Preparation and in vitro biocompatibility of PBAT and chitosan composites for novel biodegradable cardiac occluders
-
Shanshan Wang


Abstract
The biodegradable composites were prepared by melt blending of chitosan (CS) and poly(butyleneadipate-co-terephthalate) (PBAT). By utilizing Fourier transformed infrared spectroscopy, scanning electron microscopy-energy dispersive spectroscopy, mechanical properties analysis, water contact angle measuring, differential scanning calorimetry, and thermogravimetric analysis, it was demonstrated that the CS of the PBAT-CS10 composite was relatively evenly dispersed in the PBAT matrix, the mechanical properties were significantly improved, the hydrophilicity was increased, the cold crystallization temperature was increased, and a good range of melt working temperature was obtained. The PBAT-CS10 composite was used to fabricate a cardiac occluder by fused deposition modeling of three-dimensional printing, and finite element analysis, and in vitro implantation testing proved the occluder’s mechanical support and sealing function under extreme boundary conditions. In vitro degradation experiments, neutral red uptake cytotoxicity assay, CCK-8 cell proliferation detection, immunofluorescence staining of the cytoskeleton, cell apoptosis detection, and reactive oxygen species assay were all performed on the composite, confirming that it and the occluder made of it could be hydrolyzed under physiological conditions and had no adverse effects on the cell membrane, lysosome membrane, cell proliferation, cell morphology, cell apoptosis, or ROS level, and had good biocompatibility.
1 Introduction
Congenital heart disease (CHD) is one of the most prevalent birth abnormalities, with a rising global prevalence (1). Ventricular septal defect (VSD), atrial septal defect, and patent ductus arteriosus are the three most prevalent kinds of CHD, accounting for about 80% of CHD patients (2). Due to the advancements in interventional surgery and nitinol occluders, interventional closure is now the recommended treatment option for the three types of CHD (3).
While nitinol occluders have beneficial therapeutic effects (3), they have a number of disadvantages, including myocardial injury, atrioventricular block, hemolysis, thrombosis, elevated blood nickel levels, nickel allergy, incomplete endothelialization of occluders, and cytotoxicity, which impairs gene expression and cell synthesis function. Additionally, nitinol is not biodegradable and will remain a foreign substance in the body after therapy, posing lifetime safety hazards to patients (4,5,6,7,8,9,10). As a result, it is critical to seek a biodegradable cardiac occluder to replace it.
As biodegradable materials progress, there have been many research studies on biodegradable cardiac occluders worldwide. The materials used to make biodegradable occluders include polyesters, such as polylactic acid (PLA), poly ε-caprolactone (PCL), poly(p-dioxanone) (PPDO), and the copolymer of PCL and PLA (11,12,13). However, biodegradable occluders have not been used in the clinic. Therefore, the innovation of occluders still needs to be explored, and the material and production method are the key factors. The material for the preparation of biodegradable occluders should first have good biodegradability, good Young's modulus and resilience, and good processability.
Poly(butyleneadipate-co-terephthalate) (PBAT) is a compostable linear polyester with high elasticity and a low elastic modulus (14). Although PBAT is widely used in industry and daily life, such as in mulching film and packaging bags (15), there are few studies in the medical field, particularly no comprehensive research on biodegradability and biocompatibility.
Chitosan (CS) is an alkaline biological polysaccharide that has been approved by the US Food and Drug Administration as a biological material. It has been widely used in the medical field for a variety of applications, including but not limited to cartilage repair, bone tissue engineering, liver tissue engineering, vascular tissue engineering, drug delivery, wound healing, and regenerative medicine (16).
When materials are used in biomedical fields, specific performance is required for specific applications. If the performance of a single material cannot meet the requirements, composites are a good choice. Composites are heterogeneous materials containing two or more solid phases that are in close microscopic contact with one another. Melt blending is a common method to prepare composites that can gradually disperse the filler by shearing and stretching the material in the molten state using twin-screw extruders or internal mixers (17,18).
The performance of a composite can be purposely created to fit a specific application by making use of the performance characteristics of each component. CS, as a filler or enhancer agent in composites, can be used to modify the materials' mechanical properties (19), hydrophilicity (20), and biocompatibility, including blood compatibility (21), cell adhesion and proliferation (22), and angiogenesis (23).
In this work, the composites of PBAT and CS were prepared by melt blending using an internal mixer. The microstructure, mechanical properties, water contact angle, and thermal properties of the composites were evaluated. Then, we used an additive manufacturing process to create a cardiac VSD occluder (VSDO) as the entrance point for the research project on biodegradable occluders. Its function was evaluated by finite element analysis (FEA) and an in vitro implantation experiment, and its biodegradability and biocompatibility were evaluated by in vitro degradation experiments and cytotoxicity experiments, respectively.
2 Materials and methods
2.1 Preparation of PBAT–CS composites
In this study, the composites were prepared by the melt blending method. PBAT (Shanghai Macklin Biochemical Co. Ltd., Shanghai, China) was vacuum dried at 105°C for 4 h; CS (Shanghai Macklin Biochemical Co. Ltd., Shanghai, China) was vacuum-dried at 30°C for 12 h. A certain proportion of premixed PBAT and CS was placed into the internal mixer module of the RM-200C (Hapro, Harbin, China). The amount of premixed material accounted for 60% of the capacity of the internal mixer. The premixed material was blended in an internal mixer at 160°C and 30 rpm for 10 min. The blended materials were cooled to room temperature and packed for later use. According to the mass ratio of PBAT and CS, PBAT:CS = 9:1 was named PBAT-CS10, and PBAT:CS = 8:2 was named PBAT-CS20.
2.2 Characterization
2.2.1 Fourier-transformed infrared spectroscopy (FTIR)
The FTIR analysis of PBAT, CS, PBAT-CS10, and PBAT-CS20 was conducted using a Nicolet iN10MX Fourier Transform infrared spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) in the wavenumber range of 4,000–400 cm−1.
2.2.2 Scanning electron microscopy and energy dispersive spectroscopy (SEM-EDS)
The morphology of PBAT-CS10 and PBAT-CS20 composites was observed by JSM-7610F ultra-high-resolution thermal field emission scanning electron microscopy (JEOL Ltd. Tokyo, Japan) in order to assess the microstructure and determine the dispersion of CS in the PBAT matrix. After 1 h of freezing in liquid nitrogen, the samples were brittly fractured and attached to the sample table using conductive glue for platinum spraying. Following that, SEM-EDS testing was conducted.
2.2.3 Water contact angle
The PBAT, PBAT-CS10, and PBAT-CS20 were formed into 1 cm × 3 cm thin strips, and the static water contact angle was determined using the Theta Flow optical contact angle measuring instrument (Bolin, Stockholm, Sweden) to determine the hydrophilicity.
2.2.4 Mechanical property
We pressed PBAT and PBAT–CS composites to prepare the test samples with a size of 50 mm × 5 mm × 2 mm. The samples were tested using an Instron 5,300 universal testing machine (Instron, Inc., Shanghai, China) at a tensile speed rate of 10 mm·min−1.
2.2.5 Differential scanning calorimetry (DSC)
The thermal properties of the materials were determined using the DSC1 Differential Scanning Calorimeter (Mettler Toledo International Ltd., Delaware, USA). To begin, 5 mg of PBAT, PBAT-CS10, and PBAT-CS20 were taken. And then, the temperature was raised to 200°C to erase the thermal history, with a heating rate of 10°C·min−1. After 3 min, the temperature was dropped to −50°C at a pace of 10°C·min−1. Finally, the temperature was raised to 300°C at a pace of 10°C·min−1.
2.2.6 Thermogravimetric analysis (TGA)
The thermal stability of the material was determined using a TG 209 F3 thermogravimetric analyzer (Netzsch, Selb, Germany). To begin, 5 mg material samples were put in an aluminum crucible and heated at a rate of 10°C·min−1 from room temperature to 800°C in a nitrogen atmosphere. Three materials were determined, respectively: PBAT, PBAT-CS10, and PBAT-CS20.
2.3 Fabrication and functional testing of VSDO prototype
2.3.1 Design and three-dimensional (3D)-printing
We designed VSDO using SolidWorks software (Figure 1) and manufactured it using fused deposition modeling (FDM) technology. The VSDO was a double-disc with a diameter of 10 mm, spokes 1 mm thick, a waist diameter of 5 mm, and a waist height of 4 mm. According to the TGA results, the following settings were specified for 3D printing: nozzle diameter was 0.35 mm, printing temperature was 170°C, printing pressure was 0.30 MPa, printing speed was 12 mm·s−1, and layer height was 0.33 mm.

VSDO engineering drawings.
