RNA. Biophys J. 2005;88:2737–44.10.1529/biophysj.104.052811Search in Google Scholar PubMed PubMed Central
[308] Choi B, Kim T, Lee SW, Eom K. Nanomechanical characterization of amyloid fibrils using single-molecule experiments and computational simulations. J Nanomater. 2016;2016:5873695.10.1155/2016/5873695Search in Google Scholar
[309] Li H, Sun K, Li R, Fan M, Li H. Analysis and demonstration of a scaffold finite element model for cartilage tissue engineering. ACS Omega. 2020;5:32411.10.1021/acsomega.0c04378Search in Google Scholar PubMed PubMed Central
[310] Alisafaei F, Chen X, Leahy T, Janmey PA, Shenoy VB. Long-range mechanical signaling in biological systems. Soft Matter. 2021;17(2):241–53.10.1039/D0SM01442GSearch in Google Scholar PubMed PubMed Central
[311] Sanz-Herrera JA, Reina-Romo E. Continuum modeling and simulation in bone tissue engineering. Appl Sci. 2019;9(18):3674.10.3390/app9183674Search in Google Scholar
[312] Patel M, Dubey DK, Singh SP. Phenomenological models of bombyx mori silk fibroin and their mechanical behavior using molecular dynamics simulations. Mater Sci Eng C. 2020;108:110414.10.1016/j.msec.2019.110414Search in Google Scholar PubMed
[313] Solernou A, Hanson BS, Richardson RA, Welch R, Read DJ, Harlen OG, et al. Fluctuating finite element analysis (FFEA): A continuum mechanics software tool for mesoscale simulation of biomolecules. PLoS Comput Biol. 2018;14:e1005897.10.1371/journal.pcbi.1005897Search in Google Scholar PubMed PubMed Central
[314] Bruininks BMH, Souza PCT, Ingolfsson H, Marrink SJ. A molecular view on the escape of lipoplexed DNA from the endosome. ELife. 2020;9:e52012.10.7554/eLife.52012Search in Google Scholar PubMed PubMed Central
[315] Heinrich MA, Martina B, Prakash J. Nanomedicine strategies to target coronavirus. Nano Today. 2020;35:100961.10.1016/j.nantod.2020.100961Search in Google Scholar PubMed PubMed Central
[316] Yu A, Pak AJ, He P, Monje-Galvan V, Casalino L, Gaieb Z, et al. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys J. 2021;120:1097.10.1016/j.bpj.2020.10.048Search in Google Scholar PubMed PubMed Central
© 2022 Federico Fontana and Fabrizio Gelain, published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- Theoretical and experimental investigation of MWCNT dispersion effect on the elastic modulus of flexible PDMS/MWCNT nanocomposites
- Mechanical, morphological, and fracture-deformation behavior of MWCNTs-reinforced (Al–Cu–Mg–T351) alloy cast nanocomposites fabricated by optimized mechanical milling and powder metallurgy techniques
- Flammability and physical stability of sugar palm crystalline nanocellulose reinforced thermoplastic sugar palm starch/poly(lactic acid) blend bionanocomposites
- Glutathione-loaded non-ionic surfactant niosomes: A new approach to improve oral bioavailability and hepatoprotective efficacy of glutathione
- Relationship between mechano-bactericidal activity and nanoblades density on chemically strengthened glass
- In situ regulation of microstructure and microwave-absorbing properties of FeSiAl through HNO3 oxidation
- Research on a mechanical model of magnetorheological fluid different diameter particles
- Nanomechanical and dynamic mechanical properties of rubber–wood–plastic composites
- Investigative properties of CeO2 doped with niobium: A combined characterization and DFT studies
- Miniaturized peptidomimetics and nano-vesiculation in endothelin types through probable nano-disk formation and structure property relationships of endothelins’ fragments
- N/S co-doped CoSe/C nanocubes as anode materials for Li-ion batteries
- Synergistic effects of halloysite nanotubes with metal and phosphorus additives on the optimal design of eco-friendly sandwich panels with maximum flame resistance and minimum weight
- Octreotide-conjugated silver nanoparticles for active targeting of somatostatin receptors and their application in a nebulized rat model
- Controllable morphology of Bi2S3 nanostructures formed via hydrothermal vulcanization of Bi2O3 thin-film layer and their photoelectrocatalytic performances
- Development of (−)-epigallocatechin-3-gallate-loaded folate receptor-targeted nanoparticles for prostate cancer treatment
- Enhancement of the mechanical properties of HDPE mineral nanocomposites by filler particles modulation of the matrix plastic/elastic behavior
- Effect of plasticizers on the properties of sugar palm nanocellulose/cinnamon essential oil reinforced starch bionanocomposite films
- Optimization of nano coating to reduce the thermal deformation of ball screws
- Preparation of efficient piezoelectric PVDF–HFP/Ni composite films by high electric field poling
- MHD dissipative Casson nanofluid liquid film flow due to an unsteady stretching sheet with radiation influence and slip velocity phenomenon
- Effects of nano-SiO2 modification on rubberised mortar and concrete with recycled coarse aggregates
- Mechanical and microscopic properties of fiber-reinforced coal gangue-based geopolymer concrete
- Effect of morphology and size on the thermodynamic stability of cerium oxide nanoparticles: Experiment and molecular dynamics calculation
- Mechanical performance of a CFRP composite reinforced via gelatin-CNTs: A study on fiber interfacial enhancement and matrix enhancement
- A practical review over surface modification, nanopatterns, emerging materials, drug delivery systems, and their biophysiochemical properties for dental implants: Recent progresses and advances
- HTR: An ultra-high speed algorithm for cage recognition of clathrate hydrates
- Effects of microalloying elements added by in situ synthesis on the microstructure of WCu composites
