MW irradiation and ionic liquids as green tools in hydrolyses and alcoholyses
-
Nikoletta Harsági


Abstract
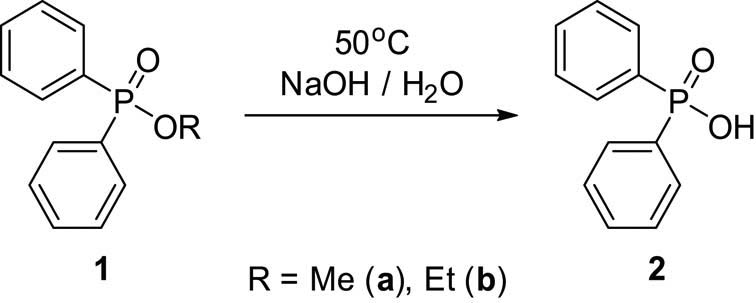
The optimized HCl-catalyzed hydrolysis of alkyl diphenylphosphinates is described. The reaction times and pseudo-first-order rate constants suggested the iPr > Me > Et ∼ Pr ∼ Bu order of reactivity in respect of the alkyl group of the phosphinates. The MW-assisted p-toluenesulfonic acid (PTSA)-catalyzed variation means a better alternative possibility due to the shorter reaction times, and the alkaline hydrolysis is another option. The transesterification of alkyl diphenylphosphinates took place only in the presence of suitable ionic liquids, such as butyl-methylimidazolium hexafluorophosphorate ([bmim][PF6]) and butyl-methylimidazolium tetrafluoroborate ([bmim][BF4]). The application of ethyl-methylimidazolium hydrosulfate ([emim][HSO4]) and butyl-methylimidazolium chloride ([bmim][Cl]) was not too efficient, as the formation of the ester was accompanied by the fission of the O–C bond resulting in the formation of Ph2P(O)OH. This surprising transformation may be utilized in the phosphinate → phosphinic acid conversion.
1 Introduction
The esters of phosphinic acids (alkyl phosphinates) prepared in most cases by the reaction of the corresponding phosphinic chloride with alcohol [1,2], or recently, by the microwave-assisted direct esterification of the phosphinic acids in the presence of an IL additive [2,3,4,5] are widely applied starting materials and intermediates in syntheses [2]. They may also be the precursors of (potentially) biologically active derivatives [6,7,8,9]. Hydrolysis of the alkyl phosphinates (and in general P-esters including dialkyl phosphonates) is an often used and rather common transformation that may be performed under acidic conditions [10,11,12,13], but also in the presence of a base [14,15,16,17]. We have recently investigated the acidic hydrolysis of a series of cyclic alkyl phosphinates in detail [18]. Optimum conditions for the acidic hydrolysis of dialkyl arylphosphonates and α-hydroxy-benzylphosphonates were also explored [19,20]. The reactivity of the starting P-esters along with the kinetics was also evaluated. In this paper, the acidic- or base-catalyzed hydrolyses of alkyl diphenylphoshinates are investigated and compared. We wished to find the optimum conditions and to characterize the processes by rate constants. The other purpose was to elaborate the transesterification of our “Ph2P(O)OR” model compounds by reaction with series of alcohols. We have had some experience on MW-assisted alcoholysis of dialkyl phosphites, which may lead to fully transesterified products via the intermediate with two different alkoxy groups [21,22]. Continuous flow accomplishments were also developed by us [23].
2 Materials and methods
2.1 General
The 31P, 13C, 1H NMR spectra were taken on a Bruker DRX-500 spectrometer operating at 202.4, 125.7, and 500 MHz, respectively. The couplings are given in Hz. LC-MS measurements were performed with an Agilent 1,200 liquid chromatography system coupled with a 6,130 quadrupole mass spectrometer equipped with an ESI ion source (Agilent Technologies, Palo Alto, CA, USA). High resolution mass spectrometric measurements were performed using a Thermo Velos Pro Orbitrap Elite hybrid mass spectrometer in positive electrospray mode.
2.2 Preparation of the starting alkyl diphenylphosphinates
The C1–C4 alkyl diphenylphosphinates (1a–e) were synthetized by the reaction of diphenylphosphinoyl chloride with the corresponding alcohols at 26°C in the presence of triethylamine in toluene (Scheme 1).
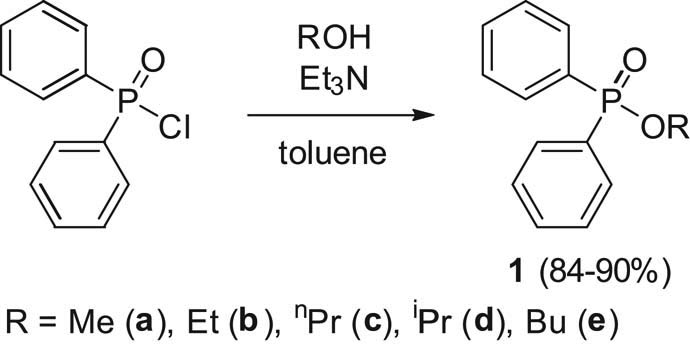
Preparation of the starting alkyl diphenylphosphinates (1a–e).
2.2.1 General procedure for the preparation of diphenyl phosphinates (1a–e)
10.5 mmol (2 mL) of diphenylphosphinic chloride was added to the mixture of 15 mL of toluene, 3 equivalents of alcohol (methanol: 1.27 mL, ethanol: 1.84 mL, propanol: 2.42 mL, isopropanol: 2.42 mL, butanol: 2.88 mL), and 1.61 mL (11.5 mmol) of triethylamine, and the reaction mixture was stirred at reflux for 2 h. After completion of the reaction, the amine salt was filtered off, the filtrate was concentrated in vacuum, and the crude product so obtained was purified by column chromatography (silica gel, hexane–ethyl acetate 7:3 as the eluent) to give the phosphinates (1a–e) in yields of 84–90%. The products were analyzed by 31P NMR spectroscopy.
2.3 General procedure for the acidic hydrolysis of diphenylphosphinates (1a–e) under conventional conditions
A mixture of 3.7 mmol of diphenylphosphinate (1a: 0.86 g, 1b: 0.91 g, 1c: 0.96 g, 1d: 0.96 g, 1e: 0.99 g), 1.0 mL (6.0 mmol) of concentrated hydrochloric acid, and 2.0 mL of water was stirred at reflux (ca. 100°C) for 4–8 h. Concentration of an aliquot part of the reaction mixture afforded Ph2P(O)OH (2) as a solid powder in yields of 90–94%, which was analyzed by 31P NMR spectroscopy and LC-MS.
2.4 General procedure for the acidic hydrolysis of diphenylphosphinates (1a–e) under MW conditions
A mixture of 1.9 mmol of diphenylphosphinate (1a: 0.43 g, 1b: 0.46 g, 1c: 0.49 g, 1d: 0.49 g, 1e: 0.51 g), 0.04 g (0.19 mmol) of PTSA, and 1.0 mL of water was irradiated in a sealed tube placed in CEM MW reactor at 160–180°C (max. 100 W) for 0.5–6 h. After evaporating the water, the residue so obtained was washed 3 times with 3 mL of water and dried. Ph2P(O)OH (2) was obtained as a solid powder in yields of 93–97%, which was analyzed by 31P NMR spectroscopy and LC-MS.
2.5 General procedure for the alkaline hydrolysis of diphenylphosphinates (1a and 1b)
A mixture of 0.43 mmol of the diphenylphosphinate (1a: 0.10 g, 1b: 0.11 g) and 0.38–0.42–0.69 mL of 5% NaOH solution (0.47–0.52–0.85 mmol of NaOH) was stirred at 50°C for 5–45 min. To liberate the free acid, hydrochloric acid was added dropwise to the Na-salt. After evaporating the water, the residue was washed 3 times with 2 mL of water and then dried. The product so obtained was analyzed by 31P NMR spectroscopy.
2.6 General procedure for the alcoholysis of diphenylphosphinates (1a–e) with pentanol
0.43 mmol of alkyl diphenylphosphinate (1a: 0.10 g, 1b: 0.11 g, 1c: 0.11 g, 1d: 0.11 g, 1e: 0.12 g,) was added to 0.70 mL (6.5 mmol) n-pentanol and 0.043 mmol ([bmim][PF6]: 9 µL, [bmim][BF4]: 6.5 µL, [bmim][Cl]: 8 µL, [emim][HSO4]: 6.5 µL) ionic liquid. The mixture was irradiated in a sealed tube placed in CEM MW reactor at 220°C (max. 100 W) for 2 h. After evaporating the excess of the pentanol and purifying the residue by column chromatography (silica gel, hexane–ethyl acetate 7:3 as the eluent), the reaction mixture was analyzed by 31P NMR spectroscopy.
2.7 General procedure for the alcoholysis of methyl diphenylphosphinate (1a) with different alcohols
0.43 mmol of methyl diphenylphosphinate (1a) (0.10 g) was added to 6.5 mmol of the alcohol (n-propanol: 0.45 mL, i-propanol 0.48 mL, n-butanol 0.56 mL, i-butanol: 0.59 mL, cyclohexanol: 0.67 mL) and 9 µL (0.043 mmol) [bmim][PF6]. The mixture was irradiated in a sealed tube placed in CEM MW reactor at 200–220°C (max. 100 W) for 2–3.5 h. After evaporating the excess of the alcohol, and purifying the residue so obtained by column chromatography (silica gel, hexane–ethyl acetate 7:3 as the eluent), the phosphinates (1c–f, h) were obtained in yields of 89–92%. The esters 1c–h were analyzed by 31P NMR spectroscopy.
