Recent progress on non-noble metal catalysts for the deoxydehydration of biomass-derived oxygenates
-
 and
and

Abstract
The utilization of renewable energy represents an effective way to address current issues associated with fossil fuels. Biomass is considered one type of renewable energy resources with abundant reserves on earth. However, the high oxygen contents and high degree of functionalization of biomass have hindered the direct exploitation of biomass for the production of fuels and chemicals. Considerable efforts have been devoted to developing effective deoxygenation methods capable of reducing the oxygen contents of biomass and its derivatives. The deoxydehydration (DODH) of biomass derivatives to generate olefins over oxophilic metal catalysts is considered a very useful approach in eliminating vicinal OH groups. In recent years, catalysts based on non-noble metals such as Mo, W, and V featuring good catalytic performance have emerged as promising alternatives to classical noble Re-based catalysts for DODH. This review aims to summarize the progress on the DODH of biomass-derived vicinal diols catalyzed by non-noble metals such as Mo, W, and V, with an emphasis on the preparation of catalysts, optimization of experimental conditions, and mechanistic studies. By surveying the performance of non-noble metal catalysts, key factors that determine the DODH activity were proposed, including the choice of reductant, the electronic and steric effects of ligand, and the interaction between solid support and metal center. The latter two could adjust the redox properties of metal centers by directly bonding with ligand or solid support.
1 Introduction
With the fast development of human society, the ever-increasing energy demand has aroused enormous concerns about the foreseen depletion of non-renewable fossil resources (coal and oil) as well as the associated climate change due to excessive CO2 emissions [1]. A shift towards renewable energies is considered a feasible approach in reducing the dependence on fossil resources and alleviating environmental issues. Considerable efforts have been devoted to developing sustainable technologies that can efficiently utilize renewable energies such as nuclear energy, geothermal energy, wind energy, and biomass [2,3,4,5]. Among these renewable energies, biomass has received particular attentions owing to its dual properties as an energy resource (e.g., burning for heat) and also a natural organic carbon resource [6,7]. The latter property renders biomass a promising carbon feedstock to partially replace fossil carbon resources [8,9]. By developing advanced methodologies that are able to unlock the potential of biomass as a carbon reservoir for the production of chemicals, fuels, and materials, the dependence on fossil resources could be reduced and the environmental issues related to the utilization of fossil carbon resources could be well addressed [10,11,12,13].
Biomass exists in the form of organic material in nature, such as wood waste, straw, corn cob, and edible carbohydrates (such as starch) [14]. Lignocellulose is the major component of various biomass resources primarily containing cellulose, hemicellulose, and lignin [15,16]. For example, wood typically contains 40–50 wt% cellulose, 10–30 wt% hemicellulose, and 20–30 wt% lignin [17]. Cellulose, a macromolecule composed of glucose unit, is the most widely distributed and most abundant polysaccharide in nature, accounting for more than 40 wt% of the carbon content in the plant kingdom [18]. Hemicellulose is a heteropolymer composed of several different types of C5 and C6 monosaccharides primarily including xylose, arabinose, and galactose [19,20]. Lignin is a biopolymer with a three-dimensional structure formed by the interconnection of three phenylpropane units through ether bonds and carbon–carbon bonds, containing abundant active groups such as aliphatic and aromatic hydroxyl groups and quinone groups [21]. In addition to lignocellulose-based biomass, vegetable oil and fat represent another category of biomass mainly comprising triglyceride, a widely used feedstock for the production of biodiesel and glycerol [22]. Despite the huge potential of biomass as an alternative carbon resource, the valorization of over-functionalized biomass into existing chemicals and fuels (mostly less functionalized) normally obtained from fossil resources faces significant challenges owing to the inherently distinct composition of biomass [23,24,25]. In contrast to fossil carbon feedstocks mainly containing hydrocarbon with very low oxygen contents, biomass is mainly composed of polyhydroxy compounds abundant in various oxygen-containing moieties [26,27,28]. Clearly, established technologies for petroleum processing/refining are inapplicable to biomass valorization [29].
Through depolymerization and subsequent upgrading reactions, biomass macromolecules can be selectively converted into a range of important platform chemicals including C5/C6 monosaccharides, glycerol, erythritol, xylitol, sorbitol, etc. (Figure 1) [30]. These highly oxygenated platform molecules usually need to undergo deoxygenation processes to meet the needs of current markets [7,31]. To this end, a series of defunctionalization approaches applicable to biomass-derived oxygenated compounds have been developed including deoxygenation [32,33], dehydration [34,35,36], hydrodeoxygenation [37,38,39], carbondeoxygenation [40], and deoxydehydration (DODH) [41,42]. Among them, DODH reaction, capable of selectively removing two adjacent hydroxyl groups in diol substrates to form a carbon–carbon double bond in the presence of a reductant (see Scheme 1), stands out as a very useful method for the deoxygenation of biomass-derived polyhydroxyl compounds [43,44]. DODH reaction not only enables the removal of two OH groups in one step but also transforms diols into various olefinic products of high value (see Figure 1) [45,46].

Typical biomass-derived platform molecules and their conversion into olefin products via DODH.
General DODH reaction of various vicinal diols into olefins. R1, R2 = H, alkyl or aryl, red = reductant, red(O) = oxidized reductant.
DODH reaction could proceed through either a non-catalytic (metal-free) or a catalytic manner. Non-catalytic DODH reaction relies on the utilization of stoichiometric deoxygenation reagents such as formic acid and orthoformates [47,48,49,50,51] to assist in the removal of OH groups. Catalytic DODH reaction has been intensively studied in recent years in terms of developing various metal catalysts, exploring different reductants, and expanding the utility of DODH in upgrading biomass-derived polyols [45,52,53,54]. The first catalytic DODH reaction was reported by Cook and Andrews employing an oxo-rhenium complex [Cp*ReO3] (Cp* = pentamethylcyclopentadienyl) as the DODH catalyst and triphenylphosphine (PPh3) as the reductant [55]. So far, a series of homogeneous and heterogeneous catalysts based on transition metals (e.g., Re, Mo, V) have been explored for DODH reaction in the context of biomass valorization [56,57,58,59,60,61,62]. Among these metals, Re-based catalysts have been the subject of numerous studies aimed at the development of diverse molecular Re complexes as well as solid Re catalysts by dispersing Re oxides on solid supports, in combination with different reductants (e.g., H2, PPh3, Na2SO3, secondary alcohol, and elemental reductants) [54,63,64,65,66]. Research advancements of metal-catalyzed DODH, in particular Re-catalyzed DODH, have been comprehensively summarized in several reviews. For example, Monbaliu and coworkers systemically summarized non-catalytic (metal-free) DODH protocols relying on deoxygenation reagents such as formic acid, tosylate/iodide, thiocarbonyldiimidazole/trimethylphosphite, and triethyl orthoformate, and metal-catalyzed DODH reactions relying on homogeneous and heterogeneous Re-, Mo-, V-, and W-based catalysts [60]. Dethlefsen et al. presented an overview of Re-catalyzed DODH reaction mainly focusing on the utilization of various Re complexes along with different reductants, substrate scope, and insights into reaction mechanism [67]. Donnelly et al. gave a short summary of catalytic DODH of biomass-derived polyols into olefins with emphasis on examples involving Re, Mo, and V complexes [68]. Very recently, Jentoft presented a complete overview of catalytic DODH reactions in aspects of reaction thermodynamics, catalyst categories, reaction mechanism, and process engineering [69].
In general, Re-based catalysts exhibit much higher catalytic activity and product selectivity than other metal catalysts [70,71,72]. However, the easily leaching, difficult recovery, and high price of Re species heavily hinder the practical viability of Re-catalyzed DODH reaction [73,74]. The development of alternative catalysts based on low-cost non-noble metals featuring improved recyclability and robustness represents an important direction to address these issues. Indeed, in parallel to the massive studies of Re catalysts, there has been some remarkable progress toward developing DODH catalysts relying on non-noble metals (primarily Mo, V, and W) [75,76,77,78]. This mini-review is dedicated to a summary of research progress in developing non-noble metal catalysts for the DODH reaction of biomass-derived polyols. Focus is placed on the key developments of non-noble metal catalysts manifesting satisfactory DODH performance and recyclability. Relevant reaction mechanism and structure–activity relationship are also discussed when available.
2 DODH reaction catalyzed by non-noble metals
Before starting the introduction of non-noble metals, we first present a brief summary of Re catalysts for the DODH of different diols in the presence of various reductants to have a benchmark for comparison. Methyltrioxorhenium MeReO3 (MTO) and CpttReO3 (Cptt = 1,3-di-tert-butylcyclopentadienyl) were selected as representative Re catalysts to structure this summary, considering the superior performance of MTO and CpttReO3 and their broad applications for different diols and reductants. As the reported non-noble metal catalysts are mainly active for model diols and simple biomass-derived polyols, the substrate scope of Re catalysts herein is mainly limited to simple diols including styrene glycol, 1,2-octanediol, 1,4-anhydroerythritol, glycerol, and erythritol.
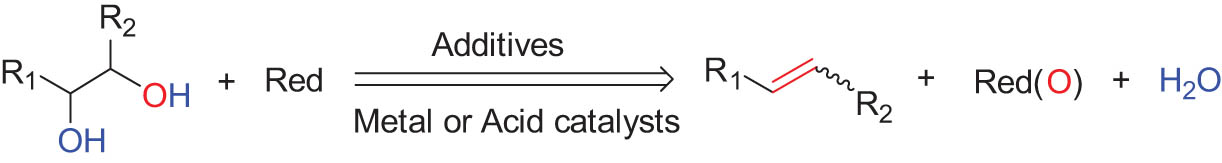
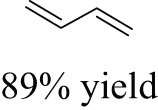
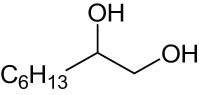
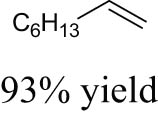
As shown in Table 1, the reactivity of diol for DODH is related to its structure. In the presence of MTO and Na2SO3, styrene glycol could be efficiently converted into styrene in 4 h in a yield of 59%, whereas much longer reaction time (21 h) was needed for the DODH of 1,2-octanediol into 1-octene under similar conditions [79]. Styrene glycol appears to possess higher reactivity toward DODH than 1,2-octanediol. However, this reactivity trend is reversed in the case of CpttReO3 and 3-octanol, probably owing to the important role of secondary alcohol as a reductant [80]. As for the other three diols (1,4-anhydroerythritol, glycerol, erythritol), 1,4-anhydroerythritol and glycerol showed very close reactivity for DODH, whereas erythritol is slightly less reactive for DODH, as indicated by the lower yields of butadiene [81]. This reactivity trend is reasonable since the complex configuration of long carbon-chain polyols requires more energy input to adapt its structure to coordinate into the metal center. The catalytic performance of Re catalysts also strongly depends on the type of reductant. Taking the DODH of 1,2-octanediol as an example, utilizing PPh3 and 3-octanol as the reductant led to a 93% yield of 1-octene [80]. In contrast, H2 only gave a very low yield of 1.2%, and no product was detected in the case of Na2SO3 probably owing to its poor solubility in organic solvents. It is difficult to summarize a general trend for the performance of these reductants since altering Re catalysts and reaction conditions can significantly influence the performance of reductants. By comparing these results, we can still conclude that both PPh3 and 3-octanol are superior to Na2SO3 and H2 for CpttReO3-catalyzed DODH reaction.
A brief summary of Re-catalyzed DODH of various diols in the presence of different reductants
| Diols | Re Catalyst | Reductant | Solvent | Temperature & time | Product & yield | Ref. |
|---|---|---|---|---|---|---|
|
MTO | Na2SO3 | Benzene | 150°C, 4 h |
|
[79] |
| CpttReO3 | 3-Octanol | 3-Octanol | 135°C, 15 h |

|
[80] | |
|
MTO | Na2SO3 | PhCl | 150°C, 21 h |
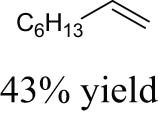
|
[79] |
| CpttReO3 | 3-Octanol | 3-Octanol | 135°C, 15 h |
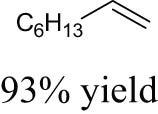
|
[80] | |
|
MTO | 3-Octanol | 3-Octanol | 170°C, 1.5 h |

|
[81] |
| MTO | H2 | THF | 150°C, 16 h |
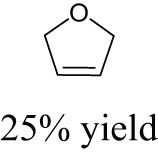
|
[82] | |
| CpttReO3 | 3-Octanol | 3-Octanol | 135°C, 15 h |
|
[80] | |
|
MTO | 3-Octanol | 3-Octanol | 170°C, 2.5 h |
|
[81] |
| CpttReO3 | 3-Octanol | 3-Octanol | 135°C, 15 h |
|
[80] | |

|
MTO | 3-Octanol | 3-Octanol | 170°C, 1.5 h |
|
[81] |
| CpttReO3 | 3-Octanol | 3-Octanol | 135°C, 15 h |

