T cell epitopes of SARS-CoV-2 spike protein and conserved surface protein of Plasmodium malariae share sequence homology
-


Abstract
Since its emergence in late 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been spreading remarkably fast worldwide. Effective countermeasures require the rapid development of data and tools to monitor its spread and better understand immunogenic profile. However, limited information is available about the tools and target of the immune responses to SARS-CoV-2. In this study, we excogitated a new approach for analyzing phylogenetic relationships by using the whole prototype proteome sequences. Phylogenetic analysis on the whole prototype proteome sequences showed that SARS-CoV-2 was a direct descendant of Bat-CoV and was closely related to Pangolin-CoV, Bat-SL-CoV, and SARS-CoV. The pairwise comparison of SARS-CoV-2 with Bat-CoV showed an unusual replacement of the motif consisting of seven amino acids (NNLDSKV) within the spike protein of SARS-CoV-2. The replaced motif in the spike protein of SARS-CoV-2 was found in 12 other species, including a conserved surface protein of a malaria-causing pathogen, Plasmodium malariae. We further identified the T and B cell epitope sequence homology of SARS-CoV-2 spike protein with conserved surface protein of P. malariae using the Immune Epitope Database and Analysis Resource (IEDB). The shared immunodominant epitopes may provide immunity against SARS-CoV-2 infection to those previously infected with P. malariae.
1 Introduction
Coronaviruses (CoVs) are the predominant cause of the common cold widely present in nature with a broad spectrum of hosts. Although the viruses frequently infect humans, the natural host of CoVs are animals, and thereby all of the CoVs for the common colds, HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1, have zoonotic origins [1]. The genomes of CoVs consist of a single-stranded, positive RNA of 26,000–32,000 base pairs and a variable number (from 6 to 11) of open reading frames [2]. Because of their considerable size with the characteristics of the RNA genomes, CoVs can frequently mutate to escape their natural hosts, causing severe diseases in humans. Outbreaks of SARS-CoV in 2003 and MERS-CoV in 2012 are well-known examples. Currently, another severe pathogenic novel coronavirus, SARS-CoV-2, has emerged and caused a global pandemic [3,4].
CoVs are RNA viruses with a positive-sense single-stranded genome, and their RNA genomes are vulnerable to natural mutations like other RNA viruses, which cause significant genetic diversity. A high degree of genetic diversity in CoVs makes it challenging to find the phylogenetic relationship of CoVs. Understanding the phylogenetic relationship of SARS-CoV-2 with other CoVs is essential for identifying its host to prevent the next outbreak. Because of the ambiguous results by genome comparison approaches, an alignment-free method called natural vector was adopted to investigate the phylogeny of SARS-CoV-2 [5]. However, the frequency of the original sequence always predominates the mutated sequence if the genomes of individuals were compared within the same species. Therefore, it is possible to construct a prototype proteome of a species by identifying a prevalent amino acid (aa) at each position of proteome after multiple alignments of individual proteomes for the analysis of the phylogenetic relationships.
CoVs contain four different structural proteins, including spike (S), envelop (E), membrane (M), and nucleocapsid (N) proteins, and S protein plays the most critical roles in viral attachment and entry [6]. The S protein first binds to the host receptor through the receptor-binding domain (RBD) in the S1 subunit and then makes entry by fusing the viral and host membranes through the S2 subunit [7,8]. Thus, the S protein of SARS-CoV-2 becomes an attractive target for the development of virus attachment inhibitors, neutralizing antibodies, and vaccines [9,10,11]. However, there is limited information available on which parts of the SARS-CoV-2 are recognized by human immune responses. Thus, the knowledge of the potential immunogenic profile of SARS-CoV-2 is of immediate relevance and would assist in vaccine design and facilitate the evaluation of vaccine candidate immunogenicity, monitoring of mutational events, and the epitope escape during transmission.
In this study, we constructed a prototype proteome sequence of SARS-CoV-2 and its related CoVs species to understand the biological characteristics and phylogenetic relationship. The phylogeny of SARS-CoV-2 and Bat-CoV prompted us to explore the distribution of unique NNLDSKV peptides on different species, including the conserved surface protein of P. malariae. Here, we used IEDB resources to find out the T and B cell epitopes of the conserved surface protein of P. malariae, which has NNLDSKV sequence homology with SARS-CoV-2 spike glycoprotein. Furthermore, we analyzed the possible shared immunogenic regions of the conserved surface protein of P. malariae and SARS-CoV-2 spike glycoprotein using immunoinformatic approaches.
2 Methods
2.1 Genome datasets
The SARS-CoV-2, Bat-CoV, Pan-CoV, Bat-SL-CoV, and SARS-CoV genome sequences were obtained from GenBank using Blastn and the GISAID (https://www.gisaid.org) databases, with data kindly deposited by the submitters (Supplementary Table S1).
2.2 Prediction of protein-coding genes in genome sequences
The protein-encoding genes of the genome sequences of each viral species were predicted by the online servers of GeneMarkS (http://exon.gatech.edu/GeneMark/genemarks.cgi) and ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) with a manual check.
2.3 In silico translation of protein-coding genes
The protein-coding genes of the genome sequences of each viral species were translated using the ExPASy protein translation tool (https://web.expasy.org/translate/).
2.4 Determination of prototype proteome sequences
To generate prototype sequences, the different proteome sequences of each viral species obtained from their genome sequences (Supplementary Table S1) were aligned using the FFT-NS-2 algorithm in MAFFT (version v7) [12]. Each amino acid position was investigated with the manual check from the corresponding multiple sequence alignment dataset, and the most prevalent amino acids were chosen for the prototype sequence. The prototype aa sequences of open reading frame ORF1a, ORF1ab, spike (S), 3a, 3b, envelope (E), membrane (M), 6, 7a, 7b, 8, Nucleocapsid (N), 9b, and 14 for all coronavirus type were determined by the following method.
Let S = (s
1, s
2, …, s
i
, …, s
n
) be an aa sequence of length n, where s
i
∈ {0, A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}. Let s[k][i] be the location of the ith occurrence of aa k. PS = (ps
1, ps
2, …, ps
i
, …, ps
n
) is a prototype aa sequence with a specific aa ps at the ith occurrence was determined by selecting k with the largest n. nk
i
2.5 Sequence similarity search of NNLDSKV motif
The spike protein of SARS-CoV-2 was overlappingly defragmented in 9 aa sequence unit. The defragmented aa sequences were used to identify the matched sequences in GenBank using Protein Blastp tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
2.6 Phylogenetic analysis, 3D homology modeling, and annotation
All ORF of SARS-CoV-2 were aligned against Bat-CoV, Pan-CoV, Bat-SL-CoV, and SARS-CoV using the FFT-NS-2 algorithm in MAFFT (version v7) [12]. Maximum likelihood phylogenies were estimated using Unipro UGENE bioinformatics toolkits [13]. To generate the 3D model, the query amino acid sequences were run in the SWISS-MODEL protein homology-modeling server to produce several best-fit homo-trimeric or monomeric protein models based on multiple template alignment [14]. The homotrimer 3D model of Bat-CoV and SARS-CoV-2 spike protein with the highest sequence identity coverage was used in this study. Furthermore, the 3D model of the NNLDSKV motif in the SARS-CoV-2 spike and P. malariae was built by the SWISS-MODEL protein homology-modeling server. The PDB sequence file of the NNLDSKV motif 3D model was then processed in UCSF CHIMERA to generate the surface structure [15].
2.7 Prediction of immunodominant epitopes of SARS-CoV-2
The T and B cell epitopes of SARS-CoV-2 from immunodominant regions were determined based on sequence-shared identities with the closely related SARS-CoV using parallel bioinformatics approaches by Grifoni et al. [16]. In this study, epitope sequences of SARS-CoV-2 spike glycoprotein were retrieved and matched with our prototype SARS-CoV-2 spike glycoprotein sequence and used to identify common immunodominant epitopes of P. malariae conserved surface protein (XP_028861348.1).
2.8 Prediction of T and B cell epitopes of the conserved surface protein of P. malariae and homology analysis with SARS-CoV-2 spike glycoprotein
The T and B cell epitopes for P. malariae conserved surface protein were determined by searching the Immune Epitope Database and Analysis Recourse (IEDB, http://www.iedb.org/) in the middle of April 2021. The prediction of B cell epitopes of P. malariae conserved surface protein was carried out using Bepipred linear epitope prediction algorithm by setting the threshold at 0.55 embedded in the B cell prediction analysis tools available in IEDB [17]. The CD4 T cell epitopes of P. malariae conserved surface protein were analyzed using the combined method by setting the threshold at 90 embedded in T cell epitope prediction tools available in IEDB [18]. The predictions of peptide binding of P. malariae conserved surface protein immunodominant T cell epitopes to MHC class I molecules were calculated by MHC-I binding predictions tool using IEDB recommended 2020.09 (NetMHCpan EL 4.1) method with selected HLA allele reference set, and outputs with lowest percentile rank were chosen from the default results (low percentile rank = good binders) [19]. The maps of the T cell epitopes identified from P. malariae conserved surface protein were analyzed based on selected sequence identity to the given parent antigen (P. malariae conserved surface protein) and nonparental sequence (SARS-CoV-2 spike glycoprotein) using ImmunomeBrowser tool available in IEDB [20]. The sequence seminaries within the T cell epitopes of SARS-CoV-2 and P. malariae conserved surface protein were analyzed using the Epitope cluster analysis tool using the threshold of 70 available in IEDB analysis resource [21]. The degree of conservancy between T cell epitopes (peptide core sequence) of P. malariae conserved surface protein and T cell epitopes of SARS-CoV-2 were calculated by the epitope conservancy analysis tool, and data were represented as identity of percentage (%) [22]. Four or five amino acid shared residues were considered significant [23].