Identification of the starting material (1a) and the ester products (1c–h), as well as the acid (2), can be found in Table 1.
31P NMR characterization of diphenylphosphinates (1a–h) and diphenylphosphinic acid (2)
| Compounds | R | δ31P NMR | |
|---|---|---|---|
| Found (solvent) | Literature (solvent) | ||
| 1a | Me | 33.3 (CDCl3) | 33.3 [24] (CDCl3) |
| 1b | Et | 31.4 (CDCl3) | 31.4 [24] (CDCl3) |
| 1c | nPr | 31.2 (CDCl3) | 31.1 [24] (CDCl3) |
| 1d | iPr | 30.0 (CDCl3) | 29.8 [24] (CDCl3) |
| 1e | Bu | 31.9 (CDCl3) | 31.1 [24] (CDCl3) |
| 1f | iBu | 31.1 (CDCl3) | 31.0 [25] (CDCl3) |
| 1g | Pent | 31.3 (CDCl3) | 31.2 [24] (CDCl3) |
| 1h | cHex | 29.8 (CDCl3) | 29.7 [24] (CDCl3) |
| 2 | H | 23.5–23.7 (acidic) 23.6–23.8 (MW) 23.4–23.6 (alkaline) (DMSO) | 23.4 [26] (DMSO) |
2.8 Use of the 31P NMR spectra during monitoring the hydrolyses and alcoholyses
The composition of the reaction mixtures was determined by integration of the areas under the corresponding peaks of the starting material and product in the 31P NMR spectra.
2.9 Curve fitting on the time – relative quantity data pairs
The acidic hydrolyses were modelled assuming pseudo-first-order kinetics. The concentration of water and hydrochloric acid was constant during the reaction. The calculated time–composition curves were fitted to the experimental data using nonlinear least-squares method. The pseudo-first-order rate constants were optimized so that the sum of squares of the residuals (i.e., the difference of the experimental and the calculated composition) be the minimal. The approximate values of the rate constants were found iteratively, using the nonlinear generalized reduced gradient method of Microsoft Excel Solver.
3 Results and discussion
3.1 Hydrolysis of alkyl diphenylphosphinates under acidic conditions on conventional heating
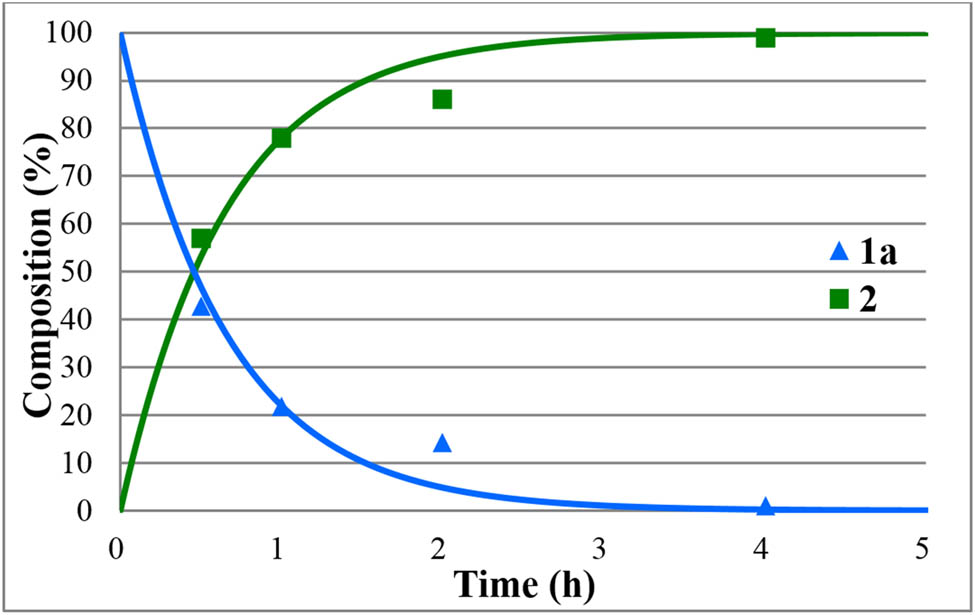
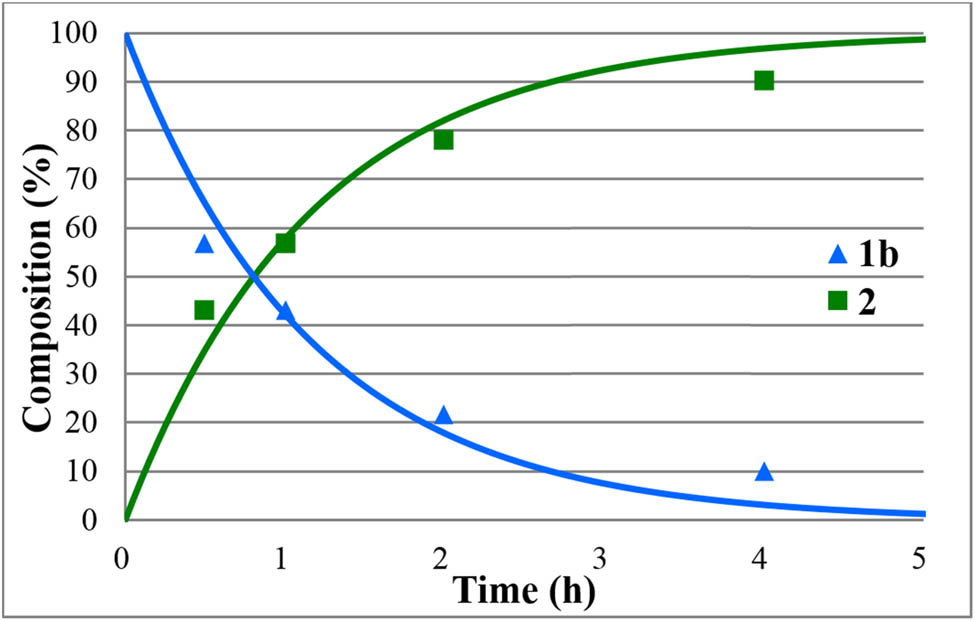
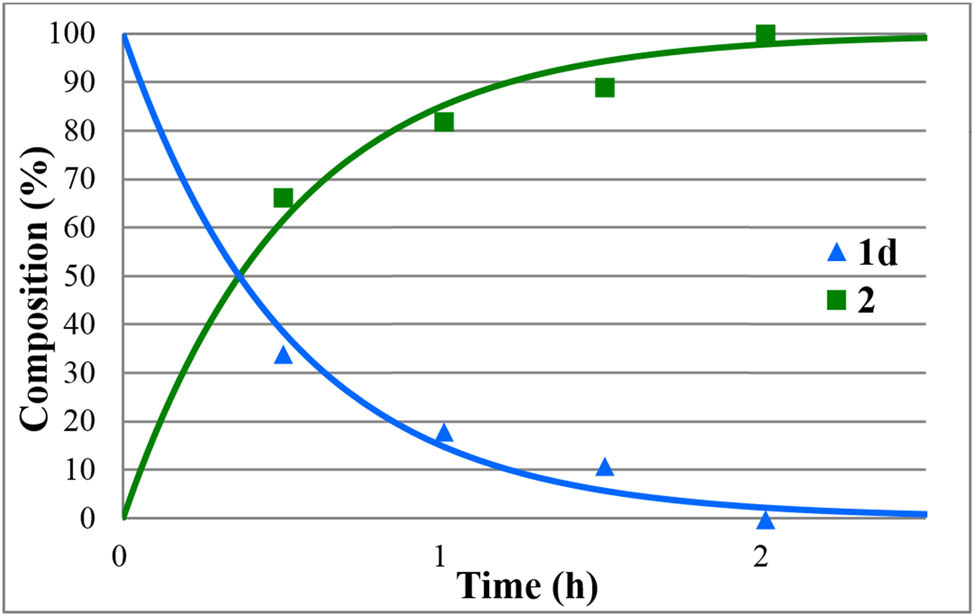
The hydrolyses of alkyl diphenylphosphinates (1a–e) were performed in water medium containing 12% of hydrochloric acid at reflux (Scheme 2). The reactions were monitored by 31P NMR. The concentration profiles for the components during the hydrolysis of the Me, Et, nPr, iPr, and Bu esters (1a–e) are shown in Figures 1–5. The curves were fit by the nonlinear least-squares method. It can be seen that the reaction time fell in the range of 3–7 h. The pseudo-first-order rate constants calculated are listed in Table 2. The hydrolysis of the methyl ester (1a) was significantly faster (t r = 4 h and k = 1.36 h−1) than that of the n-alkyl esters (1b,c,e) (t r = 6.5–7 h and k = 0.57–0.62 h−1). It is noteworthy that the iPr ester (1d) was hydrolyzed more than twice as much faster than the n Pr derivative (1c) (compare t r = 3 to 7 h, and k = 1.6 h−1 to k = 0.62 h−1). In the latter case, obviously the AAl1 mechanism operates.