|
[80] | |
|
CpttReO3 | PPh3 | PhCl | 135°C, 15 h |

|
[80] |
| 3-Octanol | 3-Octanol | 135°C, 15 h |
|
[80] | ||
| H2 | PhCl | 135°C, 16 h |
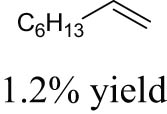
|
[80] | ||
| Na2SO3 | PhCl | 135°C, 15 h |
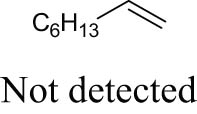
|
[80] |
2.1 Molybdenum catalyst
One prominent feature of Re related to its high DODH catalytic activity is that Re has two groups of variable oxidation states including +V to +VII and +Ⅱ to +V [83,84,85]. Similar to the multiple valences of Re, Mo also features a series of variable oxidation states ranging from +Ⅱ to +VI, rendering Mo a very promising candidate for DODH reaction. Moreover, Mo species are relatively abundant in the Earth’s crust and the prices of Mo are much lower than that of Re. Consequently, developing Mo-based catalysts represents an important direction aiming at economical and technical viable implementation of DODH reactions.
In 2013, Galindo et al. reported the first example of Mo-catalyzed DODH reaction by using molecular Mo complexes as catalysts (see Figure 2) [86]. The reaction of Mo(O)2(acac)2 with 2 equiv. of HQR ligands (HQR = 3-methyl-1-phenyl-4-alkylcarbonyl-5-pyrazolone, wherein R group = cyclohexyl (HQCy) or hexyl (HQHe)) afforded MoVI(O)2(QR)2 catalysts (see Scheme 2a). The authors used Mo(O)2(QCy)2 and Mo(O)2(QHe)2 to catalyze the DODH reaction of styrene glycol with PPh3 as a reductant (see Scheme 2b). Both catalysts afforded moderate-to-high conversions of styrene glycol but with very low styrene yields at around 10%. The authors speculated that the low olefin yields could be the inhibiting effect of the side product water. To verify this speculation, three water-removal methods including the Dean-Stark device, molecular sieve (4 Å), and Na2SO4 were applied to perform the DODH reaction of styrene glycol under anhydrous conditions. However, none of these three water-removal methods improved the olefin yields, thus excluding the detrimental effects of water formed during the reaction. In the case of 1,2-cyclooctanediol catalyzed by Mo(O)2(QCy)2, the diol was totally converted and the cyclooctene yield reached 55% at 110℃ for 18 h, corresponding to turnover number (TON) of 27.5. By contrast, the oxidized reductant (OPPh3) was generated in a much lower yield of only 28%, suggesting that the oxygen atom of the diol not only transferred to PPh3 during the reaction.
![Figure 2
Molecular structures of Mo complexes for DODH reaction. Reprinted with permission from ref. [86].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_002.jpg)
Molecular structures of Mo complexes for DODH reaction. Reprinted with permission from ref. [86].
![Scheme 2
(a) Synthesis of MoVI(O)2(QR)2 and (b) MoVI(O)2(QR)2-catalyzed DODH reaction of styrene glycol into styrene. Adapted with permission from ref. [86].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_018.jpg)
(a) Synthesis of MoVI(O)2(QR)2 and (b) MoVI(O)2(QR)2-catalyzed DODH reaction of styrene glycol into styrene. Adapted with permission from ref. [86].
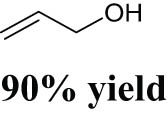
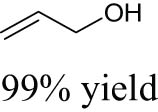
Fristrup et al. explored the performance of various Mo compounds, such as ammonium heptamolybdate (AHM, (NH4)6Mo7O24), MoO2(CH3)2(bipy) (bipy = 2,2′-bipyridine), MoO2Cl2(bipy), and Na2MoO4, for the DODH reaction of aliphatic vicinal diols into corresponding olefins [87]. Catalyst screening of different Mo compounds showed that Na2MoO4 gave the lowest olefin yield probably due to its poor solubility in dodecane solvent. AHM gave the highest yield with a 71% yield of total volatile products (of which 1-hexene accounted for 16%). Similar olefin yields of around 60% were obtained over MoO2(CH3)2(bipy) and MoO2Cl2(bipy). A plausible reason for the similar reactivities of the different Mo precursors is that these Mo compounds probably transform into the same catalytically active species when dissolving in alcohol solvent at high temperatures. It was found that the DODH reaction is accompanied by the oxidative cleavage (oxidative deformylation) of vicinal diol to generate two aldehydes as side products (see Scheme 3a), in particular the case of using diol as a reductant. These carbonyl compounds could further react with vicinal diol to generate acetals (see Scheme 3b). Given this context, the olefin yields could only be up to 50% theoretically. Consequently, if the olefin is the objective product of Mo-catalyzed DODH reaction, the sacrifice of at least 50% diols for oxidation and acetal formation would be inevitable. However, if the objective products are aldehydes and acetals, the utilization of diol as both reactant and reductant becomes attractive from an economic point of view because of the replacement of expensive reductant with diol. Through solvent screening experiments, it could be seen that the best results were obtained by using 1,5-pentanediol as the solvent (1,2-hexanediol yielding 45% 1-hexene and glycerol yielding 40% allyl alcohol). The failure to obtain more than 50% olefin yield also proves that oxidative deformylation drives Mo-catalyzed DODH reaction. The usage of 1,5-pentanediol as a solvent probably impedes the acetals formation, thereby improving the olefin yield.
![Scheme 3
(a) Oxidative deformylation of 1,2-diol into aldehydes and (b) condensation reaction of 1,2-diol with aldehyde to give acetals. Adapted with permission from ref. [87].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_019.jpg)
(a) Oxidative deformylation of 1,2-diol into aldehydes and (b) condensation reaction of 1,2-diol with aldehyde to give acetals. Adapted with permission from ref. [87].
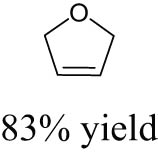
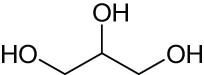
The catalytic performance of AHM for DODH was further explored by the same group [88]. As mentioned above, when the vicinal diol itself was the reductant, half of the vicinal diol was deoxydehydrated into the corresponding olefins, and the rest underwent oxidative deformylation. At the same time, side products such as aldehydes, acetals, and oligomers would affect product separation and catalyst recovery. Solvent screening experiments indicated that utilizing isopropanol as solvent could give the highest olefin yield of 49% at a full conversion of 1,2-decanediol. The substrate scope for DODH was investigated over 5 mol% AHM catalyst at 240–250℃ in the presence of isopropanol as both reductant and solvent. Experimental results showed that the corresponding olefin yields of 1,2-hexanediol and 3,4-hexanediol were 46% and 42% in the absence of base, respectively. The addition of NBu4OH further improved the olefin yields by up to 77% (1,2-hexanediol) and 69% (3,4-hexanediol). In contrast to 1,2-hexanediol and 3,4-hexanediol, 1,4-anhydroerythritol gave the similar 2,5-dihydrofuran (2,5-DHF) yields of 75% and 74%, in the absence and presence of base, respectively. The beneficial effect of NBu4OH is likely related to its basic properties blocking side reactions of diols and isopropanol. Moreover, the DODH mechanism of 1,4-anhydroerythritol was studied by density functional theory (DFT), and the corresponding free energy diagram is shown in Figure 3. The step with the highest activation energy is the oxidation of iPrOH (TS7–8, 24.1 kcal·mol−1) with concomitant reduction of MoVI glycolate to MoⅣ glycolate. The extrusion of 2,5-DHF has the second-highest activation energy (TS9–10, 13.8 kcal·mol−1). Consequently, the reduction of the formed Mo-anhydroerythritol complex prior to the olefin extrusion is the rate-limiting step. In 2018, Navarro and John used AHM to catalyze the DODH reaction of several vicinal diols such as styrene glycol and 1,2-octanediol with a variety of reductants [89]. Sodium sulfite (Na2SO3) was discovered as a benign reductant with moderate olefin yields of around 23% under relatively mild conditions (170–190℃), and its oxidation product (SO4 2−) is non-toxic and easy to recover. The olefin yields were found to decrease in the presence of pyridine, indicating that base additives could inhibit catalytic activity probably owing to the coordination of pyridine with Mo center.
![Figure 3
The free energy diagram for the catalytic DODH of 1,4-anhydroerythritol into 2,5-DHF using iPrOH as a reductant. Reprinted with permission from ref. [88].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_003.jpg)
The free energy diagram for the catalytic DODH of 1,4-anhydroerythritol into 2,5-DHF using iPrOH as a reductant. Reprinted with permission from ref. [88].
Well understanding of the reaction mechanism of Mo-catalyzed DODH is of pivotal importance in developing Mo catalysts with improved activity and selectivity as well as expanding the application of Mo-catalyzed DODH of biomass derivatives. Fristrup et al. used DFT to investigate the mechanism of Mo-catalyzed DODH of vicinal diols [85]. The catalytic cycle was proposed to include the condensation of vicinal diol with MoVI oxide, subsequent oxidative cleavage to generate MoIV complex, and the extrusion of olefin. To elucidate the details of the mechanism, the authors calculated and compared the energy barriers required to fulfill these steps based on the widely proposed catalysis cycle. It was found that diolate cleavage (olefin extrusion) occurs more readily by mononuclear MoVI complexes than by dinuclear MoVI complexes. By calculating and comparing the energy barriers of olefin extrusion and the stability of transition states, the monodiolate complex 8 more likely extrudes olefin via the [3 + 2] retro-cycloaddition reaction instead of via the bisdiolate complex 10 (see Figure 4). The above results clearly indicate that mononuclear Mo complexes could serve as active species to form a diolate intermediate with diol. Within the whole catalytic cycle, two key steps with the high energy barriers involve the cleavage of one diolate ligand in the mononuclear MoVI bisdiolate complex to form formaldehyde and acetaldehyde with a required energy barrier of 20.8 kcal·mol−1 and the subsequent extrusion of diolate to an olefin with a required energy barrier of 19.1 kcal·mol−1. Taking MoO3-catalyzed DODH of 1,2-propanediol as an example, a plausible catalytic cycle is depicted in Scheme 4: (1) the reduction of MoO3 by 1,2-propanediol to form (mononuclear) MoVI monodiolate complex 12 and water; (2) another molecule of 1,2-propanediol condensates with complex 12 to form (mononuclear) MoVI bisdiolate complex 13 and water; (3) one of the diolate ligands in complex 13 is oxidized and cleaved to form formaldehyde and acetaldehyde, subsequently forming (mononuclear) MoIV monodiolate complex 14; and (4) the extrusion of propylene from complex 14 accompanied by the regeneration of MoIV into MoO3. Verdicchio et al. investigated two possible reaction pathways for Mo-catalyzed DODH of diol in the presence of phosphane as a reductant by using DFT [90]. In the first reaction pathway, the Mo complex first condenses with diol to form a Mo-diolate intermediate, followed by reduction (eliminating one oxygen atom in Mo-diolate) and finally the extrusion of alkene. The second pathway is the same as the first one with the exception that the reduction of Mo complex occurs before the condensation of Mo with diol. In comparison to the first pathway, the relatively lower ΔG difference between reaction intermediates calculated for the second pathway (45.3 vs 55.4 kcal·mol−1) makes it a preferred mechanism for Mo-catalyzed DODH of diols using a phosphane reductant.
![Figure 4
The extrusion of alkene from monodiolate complex 8 and bisdiolate complex 10 by the [3 + 2] retro-cycloaddition mechanism. Reprinted with permission from ref. [85].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_004.jpg)
The extrusion of alkene from monodiolate complex 8 and bisdiolate complex 10 by the [3 + 2] retro-cycloaddition mechanism. Reprinted with permission from ref. [85].
![Scheme 4
Proposed reaction mechanism for Mo-catalyzed DODH accompanied by the oxidative cleavage of diol into aldehydes. Reprinted with permission from ref. [85].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_020.jpg)
Proposed reaction mechanism for Mo-catalyzed DODH accompanied by the oxidative cleavage of diol into aldehydes. Reprinted with permission from ref. [85].
Sandbrink et al. reported in 2016 that TiO2-supported Re catalyst (ReOx/TiO2) afforded superior catalytic activity and stability in DODH compared to APR/C (APR = ammonium perrhenate NH4ReO4), ReOx/C and ReOx/SiO2 [56]. Inspired by ReOx/TiO2 catalyst, it can be envisioned that Mo oxides could be supported on TiO2 to generate heterogeneous Mo catalysts. Palkovits and Okuda et al. first reported a heterogeneous Mo catalyst (MoOx/TiO2) for the DODH of 1,4-anhydroerythritol [59]. Similar to the preparation of ReOx/TiO2, MoOx/TiO2 (2b) was obtained by the reduction of (NH4)6Mo7O24·4H2O/TiO2 (2a) at 300℃ under H2 atmosphere for 3 h. In the catalytic DODH reaction of 1,4-anhydroerythritol to 2,5-DHF, MoOx/TiO2 (2b) gave 94% conversion of 1,4-anhydroerythritol and 55% yield of 2,5-DHF (see Scheme 5). One major side reaction is the dehydration of the reductant 3-octanol to 2-octene and 3-octene. As shown in Table 2, unreduced (NH4)6Mo7O24·4H2O/TiO2 (2a) showed comparable performance as that of MoOx/TiO2 (2b). The catalytic performance of catalysts 2a and 2b is higher than those of Na2MoO4·2H2O/TiO2 (1a) and MoOx/TiO2 (1b). One possible explanation is that utilizing (NH4)6Mo7O24·4H2O as the precursor led to more polymerized Mo clusters on the surface of TiO2 accounting for the higher DODH catalytic activity. It could be speculated that the catalytic performance of catalysts with polymerized Mo clusters on TiO2 is superior to that of isolated Mo species. Interestingly, the in situ mixture (2c) of TiO2 and (NH4)6Mo7O24·4H2O also gave a higher yield of 2,5-DHF than the supported catalysts 2a and 2b. In the recycling experiments, catalyst 2c showed inferior recyclability in comparison to catalyst 2b, indicating that TiO2 support plays an important role in stabilizing the catalytically active Mo species. One major side reaction is the dehydration of 3-octanol to form octene under acidic conditions. The yield of octene in the case of catalyst 2b is higher than that of catalyst 2c, indicating that the acidity of catalyst 2b is likely higher than that of catalyst 2c.
![Scheme 5
The DODH of 1,4-anhydroerythritol to 2,5-DHF over heterogeneous MoOx/TiO2 catalysts. Adapted with permission from ref. [59].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_021.jpg)
The DODH of 1,4-anhydroerythritol to 2,5-DHF over heterogeneous MoOx/TiO2 catalysts. Adapted with permission from ref. [59].
Product distribution of supported Mo catalysts and mixtures in the conversion of 1,4-anhydroerythritol to 2,5-DHF.a Reprinted with permission from ref. [59]
| Compound number | Catalyst (Mo content [%])b | X (1,4-anhydroerythritol) [%] | Y (2,5-DHF) [%] | Y (ketals) [%] | Y (octenes)c [%] |
|---|---|---|---|---|---|
| 1a | Na2MoO4·2H2O/TiO2 (4.8) | 27 | 1 | 0 | <1 |
| 1b | MoOx/TiO2 (Na2MoO4·2H2O)d (5.1) | 39 | 5 | 1 | <1 |
| 2a | (NH4)6Mo7O24·4H2O/TiO2 (4.9) | 100 | 48 | 3 | 15 |
| 2b | MoOx/TiO2 [(NH4)6Mo7O24·4H2O]e (4.7) | 94 | 55 | 9 | 48 |
| In situ mixture 1c | Na2MoO4 + TiO2 | 19 | 4 | 0 | 3 |
| In situ mixture 2c | (NH4)6Mo7O24·4H2O + TiO2 | 100 | 76 | 10 | 16 |
aConditions: 1,4-anhydroerythritol (0.4 mmol), 5 mol% Mo, 3-octanol (10 equiv. relative to substrate), 200℃, 18 h. Conversion (X) and yields (Y) determined by NMR spectroscopy. bDetermined by ICP-OES. cBased on 3-octanol. dMo-precursor: Na2MoO4·2H2O. Reduction conditions: 3 h, 300℃, 2℃·min−1. eMo precursor: (NH4)6Mo7O24·4H2O. Reduction conditions: 3 h, 300℃, 2℃·min−1.
In 2020, Xi et al. studied the reaction pathways of the DODH of 1,4-anhydroerythritol by MoOx/TiO2(101) catalyst via the first principle calculations [91]. The authors constructed the catalyst model by selecting the most stable crystal surface of anatase(101) and determining the internal atomic structure of the catalyst as MoO(2O)/3H-TiO2 by constrained thermodynamic calculations. As indicated by energy profile and microkinetic reaction model, low catalytic activity was obtained over MoO(2O)/3H-TiO2(101) at 140℃ (turnover frequency (TOF) of 1.50 × 10−6 s−1), whereas improved catalytic activity comparable to that of ReO2(2O)/6H-TiO2 was obtained (effective free energy barrier of 1.51 eV, TOF of 5.70 × 10−4 s−1) at 200℃. Mechanistically, dihydrofuran extrusion was identified as the rate-limiting step in the DODH process catalyzed by MoO(2O)/3H-TiO2. Moreover, the catalyst could regenerate by using H2 as a reductant.
The catalytic performance of molecular Mo catalysts is strongly related to the rational design of surrounding ligands connected to the Mo center. Beckerle et al. prepared a series of Mo complexes (3a–c and 4a) containing (OSSO)-type bis(phenolate) ligands (see Figure 5a) [92]. As characterized by nuclear magnetic resonance (NMR) spectroscopy and X-ray diffraction (XRD), this kind of MoVI complex is featured by octahedral spatial structures coordinated by tetradentate (OSSO) dianionic type ligands. These Mo complexes were examined for the DODH reaction of 1,4-anhydroerythritol to 2,5-DHF with the formation of by-products including ketal, furan, and octene (see Figure 5b). Catalysts 3a–c and 4a were tested under the same reaction condition (see Table 3). Catalyst 4a (see Figure 5c) gave the best catalytic performance, generating a 37% yield of 2,5-DHF. The DODH of 1,4-anhydroerythritol catalyzed by catalyst 4a at different temperatures by microwave radiation was also examined. The reaction time was shortened from 18 to 1 h by employing microwave radiation (see Table 4). At 200℃, the conversion of 1,4-anhydroerythritol and the yield of 2,5-DHF increased to 89% and 49%, respectively, with the reduced formation of by-products. Microwave radiation leads to more uniform heat distribution, thus enhancing the reaction rate and product selectivity. As the temperature decreased from 200 to 160℃, the conversion of 1,4-anhydroerythritol and the yield of 2,5-DHF also decreased significantly. The promoting effect of microwave radiation was also reported for MoO2(acac)2-catalyzed DODH of styrene glycol [93]. Under microwave radiation, styrene was generated in 51% yield at a complete conversion of styrene glycol in 40 min at 170℃ using PPh3 as a reductant. Even for the DODH of less-reactive 1,2-octanediol, the combination of MoO2(acac)2 and PPh3 still can afford a 40% yield of 1-octene in 40 min at 220℃ under microwave radiation. By contrast, a negligible amount of 1-octene (<1%) was obtained by traditional heating under the same conditions. The above results indicate that the mode of heating has an important effect on the catalytic performance of Mo catalysts.