2.3.2 FEA
The occluder should be stress–strain evaluated, and modeling software coupled with FEA is suggested, according to “YY/T 1553-2017 Cardiac Occluder-Cardiovascular Implants” (24). In the FEA, boundary conditions such as implant shape, displacement, and physiological pressure should be included. The guidelines propose that the pressure applied to one side of the cardiac occluder be regarded at the clinical maximum value of 160 mmHg (21 kPa). To evaluate the influence of mechanical properties of the composites on occlusion performance of VSDO, the friction coefficient of the contact surface of the VSDO and VSD models was set to zero. Simultaneously, the pressure exerted by the VSD on the VSDO side waist was minimized, set to zero, finally entering the boundary conditions described above into the SolidWorks for FEA.
2.3.3 In vitro occlusion test
To evaluate VSDO's functional efficacy, an in vitro simulated occlusion experiment was conducted utilizing a micro-pulse pump and a simulated extracorporeal circulation channel. The VSDO was inserted into the VSD of the simulated heart. To fill the micro-pulse pump tube, a 45% glycerin aqueous solution was utilized, and red dye was used to intersperse the 45% glycerin aqueous solution to create “simulated blood.” The temperature of the micro-pulse pump circulation was set to 37 ± 0.5°C, the left ventricular pressure was set to 80–150 mmHg (the highest value of the pump), the heart rate was set to 80 beats per minute, and the “blood” flow rate was set to 6 L·min−1. The sealing function of the VSDO was observed and recorded.
2.4 In vitro degradation experiments
2.4.1 Preparation of in vitro degradation samples
According to ISO 10993-12:2012 and ISO 10993-13:2010, we prepared PBAT-CS10 disc samples for in vitro degradation to test the biodegradability of the VSDO we fabricated (25,26). The disc sample was 10 mm in diameter and 0.3 mm thick. In accordance with ISO 13781:2017, Sorensen buffer solution (0.2 M, pH 7.4) (Shanghai Macklin Biochemical Co. Ltd., Shanghai, China) was chosen as the hydrolytic solution for a total of 24 weeks of in vitro degradation (27). The containers were put in the incubator at 37°C, and the Sorensen buffer solution was replaced once a week. The specified observation time points were 4, 8, 12, 16, 20, and 24 weeks. The disc samples were dried to a consistent weight in a 60°C vacuum oven at each time point.
2.4.2 Residual weight
At each time point, the mass of the disc samples was determined using the electronic balance ME202 (Mettler Toledo International Ltd., Delaware, USA). The measurement accuracy was 0.1 mg, and the percentages of residual weight for the samples were calculated using the following relationship:
where W 0 denotes the original weight and W r denotes the dry weight of the samples after degradation.
2.4.3 The change in molecular weight
To assess the change in molecular weight during in vitro degradation, the molecular weight of the samples was determined at each time point using gel permeation chromatography (GPC). Waters 1,525 high-performance liquid chromatography (Waters, Framingham, Massachusetts, USA) was used to determine GPC. CHCl3 was used as the mobile phase at a flowrate of 1 mL·min−1 to dissolve 2 mg of the sample and injected into an analytical instrument at a temperature of 35°C for determination. As CS is insoluble in chloroform, GPC primarily assesses the results of PBAT.
2.5 In vitro biocompatibility
2.5.1 Preparation of the material extract
The biocompatibility of the composites was assessed in this research using a variety of in vitro cell tests. The material extract is recommended for cell assays according to ISO 10993.5-2009 (28). The samples of composites were generated at the same temperature as FDM processing with a size of 25 mm × 5 mm × 0.2 mm and sterilized by ethylene oxide for use in the cytotoxicity testing.
Extract medium: DMEM high glucose medium with 10% volume fraction of calf serum and 1% volume fraction of penicillin/streptomycin (final concentration of 100 IU·mL−1 penicillin, 100 µg·mL−1 streptomycin).
To make the material extract, the quantity of extract medium needed was estimated based on the sample surface area, which was 6 cm2·mL−1. The material extract was extracted for 24 h at 37°C. The material extract was then transferred to a fresh sterile centrifugal tube and kept at 20°C. The blank medium for the blank control group was made in the same way as the extract medium but without the material.
2.5.2 Cells culture
Procell Life Science & Technology Co., Ltd. provided NIH/3T3 and BALB/c 3T3, clone A31 (Procell CL-0477) (Wuhan, Hubei, China). We incubated the cells in DMEM high-glucose medium (Solarbio, Beijing, China) (DMEM + 10% calf serum + 1% penicillin/streptomycin) at 37°C, with a volume fraction of 5% CO2 and saturated humidity conditions. We digested the cells with 0.25% trypsin (Gibco, Waltham, Massachusetts, USA) at 37°C until the cells fell off the dish, blew the collected cells to the centrifuge tube, centrifuged at 1,200 rpm to collect the cells, colonized the new culture dish according to the density of 1 × 105 cells per dish, then placed the sample in an incubator for growing, and chose a cell density of 60–70% for next step.
2.5.3 Neutral red uptake (NRU) cytotoxicity test
The impact of the composites on the capacity of the BALB/c 3T3 cells to absorb neutral red was tested using the neutral red staining solution (Beyotime Institute of Biotechnology, Jiangsu, China). After 24 h of co-culture with material extract, we removed the material extract and washed it once with phosphate buffer saline (PBS) (Beyotime Institute of biotechnology, Jiangsu, China). Then, for 30 min, we injected a sufficient quantity of neutral red staining solution to ensure that the cells were completely coated. Finally, we used a microscope (Nikon, Tokyo, Japan) to take photographs and a microtitration plate photometer SpectraMax ABS (Molecular Devices, Sunnyvale, California, USA) to measure the optical density (OD) of the solution at 550 nm. The blank control group received the same treatment as the blank medium.
2.5.4 CCK-8 cell proliferation detection
We collected logarithmic phase NIH/3T3 cells, increased the cell density to 5,000 cells per well, and increased the volume of the material extract or blank medium to 100 μL. Cells were cultivated at 37°C for 24 h in 5% CO2, and then, 10 μL CCK-8 (Solarbio, Beijing, China) solution was added to each hole and incubated for 2 h in the cell incubator. The OD was determined at 450 nm using a microtitration plate photometer.
2.5.5 Cytoskeleton immunofluorescence staining
To stain actin filaments, we utilized the Phalloidin-iFluor 555 Reagent (Abcam, Cambridge, UK). To begin, after 24 h of co-culture with the material extract or blank medium, we washed the BALB/c 3T3 cells twice with PBS and fixed them for 10 min with 4% paraformaldehyde. Second, we carefully applied the staining solution and rinsed the fixed cells twice in PBS before adding 0.1% Triton X-100 in PBS for 5 min to improve permeability. Third, we gently rinsed permeabilized cells with PBS twice, added a sufficient amount of 1× Phalloidin conjugate working solution to ensure complete coverage for 60 min, and gently rinsed cells with PBS twice to remove excess phalloidin conjugate. Finally, we sealed them with Mounting Medium and antifading (with 4’,6-diamidino-2-phenylindole (DAPI)) (Solarbio, Beijing, China) and observed the cells using a fluorescence microscope (Nikon, Tokyo, Japan) equipped with appropriate filters for Ex/Em = 556/574 nm (Phalloidin-iFluor 555) and Ex/Em = 360/460 nm (DAPI).
2.5.6 Cell apoptosis detection
The apoptosis rate caused by the composites was determined using the Annexin V-FITC apoptosis detection kit (Beyotime Institute of Biotechnology, Jiangsu, China). To begin, BALB/c 3T3 cells were co-cultured for 24 h with material extract or a blank medium. Second, cells were collected by centrifugation at 4°C for 5 min at 1,200 rpm after digestion with 0.25% trypsin without ethylenediamine tetraacetic acid (Solarbio, Beijing, China). Third, the cells were washed twice with precooled PBS and centrifuged for 5 min at 1,200 rpm at 4°C. Fourth, we discarded the PBS and added 100 μL of 1× binding buffer to resuspend the cells, followed by the addition of 5 μL of Annexin V-FITC mix and 15 min at room temperature in the dark. Finally, we added 10 μL of PI holding solution and 400 μL of 1× binding buffer to the cells, mixed well, placed the mixture on ice, and detected the cells within 5 min using flow cytometry.
2.5.7 Reactive oxygen species (ROS) assay
To precisely determine the oxidative stress level of cells caused by the composites, we utilized the ROS test kit (Beyotime Institute of Biotechnology, Jiangsu, China). To begin, BALB/c 3T3 cells were co-cultured for 24 h with material extract or a blank medium. Second, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was diluted to a concentration of 10 μmol·L−1 with a serum-free medium. Third, we withdrew the culture medium from the cells and added an appropriate volume of diluted DCFH-DA to completely cover them. Fourth, after 20 min of incubation at 37°C, the cells were washed three times with serum-free cell culture medium to completely eliminate any DCFH-DA that did not enter the cells. Fifth, we digested the cells at 37°C with 0.25% trypsin (Gibco, Waltham, Massachusetts, USA), blew the collected cells into a centrifuge tube, and centrifuged them at 1,200 rpm to collect the cells. Finally, we tested ROS-positive cells at the FITC channel using flow cytometry in CytoFlex S (Beckman, S. Kraemer Boulevard Brea, California, USA) and analyzed the data using the FlowJo software (Becton, Dickinson & Company, Franklin Lakes, New York, USA).