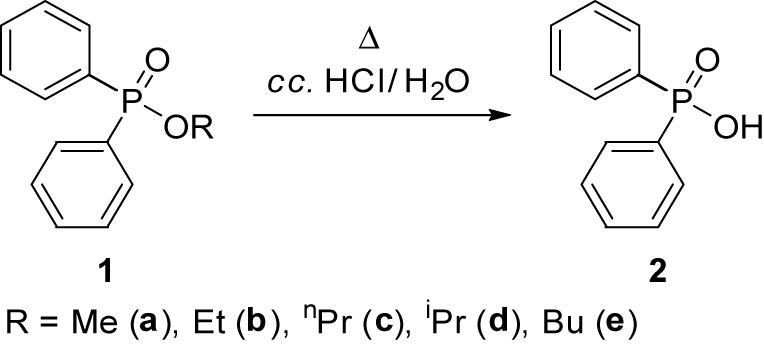
Acidic hydrolysis of diphenylphosphinates on conventional heating.
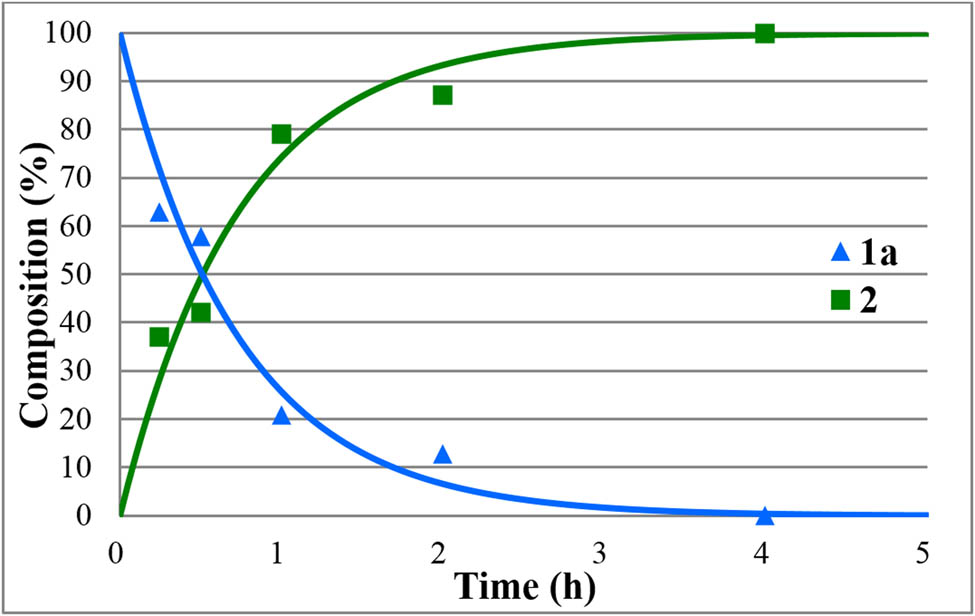
Concentration profile for the components (1a and 2) during the hydrolysis of methyl diphenylphosphinate (1a) under optimum conditions. The R 2 measure of goodness of fit is 0.941.

Concentration profile for the components (1b and 2) during the hydrolysis of ethyl diphenylphosphinate (1b) under optimum conditions. The R 2 measure of goodness of fit is 0.995.

Concentration profile for the components (1c and 2) during the hydrolysis of propyl diphenylphosphinate (1c) under optimum conditions. The R 2 measure of goodness of fit is 0.980.

Concentration profile for the components (1d and 2) during the hydrolysis of isopropyl diphenylphosphinate (1d) under optimum conditions. The R 2 measure of goodness of fit is 0.993.
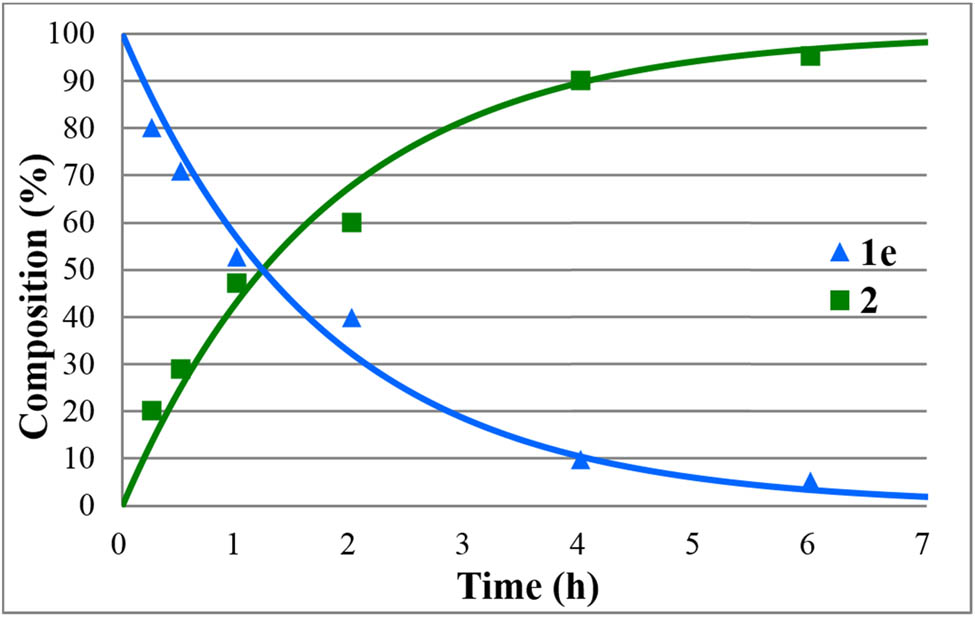
Concentration profile for the components (1e and 2) during the hydrolysis of butyl diphenylphosphinate (1e) under optimum conditions. The R 2 measure of goodness of fit is 0.958.
Pseudo-first-order rate constants (k Δ and k MW) obtained for the thermal HCl-catalyzed and MW-assisted PTSA-catalyzed hydrolyses
| Entry | R | k Δ (h−1) | k MW (h−1) |
|---|---|---|---|
| 1 | Me (1a) | 1.36 | 1.52 |
| 2 | Et (1b) | 0.62 | 0.86 |
| 3 | n Pr (1c) | 0.62 | — |
| 4 | iPr (1d) | 1.60 | 1.92 |
| 5 | n Bu (1e) | 0.57 | — |
The HCl-catalyzed hydrolyses of phosphinates 1a–e furnished diphenylphosphinic acid (2) in yields of 90–94%; however, the reaction times were in most cases (1b–d) as long as 7–8 h. The hydrolysis of the Me and iPr diphenylphosphinate (1a and 1e) was complete after 4 and 3 h, respectively.
3.2 Hydrolysis of alkyl diphenylphosphinates under acidic conditions on microwave irradiation
In the second phase of our study, the alkyl diphenylphosphinates (1a–e) were hydrolyzed under microwave (MW) irradiation. To avoid corrosion problems caused by the HCl, in these experiments PTSA served as the catalyst. The experimental data are summarized in Table 3.
MW-assisted hydrolysis of alkyl phenylphosphinates (1a–e) in the presence of PTSA catalyst
 |
|||||
|---|---|---|---|---|---|
| Entry | R | Temperature (°C) | PTSA (equiv.) | Reaction time (h) | Yield (%) |
| 1 | Me | 140 | 1 | 3.75 | |
| 2 | Me | 160 | 1 | 0.75 | |
| 3 | Me | 160 | 0.5 | 1.5 | |
| 4 | Me | 160 | 0.1 | 4 | |
| 5 | Me | 180 | 0.1 | 1.5 | 94 |
| 6 | Et | 160 | 0.1 | 6 | |
| 7 | Et | 180 | 0.1 | 2 | 96 |
| 8 | n Pr | 160 | 0.1 | 6.5 | |
| 9 | n Pr | 180 | 0.1 | 2.1 | 95 |
| 10 | iPr | 160 | 0.1 | 2 | |
| 11 | iPr | 180 | 0.1 | 0.5 | 97 |
| 12 | Bu | 160 | 0.1 | 6 | |
| 13 | Bu | 180 | 0.1 | 2.2 | 94 |
Applying 1 equivalent of PTSA, the hydrolysis of methyl phosphinate 1a was complete at 140°C after 3.75 h or at 160°C after 0.75 h (Table 3, entries 1 and 2). Using only 0.5 equivalents of PTSA at 160°C, the completion required 1.5 h (Table 3, entry 3). Decreasing the quantity of the catalyst to 0.1 equivalents, there was need for a reaction time of 4 h (Table 3, entry 4). Increasing the temperature to 180°C, an irradiation of 1.5 h was enough (Table 3, entry 5). The hydrolysis of the ethyl ester (1b) was somewhat slower; in the presence of 0.1 equivalents of PTSA at 160°C and 180°C, the completion took 6 and 2 h, respectively (Table 3, entries 6 and 7). Similar results were obtained for the hydrolysis of the n-propyl and the n-buthyl phosphinate (1c and 1e) (Table 3, entries 8, 9, 12, and 13). In the Me (1a), Et (1b), n Pr (1c), and Bu (1e) cases, the hydrolyses were about 3-times faster at 180°C than at 160°C. It is noteworthy that the hydrolysis of isopropyl diphenylphosphinate (1d) was again significantly faster: at 160°C and 180°C, the completion required 2 and 0.5 h, respectively (Table 3, entries 10 and 11). This is the consequence of the AAl1 mechanism.
A comparative thermal experiment corresponding to entry 5 of Table 3 afforded phosphinic acid 2 in a significantly lower conversion of 24% referring to the role of MWs. This observation may be the consequence of local overheatings [27] and the better MW absorbing ability of PTSA. Earlier, the beneficial effect of onium salts was demonstrated [28].
The MW-assisted hydrolyses performed at 180°C in the presence of 10% of PTSA can be regarded as robust and green affording diphenylphosphinic acid (2) practically quantitatively (in yields of 93–97%) after removing the PTSA catalyst by washing with water.