3 Results
3.1 Comparison of the whole prototype proteomes of coronaviruses to SARS-CoV-2 revealed the phylogeny without showing ambiguity
Identifying an animal vector transmitting an infectious disease to humans could be significant as much as understanding at the person-to-person transmission stage to control the disease, meaning that it is vital to identify the correct host. To overcome the current ambiguity of various genome analysis approaches, we excogitated a completely new approach comparing whole prototype proteomes of species to investigate the phylogeny of SARS-CoV-2. To construct the whole prototype proteomes of viral species related to SARS-CoV-2, we first collected all the individual genome sequences of SARS-CoV-2 as well as its related viral species by the BLAST sequence similarity search from all of the publicly available genome databases (Supplementary Table S1). The genome sequences of the individual viruses were converted into protein sequences. After in silico translation of the individual viruses, the proteomes of individual viruses of the same viral species were aligned through multiple sequence alignments (MSAs) followed by identifying the most prevalent aa in each position within the same species to determine the prototype proteome sequences for each viral species.
The prototype aa sequences of the proteomes of SARS-CoV-2 and its related species, Bat-CoV, Pan-CoV, Bat-SL-CoV, and SARS-CoV, are represented in Supplementary Figure S1 and Supplementary Table S2. As shown in Figure 1, the prototype proteome of SARS-CoV-2 has consisted of 14 proteins, similar to other beta-coronaviruses. The 5′-terminal two-thirds of the genome encodes replicase polyprotein 1ab (pp1ab) with a length of 7,096 aa and contains 15 predicted nonstructural proteins (Supplementary Table S3). The 3′ terminus encodes four structural proteins and other nonstructural proteins, including spike glycoprotein (S), ORF3a, ORF3b, envelope small membrane protein (E), membrane protein (M), ORF6, ORF7a, ORF7b, ORF8, ORF9b, nucleocapsid protein (N), and ORF14 in order.

Sequence alignment and genome organization of Bat-CoV, SARS-CoV-2, Pan-CoV, Bat-SL-CoV, and SARS-CoVs. The gene ORF1ab encodes the pp1ab protein that contains 15 predicted nonstructural proteins (nsps). The structural proteins are encoded by Spike (S), Envelope (E), and Nucleocapsid (N) genes. The protein-encoding genes of CoVs genome were predicted by GeneMarks and ORFfinder online server with a manual check.
The phylogenetic relationship of the total aa sequences of the whole prototype proteomes of the five viral species was analyzed by the neighbor-joining method using Unipro UGENE bioinformatics toolkits [13]. As shown in Figure 2a, SARS-CoV-2 is a direct descendant of Bat-CoV and did not originate from Pan-CoV, Bat-SL-CoV, and SARS-CoV. That suggests that pangolin may act as an intermediate host [24]. The sequence alignment of the prototype amino acid sequences by using MSA with MAFFT program [12] showed that the aa sequence of SARS-CoV-2 shared 98.67% sequence similarity with Bat-CoV, while 94.51, 94.35, and 82.93% of the sequences of SARS-CoV-2 were identical with that of Pan-CoV, Bat-SL-CoV, and SARS-CoV, respectively (Supplementary Table S4). The aa sequence similarities were well matched with their phylogenetic distances.

Comparison of SARS-CoV-2 and Bat-CoVs. (a) The phylogeny of SARS-CoV-2 based on the 14 ORF and receptor-binding domain (RBD) sequences of Bat-CoV, Pan-CoV, Bat-SL-CoV, and SARS-CoV. Phylogenies were estimated by the neighbor-joining method using Unipro UGENE bioinformatics toolkits. (b) Organization of genes in SARS-CoV-2 and Bat-CoVs. The distribution of mutated amino acids in SARS-CoV-2 compare to Bat-CoV are represented with red lines. (c) Homotrimer 3D model of Bat-CoV and SARS-CoV-2 spike protein constructed using the SWISS-MODEL protein homology-modeling server. Circle showing the altered surface structure in the spike region of SARS-CoV-2 for antigen binding.
3.2 Pairwise analysis of SARS-CoV-2 and Bat-CoV prototype proteome sequences identified a unique peptide (NNLDSKV) in SARS-CoV-2 spike glycoprotein
To investigate the sequence dissimilarities between SARS-CoV-2 and Bat-CoV, we performed pairwise sequence analysis between the prototype aa sequences of SARS-CoV-2 and Bat-CoV. The pairwise aa sequence analysis showed that both virus genomes encode 14 genes (Figure 2b). The aa sequence mismatches were randomly distributed except for the consecutive 7-aa alteration (439NNLDSKV445) in the spike protein. Since genetic mutation is a random process, the mutated points are expected to be distributed randomly throughout the genome. Considering the general nature of mutations, the consecutive 7-aa sequence difference in the spike protein was peculiar enough for us to speculate that an unusual event happened during the emergence of SARS-CoV-2 from Bat-CoV. 3D models of the spike proteins were made to analyze the consequence of the alteration to the SARS-CoV-2 spike protein structure. The spike protein of CoV is the surface protein that binds to a receptor on the host cell surface. The spike protein consists of three large domains: a large ectodomain, a single-pass transmembrane anchor, and a short intracellular tail [25]. The ectodomain is further divided into a receptor-binding subunit S1 and a membrane-fusion subunit S2. Coronavirus first binds to a receptor on the host cell surface through its S1 subunit and then fuses the viral and the host membranes through its S2 subunit, meaning that the S1 domain plays the most critical role in the virus’s invasion into its host [26,27]. As shown in Figure 2c, altering the consecutive 7-aa on the outer layer of the spike protein’s S1 domain did not affect the overall structural integrity of the spike protein. The 3D modeling also showed that the consecutive 7-aa alteration resulted in a new motif occupying more space in the S1 domain than its original structure by transforming a partial alpha-helical structure to a random coil. Since the S1 domain acts as the binding domain for the entry of the virus, enlargement of its space and adaptation of different conformation in the outermost layer surface of the S1 domain would endow SARS-CoV-2 to expand its host range.
The unusual genetic alteration in the spike protein between SARS-CoV-2 and Bat-CoV motivated us to investigate the unique nature of the 7-aa motif (439NNLDSKV445). To trace down its distribution, we overlappingly defragmented the aa sequence of the spike protein of SARS-CoV-2 into 7 aa units and performed a peptide sequence search in the publicly available protein databases. All of the aa sequences matched with the spike proteins of coronaviruses except for the 7-aa motif. Interestingly, the 7-aa motif (439NNLDSKV445) of SARS-CoV-2 was found in 12 other species, including P. malariae, which causes malaria to humans (Table 1). As presented in Table 1, the 7-aa motif is primarily present in the surface proteins of simple organisms. The motif’s widespread in simple organisms as a surface protein suggests that the motif plays a significant role on the surface of the organisms. It is also worth noting that the NNLDSKV motif was located at aa 449–455 of a conserved membrane-bound surface protein of P. malariae (Figure 3).
Lists of proteins having the NNLDSKV motif in nature
| Organism name | Protein name | Position | |
|---|---|---|---|
| 1 | SARS-CoV-2 | Surface glycoprotein | 439–445 |
| 2 | Plasmodium malariae | Conserved surface protein | 449–455 |
| 3 | Capsella rubella | GRIP and coiled-coil domain-containing protein 2 | 1,284–1,290 |
| 4 | Pygocentrus nattereri | Sperm-associated antigen 5 isoform X1 | 478–484 |
| 5 | Lactobacillus gigeriorum | BspA family leucine-rich repeat surface protein | 1,700–1,706 |
| 6 | Desulfuromonas sp. SDB | Hypothetical protein APR63_07190 | 580–586 |
| 7 | Campylobacter concisus | Retention module-containing protein | 1,574–1,580 |
| 8 | Nitrospira sp. | Nonribosomal peptide synthetase | 658–664 |
| 9 | Smittium simulii | Hypothetical protein BB561_003344 | 1,059–1,065 |
| 10 | Caldisericales bacterium | S8 family serine peptidase | 510–516 |
| 11 | Mizuhopecten yessoensis | Transient receptor potential cation channel subfamily M member 3-like | 1,380–1,386 |
| 12 | Bacterium ADurb.Bin132 | Bacillopeptidase F precursor | 510–516 |
| 13 | Dictyostelium purpureum | Hypothetical protein DICPUDRAFT_46686 | 241–247 |

The SARS-CoV-2 spike glycoprotein showed NNLDSKV motif identity with conserved surface protein of P. malariae. The 3D model for the NNLDSKV motif of SARS-CoV-2 and P. malariae surface protein was built by the SWISS-MODEL protein homology-modeling server, and the PDB sequence was analyzed by UCSF CHIMERA software. The orientation of 7-amino acids in the NNLDSKV motif is shown in the box with different colors.