2.5.8 Statistical analysis
The statistical significance of the difference between the values of blank control groups and material groups was determined using Student's t-test using GraphPad Prism (Version 9 for Windows, GraphPad Software, San Diego, California, USA; www.graphpad.com). In each case, p < 0.05 was considered statistically significant.
3 Results
3.1 Microstructure of nanocomposites
Figure 2 shows the FTIR spectra of CS, PBAT, PBAT-CS10, and PBAT-CS20. The characteristic band and peaks of CS (Figure 2a) were as follows: the broad peak at 3,450 cm−1 is assigned to O‒H and N‒H stretching vibration superposition, the peak at 1,660 cm−1 is assigned to C═O stretching vibration in acetyl group, and the broadband at 1,000–1,200 cm−1 is assigned to the saccharide structure of CS (29,30,31,32).

FTIR spectra of (a) CS, (b) PBAT, (c) PBAT-CS10, and (d) PBAT-CS20.
The characteristic bands of PBAT (Figure 2b) were as follows: the broad band between 2,830–3,030 cm−1 is assigned to the antisymmetric stretching vibration of CH2, the band between 1,680–1,750 cm−1 is assigned to C═O stretching vibration, and the band between 1,210–1,310 cm−1 is assigned to C‒O‒C stretching vibration in aromatic esters. Because the ester group acted as a polar substituent, π–π conjugation occurred with the benzene ring, and the ═CH out-of-plane vibration frequency of the variable angle was reduced to the vicinity of 730 cm−1 (33,34,35,36).
Figure 2c and d is the spectra of PBAT-CS10 and PBAT-CS20, respectively. The spectra of the two were generally akin to those of pure PBAT. The bands of 2,830–3,030, 1,680–1,750, 1,210–1,310, and around 730 cm−1 are all correspond to the pure PBAT matrix. Meanwhile, the absorption band at 3,400–3,500 cm−1 is the superposition of O‒H and N‒H stretching vibrations, which is the characteristic band of CS, indicating that CS has been successfully dispersed into the PBAT matrix.
Figure 3 shows the micro-morphology and element distribution of the cross-section of PBAT-CS10 and PBAT-CS20. CS particles in PBAT-CS10 had smaller sizes and were relatively evenly distributed in the PBAT matrix, forming a sea-island phase separated microstructure (Figure 3a), while CS particles in PBAT-CS20 had larger sizes and close spacing, which were prone to agglomeration (Figure 3e). Compared with PBAT, nitrogen is unique to CS. According to EDS mapping results, the N element was equally distributed throughout the composites (Figure 3b and f), showing that CS was well dispersed in the PBAT matrix.

SEM-EDS images of (a–d) PBAT-CS10 and (e–h) PBAT-CS20.
Due to the incompatibility between PBAT and CS, the SEM images reveal that the PBAT–CS composites had a heterogeneous phase distribution in their microstructure. Therefore, we took advantage of the heterogeneous distribution to design the composite as a biodegradable implant, because of which can improve cell adhesion and proliferation (37).
3.2 Mechanical properties
Table 1 shows the mechanical properties of PBAT, PBAT-CS10, and PBAT-CS20. With the increase in the CS mass ratio, the rigidity of composites increased, which was manifested by the decrease in tensile strength and elongation at break, and the increase in Young’s modulus and yield modulus. CS can increase the modulus of the composites, showing that the activation of a stress transfer mechanism across the PBAT–CS interface, demonstrating the positive interaction between CS and PBAT (38). On the other hand, CS scattered in the PBAT matrix increased steric hindrance, hence limiting the tensile performance of PBAT, and destroying the homogeneous phase. As a result, CS increased Young’s modulus while decreasing the elongation at break (19). PBAT-CS10 had better toughness than PBAT-CS20. The materials for VSDO must balance modulus and elastic, as modulus is required to maintain sealing effectiveness and elastic is required to avoid brittle fracture. In conclusion, PBAT-CS10 was chosen as the material to prepare the VSDO in the following stage due to its mechanical properties.
The mechanical properties of PBAT, PBAT-CS10, and PBAT-CS20
| Samples | Young’s modulus (MPa) | Yield strength (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|---|
| PBAT | 91.06 | 5.04 | >21.25 | >758 |
| PBAT-CS10 | 165.40 | 11.74 | 13.61 | 434 |
| PBAT-CS20 | 195.11 | 12.07 | 12.07 | 173 |
3.3 Water contact angle
Figure 4 shows the water contact angle of PBAT, PBAT-CS10, and PBAT-CS20, with average values of 92.15°, 84.59°, and 82.67°, respectively. The hydroxyl and carboxyl terminal groups of PBAT belong to hydrophilic structures, while the main chain segment of PBAT belongs to a hydrophobic structure. Under the combined action of these two factors, PBAT belonged to mildly hydrophobic material. The composites’ θ < 90° after blending the hydrophilic CS indicated that they became hydrophilic. In comparison to PLA, PCL, PPDO, and other non-hydrophilic polyester materials, increasing hydrophilicity can facilitate adhesion and proliferation of cells and tissues (39). Improved adherent proliferation is particularly advantageous as an occluder for the closure of heart defects.

Water contact angle of PBAT, PBAT-CS10, and PBAT-CS20.
3.4 Thermal properties
The secondary DSC heating curves (from 50°C to 300°C) and cooling curves (from 200°C to −50°C) of PBAT, PBAT-CS10, and PBAT-CS20 are shown in Figure 5a and b after thermal history was removed. Glass transition temperature (T g) and melting temperature (T m) values for the three materials were almost the same, owing to the fact that the composites' primary bodies remained PBAT. PBAT-CS10 and PBAT-CS20 had crystallization temperature (T c) values of 78.59°C and 80.93°C, respectively, which were much higher than PBAT's T c of 42.59°C (Table 2a). As a nucleating agent, CS distributed in the PBAT matrix decreased the interface free energy and facilitated heterogeneous crystallization of PBAT (19,40). The composites can retain the state of high elasticity and modulus over the application range of body temperature. The melting enthalpy (H m) and crystallization enthalpy (H c) values of PBAT, PBAT-CS10, and PBAT-CS20 fell as the PBAT proportion decreased, showing that the alteration in PBAT was mostly responsible for cold crystallization and melting.
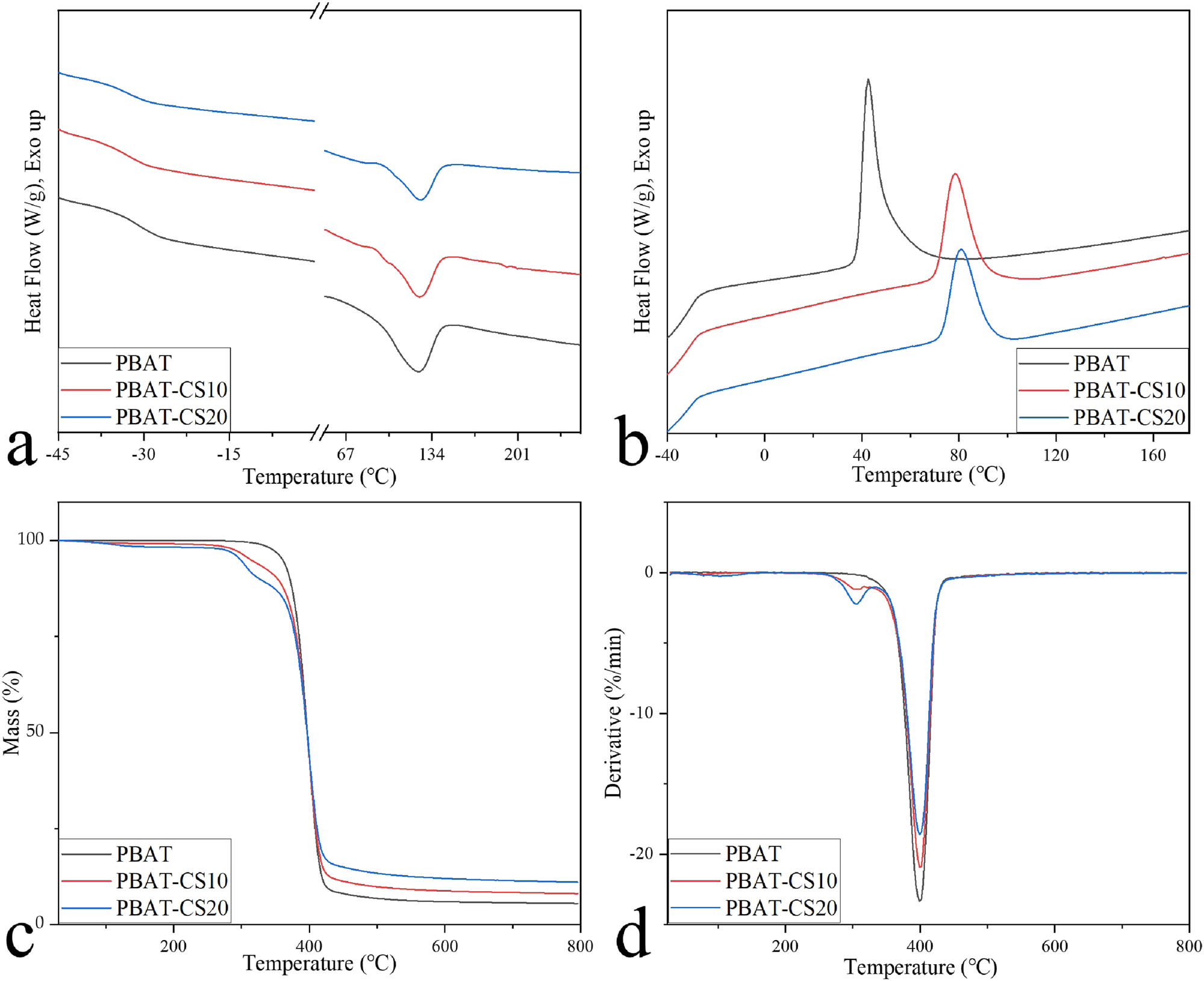
Thermal characterization of PBAT, PBAT-CS10, and PBAT-CS20: (a) DSC secondary heating curves, (b) DSC cooling curve, (c) TGA curve, and (d) DTG curve.