To determine the corresponding pseudo-first-order rate constants, the hydrolyses of phosphinates 1a, 1b, and 1d performed at 160°C in the presence of 0.1 equivalents of PTSA were monitored by 31P NMR (Figures 6–8). The rate constants are listed in Table 2.
Concentration profile for the components (1a and 2) during the hydrolysis of methyl diphenylphosphinate (1a) under MW conditions at 160°C. The R 2 measure of goodness of fit is 0.977.
Concentration profile for the components (1b and 2) during the hydrolysis of ethyl diphenylphosphinate (1b) under MW conditions at 160°C. The R 2 measure of goodness of fit is 0.903.
Concentration profile for the components (1d and 2) during the hydrolysis of isopropyl diphenylphosphinate (1d) under MW conditions at 160°C. The R 2 measure of goodness of fit is 0.896.
It can be seen that the rate constants obtained under MW conditions at 160°C applying 0.1 equivalents of PTSA were significantly higher than those determined on conventional heating at 100°C in the presence of 12% aqueous HCl.
As a comparison, the MW-assisted PTSA-catalyzed hydrolyses seem to be more advantageous than the HCl-promoted conversions on conventional heating, as the reaction times were significantly shorter, especially for the hydrolyses of the esters with n-alkyl substituent (ca. 5 h at 160°C vs 7–8 h at 100°C).
3.3 Alkaline hydrolysis of alkyl diphenylphosphinates
Beside acidic hydrolysis, the alkaline version is also a good option to convert P-esters to P-acids. The methyl and ethyl diphenylphosphinates (1a and 1b) were hydrolyzed at 50°C using aqueous 5% NaOH solution in different portions (Table 4). Applying 1.1, 1.2, and 2 equivalents of NaOH in reaction with methyl diphenylphosphinate (1a), the hydrolysis was complete after 45, 35, and 15 min, respectively (Table 4, entries 1–3). It is noteworthy that the conversion of the ethyl ester (1b) to diphenylphosphinic acid (2) was much slower under similar conditions and took 7 and 4 h using 1.2 equivalents and 2 equivalents of NaOH, respectively (Table 4, entries 4 and 5). The sensitivity of the basic hydrolysis on the substituents is in accord with earlier observations [29]. Two selected cases marked by entries 1 and 4 of Table 4 were monitored by 31P NMR. The resulting curves are shown in Figures 9 and 10, respectively. The second order rate constants were obtained as 4.31 and 0.31 dm3/mol h, respectively.
Alkaline hydrolysis of alkyl diphenylphosphinates (1a–b) under different conditions
 |
|||||
|---|---|---|---|---|---|
| Entry | R | NaOH (equiv.) | Time | Conversion to 2 (%) | k (dm3/mol h) |
| 1 | Me | 1.1 | 45 min | 98 | 4.31 |
| 2 | Me | 1.2 | 35 min | 100 | — |
| 3 | Me | 2 | 15 min | 100 | — |
| 4 | Et | 1.2 | 7 h | 100 | 0.31 |
| 5 | Et | 2 | 4 h | 98 | — |
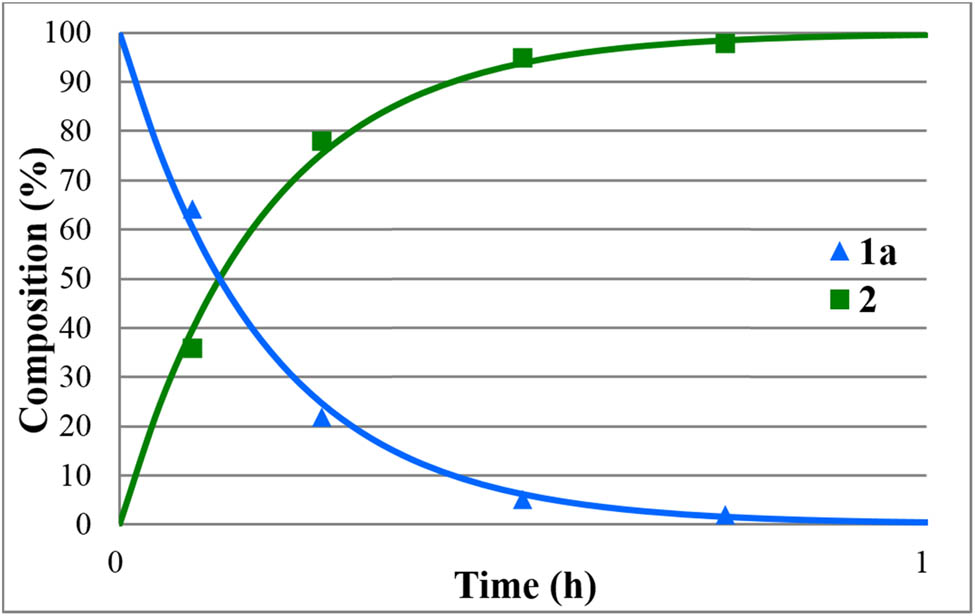
Concentration profile for the components (1a and 2) during the alkaline hydrolysis of methyl diphenylphosphinate (1a) at 50°C, in the presence of 1.1 equiv. NaOH. The R 2 measure of goodness of fit is 0.991.
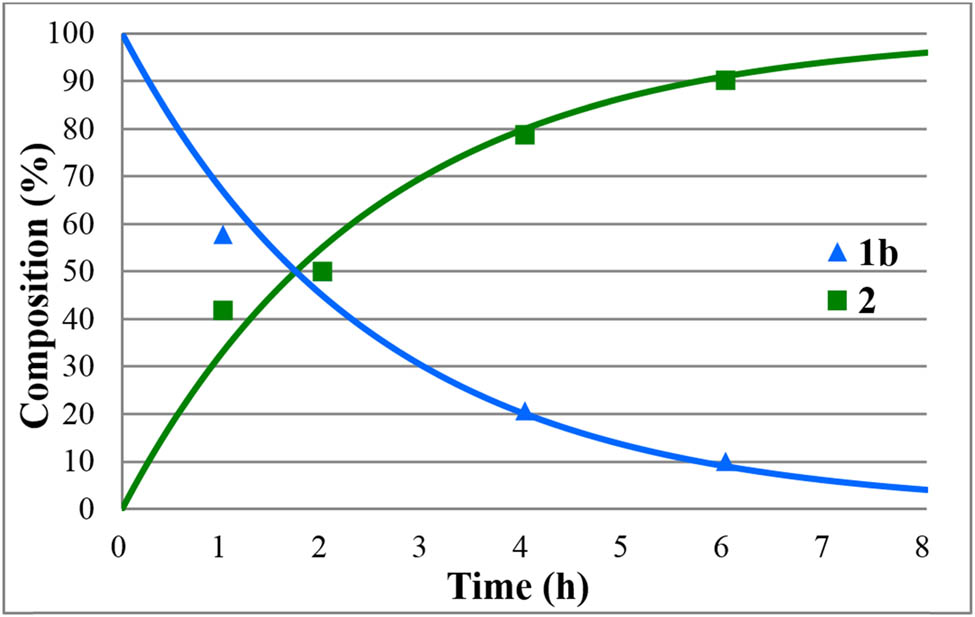
Concentration profile for the components (1b and 2) during the alkaline hydrolysis of ethyl diphenylphosphinate (1b) at 50°C, in the presence of 1.2 equiv. NaOH. The R 2 measure of goodness of fit is 0.931.
The alkaline hydrolysis is an alternative possibility to the option performed in the presence of an acid. It required a lower temperature of 50°C, but the rate was sensitive to the nature of the alkyl substituent: the hydrolysis of the methyl diphenylphosphinate (1a) was complete after 1 h, while that of the ethyl ester (1b) required a prolonged reaction time of 8 h. Moreover, after the hydrolysis, there was need to liberate the free acid (2) by an acidic treatment.
3.4 The alcoholysis of alkyl diphenylphosphinates
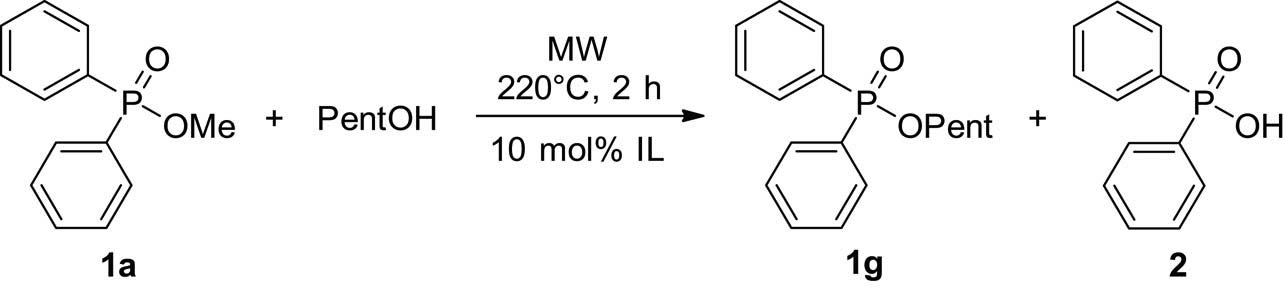
Phosphinic esters may also participate in alcoholysis reactions. The first model reaction was the transesterification of methyl diphenylphosphinate (1a) with pentanol. The results are summarized in Table 5. As can be seen, at 220°C, there was no reaction between phosphinate 1a and PentOH after a 2 h MW irradiation (Table 5, entry 1). On the basis of our earlier experiences, ionic liquid additives promoted the direct esterification of P-acids [4,5,30]. For this, the alcoholysis was also attempted in the presence of 10 mol% of the selected ionic liquids. Applying 10 mol% of [bmim][BF4] or [bmim][PF6] as an additive at 220°C, the conversion was 63% and 100%, respectively (Table 5, entries 2 and 3), meaning that the use of 10 mol% [bmim][PF6] is the method of choice for an efficient transesterification. The latter reaction was monitored by 31P NMR (Figure 11). The pseudo-first-order rate constant was obtained as 1.47 h−1. However, carrying out the model reaction in the presence of [bmim][Cl] and [emim][HSO4], surprisingly a side reaction comprising the fission of the ester group to the acid function predominated (Table 5, entries 4 and 5). The [emim][HSO4] additive was so efficient that the ratio of the ester (1g) and acid (2) was 18:82. In other words, instead of transesterification, the fission of the ester function took place with a selectivity of 82%. This novel transformation can be regarded as an alternative for hydrolysis and will be studied in detail in due course.