3.3 The predicted T cell epitopes of the conserved surface protein of P. malariae contain the unique “NNLDSKV” peptide
The homology and unique nature of the NNLDSKV motif of SARS-CoV-2 and P. malariae conserved surface protein let us to investigate the immunogenic nature of NNLDSKV in the conserved surface protein of P. malariae T and B cell epitopes. To determine potential T cell epitopes of P. malariae conserved surface protein, we used CD4 T cell epitope immunogenicity prediction tool using a combined method embedded in IEDB analysis resource. A total of 801 T cell immunodominant epitopes were identified by defining the threshold at 90%. The response frequency (RF) of T cell epitopes was shown by mapping the epitopes against the parental P. malariae conserved surface protein sequence (Figure 4a). Surprisingly, mapping of T cell epitopes of P. malariae conserved surface protein against SARS-CoV-2 spike glycoprotein sequence identified two T cell epitopes, 421LNDEQWNNLDSKVLN435 and 426WNNLDSKVLNYEQDN440, which showed 97.72 and 98.60% of CD4 T cell immunogenicity and contains peptide core “NNLDSKV” motif (Figure 4b).

T cell immunodominant regions based on the conserved surface protein of P. malariae. (a) Specific T cell epitope mapping response frequency score (RF) for each epitope position from P. malariae conserved surface protein. (b) Mapping of T cell epitopes of the conserved surface protein of P. malariae against SARS-CoV-2 spike glycoprotein. (c and d) Cluster analysis of epitopes of SARS-CoV-2 spike glycoprotein and surface protein of P. malariae for identification sequence homology. (d) Mapping of B cell epitopes from a conserved surface protein of P. malariae.
We separately performed the cluster analysis of T cell epitopes of SARS-CoV-2 and P. malariae conserved surface protein to investigate the sequence similarity within the population. The results showed that only two T cell epitopes of SARS-CoV-2 showed sequence similarity by making one cluster while 17 epitopes of P. malariae conserved surface protein divided into two different epitope clusters (Figure 4c). To define the B cell epitopes of P. malariae conserved surface protein, we used the B cell epitope prediction tools provided with IEDB. Using Bepipred method and a threshold of 0.55, the target protein had the highest 122 B cell episodes (Figure 4d). However, the B cell epitopes of P. malariae conserved surface protein did not show any identity for mapping against the SARS-CoV-2 spike glycoprotein sequence.
3.4 The T cell epitopes of SARS-CoV-2 spike glycoprotein showed significant sequence homology with T cell epitopes of the conserved surface protein of P. malariae
The SARS-CoV-2 T and B cell epitopes identified by Grifoni et al. [16] were used to investigate the presence of immunodominant epitopes in P. malariae conserved surface protein. Before homology analysis, all the T and B cell epitopes of SARS-CoV-2 spike glycoprotein from the previous study were aligned against our prototype SARS-CoV-2 spike glycoprotein sequence, and the aligned sequence showed 100% sequence identity after analysis. The tested (421–435), (426–440), and (841–855) T cell epitope residues of P. malariae conserved surface protein showed 44.44, 55.56, and 44.44% homology with (101–118), (304–321), and (440–457) T cell epitope residues of SARS-CoV-2 spike glycoprotein (Table 2). Due to the phylogenetic distance between these two organisms, four or five shared amino acids in a single immunodominant epitope would be considered significant [23]. Surprisingly, two T cell epitopes of SARS-CoV-2 spike glycoprotein (101IRGWIFGTTLDSKTQSLL118 and 440 NLDSKVGGNYNYLYRLFR457) shared sequence homology with P. malariae conserved surface protein T cell epitope by NNLDSKV motif. Conversely, all tested B cell epitopes share no significant homology with SARS-CoV-2 according to the reference sequences of the previous study [16]. In addition to sequence homology, we observed the binding affinity of three different T cell epitopes of P. malariae conserved surface protein to the 12 most frequent HLA class I alleles in the worldwide population and ranked based on the lowest percentile score, which has the higher binding ability (Table 2).
Experimental T cell immunodominant epitopes from a conserved surface protein of P. malariae sharing homology with T cell epitopes of SARS-CoV-2 spike protein
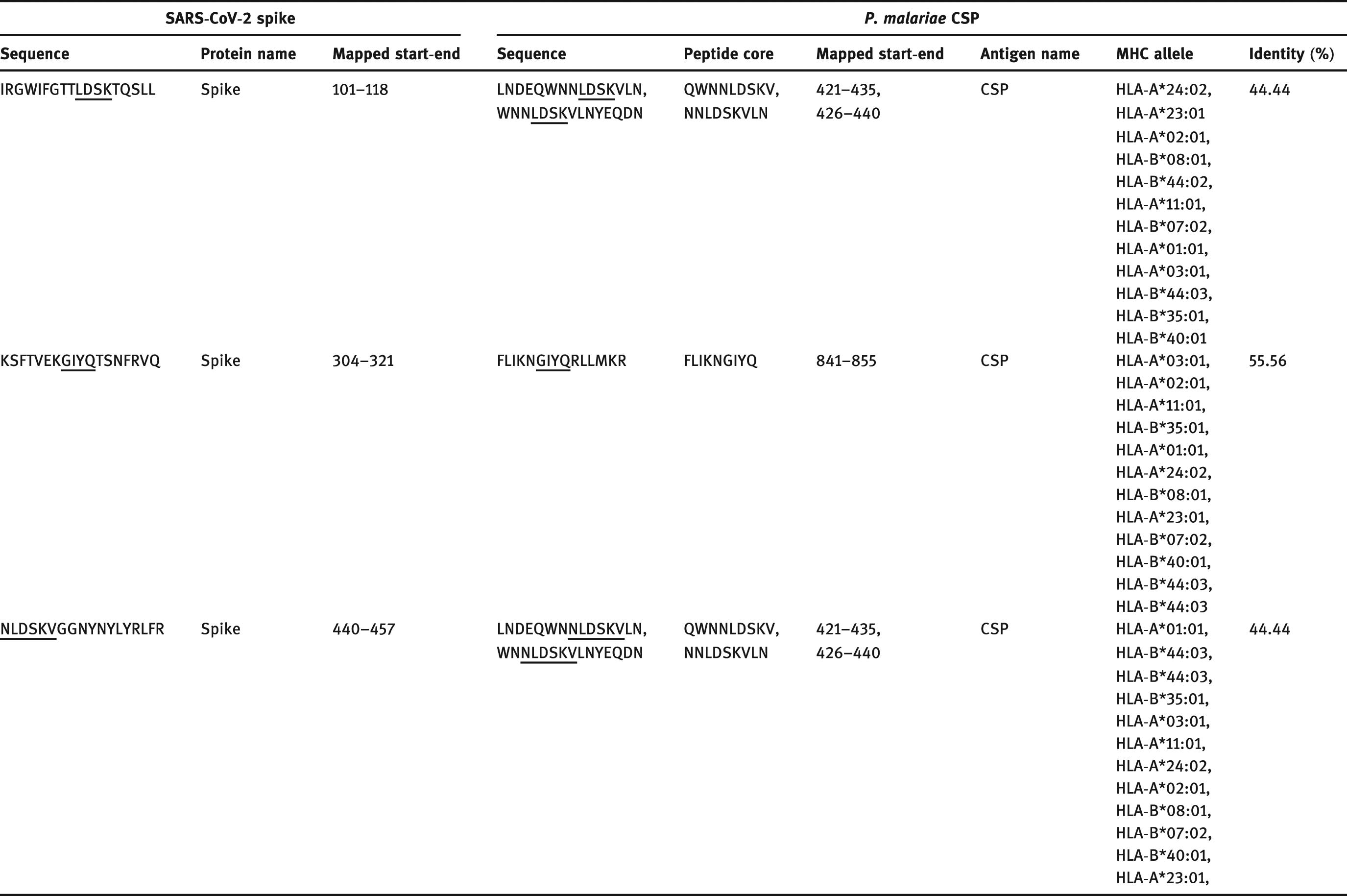 |
Note: CSP, conserved surface protein; MHC, major histocompatibility complex.
