The entire chloroplast genome sequence of Asparagus setaceus (Kunth) Jessop: Genome structure, gene composition, and phylogenetic analysis in Asparagaceae
-
Quan Kuang


Abstract
Asparagus setaceus (Kunth) Jessop is a horticultural plant of the genus Asparagus. Herein, the whole chloroplast (cp) genome of A. setaceus was sequenced with PacBio and Illumina sequencing systems. The cp genome shows a characteristic quadripartite structure with 158,076 bp. In total, 135 genes were annotated, containing 89 protein-coding, 38 tRNA, and 8 rRNA genes. Contrast with the previous cp genome of A. setaceus registered in NCBI, we identified 7 single-nucleotide polymorphisms and 15 indels, mostly situated in noncoding areas. Meanwhile, 36 repeat structures and 260 simple sequence repeats were marked out. A bias for A/T-ending codons was shown in this cp genome. Furthermore, we predicted 78 RNA-editing sites in 29 genes, which were all for C-to-U transitions. And it was also proven that positive selection was exerted on the rpoC1 gene of A. setaceus with the K a/K s data. Meanwhile, a conservative gene order and highly similar sequences of protein-coding genes were revealed within Asparagus species. Phylogenetic tree analysis indicated that A. setaceus was a sister to Asparagus cochinchinensis. Taken together, our released genome provided valuable information for the gene composition, genetics comparison, and the phylogeny studies of A. setaceus.
1 Introduction
Chloroplast (cp) is the organ of photosynthesis in plant cells. The cp genome plays a key role in plant evolution, growth, and development [1]. In general, its genome is a circular double-stranded DNA molecule with a length of several hundred kilobases (kb). In its structure composition, the cp genome is mainly made up of four independent regions, namely consisting of a large single copy (LSC) region, two separate inverted repeat (IRa/IRb) regions, and a small single copy (SSC) region [2]. Based on the characteristics of its small genome size, conserved genome structure, and gene composition, the cp genomic sequences have supplied abundant data that are helpful for resolving the phylogenetic relationship in plant taxonomy [3].
Asparagus setaceus (common name: Asparagus fern) is a useful ornamental plant affiliated with the genus Asparagus in Asparagaceae. This genus includes both hermaphrodite and dioecious plants, which can be considered as an ideal genus for the sex chromosome origin and its phylogenetic relationship studies [4]. As a representative plant in this genus, A. setaceus is a hermaphrodite plant with a mall genome size, which can be used for the sex chromosome evolution analysis and species identification of the genus Asparagus [5]. Meanwhile, A. setaceus has also been proved to be used in Chinese traditional medicine [6]. Considering A. setaceus as a closely wild relative species of the most economical vegetable Asparagus officinalis in the same genus, it showed strong disease resistance such as rust dot commonly caused by Puccinia asparagi [7]. Moreover, we can utilize this important agronomical trait of A. setaceus to improve the cultivars of A. officinalis by molecular breeding technologies. Therefore, A. setaceus shows much importance in our scientific field and horticultural decoration value in ordinary lives for its intrinsic properties.
However, in spite of its great value, there were few genomic resources for A. setaceus. Up to now, limited systematic and comprehensive comparative studies of the cp genome was reported in this species, although only one assembled genome of A. setaceus has been registered in NCBI (GenBank accession number: NC_047458.1), but this reported genome was released without further sequence analysis. Within the genus, there are five cp genomes released in GenBank (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/Asparagus), thus providing valuable genetic information for genomics and phylogeny comparative analysis. In this research, the entire cp genome of A. setaceus was de novo sequenced with Illumina and PacBio sequencing technologies. In addition to gene annotation and genome characteristics analysis, we have identified a large number of single-nucleotide polymorphism (SNP) and insertion and deletions (Indels) between our new reported genome and the precious assembly registered in NCBI. Moreover, genomic comparison analysis was carried out with the registered cp genomes of Asparagus species, which were useful for the phylogenetic reconstruction, genomic information analysis, and evolutionary research in the genus Asparagus.