Thermal properties of PBAT, PBAT-CS10, and PBAT-CS20
| a | |||||||
|---|---|---|---|---|---|---|---|
| T g (°C) | T c (°C) | ΔH c (J‧g−1) | T m (°C) | ΔH m (J‧g−1) | T d (°C) | ΔT (°C) | |
| PBAT | −30.73 | 42.59 | 20.89 | 123.07 | 17.60 | 360.72 | 237.65 |
| PBAT-CS10 | −32.37 | 78.59 | 14.25 | 124.24 | 12.55 | 314.42 | 190.18 |
| PBAT-CS20 | −31.78 | 80.93 | 12.87 | 125.25 | 9.91 | 298.99 | 173.74 |
| b | ||||||
|---|---|---|---|---|---|---|
| D max1 (%‧min−1) | D max2 (%‧min−1) | D max3 (%‧min−1) | T max1 (°C) | T max2 (°C) | T max3 (°C) | |
| PBAT | — | — | 23.35 | — | — | 399.31 |
| PBAT-CS10 | 0.13 | 1.18 | 20.99 | 62.33 | 309.28 | 399.31 |
| PBAT-CS20 | 0.24 | 2.24 | 18.60 | 103.49 | 304.13 | 399.31 |
Figure 5c and d shows the TGA curve and derivative thermogravimetry (DTG) curve of PBAT, PBAT-CS10, and PBAT-CS20, respectively. In the DTG figure, the curve peaks of PBAT-CS10 and PBAT-CS20 were different from those of PBAT, and there were two more peaks, D max1 and D max2, in the range of 50–150°C and 250–330°C (Table 2b), which were caused by the removal of water in the crystallization and thermal decomposition of CS, respectively (41). The ratio of D max1 to D max2 was about 1:2, corresponding to the mass proportion of CS in the composites. The T max3 of the three materials was basically the same, and their D max3 ratio was the same as the content ratio of PBAT, indicating that this was caused by the thermal decomposition of PBAT (42,43).
The TGA test was conducted to determine a safe processing temperature range for the composites in the FDM, ensuring that the composition did not change considerably during processing. When the thermal weight loss of a polymer exceeds 5%, it shows that its decomposition is evident. Therefore, the initial decomposition temperature (T d) of 5% is the theoretical maximum processing temperature for polymers. ΔT was the temperature range from T m to T d, representing the temperature range of material processing in the melting state, which corresponded to the theoretical temperature range for thermal processing of composites, which was approximately 125–300°C. When the temperature was less than 250°C, the DTG curve indicated that the three curves were nearly parallel, showing that CS was relatively stable below 250°C. To summarize, investigating an optimal temperature range for FDM between 125°C and 250°C can ensure that no obvious change in the characteristics of composites.
3.5 FEA, fabrication, and in vitro test of VSDO
Prior to fabricating the VSDO by 3D printing, an FEA was conducted to determine the VSDO's stress and strain under boundary conditions. The cloud images of VSDO stress, strain, and displacement relative to VSD under boundary conditions are shown in Figure 6b–g. The maximum stress change at the root inside the left disk was 6.606 × 104 Pa, which was significantly less than the yield strength of PBAT-CS10 of 11.74 MPa, indicating that VSDO fabricated with PBAT-CS10 could still maintain elastic deformation under boundary conditions and could rebound with a change in ventricular pressure. The equivalent strain (ESTRN) was 3.298 × 104, which was similarly found at the root of the proximal left disk, indicating that the maximum strain was approximately 0.03%. At the center of the left disk, the maximum displacement of VSDO relative to VSD was approximately 6.163 × 10−4 mm. The VSDO can be placed firmly in the VSD due to the little displacement. According to the FEA results under the boundary conditions, it demonstrated that the mechanical properties of PBAT-CS10 were fully qualified for the VSDO sealing function.
Then, we tested the effect of real occlusion by inserting VSDO into a heart model with VSD (Figure 6a). The VSD in the heart model has a diameter of 5 mm and a waist height of 4 mm. When the left ventricular pressure varied between 85 and 150 mmHg, the heart rate was 80 beats per minute, the temperature was 37 ± 0.5°C, and the flow rate was 6 L·min−1; the VSDO exhibited a stable structure with no visible deformation, which proved an optimum sealing function.
Previously, the sealing function of the occluders was evaluated mainly through animal implantation (12,44). While this approach is capable of simulating the complicated internal environment of bodies, it does not allow for the evaluation of functionality under extreme situations. We can more clearly analyze the sealing function under extreme settings using FEA boundary conditions and in vitro simulated sealing conditions, which is superior to animal tests in quantitative analysis (Figure 6).

In vitro implantation test and FEA of VSDO: (a) in vitro implantation test of VSDO; (b and c) cloud image of VSDO strain under boundary conditions; (d and e) cloud image of VSDO stress under boundary conditions; and (f and g) cloud image of VSDO relative to VSD displacement under boundary conditions.
3.6 In vitro degradation
The residual weight curve of the PBAT-CS10 samples for 24 weeks of degradation in Sorensen buffer solution at 37°C is shown in Figure 7. The solution was replaced once a week to imitate the metabolic milieu of the dynamic circulation of the human body. The sample weight did not drop over the first 4 weeks but slightly rose, with an average value of 100.40%. The weight steadily declined over time, from 95.58% at 8 weeks to 85.83% at 24 weeks.

(a) Residual weight of PBAT-CS10 samples in 24 weeks; (b) M n, M w, and PDI changes in PBAT-CS10 samples in 24 weeks.
The variations in number-average molecular weight (M n), weight-average molecular weight (M w), and polydispersity index (PDI) (red curve) of the PBAT in the PBAT-CS10 composites during 24 weeks are shown in Figure 7b. With an increase in time, the Mn and Mw of the samples decreased gradually from 20,530 and 45,506 g·mol−1 to 6,690 and 15,869 g·mol−1 for 24 weeks, respectively. PDI increased slightly from 2.217 at the start to 2.372 after 24 weeks, which indicated that there was no obvious difference in the degradation rate of PBAT between the surface layer and the deep layer.
As a commonly used industrial polymer, PBAT has been well investigated for composting degradation, but its biodegradability in the human physiological milieu is still unknown. Evaluation of the PBAT-CS10 composite's biodegradability is required for its usage as a biodegradable implant material, such as the biodegradable cardiac occluder we fabricated. In accordance with ISO 10993-13:2010, in vitro degradation experiments of PBAT-CS10 composites were conducted, and the findings revealed that PBAT can be hydrolyzed in a physiological environment (26). The VSDO prepared by PBAT-CS10 composite was a biodegradable cardiac occluder.
3.7 In vitro biocompatibility
To comprehensively evaluate the in vitro biocompatibility of the PBAT-CS10 composites, the NRU cytotoxicity assay, CCK-8 cell proliferation assay, cytoskeleton immunofluorescence staining, cell apoptosis assay, and ROS assay were performed.
When a variety of adverse conditions cause brittle changes to the cell surface or sensitive lysosomal membrane, or growth rate and quantity are lowered, NRU by the cells can be diminished (45). The CCK-8 assay assesses cell proliferation activity by determining the OD of formazan produced by cells. The cytotoxicity of NRU is depicted in Figure 8a and b. There were no significant variations in cell size, morphology, or NRU capacity between the material and blank control groups, as shown in Figure 8a. The quantitative comparison of the OD values of NRU capacity between the two groups is shown in Figure 8b, and the comparison of the OD values in the CCK-8 test is shown in Figure 8d, both of which showed no significant differences (p > 0.05), indicating that the VSDO made of PBAT-CS10 had no significant toxicity to the cell membrane, lysosomal membrane, or cell proliferation. One study using the CCK-8 experiment revealed that CS-enhanced PLGA material has good biocompatibility (46). However, as far as we know, there are no reports on the effects of PBAT–CS composites on cell membrane, cell lysosome membrane, or cell proliferation activity.