4 Discussion
RNA genomes, as in the case of CoVs, are prone to be naturally mutated and thus have a wide range of genetic variation [28,29,30]. Because of the genetic variation between individual strains, it is natural to find that phylogenetic analyses generate different results if different kinds of individual genome sequences were used, explaining current discordant conclusions on the origin of SARS-CoV-2. Since genetic mutation or recombination occurs on an individual basis and these events cannot occur at the same position repeatedly, the prototype sequence of the genome within a species is always the most prevalent base among the members. Also, the degeneracy of codons is present in nucleotide sequences but not in protein aa sequences. Since degenerate codons are not affected by natural selection, there are more genetic variations present in degenerate codons. Considering these natural variabilities of nucleotide sequences in RNA genomes, we believe that our approach of the whole prototype proteome analysis using aa sequence could generate a much more precise result than current approaches in phylogenetic analysis.
The clinical presentation of COVID-19 significantly differs from that of other coronaviruses [31]. The acquisition of the P. malariae gene in the spike protein seems to explain why. The malaria-causing Plasmodium species infect liver cells and erythrocytes to promote blood clotting and damages in the heart, liver, or kidney. It is fascinating to note that, unlike other coronaviral infections, symptoms similar to the case of the P. malariae infection were reported in COVID-19 [33,34,35,36,37]. Red cell distribution width (RDW) of erythrocytes has been reported to be enlarged after the malarial invasion because the growth of the malaria parasite causes the cells to be enlarged [32,33]. It was also observed in COVID-19 that elevated RDW is correlated with the increased mortality risk in COVID-19 [34]. The enlargement of erythrocytes in COVID-19 is not the only factor correlated with malaria infection. Clinical presentations such as blood clotting and damages in the heart, liver, or kidney also were observed in COVID-19 patients [35,36,37]. Considering that, the investigation of different kinds of antimalaria drugs for COVID-19 treatment needs to be pursued.
Several studies have reported the relationship of the ABO blood group system to susceptibility and resistance of excessive Plasmodium parasite invasion in severe malaria [38,39]. Individuals with A, B, or AB blood groups are more susceptible to the malarial parasite than O blood group. In agreement with our results, ABO blood group system is also associated with COVID-19, as in malaria. A recent meta-analysis showed that A, B, or AB blood groups are more susceptible to SARS-CoV-2 than O blood group [40,41]. Furthermore, comparative analysis between COVID-19 and malaria showed that malaria-free countries have much higher rates of infectivity and fatality to SARS-CoV-2 compared to malaria-endemic countries [42]. The association between malaria and COVID-19 has suggested that low COVID-19 cases in malaria-endemic countries could be due to the anti-malaria immunity, which provides heterogeneous protection against SARS-CoV-2 [40]. Despite the link between malaria and COVID-19, the reason has not been known. This work suggests that the NNLDSKV motif could be the missing link of malaria and COVID-19. The apparent immunodominant T cell epitope conservation between SARS-CoV-2 spike glycoprotein and conserved surface protein of P. malariae may provide immunity against SARS-CoV-2 infection to those previously infected with Plasmodium. With that in mind, the possible significance of said motif should be considered during the development of both COVID-19 and malaria vaccines.
According to the current COVID-19 outbreak statistics (https://www.worldometers.info/coronavirus/), a racial background affects the morbidity and mortality of COVID-19. It seems that the morbidity and mortality by COVID-19 are much more severe in the Western world than in Eastern countries. Contrary to the Western, multisystem inflammatory syndrome in children was never reported, and the morbidity and mortality by COVID-19 are much milder in Eastern countries. It would be interesting to investigate the role of the NNLDSKV motif among different racial backgrounds, including ACE-2, since the genetic polymorphism of ACE-2 is known to differ among different ethnic and racial groups [43].
-
Funding information: This work was supported by JINIS BDRD Research Institute, JINIS Biopharmaceuticals Inc. (Wanju, South Korea).
-
Conflict of interest: The authors state no conflict of interest.
-
Data availability statement: All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
[1] Ye ZW, Yuan S, Yuen KS, Fung SY, Chan CP, Jin DY. Zoonotic origins of human coronaviruses. Int J Biol Sci. 2020;16:1686–97.10.7150/ijbs.45472Search in Google Scholar
[2] Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, et al. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11:E59.10.3390/v11010059Search in Google Scholar
[3] da Costa VG, Moreli ML, Saivish MV. The emergence of SARS, MERS and novel SARS-2 coronaviruses in the 21st century. Arch Virol. 2020;165:1517–26.10.1007/s00705-020-04628-0Search in Google Scholar
[4] Morens DM, Daszak P, Taubenberger JK. Escaping Pandora’s box – another novel coronavirus. N Engl J Med. 2020;382:1293–5.10.1056/NEJMp2002106Search in Google Scholar
[5] Dong R, Pei S, Yin C, He RL, Yau SS. Analysis of the hosts and transmission paths of SARS-CoV-2 in the COVID-19 outbreak. Genes. 2020;11:E637.10.3390/genes11060637Search in Google Scholar
[6] Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17:613–20.10.1038/s41423-020-0400-4Search in Google Scholar
[7] Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–47.10.1016/S0140-6736(04)15788-7Search in Google Scholar
[8] Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–8.10.1126/science.1116480Search in Google Scholar PubMed
[9] Du L, Yang Y, Zhou Y, Lu L, Li F, Jiang S. MERS-CoV spike protein: a key target for antivirals. Expert Opin Ther Targets. 2017;21:131–43.10.1080/14728222.2017.1271415Search in Google Scholar PubMed PubMed Central
[10] Wang D, Mai J, Zhou W, Yu W, Zhan Y, Wang N, et al. Immunoinformatic analysis of T- and B-cell epitopes for SARS-CoV-2 vaccine design. Vaccines. 2020;8:355.10.3390/vaccines8030355Search in Google Scholar PubMed PubMed Central
[11] Du L, Tai W, Yang Y, Zhao G, Zhu Q, Sun S, et al. Introduction of neutralizing immunogenicity index to the rational design of MERS coronavirus subunit vaccines. Nat Commun. 2016;7:13473.10.1038/ncomms13473Search in Google Scholar PubMed PubMed Central
[12] Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66.10.1093/nar/gkf436Search in Google Scholar PubMed PubMed Central
[13] Okonechnikov K, Golosova O, Fursov M. The UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28:1166–7.10.1093/bioinformatics/bts091Search in Google Scholar PubMed
[14] Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5.10.1093/nar/gkg520Search in Google Scholar PubMed PubMed Central
[15] Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.10.1002/jcc.20084Search in Google Scholar PubMed
[16] Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A sequence Homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microb. 2020;27:671–80.e2.10.1016/j.chom.2020.03.002Search in Google Scholar PubMed PubMed Central
[17] Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2.10.1186/1745-7580-2-2Search in Google Scholar PubMed PubMed Central
[18] Paul S, Lindestam Arlehamn CS, Scriba TJ, Dillon MB, Oseroff C, Hinz D, et al. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 2015;422:28–34.10.1145/2649387.2660842Search in Google Scholar
[19] Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–54.10.1093/nar/gkaa379Search in Google Scholar PubMed PubMed Central
[20] Dhanda SK, Vita R, Ha B, Grifoni A, Peters B, Sette A. ImmunomeBrowser: a tool to aggregate and visualize complex and heterogeneous epitopes in reference proteins. Bioinformatics. 2018;34:3931–3.10.1093/bioinformatics/bty463Search in Google Scholar PubMed PubMed Central
[21] Dhanda SK, Vaughan K, Schulten V, Grifoni A, Weiskopf D, Sidney J, et al. Development of a novel clustering tool for linear peptide sequences. Immunology. 2018;155:331–45.10.1111/imm.12984Search in Google Scholar PubMed PubMed Central
[22] Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007;8:361.10.1186/1471-2105-8-361Search in Google Scholar PubMed PubMed Central