2 Materials and methods
2.1 DNA extraction and sequencing
The plant material of A. setaceus came from the Department of Biological Technology of Nanchang Normal University (115°27′E, 28°09′N). The genomic DNA of its tender fascicled cladodes was extracted by the improved cetyltrimethyl-ammonium bromide method, using the Qiagen genomic DNA extraction kit (Qiagen, CA, USA) [8]. Based on the manufacturer’s procedure, two libraries with the insert size of 350 bp and 20 kb were constructed individually and then sequenced on an Illumina HiSeq PE150 and a PacBio Sequel sequencing platform at Genepioneer Biotechnologies (Nanjing, China).
2.2 Cp genome assembly and annotation
The clean data obtained from the third-generation PacBio sequencing were spliced with Canu software, which included the process of error correction, modification, and assembly [9]. The contigs with coverage >10 were selected for homology search, the cp sequence was determined, and these contigs were screened. Taking the published cp genome sequences of A. officinalis (NC_034777.1) and A. setaceus (NC_047458.1) in NCBI as a reference genome, the cp data in the whole genome of the sample were isolated by Blastn search and its cp-related reads were assembled with the software Canu. To solve the problem of assembly accuracy in this third-generation sequenced genome, Nextpolish software was used in this study to polish the assembled genome combined with the second-generation Illumina sequencing data [10]. The Illumina reads were assembled with SOAPdenovo2 [11]. The software PGA was used for its annotation [12]. The annotated gene sequence was visualized in Geneious 11.0.3 software [13]. And the annotation was manually corrected to obtain the final result and submitted to GenBank with the serial login number of MT712152.1. Using online OGDRAW1.3.1 software mapped the whole cp genome of A. setaceus [14]. In addition, the indels and SNPs detected between the two cp genomes (MT712152.1 and NC_047458.1) were verified by PCR amplification and direct DNA product sequencing (primers used are listed in Table S1). The PCR system was 10 μL, including 1 μL of each forward and reverse primer, 1 μL of genomic DNA (100 ng/μL), 5 μL of 2× EasyTaq® PCR SuperMix (+dye), and 2 μL of deionized water. The PCR procedure was as follows: pre-denaturation at 95°C for 4 min; 35 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s; and 72°C for 5 min.
2.3 Comparative analysis in cp genome
Based on the Python script prepared by the research group, we counted the cp genome size, LSC, SSC, and IR region size, GC content, total gene number, and gene copy number. Compared with the prior deposited cp genome, the boundary difference of LSC, IR, and SSC regions was determined among five Asparagus plants using Mummer 3.0 [15]. Then, the boundaries of LSC, SSC, and IR regions of cp genomes in five Asparagus species were visualized by using the SVG module of Perl language, including the expansion and contraction of LSC, IR, and SSC regions, and the gene differences located on the boundaries.
2.4 Genome repeats and variation sites
The simple sequence repeat (SSR) sequence with repeat units of 1–6 bases in cp genome was marked out by using the script MISA written in Perl language [16]. The long segment repeats were detected by Reputer in the cp genome [17]. The specific parameter settings containing four types were as follows: forward, reverse, complementary, and palindrome; the shortest repeat unit contained at least 30 bp; and repeat sequence similarity was at least 90%. The cp genome sequences were compared by Mafft software [18]. Based on the comparison results, the mining and visualization of variable outliers were carried out by using Dnasp 6, and the parameters were set by default value [19].
2.5 Phylogenetic tree reconstruction
We downloaded the cp genome sequences of 25 species from NCBI (https://www.ncbi. nlm.nih.gov/genome/browse#!/organelles/) in Asparagaceae. Taking Allium chinense (NC_043922.1) in the Amaryllidaceae family as the out-group, the total genome sequences in this analysis were compared by Mafft software [18]. The comparison was further optimized by Trimal software to adjust the calculative results [20]. According to the trimmed comparison results, the phylogenetic tree of A. setaceus with the maximum likelihood (ML) algorithm was reconstructed using RAxML version 8.0 with the GTRGAMMA model [21]. The bootstrap value was set to 1,000 replicates.