- A highly sensitive nanobiosensor based on aptamer-conjugated graphene-decorated rhodium nanoparticles for detection of HER2-positive circulating tumor cells
- Progressive collapse performance of shear strengthened RC frames by nano CFRP
- Core–shell heterostructured composites of carbon nanotubes and imine-linked hyperbranched polymers as metal-free Li-ion anodes
- A Galerkin strategy for tri-hybridized mixture in ethylene glycol comprising variable diffusion and thermal conductivity using non-Fourier’s theory
- Simple models for tensile modulus of shape memory polymer nanocomposites at ambient temperature
- Preparation and morphological studies of tin sulfide nanoparticles and use as efficient photocatalysts for the degradation of rhodamine B and phenol
- Polyethyleneimine-impregnated activated carbon nanofiber composited graphene-derived rice husk char for efficient post-combustion CO2 capture
- Electrospun nanofibers of Co3O4 nanocrystals encapsulated in cyclized-polyacrylonitrile for lithium storage
- Pitting corrosion induced on high-strength high carbon steel wire in high alkaline deaerated chloride electrolyte
- Formulation of polymeric nanoparticles loaded sorafenib; evaluation of cytotoxicity, molecular evaluation, and gene expression studies in lung and breast cancer cell lines
- Engineered nanocomposites in asphalt binders
- Influence of loading voltage, domain ratio, and additional load on the actuation of dielectric elastomer
- Thermally induced hex-graphene transitions in 2D carbon crystals
- The surface modification effect on the interfacial properties of glass fiber-reinforced epoxy: A molecular dynamics study
- Molecular dynamics study of deformation mechanism of interfacial microzone of Cu/Al2Cu/Al composites under tension
- Nanocolloid simulators of luminescent solar concentrator photovoltaic windows
- Compressive strength and anti-chloride ion penetration assessment of geopolymer mortar merging PVA fiber and nano-SiO2 using RBF–BP composite neural network
- Effect of 3-mercapto-1-propane sulfonate sulfonic acid and polyvinylpyrrolidone on the growth of cobalt pillar by electrodeposition
- Dynamics of convective slippery constraints on hybrid radiative Sutterby nanofluid flow by Galerkin finite element simulation
- Preparation of vanadium by the magnesiothermic self-propagating reduction and process control
- Microstructure-dependent photoelectrocatalytic activity of heterogeneous ZnO–ZnS nanosheets
- Cytotoxic and pro-inflammatory effects of molybdenum and tungsten disulphide on human bronchial cells
- Improving recycled aggregate concrete by compression casting and nano-silica
- Chemically reactive Maxwell nanoliquid flow by a stretching surface in the frames of Newtonian heating, nonlinear convection and radiative flux: Nanopolymer flow processing simulation
- Nonlinear dynamic and crack behaviors of carbon nanotubes-reinforced composites with various geometries
- Biosynthesis of copper oxide nanoparticles and its therapeutic efficacy against colon cancer
- Synthesis and characterization of smart stimuli-responsive herbal drug-encapsulated nanoniosome particles for efficient treatment of breast cancer
- Homotopic simulation for heat transport phenomenon of the Burgers nanofluids flow over a stretching cylinder with thermal convective and zero mass flux conditions
- Incorporation of copper and strontium ions in TiO2 nanotubes via dopamine to enhance hemocompatibility and cytocompatibility
- Mechanical, thermal, and barrier properties of starch films incorporated with chitosan nanoparticles
- Mechanical properties and microstructure of nano-strengthened recycled aggregate concrete
- Glucose-responsive nanogels efficiently maintain the stability and activity of therapeutic enzymes
- Tunning matrix rheology and mechanical performance of ultra-high performance concrete using cellulose nanofibers
- Flexible MXene/copper/cellulose nanofiber heat spreader films with enhanced thermal conductivity
- Promoted charge separation and specific surface area via interlacing of N-doped titanium dioxide nanotubes on carbon nitride nanosheets for photocatalytic degradation of Rhodamine B
- Elucidating the role of silicon dioxide and titanium dioxide nanoparticles in mitigating the disease of the eggplant caused by Phomopsis vexans, Ralstonia solanacearum, and root-knot nematode Meloidogyne incognita
- An implication of magnetic dipole in Carreau Yasuda liquid influenced by engine oil using ternary hybrid nanomaterial
- Robust synthesis of a composite phase of copper vanadium oxide with enhanced performance for durable aqueous Zn-ion batteries
- Tunning self-assembled phases of bovine serum albumin via hydrothermal process to synthesize novel functional hydrogel for skin protection against UVB
- A comparative experimental study on damping properties of epoxy nanocomposite beams reinforced with carbon nanotubes and graphene nanoplatelets
- Lightweight and hydrophobic Ni/GO/PVA composite aerogels for ultrahigh performance electromagnetic interference shielding
- Research on the auxetic behavior and mechanical properties of periodically rotating graphene nanostructures
- Repairing performances of novel cement mortar modified with graphene oxide and polyacrylate polymer
- Closed-loop recycling and fabrication of hydrophilic CNT films with high performance
- Design of thin-film configuration of SnO2–Ag2O composites for NO2 gas-sensing applications
- Study on stress distribution of SiC/Al composites based on microstructure models with microns and nanoparticles
- PVDF green nanofibers as potential carriers for improving self-healing and mechanical properties of carbon fiber/epoxy prepregs