![Figure 5
(a) Synthesis of MoVI complexes 3a–c and 4a containing (OSSO)-type ligands; (b) DODH reaction of 1,4-anhydroerythritol into 2,5-DHF; and (c) the molecular structure of catalyst 4a. Adapted with permission from ref. [92].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_005.jpg)
(a) Synthesis of MoVI complexes 3a–c and 4a containing (OSSO)-type ligands; (b) DODH reaction of 1,4-anhydroerythritol into 2,5-DHF; and (c) the molecular structure of catalyst 4a. Adapted with permission from ref. [92].
Reaction products of DODH of 1,4-anhydroerythritol. Reprinted with permission from ref. [92]
| Run | Catalyst | Conversion [%] | Yield of | |||
|---|---|---|---|---|---|---|
| 15 [%] | 16 [%] | 17 [%] | 18a [%] | |||
| 1 | 3a | 21 | 3 | 4 | <1 | 3 |
| 2 | 3b | 41 | <1 | 18 | <1 | 6 |
| 3 | 3c | 35 | 3 | 8 | <1 | 6 |
| 4 | 4a | 74 | 37 | 30 | 2 | 14 |
| 5b | 4a | 91 | 47 | 17 | 1 | 20 |
| 6c | 4a | 81 | 57 | 17 | 4 | 5 |
5 mol% Mo and 10 equiv. of 3-octanol relative to substrate, 200°C, 18 h reaction time; conversion and yield determined by NMR spectroscopy. aBased on 3-octanol. b5 Equiv. of 3-octanol. c20 Equiv. of 3-octanol.
DODH of 1,4-anhydroerythritol under microwave irradiation. Reprinted with permission from ref. [92]
| Run | Catalyst | T [℃] | Equiv. 3-octanol | Conversion [%] | Yield of | |||
|---|---|---|---|---|---|---|---|---|
| 15 [%] | 16 [%] | 17 [%] | 18a [%] | |||||
| 7 | 4a | 200 | 10 | 89 | 49 | 12 | 1 | 10 |
| 8 | 4a | 180 | 10 | 50 | 17 | 7 | <1 | 1 |
| 9 | 4a | 160 | 10 | 33 | 3 | 3 | <1 | <1 |
5 mol% Mo and 10 equiv. of 3-octanol relative to substrate, 1 h reaction time; conversion and yield determined by NMR spectroscopy. aBased on 3-octanol.
In addition to OSSO-type ligands, MoVI complexes consisting of ONNO-type salan ligands were examined for the DODH reaction of styrene glycol (see Figure 6a) [94]. The tetradentate salan ligand coordinates with the Mo center via two amine nitrogen atoms and two phenolic oxygen atoms. The skeleton region of the ligand is formed by a diamine linking to two benzyl rings. Using diamines with different structures (e.g., 1,2-ethylene diamine, 1,2-propyl diamine, and 1,2-phenylene diamine) could change the skeleton flexibility of salan-type ligands. The Mo complexes with ethylene and 1,2-propyl backbones afforded similar yields of alkene (47% for tBu,tBuL1 MoO2 and 48% for tBu,tBuL2 MoO2), whereas 1,2-phenylene backbone afforded a much lower yield of 12%. It was speculated that the Mo center needs to adapt its geometries to meet the required spatial states for effective coordination with OH moieties. Therefore, ligands with a more flexible skeleton would make the Mo catalyst more active. The effect of changing ortho- and para-substituents of the salan ligands on the DODH activity was studied. Among examined substituents (i.e., methoxy, nitro, chloride, tert-butyl), the di-tert-butyl substituted complexes, tBu,tBuL1 MoO2 and tBu,tBuL2 MoO2, showed highest DODH activity (see Figure 6b) with olefin yields of approximately 48%, slightly higher than the unsubstituted Mo complexes (45% yield). It could be concluded that the DODH activity of MoVI complexes is strongly influenced by the electron-donating properties of substituents rather than the space size of substituents. In addition, ligands with secondary amine structures on benzyl groups are more effective than those with tertiary amine structures because of the electron-donating effect of the alkyl group making the Mo complex more susceptible to dimerization.
![Figure 6
(a) Typical structure of salan ligand and (b) the structures of tBu,tBuL1 MoO2 (left) and tBu,tBuL2 MoO2 (right). Adapted with permission from ref. [94].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_006.jpg)
(a) Typical structure of salan ligand and (b) the structures of tBu,tBuL1 MoO2 (left) and tBu,tBuL2 MoO2 (right). Adapted with permission from ref. [94].
To find out the appropriate ligands for Mo catalysts, the influence of different types of ligands on the catalytic activity of Mo complexes needs to be investigated in depth. In 2018, Stalpaert and De Vos tested a range of Mo complexes with different spatial and electronic properties of β-diketone ligands (see Figure 7a) [95]. When excessive β-diketones were added into the DODH reaction with bis-acetylacetonato-dioxo-Mo (MoVIO2(acac)2) as a catalyst, β-diketones tended to replace the acetylacetone ligands of MoVIO2(acac)2 to form new Mo complexes. According to the experimental results (see Figure 7b), the catalytic systems added bulky dibenzoylmethane and 2,2,6,6-tetramethylheptanedione (TMHDH) exhibited superior activity, with TMHDH giving the highest yield of 1-hexene (36%). As indicated by the electrospray ionization mass spectrometry of the post-reaction liquid sample, Mo in monomer and oligomer forms is only detected in the TMHDH-added sample, probably because of the bulky β-diketone ligands preventing the oligomerization of Mo. As a result, Mo complexes remain stable during the reaction without precipitating and the loss of activity. This stabilizing effect of TMDHH for the Mo center provides a new idea in designing robust Mo catalysts for DODH. Furthermore, the electron donor TMHDH increases the electron density of Mo centers, which can facilitate the electron transfer between Mo and vicinal diol, speeding up the rate-controlled olefin extrusion step. These results suggest that the large size and electron-donating properties of TMHDH could effectively improve the catalyst stability and reaction rate. The olefin yields increased with the electron-donating ability increasing in the order of hexafluoroacetylacetone, trifluoroacetylacetone, acetylacetonate, and TMHDH (see Figure 7b). At 4 equiv. of TMHDH, a high 1-hexene yield of up to 93% with the highest TOF (4.7 h−1) was achieved. The high boiling point (202℃) of TMDHH and the volatile DODH products in the reaction mixture were separated by evaporation. The catalytic system of TMHDH and MoO2(acac)2 showed good universality with applications for different reductants, substrates (cyclohexanediol, styrene glycol, diethyl tartrate, erythritol, etc.), and solvents.
![Figure 7
(a) Mo complexes with different β-diketone ligands and (b) effect of the addition of different β-diketones to the reaction mixture on the Mo-catalyzed DODH of 1,2-hexanediol to 1-hexene. Adapted with permission from ref. [95].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_007.jpg)
(a) Mo complexes with different β-diketone ligands and (b) effect of the addition of different β-diketones to the reaction mixture on the Mo-catalyzed DODH of 1,2-hexanediol to 1-hexene. Adapted with permission from ref. [95].
In 2019, Lu et al. reported another bulky ligand of 8-hydroxyquinoline (8-HQ) with electron-donating properties for Mo catalysts [96]. The 8-HQ Mo complex (Mo-8-HQ) can be considered a single-site Mo catalyst. Mo-8-HQ was synthesized by a ligand exchange method, that is, the pyridine nitrogen and hydroxyl oxygen in 8-HQ coordinated with Mo atom, thereby replacing the acetylacetone ligand of MoO2(acac)2. Based on Fourier transform infrared spectroscopy and XRD, the structure of Mo-8-HQ complex contained a MoO2 (cis) center coordinating with two 8-HQ molecules. The authors speculated that the electron-donating pyridine nitrogen coordinated to the Mo center, making the Mo atom in the Mo-8-HQ in an electron-rich state. Regular diffraction spots were found by the selected area electron diffraction diagram, indicating the single-crystal characteristics of Mo-8-HQ. The scanning electron microscopy image showed that Mo-8-HQ particles appear as multilayered stacked prisms. Each structural unit of Mo-8-HQ forms layers through π–π stacking interaction, with the layers connected by hydrogen bonds, resulting in a stable prismatic single crystal structure (see Figure 8a). In the DODH of tartaric acid (TA) over a series of Mo catalysts, Mo-8-HQ gave the highest yield of dibutyl fumarate (DBFA) (about 74%). Using the dibutyl tartrate as the substrate, the yield of DBFA reached 86%, corresponding to a TON of 8.6. Moreover, no other side products were found for Mo-8-HQ, indicating its high selectivity in the DODH reaction. The low activity of other Mo catalysts could be a result of the aggregation of Mo species, low solubility, and loose coordination structure of Mo under reaction conditions. Comparing the performance of MTO and Mo-8-HQ at different molar concentrations under the same experimental conditions (see Figure 8b), the DBFA yield of Mo-8-HQ at 1 mol% was lower, but DBFA yields of both catalysts were similar when the metal content exceeded 5 mol%. It can be concluded that the catalytic performance of Mo-8-HQ with high Mo content is comparable to that of Re catalysts. One prominent feature of Mo-8-HQ is that after the reaction, it can be separated from the reaction mixture as a solid power for the next run with good catalytic activity. The good recyclability could be attributed to the stabilized Mo centers by the strong coordination ability of 8-HQ. Besides, owing to the presence of π–π stacking and hydrogen bonding effects in Mo-8-HQ, the catalyst structure remains unchanged after the reaction. Deuterium isotope experiments showed that the Mo-8-HQ-catalyzed DODH reaction proceeds by the synergistic cleavage of two adjacent C–O bonds of the substrate. The detailed steps of the reaction mechanism are shown in Scheme 6. The electron-rich Mo centers could facilitate the oxidation of MoIV-diolate intermediate by losing two electrons to form MoⅥ, thereby accelerating the extrusion of the olefins.
![Figure 8
(a) Possible Mo-8-HQ stacking structure. The Mo, oxygen, carbon, and nitrogen atoms are denoted in black, red, grey, and blue, respectively; (b) DODH of TA catalyzed by different molar ratios of MTO and Mo-8-HQ catalysts at 160℃ in air for 12 h. Adapted with permission from ref. [96].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_008.jpg)
(a) Possible Mo-8-HQ stacking structure. The Mo, oxygen, carbon, and nitrogen atoms are denoted in black, red, grey, and blue, respectively; (b) DODH of TA catalyzed by different molar ratios of MTO and Mo-8-HQ catalysts at 160℃ in air for 12 h. Adapted with permission from ref. [96].
![Scheme 6
Proposed reaction mechanism for the DODH of TA catalyzed by Mo-8-HQ. Reprinted with permission from ref. [96].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_022.jpg)
Proposed reaction mechanism for the DODH of TA catalyzed by Mo-8-HQ. Reprinted with permission from ref. [96].
Tran and Kilyanek synthesized a cis-dioxo Mo complex using a dianionic ONO pincer ligand for the DODH reaction of aromatic and aliphatic diols [97]. The synthesis method is shown in Scheme 7a, where the addition of triphenylphosphine oxide (OPPh3) produces a six-coordinate complex 20 and without additional ligands produces a five-coordinate complex 21 (see Figure 9). The hexa-coordinated complex 20 readily loses the labile ligand OPPh3 to form the penta-coordinated dioxygen MoVI species. Complex 20 with an octahedral cis-dioxo-bipyramidal Mo center (see Figure 9) is structurally similar to that of the most of hexa-coordinated cis-dioxo-MoVI complexes. Both complexes 20 and 21 have shown good catalytic activity in the DODH of styrene glycol, 1,2-octanediol, and hydrobenzoin. The DODH reaction of 1,2-octanediol catalyzed by complex 20 using PPh3 as reductant gave a 59% yield of 1-octene accompanied by the formation of deformylation products (6% yield). In the case of styrene glycol, styrene and deformylation products (aldehydes) were produced in moderate overall yields (30–50%) at a molar ratio of around 3/1 using different reductants. These results indicated that Mo-catalyzed conversion of diols followed two competitive reaction pathways including DODH to form olefins and deformylation to form aldehydes, as shown in Scheme 7b. At lower temperatures, complexes 20 and 21 showed higher activity in catalyzing the DODH of aromatic vicinal diols than aliphatic vicinal diols. This may be due to the partial delocalization of the π electrons of the C═C bond into the adjacent aromatic ring, which reduces the total energy of the orbital, resulting in a lower barrier to the aromatic olefin extrusion. The addition of exogenous OPPh3 to the DODH of styrene glycol catalyzed by complexes 20 and 21 resulted in lower product yields, suggesting that OPPh3 had an inhibitory effect on product formation. It was therefore speculated that the dissociation of OPPh3 from Mo complexes was required for the formation of catalytically active species.
![Scheme 7
(a) Synthesis process of ONO-Mo complexes and (b) possible reaction pathways involved in the conversion of diols over Mo complexes. Adapted with permission from ref. [97].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_023.jpg)
(a) Synthesis process of ONO-Mo complexes and (b) possible reaction pathways involved in the conversion of diols over Mo complexes. Adapted with permission from ref. [97].
![Figure 9
Thermal ellipsoid plots of complexes 20 (left) and 21 (right). Reprinted with permission from ref. [97].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_009.jpg)
Thermal ellipsoid plots of complexes 20 (left) and 21 (right). Reprinted with permission from ref. [97].
In 2020, Siu et al. reported dioxo-Mo complexes containing amine bisphenolate ligands as DODH catalysts, focusing on the influence of the electronic and spatial properties of the bisphenolate ligands on the catalyst performance [98]. The authors prepared several dioxo-Mo complexes by the ligand exchange reaction of MoO2(acac)2 and different bisphenolate ligands containing the pendant arm of various amine donors (see Scheme 8a). These complexes were examined in the DODH reaction of styrene glycol with PPh3 as reductant and toluene as solvent at 170℃ (see Scheme 8b). The catalytic performance of bisphenolate ligands with different structure were compared to understand structure–activity relationship. Complex 5a containing a pyridine pendant arm showed the lowest styrene yield (5%). This may be due to the strong coordination of the pyridine N atom on the pendant arm of 5a to the Mo center, occupying the binding sties of Mo centers and thus reducing the catalytic activity. The styrene yields of 5c and 5d were higher than 5a (31% and 34%, respectively), indicating that the side arms with weakly coordinated and uncoordinated properties could enhance the catalytic activity of Mo complexes. Compared with the two phenol arms in complex 5a, complex 6b with one less phenol arm showed a much higher yield of styrene (32%), indicating that removing the phenol arm had a similar beneficial effect as that of reducing the coordination abilities of the pendant arm. Solvent screening experiments using complex 5d showed similar moderate styrene yields (30–34%) in the case of aromatic solvents (trimethylbenzene, xylene, toluene, etc.) and comparatively lower styrene yields (about 20%) were obtained for polar solvents such as N,N-dimethylformamide and N,N′-dimethylpropyleneurea (DMPU). A small amount of benzaldehyde was always found as a side product (1–13% yield) resulting from the deformylation of styrene glycol, and the highest yield of benzaldehyde was obtained in DMPU.
![Scheme 8
(a) Synthesis process of Mo bisphenolate complexes and (b) Mo-catalyzed DODH of styrene glycol. Adapted with permission from ref. [98].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_024.jpg)
(a) Synthesis process of Mo bisphenolate complexes and (b) Mo-catalyzed DODH of styrene glycol. Adapted with permission from ref. [98].
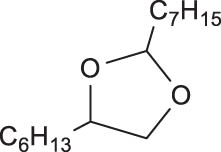
Oxo-rhenium complexes (CpReO3) containing versatile substituted cyclopentadienyl (Cp) groups as ligands have been extensively investigated for DODH. Similarly, Cp-based Mo complexes are expected to be promising catalyst candidates for DODH reaction. In 2020, Li et al. explored a series of Cp-based Mo complexes including [Cp*MoO2]2O (Cp* = 1,2,3,4,5-pentamethylcyclopentadienyl), [CpttMoO2]2O, and NBu4[Cp*MoO3] for the DODH reaction of 1,2-octanediol with PPh3 as the reductant and anisole as the solvent [99]. The DODH reaction of 1,2-octanediol was carried out under the optimal conditions of 5 mL anisole, 1.1 equiv. of PPh3, 2 mol% [Cp*MoO2]2O and 200℃, affording 55% olefin yield at complete conversion of 1,2-octanediol after 15 h. The experimental results show that the [Cp*MoO2]2O gave the highest yield and selectivity of 1-octene (Table 5, Run 1). Both the catalyst NBu4[Cp*MoO3] and the in situ mixed catalyst prepared by NBu4OH and [Cp*MoO2]2O gave the same olefin yields (38%) at full conversion (>99%). Under the reaction conditions, two possible reaction pathways were proposed for [Cp*MoO2]2O-catalyed DODH reaction, either undergoing reduction to form [Cp*MoO2]2 or direct reaction with diols to form asymmetric diolate. Mechanistic studies revealed that the catalytic activity of [Cp*MoO2]2 is very low, despite being detected during the reaction, thus excluding the possibility of [Cp*MoO2]2O reduction to form [Cp*MoO2]2 as the active species that participate in the catalytic cycle. Hence, the authors proposed a likely reaction mechanism beginning with the condensation of diol with [Cp*MoO2]2O to form a diolate intermediate followed by reduction and olefin extrusion (see Scheme 9).
Product distribution of DODH of 1,2-octanediol catalyzed by using different Mo catalysts.a Reprinted with permission from ref. [99]
| Run | [Mo] |