3 Results
3.1 Cp genome characteristics
The cp genome of A. setaceus exhibited a quadripartite structure with a conserved genome arrangement (Figure 1). The cp genome size is 158,076 bp, including a pair of IRs (IRa and IRb, 55,160 bp in total) separated by a LSC region (84,264 bp) and a SSC region (18,652 bp). The GC content of the genome is 37.48%. And the GC content in the IR region (42.6%) was higher than that in LSC (35.45%) and SSC (31.47%), which was in accordance with previous studies [22]. The distribution of four rRNAs in IR region was an important reason for the high GC content in this part [23]. In addition, 135 genes were annotated in A. setaceus cp genome, composing of 38 tRNA, 8 rRNA, and 89 protein-coding genes (Table 1). It is reported that introns can regulate the gene transcription rate, which played a vital role in gene structure and function [24]. Statistics showed that 17 genes owned introns in the cp genome of A. setaceus. Among them, 10 protein-coding genes and 5 tRNA genes contained 1 intron, and 2 protein-coding genes (ycf3 and clpP) included 2 introns. Furthermore, rps12 was a trans-spliced gene, with 5′ end in the LSC region and 3′ end in the IR region. The length of the introns ranged from 222 to 1,122 bp, among which the intron of petb gene was the shortest with the size of 222 bp. And the intron of ndhA gene was the longest, which was 1,122 bp in size (Table 2). In addition, the number and type of introns contained in A. setaceus were consistent with A. officinalis, indicating a highly conserved cp genome of the genus Asparagus [25]. And the complete cp genome with gene annotations has been registered under GenBank accession number MT712152.1 for A. setaceus.
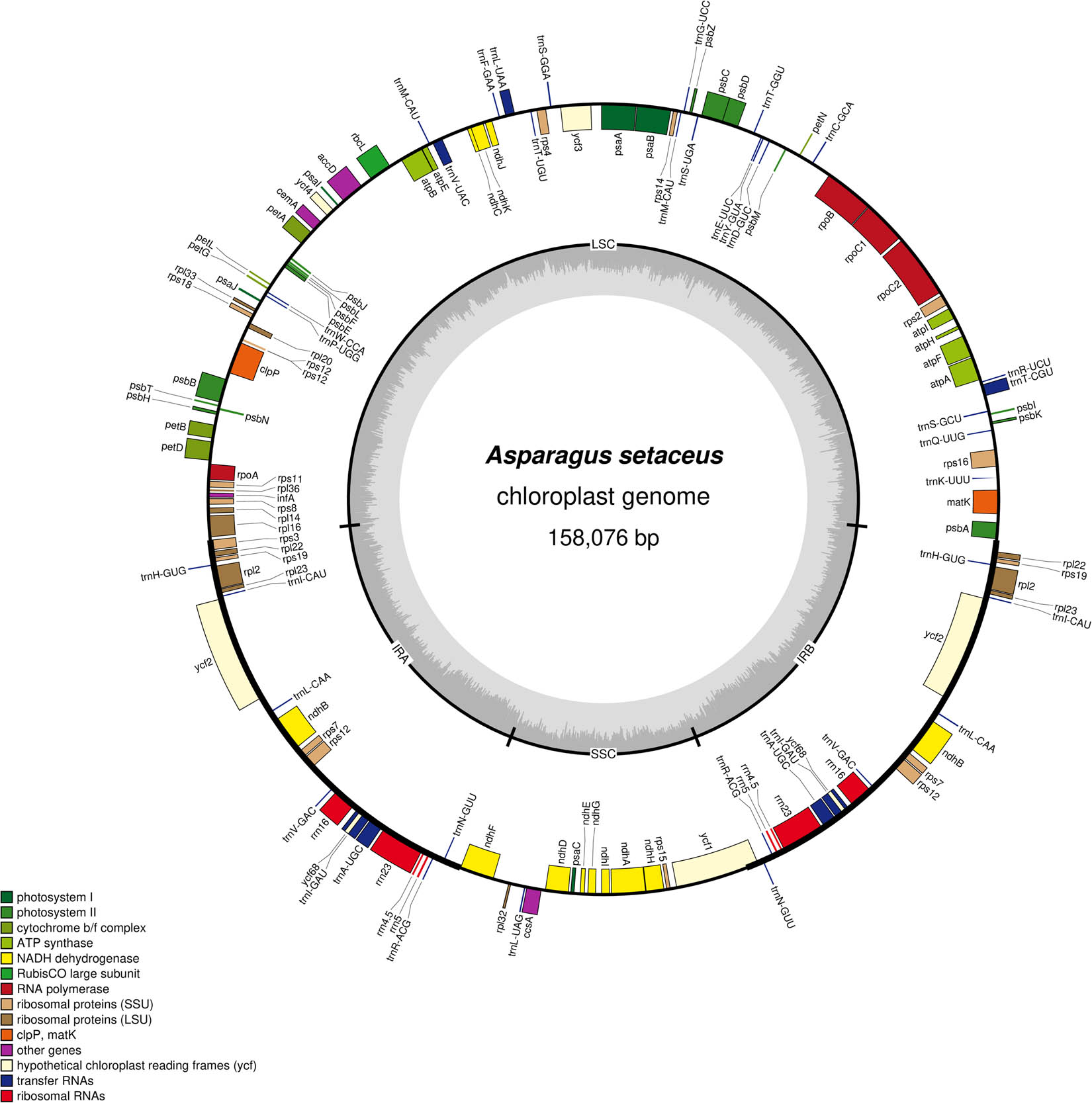
The cp genome map of A. setaceus. Note: The genes in the outer ring were arranged clockwise and the genes in the inner ring were arranged counterclockwise in the cp genome. Different colors represented different functions of genes. In the inner cycle, dark gray represented GC content; light gray represented AT content.