- Osteogenesis capability of three-dimensionally printed poly(lactic acid)-halloysite nanotube scaffolds containing strontium ranelate
- Silver nanoparticles induce mitochondria-dependent apoptosis and late non-canonical autophagy in HT-29 colon cancer cells
- Preparation and bonding mechanisms of polymer/metal hybrid composite by nano molding technology
- Damage self-sensing and strain monitoring of glass-reinforced epoxy composite impregnated with graphene nanoplatelet and multiwalled carbon nanotubes
- Thermal analysis characterisation of solar-powered ship using Oldroyd hybrid nanofluids in parabolic trough solar collector: An optimal thermal application
- Pyrene-functionalized halloysite nanotubes for simultaneously detecting and separating Hg(ii) in aqueous media: A comprehensive comparison on interparticle and intraparticle excimers
- Fabrication of self-assembly CNT flexible film and its piezoresistive sensing behaviors
- Thermal valuation and entropy inspection of second-grade nanoscale fluid flow over a stretching surface by applying Koo–Kleinstreuer–Li relation
- Mechanical properties and microstructure of nano-SiO2 and basalt-fiber-reinforced recycled aggregate concrete
- Characterization and tribology performance of polyaniline-coated nanodiamond lubricant additives
- Combined impact of Marangoni convection and thermophoretic particle deposition on chemically reactive transport of nanofluid flow over a stretching surface
- Spark plasma extrusion of binder free hydroxyapatite powder
- An investigation on thermo-mechanical performance of graphene-oxide-reinforced shape memory polymer
- Effect of nanoadditives on the novel leather fiber/recycled poly(ethylene-vinyl-acetate) polymer composites for multifunctional applications: Fabrication, characterizations, and multiobjective optimization using central composite design
- Design selection for a hemispherical dimple core sandwich panel using hybrid multi-criteria decision-making methods
- Improving tensile strength and impact toughness of plasticized poly(lactic acid) biocomposites by incorporating nanofibrillated cellulose
- Green synthesis of spinel copper ferrite (CuFe2O4) nanoparticles and their toxicity
- The effect of TaC and NbC hybrid and mono-nanoparticles on AA2024 nanocomposites: Microstructure, strengthening, and artificial aging
- Excited-state geometry relaxation of pyrene-modified cellulose nanocrystals under UV-light excitation for detecting Fe3+
- Effect of CNTs and MEA on the creep of face-slab concrete at an early age
- Effect of deformation conditions on compression phase transformation of AZ31
- Application of MXene as a new generation of highly conductive coating materials for electromembrane-surrounded solid-phase microextraction
- A comparative study of the elasto-plastic properties for ceramic nanocomposites filled by graphene or graphene oxide nanoplates
- Encapsulation strategies for improving the biological behavior of CdS@ZIF-8 nanocomposites
- Biosynthesis of ZnO NPs from pumpkin seeds’ extract and elucidation of its anticancer potential against breast cancer
- Preliminary trials of the gold nanoparticles conjugated chrysin: An assessment of anti-oxidant, anti-microbial, and in vitro cytotoxic activities of a nanoformulated flavonoid
- Effect of micron-scale pores increased by nano-SiO2 sol modification on the strength of cement mortar
- Fractional simulations for thermal flow of hybrid nanofluid with aluminum oxide and titanium oxide nanoparticles with water and blood base fluids
- The effect of graphene nano-powder on the viscosity of water: An experimental study and artificial neural network modeling
- Development of a novel heat- and shear-resistant nano-silica gelling agent
- Characterization, biocompatibility and in vivo of nominal MnO2-containing wollastonite glass-ceramic
- Entropy production simulation of second-grade magnetic nanomaterials flowing across an expanding surface with viscidness dissipative flux
- Enhancement in structural, morphological, and optical properties of copper oxide for optoelectronic device applications
- Aptamer-functionalized chitosan-coated gold nanoparticle complex as a suitable targeted drug carrier for improved breast cancer treatment
- Performance and overall evaluation of nano-alumina-modified asphalt mixture
- Analysis of pure nanofluid (GO/engine oil) and hybrid nanofluid (GO–Fe3O4/engine oil): Novel thermal and magnetic features
- Synthesis of Ag@AgCl modified anatase/rutile/brookite mixed phase TiO2 and their photocatalytic property
- Mechanisms and influential variables on the abrasion resistance hydraulic concrete
- Synergistic reinforcement mechanism of basalt fiber/cellulose nanocrystals/polypropylene composites
- Achieving excellent oxidation resistance and mechanical properties of TiB2–B4C/carbon aerogel composites by quick-gelation and mechanical mixing
- Microwave-assisted sol–gel template-free synthesis and characterization of silica nanoparticles obtained from South African coal fly ash
- Pulsed laser-assisted synthesis of nano nickel(ii) oxide-anchored graphitic carbon nitride: Characterizations and their potential antibacterial/anti-biofilm applications
- Effects of nano-ZrSi2 on thermal stability of phenolic resin and thermal reusability of quartz–phenolic composites
- Benzaldehyde derivatives on tin electroplating as corrosion resistance for fabricating copper circuit
- Mechanical and heat transfer properties of 4D-printed shape memory graphene oxide/epoxy acrylate composites