Alcoholysis of methyl diphenylphosphinate (1a) with pentanol under MW conditions in the presence of different ILs
 |
||||
|---|---|---|---|---|
| Entry | IL | Compositiona | ||
| 1a (%) | 1g (%) | 2 (%) | ||
| 1 | — | 99 | 1 | 0 |
| 2 | [bmim][BF4] | 37 | 63 | 0 |
| 3 | [bmim][PF6] | 0 | 100 | 0 |
| 4 | [bmim][Cl] | 42 | 2 | 56 |
| 5 | [emim][HSO4]b | 0 | 18 | 82 |
- a
On the basis of relative 31P NMR intensities.
- b
The reaction time was 130 min.
![Figure 11
Concentration profile for the components (1a and 1g) during the alcoholysis of methyl diphenylphosphinate (1a) with pentanol under MW condition at 220°C, in the presence of 10% of [bmim][PF6] monitored by LC-MS. The R
2 measure of goodness of fit is 0.938 (k = 1.31 h−1).](/document/doi/10.1515/gps-2021-0001/asset/graphic/j_gps-2021-0001_fig_011.jpg)
Concentration profile for the components (1a and 1g) during the alcoholysis of methyl diphenylphosphinate (1a) with pentanol under MW condition at 220°C, in the presence of 10% of [bmim][PF6] monitored by LC-MS. The R 2 measure of goodness of fit is 0.938 (k = 1.31 h−1).
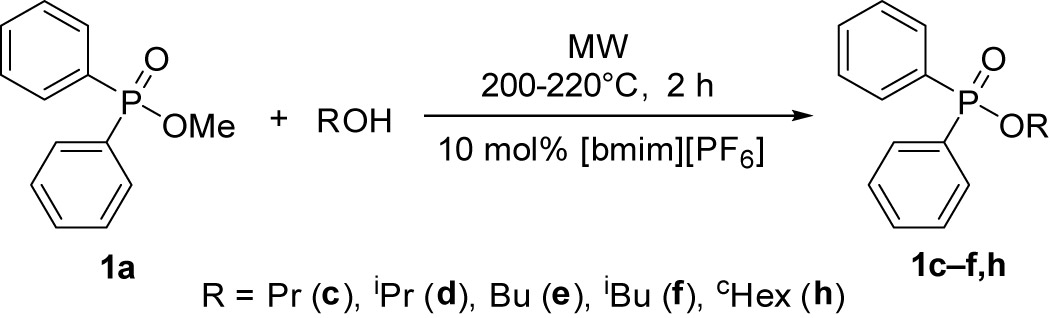
In the next stage, methyl diphenylphosphinate (1a) was reacted with different alcohols under MW irradiation in the presence of 10 mol% of [bmim][PF6] (Table 6). The alcoholysis of phosphinate 1a with n-propyl alcohol was complete after a 2 h heating at 200°C (Table 6, entry 1). At the same time, the similar reaction with i-propyl alcohol was not successful, as the product (1d) formed decomposed almost quantitatively under the conditions of the reaction (Table 6, entry 2). The unstability of i-propyl diphenylphosphinate (1d) on heating has been described [31]. Completion of the transesterification of phosphinate 1a with n- and i-butyl alcohol required 2 h at 220°C (Table 6, entries 3 and 4). The similar reaction of ester 1a with cyclohexanol required a longer reaction time of 3.5 h at 220°C (Table 6, entry 5). This is the consequence of steric hindrance. After flash column chromatography, the phosphinates were obtained in yields of 89–92%.
Alcoholysis of methyl diphenylphosphinate with different alcohols under MW conditions in the presence of 10 mol% of [bmim][PF6]
 |
||||||
|---|---|---|---|---|---|---|
| Entry | R | T (°C) | Time (h) | Compositiona | Yield (%) | |
| 1a (%) | 1c–h (%) | 1c–h | ||||
| 1 | n Pr | 200 | 2 | 5 (1c) | 95 | 89 |
| 2 | iPr | 200b | 3 | 3 (1d) | 16 | — |
| 3 | Bu | 220 | 2 | 1 (1e) | 99 | 92 |
| 4 | iBu | 220 | 2 | 1 (1f) | 99 | 90 |
| 5 | cHex | 220 | 3.5 | 3 (1h) | 97 | 90 |
- a
On the basis of relative 31P NMR intensities.
- b
81% of Ph2P(O)OH (2) was present in the mixture (δ P = 22.7 (DMSO), δ P[26] = 23.4 (DMSO); M + H = 219).
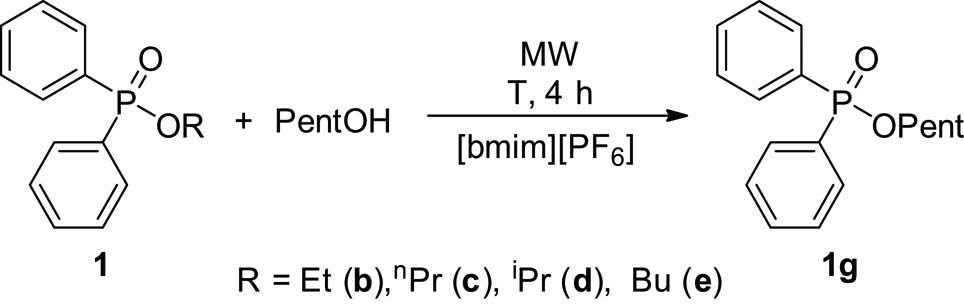
Finally, different alkyl phosphinates (1b–e) were subjected to alcoholysis with n-pentanol in the presence of 10 mol% of [bmim][PF6]. The case starting from the methyl ester (1a) is recalled (Table 5, entry 3). The alcoholysis of the ethyl phosphinate 1b hardly proceeded after a treatment at 220°C for 4 h (Table 7, entry 1). However, after an irradiation at 225°C for 4 h in the presence of 20 mol% of the additive, the conversion was 97% (Table 7, entry 2). Alcoholyses of the n-propyl and n-butyl esters (1c and 1e) at 225°C remained incomplete after 4 h, when only 10 mol% of the additive was used. The conversions were ca. 43% (Table 7, entries 3 and 6). Repeating the reaction at 225°C using 20 mol% of the additive, the conversion amounted to 80% (Table 7, entry 7). As regards the transesterification of i-propyl phosphinate 1d, it was faster than that of the n-propyl derivative (1c) (Table 7, entries 4 and 5 vs entry 3). Performing the reaction at 220°C for 4 h, only 16% starting material (1d) remained in the mixture, while at 225°C all 1d was consumed. This surprising result can be explained assuming a different mechanism. In the case under discussion, the i-propyl ester (1d) is converted to the corresponding acid (2), and the latter species take part in a direct MW-assisted and IL-catalyzed esterification. This was confirmed by the fact that some Ph2P(O)OH could be detected in the crude mixtures (see footnotes “b” and “c” of entries 4 and 5 of Table 7). The reactivity of the ethyl, n-propyl, and n-butyl phosphinates 1a, 1b, and 1e seemed to be comparable (see entries 1, 3, and 6, as well as entries 2 and 7 of Table 7).
Alcoholysis of alkyl diphenylphosphinates with pentanol under MW conditions in the presence of [bmim][PF6] as the catalyst
 |
|||||
|---|---|---|---|---|---|
| Entry | R | T (°C) | IL (mol%) | Compositiona | |
| 1b–e (%) | 1g (%) | ||||
| 1 | Et | 220 | 10 | 81 (1b) | 19 |
| 2 | Et | 225 | 20 | 3 (1b) | 97 |
| 3 | n Pr | 225 | 10 | 56 (1c) | 44 |
| 4 | iPr | 220 | 10 | 16 (1d) | 70b |
| 5 | iPr | 225 | 10 | 0 (1d) | 77c |
| 6 | Bu | 225 | 10 | 58 (1e) | 42 |
| 7 | Bu | 225 | 20 | 20 (1e) | 80 |
- a
On the basis of relative 31P NMR intensities.
- b
There was 14% of Ph2P(O)OH in the crude mixture.
- c
There was 23% of Ph2P(O)OH in the crude mixture.