|

|
|
Conversion [%] | Mass balancec [%] |
|---|---|---|---|---|---|---|
| 1 | [Cp*MoO2]2O | 55 | 6 | 6 | >99 | 79 |
| 2 | NBu4[Cp*MoO3] | 38 | 7 | 8 | >99 | 68 |
| 3b | [Cp*MoO2]2O | 38 | 11 | 4 | >99 | 68 |
aReaction conditions: 1,2-octanediol (0.5 mmol), PPh3 (0.55 mmol, 1.1 equiv.), [Mo] (4 mol% on basis of Mo atom), anisole (5 mL), 200°C, 15 h, N2, closed pressure tube. Reported values are the average of three independent runs. Products were determined by gas chromatography using mesitylene (0.5 mmol) as an internal standard. b6 mol% of NBu4OH was added as a 1 M solution of NBu4OH in methanol. cMass balance = ([yield (1-octene) + 2 × yield (2,4-dihexyl-1,3-dioxolane) + 2 × yield (2-heptyl-4-hexyl-1,3-dioxolane)]/conversion) × 100%.
![Scheme 9
Possible mechanism of DODH reaction catalyzed by [Cp*MoO2]2O. Red. = PPh3 or diol, ORed. = OPPh3 or carbonyl compound. Reprinted with permission from ref. [99].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_025.jpg)
Possible mechanism of DODH reaction catalyzed by [Cp*MoO2]2O. Red. = PPh3 or diol, ORed. = OPPh3 or carbonyl compound. Reprinted with permission from ref. [99].
Hacatrjan et al. reported a heterogeneous Mo catalyst MoOx-Au/TiO2 for DODH reaction [100]. MoOx-Au/TiO2 is capable of selectively catalyzing the conversion of 1,4-anhydroerythritol to 2,5-DHF with H2 as an economical reductant. The catalyst was synthesized by the combination of the Mo compound (NH4)6Mo7O24·4H2O with Au precursor as promoter dispersed on P-25 TiO2 support. First, TiO2-supported Au (dpAu/TiO2) was prepared by a deposition–precipitation method and then by impregnating dpAu/TiO2 with (NH4)6Mo7O24·4H2O aqueous solution to prepare MoOx-dpAu/TiO2 catalyst. As a control, MoOx-impAu/TiO2 catalyst was prepared by the impregnation method. Compared with MoOx-impAu/TiO2 catalyst, MoOx-dpAu/TiO2 catalyst showed higher catalytic activity with 67% conversion of 1,4-anhydroerythritol and 96% selectivity towards 2,5-DHF. MoOx-dpAu/TiO2 has a high Au dispersion and 3–5 nm Au particle size, whereas MoOx-impAu/TiO2 has a relatively low Au dispersion and 25–150 nm Au particle size (see Figure 10a). Small Au particles were believed to exhibit stronger hydrogen adsorption and spillover capability than large Au particles, accounting for the difference in catalytic activity for MoOx-dpAu/TiO2 and MoOx-impAu/TiO2. It was found that the MoOx-dpAu/TiO2 (P-25) with 1 wt% Mo and 0.3 wt% Au gave the best catalytic performance. The MoOx-dpAu/TiO2 (P-25) after the reaction was collected via the “regeneration” method. The organic deposits on the catalyst surface after the reaction led to the catalyst deactivation, and the calcination step in the “regeneration” method removed them sufficiently. Therefore, it is possible to obtain activity-maintained MoOx-dpAu/TiO2(P-25) by the “regeneration” method. The H2-temperature programmed reduction (TPR) profiles in Figure 10b display that dpAu/TiO2 (P-25) shows only very small hydrogen consumption below 500℃, MoOx/TiO2 (P-25) shows hydrogen consumption at 400℃, and MoOx-dpAu/TiO2 (P-25) shows hydrogen consumption below 190℃ (corresponding to the reduction of MoVI–MoIV). It is suggested that Au nanoparticles on the surface of MoOx-dpAu/TiO2 (P-25) catalyst promote the Mo reduction at lower temperatures. As indicated by various characterization methods including XRD, STEM, H2-TPR and X-ray absorption fine structure, well-dispersed MoIV oxide clusters on the TiO2 particles surface are proposed as the active sites for DODH reaction. As depicted in Figure 10c, the reaction mechanism was divided into four steps: (1) the first coordination of MoIV with 1,4-anhydroerythritol to give a diolate specie and two molecules of water; (2) the simultaneous cleavage of two C–O bonds to form 2,5-DHF and MoIV–MoVI specie; (3) the transformation of MoIV–MoVI specie into MoV–MoV specie in a dynamic equilibrium; and (4) the reduction of MoV–MoV specie into initial MoIV oxide cluster specie by the active hydrogen species resulting from the H2 activation on Au particles.
![Figure 10
(a) TEM images of MoOx-impAu/TiO2 (P-25) (left) and MoOx-dpAu/TiO2 (P-25) (right) (Au 0.3 wt%, Mo 1.0 wt%) after reaction; (b) H2-TPR profiles of different Au catalysts; and (c) the catalytic mechanism of MoOx-dpAu/TiO2 (P-25) for the DODH of 1,4-anhydroerythritol. Reprinted with permission from ref. [100].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_010.jpg)
(a) TEM images of MoOx-impAu/TiO2 (P-25) (left) and MoOx-dpAu/TiO2 (P-25) (right) (Au 0.3 wt%, Mo 1.0 wt%) after reaction; (b) H2-TPR profiles of different Au catalysts; and (c) the catalytic mechanism of MoOx-dpAu/TiO2 (P-25) for the DODH of 1,4-anhydroerythritol. Reprinted with permission from ref. [100].
Typically, transition-metal-catalyzed DODH reactions generally involve three steps: (1) the condensation of diol with MOx (M = Re, Mo, V, etc.) catalyst to form a diolate intermediate accompanied by the formation of one molecule of water; (2) the cleavage of two C–O bonds of diolate intermediate to give olefin products; and (3) the reduction of one M═O bond of the oxidized MOx catalyst to recover the oxidation state of MOx. In 2020, Xi and Heyden reported novel heterogeneous atom-pair catalysts for DODH including Ru2/MoS2, Ir2/MoS2, Ru3/MoS2, and Ir3/MoS2 by supporting transition metals Ru and Ir onto MoS2 [101]. Different from the above three-step mechanism, this type of catalyst removes the vicinal OH group by another mechanism, namely, the cleavage of the C–O bond of vicinal diol is triggered by the first activation of the C–H bond over Ru or Ir sites. The basal planes of the MoS2 support are susceptible to the formation of multiple sulfur vacancies by hydrogen annealing, electrochemical desulfurization, or H2O2 chemical etching (see Figure 11a), and the sulfur vacancies can serve as anchoring sites for transition metals. Experimental and computational results have shown that the sulfur vacancies of MoS2 are capable of stabilizing the supported Ru, Ir metal monoatoms, and also Ru2, Ru3, Ir2, and Ir3 atom-pair. Compared with monoatoms, sulfur vacancies stabilize metal atom-pair better [102]. Through kinetic modeling studies, Ru2/MoS2, Ir2/MoS2, and Ru3/MoS2 are found to catalyze the DODH of ethylene glycol efficiently by the C–H activation mechanism with predicted TOFs exceeding 1 s−1, whereas Ir1/MoS2 and Ru1/MoS2 showed inferior activity. The synergic effects of Ru or Ir atoms with MoS2 result in the excellent catalytic activity for DODH. The exergonic adsorption of ethylene glycol on the transition metal sites reduces the overall barrier effectively, and the C–H activation reduces the energy barrier for the subsequent C–O bond cleavage. A conventional bifunctional catalyst of ReOx-Pd/CeO2 activates H2 via the catalysis of active Pd species, whereas atom-pair catalysts activate H2 via Ru or Ir atomic clusters without the participation of a second metal component. Overall, the C–H activation pathway is superior to the C–OH direct cleavage pathway in the case of MoS2-supported atom-pair catalysts (see Figure 11b).
![Figure 11
(a) Configuration of a (4 × 4) MoS2 supercell with two S vacancies. The position of S vacancy is marked by red dashed circle; (b) comparison of the effective activation free energies of the C–OH bond cleavage by direct chemical bond-cleavage mechanism and C–H bond activation mechanism. Reprinted with permission from ref. [101].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_011.jpg)
(a) Configuration of a (4 × 4) MoS2 supercell with two S vacancies. The position of S vacancy is marked by red dashed circle; (b) comparison of the effective activation free energies of the C–OH bond cleavage by direct chemical bond-cleavage mechanism and C–H bond activation mechanism. Reprinted with permission from ref. [101].
2.2 Tungsten catalyst
W holds great potential to catalyze DODH considering that the nature of W is very similar to that of Mo, a widely studied active component for DODH catalysts. However, the successful application of W-based catalysts for DODH is rare. Sharpless et al. reported that K2WCl6 can be used to facilitate the DODH reaction of diols [76]. Under a nitrogen atmosphere, the conversion of diols to olefin proceeded smoothly using THF as a solvent in the presence of methyl lithium (MeLi) and K2WCl6. The diol first reacts with 2 equiv. of MeLi to form the dilithium intermediate 22, followed by adding 2 equiv. of K2WCl6 for catalysis and reflux into the corresponding olefin (see Scheme 10). The number of substituents in the vicinal diol has an effect on the reaction efficiency. The reaction time for tetra-substituted vicinal diols was 5–9 h with the olefin yields of 72–74%, while the reaction time for mono-, di-, and tri-substituted vicinal diols was up to 2–4 days with the olefin yields of 36–66%. Strictly speaking, K2WCl6 cannot be considered a catalyst despite its DODH ability, since the oxidized WⅥ species (K2WCl4O2) needs another reduction step to regenerate the initial K2WCl6. In addition to K2WCl6, oxygenated W compounds have also been explored for DODH including WO3, Pd-WOx, and oxide-supported tungstate [103,104]. However, these W oxides showed negligible activities in the DODH of diol substrates into olefins with the considerable formation of side products. To open the potential of W species for DODH, more studies are needed to investigate the interaction of diols with W species as well as the effect of different ligands/atoms connected to W center.
![Scheme 10
The DODH of vicinal diol into olefin via an intermediate of vicinal dialkoxide promoted by methyllithium and K2WCl6 in sequence. Reprinted with permission from ref. [76].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_026.jpg)
The DODH of vicinal diol into olefin via an intermediate of vicinal dialkoxide promoted by methyllithium and K2WCl6 in sequence. Reprinted with permission from ref. [76].
2.3 Vanadium catalyst
In 2013, Chapman and Nicholas first reported that V oxide compounds could serve as catalysts for DODH reaction (see Figure 12b) [105]. Various V catalysts including VO3 − and dioxovanadate derivatives along with reductants (e.g., PPh3, sodium sulfite) were screened for the DODH reaction of styrene glycol and 1,2-octanediol. As indicated by experimental results, the combination of N,O,O-tridentate V complex [n-Bu4N](dipic)VO2 (dipic = 2,6-pyridinedicarboxylate) (see Figure 12a) with PPh3 or sodium sulfite as reductant showed the highest DODH activity with the conversions of both styrene glycol and 1,2-octanediol up to 100%. The highest yield of 1-octene (97%) was obtained when using PPh3 as a reductant in PhCl after 48 h without the formation of olefin isomers. Despite the high yield of 1-octene, the TON of [n-Bu4N](dipic)VO2 catalyst was estimated to be only 9.7. This very low activity is a result of the applied high catalyst loading (10 mol%). The catalytic activity of [n-Bu4N](dipic)VO2 in the DODH of various diols with different stereoselectivity was examined using PPh3 or Na2SO3 as a reductant. The combination of [n-Bu4N](dipic)VO2 with PPh3 can effectively catalyze the DODH of styrene glycol, 1,2-octanediol, 1,2-hexanediol, and diethyl tartrate, with excellent conversions (>90%) and olefin yields (>80%) at 170℃. However, the DODH of cis-1,2-cyclohexanediol afforded a relatively low yield of cyclohexene (15%), which may be caused by spatial/conformational effects during diolate formation/cleavage or by competitive cis–trans isomerization of glycol. The possible V-catalyzed pathways were hypothesized based on previously reported reaction pathways for the oxorhenium catalytic system [106]. As shown in Figure 12c, there are two possible pathways for the catalytic reaction: (path A) the condensation of vicinal diol with complex 23 to form complex 24 followed by reduction by PPh3 to form diolate 25 and finally the extrusion of olefin from diolate 25 to regenerate complex 23 and (path B) the reduction of complex 23 by PPh3 to form complex 26 followed by condensation with a vicinal diol to form diolate 25 and finally the extrusion of olefin from diolate 25 to regenerate complex 23.
VO2; (b) DODH of vicinal diol catalyzed by V oxides; and (c) possible reaction pathways for [n-Bu4N](dipic)VO2-catalyzed DODH. Adapted with permission from ref. [105,106].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_012.jpg)
In 2016, Galindo et al. further investigated above-mentioned two reaction pathways of [n-Bu4N](dipic)VO2-catalyzed DODH reaction by DFT calculations [107]. Figure 13 shows the relative Gibbs free energy diagrams for reaction intermediates and transition states of two catalytic processes in benzene. By comparison, the activation energy for vicinal diol condensation in path B is 13.4 kcal·mol−1, which is much lower than that of this step in path A (39.8 kcal·mol−1). The activation energies of the other two stages (i.e., reduction and olefin extrusion) are similar. Olefin extrusion was identified as the rate-determining step for both pathways with an activation energy of up to 45.1 kcal·mol−1. The main difference between the two catalytic pathways is the activation energy of the vicinal diol condensation step. In path A, the vicinal diol condenses with the coordination-saturated VV complex through the transition state TS(2–3), while in path B, the vicinal diol condenses with the coordination-unsaturated VIII complex (generated from the reduction of VV complex) through the transition state TS(12–13)t. The V-catalyzed DODH reaction was believed to follow path B by comparing the activation energies of vicinal diol condensation in two catalytic cycles.
![Figure 13
Relative Gibbs free energy diagrams for the intermediates and transition states of path A and path B. Reprinted with permission from ref. [107].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_013.jpg)
Relative Gibbs free energy diagrams for the intermediates and transition states of path A and path B. Reprinted with permission from ref. [107].
Based on the report by Galindo et al., Poutas et al. proposed a new reaction mechanism for [n-Bu4N](dipic)VO2-catalyzed DODH in the same year [108]. To simplify the reaction pathway and reduce computational workload, the authors replaced PPh3 with trimethylphosphine (PMe3) as the reductant and glycerol with glycol as the substrate. As reported by Galindo et al., the reaction was believed to follow path B via the condensation of reduced V complex with diol. Faza et al. proposed a similar reaction mechanism except that the cleavage of diolate 6 (the condensation product of diol with VIII complex) to form olefin prefers to proceed via a free radical mechanism with an activation energy of 34.1 kcal·mol−1, rather than by synergistic [3 + 2] retro-cycloaddition with a much higher activation energy of 45.1 kcal·mol−1, as reported by Galindo et al. Specific reaction steps were proposed based on DFT calculations (see Figure 14): (1) the reduction of [n-Bu4N](dipic)VVO2 (complex 1) by PMe3 followed by condensation with glycol to form [VIII(dipic)(–O–CH2CH2–O–)] (complex 6); (2) the homolytic cleavage of one of the C–O bonds of diolate 6 (with an energy barrier of 12.5 kcal·mol−1) to produce the biradical intermediate 7; and (3) the subsequent elimination of olefin from intermediate 7 probably via the [2 + 2] retro-cycloaddition reaction with the regeneration of complex 1. The cleavage of the C–O bond of diolate 6 to form biradical is the rate-determining step with an energy barrier of 34.1 kcal·mol−1. In addition, the authors proposed that the stabilization of biradical species 7 could facilitate the C–O bond cleavage of diolate 6, thus possibly increasing the reaction rate. The replacement of oxygen atom with the sulfur atom for the pyridine ligand of V complex was found to stabilize the biradical species 7, resulting in an energy barrier of only 23.0 kcal·mol−1 for the conversion of diolate 6 to intermediate 7.
VO2. Reprinted with permission from ref. [108].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_014.jpg)
Proposed DODH reaction mechanism catalyzed by [n-Bu4N](dipic)VO2. Reprinted with permission from ref. [108].
Inspired by the multiple reaction pathways reported for MeReO3-catalyzed DODH reaction [81,84,109], Jiang et al. systemically studied three possible pathways of DODH catalyzed by [n-Bu4N](dipic)VO2 by DFT calculations [110]. As shown in Scheme 11a, neither path B nor path C is considered possible for V-catalyzed DODH, owing to the high energy barriers required for the reduction of oxovanadium catalyst (>40 kcal·mol−1) as well as olefin extrusion (>40 kcal·mol−1). The reaction prefers to proceed via path A through condensation/reduction/olefin extrusion steps with a lower free energy barrier of 33.7 kcal·mol−1 for olefin production. Distinct from the aforementioned pathways for V-catalyzed DODH, the authors have pointed out the important roles of spin crossover related to reaction intermediates in promoting the reduction of V diolate and olefin extrusion, two key steps in path A. A complete catalytic cycle of path A is depicted in Scheme 11b. First, LVO2 (i.e. [(dipic)VO2]−) condenses with a vicinal diol to form VV diolate. Subsequently, the VV diolate is reduced by PPh3 to form triplet VIII diolate by spin crossover. The cleavage of a C–O bond of VIII diolate generates a carbon radical, followed by a second spin crossover to form a singlet alkyl VV cyclic intermediate. Finally, this cyclic intermediate undergoes synergistic cleavage of V–C and C–O bonds to yield olefin and complex LVO2. The total free energy barriers for the reduction of VV-diolate by PPh3 (34.3 kcal·mol−1) and the extrusion of olefin (33.7 kcal·mol−1) are higher than other steps in path A, indicating that both the reduction of VV diolate and olefin extrusion act as the rate-determining steps.
![Scheme 11
(a) Possible mechanism of DODH reaction catalyzed by a V complex and (b) Proposed reaction mechanism of V-catalyzed DODH according to DFT. Adapted with permission from ref. [110].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_027.jpg)
(a) Possible mechanism of DODH reaction catalyzed by a V complex and (b) Proposed reaction mechanism of V-catalyzed DODH according to DFT. Adapted with permission from ref. [110].
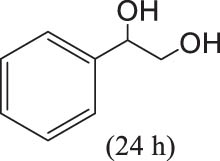
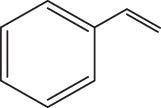
In 2016, Gopaladasu and Nicholas explored the catalytic activity of four Z+[LVO2]-type oxovanadium complexes (see Figure 15a) for the DODH of vicinal diol to olefin in the presence of different reductants. Hydroxyquinoline-V complex (complex 28), triazene-hydroxylamine derivative (complex 29), salicylaldehyde hydrazide complex (complex 30), and [n-Bu4N](dipic)VO2 (complex 27) have similar electronic properties and spatial structures, that is, they all have ONO-type or ONNO-type chelating ligands, five- or six-coordinated geometries, and negatively charged complex ions. The DODH experiments of styrene glycol as a model substrate were investigated for these four catalysts in combination with different reductants (Na2SO3, PPh3, H2, CO). Compared with other catalyst/reductant systems, the catalytic systems of [Bu4N](Salhyd)VO2/H2 (20 atm) and [Bu4N](Salhyd)VO2/CO (20 atm) showed superior catalytic activity with styrene yields of 33% and 48%, respectively. The combination of complex 28 with all reductants showed very low catalytic performance. This may be due to the fact that complex 28 and styrene glycol tend to generate unreactive resting-state intermediate 31 in the reaction (see Figure 15b). Subsequently, the less reactive aliphatic vicinal diols (1,2-hexanediol and (+)-diethyl tartrate) were tested using complex 30 and different reductants at 180℃. As shown in Table 6, the olefin yield and selectivity of 1,2-hexanediol and (+)-diethyl tartrate are similar or even better compared with styrene glycol, probably due to the low tendency of these less reactive aliphatic diols and olefin products to undergo side reactions. Among examined reductants, CO always gave the highest conversions and yields. The catalytic performance of the combined system of complex 30 with CO is comparable or even higher performance than that of most LReOx/reductant catalytic systems (Table 6) [61].
![Figure 15
(a) The molecular structure of oxovanadium complexes 27–30 and (b) complex 28-catalyzed DODH of styrene glycol at room temperature. Adapted with permission from ref. [61].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_015.jpg)
(a) The molecular structure of oxovanadium complexes 27–30 and (b) complex 28-catalyzed DODH of styrene glycol at room temperature. Adapted with permission from ref. [61].
The DODH reaction for different substrates catalyzed by complex 30.a Reprinted with permission from ref. [61]
| Run | Substrate | Reductant | Solvent | Product | Conv. (%) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |
|
Na2SO3 | PhH |
|
100 | 27 |
| 2 | Benzyl alcohol | 100 | 15 | |||
| 3 | H2 | 100 | 33 | |||
| 4 | CO | 100 | 48 | |||
| 5 |

|
Na2SO3 | PhH |

|
45 | 31 |
| 6 | Benzyl alcohol | 25 | 20 | |||
| 7 | H2 | 50 | 38 | |||
| 8 | CO | 80 | 67 | |||
| 9 |