- Coupling the vanadium-induced amorphous/crystalline NiFe2O4 with phosphide heterojunction toward active oxygen evolution reaction catalysts
- Graphene-oxide-reinforced cement composites mechanical and microstructural characteristics at elevated temperatures
- Gray correlation analysis of factors influencing compressive strength and durability of nano-SiO2 and PVA fiber reinforced geopolymer mortar
- Preparation of layered gradient Cu–Cr–Ti alloy with excellent mechanical properties, thermal stability, and electrical conductivity
- Recovery of Cr from chrome-containing leather wastes to develop aluminum-based composite material along with Al2O3 ceramic particles: An ingenious approach
- Mechanisms of the improved stiffness of flexible polymers under impact loading
- Anticancer potential of gold nanoparticles (AuNPs) using a battery of in vitro tests
- Review Articles
- Proposed approaches for coronaviruses elimination from wastewater: Membrane techniques and nanotechnology solutions
- Application of Pickering emulsion in oil drilling and production
- The contribution of microfluidics to the fight against tuberculosis
- Graphene-based biosensors for disease theranostics: Development, applications, and recent advancements
- Synthesis and encapsulation of iron oxide nanorods for application in magnetic hyperthermia and photothermal therapy
- Contemporary nano-architectured drugs and leads for ανβ3 integrin-based chemotherapy: Rationale and retrospect
- State-of-the-art review of fabrication, application, and mechanical properties of functionally graded porous nanocomposite materials
- Insights on magnetic spinel ferrites for targeted drug delivery and hyperthermia applications
- A review on heterogeneous oxidation of acetaminophen based on micro and nanoparticles catalyzed by different activators
- Early diagnosis of lung cancer using magnetic nanoparticles-integrated systems
- Advances in ZnO: Manipulation of defects for enhancing their technological potentials
- Efficacious nanomedicine track toward combating COVID-19
- A review of the design, processes, and properties of Mg-based composites
- Green synthesis of nanoparticles for varied applications: Green renewable resources and energy-efficient synthetic routes
- Two-dimensional nanomaterial-based polymer composites: Fundamentals and applications
- Recent progress and challenges in plasmonic nanomaterials
- Apoptotic cell-derived micro/nanosized extracellular vesicles in tissue regeneration
- Electronic noses based on metal oxide nanowires: A review
- Framework materials for supercapacitors
- An overview on the reproductive toxicity of graphene derivatives: Highlighting the importance
- Antibacterial nanomaterials: Upcoming hope to overcome antibiotic resistance crisis
- Research progress of carbon materials in the field of three-dimensional printing polymer nanocomposites
- A review of atomic layer deposition modelling and simulation methodologies: Density functional theory and molecular dynamics
- Recent advances in the preparation of PVDF-based piezoelectric materials
- Recent developments in tensile properties of friction welding of carbon fiber-reinforced composite: A review
- Comprehensive review of the properties of fly ash-based geopolymer with additive of nano-SiO2
- Perspectives in biopolymer/graphene-based composite application: Advances, challenges, and recommendations
- Graphene-based nanocomposite using new modeling molecular dynamic simulations for proposed neutralizing mechanism and real-time sensing of COVID-19
- Nanotechnology application on bamboo materials: A review
- Recent developments and future perspectives of biorenewable nanocomposites for advanced applications
- Nanostructured lipid carrier system: A compendium of their formulation development approaches, optimization strategies by quality by design, and recent applications in drug delivery
- 3D printing customized design of human bone tissue implant and its application
- Design, preparation, and functionalization of nanobiomaterials for enhanced efficacy in current and future biomedical applications
- A brief review of nanoparticles-doped PEDOT:PSS nanocomposite for OLED and OPV
- Nanotechnology interventions as a putative tool for the treatment of dental afflictions
- Recent advancements in metal–organic frameworks integrating quantum dots (QDs@MOF) and their potential applications
- A focused review of short electrospun nanofiber preparation techniques for composite reinforcement
- Microstructural characteristics and nano-modification of interfacial transition zone in concrete: A review
- Latest developments in the upconversion nanotechnology for the rapid detection of food safety: A review
- Strategic applications of nano-fertilizers for sustainable agriculture: Benefits and bottlenecks
- Molecular dynamics application of cocrystal energetic materials: A review
- Synthesis and application of nanometer hydroxyapatite in biomedicine
- Cutting-edge development in waste-recycled nanomaterials for energy storage and conversion applications
- Biological applications of ternary quantum dots: A review
- Nanotherapeutics for hydrogen sulfide-involved treatment: An emerging approach for cancer therapy
- Application of antibacterial nanoparticles in orthodontic materials
- Effect of natural-based biological hydrogels combined with growth factors on skin wound healing
- Nanozymes – A route to overcome microbial resistance: A viewpoint
- Recent developments and applications of smart nanoparticles in biomedicine
- Contemporary review on carbon nanotube (CNT) composites and their impact on multifarious applications