(a) Comparison under the microscope in the NRU test; (b) effect of material group and blank control group on NRU in BALB/c 3T3 cells (p > 0.05); (c) comparison of cytoskeleton staining between material group and blank control group; and (d) effect of material group and blank control group on NIH 3T3 cell proliferation (p > 0.05).
The cytoskeleton and nucleus of the cells in the two groups were stained with phalloidin-ifluor 555 and DAPI, respectively. Figure 8c illustrates the results. The cytoskeleton morphology and nuclear structure of the two groups were normal, and there was no significant difference between them, showing that the PBAT-CS10 had no influence on cell morphology. This is consistent with some reports on the biocompatibility of CS (47,48).
To differentiate apoptotic cells from healthy cells, both groups of cells were stained with Annexin V-FITC and PI. Figure 9a shows the flow cytometry picture of cells following staining in the two groups. Comparative quantitative analysis (Figure 9b) reveals that there is no statistically significant difference between the two groups (p > 0.05), showing that PBAT-CS10 does not significantly induce cell apoptosis, which was consistent with previous research on CS (49).

(a and b) Comparison of cell apoptosis between material group and blank control group (p > 0.05); (c and d) comparison of ROS level between material group and blank control group (p > 0.05).
The influence of the material group on ROS production in cells was determined by counting ROS-positive cells. Figure 9c compares flow cytometry pictures from the two groups, whereas Figure 9d compares the number of ROS positive cells across the two groups, revealing no significant difference (p > 0.05) and demonstrating that PBAT-CS10 had no effect on the ROS level of cells.
As part of biocompatibility research on materials for medical implants, it is necessary to assess their biosafety using in vitro cytotoxicity assays. The cytotoxicity of PBAT-CS10 composites for VSDO was determined by the effects on the cell membrane, lysosome membrane, proliferation, morphology, apoptosis, and ROS level in mouse fibroblasts. The results established that PBAT-CS10 is not cytotoxic, implying that it has a high degree of biocompatibility, giving a theoretical foundation for further experimental investigation.
4 Conclusions
The mechanical properties, hydrophilicity, and cold crystallization temperature of PBAT were enhanced in this work by utilizing CS as a reinforcing agent. PBAT–CS composites had a good thermal processing temperature range of 125–250℃. It was shown via FEA and an in vitro occlusion test that VSDO fabricated by using the PBAT-CS10 composite and FDM could offer an adequate sealing function and mechanical stability under extreme boundary conditions (left disk pressure was 160 mmHg, waist lateral pressure was zero, and friction coefficient was zero). The VSDO can be hydrolyzed gradually in the in vitro degradation experiment and showed no significant adverse effects on cell membrane, lysosome membrane, cell proliferation, cell morphology, cell apoptosis, and intracellular peroxide of mouse fibroblasts, indicating that PBAT-CS10 and the occluder made of it had good biodegradability and biocompatibility.
Acknowledgment
The authors thank the research funding below.
-
Funding information: This study was supported by The National Natural Science Foundation of China (NSFC) (Grant Nos. 81770315 and 81570287) and Taishan Scholars Program (2019).
-
Author contributions: Quansheng Xing: conceptualization, methodology, software, formal analysis, resources, writing – review and editing, supervision, project administration, funding acquisition; Shanshan Wang: methodology, software, validation, formal analysis, investigation, resources, data curation, writing – original draft, visualization.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: All data generated or analyzed during this study are included in this published article.
References
(1) Liu Y, Chen S, Zühlke L, Babu-Narayan SV, Black GC, Choy M-K, et al. Global prevalence of congenital heart disease in school-age children: a meta-analysis and systematic review. BMC Cardiovasc Disord. 2020;20(1):1–10. 10.1186/s12872-020-01781-x. Search in Google Scholar
(2) Ministry of Health of the People's Republic of China. Report on prevention and treatment of birth defects in China. Ministry of Health of the People's Republic of China: Beijing, China, 2012.Search in Google Scholar
(3) Yang S, Chen S. Pediatric cardiology. People’s Medical Publishing House: Beijing, 2012.Search in Google Scholar
(4) Zhou Y, Chen F, Huang X, Zhao X, Wu H, Bai Y, et al. A new coated nitinol occluder for transcatheter closure of ventricular septal defects in a canine model. Biomed Res Int. 2013;2013:1–7. 10.1155/2013/507919.Search in Google Scholar PubMed PubMed Central
(5) Matkar SS, Wrischnik LA, Jones PR, Hellmann-Blumberg U. Two closely related nickel complexes have different effects on dna damage and cell viability. Biochem Biophys Res Commun. 2006;343(3):754–61. 10.1016/j.bbrc.2006.03.019.Search in Google Scholar PubMed
(6) Elshahawy WM, Watanabe I, Kramer P. In Vitro cytotoxicity evaluation of elemental ions released from different prosthodontic materials. Dent Mater. 2009;25(12):1551–5. 10.1016/j.dental.2009.07.008.Search in Google Scholar PubMed
(7) Lü X, Lu H, Zhao L, Yang Y, Lu Z. Genome-wide pathways analysis of nickel ion-induced differential genes expression in fibroblasts. Biomaterials. 2010;31(8):1965–73. 10.1016/j.biomaterials.2009.12.044.Search in Google Scholar PubMed
(8) Lü X, Bao X, Huang Y, Qu Y, Lu H, Lu Z. Mechanisms of cytotoxicity of nickel ions based on gene expression profiles. Biomaterials. 2009;30(2):141–8. 10.1016/j.biomaterials.2008.09.011.Search in Google Scholar PubMed
(9) Wertman B, Azarbal B, Riedl M, Tobis J. Adverse events associated with nickel allergy in patients undergoing percutaneous atrial septal defect or patent foramen ovale closure. J Am Coll Cardiol. 2006;47(6):1226–7. 10.1016/j.jacc.2005.12.017.Search in Google Scholar PubMed
(10) Chao Y, Guo-hong Z, Hui-shen W, Zhi-wei Z, Shu-shui W, Xu-xia L. Changes of blood nickle levels before and after transcatheter closure for atrial septal defect in children. South China J Cardiovasc Dis. 2008;14(5):327–9.Search in Google Scholar
(11) Huang Y, Wong YS, Wu J, Kong JF, Chan JN, Khanolkar L, et al. The mechanical behavior and biocompatibility of polymer blends for patent ductus arteriosus (PDA) occlusion device. J Mech Behav Biomed Mater. 2014;36:143–60. 10.1016/j.jmbbm.2014.04.012.Search in Google Scholar PubMed
(12) Li BN, Xie YM, Xie ZF, Chen XM, Zhang G, Zhang DY, et al. Study of biodegradable occluder of atrial septal defect in a porcine model. Catheter Cardiovasc Interv. 2018;93:1–8. 10.1002/ccd.27852.Search in Google Scholar PubMed PubMed Central
(13) Zhu YF, Huang XM, Cao J, Hu JQ, Bai Y, Jiang HB, et al. Animal experimental study of the fully biodegradable atrial septal defect (ASD) occluder. J Biomed Biotechnol. 2012;2012:735989. 10.1155/2012/735989.Search in Google Scholar PubMed PubMed Central
(14) Pinheiro IF, Ferreira FV, Souza DHS, Gouveia RF, Lona LMF, Morales AR, et al. Mechanical, rheological and degradation properties of PBAT nanocomposites reinforced by functionalized cellulose nanocrystals. Eur Polym J. 2017;97:356–65. 10.1016/j.eurpolymj.2017.10.026.Search in Google Scholar
(15) Ting Z, Caili Z, Xinyu S, Yunxuan W. Research progress in preparation and applications of PBAT films. CHINA Plast. 2021;35(7):116–25.Search in Google Scholar
(16) Kołodziejska M, Jankowska K, Klak M, Wszoła M. Chitosan as an underrated polymer in modern tissue engineering. Nanomaterials. 2021;11:3019. 10.3390/nano11113019.Search in Google Scholar PubMed PubMed Central
(17) Toh M, Gondoh T, Mori T, Mishima M. Mixing characteristics of an internal mixer: uniformity of mixed rubber. J Appl Polym Sci. 2005;95(1):166–72. 10.1002/app.20816.Search in Google Scholar
(18) Verilhac K, Desse M, Carrot C, Fenouillot F. Mixing of polar and nonpolar molten olefinic copolymer with polar liquids in conditions of very low viscosity ratio: Shear dominated flows. J Rheol. 2016;60(6):1121–35. 10.1122/1.4960335.Search in Google Scholar
(19) Karpova SG, Ol’khov AA, Iordanskii AL, Lomakin SM, Shilkina NS, Popov AA. Structural dynamic properties of nonwoven composite mixtures based on ultrafine tissues of poly(3-Hydroxybutyrate) with Chitosan. Russ J Phys Chem B. 2016;10(4):687–98. 10.1134/S1990793116040230.Search in Google Scholar
(20) Nasution H, Olaiya NG, Haafiz MKM, Abdullah CK, Bakar SA, Olaiya FG, et al. The role of amphiphilic chitosan in hybrid nanocellulose-reinforced polylactic acid biocomposite. Polym Adv Technol. 2021;32(9):3446–57. 10.1002/pat.5355.Search in Google Scholar