|
Na2SO3 | PhCl |
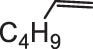
|
100 | 75 |
| 10 | Benzyl alcohol | 66 | 48 | |||
| 11 | H2 | 54 | 33 | |||
| 12 | CO | 100 | 97 |
aReaction conditions: 1.0 mmol glycol, 1.5 mmol reductant or (20 atm H2 or CO), and 10 mol% catalyst in 5 mL of solvent heated at 160–180°C (oil bath) in a sealed thick-walled glass tube or stainless-steel reactor.
In 2017, Kwok et al. reported silica-supported vanadia as heterogeneous V-based DODH catalysts [111]. As shown by NH3-TPD (see Figure 16a), V/SiO2 shows high intensity peaks at the lower temperature range (from 120 to 150℃) compared to other acid catalysts, indicating that V/SiO2 is featured by weak acidic sites. With the increase of V loading from 1 to 5 wt%, the acid site density increases from 1.64 to 7.12 sites·nm−2. According to XRD, UV–visible, and Raman spectra, 1V/SiO2 and 5V/SiO2 mainly contain monomeric VO4 species, tetrahedrally coordinated polymeric VOx species, and crystalline V2O5 with smaller grain sizes. By contrast, 10V/SiO2 mainly contains a bulk-like octahedrally coordinated V2O5 phase. The DODH of 2,3-butanediol to butene was used to evaluate the catalytic performance of these V/SiO2 catalysts in a plug flow reactor. Among these catalysts, 5V/SiO2 showed the best catalytic performance with a butene yield of 45.2% at complete conversion of 2,3-butanediol at 500℃. The excellent catalytic activity of 5V/SiO2 could be attributed to the presence of weak acid sites and surface polymeric VOx species. Under the bifunctional catalysis of weak acid sites and polymeric VOx species, 2,3-butanediol could be converted to butene mainly through dehydration and transfer hydrogenation steps, as shown in Figure 16b. Noteworthy, this dehydration-hydrogenation-dehydration pathway is significantly different from above-mentioned V-catalyzed DODH following sequential reduction-condensation-extrusion or condensation-reduction-extrusion steps.
![Figure 16
(a) NH3-TPD profile of various V-based catalysts and (b) proposed reaction mechanism for the conversion of 2,3-butanediol into butene catalyzed by 5V/SiO2. Reprinted with permission from ref. [111].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_016.jpg)
(a) NH3-TPD profile of various V-based catalysts and (b) proposed reaction mechanism for the conversion of 2,3-butanediol into butene catalyzed by 5V/SiO2. Reprinted with permission from ref. [111].
Petersen et al. reported the DODH reaction of glycerol to allyl alcohol catalyzed by a series of high-valence V catalysts containing terminal oxo ligands in a reactive distillation setup [112]. Ammonium metavanadate (NH4VO3) was discovered as the most effective catalyst, affording a 22% yield of allyl alcohol and also a 3.7% yield of acrolein at 275℃. The catalytic activity of V2O5, V(acac)3, and VO(acac)2 (acac = acetylacetonate) was comparable to that of NH4VO3. Lower yields of allyl alcohol were obtained over NaVO3 (14%) and V2O4 (6%) probably due to the poor solubility of NaVO3 and V2O4 in glycerol. As revealed by the control experiment using deuterium-labelled glycerol as substrate, acrolein was believed to arise from two consecutive dehydration steps of glycerol (see Scheme 12a), while the formation of allyl alcohol likely follows the V-catalyzed DODH pathway via the extrusion of olefin from a metal-diolate intermediate (see Scheme 12b). Glycerol serves as a reductant for V-catalyzed DODH of glycerol to allyl alcohol.
![Scheme 12
Two possible pathways proposed for the formation of allyl alcohol from glycerol including (a) acid-catalyzed dehydration with hydrogenation and (b) V-catalyzed DODH. Reprinted with permission from ref. [112].](/document/doi/10.1515/gps-2023-0129/asset/graphic/j_gps-2023-0129_fig_028.jpg)
Two possible pathways proposed for the formation of allyl alcohol from glycerol including (a) acid-catalyzed dehydration with hydrogenation and (b) V-catalyzed DODH. Reprinted with permission from ref. [112].
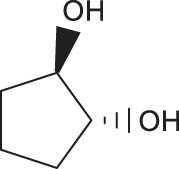
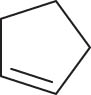
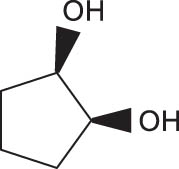
In general, transition metal-catalyzed DODH reaction is applicable to linear vicinal diols and cyclic cis-diols but not applicable to cyclic trans-diols [113]. Aksanoglu et al. studied a V-catalyzed DODH reaction for the transformation of cyclic trans-diol substrates [62]. Experiments were carried out in normal light conditions or dark conditions using [n-Bu4N](dipic)VO2 as the catalyst and PPh3 as the reductant (see Table 7). The experimental results show that trans-1,2-cyclopentanediol and trans-1,2-cyclohexanediol could be successfully converted to corresponding olefins in yields of around 20% via V-catalyzed DODH reaction. Compared with the normal experiment under light conditions, the production of olefin was significantly inhibited (below 10%) under dark conditions. The strong dependence of olefin yields on light indicates that light provides additional energy to promote the spin-crossover of V during DODH, and the triplet state of V is probably involved in the DODH. According to computational results of the reaction mechanism, the DODH of five-membered ring trans-diol substrates was proposed to proceed through a two-step extrusion of olefin on the triplet state of V, while the involvement of both triplet and singlet states of V during the stepwise cleavage of C–O bonds was predicted to be possible for the DODH of six-membered ring trans-diol.
The DODH of substrate vicinal diols catalyzed by oxovanadium catalyst in normal light and dark conditions.a Reprinted with permission from ref. [62]

|
||||
|---|---|---|---|---|
| Substrate | Product | Yield (%) lightb | Darkc | Fold Δ d |
|
|
20 | 6 | 3.3 |
|
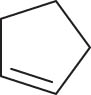
|
18 | 4 | 4.5 |
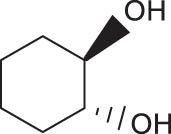
|