- Interfacial interactions and reinforcing mechanisms of cellulose and chitin nanomaterials and starch derivatives for cement and concrete strength and durability enhancement: A review
- Diamond-like carbon films for tribological modification of rubber
- Layered double hydroxides (LDHs) modified cement-based materials: A systematic review
- Recent research progress and advanced applications of silica/polymer nanocomposites
- Modeling of supramolecular biopolymers: Leading the in silico revolution of tissue engineering and nanomedicine
- Recent advances in perovskites-based optoelectronics
- Biogenic synthesis of palladium nanoparticles: New production methods and applications
- A comprehensive review of nanofluids with fractional derivatives: Modeling and application
- Electrospinning of marine polysaccharides: Processing and chemical aspects, challenges, and future prospects
- Electrohydrodynamic printing for demanding devices: A review of processing and applications
- Rapid Communications
- Structural material with designed thermal twist for a simple actuation
- Recent advances in photothermal materials for solar-driven crude oil adsorption
Articles in the same Issue
- Research Articles
- Theoretical and experimental investigation of MWCNT dispersion effect on the elastic modulus of flexible PDMS/MWCNT nanocomposites
- Mechanical, morphological, and fracture-deformation behavior of MWCNTs-reinforced (Al–Cu–Mg–T351) alloy cast nanocomposites fabricated by optimized mechanical milling and powder metallurgy techniques
- Flammability and physical stability of sugar palm crystalline nanocellulose reinforced thermoplastic sugar palm starch/poly(lactic acid) blend bionanocomposites
- Glutathione-loaded non-ionic surfactant niosomes: A new approach to improve oral bioavailability and hepatoprotective efficacy of glutathione
- Relationship between mechano-bactericidal activity and nanoblades density on chemically strengthened glass
- In situ regulation of microstructure and microwave-absorbing properties of FeSiAl through HNO3 oxidation
- Research on a mechanical model of magnetorheological fluid different diameter particles
- Nanomechanical and dynamic mechanical properties of rubber–wood–plastic composites
- Investigative properties of CeO2 doped with niobium: A combined characterization and DFT studies
- Miniaturized peptidomimetics and nano-vesiculation in endothelin types through probable nano-disk formation and structure property relationships of endothelins’ fragments
- N/S co-doped CoSe/C nanocubes as anode materials for Li-ion batteries
- Synergistic effects of halloysite nanotubes with metal and phosphorus additives on the optimal design of eco-friendly sandwich panels with maximum flame resistance and minimum weight
- Octreotide-conjugated silver nanoparticles for active targeting of somatostatin receptors and their application in a nebulized rat model
- Controllable morphology of Bi2S3 nanostructures formed via hydrothermal vulcanization of Bi2O3 thin-film layer and their photoelectrocatalytic performances
- Development of (−)-epigallocatechin-3-gallate-loaded folate receptor-targeted nanoparticles for prostate cancer treatment
- Enhancement of the mechanical properties of HDPE mineral nanocomposites by filler particles modulation of the matrix plastic/elastic behavior
- Effect of plasticizers on the properties of sugar palm nanocellulose/cinnamon essential oil reinforced starch bionanocomposite films
- Optimization of nano coating to reduce the thermal deformation of ball screws
- Preparation of efficient piezoelectric PVDF–HFP/Ni composite films by high electric field poling
- MHD dissipative Casson nanofluid liquid film flow due to an unsteady stretching sheet with radiation influence and slip velocity phenomenon
- Effects of nano-SiO2 modification on rubberised mortar and concrete with recycled coarse aggregates
- Mechanical and microscopic properties of fiber-reinforced coal gangue-based geopolymer concrete
- Effect of morphology and size on the thermodynamic stability of cerium oxide nanoparticles: Experiment and molecular dynamics calculation
- Mechanical performance of a CFRP composite reinforced via gelatin-CNTs: A study on fiber interfacial enhancement and matrix enhancement
- A practical review over surface modification, nanopatterns, emerging materials, drug delivery systems, and their biophysiochemical properties for dental implants: Recent progresses and advances
- HTR: An ultra-high speed algorithm for cage recognition of clathrate hydrates
- Effects of microalloying elements added by in situ synthesis on the microstructure of WCu composites
- A highly sensitive nanobiosensor based on aptamer-conjugated graphene-decorated rhodium nanoparticles for detection of HER2-positive circulating tumor cells
- Progressive collapse performance of shear strengthened RC frames by nano CFRP
- Core–shell heterostructured composites of carbon nanotubes and imine-linked hyperbranched polymers as metal-free Li-ion anodes
- A Galerkin strategy for tri-hybridized mixture in ethylene glycol comprising variable diffusion and thermal conductivity using non-Fourier’s theory
- Simple models for tensile modulus of shape memory polymer nanocomposites at ambient temperature
- Preparation and morphological studies of tin sulfide nanoparticles and use as efficient photocatalysts for the degradation of rhodamine B and phenol
- Polyethyleneimine-impregnated activated carbon nanofiber composited graphene-derived rice husk char for efficient post-combustion CO2 capture
- Electrospun nanofibers of Co3O4 nanocrystals encapsulated in cyclized-polyacrylonitrile for lithium storage