(21) Xu L, Zhang X, Zhu C, Zhang Y, Fu C, Yang B, et al. Nonionic polymer cross-linked chitosan hydrogel: preparation and bioevaluation. Biomater J Sci Polym. 2013;24:1564–74. 10.1080/09205063.2013.781934.Search in Google Scholar PubMed
(22) Jabbari F, Hesaraki S, Houshmand B. The Physical, mechanical, and biological properties of silk fibroin/chitosan/reduced graphene oxide composite membranes for guided bone regeneration. Biomater J Sci Polym. 2019;30:1779–802. 10.1080/09205063.2019.1666235.Search in Google Scholar PubMed
(23) Fang QQ, Wang XF, Zhao WY, Shi BH, Lou D, Chen CY, et al. Development of a chitosan-vaseline gauze dressing with wound-healing properties in murine models. Am J Trop Med Hyg. 2020;102(2):468–75. 10.4269/ajtmh.19-0387.Search in Google Scholar PubMed PubMed Central
(24) YY/T 1553. Cardiovascular implants cardiac occluder. Beijing, China. 2017;2017.Search in Google Scholar
(25) ISO 10993-12. Biological evaluation of medical devices part 12: sample preparation and reference materials. Geneva, Switzerland: 2012.Search in Google Scholar
(26) ISO 10993-13. Biological evaluation of medical devices part 13: identification and quantification of degradation products from polymeric medical devices. Geneva, Switzerland: 2010.Search in Google Scholar
(27) ISO 13781. Implants for surgery - homopolymers, copolymers and blends on poly(lactide) - in vitro degradation testing. Geneva, Switzerland: 2017.Search in Google Scholar
(28) ISO 10993-5. Biological evaluation of medical devices part 5: tests for in vitro cytotoxicity. Geneva, Switzerland: 2009.Search in Google Scholar
(29) Ren D-w, Wang Y-l, Bao D-c, Xie W-y, Ma X-j. FTIR analysis of the enzymatic degradation of chitosan blend membranes. Spectrosc Spectr Anal. 2006;26(10):1825–8.Search in Google Scholar
(30) Kaczmarek H, Zawadzki J. Chitosan pyrolysis and adsorption properties of chitosan and its carbonizate. Carbohydr Res. 2010;345(7):941–7. 10.1016/j.carres.2010.02.024.Search in Google Scholar PubMed
(31) Ibrahim M, Osman O, Mahmoud AA. Spectroscopic analyses of cellulose and chitosan: FTIR and modeling approach. J Comput Theor Nanosci. 2011;8:117–23. 10.1166/jctn.2011.1668.Search in Google Scholar
(32) Boonsongrit Y, Mueller BW, Mitrevej A. Characterization of drug-chitosan interaction by 1h nmr, ftir and isothermal titration calorimetry. Eur J Pharm Biopharm. 2008;69:388–95. 10.1016/j.ejpb.2007.11.008.Search in Google Scholar PubMed
(33) Chen R, Huang X, Wang M, Jin G. Evaluation and comparison of mid-infrared, raman and near-infrared spectroscopies for characterization and determination of the compositions in fully biodegradable poly(Lactic Acid)/poly(Propylene Carbonate)/poly(Butylene Adipate-co-Terephthalate) blends. J Macromol Sci Part A Pure Appl Chem. 2016;53(6):354–61. 10.1080/10601325.2016.1166001.Search in Google Scholar
(34) Cai Y, Lv J, Feng J, Liu Y, Wang Z, Zhao M, et al. Discrimination of poly(Butylenes Adipate-co-Terephthalate) and poly(Ethylene Terephthalate) with fourier transform infrared microscope and raman spectroscope. Spectrosc Lett. 2012;45(4):280–4. 10.1080/00387010.2011.610420.Search in Google Scholar
(35) Nobrega MM, Olivato JB, Müller CMO, Yamashita F. Biodegradable starch-based films containing saturated fatty acids: thermal, infrared and raman spectroscopic characterization. Polimeros. 2012;22(5):475–80. 10.1590/S0104-14282012005000068.Search in Google Scholar
(36) DB46/T519-2020 Biodegradable Plastics Products - Rapid Test Method Using Infrared/Raman Fingerprint Spectra; Hainan Market Supervision Administration: Hainan, China, 2020.Search in Google Scholar
(37) Du L, Yang X, Li W, Luo X, Wu H, Zhang J, et al. Construction of physical crosslink-based chitosan/liquid crystal composite hydrogel and evaluation on their cytocompatibility regen. Biomater. 2017;4(1):39–45. 10.1093/rb/rbw035.Search in Google Scholar PubMed PubMed Central
(38) Taurino R, Sciancalepore C, Collini L, Bondi M, Bondioli F. Functionalization of PVC by chitosan addition: compound stability and tensile properties. Compos Part B Eng. 2018;149:240–7. 10.1016/j.compositesb.2018.05.021.Search in Google Scholar
(39) Kim HS, Kumbar SG, Nukavarapu SP. Biomaterial-directed cell behavior for tissue engineering. Curr Opin Biomed Eng. 2021;17:100260. 10.1016/j.cobme.2020.100260.Search in Google Scholar PubMed PubMed Central
(40) Arul Jeya Kumar A, Srinivasan V. Thermal characteristics of chitosan dispersed poly lactic acid/basalt hybrid composites. Mater Express. 2016;6(4):337–43. 10.1166/mex.2016.1310.Search in Google Scholar
(41) Hongqin Yan, Quan Feng, Xiang Peng, Thermal XieFei. Stability and combustion behavior of chitosan fiber. J Text Research. 2015;36(10):12–6.Search in Google Scholar
(42) Cosate de Andrade MF, Strauss M, Morales AR. Toward greener polymeric blends: study of PBAT/thermoplastic whey protein isolate/beeswax blends. J Polym Env. 2019;27(10):2131–43. 10.1007/s10924-019-01502-2.Search in Google Scholar
(43) de Souza AG, Komatsu LGH, Barbosa RFS, Parra DF, Rosa DS. The effect of ZnO nanoparticles as Ag-carrier in PBAT for antimicrobial films. Polym Bull. 2022;79(6):4031–48. 10.1007/s00289-021-03681-2.Search in Google Scholar
(44) Lu W, Ouyang W, Wang S, Liu Y, Zhang F, Wang W, et al. A novel totally biodegradable device for effective atrial septal defect closure: A 2-year study in sheep. J Interv Cardiol. 2018;31(6):841–8. 10.1111/joic.12550.Search in Google Scholar PubMed
(45) Guidance Document on Using In Vitro Data to Estimate In Vivo Starting Doses for Acute Toxicity. National Institutes of Enviromental Health Sciences: U.S., 2001.Search in Google Scholar
(46) Kim SJ, Yang DH, Chun HJ, Chae GT, Jang JW, Shim YB. Evaluations of chitosan/poly(D,L-Lactic-co-Glycolic Acid) composite fibrous scaffold for tissue engineering applications. Macromol Res. 2013;21(8):931–9. 10.1007/s13233-013-1110-x.Search in Google Scholar
(47) Domingues RMA, Chiera S, Gershovich P, Motta A, Reis RL, Gomes ME. Enhancing the biomechanical performance of anisotropic nanofibrous scaffolds in tendon tissue engineering: reinforcement with cellulose nanocrystals. Adv Healthc Mater. 2016;5:1364–75. 10.1002/adhm.201501048.Search in Google Scholar PubMed
(48) Carvalho DN, López-Cebral R, Sousa RO, Alves AL, Reys LL, Silva SS, et al. marine collagen-chitosan-fucoidan cryogels as cell-laden biocomposites envisaging tissue engineering. Biomed Mater. 2020;15(5):055030. 10.1088/1748-605X/ab9f04.Search in Google Scholar PubMed
(49) Ahlawat J, Deemer EM, Narayan M. Chitosan nanoparticles rescue rotenone-mediated cell death. Mater (Basel). 2019;12(7):1176. 10.3390/ma12071176.Search in Google Scholar PubMed PubMed Central
© 2022 Shanshan Wang and Quansheng Xing, published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- The effect of isothermal crystallization on mechanical properties of poly(ethylene 2,5-furandicarboxylate)
- The effect of different structural designs on impact resistance to carbon fiber foam sandwich structures
- Hyper-crosslinked polymers with controlled multiscale porosity for effective removal of benzene from cigarette smoke
- The HDPE composites reinforced with waste hybrid PET/cotton fibers modified with the synthesized modifier
- Effect of polyurethane/polyvinyl alcohol coating on mechanical properties of polyester harness cord
- Fabrication of flexible conductive silk fibroin/polythiophene membrane and its properties
- Development, characterization, and in vitro evaluation of adhesive fibrous mat for mucosal propranolol delivery
- Fused deposition modeling of polypropylene-aluminium silicate dihydrate microcomposites
- Preparation of highly water-resistant wood adhesives using ECH as a crosslinking agent
- Chitosan-based antioxidant films incorporated with root extract of Aralia continentalis Kitagawa for active food packaging applications
- Molecular dynamics simulation of nonisothermal crystallization of a single polyethylene chain and short polyethylene chains based on OPLS force field
- Synthesis and properties of polyurethane acrylate oligomer based on polycaprolactone diol
- Preparation and electroactuation of water-based polyurethane-based polyaniline conductive composites
- Rapeseed oil gallate-amide-urethane coating material: Synthesis and evaluation of coating properties
- Synthesis and properties of tetrazole-containing polyelectrolytes based on chitosan, starch, and arabinogalactan
- Preparation and properties of natural rubber composite with CoFe2O4-immobilized biomass carbon
- A lightweight polyurethane-carbon microsphere composite foam for electromagnetic shielding
- Effects of chitosan and Tween 80 addition on the properties of nanofiber mat through the electrospinning
- Effects of grafting and long-chain branching structures on rheological behavior, crystallization properties, foaming performance, and mechanical properties of polyamide 6