[23] Iesa MAM, Osman MEM, Hassan MA, Dirar AIA, Abuzeid N, Mancuso JJ, et al. SARS-CoV-2 and Plasmodium falciparum common immunodominant regions may explain low COVID-19 incidence in the malaria-endemic belt. New Microbes New Infect. 2020;38:100817.10.1016/j.nmni.2020.100817Search in Google Scholar PubMed PubMed Central
[24] Zhao J, Cui W, Tian BP. The Potential intermediate hosts for SARS-CoV-2. Front Microbiol. 2020;11:580137.10.3389/fmicb.2020.580137Search in Google Scholar PubMed PubMed Central
[25] Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–20.10.1038/s41586-020-2180-5Search in Google Scholar PubMed
[26] Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620.10.1038/s41467-020-15562-9Search in Google Scholar PubMed PubMed Central
[27] Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30:1578.10.1016/j.cub.2020.03.063Search in Google Scholar PubMed PubMed Central
[28] Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 1997;51:151–78.10.1146/annurev.micro.51.1.151Search in Google Scholar PubMed
[29] Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16:e3000003.10.1371/journal.pbio.3000003Search in Google Scholar PubMed PubMed Central
[30] Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18:179.10.1186/s12967-020-02344-6Search in Google Scholar PubMed PubMed Central
[31] Lescure FX, Bouadma L, Nguyen D, Parisey M, Wicky PH, Behillil S, et al. Clinical and virological data of the first cases of COVID-19 in Europe: a case series. The Lancet Infect Dis. 2020;20:697–706.10.1016/S1473-3099(20)30200-0Search in Google Scholar
[32] Jairajpuri ZS, Rana S, Hassan MJ, Nabi F, Jetley S. An analysis of hematological parameters as a diagnostic test for malaria in patients with acute febrile illness: an institutional experience. Oman Med J. 2014;29:12–7.10.5001/omj.2014.04Search in Google Scholar PubMed PubMed Central
[33] Koltas IS, Demirhindi H, Hazar S, Ozcan K. Supportive presumptive diagnosis of Plasmodium vivax malaria. Thrombocytopenia and red cell distribution width. Saudi Med J. 2007;28:535–9.Search in Google Scholar
[34] Foy BH, Carlson JCT, Reinertsen E, Padros I, Valls R, Pallares Lopez R, et al. Association of red blood cell distribution width with mortality risk in hospitalized adults with SARS-CoV-2 infection. JAMA Netw Open. 2020;3:e2022058.10.1001/jamanetworkopen.2020.22058Search in Google Scholar PubMed PubMed Central
[35] Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116:1666–87.10.1093/cvr/cvaa106Search in Google Scholar PubMed PubMed Central
[36] Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020;191:9–14.10.1016/j.thromres.2020.04.024Search in Google Scholar PubMed PubMed Central
[37] Rismanbaf A, Zarei S. Liver and kidney injuries in COVID-19 and their effects on drug therapy; a letter to editor. Arch Acad Emerg Med. 2020;8:e17.Search in Google Scholar
[38] Degarege A, Gebrezgi MT, Ibanez G, Wahlgren M, Madhivanan P. Effect of the ABO blood group on susceptibility to severe malaria: a systematic review and meta-analysis. Blood rev. 2019;33:53–62.10.1016/j.blre.2018.07.002Search in Google Scholar PubMed
[39] Goel S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri RR, et al. RIFINs are adhesins implicated in severe plasmodium falciparum malaria. Nat med. 2015;21:314–7.10.1038/nm.3812Search in Google Scholar PubMed
[40] Zhao J, Yang Y, Huang H, Li D, Gu D, Lu X, et al. Relationship between the ABO blood group and the COVID-19 susceptibility. Clin Infect Dis. 2020;ciaa1150.10.1101/2020.03.11.20031096Search in Google Scholar
[41] Zietz M, Zucker J, Tatonetti NP. Associations between blood type and COVID-19 infection, intubation, and death. Nat Commun. 2020;11:5761.10.1038/s41467-020-19623-xSearch in Google Scholar PubMed PubMed Central
[42] Muneer A, Kumari K, Tripathi M, Srivastava R, Mohmmed A, Rathore S. Comparative analyses revealed reduced spread of COVID-19 in malaria endemic countries. MedRxiv. 2020. 10.1101/2020.05.11.20097923.Search in Google Scholar
[43] Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. 2020;92:1580–6. 10.1002/jmv.25832.Search in Google Scholar PubMed PubMed Central
© 2021 Md. Mehedi Hassan et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Biomedical Sciences
- Research progress on the mechanism of orexin in pain regulation in different brain regions
- Adriamycin-resistant cells are significantly less fit than adriamycin-sensitive cells in cervical cancer
- Exogenous spermidine affects polyamine metabolism in the mouse hypothalamus
- Iris metastasis of diffuse large B-cell lymphoma misdiagnosed as primary angle-closure glaucoma: A case report and review of the literature
- LncRNA PVT1 promotes cervical cancer progression by sponging miR-503 to upregulate ARL2 expression
- Two new inflammatory markers related to the CURB-65 score for disease severity in patients with community-acquired pneumonia: The hypersensitive C-reactive protein to albumin ratio and fibrinogen to albumin ratio
- Circ_0091579 enhances the malignancy of hepatocellular carcinoma via miR-1287/PDK2 axis
- Silencing XIST mitigated lipopolysaccharide (LPS)-induced inflammatory injury in human lung fibroblast WI-38 cells through modulating miR-30b-5p/CCL16 axis and TLR4/NF-κB signaling pathway
- Protocatechuic acid attenuates cerebral aneurysm formation and progression by inhibiting TNF-alpha/Nrf-2/NF-kB-mediated inflammatory mechanisms in experimental rats
- ABCB1 polymorphism in clopidogrel-treated Montenegrin patients
- Metabolic profiling of fatty acids in Tripterygium wilfordii multiglucoside- and triptolide-induced liver-injured rats
- miR-338-3p inhibits cell growth, invasion, and EMT process in neuroblastoma through targeting MMP-2
- Verification of neuroprotective effects of alpha-lipoic acid on chronic neuropathic pain in a chronic constriction injury rat model
- Circ_WWC3 overexpression decelerates the progression of osteosarcoma by regulating miR-421/PDE7B axis
- Knockdown of TUG1 rescues cardiomyocyte hypertrophy through targeting the miR-497/MEF2C axis
- MiR-146b-3p protects against AR42J cell injury in cerulein-induced acute pancreatitis model through targeting Anxa2
- miR-299-3p suppresses cell progression and induces apoptosis by downregulating PAX3 in gastric cancer
- Diabetes and COVID-19
- Discovery of novel potential KIT inhibitors for the treatment of gastrointestinal stromal tumor
- TEAD4 is a novel independent predictor of prognosis in LGG patients with IDH mutation
- circTLK1 facilitates the proliferation and metastasis of renal cell carcinoma by regulating miR-495-3p/CBL axis
- microRNA-9-5p protects liver sinusoidal endothelial cell against oxygen glucose deprivation/reperfusion injury
- Long noncoding RNA TUG1 regulates degradation of chondrocyte extracellular matrix via miR-320c/MMP-13 axis in osteoarthritis
- Duodenal adenocarcinoma with skin metastasis as initial manifestation: A case report
- Effects of Loofah cylindrica extract on learning and memory ability, brain tissue morphology, and immune function of aging mice
- Recombinant Bacteroides fragilis enterotoxin-1 (rBFT-1) promotes proliferation of colorectal cancer via CCL3-related molecular pathways
- Blocking circ_UBR4 suppressed proliferation, migration, and cell cycle progression of human vascular smooth muscle cells in atherosclerosis
- Gene therapy in PIDs, hemoglobin, ocular, neurodegenerative, and hemophilia B disorders
- Downregulation of circ_0037655 impedes glioma formation and metastasis via the regulation of miR-1229-3p/ITGB8 axis
- Vitamin D deficiency and cardiovascular risk in type 2 diabetes population
- Circ_0013359 facilitates the tumorigenicity of melanoma by regulating miR-136-5p/RAB9A axis
- Mechanisms of circular RNA circ_0066147 on pancreatic cancer progression
- lncRNA myocardial infarction-associated transcript (MIAT) knockdown alleviates LPS-induced chondrocytes inflammatory injury via regulating miR-488-3p/sex determining region Y-related HMG-box 11 (SOX11) axis
- Identification of circRNA circ-CSPP1 as a potent driver of colorectal cancer by directly targeting the miR-431/LASP1 axis
- Hyperhomocysteinemia exacerbates ischemia-reperfusion injury-induced acute kidney injury by mediating oxidative stress, DNA damage, JNK pathway, and apoptosis
- Potential prognostic markers and significant lncRNA–mRNA co-expression pairs in laryngeal squamous cell carcinoma
- Gamma irradiation-mediated inactivation of enveloped viruses with conservation of genome integrity: Potential application for SARS-CoV-2 inactivated vaccine development
- ADHFE1 is a correlative factor of patient survival in cancer
- The association of transcription factor Prox1 with the proliferation, migration, and invasion of lung cancer
- Is there a relationship between the prevalence of autoimmune thyroid disease and diabetic kidney disease?