Gene annotation and classification in A. setaceus cp genome
| Category | Gene group | Gene name |
|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of NADH dehydrogenase | ndhA*, ndhB*(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB*, petD*, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Subunits photochlorophyllide reductase | — | |
| Self-replication | Proteins of large ribosomal subunit | rpl14, rpl16*, rpl2*(2), rpl20, rpl22(2), rpl23(2), rpl32, rpl33, rpl36 |
| Proteins of small ribosomal subunit | rps11, rps12**(2), rps14, rps15, rps16*, rps18, rps19(2), rps2, rps3, rps4, rps7(2), rps8 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1*, rpoC2 | |
| Ribosomal RNAs | rrn16(2), rrn23(2), rrn4.5(2), rrn5(2) | |
| Transfer RNAs | trnA-UGC*(2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-UCC, trnH-GUG(2), trnI-CAU(2), trnI-GAU*(2), trnK-UUU, trnL-CAA(2), trnL-UAA*, trnL-UAG, trnM-CAU(2), trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-CGU*, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-UAC*, trnW-CCA, trnY-GUA | |
| Other genes | Maturase | matK |
| Protease | clpP** | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor | infA | |
| other | — | |
| Genes of unknown function | Conserved hypothetical cp ORF | ycf1, ycf2(2), ycf3**, ycf4, ycf68(2) |
Notes: Gene* (gene with one introns); Gene** (gene with two introns); Gene (2) (gene with two copies).
The number and length of exons and introns in the cp genome of A. setaceus.
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| rps16 | LSC | 44 | 927 | 136 | ||
| trnT-CGU | LSC | 34 | 671 | 45 | ||
| atpF | LSC | 159 | 841 | 411 | ||
| rpoC1 | LSC | 438 | 740 | 1,632 | ||
| ycf3 | LSC | 129 | 750 | 228 | 718 | 153 |
| trnL-UAA | LSC | 35 | 527 | 50 | ||
| trnV-UAC | LSC | 39 | 585 | 46 | ||
| rps12 | IRa | 120 | — | 232 | 544 | 26 |
| clpP | LSC | 69 | 665 | 291 | 812 | 252 |
| petB | LSC | 6 | 222 | 642 | ||
| petD | LSC | 6 | 728 | 477 | ||
| rpl16 | LSC | 8 | 960 | 412 | ||
| rpl2 | IRb | 384 | 664 | 432 | ||
| ndhB | IRb | 777 | 699 | 756 | ||
| rps12 | IRb | 232 | — | 26 | 544 | 114 |
| trnI-GAU | IRb | 42 | 936 | 35 | ||
| trnA-UGC | IRb | 38 | 815 | 35 | ||
| ndhA | SSC | 558 | 1,122 | 540 | ||
| trnA-UGC | IRa | 38 | 815 | 35 | ||
| trnI-GAU | IRa | 42 | 936 | 35 | ||
| ndhB | IRa | 777 | 699 | 756 | ||
| rpl2 | IRa | 384 | 664 | 432 |
3.2 Genome variation
Contrast our new assembled genome with the prior registered genome in GenBank (NC_047458.1), we detected a number of variations including 7 SNPs (6 transversions and 1 transitions) and 16 indels (from 1 to 3 bp) between the two genomes. To further confirm the existence of these mutation sites, 23 pairs of primers were further designed to verify the existence of these mutation sites (Table S1). Among the variations, 2 SNPs and 13 indels were found in the LSC regions, 2 SNPs and 1 indels were marked out within the SSC region, 1 SNP and 1 indel were detected in the IRa region, and 2 SNPs and 1 indel were checked in the IRb region (Table 3). And nearly all the variations were positioned in noncoding regions consisting of intergenic spacer (IGS) and intron sequences, except two variations that were found in the rpoC1 and rps15 genes. From the above results, we can conclude that the variation in LSC region was the largest (65.22%), the variation in IR region was the second (21.74%), and the variation in SSC region was the smallest (13.04%) in A. setaceus cp genome. It was also found that the variation in noncoding region sequence (91.3%) was much greater than that in the coding region (8.7%).