|
14 | 6 | 2.3 |
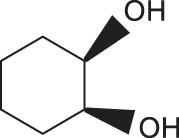
|

|
16 | 3 | 5.3 |
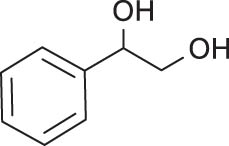 e
e
|

|
36 | 14 | 2.6 |
aReaction conditions: 0.1 mmol substrate, 0.15 mmol PPh3, and 10 mol% of the catalyst. Reaction time is 72 h unless otherwise stated. Yields are determined by 1H NMR spectroscopy using mesitylene as the internal standard and based on an average of three repeats. bOlefin yield when the reaction was carried out in normal light. cOlefin yield in the dark. dFold difference (fold Δ) is calculated as (yield in light)/(yield in dark). eReaction time is 24 h.
3 Summary and outlook
The unique capability of DODH to remove two adjacent OH groups from diols in one step to generate olefins renders it a very useful deoxygenation approach in the field of biomass valorization. DODH has found wide applications in the transformation of various biomass-derived polyols into valuable chemicals containing double bonds. Numerous oxorhenium compounds have been extensively explored as homogeneous and heterogonous catalysts for DODH showing excellent catalytic performances. The rarity and high cost of rhenium have stimulated great interest in searching for alternative DODH catalysts based on cheap non-noble metals. So far, a range of non-noble metals primarily including Mo, W, and V have been explored as catalytically active centers to replace Re for DODH. The application of W-containing compounds as DODH catalysts seems very challenging with only one successful demonstration of K2WCl6. It should be mentioned that K2WCl6 actually serves as a reagent rather than a catalyst, since it cannot recover to initial state and participate in the next catalytic cycle. Mo- and V-containing compounds present very promising DODH activities among these non-noble metals due to their variable oxidation states and oxyphilic properties similar to Re. A variety of Mo- and V-based complexes with different ligands bearing N, S, and O binding sites have been reported as efficient DODH catalysts. In contrast, only a few attempts have explored the utilization of Mo and V oxides immobilized on solid supports as heterogeneous DODH catalysts. Compared to Re catalysts, Mo and V catalysts usually necessitate higher reaction temperatures (160–220℃), and their catalytic activities still cannot rival that of Re catalysts. The substrate scope of Mo and V catalysts is predominantly limited to model diol compounds (e.g., styrene glycol, alkanediol) and simple diols like glycerol and 1,4-anhydroerythritol. Computational studies have shown that the catalytic mechanism of DODH generally consists of three stages: condensation of diol with the catalyst to form metal-glycolate, reduction of the oxo-metal, and olefin extrusion from the reduced metal-glycolate. The rate-limiting step is attributed to either the olefin extrusion or the reduction of oxo-metal depending on the choice of catalysts and reductants. The reduction of oxo-metal catalyst is often proposed as the rate-limiting step when alcohols are used as reductants.
Despite great advancements made in developing non-noble metal DODH catalysts, there exists a lot of room for further improvements in the aspects of catalyst activity, recyclability, and reductant. Secondary alcohols or diols themselves were frequently used as reductants for Mo- and V-catalyzed DODH. These alcohol reductants could lead to side reactions such as acetalization, esterification, and etherification, thus reducing the formation of olefin products. H2 is an ideal option for reductants considering that H2 only produces water as by product. Further studies should focus on the utilization of H2 to reduce Mo and V catalysts. This in turn requires the rational design of Mo and V catalysts to enable the reduction of high-valence Mo and V species by H2 but not lead to over-reduction to form inactive Mo and V species. The yields of olefin products obtained from Mo- and V-catalyzed DODH remain at low levels especially when using complex diol substrates. The stability of supported Mo and V oxides remains problematic due to the readily leaching of metal species. One possible reason could be the applied high reaction temperatures for Mo and V catalysts which facilitate metal leaching. Further studies are required to understand the structure–activity relationships of Mo- and V-catalyzed DODH including the electronic and steric effects of organic ligands and solid supports, as well as the deactivation/leaching mechanism. These fundamental understandings could guide the design of Mo and V catalysts with enhanced activity and stability. In addition to Mo and V, exploring other oxyphilic non-noble metals such as Mn and Nb by developing suitable organic ligands or solid supports to tune the interaction of metal centers with OH groups represents an important direction deserving further research efforts.
-
Funding information: This research was funded by the National Natural Science Foundation of China (No. 22208142) and the Natural Science Foundation of Jiangsu Province (No. BK20220347).
-
Author contributions: Xiyan Geng: Writing – original draft, Writing – review & editing; Mengyu Xu, Jiaxiang Zhang, Cui Yang, and Zheng Fang: Writing – review & editing; Ruiyan Sun and Kai Guo: Conceptualization, Funding acquisition, Writing – review & editing.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
[1] Olabi AG, Abdelkareem MA. Renewable energy and climate change. Renew Sustain Energy Rev. 2022;158:112111. 10.1016/j.rser.2022.112111.Search in Google Scholar
[2] Rahman A, Farrok O, Haque MM. Environmental impact of renewable energy source based electrical power plants: Solar, wind, hydroelectric, biomass, geothermal, tidal, ocean, and osmotic. Renew Sustain Energy Rev. 2022;161:112279. 10.1016/j.rser.2022.112279.Search in Google Scholar
[3] Anderson A, Rezaie B. Geothermal technology: Trends and potential role in a sustainable future. Appl Energy. 2019;248:18–34. 10.1016/j.apenergy.2019.04.102.Search in Google Scholar
[4] Zwarteveen JW, Figueira C, Zawwar I, Angus A. Barriers and drivers of the global imbalance of wind energy diffusion: A meta-analysis from a wind power Original Equipment Manufacturer perspective. J Clean Prod. 2021;290:125636. 10.1016/j.jclepro.2020.125636.Search in Google Scholar
[5] Holechek JL, Geli HME, Sawalhah MN, Valdez R. A global assessment: Can renewable energy replace fossil fuels by 2050. Sustainability. 2022;14(8):4792. 10.3390/su14084792.Search in Google Scholar
[6] He MY, Sun YH, Han BX. Green carbon science: efficient carbon resource processing, utilization, and recycling towards carbon neutrality. Angew Chem Int Ed. 2022;61(15):e202112835. 10.1002/anie.202112835.Search in Google Scholar PubMed
[7] Velvizhi G, Balakumar K, Shetti NP, Ahmad E, Pant KK, Aminabhavi TM. Integrated biorefinery processes for conversion of lignocellulosic biomass to value added materials: Paving a path towards circular economy. Bioresour Technol. 2022;343:126151. 10.1016/j.biortech.2021.126151.Search in Google Scholar PubMed
[8] He MY, Zhang K, Guan YJ, Sun YH, Han BX. Green carbon science: fundamental aspects. Natl Sci Rev. 2023;10(9):nwad046. 10.1093/nsr/nwad046.Search in Google Scholar PubMed PubMed Central
[9] Xu YX, Wang L, Shi ZL, Su N, Li C, Huang YP, et al. Peroxide-mediated selective conversion of biomass polysaccharides over high entropy sulfides via solar energy catalysis. Energy Environ Sci. 2023;16(4):1531–9. 10.1039/d2ee03357g.Search in Google Scholar
[10] Li N, Zong MH. (Chemo)biocatalytic upgrading of biobased furanic platforms to chemicals, fuels, and materials: A comprehensive review. ACS Catal. 2022;12(16):10080–114. 10.1021/acscatal.2c02912.Search in Google Scholar
[11] Deng WP, Feng YC, Fu J, Guo HW, Guo Y, Han BX, et al. Catalytic conversion of lignocellulosic biomass into chemicals and fuels. Green Energy Environ. 2023;8(1):10–114. 10.1016/j.gee.2022.07.003.Search in Google Scholar
[12] Ashokkumar V, Venkatkarthick R, Jayashree S, Chuetor S, Dharmaraj S, Kumar G, et al. Recent advances in lignocellulosic biomass for biofuels and value-added bioproducts-A critical review. Bioresour Technol. 2022;344:126195. 10.1016/j.biortech.2021.126195.Search in Google Scholar PubMed
[13] Pileidis FD, Titirici MM. Levulinic Acid biorefineries: new challenges for efficient utilization of biomass. ChemSusChem. 2016;9(6):562–82. 10.1002/cssc.201501405.Search in Google Scholar PubMed
[14] Zeng J, Zeng H, Wang Z. Review on technology of making biofuel from food waste. Int J Energy Res. 2022;46(8):10301–19. 10.1002/er.7868.Search in Google Scholar
[15] Hoang AT, Ong HC, Fattah IMR, Chong CT, Cheng CK, Sakthivel R, et al. Progress on the lignocellulosic biomass pyrolysis for biofuel production toward environmental sustainability. Fuel Process Technol. 2021;223:106997. 10.1016/j.fuproc.2021.106997.Search in Google Scholar
[16] Kaloudas D, Pavlova N, Penchovsky R. Lignocellulose, algal biomass, biofuels and biohydrogen: a review. Environ Chem Lett. 2021;19(4):2809–24. 10.1007/s10311-021-01213-y.Search in Google Scholar
[17] Berglund J, Mikkelsen D, Flanagan BM, Dhital S, Gaunitz S, Henriksson G, et al. Wood hemicelluloses exert distinct biomechanical contributions to cellulose fibrillar networks. Nat Commun. 2020;11(1):4692. 10.1038/s41467-020-18390-z.Search in Google Scholar PubMed PubMed Central
[18] Xu ZM, Luo JY, Huang YB. Recent advances in the chemical valorization of cellulose and its derivatives into ester compounds. Green Chem. 2022;24(10):3895–921. 10.1039/d2gc00377e.Search in Google Scholar
[19] Luo Y, Li Z, Li X, Liu X, Fan J, Clark JH, et al. The production of furfural directly from hemicellulose in lignocellulosic biomass: A review. Catal Today. 2019;319:14–24. 10.1016/j.cattod.2018.06.042.Search in Google Scholar
[20] Lu YC, He Q, Fan GZ, Cheng QP, Song GS. Extraction and modification of hemicellulose from lignocellulosic biomass: A review. Green Process Synth. 2021;10(1):779–804. 10.1515/gps-2021-0065.Search in Google Scholar
[21] Yoo CG, Meng X, Pu Y, Ragauskas AJ. The critical role of lignin in lignocellulosic biomass conversion and recent pretreatment strategies: A comprehensive review. Bioresour Technol. 2020;301:122784. 10.1016/j.biortech.2020.122784.Search in Google Scholar PubMed
[22] Sawangkeaw R, Ngamprasertsith S. A review of lipid-based biomasses as feedstocks for biofuels production. Renew Sustain Energy Rev. 2013;25:97–108. 10.1016/j.rser.2013.04.007.Search in Google Scholar
[23] Singh NB, Kumar A, Rai S. Potential production of bioenergy from biomass in an Indian perspective. Renew Sustain Energy Rev. 2014;39:65–78. 10.1016/j.rser.2014.07.110.Search in Google Scholar
[24] Shamsul NS, Kamarudin SK, Rahman NA, Kofli NT. An overview on the production of bio-methanol as potential renewable energy. Renew Sustain Energy Rev. 2014;33:578–88. 10.1016/j.rser.2014.02.024.Search in Google Scholar
[25] Yang Z, Wu Y, Zhang Z, Li H, Li X, Egorov RI, et al. Recent advances in co-thermochemical conversions of biomass with fossil fuels focusing on the synergistic effects. Renew Sustain Energy Rev. 2019;103:384–98. 10.1016/j.rser.2018.12.047.Search in Google Scholar
[26] Alper K, Tekin K, Karagoz S, Ragauskas AJ. Sustainable energy and fuels from biomass: a review focusing on hydrothermal biomass processing. Sustain Energy Fuels. 2020;4(9):4390–414. 10.1039/d0se00784f.Search in Google Scholar
[27] Luo WH, Cao WX, Bruijnincx PCA, Lin L, Wang AQ, Zhang T. Zeolite-supported metal catalysts for selective hydrodeoxygenation of biomass-derived platform molecules. Green Chem. 2019;21(14):3744–68. 10.1039/c9gc01216h.Search in Google Scholar
[28] Shi H. Valorization of biomass-derived small oxygenates: kinetics, mechanisms and site requirements of H2-involved hydrogenation and deoxygenation pathways over heterogeneous catalysts. ChemCatChem. 2019;11(7):1824–77. 10.1002/cctc.201801828.Search in Google Scholar
[29] Dapsens PY, Mondelli C, Pérez-Ramírez J. Biobased chemicals from Conception toward industrial reality: Lessons learned and to be learned. ACS Catal. 2012;2(7):1487–99. 10.1021/cs300124m.Search in Google Scholar
[30] Gallezot P. Conversion of biomass to selected chemical products. Chem Soc Rev. 2012;41(4):1538–58. 10.1039/C1CS15147A.Search in Google Scholar PubMed
[31] Xi Y, Yang W, Ammal SC, Lauterbach J, Pagan-Torres Y, Heyden A. Mechanistic study of the ceria supported, re-catalyzed deoxydehydration of vicinal OH groups. Catal Sci Technol. 2018;8(22):5750–62. 10.1039/C8CY01782D.Search in Google Scholar
[32] Sousa SCA, Fernandes AC. Efficient deoxygenation methodologies catalyzed by oxo-molybdenum and oxo-rhenium complexes. Coord Chem Rev. 2015:284:67–92. 10.1016/j.ccr.2014.09.008.Search in Google Scholar
[33] da Costa AAF, Pires LHD, Padron DR, Balu AM, da Rocha GN, Luque R, et al. Recent advances on catalytic deoxygenation of residues for bio-oil production: An overview. Mol Catal. 2022;518:112052. 10.1016/j.mcat.2021.112052.Search in Google Scholar
[34] Lima S, Antunes MM, Pillinger M, Valente AA. Ionic liquids as tools for the acid-catalyzed hydrolysis/dehydration of saccharides to furanic aldehydes. ChemCatChem. 2011;3(11):1686–706. 10.1002/cctc.201100105.Search in Google Scholar
[35] Korstanje TJ, de Waard EF, Jastrzebski JTBH, Klein Gebbink RJM. Rhenium-catalyzed dehydration of nonbenzylic and terpene alcohols to olefins. ACS Catal. 2012;2(10):2173–81. 10.1021/cs300455w.Search in Google Scholar
[36] Wang H, Kong Q, Wang Y, Deng T, Chen C, Hou X, et al. Graphene oxide catalyzed dehydration of fructose into 5-Hydroxymethylfurfural with isopropanol as cosolvent. ChemCatChem. 2014;6(3):728–32. 10.1002/cctc.201301067.Search in Google Scholar
[37] Larsen DB, Petersen AR, Dethlefsen JR, Teshome A, Fristrup P. Mechanistic Investigation of Molybdate-Catalysed Transfer Hydrodeoxygenation. Chem Eur J. 2016;22(46):16621–31. 10.1002/chem.201603028.Search in Google Scholar PubMed
[38] Wei HY, Wang ZY, Li H. Sustainable biomass hydrodeoxygenation in biphasic systems. Green Chem. 2022;24(5):1930–50. 10.1039/d1gc03836b.Search in Google Scholar
[39] Mamun O, Walker E, Faheem M, Bond JQ, Heyden A. Theoretical investigation of the hydrodeoxygenation of levulinic acid to γ-Valerolactone over Ru(0001). ACS Catal. 2017;7(1):215–28. 10.1021/acscatal.6b02548.Search in Google Scholar
[40] Coskun T, Conifer CM, Stevenson LC, Britovsek GJP. Carbodeoxygenation of biomass: The carbonylation of glycerol and higher polyols to monocarboxylic acids. Chem Eur J. 2013;19(21):6840–4. 10.1002/chem.201203069.Search in Google Scholar PubMed
[41] Raju S, Moret ME, Gebbink R. Rhenium-catalyzed dehydration and deoxydehydration of alcohols and polyols: opportunities for the formation of olefins from biomass. ACS Catal. 2015;5(1):281–300. 10.1021/cs501511x.Search in Google Scholar
[42] Tazawa S, Ota N, Tamura M, Nakagawa Y, Okumura K, Tomishige K. Deoxydehydration with molecular hydrogen over ceria-supported rhenium catalyst with gold promoter. ACS Catal. 2016;6(10):6393–7. 10.1021/acscatal.6b01864.Search in Google Scholar
[43] Jang JH, Hopper JT, Ro I, Christopher P, Abu-Omar MM. One-step production of renewable adipic acid esters from mucic acid over an Ir–ReOx/C catalyst with low Ir loading. Catal Sci Technol. 2023;13(3):714–25. 10.1039/D2CY01144A.Search in Google Scholar
[44] Li X, Zhang Y. Highly selective deoxydehydration of tartaric acid over supported and unsupported rhenium catalysts with modified acidities. ChemSusChem. 2016;9(19):2774–8. 10.1002/cssc.201600865.Search in Google Scholar PubMed
[45] Asada D, Ikeda T, Muraoka K, Nakagawa Y, Tomishige K, Nakayama A. Density functional theory study of deoxydehydration reaction by TiO2-supported monomeric and dimeric molybdenum oxide catalysts. J Phys Chem C. 2022;126(48):20375–87. 10.1021/acs.jpcc.2c06018.Search in Google Scholar
[46] Yamaguchi K, Nakagawa Y, Li C, Yabushita M, Tomishige K. Utilization of Ni as a non-noble-metal co-catalyst for ceria-supported rhenium oxide in combination of deoxydehydration and hydrogenation of vicinal diols. ACS Catal. 2022;12(20):12582–95. 10.1021/acscatal.2c03042.Search in Google Scholar
[47] Arceo E, Marsden P, Bergman RG, Ellman JA. An efficient didehydroxylation method for the biomass-derived polyols glycerol and erythritol. Mechanistic studies of a formic acid-mediated deoxygenation. Chem Commun. 2009;23:3357–9. 10.1039/b907746d.Search in Google Scholar PubMed
[48] Li XK, Zhang YG. Highly efficient process for the conversion of glycerol to acrylic acid via gas phase catalytic oxidation of an allyl alcohol intermediate. ACS Catal. 2016;6(1):143–50. 10.1021/acscatal.5b01843.Search in Google Scholar
[49] Sun RY, Zheng MY, Li XS, Pang JF, Wang AQ, Wang XD, et al. Production of renewable 1,3-pentadiene from xylitol via formic acid-mediated deoxydehydration and palladium-catalyzed deoxygenation reactions. Green Chem. 2017;19(3):638–42. 10.1039/c6gc02868c.Search in Google Scholar
[50] Tshibalonza NN, Monbaliu JCM. Revisiting the deoxydehydration of glycerol towards allyl alcohol under continuous-flow conditions. Green Chem. 2017;19(13):3006–13. 10.1039/c7gc00657h.Search in Google Scholar
[51] Tshibalonza NN, Gerardy R, Alsafra Z, Eppe G, Monbaliu JCM. A versatile biobased continuous flow strategy for the production of 3-butene-1,2-diol and vinyl ethylene carbonate from erythritol. Green Chem. 2018;20(22):5147–57. 10.1039/c8gc02468e.Search in Google Scholar
[52] Deng W, Yan L, Wang B, Zhang Q, Song H, Wang S, et al. Efficient catalysts for the green synthesis of adipic acid from biomass. Angew Chem Int Ed. 2021;60(9):4712–9. 10.1002/anie.202013843.Search in Google Scholar PubMed
[53] Petersen AR, Fristrup P. New motifs in deoxydehydration: beyond the realms of Rhenium. Chem Eur J. 2017;23(43):10235–43. 10.1002/chem.201701153.Search in Google Scholar PubMed
[54] Michael McClain J II, Nicholas KM. Elemental reductants for the deoxydehydration of glycols. ACS Catal. 2014;4(7):2109–12. 10.1021/cs500461v.Search in Google Scholar
[55] Cook GK, Andrews MA. Toward nonoxidative routes to oxygenated organics: stereospecific deoxydehydration of diols and polyols to alkenes and allylic alcohols catalyzed by the metal oxo complex (C5Me5)ReO3. J Am Chem Soc. 1996;118(39):9448–9. 10.1021/ja9620604.Search in Google Scholar
[56] Sandbrink L, Klindtworth E, Islam HU, Beale AM, Palkovits R. ReOx/TiO2: A recyclable solid catalyst for deoxydehydration. ACS Catal. 2016;6(2):677–80. 10.1021/acscatal.5b01936.Search in Google Scholar
[57] Ota N, Tamura M, Nakagawa Y, Okumura K, Tomishige K. Performance, structure, and mechanism of ReOx–Pd/CeO2 catalyst for simultaneous removal of vicinal OH groups with H2. ACS Catal. 2016;6(5):3213–26. 10.1021/acscatal.6b00491.Search in Google Scholar
[58] Nakagawa Y, Tazawa S, Wang TM, Tamura M, Hiyoshi N, Okumura K, et al. Mechanistic study of hydrogen-driven deoxydehydration over ceria-supported rhenium catalyst promoted by Au nanoparticles. ACS Catal. 2018;8(1):584–95. 10.1021/acscatal.7b02879.Search in Google Scholar
[59] Sandbrink L, Beckerle K, Meiners I, Liffmann R, Rahimi K, Okuda J, et al. Supported molybdenum catalysts for the deoxydehydration of 1,4-anhydroerythritol into 2,5-dihydrofuran. ChemSusChem. 2017;10(7):1375–9. 10.1002/cssc.201700010.Search in Google Scholar PubMed
[60] Tshibalonza NN, Monbaliu JCM. The deoxydehydration (DODH) reaction: a versatile technology for accessing olefins from bio-based polyols. Green Chem. 2020;22(15):4801–48. 10.1039/d0gc00689k.Search in Google Scholar
[61] Gopaladasu TV, Nicholas KM. Carbon monoxide (CO)- and hydrogen-driven, Vanadium-catalyzed deoxydehydration of glycols. ACS Catal. 2016;6(3):1901–4. 10.1021/acscatal.5b02667.Search in Google Scholar
[62] Aksanoglu E, Lim YH, Bryce RA. Direct deoxydehydration of cyclic trans-diol substrates: an experimental and computational study of the reaction mechanism of Vanadium(V)-based catalysis. ChemSusChem. 2021;14(6):1545–53. 10.1002/cssc.202002594.Search in Google Scholar PubMed PubMed Central
[63] Sharkey BE, Jentoft FC. Fundamental Insights into deactivation by leaching during rhenium-catalyzed deoxydehydration. ACS Catal. 2019;9(12):11317–28. 10.1021/acscatal.9b02806.Search in Google Scholar
[64] Vkuturi S, Chapman G, Ahmad I, Nicholas KM. Rhenium-catalyzed deoxydehydration of glycols by sulfite. Inorg Chem. 2010;49(11):4744–6. 10.1021/ic100467p.Search in Google Scholar PubMed
[65] Larson RT, Samant A, Chen J, Lee W, Bohn MA, Ohlmann DM, et al. Hydrogen gas-mediated deoxydehydration/hydrogenation of sugar acids: Catalytic conversion of glucarates to adipates. J Am Chem Soc. 2017;139(40):14001–4. 10.1021/jacs.7b07801.Search in Google Scholar PubMed
[66] Boucher-Jacobs C, Nicholas KM. Catalytic deoxydehydration of glycols with alcohol reductants. ChemSusChem. 2013;6(4):597–9. 10.1002/cssc.201200781.Search in Google Scholar PubMed