- Study on the interfacial interaction between ammonium perchlorate and hydroxyl-terminated polybutadiene in solid propellants by molecular dynamics simulation
- Study on the self-assembly of aromatic antimicrobial peptides based on different PAF26 peptide sequences
- Effects of high polyamic acid content and curing process on properties of epoxy resins
- Experiment and analysis of mechanical properties of carbon fiber composite laminates under impact compression
- A machine learning investigation of low-density polylactide batch foams
- A comparison study of hyaluronic acid hydrogel exquisite micropatterns with photolithography and light-cured inkjet printing methods
- Multifunctional nanoparticles for targeted delivery of apoptin plasmid in cancer treatment
- Thermal stability, mechanical, and optical properties of novel RTV silicone rubbers using octa(dimethylethoxysiloxy)-POSS as a cross-linker
- Preparation and applications of hydrophilic quaternary ammonium salt type polymeric antistatic agents
- Coefficient of thermal expansion and mechanical properties of modified fiber-reinforced boron phenolic composites
- Synergistic effects of PEG middle-blocks and talcum on crystallizability and thermomechanical properties of flexible PLLA-b-PEG-b-PLLA bioplastic
- A poly(amidoxime)-modified MOF macroporous membrane for high-efficient uranium extraction from seawater
- Simultaneously enhance the fire safety and mechanical properties of PLA by incorporating a cyclophosphazene-based flame retardant
- Fabrication of two multifunctional phosphorus–nitrogen flame retardants toward improving the fire safety of epoxy resin
- The role of natural rubber endogenous proteins in promoting the formation of vulcanization networks
- The impact of viscoelastic nanofluids on the oil droplet remobilization in porous media: An experimental approach
- A wood-mimetic porous MXene/gelatin hydrogel for electric field/sunlight bi-enhanced uranium adsorption
- Fabrication of functional polyester fibers by sputter deposition with stainless steel
- Facile synthesis of core–shell structured magnetic Fe3O4@SiO2@Au molecularly imprinted polymers for high effective extraction and determination of 4-methylmethcathinone in human urine samples
- Interfacial structure and properties of isotactic polybutene-1/polyethylene blends
- Toward long-live ceramic on ceramic hip joints: In vitro investigation of squeaking of coated hip joint with layer-by-layer reinforced PVA coatings
- Effect of post-compaction heating on characteristics of microcrystalline cellulose compacts
- Polyurethane-based retanning agents with antimicrobial properties
- Preparation of polyamide 12 powder for additive manufacturing applications via thermally induced phase separation
- Polyvinyl alcohol/gum Arabic hydrogel preparation and cytotoxicity for wound healing improvement
- Synthesis and properties of PI composite films using carbon quantum dots as fillers
- Effect of phenyltrimethoxysilane coupling agent (A153) on simultaneously improving mechanical, electrical, and processing properties of ultra-high-filled polypropylene composites
- High-temperature behavior of silicone rubber composite with boron oxide/calcium silicate
- Lipid nanodiscs of poly(styrene-alt-maleic acid) to enhance plant antioxidant extraction
- Study on composting and seawater degradation properties of diethylene glycol-modified poly(butylene succinate) copolyesters
- A ternary hybrid nucleating agent for isotropic polypropylene: Preparation, characterization, and application
- Facile synthesis of a triazine-based porous organic polymer containing thiophene units for effective loading and releasing of temozolomide
- Preparation and performance of retention and drainage aid made of cationic spherical polyelectrolyte brushes
- Preparation and properties of nano-TiO2-modified photosensitive materials for 3D printing
- Mechanical properties and thermal analysis of graphene nanoplatelets reinforced polyimine composites
- Preparation and in vitro biocompatibility of PBAT and chitosan composites for novel biodegradable cardiac occluders
- Fabrication of biodegradable nanofibers via melt extrusion of immiscible blends
- Epoxy/melamine polyphosphate modified silicon carbide composites: Thermal conductivity and flame retardancy analyses
- Effect of dispersibility of graphene nanoplatelets on the properties of natural rubber latex composites using sodium dodecyl sulfate
- Preparation of PEEK-NH2/graphene network structured nanocomposites with high electrical conductivity
- Preparation and evaluation of high-performance modified alkyd resins based on 1,3,5-tris-(2-hydroxyethyl)cyanuric acid and study of their anticorrosive properties for surface coating applications
- A novel defect generation model based on two-stage GAN
- Thermally conductive h-BN/EHTPB/epoxy composites with enhanced toughness for on-board traction transformers
- Conformations and dynamic behaviors of confined wormlike chains in a pressure-driven flow
- Mechanical properties of epoxy resin toughened with cornstarch
- Optoelectronic investigation and spectroscopic characteristics of polyamide-66 polymer
- Novel bridged polysilsesquioxane aerogels with great mechanical properties and hydrophobicity
- Zeolitic imidazolate frameworks dispersed in waterborne epoxy resin to improve the anticorrosion performance of the coatings
- Fabrication of silver ions aramid fibers and polyethylene composites with excellent antibacterial and mechanical properties
- Thermal stability and optical properties of radiation-induced grafting of methyl methacrylate onto low-density polyethylene in a solvent system containing pyridine
- Preparation and permeation recognition mechanism of Cr(vi) ion-imprinted composite membranes
- Oxidized hyaluronic acid/adipic acid dihydrazide hydrogel as cell microcarriers for tissue regeneration applications
- Study of the phase-transition behavior of (AB)3 type star polystyrene-block-poly(n-butylacrylate) copolymers by the combination of rheology and SAXS
- A new insight into the reaction mechanism in preparation of poly(phenylene sulfide)
- Modified kaolin hydrogel for Cu2+ adsorption
- Thyme/garlic essential oils loaded chitosan–alginate nanocomposite: Characterization and antibacterial activities
- Thermal and mechanical properties of poly(lactic acid)/poly(butylene adipate-co-terephthalate)/calcium carbonate composite with single continuous morphology
- Review Articles
- The use of chitosan as a skin-regeneration agent in burns injuries: A review
- State of the art of geopolymers: A review
- Mechanical, thermal, and tribological characterization of bio-polymeric composites: A comprehensive review
- The influence of ionic liquid pretreatment on the physicomechanical properties of polymer biocomposites: A mini-review
- Influence of filler material on properties of fiber-reinforced polymer composites: A review
- Rapid Communications
- Pressure-induced flow processing behind the superior mechanical properties and heat-resistance performance of poly(butylene succinate)
- RAFT polymerization-induced self-assembly of semifluorinated liquid-crystalline block copolymers
- RAFT polymerization-induced self-assembly of poly(ionic liquids) in ethanol
- Topical Issue: Recent advances in smart polymers and their composites: Fundamentals and applications (Guest Editors: Shaohua Jiang and Chunxin Ma)
- Fabrication of PANI-modified PVDF nanofibrous yarn for pH sensor
- Shape memory polymer/graphene nanocomposites: State-of-the-art
- Recent advances in dynamic covalent bond-based shape memory polymers
- Construction of esterase-responsive hyperbranched polyprodrug micelles and their antitumor activity in vitro
- Regenerable bacterial killing–releasing ultrathin smart hydrogel surfaces modified with zwitterionic polymer brushes
Articles in the same Issue
- Research Articles
- The effect of isothermal crystallization on mechanical properties of poly(ethylene 2,5-furandicarboxylate)
- The effect of different structural designs on impact resistance to carbon fiber foam sandwich structures
- Hyper-crosslinked polymers with controlled multiscale porosity for effective removal of benzene from cigarette smoke
- The HDPE composites reinforced with waste hybrid PET/cotton fibers modified with the synthesized modifier
- Effect of polyurethane/polyvinyl alcohol coating on mechanical properties of polyester harness cord
- Fabrication of flexible conductive silk fibroin/polythiophene membrane and its properties
- Development, characterization, and in vitro evaluation of adhesive fibrous mat for mucosal propranolol delivery
- Fused deposition modeling of polypropylene-aluminium silicate dihydrate microcomposites
- Preparation of highly water-resistant wood adhesives using ECH as a crosslinking agent
- Chitosan-based antioxidant films incorporated with root extract of Aralia continentalis Kitagawa for active food packaging applications