SNP and Indel difference between the new registered MT712152.1 and the previous NC_047458.1 of A. setaceus cp genome in NCBI
| No. | Type | Region | Positiona | Location | MT712152.1 | NC_047458.1 | Gene |
|---|---|---|---|---|---|---|---|
| 1 | Indel | LSC | 3,363 | trnK-UUU-intron (Nb) | C | CTT | |
| 2 | Indel | LSC | 4,934 | trnK-UUU-rps16 (N) | C | CC | |
| 3 | Indel | LSC | 8,509 | trnS-GCU (N) | T | TAA | |
| 4 | Indel | LSC | 8,654 | trnS-GCU (N) | G | GA | |
| 5 | SNP | LSC | 22,410 | rpoC1(Cc) (N) | T | G | rpoC1 |
| 6 | Indel | LSC | 28,007–28,008 | trnC-GCA (N) | TA | T | |
| 7 | SNP | LSC | 42,876 | psaA (N) | G | C | |
| 8 | Indel | LSC | 60,326–60,326 | ycf4-intron (N) | C | CT | |
| 9 | Indel | LSC | 60,848 | ycf4-intron (N) | T | TAA | |
| 10 | Indel | LSC | 70,764 | rpl20-rps12 (N) | C | CA | |
| 11 | Indel | LSC | 71,986 | rps12-intron (N) | G | GA | |
| 12 | Indel | LSC | 73,101–73,102 | clpP-intron (N) | GC | G | |
| 13 | Indel | LSC | 77,858 | petB-intron(N) | G | GA | |
| 14 | Indel | LSC | 78,673–78,674 | petD-intron (N) | CG | C | |
| 15 | Indel | LSC | 82,133 | rpl14-rpl16 (N) | T | TAA | |
| 16 | SNP | IRA | 107,283 | trnA-UGC-rrn23 (N) | G | A | |
| 17 | Indel | IRA | 110,166 | trnN-GUU-intron (N) | C | CTT | |
| 18 | SNP | SSC | 114,478 | ycf1-intron (N) | C | T | |
| 19 | Indel | SSC | 114,489 | ycf1-intron (N) | G | GA | |
| 20 | SNP | SSC | 126,686 | rps15-intron (N) | T | G | rps15 |
| 21 | Indel | IRB | 132,569 | rps15-intron (N) | T | TAA | |
| 22 | SNP | IRB | 133,089 | trnA-UGC-trnV-GAC (N) | A | C | |
| 23 | SNP | IRB | 141,598 | trnV-GAC-intron (N) | A | C |
aNucleotide position is referenced to our new assembly. bN, noncoding sequences including IGS region and intron. cCoding sequences.
3.3 Codon usage bias and RNA editing sties predication
The relative synonymous codon usage (RSCU) was calculated in the cp genome of A. setaceus with Codon W1.4.2 (https://sourceforge.net/projects/codonw/files/OldFiles/CodonWSourceCode_1_4_2.tar.gz/download). Using the method proposed by Wright [26], 52 (coding sequence, CDs) sequences meeting the requirements were finally selected from 89 CDs annotated in the cp genome of A. setaceus for further analysis. According to the analysis results, leucine (2757, 10.31%) was the largest number amino acid among the proteins encoded by the cp genes, followed by Ile (2287, 8.55%) and Ser (2094, 7.83%). Cysteine (316, 1.18%) was the least abundant amino acid among the proteins encoded by the cp genes in A. setaceus genome (Figure 2). Leucine and isoleucine were the most commonly observed amino acids in the cp genome proteins. And usage of the codon UGG (tryptophan) had no bias (RSCU = 1). All preferred relative synonymous codons (RSCU > 1) ended with A or U.