[67] Dethlefsen JR, Fristrup P. Rhenium-catalyzed deoxydehydration of diols and polyols. ChemSusChem. 2015;8(5):767–75. 10.1002/cssc.201402987.Search in Google Scholar PubMed
[68] Donnelly LJ, Thomas SP, Love JB. Recent advances in the deoxydehydration of Vicinal Diols and Polyols. Chem Asian J. 2019;14(21):3782–90. 10.1002/asia.201901274.Search in Google Scholar PubMed
[69] Jentoft FC. Transition metal-catalyzed deoxydehydration: missing pieces of the puzzle. Catal Sci Technol. 2022;12(21):6308–58. 10.1039/d1cy02083h.Search in Google Scholar
[70] Tomishige K, Nakagawa Y, Tamura M. Taming heterogeneous rhenium catalysis for the production of biomass-derived chemicals. Chin Chem Lett. 2020;31(5):1071–7. 10.1016/j.cclet.2019.07.014.Search in Google Scholar
[71] Li J, Lutz M JM, Klein Gebbink R. N-donor ligand supported “ReO2 +”: A pre-catalyst for the deoxydehydration of diols and polyols. Catalysts. 2020;10(7):754. 10.3390/catal10070754.Search in Google Scholar
[72] Mascitti A, Scioli G, Tonucci L, Canale V, Germani R, Di Profio P, et al. First evidence of the double-bond formation by deoxydehydration of glycerol and 1,2-propanediol in ionic liquids. ACS Omega. 2022;7(32):27980–90. 10.1021/acsomega.2c01803.Search in Google Scholar PubMed PubMed Central
[73] Jang JH, Ro I, Christopher P, Abu-Omar MM. A Heterogeneous Pt-ReOx/C Catalyst for Making Renewable Adipates in One Step from Sugar Acids. ACS Catal. 2021;11(1):95–109. 10.1021/acscatal.0c04158.Search in Google Scholar
[74] Cao J, Tamura M, Hosaka R, Nakayama A, Hasegawa J-y, Nakagawa Y, et al. Mechanistic study on deoxydehydration and hydrogenation of methyl glycosides to dideoxy sugars over a ReOx–Pd/CeO2 catalyst. ACS Catal. 2020;10(20):12040–51. 10.1021/acscatal.0c02309.Search in Google Scholar
[75] Sebastian J, Mebrahtu C, Palkovits R. Influence and stability of the surface density of MoOx on TiO2 in deoxydehydration: structure–activity correlations. Catal Sci Technol. 2023;13(4):1087–97. 10.1039/D2CY01854C.Search in Google Scholar
[76] Sharpless KB, Flood TC. Direct deoxygenation of vicinal diols with tungsten(IV). A new olefin synthesis. J Chem Soc, Chem Commun. 1972;7:370–1. 10.1039/C39720000370.Search in Google Scholar
[77] McMurry JE, Fleming MP. Improved procedures for the reductive coupling of carbonyls to olefins and for the reduction of diols to olefins. J Org Chem. 1976;41(5):896–7. 10.1021/jo00867a038.Search in Google Scholar
[78] Lan H, Yao Q, Zhou Y, Zhang B, Jiang Y. Direct conversion of gas-glycerol to Allyl alcohol over V, Ti or Nb modified MoFe/KIT-6 oxide catalysts. Mol Catal. 2020;498:111279. 10.1016/j.mcat.2020.111279.Search in Google Scholar
[79] Ahmad I, Chapman G, Nicholas KM. Sulfite-driven, oxorhenium-catalyzed deoxydehydration of glycols. Organometallics. 2011;30(10):2810–8. 10.1021/om2001662.Search in Google Scholar
[80] Li J, Lutz M, Otte M, Klein, Gebbink RJM. A Cptt-Based trioxo-rhenium catalyst for the deoxydehydration of diols and polyols. ChemCatChem. 2018;10(20):4755–60. 10.1002/cctc.201801151.Search in Google Scholar PubMed PubMed Central
[81] Shiramizu M, Toste FD. Deoxygenation of biomass-derived feedstocks: oxorhenium-catalyzed deoxydehydration of sugars and sugar alcohols. Angew Chem Int Ed. 2012;51(32):8082–6. 10.1002/anie.201203877.Search in Google Scholar PubMed
[82] Ziegler JE, Zdilla MJ, Evans AJ, Abu-Omar MM. H2-driven deoxygenation of epoxides and diols to alkenes catalyzed by methyltrioxorhenium. Inorg Chem. 2009;48(21):9998–10000. 10.1021/ic901792b.Search in Google Scholar PubMed
[83] Liu S, Senocak A, Smeltz JL, Yang LN, Wegenhart B, Yi J, et al. Mechanism of MTO-catalyzed deoxydehydration of diols to alkenes using sacrificial alcohols. Organometallics. 2013;32(11):3210–9. 10.1021/om400127z.Search in Google Scholar
[84] Qu S, Dang Y, Wen M, Wang Z-X. Mechanism of the methyltrioxorhenium-catalyzed deoxydehydration of polyols: A new pathway revealed. Chem Eur J. 2013;19(12):3827–32. 10.1002/chem.201204001.Search in Google Scholar PubMed
[85] Lupp D, Christensen NJ, Dethlefsen JR, Fristrup P. DFT Study of the molybdenum-catalyzed deoxydehydration of vicinal diols. Chem Eur J. 2015;21(8):3435–42. 10.1002/chem.201405473.Search in Google Scholar PubMed
[86] Hills L, Moyano R, Montilla F, Pastor A, Galindo A, Alvarez E, et al. Dioxomolybdenum(VI) complexes with acylpyrazolonate ligands: Synthesis, structures, and catalytic properties. Eur J Inorg Chem. 2013;19:3352–61. 10.1002/ejic.201300098.Search in Google Scholar
[87] Dethlefsen JR, Lupp D, Oh BC, Fristrup P. Molybdenum-catalyzed deoxydehydration of vicinal diols. ChemSusChem. 2014;7(2):425–8. 10.1002/cssc.201300945.Search in Google Scholar PubMed
[88] Dethlefsen JR, Lupp D, Teshome A, Nielsen LB, Fristrup P. Molybdenum-catalyzed conversion of diols and biomass-derived polyols to alkenes using isopropyl alcohol as reductant and solvent. ACS Catal. 2015;5(6):3638–47. 10.1021/acscatal.5b00427.Search in Google Scholar
[89] Navarro CA, John A. Deoxydehydration using a commercial catalyst and readily available reductant. Inorg Chem Commun. 2019;99:145–8. 10.1016/j.inoche.2018.11.015.Search in Google Scholar
[90] Verdicchio F, Galindo A. Theoretical studies on the mechanism of molybdenum-catalysed deoxydehydration of diols. Dalton Trans. 2023;52(18):5935–42. 10.1039/D3DT00340J.Search in Google Scholar PubMed
[91] Xi YJ, Lauterbach J, Pagan-Torres Y, Heyden A. Deoxydehydration of 1,4-anhydroerythritol over anatase TiO2(101)-supported ReOx and MoOx. Catal Sci Technol. 2020;10(11):3731–8. 10.1039/d0cy00434k.Search in Google Scholar
[92] Beckerle K, Sauer A, Spaniol TP, Okuda J. Bis(phenolato)molybdenum complexes as catalyst precursors for the deoxydehydration of biomass-derived polyols. Polyhedron. 2016;116:105–10. 10.1016/j.poly.2016.03.053.Search in Google Scholar
[93] Lam PM, John A. Molybdenum catalyzed deoxydehydration of aliphatic glycols under microwave irradiation. J Organomet Chem. 2023;992:122705. 10.1016/j.jorganchem.2023.122705.Search in Google Scholar
[94] Wagner N. A Preliminary Study of Molybdenum Salan Complexes as Catalysts for Deoxydehydration of Vicinal Diols. Pomona: California State Polytechnic University; 2019.Search in Google Scholar
[95] Stalpaert M, De Vos D. Stabilizing effect of bulky β-diketones on homogeneous Mo catalysts for deoxydehydration. ACS Sustain Chem Eng. 2018;6(9):12197–204. 10.1021/acssuschemeng.8b02532.Search in Google Scholar
[96] Jiang HF, Lu R, Si XQ, Luo XL, Xu J, Lu F. Single-Site Molybdenum Catalyst for the Synthesis of Fumarate. ChemCatChem. 2019;11(17):4291–6. 10.1002/cctc.201900332.Search in Google Scholar
[97] Tran R, Kilyanek SM. Deoxydehydration of polyols catalyzed by a molybdenum dioxo-complex supported by a dianionic ONO pincer ligand. Dalton Trans. 2019;48(43):16304–11. 10.1039/c9dt03759d.Search in Google Scholar PubMed
[98] Siu TC, Silva I, Lunn MJ, John A. Influence of the pendant arm in deoxydehydration catalyzed by dioxomolybdenum complexes supported by amine bisphenolate ligands. New J Chem. 2020;44(23):9933–41. 10.1039/d0nj02151b.Search in Google Scholar
[99] Li J, Lutz M, Gebbink R. A Cp-based molybdenum catalyst for the deoxydehydration of biomass-derived diols. ChemCatChem. 2020;12(24):6356–65. 10.1002/cctc.202001115.Search in Google Scholar
[100] Hacatrjan S, Liu LJ, Gan JX, Nakagawa Y, Cao J, Yabushita M, et al. Titania-supported molybdenum oxide combined with Au nanoparticles as a hydrogen-driven deoxydehydration catalyst of diol compounds. Catal Sci Technol. 2022;12(7):2146–61. 10.1039/d1cy02144c.Search in Google Scholar
[101] Xi YJ, Heyden A. Highly efficient deoxydehydration and hydrodeoxygenation on MoS2-supported transition-metal atoms through a C-H activation mechanism. ACS Catal. 2020;10(19):11346–55. 10.1021/acscatal.0c02669.Search in Google Scholar
[102] Liu GL, Robertson AW, Li MMJ, Kuo WCH, Darby MT, Muhieddine MH, et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat Chem. 2017;9(8):810–6. 10.1038/nchem.2740.Search in Google Scholar PubMed
[103] Amada Y, Ota N, Tamura M, Nakagawa Y, Tomishige K. Selective hydrodeoxygenation of cyclic vicinal diols to cyclic alcohols over tungsten oxide–palladium catalysts. ChemSusChem. 2014;7(8):2185–92. 10.1002/cssc.201402188.Search in Google Scholar PubMed
[104] Sharkey BE, Denning AL, Jentoft FC, Gangadhara R, Gopaladasu TV, Nicholas KM. New solid oxo-rhenium and oxo-molybdenum catalysts for the deoxydehydration of glycols to olefins. Catal Today. 2018;310:86–93. 10.1016/j.cattod.2017.05.090.Search in Google Scholar
[105] Chapman G, Nicholas KM. Vanadium-catalyzed deoxydehydration of glycols. Chem Commun. 2013;49(74):8199–201. 10.1039/c3cc44656e.Search in Google Scholar PubMed
[106] Bi SW, Wang JY, Liu LJ, Li P, Lin ZY. Mechanism of the MeReO3-catalyzed deoxygenation of epoxides. Organometallics. 2012;31(17):6139–47. 10.1021/om300485w.Search in Google Scholar
[107] Galindo A. DFT studies on the mechanism of the vanadium-catalyzed deoxydehydration of diols. Inorg Chem. 2016;55(5):2284–9. 10.1021/acs.inorgchem.5b02649.Search in Google Scholar PubMed
[108] Poutas LCD, Reis MC, Sanz R, Lopez CS, Faza ON. A radical mechanism for the vanadium-catalyzed deoxydehydration of glycols. Inorg Chem. 2016;55(21):11372–82. 10.1021/acs.inorgchem.6b01916.Search in Google Scholar PubMed
[109] Yi J, Liu S, Abu-Omar MM. Rhenium-catalyzed transfer hydrogenation and deoxygenation of biomass-derived polyols to small and useful organics. ChemSusChem. 2012;5(8):1401–4. 10.1002/cssc.201200138.Search in Google Scholar PubMed
[110] Jiang YY, Jiang JL, Fu Y. Mechanism of vanadium-catalyzed deoxydehydration of vicinal diols: Spin-crossover-involved processes. Organometallics. 2016;35(19):3388–96. 10.1021/acs.organomet.6b00602.Search in Google Scholar
[111] Kwok KM, Choong CKS, Ong DSW, Ng JCQ, Gwie CG, Chen L, et al. Hydrogen-free gas-phase deoxydehydration of 2,3-butanediol to butene on silica-supported vanadium catalysts. ChemCatChem. 2017;9(13):2443–7. 10.1002/cctc.201700301.Search in Google Scholar
[112] Petersen AR, Nielsen LB, Dethlefsen JR, Fristrup P. Vanadium-catalyzed deoxydehydration of glycerol without an external reductant. ChemCatChem. 2018;10(4):769–78. 10.1002/cctc.201701049.Search in Google Scholar
[113] Raju S, Jastrzebski J, Lutz M, Gebbink R. Catalytic deoxydehydration of diols to olefins by using a bulky cyclopentadiene-based trioxorhenium catalyst. ChemSusChem. 2013;6(9):1673–80. 10.1002/cssc.201300364.Search in Google Scholar PubMed
© 2023 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- Value-added utilization of coal fly ash and recycled polyvinyl chloride in door or window sub-frame composites
- High removal efficiency of volatile phenol from coking wastewater using coal gasification slag via optimized adsorption and multi-grade batch process
- Evolution of surface morphology and properties of diamond films by hydrogen plasma etching
- Removal efficiency of dibenzofuran using CuZn-zeolitic imidazole frameworks as a catalyst and adsorbent
- Rapid and efficient microwave-assisted extraction of Caesalpinia sappan Linn. heartwood and subsequent synthesis of gold nanoparticles
- The catalytic characteristics of 2-methylnaphthalene acylation with AlCl3 immobilized on Hβ as Lewis acid catalyst
- Biodegradation of synthetic PVP biofilms using natural materials and nanoparticles
- Rutin-loaded selenium nanoparticles modulated the redox status, inflammatory, and apoptotic pathways associated with pentylenetetrazole-induced epilepsy in mice
- Optimization of apigenin nanoparticles prepared by planetary ball milling: In vitro and in vivo studies
- Synthesis and characterization of silver nanoparticles using Origanum onites leaves: Cytotoxic, apoptotic, and necrotic effects on Capan-1, L929, and Caco-2 cell lines
- Exergy analysis of a conceptual CO2 capture process with an amine-based DES
- Construction of fluorescence system of felodipine–tetracyanovinyl–2,2′-bipyridine complex
- Excellent photocatalytic degradation of rhodamine B over Bi2O3 supported on Zn-MOF nanocomposites under visible light
- Optimization-based control strategy for a large-scale polyhydroxyalkanoates production in a fed-batch bioreactor using a coupled PDE–ODE system
- Effectiveness of pH and amount of Artemia urumiana extract on physical, chemical, and biological attributes of UV-fabricated biogold nanoparticles
- Geranium leaf-mediated synthesis of silver nanoparticles and their transcriptomic effects on Candida albicans
- Synthesis, characterization, anticancer, anti-inflammatory activities, and docking studies of 3,5-disubstituted thiadiazine-2-thiones
- Synthesis and stability of phospholipid-encapsulated nano-selenium
- Putative anti-proliferative effect of Indian mustard (Brassica juncea) seed and its nano-formulation
- Enrichment of low-grade phosphorites by the selective leaching method
- Electrochemical analysis of the dissolution of gold in a copper–ethylenediamine–thiosulfate system
- Characterisation of carbonate lake sediments as a potential filler for polymer composites
- Evaluation of nano-selenium biofortification characteristics of alfalfa (Medicago sativa L.)
- Quality of oil extracted by cold press from Nigella sativa seeds incorporated with rosemary extracts and pretreated by microwaves
- Heteropolyacid-loaded MOF-derived mesoporous zirconia catalyst for chemical degradation of rhodamine B
- Recovery of critical metals from carbonatite-type mineral wastes: Geochemical modeling investigation of (bio)hydrometallurgical leaching of REEs
- Photocatalytic properties of ZnFe-mixed oxides synthesized via a simple route for water remediation
- Attenuation of di(2-ethylhexyl)phthalate-induced hepatic and renal toxicity by naringin nanoparticles in a rat model
- Novel in situ synthesis of quaternary core–shell metallic sulfide nanocomposites for degradation of organic dyes and hydrogen production
- Microfluidic steam-based synthesis of luminescent carbon quantum dots as sensing probes for nitrite detection
- Transformation of eggshell waste to egg white protein solution, calcium chloride dihydrate, and eggshell membrane powder
- Preparation of Zr-MOFs for the adsorption of doxycycline hydrochloride from wastewater
- Green nanoarchitectonics of the silver nanocrystal potential for treating malaria and their cytotoxic effects on the kidney Vero cell line
- Carbon emissions analysis of producing modified asphalt with natural asphalt
- An efficient and green synthesis of 2-phenylquinazolin-4(3H)-ones via t-BuONa-mediated oxidative condensation of 2-aminobenzamides and benzyl alcohols under solvent- and transition metal-free conditions
- Chitosan nanoparticles loaded with mesosulfuron methyl and mesosulfuron methyl + florasulam + MCPA isooctyl to manage weeds of wheat (Triticum aestivum L.)
- Synergism between lignite and high-sulfur petroleum coke in CO2 gasification
- Facile aqueous synthesis of ZnCuInS/ZnS–ZnS QDs with enhanced photoluminescence lifetime for selective detection of Cu(ii) ions
- Rapid synthesis of copper nanoparticles using Nepeta cataria leaves: An eco-friendly management of disease-causing vectors and bacterial pathogens
- Study on the photoelectrocatalytic activity of reduced TiO2 nanotube films for removal of methyl orange
- Development of a fuzzy logic model for the prediction of spark-ignition engine performance and emission for gasoline–ethanol blends
- Micro-impact-induced mechano-chemical synthesis of organic precursors from FeC/FeN and carbonates/nitrates in water and its extension to nucleobases
- Green synthesis of strontium-doped tin dioxide (SrSnO2) nanoparticles using the Mahonia bealei leaf extract and evaluation of their anticancer and antimicrobial activities
- A study on the larvicidal and adulticidal potential of Cladostepus spongiosus macroalgae and green-fabricated silver nanoparticles against mosquito vectors
- Catalysts based on nickel salt heteropolytungstates for selective oxidation of diphenyl sulfide
- Powerful antibacterial nanocomposites from Corallina officinalis-mediated nanometals and chitosan nanoparticles against fish-borne pathogens
- Removal behavior of Zn and alkalis from blast furnace dust in pre-reduction sinter process
- Environmentally friendly synthesis and computational studies of novel class of acridinedione integrated spirothiopyrrolizidines/indolizidines
- The mechanisms of inhibition and lubrication of clean fracturing flowback fluids in water-based drilling fluids
- Adsorption/desorption performance of cellulose membrane for Pb(ii)
- A one-pot, multicomponent tandem synthesis of fused polycyclic pyrrolo[3,2-c]quinolinone/pyrrolizino[2,3-c]quinolinone hybrid heterocycles via environmentally benign solid state melt reaction
- Green synthesis of silver nanoparticles using durian rind extract and optical characteristics of surface plasmon resonance-based optical sensor for the detection of hydrogen peroxide
- Electrochemical analysis of copper-EDTA-ammonia-gold thiosulfate dissolution system
- Characterization of bio-oil production by microwave pyrolysis from cashew nut shells and Cassia fistula pods
- Green synthesis methods and characterization of bacterial cellulose/silver nanoparticle composites
- Photocatalytic research performance of zinc oxide/graphite phase carbon nitride catalyst and its application in environment
- Effect of phytogenic iron nanoparticles on the bio-fortification of wheat varieties
- In vitro anti-cancer and antimicrobial effects of manganese oxide nanoparticles synthesized using the Glycyrrhiza uralensis leaf extract on breast cancer cell lines
- Preparation of Pd/Ce(F)-MCM-48 catalysts and their catalytic performance of n-heptane isomerization
- Green “one-pot” fluorescent bis-indolizine synthesis with whole-cell plant biocatalysis
- Silica-titania mesoporous silicas of MCM-41 type as effective catalysts and photocatalysts for selective oxidation of diphenyl sulfide by H2O2
- Biosynthesis of zinc oxide nanoparticles from molted feathers of Pavo cristatus and their antibiofilm and anticancer activities
- Clean preparation of rutile from Ti-containing mixed molten slag by CO2 oxidation
- Synthesis and characterization of Pluronic F-127-coated titanium dioxide nanoparticles synthesized from extracts of Atractylodes macrocephala leaf for antioxidant, antimicrobial, and anticancer properties
- Effect of pretreatment with alkali on the anaerobic digestion characteristics of kitchen waste and analysis of microbial diversity
- Ameliorated antimicrobial, antioxidant, and anticancer properties by Plectranthus vettiveroides root extract-mediated green synthesis of chitosan nanoparticles
- Microwave-accelerated pretreatment technique in green extraction of oil and bioactive compounds from camelina seeds: Effectiveness and characterization
- Studies on the extraction performance of phorate by aptamer-functionalized magnetic nanoparticles in plasma samples
- Investigation of structural properties and antibacterial activity of AgO nanoparticle extract from Solanum nigrum/Mentha leaf extracts by green synthesis method
- Green fabrication of chitosan from marine crustaceans and mushroom waste: Toward sustainable resource utilization
- Synthesis, characterization, and evaluation of nanoparticles of clodinofop propargyl and fenoxaprop-P-ethyl on weed control, growth, and yield of wheat (Triticum aestivum L.)
- The enhanced adsorption properties of phosphorus from aqueous solutions using lanthanum modified synthetic zeolites
- Separation of graphene oxides of different sizes by multi-layer dialysis and anti-friction and lubrication performance
- Visible-light-assisted base-catalyzed, one-pot synthesis of highly functionalized cinnolines
- The experimental study on the air oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid with Co–Mn–Br system
- Highly efficient removal of tetracycline and methyl violet 2B from aqueous solution using the bimetallic FeZn-ZIFs catalyst
- A thermo-tolerant cellulase enzyme produced by Bacillus amyloliquefaciens M7, an insight into synthesis, optimization, characterization, and bio-polishing activity
- Exploration of ketone derivatives of succinimide for their antidiabetic potential: In vitro and in vivo approaches
- Ultrasound-assisted green synthesis and in silico study of 6-(4-(butylamino)-6-(diethylamino)-1,3,5-triazin-2-yl)oxypyridazine derivatives
- A study of the anticancer potential of Pluronic F-127 encapsulated Fe2O3 nanoparticles derived from Berberis vulgaris extract
- Biogenic synthesis of silver nanoparticles using Consolida orientalis flowers: Identification, catalytic degradation, and biological effect
- Initial assessment of the presence of plastic waste in some coastal mangrove forests in Vietnam
- Adsorption synergy electrocatalytic degradation of phenol by active oxygen-containing species generated in Co-coal based cathode and graphite anode
- Antibacterial, antifungal, antioxidant, and cytotoxicity activities of the aqueous extract of Syzygium aromaticum-mediated synthesized novel silver nanoparticles