- Immunoregulatory function of Dictyophora echinovolvata spore polysaccharides in immunocompromised mice induced by cyclophosphamide
- T cell epitopes of SARS-CoV-2 spike protein and conserved surface protein of Plasmodium malariae share sequence homology
- Anti-obesity effect and mechanism of mesenchymal stem cells influence on obese mice
- Long noncoding RNA HULC contributes to paclitaxel resistance in ovarian cancer via miR-137/ITGB8 axis
- Glucocorticoids protect HEI-OC1 cells from tunicamycin-induced cell damage via inhibiting endoplasmic reticulum stress
- Prognostic value of the neutrophil-to-lymphocyte ratio in acute organophosphorus pesticide poisoning
- Gastroprotective effects of diosgenin against HCl/ethanol-induced gastric mucosal injury through suppression of NF-κβ and myeloperoxidase activities
- Silencing of LINC00707 suppresses cell proliferation, migration, and invasion of osteosarcoma cells by modulating miR-338-3p/AHSA1 axis
- Successful extracorporeal membrane oxygenation resuscitation of patient with cardiogenic shock induced by phaeochromocytoma crisis mimicking hyperthyroidism: A case report
- Effects of miR-185-5p on replication of hepatitis C virus
- Lidocaine has antitumor effect on hepatocellular carcinoma via the circ_DYNC1H1/miR-520a-3p/USP14 axis
- Primary localized cutaneous nodular amyloidosis presenting as lymphatic malformation: A case report
- Multimodal magnetic resonance imaging analysis in the characteristics of Wilson’s disease: A case report and literature review
- Therapeutic potential of anticoagulant therapy in association with cytokine storm inhibition in severe cases of COVID-19: A case report
- Neoadjuvant immunotherapy combined with chemotherapy for locally advanced squamous cell lung carcinoma: A case report and literature review
- Rufinamide (RUF) suppresses inflammation and maintains the integrity of the blood–brain barrier during kainic acid-induced brain damage
- Inhibition of ADAM10 ameliorates doxorubicin-induced cardiac remodeling by suppressing N-cadherin cleavage
- Invasive ductal carcinoma and small lymphocytic lymphoma/chronic lymphocytic leukemia manifesting as a collision breast tumor: A case report and literature review
- Clonal diversity of the B cell receptor repertoire in patients with coronary in-stent restenosis and type 2 diabetes
- CTLA-4 promotes lymphoma progression through tumor stem cell enrichment and immunosuppression
- WDR74 promotes proliferation and metastasis in colorectal cancer cells through regulating the Wnt/β-catenin signaling pathway
- Down-regulation of IGHG1 enhances Protoporphyrin IX accumulation and inhibits hemin biosynthesis in colorectal cancer by suppressing the MEK-FECH axis
- Curcumin suppresses the progression of gastric cancer by regulating circ_0056618/miR-194-5p axis
- Scutellarin-induced A549 cell apoptosis depends on activation of the transforming growth factor-β1/smad2/ROS/caspase-3 pathway
- lncRNA NEAT1 regulates CYP1A2 and influences steroid-induced necrosis
- A two-microRNA signature predicts the progression of male thyroid cancer
- Isolation of microglia from retinas of chronic ocular hypertensive rats
- Changes of immune cells in patients with hepatocellular carcinoma treated by radiofrequency ablation and hepatectomy, a pilot study
- Calcineurin Aβ gene knockdown inhibits transient outward potassium current ion channel remodeling in hypertrophic ventricular myocyte
- Aberrant expression of PI3K/AKT signaling is involved in apoptosis resistance of hepatocellular carcinoma
- Clinical significance of activated Wnt/β-catenin signaling in apoptosis inhibition of oral cancer
- circ_CHFR regulates ox-LDL-mediated cell proliferation, apoptosis, and EndoMT by miR-15a-5p/EGFR axis in human brain microvessel endothelial cells
- Resveratrol pretreatment mitigates LPS-induced acute lung injury by regulating conventional dendritic cells’ maturation and function
- Ubiquitin-conjugating enzyme E2T promotes tumor stem cell characteristics and migration of cervical cancer cells by regulating the GRP78/FAK pathway
- Carriage of HLA-DRB1*11 and 1*12 alleles and risk factors in patients with breast cancer in Burkina Faso
- Protective effect of Lactobacillus-containing probiotics on intestinal mucosa of rats experiencing traumatic hemorrhagic shock
- Glucocorticoids induce osteonecrosis of the femoral head through the Hippo signaling pathway
- Endothelial cell-derived SSAO can increase MLC20 phosphorylation in VSMCs
- Downregulation of STOX1 is a novel prognostic biomarker for glioma patients
- miR-378a-3p regulates glioma cell chemosensitivity to cisplatin through IGF1R
- The molecular mechanisms underlying arecoline-induced cardiac fibrosis in rats
- TGF-β1-overexpressing mesenchymal stem cells reciprocally regulate Th17/Treg cells by regulating the expression of IFN-γ
- The influence of MTHFR genetic polymorphisms on methotrexate therapy in pediatric acute lymphoblastic leukemia
- Red blood cell distribution width-standard deviation but not red blood cell distribution width-coefficient of variation as a potential index for the diagnosis of iron-deficiency anemia in mid-pregnancy women
- Small cell neuroendocrine carcinoma expressing alpha fetoprotein in the endometrium
- Superoxide dismutase and the sigma1 receptor as key elements of the antioxidant system in human gastrointestinal tract cancers
- Molecular characterization and phylogenetic studies of Echinococcus granulosus and Taenia multiceps coenurus cysts in slaughtered sheep in Saudi Arabia
- ITGB5 mutation discovered in a Chinese family with blepharophimosis-ptosis-epicanthus inversus syndrome
- ACTB and GAPDH appear at multiple SDS-PAGE positions, thus not suitable as reference genes for determining protein loading in techniques like Western blotting
- Facilitation of mouse skin-derived precursor growth and yield by optimizing plating density
- 3,4-Dihydroxyphenylethanol ameliorates lipopolysaccharide-induced septic cardiac injury in a murine model
- Downregulation of PITX2 inhibits the proliferation and migration of liver cancer cells and induces cell apoptosis
- Expression of CDK9 in endometrial cancer tissues and its effect on the proliferation of HEC-1B
- Novel predictor of the occurrence of DKA in T1DM patients without infection: A combination of neutrophil/lymphocyte ratio and white blood cells
- Investigation of molecular regulation mechanism under the pathophysiology of subarachnoid hemorrhage
- miR-25-3p protects renal tubular epithelial cells from apoptosis induced by renal IRI by targeting DKK3
- Bioengineering and Biotechnology
- Green fabrication of Co and Co3O4 nanoparticles and their biomedical applications: A review
- Agriculture
- Effects of inorganic and organic selenium sources on the growth performance of broilers in China: A meta-analysis
- Crop-livestock integration practices, knowledge, and attitudes among smallholder farmers: Hedging against climate change-induced shocks in semi-arid Zimbabwe
- Food Science and Nutrition
- Effect of food processing on the antioxidant activity of flavones from Polygonatum odoratum (Mill.) Druce
- Vitamin D and iodine status was associated with the risk and complication of type 2 diabetes mellitus in China
- Diversity of microbiota in Slovak summer ewes’ cheese “Bryndza”
- Comparison between voltammetric detection methods for abalone-flavoring liquid
- Composition of low-molecular-weight glutenin subunits in common wheat (Triticum aestivum L.) and their effects on the rheological properties of dough
- Application of culture, PCR, and PacBio sequencing for determination of microbial composition of milk from subclinical mastitis dairy cows of smallholder farms
- Investigating microplastics and potentially toxic elements contamination in canned Tuna, Salmon, and Sardine fishes from Taif markets, KSA
- From bench to bar side: Evaluating the red wine storage lesion
- Establishment of an iodine model for prevention of iodine-excess-induced thyroid dysfunction in pregnant women
- Plant Sciences
- Characterization of GMPP from Dendrobium huoshanense yielding GDP-D-mannose
- Comparative analysis of the SPL gene family in five Rosaceae species: Fragaria vesca, Malus domestica, Prunus persica, Rubus occidentalis, and Pyrus pyrifolia
- Identification of leaf rust resistance genes Lr34 and Lr46 in common wheat (Triticum aestivum L. ssp. aestivum) lines of different origin using multiplex PCR
- Investigation of bioactivities of Taxus chinensis, Taxus cuspidata, and Taxus × media by gas chromatography-mass spectrometry
- Morphological structures and histochemistry of roots and shoots in Myricaria laxiflora (Tamaricaceae)
- Transcriptome analysis of resistance mechanism to potato wart disease
- In silico analysis of glycosyltransferase 2 family genes in duckweed (Spirodela polyrhiza) and its role in salt stress tolerance
- Comparative study on growth traits and ions regulation of zoysiagrasses under varied salinity treatments
- Role of MS1 homolog Ntms1 gene of tobacco infertility
- Biological characteristics and fungicide sensitivity of Pyricularia variabilis
- In silico/computational analysis of mevalonate pyrophosphate decarboxylase gene families in Campanulids
- Identification of novel drought-responsive miRNA regulatory network of drought stress response in common vetch (Vicia sativa)
- How photoautotrophy, photomixotrophy, and ventilation affect the stomata and fluorescence emission of pistachios rootstock?