- Molecular dynamics simulation of nonisothermal crystallization of a single polyethylene chain and short polyethylene chains based on OPLS force field
- Synthesis and properties of polyurethane acrylate oligomer based on polycaprolactone diol
- Preparation and electroactuation of water-based polyurethane-based polyaniline conductive composites
- Rapeseed oil gallate-amide-urethane coating material: Synthesis and evaluation of coating properties
- Synthesis and properties of tetrazole-containing polyelectrolytes based on chitosan, starch, and arabinogalactan
- Preparation and properties of natural rubber composite with CoFe2O4-immobilized biomass carbon
- A lightweight polyurethane-carbon microsphere composite foam for electromagnetic shielding
- Effects of chitosan and Tween 80 addition on the properties of nanofiber mat through the electrospinning
- Effects of grafting and long-chain branching structures on rheological behavior, crystallization properties, foaming performance, and mechanical properties of polyamide 6
- Study on the interfacial interaction between ammonium perchlorate and hydroxyl-terminated polybutadiene in solid propellants by molecular dynamics simulation
- Study on the self-assembly of aromatic antimicrobial peptides based on different PAF26 peptide sequences
- Effects of high polyamic acid content and curing process on properties of epoxy resins
- Experiment and analysis of mechanical properties of carbon fiber composite laminates under impact compression
- A machine learning investigation of low-density polylactide batch foams
- A comparison study of hyaluronic acid hydrogel exquisite micropatterns with photolithography and light-cured inkjet printing methods
- Multifunctional nanoparticles for targeted delivery of apoptin plasmid in cancer treatment
- Thermal stability, mechanical, and optical properties of novel RTV silicone rubbers using octa(dimethylethoxysiloxy)-POSS as a cross-linker
- Preparation and applications of hydrophilic quaternary ammonium salt type polymeric antistatic agents
- Coefficient of thermal expansion and mechanical properties of modified fiber-reinforced boron phenolic composites
- Synergistic effects of PEG middle-blocks and talcum on crystallizability and thermomechanical properties of flexible PLLA-b-PEG-b-PLLA bioplastic
- A poly(amidoxime)-modified MOF macroporous membrane for high-efficient uranium extraction from seawater
- Simultaneously enhance the fire safety and mechanical properties of PLA by incorporating a cyclophosphazene-based flame retardant
- Fabrication of two multifunctional phosphorus–nitrogen flame retardants toward improving the fire safety of epoxy resin
- The role of natural rubber endogenous proteins in promoting the formation of vulcanization networks
- The impact of viscoelastic nanofluids on the oil droplet remobilization in porous media: An experimental approach
- A wood-mimetic porous MXene/gelatin hydrogel for electric field/sunlight bi-enhanced uranium adsorption
- Fabrication of functional polyester fibers by sputter deposition with stainless steel
- Facile synthesis of core–shell structured magnetic Fe3O4@SiO2@Au molecularly imprinted polymers for high effective extraction and determination of 4-methylmethcathinone in human urine samples
- Interfacial structure and properties of isotactic polybutene-1/polyethylene blends
- Toward long-live ceramic on ceramic hip joints: In vitro investigation of squeaking of coated hip joint with layer-by-layer reinforced PVA coatings
- Effect of post-compaction heating on characteristics of microcrystalline cellulose compacts
- Polyurethane-based retanning agents with antimicrobial properties
- Preparation of polyamide 12 powder for additive manufacturing applications via thermally induced phase separation
- Polyvinyl alcohol/gum Arabic hydrogel preparation and cytotoxicity for wound healing improvement
- Synthesis and properties of PI composite films using carbon quantum dots as fillers
- Effect of phenyltrimethoxysilane coupling agent (A153) on simultaneously improving mechanical, electrical, and processing properties of ultra-high-filled polypropylene composites
- High-temperature behavior of silicone rubber composite with boron oxide/calcium silicate
- Lipid nanodiscs of poly(styrene-alt-maleic acid) to enhance plant antioxidant extraction
- Study on composting and seawater degradation properties of diethylene glycol-modified poly(butylene succinate) copolyesters
- A ternary hybrid nucleating agent for isotropic polypropylene: Preparation, characterization, and application
- Facile synthesis of a triazine-based porous organic polymer containing thiophene units for effective loading and releasing of temozolomide
- Preparation and performance of retention and drainage aid made of cationic spherical polyelectrolyte brushes
- Preparation and properties of nano-TiO2-modified photosensitive materials for 3D printing
- Mechanical properties and thermal analysis of graphene nanoplatelets reinforced polyimine composites
- Preparation and in vitro biocompatibility of PBAT and chitosan composites for novel biodegradable cardiac occluders
- Fabrication of biodegradable nanofibers via melt extrusion of immiscible blends
- Epoxy/melamine polyphosphate modified silicon carbide composites: Thermal conductivity and flame retardancy analyses
- Effect of dispersibility of graphene nanoplatelets on the properties of natural rubber latex composites using sodium dodecyl sulfate
- Preparation of PEEK-NH2/graphene network structured nanocomposites with high electrical conductivity
- Preparation and evaluation of high-performance modified alkyd resins based on 1,3,5-tris-(2-hydroxyethyl)cyanuric acid and study of their anticorrosive properties for surface coating applications
- A novel defect generation model based on two-stage GAN
- Thermally conductive h-BN/EHTPB/epoxy composites with enhanced toughness for on-board traction transformers
- Conformations and dynamic behaviors of confined wormlike chains in a pressure-driven flow
- Mechanical properties of epoxy resin toughened with cornstarch
- Optoelectronic investigation and spectroscopic characteristics of polyamide-66 polymer
- Novel bridged polysilsesquioxane aerogels with great mechanical properties and hydrophobicity
- Zeolitic imidazolate frameworks dispersed in waterborne epoxy resin to improve the anticorrosion performance of the coatings
- Fabrication of silver ions aramid fibers and polyethylene composites with excellent antibacterial and mechanical properties
- Thermal stability and optical properties of radiation-induced grafting of methyl methacrylate onto low-density polyethylene in a solvent system containing pyridine
- Preparation and permeation recognition mechanism of Cr(vi) ion-imprinted composite membranes
- Oxidized hyaluronic acid/adipic acid dihydrazide hydrogel as cell microcarriers for tissue regeneration applications
- Study of the phase-transition behavior of (AB)3 type star polystyrene-block-poly(n-butylacrylate) copolymers by the combination of rheology and SAXS
- A new insight into the reaction mechanism in preparation of poly(phenylene sulfide)
- Modified kaolin hydrogel for Cu2+ adsorption
- Thyme/garlic essential oils loaded chitosan–alginate nanocomposite: Characterization and antibacterial activities
- Thermal and mechanical properties of poly(lactic acid)/poly(butylene adipate-co-terephthalate)/calcium carbonate composite with single continuous morphology
- Review Articles
- The use of chitosan as a skin-regeneration agent in burns injuries: A review
- State of the art of geopolymers: A review
- Mechanical, thermal, and tribological characterization of bio-polymeric composites: A comprehensive review
- The influence of ionic liquid pretreatment on the physicomechanical properties of polymer biocomposites: A mini-review
- Influence of filler material on properties of fiber-reinforced polymer composites: A review
- Rapid Communications
- Pressure-induced flow processing behind the superior mechanical properties and heat-resistance performance of poly(butylene succinate)
- RAFT polymerization-induced self-assembly of semifluorinated liquid-crystalline block copolymers
- RAFT polymerization-induced self-assembly of poly(ionic liquids) in ethanol
- Topical Issue: Recent advances in smart polymers and their composites: Fundamentals and applications (Guest Editors: Shaohua Jiang and Chunxin Ma)
- Fabrication of PANI-modified PVDF nanofibrous yarn for pH sensor
- Shape memory polymer/graphene nanocomposites: State-of-the-art
- Recent advances in dynamic covalent bond-based shape memory polymers
- Construction of esterase-responsive hyperbranched polyprodrug micelles and their antitumor activity in vitro
- Regenerable bacterial killing–releasing ultrathin smart hydrogel surfaces modified with zwitterionic polymer brushes