4 Conclusions
In summary, from among the three possibilities of the hydrolysis of alkyl diphenylphosphinates, the MW-assisted and PTSA-catalyzed method seems to be the best due to the shorter reaction times, but the HCl-promoted option on conventional heating, as well as the alkaline hydrolysis, may also be applied as these are also efficient, but, with one exception, are somewhat slower. The reactivity of the alkyl phosphinates was also mapped and rate constants were determined. In case of the i-propyl ester, the AAl1 mechanism was substantiated. The alcoholyses of alkyl diphenylphosphinates may be performed under the effect of MWs in the presence of [bmim][PF6] as the catalyst. The application of [bmim][HSO4] led to the fission of the ester moiety to the acid function. This novel reactivity will be explored in due course.
Acknowledgment
The research was supported by the National Research, Development and Innovation Office (K134318). N. Z. K. is grateful for the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00130/19/7) and ÚNKP-20-5-BME-329 New National Excellence Program of the Ministry of Human Capacities.
References
[1] Quin LD. A guide to organophosphorus chemistry. New York: Wiley; 2000.Suche in Google Scholar
[2] Kiss NZ, Keglevich G. An overview of the synthesis of phosphinates and phosphinic amides. Curr Org Chem. 2014;18:2673–90. 10.2174/1385272819666140829011741.Suche in Google Scholar
[3] Kiss NZ, Keglevich G. Methods for the preparation of phosphinates and phosphonates with a focus on recent advances. In: Keglevich G, editor. Berlin: De Gruyter; 2018. p. 35–52.10.1515/9783110535839-002Suche in Google Scholar
[4] Kiss NZ, Keglevich G. Microwave-assisted direct esterification of cyclic phosphinic acids in the presence of ionic liquids. Tetrahedron Lett. 2016;57:971–4. 10.1016/j.tetlet.2016.01.044.Suche in Google Scholar
[5] Kiss NZ, Keglevich G. Direct esterification of phosphinic and phosphonic acids enhanced by ionic liquid additives. Pure Appl Chem. 2019;91:59–65. 10.1515/pac-2018-1008.Suche in Google Scholar
[6] Gavande N, Yamamoto I, Salam NK, Ai TH, Burden PM, Johnston GAR, et al. Novel cyclic phosphinic acids as GABAC receptor antagonists: design, synthesis, and pharmacology. ACS Med Chem Lett. 2011;2:11–6. 10.1021/ml1001344.Suche in Google Scholar PubMed PubMed Central
[7] Reiter LA, Jones BP. Amide-assisted hydrolysis of β-carboxamido-substituted phosphinic acid esters metal ions, and appropriately substituted phosphinic responsible for promoting the cleavage of the phosphinic acid esters. J Org Chem. 1997;62:2808–12. 10.1021/jo962275w.Suche in Google Scholar PubMed
[8] Yang Y, Coward JK. Synthesis of p-aminophenyl aryl H-phosphinic acids and esters via cross-coupling reactions: elaboration to phosphinic acid pseudopeptide analogues of pteroyl glutamic acid and related antifolates. J Org Chem. 2007;72:5748–58. 10.1021/jo0707840.Suche in Google Scholar PubMed
[9] Hall RG. The role of phosphorus in crop protection: commercial and experimental weed control agents. Phosphorus Sulfur Silicon Relat Elem. 2008;183:258–65. 10.1080/10426500701734216.Suche in Google Scholar
[10] Bunnett JF, Edwards JO, Wells DV, Brass HJ, Curci R. The hydrolysis of methyl methylarylphosphinates in perchloric acid solution. J Org Chem. 1973;38:2703–7. 10.1021/jo00955a028.Suche in Google Scholar
[11] Haake P, Hurst G. Reactions of phosphinates. The acid-catalyzed and acid-inhibited hydrolysis of p-nitrophenyl diphenylphosphinate. J Am Chem Soc. 1966;88:2544–50. 10.1021/ja00963a033.Suche in Google Scholar
[12] Tcarkova KV, Artyushin OI, Bondarenko NA. Synthetic routes to bis(3-aminophenyl) phosphinic acid. Phosphorus Sulfur Silicon Relat Elem. 2016;191:1520–2. 10.1080/10426507.2016.1212347.Suche in Google Scholar
[13] Desai J, Wang Y, Wang K, Malwal SR, Oldfield E. Isoprenoid biosynthesis inhibitors targeting bacterial cell growth. Chem Med Chem. 2016;11:2205–15. 10.1002/cmdc.201600343.Suche in Google Scholar PubMed PubMed Central
[14] Lin Y, Liu JT. Convenient synthesis of β-allenic α-difluoromethylenephosphonic acid monoesters: potential synthons for cyclic phosphate mimics. Chin Chem Lett. 2007;18:33–6. 10.1016/j.cclet.2006.11.029.Suche in Google Scholar
[15] Wróblewski AE, Verkade JG. 1-Oxo-2-oxa-1-phosphabicyclo[2.2.2]octane: a new mechanistic probe for the basic hydrolysis of phosphate esters. J Am Chem Soc. 1996;118:10168–74. 10.1021/ja9611147.Suche in Google Scholar
[16] Haake PC, Westheimer FH. Hydrolysis and exchange in esters of phosphoric acid. J Am Chem Soc. 1961;83:1102–9. 10.1021/ja01466a025.Suche in Google Scholar
[17] Zhang X, Glunz PW, Johnson JA, Jiang W, Jacutin-Porte S, Ladziata V, et al. Discovery of a highly potent, selective, and orally bioavailable macrocyclic inhibitor of blood coagulation factor VIIa-tissue factor complex. J Med Chem. 2016;59:7125–37. 10.1021/acs.jmedchem.6b00469.Suche in Google Scholar PubMed
[18] Keglevich G, Rádai Z, Harsági N, Szigetvári Á, Kiss NZ. A study on the acidic hydrolysis of cyclic phosphinates: 1-alkoxy-3-phospholene 1-oxides, 1-ethoxy-3-methylphospholane 1-oxide, and 1-ethoxy-3-methyl-1,2,3,4,5,6-hexahydrophosphinine 1-oxide. Heteroat Chem. 2017;28:e21394. 10.1002/hc.21394.Suche in Google Scholar
[19] Harsági N, Rádai Z, Kiss NZ, Szigetvári A, Keglevich G. Two step acidic hydrolysis of dialkyl arylphosphonates. Mendeleev Commun. 2020;30:38–9. 10.1016/j.mencom.2020.01.012.Suche in Google Scholar
[20] Harsági N, Rádai Z, Szigetvári Á, Kóti J, Keglevich G. Optimization and a kinetic study on the acidic hydrolysis of dialkyl α-hydroxybenzylphosphonates. Molecules. 2020;25:3793. 10.3390/molecules25173793.Suche in Google Scholar PubMed PubMed Central
[21] Bálint E, Tajti A, Drahos L, Ilia G, Keglevich G. Alcoholysis of dialkyl phosphites under microwave conditions. Curr Org Chem. 2013;17:555–62. 10.2174/1385272811317050010.Suche in Google Scholar
[22] Tajti A, Bálint E, Keglevich G. Synthesis of ethyl octyl α-aminophosphonate derivatives. Curr Org Synth. 2015;13:638–45. 10.2174/1570179413666151218202757.Suche in Google Scholar
[23] Kiss NZ, Henyecz R, Keglevich G. Continuous flow esterification of a H-phosphinic acid, and transesterification of H-phosphinates and H-phosphonates under microwave conditions. Molecules. 2020;25:719. 10.3390/molecules25030719.Suche in Google Scholar PubMed PubMed Central
[24] Ou Y, Huang Y, He Z, Yu G, Huo Y, Li X, et al. A phosphoryl radical-initiated Atherton–Todd-type reaction under open air. Chem Commun. 2020;56:1357–60. 10.1039/c9cc09407e.Suche in Google Scholar PubMed
[25] Keglevich G, Jablonkai E, Balázs LB. A “green” variation of the Hirao reaction: the P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides with bromoarenes using P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions. RSC Adv. 2014;4:22808–16. 10.1039/c4ra03292f.Suche in Google Scholar
[26] Gholivand K, Fallah N, Ebrahimi Valmoozi AA, Gholami A, Dusek M, Eigner V, et al. Synthesis and structural characterization of phosphinate coordination polymers with tin(IV) and copper(II). J Mol Struct. 2020;1202:127369. 10.1016/j.molstruc.2019.127369.Suche in Google Scholar
[27] Keglevich G, Kiss NZ, Mucsi Z, Körtvélyesi T. Insights into a surprising reaction: The microwave-assisted direct esterification of phosphinic acids. Org Biomol Chem. 2012;10:2011–8. 10.1039/c2ob06972e.Suche in Google Scholar PubMed
[28] Hohmann E, Keglevich G, Greiner I. The effect of onium salt additives on the Diels Alder reactions of a 1-phenyl-1,2-dihydrophosphinine oxide under microwave conditions. Phosphorus Sulfur Silicon. 2007;182:2351–7. 10.1080/10426500701441473.Suche in Google Scholar
[29] Mabey W, Mill T. Critical review of hydrolysis of organic compounds in water under environmental conditions. J Phys Chem Ref Data. 1978;7:383–415. 10.1063/1.555572.Suche in Google Scholar
[30] Henyecz R, Kiss A, Mórocz V, Kiss NZ, Keglevich G. Synthesis of phosphonates from phenylphosphonic acid and its monoesters. Synth Commun. 2019;49:2642–50. 10.1080/00397911.2019.1637894.Suche in Google Scholar
[31] Haake P, Diebert CE. Phosphinic acids and derivates. Pyrolytic elimination in phosphinate esters. J Am Chem Soc. 1971;93:6931–7. 10.1021/ja00754a040.Suche in Google Scholar
© 2021 Nikoletta Harsági et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Research Articles
- MW irradiation and ionic liquids as green tools in hydrolyses and alcoholyses
- Effect of CaO on catalytic combustion of semi-coke
- Studies of Penicillium species associated with blue mold disease of grapes and management through plant essential oils as non-hazardous botanical fungicides
- Development of leftover rice/gelatin interpenetrating polymer network films for food packaging
- Potent antibacterial action of phycosynthesized selenium nanoparticles using Spirulina platensis extract
- Green synthesized silver and copper nanoparticles induced changes in biomass parameters, secondary metabolites production, and antioxidant activity in callus cultures of Artemisia absinthium L.