- Pitting corrosion induced on high-strength high carbon steel wire in high alkaline deaerated chloride electrolyte
- Formulation of polymeric nanoparticles loaded sorafenib; evaluation of cytotoxicity, molecular evaluation, and gene expression studies in lung and breast cancer cell lines
- Engineered nanocomposites in asphalt binders
- Influence of loading voltage, domain ratio, and additional load on the actuation of dielectric elastomer
- Thermally induced hex-graphene transitions in 2D carbon crystals
- The surface modification effect on the interfacial properties of glass fiber-reinforced epoxy: A molecular dynamics study
- Molecular dynamics study of deformation mechanism of interfacial microzone of Cu/Al2Cu/Al composites under tension
- Nanocolloid simulators of luminescent solar concentrator photovoltaic windows
- Compressive strength and anti-chloride ion penetration assessment of geopolymer mortar merging PVA fiber and nano-SiO2 using RBF–BP composite neural network
- Effect of 3-mercapto-1-propane sulfonate sulfonic acid and polyvinylpyrrolidone on the growth of cobalt pillar by electrodeposition
- Dynamics of convective slippery constraints on hybrid radiative Sutterby nanofluid flow by Galerkin finite element simulation
- Preparation of vanadium by the magnesiothermic self-propagating reduction and process control
- Microstructure-dependent photoelectrocatalytic activity of heterogeneous ZnO–ZnS nanosheets
- Cytotoxic and pro-inflammatory effects of molybdenum and tungsten disulphide on human bronchial cells
- Improving recycled aggregate concrete by compression casting and nano-silica
- Chemically reactive Maxwell nanoliquid flow by a stretching surface in the frames of Newtonian heating, nonlinear convection and radiative flux: Nanopolymer flow processing simulation
- Nonlinear dynamic and crack behaviors of carbon nanotubes-reinforced composites with various geometries
- Biosynthesis of copper oxide nanoparticles and its therapeutic efficacy against colon cancer
- Synthesis and characterization of smart stimuli-responsive herbal drug-encapsulated nanoniosome particles for efficient treatment of breast cancer
- Homotopic simulation for heat transport phenomenon of the Burgers nanofluids flow over a stretching cylinder with thermal convective and zero mass flux conditions
- Incorporation of copper and strontium ions in TiO2 nanotubes via dopamine to enhance hemocompatibility and cytocompatibility
- Mechanical, thermal, and barrier properties of starch films incorporated with chitosan nanoparticles
- Mechanical properties and microstructure of nano-strengthened recycled aggregate concrete
- Glucose-responsive nanogels efficiently maintain the stability and activity of therapeutic enzymes
- Tunning matrix rheology and mechanical performance of ultra-high performance concrete using cellulose nanofibers
- Flexible MXene/copper/cellulose nanofiber heat spreader films with enhanced thermal conductivity
- Promoted charge separation and specific surface area via interlacing of N-doped titanium dioxide nanotubes on carbon nitride nanosheets for photocatalytic degradation of Rhodamine B
- Elucidating the role of silicon dioxide and titanium dioxide nanoparticles in mitigating the disease of the eggplant caused by Phomopsis vexans, Ralstonia solanacearum, and root-knot nematode Meloidogyne incognita
- An implication of magnetic dipole in Carreau Yasuda liquid influenced by engine oil using ternary hybrid nanomaterial
- Robust synthesis of a composite phase of copper vanadium oxide with enhanced performance for durable aqueous Zn-ion batteries
- Tunning self-assembled phases of bovine serum albumin via hydrothermal process to synthesize novel functional hydrogel for skin protection against UVB
- A comparative experimental study on damping properties of epoxy nanocomposite beams reinforced with carbon nanotubes and graphene nanoplatelets
- Lightweight and hydrophobic Ni/GO/PVA composite aerogels for ultrahigh performance electromagnetic interference shielding
- Research on the auxetic behavior and mechanical properties of periodically rotating graphene nanostructures
- Repairing performances of novel cement mortar modified with graphene oxide and polyacrylate polymer
- Closed-loop recycling and fabrication of hydrophilic CNT films with high performance
- Design of thin-film configuration of SnO2–Ag2O composites for NO2 gas-sensing applications
- Study on stress distribution of SiC/Al composites based on microstructure models with microns and nanoparticles
- PVDF green nanofibers as potential carriers for improving self-healing and mechanical properties of carbon fiber/epoxy prepregs
- Osteogenesis capability of three-dimensionally printed poly(lactic acid)-halloysite nanotube scaffolds containing strontium ranelate
- Silver nanoparticles induce mitochondria-dependent apoptosis and late non-canonical autophagy in HT-29 colon cancer cells
- Preparation and bonding mechanisms of polymer/metal hybrid composite by nano molding technology
- Damage self-sensing and strain monitoring of glass-reinforced epoxy composite impregnated with graphene nanoplatelet and multiwalled carbon nanotubes
- Thermal analysis characterisation of solar-powered ship using Oldroyd hybrid nanofluids in parabolic trough solar collector: An optimal thermal application
- Pyrene-functionalized halloysite nanotubes for simultaneously detecting and separating Hg(ii) in aqueous media: A comprehensive comparison on interparticle and intraparticle excimers
- Fabrication of self-assembly CNT flexible film and its piezoresistive sensing behaviors
- Thermal valuation and entropy inspection of second-grade nanoscale fluid flow over a stretching surface by applying Koo–Kleinstreuer–Li relation