The RSCU histogram in the cp genome of A. setaceus. Note: the Y-axis represents the value of RSCU, the X-axis represents the type of amino acids, and the following block represents the codon encoding each amino acid.
To gain insights into the RNA-editing sites in A. setaceus, 78 RNA editing sites of 29 cp genes were calculated with the PREP suite [27]. The result showed the number of editing sites was from 1 to 26, of which ndhB contained the largest number of editing sites. And most genes had one site, accounting for 46.67%. There were two types of editing sites, U–C and C–U, which were 16.67% and 83.33% respectively. Among the variation types of amino acids, the maximum number of serine (S)–leucine (L) was 37, accounting for 30.83% (Figure 3). It was seen that the amino acid conversion from S to L was the most frequent type. As previously reported, the conversion from S to L became more frequent along with the number increasing of amino acids [28]. This finding indicated that the amino acid conversion was essential in RNA editing during the evolutionary process.
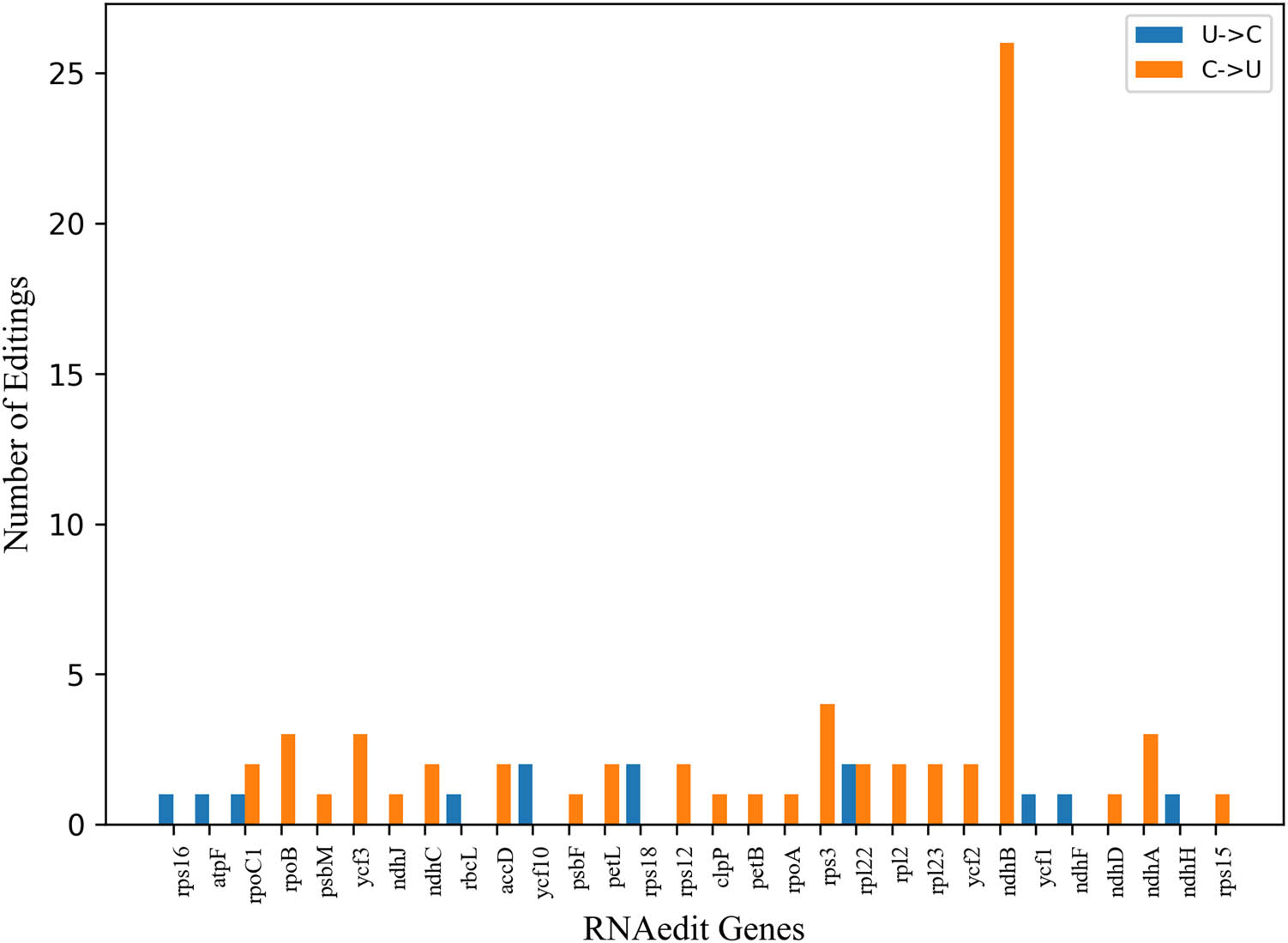
The value of RNA edit genes in the cp genome of A. setaceus. Note: Abscissa refers to different gene types; the ordinate refers to the number of RNA editing sites.