- Apoplastic histochemical features of plant root walls that may facilitate ion uptake and retention
- Ecology and Environmental Sciences
- The impact of sewage sludge on the fungal communities in the rhizosphere and roots of barley and on barley yield
- Domestication of wild animals may provide a springboard for rapid variation of coronavirus
- Response of benthic invertebrate assemblages to seasonal and habitat condition in the Wewe River, Ashanti region (Ghana)
- Molecular record for the first authentication of Isaria cicadae from Vietnam
- Twig biomass allocation of Betula platyphylla in different habitats in Wudalianchi Volcano, northeast China
- Animal Sciences
- Supplementation of probiotics in water beneficial growth performance, carcass traits, immune function, and antioxidant capacity in broiler chickens
- Predators of the giant pine scale, Marchalina hellenica (Gennadius 1883; Hemiptera: Marchalinidae), out of its natural range in Turkey
- Honey in wound healing: An updated review
- NONMMUT140591.1 may serve as a ceRNA to regulate Gata5 in UT-B knockout-induced cardiac conduction block
- Radiotherapy for the treatment of pulmonary hydatidosis in sheep
- Retraction
- Retraction of “Long non-coding RNA TUG1 knockdown hinders the tumorigenesis of multiple myeloma by regulating microRNA-34a-5p/NOTCH1 signaling pathway”
- Special Issue on Reuse of Agro-Industrial By-Products
- An effect of positional isomerism of benzoic acid derivatives on antibacterial activity against Escherichia coli
- Special Issue on Computing and Artificial Techniques for Life Science Applications - Part II
- Relationship of Gensini score with retinal vessel diameter and arteriovenous ratio in senile CHD
- Effects of different enantiomers of amlodipine on lipid profiles and vasomotor factors in atherosclerotic rabbits
- Establishment of the New Zealand white rabbit animal model of fatty keratopathy associated with corneal neovascularization
- lncRNA MALAT1/miR-143 axis is a potential biomarker for in-stent restenosis and is involved in the multiplication of vascular smooth muscle cells
Articles in the same Issue
- Biomedical Sciences
- Research progress on the mechanism of orexin in pain regulation in different brain regions
- Adriamycin-resistant cells are significantly less fit than adriamycin-sensitive cells in cervical cancer
- Exogenous spermidine affects polyamine metabolism in the mouse hypothalamus
- Iris metastasis of diffuse large B-cell lymphoma misdiagnosed as primary angle-closure glaucoma: A case report and review of the literature
- LncRNA PVT1 promotes cervical cancer progression by sponging miR-503 to upregulate ARL2 expression
- Two new inflammatory markers related to the CURB-65 score for disease severity in patients with community-acquired pneumonia: The hypersensitive C-reactive protein to albumin ratio and fibrinogen to albumin ratio
- Circ_0091579 enhances the malignancy of hepatocellular carcinoma via miR-1287/PDK2 axis
- Silencing XIST mitigated lipopolysaccharide (LPS)-induced inflammatory injury in human lung fibroblast WI-38 cells through modulating miR-30b-5p/CCL16 axis and TLR4/NF-κB signaling pathway
- Protocatechuic acid attenuates cerebral aneurysm formation and progression by inhibiting TNF-alpha/Nrf-2/NF-kB-mediated inflammatory mechanisms in experimental rats
- ABCB1 polymorphism in clopidogrel-treated Montenegrin patients
- Metabolic profiling of fatty acids in Tripterygium wilfordii multiglucoside- and triptolide-induced liver-injured rats
- miR-338-3p inhibits cell growth, invasion, and EMT process in neuroblastoma through targeting MMP-2
- Verification of neuroprotective effects of alpha-lipoic acid on chronic neuropathic pain in a chronic constriction injury rat model
- Circ_WWC3 overexpression decelerates the progression of osteosarcoma by regulating miR-421/PDE7B axis
- Knockdown of TUG1 rescues cardiomyocyte hypertrophy through targeting the miR-497/MEF2C axis
- MiR-146b-3p protects against AR42J cell injury in cerulein-induced acute pancreatitis model through targeting Anxa2
- miR-299-3p suppresses cell progression and induces apoptosis by downregulating PAX3 in gastric cancer
- Diabetes and COVID-19
- Discovery of novel potential KIT inhibitors for the treatment of gastrointestinal stromal tumor
- TEAD4 is a novel independent predictor of prognosis in LGG patients with IDH mutation
- circTLK1 facilitates the proliferation and metastasis of renal cell carcinoma by regulating miR-495-3p/CBL axis
- microRNA-9-5p protects liver sinusoidal endothelial cell against oxygen glucose deprivation/reperfusion injury
- Long noncoding RNA TUG1 regulates degradation of chondrocyte extracellular matrix via miR-320c/MMP-13 axis in osteoarthritis
- Duodenal adenocarcinoma with skin metastasis as initial manifestation: A case report
- Effects of Loofah cylindrica extract on learning and memory ability, brain tissue morphology, and immune function of aging mice
- Recombinant Bacteroides fragilis enterotoxin-1 (rBFT-1) promotes proliferation of colorectal cancer via CCL3-related molecular pathways
- Blocking circ_UBR4 suppressed proliferation, migration, and cell cycle progression of human vascular smooth muscle cells in atherosclerosis
- Gene therapy in PIDs, hemoglobin, ocular, neurodegenerative, and hemophilia B disorders
- Downregulation of circ_0037655 impedes glioma formation and metastasis via the regulation of miR-1229-3p/ITGB8 axis
- Vitamin D deficiency and cardiovascular risk in type 2 diabetes population
- Circ_0013359 facilitates the tumorigenicity of melanoma by regulating miR-136-5p/RAB9A axis
- Mechanisms of circular RNA circ_0066147 on pancreatic cancer progression
- lncRNA myocardial infarction-associated transcript (MIAT) knockdown alleviates LPS-induced chondrocytes inflammatory injury via regulating miR-488-3p/sex determining region Y-related HMG-box 11 (SOX11) axis
- Identification of circRNA circ-CSPP1 as a potent driver of colorectal cancer by directly targeting the miR-431/LASP1 axis
- Hyperhomocysteinemia exacerbates ischemia-reperfusion injury-induced acute kidney injury by mediating oxidative stress, DNA damage, JNK pathway, and apoptosis
- Potential prognostic markers and significant lncRNA–mRNA co-expression pairs in laryngeal squamous cell carcinoma
- Gamma irradiation-mediated inactivation of enveloped viruses with conservation of genome integrity: Potential application for SARS-CoV-2 inactivated vaccine development
- ADHFE1 is a correlative factor of patient survival in cancer
- The association of transcription factor Prox1 with the proliferation, migration, and invasion of lung cancer
- Is there a relationship between the prevalence of autoimmune thyroid disease and diabetic kidney disease?