- Mechanical properties and microstructure of nano-SiO2 and basalt-fiber-reinforced recycled aggregate concrete
- Characterization and tribology performance of polyaniline-coated nanodiamond lubricant additives
- Combined impact of Marangoni convection and thermophoretic particle deposition on chemically reactive transport of nanofluid flow over a stretching surface
- Spark plasma extrusion of binder free hydroxyapatite powder
- An investigation on thermo-mechanical performance of graphene-oxide-reinforced shape memory polymer
- Effect of nanoadditives on the novel leather fiber/recycled poly(ethylene-vinyl-acetate) polymer composites for multifunctional applications: Fabrication, characterizations, and multiobjective optimization using central composite design
- Design selection for a hemispherical dimple core sandwich panel using hybrid multi-criteria decision-making methods
- Improving tensile strength and impact toughness of plasticized poly(lactic acid) biocomposites by incorporating nanofibrillated cellulose
- Green synthesis of spinel copper ferrite (CuFe2O4) nanoparticles and their toxicity
- The effect of TaC and NbC hybrid and mono-nanoparticles on AA2024 nanocomposites: Microstructure, strengthening, and artificial aging
- Excited-state geometry relaxation of pyrene-modified cellulose nanocrystals under UV-light excitation for detecting Fe3+
- Effect of CNTs and MEA on the creep of face-slab concrete at an early age
- Effect of deformation conditions on compression phase transformation of AZ31
- Application of MXene as a new generation of highly conductive coating materials for electromembrane-surrounded solid-phase microextraction
- A comparative study of the elasto-plastic properties for ceramic nanocomposites filled by graphene or graphene oxide nanoplates
- Encapsulation strategies for improving the biological behavior of CdS@ZIF-8 nanocomposites
- Biosynthesis of ZnO NPs from pumpkin seeds’ extract and elucidation of its anticancer potential against breast cancer
- Preliminary trials of the gold nanoparticles conjugated chrysin: An assessment of anti-oxidant, anti-microbial, and in vitro cytotoxic activities of a nanoformulated flavonoid
- Effect of micron-scale pores increased by nano-SiO2 sol modification on the strength of cement mortar
- Fractional simulations for thermal flow of hybrid nanofluid with aluminum oxide and titanium oxide nanoparticles with water and blood base fluids
- The effect of graphene nano-powder on the viscosity of water: An experimental study and artificial neural network modeling
- Development of a novel heat- and shear-resistant nano-silica gelling agent
- Characterization, biocompatibility and in vivo of nominal MnO2-containing wollastonite glass-ceramic
- Entropy production simulation of second-grade magnetic nanomaterials flowing across an expanding surface with viscidness dissipative flux
- Enhancement in structural, morphological, and optical properties of copper oxide for optoelectronic device applications
- Aptamer-functionalized chitosan-coated gold nanoparticle complex as a suitable targeted drug carrier for improved breast cancer treatment
- Performance and overall evaluation of nano-alumina-modified asphalt mixture
- Analysis of pure nanofluid (GO/engine oil) and hybrid nanofluid (GO–Fe3O4/engine oil): Novel thermal and magnetic features
- Synthesis of Ag@AgCl modified anatase/rutile/brookite mixed phase TiO2 and their photocatalytic property
- Mechanisms and influential variables on the abrasion resistance hydraulic concrete
- Synergistic reinforcement mechanism of basalt fiber/cellulose nanocrystals/polypropylene composites
- Achieving excellent oxidation resistance and mechanical properties of TiB2–B4C/carbon aerogel composites by quick-gelation and mechanical mixing
- Microwave-assisted sol–gel template-free synthesis and characterization of silica nanoparticles obtained from South African coal fly ash
- Pulsed laser-assisted synthesis of nano nickel(ii) oxide-anchored graphitic carbon nitride: Characterizations and their potential antibacterial/anti-biofilm applications
- Effects of nano-ZrSi2 on thermal stability of phenolic resin and thermal reusability of quartz–phenolic composites
- Benzaldehyde derivatives on tin electroplating as corrosion resistance for fabricating copper circuit
- Mechanical and heat transfer properties of 4D-printed shape memory graphene oxide/epoxy acrylate composites
- Coupling the vanadium-induced amorphous/crystalline NiFe2O4 with phosphide heterojunction toward active oxygen evolution reaction catalysts
- Graphene-oxide-reinforced cement composites mechanical and microstructural characteristics at elevated temperatures
- Gray correlation analysis of factors influencing compressive strength and durability of nano-SiO2 and PVA fiber reinforced geopolymer mortar
- Preparation of layered gradient Cu–Cr–Ti alloy with excellent mechanical properties, thermal stability, and electrical conductivity
- Recovery of Cr from chrome-containing leather wastes to develop aluminum-based composite material along with Al2O3 ceramic particles: An ingenious approach
- Mechanisms of the improved stiffness of flexible polymers under impact loading
- Anticancer potential of gold nanoparticles (AuNPs) using a battery of in vitro tests
- Review Articles
- Proposed approaches for coronaviruses elimination from wastewater: Membrane techniques and nanotechnology solutions
- Application of Pickering emulsion in oil drilling and production
- The contribution of microfluidics to the fight against tuberculosis