- Gold nanoparticles from Celastrus hindsii and HAuCl4: Green synthesis, characteristics, and their cytotoxic effects on HeLa cells
- Green synthesis of silver nanoparticles using Tropaeolum majus: Phytochemical screening and antibacterial studies
- One-step preparation of metal-free phthalocyanine with controllable crystal form
- In vitro and in vivo applications of Euphorbia wallichii shoot extract-mediated gold nanospheres
- Fabrication of green ZnO nanoparticles using walnut leaf extract to develop an antibacterial film based on polyethylene–starch–ZnO NPs
- Preparation of Zn-MOFs by microwave-assisted ball milling for removal of tetracycline hydrochloride and Congo red from wastewater
- Feasibility of fly ash as fluxing agent in mid- and low-grade phosphate rock carbothermal reduction and its reaction kinetics
- Three combined pretreatments for reactive gasification feedstock from wet coffee grounds waste
- Biosynthesis and antioxidation of nano-selenium using lemon juice as a reducing agent
- Combustion and gasification characteristics of low-temperature pyrolytic semi-coke prepared through atmosphere rich in CH4 and H2
- Microwave-assisted reactions: Efficient and versatile one-step synthesis of 8-substituted xanthines and substituted pyrimidopteridine-2,4,6,8-tetraones under controlled microwave heating
- New approach in process intensification based on subcritical water, as green solvent, in propolis oil in water nanoemulsion preparation
- Continuous sulfonation of hexadecylbenzene in a microreactor
- Synthesis, characterization, biological activities, and catalytic applications of alcoholic extract of saffron (Crocus sativus) flower stigma-based gold nanoparticles
- Foliar applications of plant-based titanium dioxide nanoparticles to improve agronomic and physiological attributes of wheat (Triticum aestivum L.) plants under salinity stress
- Simultaneous leaching of rare earth elements and phosphorus from a Chinese phosphate ore using H3PO4
- Silica extraction from bauxite reaction residue and synthesis water glass
- Metal–organic framework-derived nanoporous titanium dioxide–heteropoly acid composites and its application in esterification
- Highly Cr(vi)-tolerant Staphylococcus simulans assisting chromate evacuation from tannery effluent
- A green method for the preparation of phoxim based on high-boiling nitrite
- Silver nanoparticles elicited physiological, biochemical, and antioxidant modifications in rice plants to control Aspergillus flavus
- Mixed gel electrolytes: Synthesis, characterization, and gas release on PbSb electrode
- Supported on mesoporous silica nanospheres, molecularly imprinted polymer for selective adsorption of dichlorophen
- Synthesis of zeolite from fly ash and its adsorption of phosphorus in wastewater
- Development of a continuous PET depolymerization process as a basis for a back-to-monomer recycling method
- Green synthesis of ZnS nanoparticles and fabrication of ZnS–chitosan nanocomposites for the removal of Cr(vi) ion from wastewater
- Synthesis, surface modification, and characterization of Fe3O4@SiO2 core@shell nanostructure
- Antioxidant potential of bulk and nanoparticles of naringenin against cadmium-induced oxidative stress in Nile tilapia, Oreochromis niloticus
- Variability and improvement of optical and antimicrobial performances for CQDs/mesoporous SiO2/Ag NPs composites via in situ synthesis
- Green synthesis of silver nanoparticles: Characterization and its potential biomedical applications
- Green synthesis, characterization, and antimicrobial activity of silver nanoparticles prepared using Trigonella foenum-graecum L. leaves grown in Saudi Arabia
- Intensification process in thyme essential oil nanoemulsion preparation based on subcritical water as green solvent and six different emulsifiers
- Synthesis and biological activities of alcohol extract of black cumin seeds (Bunium persicum)-based gold nanoparticles and their catalytic applications
- Digera muricata (L.) Mart. mediated synthesis of antimicrobial and enzymatic inhibitory zinc oxide bionanoparticles
- Aqueous synthesis of Nb-modified SnO2 quantum dots for efficient photocatalytic degradation of polyethylene for in situ agricultural waste treatment
- Study on the effect of microwave roasting pretreatment on nickel extraction from nickel-containing residue using sulfuric acid
- Green nanotechnology synthesized silver nanoparticles: Characterization and testing its antibacterial activity
- Phyto-fabrication of selenium nanorods using extract of pomegranate rind wastes and their potentialities for inhibiting fish-borne pathogens
- Hydrophilic modification of PVDF membranes by in situ synthesis of nano-Ag with nano-ZrO2
- Paracrine study of adipose tissue-derived mesenchymal stem cells (ADMSCs) in a self-assembling nano-polypeptide hydrogel environment
- Study of the corrosion-inhibiting activity of the green materials of the Posidonia oceanica leaves’ ethanolic extract based on PVP in corrosive media (1 M of HCl)
- Callus-mediated biosynthesis of Ag and ZnO nanoparticles using aqueous callus extract of Cannabis sativa: Their cytotoxic potential and clinical potential against human pathogenic bacteria and fungi
- Ionic liquids as capping agents of silver nanoparticles. Part II: Antimicrobial and cytotoxic study
- CO2 hydrogenation to dimethyl ether over In2O3 catalysts supported on aluminosilicate halloysite nanotubes
- Corylus avellana leaf extract-mediated green synthesis of antifungal silver nanoparticles using microwave irradiation and assessment of their properties
- Novel design and combination strategy of minocycline and OECs-loaded CeO2 nanoparticles with SF for the treatment of spinal cord injury: In vitro and in vivo evaluations
- Fe3+ and Ce3+ modified nano-TiO2 for degradation of exhaust gas in tunnels
- Analysis of enzyme activity and microbial community structure changes in the anaerobic digestion process of cattle manure at sub-mesophilic temperatures
- Synthesis of greener silver nanoparticle-based chitosan nanocomposites and their potential antimicrobial activity against oral pathogens
- Baeyer–Villiger co-oxidation of cyclohexanone with Fe–Sn–O catalysts in an O2/benzaldehyde system
- Increased flexibility to improve the catalytic performance of carbon-based solid acid catalysts
- Study on titanium dioxide nanoparticles as MALDI MS matrix for the determination of lipids in the brain
- Green-synthesized silver nanoparticles with aqueous extract of green algae Chaetomorpha ligustica and its anticancer potential
- Curcumin-removed turmeric oleoresin nano-emulsion as a novel botanical fungicide to control anthracnose (Colletotrichum gloeosporioides) in litchi
- Antibacterial greener silver nanoparticles synthesized using Marsilea quadrifolia extract and their eco-friendly evaluation against Zika virus vector, Aedes aegypti
- Optimization for simultaneous removal of NH3-N and COD from coking wastewater via a three-dimensional electrode system with coal-based electrode materials by RSM method
- Effect of Cu doping on the optical property of green synthesised l-cystein-capped CdSe quantum dots
- Anticandidal potentiality of biosynthesized and decorated nanometals with fucoidan
- Biosynthesis of silver nanoparticles using leaves of Mentha pulegium, their characterization, and antifungal properties
- A study on the coordination of cyclohexanocucurbit[6]uril with copper, zinc, and magnesium ions
- Ultrasound-assisted l-cysteine whole-cell bioconversion by recombinant Escherichia coli with tryptophan synthase
- Green synthesis of silver nanoparticles using aqueous extract of Citrus sinensis peels and evaluation of their antibacterial efficacy
- Preparation and characterization of sodium alginate/acrylic acid composite hydrogels conjugated to silver nanoparticles as an antibiotic delivery system
- Synthesis of tert-amylbenzene for side-chain alkylation of cumene catalyzed by a solid superbase
- Punica granatum peel extracts mediated the green synthesis of gold nanoparticles and their detailed in vivo biological activities
- Simulation and improvement of the separation process of synthesizing vinyl acetate by acetylene gas-phase method
- Review Articles
- Carbon dots: Discovery, structure, fluorescent properties, and applications
- Potential applications of biogenic selenium nanoparticles in alleviating biotic and abiotic stresses in plants: A comprehensive insight on the mechanistic approach and future perspectives
- Review on functionalized magnetic nanoparticles for the pretreatment of organophosphorus pesticides
- Extraction and modification of hemicellulose from lignocellulosic biomass: A review
- Topical Issue: Recent advances in deep eutectic solvents: Fundamentals and applications (Guest Editors: Santiago Aparicio and Mert Atilhan)
- Delignification of unbleached pulp by ternary deep eutectic solvents
- Removal of thiophene from model oil by polyethylene glycol via forming deep eutectic solvents
- Valorization of birch bark using a low transition temperature mixture composed of choline chloride and lactic acid
- Topical Issue: Flow chemistry and microreaction technologies for circular processes (Guest Editor: Gianvito Vilé)
- Stille, Heck, and Sonogashira coupling and hydrogenation catalyzed by porous-silica-gel-supported palladium in batch and flow
- In-flow enantioselective homogeneous organic synthesis
Artikel in diesem Heft
- Research Articles
- MW irradiation and ionic liquids as green tools in hydrolyses and alcoholyses
- Effect of CaO on catalytic combustion of semi-coke
- Studies of Penicillium species associated with blue mold disease of grapes and management through plant essential oils as non-hazardous botanical fungicides
- Development of leftover rice/gelatin interpenetrating polymer network films for food packaging
- Potent antibacterial action of phycosynthesized selenium nanoparticles using Spirulina platensis extract
- Green synthesized silver and copper nanoparticles induced changes in biomass parameters, secondary metabolites production, and antioxidant activity in callus cultures of Artemisia absinthium L.