- Immunoregulatory function of Dictyophora echinovolvata spore polysaccharides in immunocompromised mice induced by cyclophosphamide
- T cell epitopes of SARS-CoV-2 spike protein and conserved surface protein of Plasmodium malariae share sequence homology
- Anti-obesity effect and mechanism of mesenchymal stem cells influence on obese mice
- Long noncoding RNA HULC contributes to paclitaxel resistance in ovarian cancer via miR-137/ITGB8 axis
- Glucocorticoids protect HEI-OC1 cells from tunicamycin-induced cell damage via inhibiting endoplasmic reticulum stress
- Prognostic value of the neutrophil-to-lymphocyte ratio in acute organophosphorus pesticide poisoning
- Gastroprotective effects of diosgenin against HCl/ethanol-induced gastric mucosal injury through suppression of NF-κβ and myeloperoxidase activities
- Silencing of LINC00707 suppresses cell proliferation, migration, and invasion of osteosarcoma cells by modulating miR-338-3p/AHSA1 axis
- Successful extracorporeal membrane oxygenation resuscitation of patient with cardiogenic shock induced by phaeochromocytoma crisis mimicking hyperthyroidism: A case report
- Effects of miR-185-5p on replication of hepatitis C virus
- Lidocaine has antitumor effect on hepatocellular carcinoma via the circ_DYNC1H1/miR-520a-3p/USP14 axis
- Primary localized cutaneous nodular amyloidosis presenting as lymphatic malformation: A case report
- Multimodal magnetic resonance imaging analysis in the characteristics of Wilson’s disease: A case report and literature review
- Therapeutic potential of anticoagulant therapy in association with cytokine storm inhibition in severe cases of COVID-19: A case report
- Neoadjuvant immunotherapy combined with chemotherapy for locally advanced squamous cell lung carcinoma: A case report and literature review
- Rufinamide (RUF) suppresses inflammation and maintains the integrity of the blood–brain barrier during kainic acid-induced brain damage
- Inhibition of ADAM10 ameliorates doxorubicin-induced cardiac remodeling by suppressing N-cadherin cleavage
- Invasive ductal carcinoma and small lymphocytic lymphoma/chronic lymphocytic leukemia manifesting as a collision breast tumor: A case report and literature review
- Clonal diversity of the B cell receptor repertoire in patients with coronary in-stent restenosis and type 2 diabetes
- CTLA-4 promotes lymphoma progression through tumor stem cell enrichment and immunosuppression
- WDR74 promotes proliferation and metastasis in colorectal cancer cells through regulating the Wnt/β-catenin signaling pathway
- Down-regulation of IGHG1 enhances Protoporphyrin IX accumulation and inhibits hemin biosynthesis in colorectal cancer by suppressing the MEK-FECH axis
- Curcumin suppresses the progression of gastric cancer by regulating circ_0056618/miR-194-5p axis
- Scutellarin-induced A549 cell apoptosis depends on activation of the transforming growth factor-β1/smad2/ROS/caspase-3 pathway
- lncRNA NEAT1 regulates CYP1A2 and influences steroid-induced necrosis
- A two-microRNA signature predicts the progression of male thyroid cancer
- Isolation of microglia from retinas of chronic ocular hypertensive rats
- Changes of immune cells in patients with hepatocellular carcinoma treated by radiofrequency ablation and hepatectomy, a pilot study
- Calcineurin Aβ gene knockdown inhibits transient outward potassium current ion channel remodeling in hypertrophic ventricular myocyte
- Aberrant expression of PI3K/AKT signaling is involved in apoptosis resistance of hepatocellular carcinoma
- Clinical significance of activated Wnt/β-catenin signaling in apoptosis inhibition of oral cancer
- circ_CHFR regulates ox-LDL-mediated cell proliferation, apoptosis, and EndoMT by miR-15a-5p/EGFR axis in human brain microvessel endothelial cells
- Resveratrol pretreatment mitigates LPS-induced acute lung injury by regulating conventional dendritic cells’ maturation and function
- Ubiquitin-conjugating enzyme E2T promotes tumor stem cell characteristics and migration of cervical cancer cells by regulating the GRP78/FAK pathway
- Carriage of HLA-DRB1*11 and 1*12 alleles and risk factors in patients with breast cancer in Burkina Faso
- Protective effect of Lactobacillus-containing probiotics on intestinal mucosa of rats experiencing traumatic hemorrhagic shock
- Glucocorticoids induce osteonecrosis of the femoral head through the Hippo signaling pathway
- Endothelial cell-derived SSAO can increase MLC20 phosphorylation in VSMCs
- Downregulation of STOX1 is a novel prognostic biomarker for glioma patients
- miR-378a-3p regulates glioma cell chemosensitivity to cisplatin through IGF1R
- The molecular mechanisms underlying arecoline-induced cardiac fibrosis in rats
- TGF-β1-overexpressing mesenchymal stem cells reciprocally regulate Th17/Treg cells by regulating the expression of IFN-γ
- The influence of MTHFR genetic polymorphisms on methotrexate therapy in pediatric acute lymphoblastic leukemia
- Red blood cell distribution width-standard deviation but not red blood cell distribution width-coefficient of variation as a potential index for the diagnosis of iron-deficiency anemia in mid-pregnancy women
- Small cell neuroendocrine carcinoma expressing alpha fetoprotein in the endometrium
- Superoxide dismutase and the sigma1 receptor as key elements of the antioxidant system in human gastrointestinal tract cancers
- Molecular characterization and phylogenetic studies of Echinococcus granulosus and Taenia multiceps coenurus cysts in slaughtered sheep in Saudi Arabia
- ITGB5 mutation discovered in a Chinese family with blepharophimosis-ptosis-epicanthus inversus syndrome
- ACTB and GAPDH appear at multiple SDS-PAGE positions, thus not suitable as reference genes for determining protein loading in techniques like Western blotting
- Facilitation of mouse skin-derived precursor growth and yield by optimizing plating density
- 3,4-Dihydroxyphenylethanol ameliorates lipopolysaccharide-induced septic cardiac injury in a murine model
- Downregulation of PITX2 inhibits the proliferation and migration of liver cancer cells and induces cell apoptosis
- Expression of CDK9 in endometrial cancer tissues and its effect on the proliferation of HEC-1B
- Novel predictor of the occurrence of DKA in T1DM patients without infection: A combination of neutrophil/lymphocyte ratio and white blood cells
- Investigation of molecular regulation mechanism under the pathophysiology of subarachnoid hemorrhage
- miR-25-3p protects renal tubular epithelial cells from apoptosis induced by renal IRI by targeting DKK3
- Bioengineering and Biotechnology
- Green fabrication of Co and Co3O4 nanoparticles and their biomedical applications: A review
- Agriculture
- Effects of inorganic and organic selenium sources on the growth performance of broilers in China: A meta-analysis
- Crop-livestock integration practices, knowledge, and attitudes among smallholder farmers: Hedging against climate change-induced shocks in semi-arid Zimbabwe
- Food Science and Nutrition
- Effect of food processing on the antioxidant activity of flavones from Polygonatum odoratum (Mill.) Druce
- Vitamin D and iodine status was associated with the risk and complication of type 2 diabetes mellitus in China
- Diversity of microbiota in Slovak summer ewes’ cheese “Bryndza”
- Comparison between voltammetric detection methods for abalone-flavoring liquid
- Composition of low-molecular-weight glutenin subunits in common wheat (Triticum aestivum L.) and their effects on the rheological properties of dough
- Application of culture, PCR, and PacBio sequencing for determination of microbial composition of milk from subclinical mastitis dairy cows of smallholder farms
- Investigating microplastics and potentially toxic elements contamination in canned Tuna, Salmon, and Sardine fishes from Taif markets, KSA
- From bench to bar side: Evaluating the red wine storage lesion
- Establishment of an iodine model for prevention of iodine-excess-induced thyroid dysfunction in pregnant women
- Plant Sciences
- Characterization of GMPP from Dendrobium huoshanense yielding GDP-D-mannose
- Comparative analysis of the SPL gene family in five Rosaceae species: Fragaria vesca, Malus domestica, Prunus persica, Rubus occidentalis, and Pyrus pyrifolia
- Identification of leaf rust resistance genes Lr34 and Lr46 in common wheat (Triticum aestivum L. ssp. aestivum) lines of different origin using multiplex PCR
- Investigation of bioactivities of Taxus chinensis, Taxus cuspidata, and Taxus × media by gas chromatography-mass spectrometry
- Morphological structures and histochemistry of roots and shoots in Myricaria laxiflora (Tamaricaceae)
- Transcriptome analysis of resistance mechanism to potato wart disease
- In silico analysis of glycosyltransferase 2 family genes in duckweed (Spirodela polyrhiza) and its role in salt stress tolerance
- Comparative study on growth traits and ions regulation of zoysiagrasses under varied salinity treatments
- Role of MS1 homolog Ntms1 gene of tobacco infertility
- Biological characteristics and fungicide sensitivity of Pyricularia variabilis
- In silico/computational analysis of mevalonate pyrophosphate decarboxylase gene families in Campanulids
- Identification of novel drought-responsive miRNA regulatory network of drought stress response in common vetch (Vicia sativa)
- How photoautotrophy, photomixotrophy, and ventilation affect the stomata and fluorescence emission of pistachios rootstock?
- Apoplastic histochemical features of plant root walls that may facilitate ion uptake and retention
- Ecology and Environmental Sciences
- The impact of sewage sludge on the fungal communities in the rhizosphere and roots of barley and on barley yield
- Domestication of wild animals may provide a springboard for rapid variation of coronavirus
- Response of benthic invertebrate assemblages to seasonal and habitat condition in the Wewe River, Ashanti region (Ghana)
- Molecular record for the first authentication of Isaria cicadae from Vietnam
- Twig biomass allocation of Betula platyphylla in different habitats in Wudalianchi Volcano, northeast China
- Animal Sciences
- Supplementation of probiotics in water beneficial growth performance, carcass traits, immune function, and antioxidant capacity in broiler chickens
- Predators of the giant pine scale, Marchalina hellenica (Gennadius 1883; Hemiptera: Marchalinidae), out of its natural range in Turkey
- Honey in wound healing: An updated review
- NONMMUT140591.1 may serve as a ceRNA to regulate Gata5 in UT-B knockout-induced cardiac conduction block
- Radiotherapy for the treatment of pulmonary hydatidosis in sheep
- Retraction
- Retraction of “Long non-coding RNA TUG1 knockdown hinders the tumorigenesis of multiple myeloma by regulating microRNA-34a-5p/NOTCH1 signaling pathway”
- Special Issue on Reuse of Agro-Industrial By-Products
- An effect of positional isomerism of benzoic acid derivatives on antibacterial activity against Escherichia coli
- Special Issue on Computing and Artificial Techniques for Life Science Applications - Part II
- Relationship of Gensini score with retinal vessel diameter and arteriovenous ratio in senile CHD
- Effects of different enantiomers of amlodipine on lipid profiles and vasomotor factors in atherosclerotic rabbits
- Establishment of the New Zealand white rabbit animal model of fatty keratopathy associated with corneal neovascularization
- lncRNA MALAT1/miR-143 axis is a potential biomarker for in-stent restenosis and is involved in the multiplication of vascular smooth muscle cells