- Graphene-based biosensors for disease theranostics: Development, applications, and recent advancements
- Synthesis and encapsulation of iron oxide nanorods for application in magnetic hyperthermia and photothermal therapy
- Contemporary nano-architectured drugs and leads for ανβ3 integrin-based chemotherapy: Rationale and retrospect
- State-of-the-art review of fabrication, application, and mechanical properties of functionally graded porous nanocomposite materials
- Insights on magnetic spinel ferrites for targeted drug delivery and hyperthermia applications
- A review on heterogeneous oxidation of acetaminophen based on micro and nanoparticles catalyzed by different activators
- Early diagnosis of lung cancer using magnetic nanoparticles-integrated systems
- Advances in ZnO: Manipulation of defects for enhancing their technological potentials
- Efficacious nanomedicine track toward combating COVID-19
- A review of the design, processes, and properties of Mg-based composites
- Green synthesis of nanoparticles for varied applications: Green renewable resources and energy-efficient synthetic routes
- Two-dimensional nanomaterial-based polymer composites: Fundamentals and applications
- Recent progress and challenges in plasmonic nanomaterials
- Apoptotic cell-derived micro/nanosized extracellular vesicles in tissue regeneration
- Electronic noses based on metal oxide nanowires: A review
- Framework materials for supercapacitors
- An overview on the reproductive toxicity of graphene derivatives: Highlighting the importance
- Antibacterial nanomaterials: Upcoming hope to overcome antibiotic resistance crisis
- Research progress of carbon materials in the field of three-dimensional printing polymer nanocomposites
- A review of atomic layer deposition modelling and simulation methodologies: Density functional theory and molecular dynamics
- Recent advances in the preparation of PVDF-based piezoelectric materials
- Recent developments in tensile properties of friction welding of carbon fiber-reinforced composite: A review
- Comprehensive review of the properties of fly ash-based geopolymer with additive of nano-SiO2
- Perspectives in biopolymer/graphene-based composite application: Advances, challenges, and recommendations
- Graphene-based nanocomposite using new modeling molecular dynamic simulations for proposed neutralizing mechanism and real-time sensing of COVID-19
- Nanotechnology application on bamboo materials: A review
- Recent developments and future perspectives of biorenewable nanocomposites for advanced applications
- Nanostructured lipid carrier system: A compendium of their formulation development approaches, optimization strategies by quality by design, and recent applications in drug delivery
- 3D printing customized design of human bone tissue implant and its application
- Design, preparation, and functionalization of nanobiomaterials for enhanced efficacy in current and future biomedical applications
- A brief review of nanoparticles-doped PEDOT:PSS nanocomposite for OLED and OPV
- Nanotechnology interventions as a putative tool for the treatment of dental afflictions
- Recent advancements in metal–organic frameworks integrating quantum dots (QDs@MOF) and their potential applications
- A focused review of short electrospun nanofiber preparation techniques for composite reinforcement
- Microstructural characteristics and nano-modification of interfacial transition zone in concrete: A review
- Latest developments in the upconversion nanotechnology for the rapid detection of food safety: A review
- Strategic applications of nano-fertilizers for sustainable agriculture: Benefits and bottlenecks
- Molecular dynamics application of cocrystal energetic materials: A review
- Synthesis and application of nanometer hydroxyapatite in biomedicine
- Cutting-edge development in waste-recycled nanomaterials for energy storage and conversion applications
- Biological applications of ternary quantum dots: A review
- Nanotherapeutics for hydrogen sulfide-involved treatment: An emerging approach for cancer therapy
- Application of antibacterial nanoparticles in orthodontic materials
- Effect of natural-based biological hydrogels combined with growth factors on skin wound healing
- Nanozymes – A route to overcome microbial resistance: A viewpoint
- Recent developments and applications of smart nanoparticles in biomedicine
- Contemporary review on carbon nanotube (CNT) composites and their impact on multifarious applications
- Interfacial interactions and reinforcing mechanisms of cellulose and chitin nanomaterials and starch derivatives for cement and concrete strength and durability enhancement: A review
- Diamond-like carbon films for tribological modification of rubber
- Layered double hydroxides (LDHs) modified cement-based materials: A systematic review
- Recent research progress and advanced applications of silica/polymer nanocomposites
- Modeling of supramolecular biopolymers: Leading the in silico revolution of tissue engineering and nanomedicine
- Recent advances in perovskites-based optoelectronics
- Biogenic synthesis of palladium nanoparticles: New production methods and applications
- A comprehensive review of nanofluids with fractional derivatives: Modeling and application
- Electrospinning of marine polysaccharides: Processing and chemical aspects, challenges, and future prospects
- Electrohydrodynamic printing for demanding devices: A review of processing and applications
- Rapid Communications
- Structural material with designed thermal twist for a simple actuation
- Recent advances in photothermal materials for solar-driven crude oil adsorption