- Synthesis of a silica matrix with ZnO nanoparticles for the fabrication of a recyclable photodegradation system to eliminate methylene blue dye
- Natural polymer fillers instead of dye and pigments: Pumice and scoria in PDMS fluid and elastomer composites
- Study on the preparation of glycerylphosphorylcholine by transesterification under supported sodium methoxide
- Wireless network handheld terminal-based green ecological sustainable design evaluation system: Improved data communication and reduced packet loss rate
- The optimization of hydrogel strength from cassava starch using oxidized sucrose as a crosslinking agent
- Green synthesis of silver nanoparticles using Saccharum officinarum leaf extract for antiviral paint
- Study on the reliability of nano-silver-coated tin solder joints for flip chips
- Environmentally sustainable analytical quality by design aided RP-HPLC method for the estimation of brilliant blue in commercial food samples employing a green-ultrasound-assisted extraction technique
- Anticancer and antimicrobial potential of zinc/sodium alginate/polyethylene glycol/d-pinitol nanocomposites against osteosarcoma MG-63 cells
- Nanoporous carbon@CoFe2O4 nanocomposite as a green absorbent for the adsorptive removal of Hg(ii) from aqueous solutions
- Characterization of silver sulfide nanoparticles from actinobacterial strain (M10A62) and its toxicity against lepidopteran and dipterans insect species
- Phyto-fabrication and characterization of silver nanoparticles using Withania somnifera: Investigating antioxidant potential
- Effect of e-waste nanofillers on the mechanical, thermal, and wear properties of epoxy-blend sisal woven fiber-reinforced composites
- Magnesium nanohydroxide (2D brucite) as a host matrix for thymol and carvacrol: Synthesis, characterization, and inhibition of foodborne pathogens
- Synergistic inhibitive effect of a hybrid zinc oxide-benzalkonium chloride composite on the corrosion of carbon steel in a sulfuric acidic solution
- Review Articles
- Role and the importance of green approach in biosynthesis of nanopropolis and effectiveness of propolis in the treatment of COVID-19 pandemic
- Gum tragacanth-mediated synthesis of metal nanoparticles, characterization, and their applications as a bactericide, catalyst, antioxidant, and peroxidase mimic
- Green-processed nano-biocomposite (ZnO–TiO2): Potential candidates for biomedical applications
- Reaction mechanisms in microwave-assisted lignin depolymerisation in hydrogen-donating solvents
- Recent progress on non-noble metal catalysts for the deoxydehydration of biomass-derived oxygenates
- Rapid Communication
- Phosphorus removal by iron–carbon microelectrolysis: A new way to achieve phosphorus recovery
- Special Issue: Biomolecules-derived synthesis of nanomaterials for environmental and biological applications (Guest Editors: Arpita Roy and Fernanda Maria Policarpo Tonelli)
- Biomolecules-derived synthesis of nanomaterials for environmental and biological applications
- Nano-encapsulated tanshinone IIA in PLGA-PEG-COOH inhibits apoptosis and inflammation in cerebral ischemia/reperfusion injury
- Green fabrication of silver nanoparticles using Melia azedarach ripened fruit extract, their characterization, and biological properties
- Green-synthesized nanoparticles and their therapeutic applications: A review
- Antioxidant, antibacterial, and cytotoxicity potential of synthesized silver nanoparticles from the Cassia alata leaf aqueous extract
- Green synthesis of silver nanoparticles using Callisia fragrans leaf extract and its anticancer activity against MCF-7, HepG2, KB, LU-1, and MKN-7 cell lines
- Algae-based green AgNPs, AuNPs, and FeNPs as potential nanoremediators
- Green synthesis of Kickxia elatine-induced silver nanoparticles and their role as anti-acetylcholinesterase in the treatment of Alzheimer’s disease
- Phytocrystallization of silver nanoparticles using Cassia alata flower extract for effective control of fungal skin pathogens
- Antibacterial wound dressing with hydrogel from chitosan and polyvinyl alcohol from the red cabbage extract loaded with silver nanoparticles
- Leveraging of mycogenic copper oxide nanostructures for disease management of Alternaria blight of Brassica juncea
- Nanoscale molecular reactions in microbiological medicines in modern medical applications
- Synthesis and characterization of ZnO/β-cyclodextrin/nicotinic acid nanocomposite and its biological and environmental application
- Green synthesis of silver nanoparticles via Taxus wallichiana Zucc. plant-derived Taxol: Novel utilization as anticancer, antioxidation, anti-inflammation, and antiurolithic potential
- Recyclability and catalytic characteristics of copper oxide nanoparticles derived from bougainvillea plant flower extract for biomedical application
- Phytofabrication, characterization, and evaluation of novel bioinspired selenium–iron (Se–Fe) nanocomposites using Allium sativum extract for bio-potential applications
- Erratum
- Erratum to “Synthesis, characterization, and evaluation of nanoparticles of clodinofop propargyl and fenoxaprop-P-ethyl on weed control, growth, and yield of wheat (Triticum aestivum L.)”
Articles in the same Issue
- Research Articles
- Value-added utilization of coal fly ash and recycled polyvinyl chloride in door or window sub-frame composites
- High removal efficiency of volatile phenol from coking wastewater using coal gasification slag via optimized adsorption and multi-grade batch process
- Evolution of surface morphology and properties of diamond films by hydrogen plasma etching
- Removal efficiency of dibenzofuran using CuZn-zeolitic imidazole frameworks as a catalyst and adsorbent
- Rapid and efficient microwave-assisted extraction of Caesalpinia sappan Linn. heartwood and subsequent synthesis of gold nanoparticles
- The catalytic characteristics of 2-methylnaphthalene acylation with AlCl3 immobilized on Hβ as Lewis acid catalyst
- Biodegradation of synthetic PVP biofilms using natural materials and nanoparticles
- Rutin-loaded selenium nanoparticles modulated the redox status, inflammatory, and apoptotic pathways associated with pentylenetetrazole-induced epilepsy in mice
- Optimization of apigenin nanoparticles prepared by planetary ball milling: In vitro and in vivo studies
- Synthesis and characterization of silver nanoparticles using Origanum onites leaves: Cytotoxic, apoptotic, and necrotic effects on Capan-1, L929, and Caco-2 cell lines
- Exergy analysis of a conceptual CO2 capture process with an amine-based DES
- Construction of fluorescence system of felodipine–tetracyanovinyl–2,2′-bipyridine complex
- Excellent photocatalytic degradation of rhodamine B over Bi2O3 supported on Zn-MOF nanocomposites under visible light
- Optimization-based control strategy for a large-scale polyhydroxyalkanoates production in a fed-batch bioreactor using a coupled PDE–ODE system
- Effectiveness of pH and amount of Artemia urumiana extract on physical, chemical, and biological attributes of UV-fabricated biogold nanoparticles
- Geranium leaf-mediated synthesis of silver nanoparticles and their transcriptomic effects on Candida albicans
- Synthesis, characterization, anticancer, anti-inflammatory activities, and docking studies of 3,5-disubstituted thiadiazine-2-thiones
- Synthesis and stability of phospholipid-encapsulated nano-selenium
- Putative anti-proliferative effect of Indian mustard (Brassica juncea) seed and its nano-formulation
- Enrichment of low-grade phosphorites by the selective leaching method
- Electrochemical analysis of the dissolution of gold in a copper–ethylenediamine–thiosulfate system
- Characterisation of carbonate lake sediments as a potential filler for polymer composites
- Evaluation of nano-selenium biofortification characteristics of alfalfa (Medicago sativa L.)
- Quality of oil extracted by cold press from Nigella sativa seeds incorporated with rosemary extracts and pretreated by microwaves
- Heteropolyacid-loaded MOF-derived mesoporous zirconia catalyst for chemical degradation of rhodamine B
- Recovery of critical metals from carbonatite-type mineral wastes: Geochemical modeling investigation of (bio)hydrometallurgical leaching of REEs
- Photocatalytic properties of ZnFe-mixed oxides synthesized via a simple route for water remediation
- Attenuation of di(2-ethylhexyl)phthalate-induced hepatic and renal toxicity by naringin nanoparticles in a rat model
- Novel in situ synthesis of quaternary core–shell metallic sulfide nanocomposites for degradation of organic dyes and hydrogen production
- Microfluidic steam-based synthesis of luminescent carbon quantum dots as sensing probes for nitrite detection
- Transformation of eggshell waste to egg white protein solution, calcium chloride dihydrate, and eggshell membrane powder
- Preparation of Zr-MOFs for the adsorption of doxycycline hydrochloride from wastewater
- Green nanoarchitectonics of the silver nanocrystal potential for treating malaria and their cytotoxic effects on the kidney Vero cell line
- Carbon emissions analysis of producing modified asphalt with natural asphalt
- An efficient and green synthesis of 2-phenylquinazolin-4(3H)-ones via t-BuONa-mediated oxidative condensation of 2-aminobenzamides and benzyl alcohols under solvent- and transition metal-free conditions
- Chitosan nanoparticles loaded with mesosulfuron methyl and mesosulfuron methyl + florasulam + MCPA isooctyl to manage weeds of wheat (Triticum aestivum L.)
- Synergism between lignite and high-sulfur petroleum coke in CO2 gasification
- Facile aqueous synthesis of ZnCuInS/ZnS–ZnS QDs with enhanced photoluminescence lifetime for selective detection of Cu(ii) ions
- Rapid synthesis of copper nanoparticles using Nepeta cataria leaves: An eco-friendly management of disease-causing vectors and bacterial pathogens
- Study on the photoelectrocatalytic activity of reduced TiO2 nanotube films for removal of methyl orange
- Development of a fuzzy logic model for the prediction of spark-ignition engine performance and emission for gasoline–ethanol blends
- Micro-impact-induced mechano-chemical synthesis of organic precursors from FeC/FeN and carbonates/nitrates in water and its extension to nucleobases
- Green synthesis of strontium-doped tin dioxide (SrSnO2) nanoparticles using the Mahonia bealei leaf extract and evaluation of their anticancer and antimicrobial activities
- A study on the larvicidal and adulticidal potential of Cladostepus spongiosus macroalgae and green-fabricated silver nanoparticles against mosquito vectors
- Catalysts based on nickel salt heteropolytungstates for selective oxidation of diphenyl sulfide
- Powerful antibacterial nanocomposites from Corallina officinalis-mediated nanometals and chitosan nanoparticles against fish-borne pathogens
- Removal behavior of Zn and alkalis from blast furnace dust in pre-reduction sinter process
- Environmentally friendly synthesis and computational studies of novel class of acridinedione integrated spirothiopyrrolizidines/indolizidines
- The mechanisms of inhibition and lubrication of clean fracturing flowback fluids in water-based drilling fluids
- Adsorption/desorption performance of cellulose membrane for Pb(ii)
- A one-pot, multicomponent tandem synthesis of fused polycyclic pyrrolo[3,2-c]quinolinone/pyrrolizino[2,3-c]quinolinone hybrid heterocycles via environmentally benign solid state melt reaction
- Green synthesis of silver nanoparticles using durian rind extract and optical characteristics of surface plasmon resonance-based optical sensor for the detection of hydrogen peroxide
- Electrochemical analysis of copper-EDTA-ammonia-gold thiosulfate dissolution system
- Characterization of bio-oil production by microwave pyrolysis from cashew nut shells and Cassia fistula pods
- Green synthesis methods and characterization of bacterial cellulose/silver nanoparticle composites
- Photocatalytic research performance of zinc oxide/graphite phase carbon nitride catalyst and its application in environment
- Effect of phytogenic iron nanoparticles on the bio-fortification of wheat varieties
- In vitro anti-cancer and antimicrobial effects of manganese oxide nanoparticles synthesized using the Glycyrrhiza uralensis leaf extract on breast cancer cell lines
- Preparation of Pd/Ce(F)-MCM-48 catalysts and their catalytic performance of n-heptane isomerization
- Green “one-pot” fluorescent bis-indolizine synthesis with whole-cell plant biocatalysis
- Silica-titania mesoporous silicas of MCM-41 type as effective catalysts and photocatalysts for selective oxidation of diphenyl sulfide by H2O2
- Biosynthesis of zinc oxide nanoparticles from molted feathers of Pavo cristatus and their antibiofilm and anticancer activities
- Clean preparation of rutile from Ti-containing mixed molten slag by CO2 oxidation
- Synthesis and characterization of Pluronic F-127-coated titanium dioxide nanoparticles synthesized from extracts of Atractylodes macrocephala leaf for antioxidant, antimicrobial, and anticancer properties
- Effect of pretreatment with alkali on the anaerobic digestion characteristics of kitchen waste and analysis of microbial diversity
- Ameliorated antimicrobial, antioxidant, and anticancer properties by Plectranthus vettiveroides root extract-mediated green synthesis of chitosan nanoparticles
- Microwave-accelerated pretreatment technique in green extraction of oil and bioactive compounds from camelina seeds: Effectiveness and characterization
- Studies on the extraction performance of phorate by aptamer-functionalized magnetic nanoparticles in plasma samples
- Investigation of structural properties and antibacterial activity of AgO nanoparticle extract from Solanum nigrum/Mentha leaf extracts by green synthesis method
- Green fabrication of chitosan from marine crustaceans and mushroom waste: Toward sustainable resource utilization
- Synthesis, characterization, and evaluation of nanoparticles of clodinofop propargyl and fenoxaprop-P-ethyl on weed control, growth, and yield of wheat (Triticum aestivum L.)
- The enhanced adsorption properties of phosphorus from aqueous solutions using lanthanum modified synthetic zeolites
- Separation of graphene oxides of different sizes by multi-layer dialysis and anti-friction and lubrication performance
- Visible-light-assisted base-catalyzed, one-pot synthesis of highly functionalized cinnolines
- The experimental study on the air oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid with Co–Mn–Br system
- Highly efficient removal of tetracycline and methyl violet 2B from aqueous solution using the bimetallic FeZn-ZIFs catalyst
- A thermo-tolerant cellulase enzyme produced by Bacillus amyloliquefaciens M7, an insight into synthesis, optimization, characterization, and bio-polishing activity
- Exploration of ketone derivatives of succinimide for their antidiabetic potential: In vitro and in vivo approaches
- Ultrasound-assisted green synthesis and in silico study of 6-(4-(butylamino)-6-(diethylamino)-1,3,5-triazin-2-yl)oxypyridazine derivatives
- A study of the anticancer potential of Pluronic F-127 encapsulated Fe2O3 nanoparticles derived from Berberis vulgaris extract
- Biogenic synthesis of silver nanoparticles using Consolida orientalis flowers: Identification, catalytic degradation, and biological effect
- Initial assessment of the presence of plastic waste in some coastal mangrove forests in Vietnam
- Adsorption synergy electrocatalytic degradation of phenol by active oxygen-containing species generated in Co-coal based cathode and graphite anode
- Antibacterial, antifungal, antioxidant, and cytotoxicity activities of the aqueous extract of Syzygium aromaticum-mediated synthesized novel silver nanoparticles
- Synthesis of a silica matrix with ZnO nanoparticles for the fabrication of a recyclable photodegradation system to eliminate methylene blue dye
- Natural polymer fillers instead of dye and pigments: Pumice and scoria in PDMS fluid and elastomer composites
- Study on the preparation of glycerylphosphorylcholine by transesterification under supported sodium methoxide
- Wireless network handheld terminal-based green ecological sustainable design evaluation system: Improved data communication and reduced packet loss rate
- The optimization of hydrogel strength from cassava starch using oxidized sucrose as a crosslinking agent
- Green synthesis of silver nanoparticles using Saccharum officinarum leaf extract for antiviral paint
- Study on the reliability of nano-silver-coated tin solder joints for flip chips
- Environmentally sustainable analytical quality by design aided RP-HPLC method for the estimation of brilliant blue in commercial food samples employing a green-ultrasound-assisted extraction technique
- Anticancer and antimicrobial potential of zinc/sodium alginate/polyethylene glycol/d-pinitol nanocomposites against osteosarcoma MG-63 cells
- Nanoporous carbon@CoFe2O4 nanocomposite as a green absorbent for the adsorptive removal of Hg(ii) from aqueous solutions
- Characterization of silver sulfide nanoparticles from actinobacterial strain (M10A62) and its toxicity against lepidopteran and dipterans insect species
- Phyto-fabrication and characterization of silver nanoparticles using Withania somnifera: Investigating antioxidant potential
- Effect of e-waste nanofillers on the mechanical, thermal, and wear properties of epoxy-blend sisal woven fiber-reinforced composites
- Magnesium nanohydroxide (2D brucite) as a host matrix for thymol and carvacrol: Synthesis, characterization, and inhibition of foodborne pathogens
- Synergistic inhibitive effect of a hybrid zinc oxide-benzalkonium chloride composite on the corrosion of carbon steel in a sulfuric acidic solution
- Review Articles
- Role and the importance of green approach in biosynthesis of nanopropolis and effectiveness of propolis in the treatment of COVID-19 pandemic
- Gum tragacanth-mediated synthesis of metal nanoparticles, characterization, and their applications as a bactericide, catalyst, antioxidant, and peroxidase mimic
- Green-processed nano-biocomposite (ZnO–TiO2): Potential candidates for biomedical applications
- Reaction mechanisms in microwave-assisted lignin depolymerisation in hydrogen-donating solvents
- Recent progress on non-noble metal catalysts for the deoxydehydration of biomass-derived oxygenates
- Rapid Communication
- Phosphorus removal by iron–carbon microelectrolysis: A new way to achieve phosphorus recovery
- Special Issue: Biomolecules-derived synthesis of nanomaterials for environmental and biological applications (Guest Editors: Arpita Roy and Fernanda Maria Policarpo Tonelli)
- Biomolecules-derived synthesis of nanomaterials for environmental and biological applications
- Nano-encapsulated tanshinone IIA in PLGA-PEG-COOH inhibits apoptosis and inflammation in cerebral ischemia/reperfusion injury
- Green fabrication of silver nanoparticles using Melia azedarach ripened fruit extract, their characterization, and biological properties
- Green-synthesized nanoparticles and their therapeutic applications: A review
- Antioxidant, antibacterial, and cytotoxicity potential of synthesized silver nanoparticles from the Cassia alata leaf aqueous extract
- Green synthesis of silver nanoparticles using Callisia fragrans leaf extract and its anticancer activity against MCF-7, HepG2, KB, LU-1, and MKN-7 cell lines
- Algae-based green AgNPs, AuNPs, and FeNPs as potential nanoremediators
- Green synthesis of Kickxia elatine-induced silver nanoparticles and their role as anti-acetylcholinesterase in the treatment of Alzheimer’s disease
- Phytocrystallization of silver nanoparticles using Cassia alata flower extract for effective control of fungal skin pathogens
- Antibacterial wound dressing with hydrogel from chitosan and polyvinyl alcohol from the red cabbage extract loaded with silver nanoparticles
- Leveraging of mycogenic copper oxide nanostructures for disease management of Alternaria blight of Brassica juncea
- Nanoscale molecular reactions in microbiological medicines in modern medical applications
- Synthesis and characterization of ZnO/β-cyclodextrin/nicotinic acid nanocomposite and its biological and environmental application
- Green synthesis of silver nanoparticles via Taxus wallichiana Zucc. plant-derived Taxol: Novel utilization as anticancer, antioxidation, anti-inflammation, and antiurolithic potential
- Recyclability and catalytic characteristics of copper oxide nanoparticles derived from bougainvillea plant flower extract for biomedical application
- Phytofabrication, characterization, and evaluation of novel bioinspired selenium–iron (Se–Fe) nanocomposites using Allium sativum extract for bio-potential applications
- Erratum
- Erratum to “Synthesis, characterization, and evaluation of nanoparticles of clodinofop propargyl and fenoxaprop-P-ethyl on weed control, growth, and yield of wheat (Triticum aestivum L.)”