- Gold nanoparticles from Celastrus hindsii and HAuCl4: Green synthesis, characteristics, and their cytotoxic effects on HeLa cells
- Green synthesis of silver nanoparticles using Tropaeolum majus: Phytochemical screening and antibacterial studies
- One-step preparation of metal-free phthalocyanine with controllable crystal form
- In vitro and in vivo applications of Euphorbia wallichii shoot extract-mediated gold nanospheres
- Fabrication of green ZnO nanoparticles using walnut leaf extract to develop an antibacterial film based on polyethylene–starch–ZnO NPs
- Preparation of Zn-MOFs by microwave-assisted ball milling for removal of tetracycline hydrochloride and Congo red from wastewater
- Feasibility of fly ash as fluxing agent in mid- and low-grade phosphate rock carbothermal reduction and its reaction kinetics
- Three combined pretreatments for reactive gasification feedstock from wet coffee grounds waste
- Biosynthesis and antioxidation of nano-selenium using lemon juice as a reducing agent
- Combustion and gasification characteristics of low-temperature pyrolytic semi-coke prepared through atmosphere rich in CH4 and H2
- Microwave-assisted reactions: Efficient and versatile one-step synthesis of 8-substituted xanthines and substituted pyrimidopteridine-2,4,6,8-tetraones under controlled microwave heating
- New approach in process intensification based on subcritical water, as green solvent, in propolis oil in water nanoemulsion preparation
- Continuous sulfonation of hexadecylbenzene in a microreactor
- Synthesis, characterization, biological activities, and catalytic applications of alcoholic extract of saffron (Crocus sativus) flower stigma-based gold nanoparticles
- Foliar applications of plant-based titanium dioxide nanoparticles to improve agronomic and physiological attributes of wheat (Triticum aestivum L.) plants under salinity stress
- Simultaneous leaching of rare earth elements and phosphorus from a Chinese phosphate ore using H3PO4
- Silica extraction from bauxite reaction residue and synthesis water glass
- Metal–organic framework-derived nanoporous titanium dioxide–heteropoly acid composites and its application in esterification
- Highly Cr(vi)-tolerant Staphylococcus simulans assisting chromate evacuation from tannery effluent
- A green method for the preparation of phoxim based on high-boiling nitrite
- Silver nanoparticles elicited physiological, biochemical, and antioxidant modifications in rice plants to control Aspergillus flavus
- Mixed gel electrolytes: Synthesis, characterization, and gas release on PbSb electrode
- Supported on mesoporous silica nanospheres, molecularly imprinted polymer for selective adsorption of dichlorophen
- Synthesis of zeolite from fly ash and its adsorption of phosphorus in wastewater
- Development of a continuous PET depolymerization process as a basis for a back-to-monomer recycling method
- Green synthesis of ZnS nanoparticles and fabrication of ZnS–chitosan nanocomposites for the removal of Cr(vi) ion from wastewater
- Synthesis, surface modification, and characterization of Fe3O4@SiO2 core@shell nanostructure
- Antioxidant potential of bulk and nanoparticles of naringenin against cadmium-induced oxidative stress in Nile tilapia, Oreochromis niloticus
- Variability and improvement of optical and antimicrobial performances for CQDs/mesoporous SiO2/Ag NPs composites via in situ synthesis
- Green synthesis of silver nanoparticles: Characterization and its potential biomedical applications
- Green synthesis, characterization, and antimicrobial activity of silver nanoparticles prepared using Trigonella foenum-graecum L. leaves grown in Saudi Arabia
- Intensification process in thyme essential oil nanoemulsion preparation based on subcritical water as green solvent and six different emulsifiers
- Synthesis and biological activities of alcohol extract of black cumin seeds (Bunium persicum)-based gold nanoparticles and their catalytic applications
- Digera muricata (L.) Mart. mediated synthesis of antimicrobial and enzymatic inhibitory zinc oxide bionanoparticles
- Aqueous synthesis of Nb-modified SnO2 quantum dots for efficient photocatalytic degradation of polyethylene for in situ agricultural waste treatment
- Study on the effect of microwave roasting pretreatment on nickel extraction from nickel-containing residue using sulfuric acid
- Green nanotechnology synthesized silver nanoparticles: Characterization and testing its antibacterial activity
- Phyto-fabrication of selenium nanorods using extract of pomegranate rind wastes and their potentialities for inhibiting fish-borne pathogens
- Hydrophilic modification of PVDF membranes by in situ synthesis of nano-Ag with nano-ZrO2
- Paracrine study of adipose tissue-derived mesenchymal stem cells (ADMSCs) in a self-assembling nano-polypeptide hydrogel environment
- Study of the corrosion-inhibiting activity of the green materials of the Posidonia oceanica leaves’ ethanolic extract based on PVP in corrosive media (1 M of HCl)
- Callus-mediated biosynthesis of Ag and ZnO nanoparticles using aqueous callus extract of Cannabis sativa: Their cytotoxic potential and clinical potential against human pathogenic bacteria and fungi
- Ionic liquids as capping agents of silver nanoparticles. Part II: Antimicrobial and cytotoxic study
- CO2 hydrogenation to dimethyl ether over In2O3 catalysts supported on aluminosilicate halloysite nanotubes
- Corylus avellana leaf extract-mediated green synthesis of antifungal silver nanoparticles using microwave irradiation and assessment of their properties
- Novel design and combination strategy of minocycline and OECs-loaded CeO2 nanoparticles with SF for the treatment of spinal cord injury: In vitro and in vivo evaluations
- Fe3+ and Ce3+ modified nano-TiO2 for degradation of exhaust gas in tunnels
- Analysis of enzyme activity and microbial community structure changes in the anaerobic digestion process of cattle manure at sub-mesophilic temperatures
- Synthesis of greener silver nanoparticle-based chitosan nanocomposites and their potential antimicrobial activity against oral pathogens
- Baeyer–Villiger co-oxidation of cyclohexanone with Fe–Sn–O catalysts in an O2/benzaldehyde system
- Increased flexibility to improve the catalytic performance of carbon-based solid acid catalysts
- Study on titanium dioxide nanoparticles as MALDI MS matrix for the determination of lipids in the brain
- Green-synthesized silver nanoparticles with aqueous extract of green algae Chaetomorpha ligustica and its anticancer potential
- Curcumin-removed turmeric oleoresin nano-emulsion as a novel botanical fungicide to control anthracnose (Colletotrichum gloeosporioides) in litchi
- Antibacterial greener silver nanoparticles synthesized using Marsilea quadrifolia extract and their eco-friendly evaluation against Zika virus vector, Aedes aegypti
- Optimization for simultaneous removal of NH3-N and COD from coking wastewater via a three-dimensional electrode system with coal-based electrode materials by RSM method
- Effect of Cu doping on the optical property of green synthesised l-cystein-capped CdSe quantum dots
- Anticandidal potentiality of biosynthesized and decorated nanometals with fucoidan
- Biosynthesis of silver nanoparticles using leaves of Mentha pulegium, their characterization, and antifungal properties
- A study on the coordination of cyclohexanocucurbit[6]uril with copper, zinc, and magnesium ions
- Ultrasound-assisted l-cysteine whole-cell bioconversion by recombinant Escherichia coli with tryptophan synthase
- Green synthesis of silver nanoparticles using aqueous extract of Citrus sinensis peels and evaluation of their antibacterial efficacy
- Preparation and characterization of sodium alginate/acrylic acid composite hydrogels conjugated to silver nanoparticles as an antibiotic delivery system
- Synthesis of tert-amylbenzene for side-chain alkylation of cumene catalyzed by a solid superbase
- Punica granatum peel extracts mediated the green synthesis of gold nanoparticles and their detailed in vivo biological activities
- Simulation and improvement of the separation process of synthesizing vinyl acetate by acetylene gas-phase method
- Review Articles
- Carbon dots: Discovery, structure, fluorescent properties, and applications
- Potential applications of biogenic selenium nanoparticles in alleviating biotic and abiotic stresses in plants: A comprehensive insight on the mechanistic approach and future perspectives
- Review on functionalized magnetic nanoparticles for the pretreatment of organophosphorus pesticides
- Extraction and modification of hemicellulose from lignocellulosic biomass: A review
- Topical Issue: Recent advances in deep eutectic solvents: Fundamentals and applications (Guest Editors: Santiago Aparicio and Mert Atilhan)
- Delignification of unbleached pulp by ternary deep eutectic solvents
- Removal of thiophene from model oil by polyethylene glycol via forming deep eutectic solvents
- Valorization of birch bark using a low transition temperature mixture composed of choline chloride and lactic acid
- Topical Issue: Flow chemistry and microreaction technologies for circular processes (Guest Editor: Gianvito Vilé)
- Stille, Heck, and Sonogashira coupling and hydrogenation catalyzed by porous-silica-gel-supported palladium in batch and flow
- In-flow enantioselective homogeneous organic synthesis