3.4 Repeat sequence analysis
Long repeats greater than or equal to 30 bp were considered playing a key role in genome rearrangement [29,30]. In A. setaceus, there were 36 repeats including 13 forward repeats, 2 reverse repeats, and 21 palindrome repeats, without complementary repeats (Table 4). The length distribution was mainly 30–56 bp in the repeat sequence; the longest repeat was 27,580 bp, positioned in the IR region; and the shortest repeat was 30 bp, containing 12 sites. According to the quadripartite structure in the cp genome, IR regions had the most repeats (16, 44.45%), followed by LSC region (12, 33.33%), SSC region (6, 16.67%), and the overhanging junction region (2, 5.55%). Based on the classification of gene structure, a majority of the repeat sites were located in IGS regions, in which the ycf2-IGS area contained the most numbers of repeat sites (4, 11.11%). And only a few types of genes (ycf1, ycf2, ycf3, psaB; psaA, trnS-GCU, trnS-GGA, atpF, trnS-UGA, trnS-GGA, trnT-CGU, trnG-UCC) possessed repeat elements, and ycf2 had the highest number of repeat sites (11, 30.56%).
Repeat sequences in the A. setaceus cp genome
| ID | Repeat I start | Repeat II start | Type | Size (bp) | Distance | E-value | Gene | Region |
|---|---|---|---|---|---|---|---|---|
| 1 | 84,265 | 130,497 | P | 27,580 | 0 | 0.00 × 1000 | — | IR |
| 2 | 29,819 | 29,819 | P | 56 | 0 | 1.35 × 10−24 | IGS | LSC; LSC |
| 3 | 90,646 | 90,667 | F | 49 | −1 | 3.26 × 10−18 | ycf2; ycf2 | IRb; IRb |
| 4 | 90,646 | 151,626 | P | 49 | −1 | 3.26 × 10−18 | ycf2; ycf2 | IRb; IRa |
| 5 | 90,667 | 151,647 | P | 49 | −1 | 3.26 × 10−18 | ycf2; ycf2 | IRb; IRa |
| 6 | 151,626 | 151,647 | F | 49 | −1 | 3.26 × 10−18 | ycf2; ycf2 | IRa; IRa |
| 7 | 39,149 | 41,373 | F | 47 | −3 | 1.55 × 10−13 | psaB; psaA | LSC; LSC |
| 8 | 44,084 | 100,421 | F | 39 | −3 | 5.74 × 10−9 | ycf3; IGS | LSC; IRb |
| 9 | 44,084 | 141,882 | P | 39 | −3 | 5.74 × 10−9 | ycf3; IGS | LSC; IRa |
| 10 | 125,629 | 125,629 | P | 39 | −3 | 5.74 × 10−9 | IGS | SSC; SSC |
| 11 | 126,988 | 126,988 | P | 39 | −3 | 5.74 × 10−9 | ycf1; ycf1 | SSC; SSC |
| 12 | 129,357 | 129,372 | F | 39 | −3 | 5.74 × 10−9 | ycf1; ycf1 | SSC; SSC |
| 13 | 8786 | 8786 | P | 38 | 0 | 9.30 × 10−14 | IGS | LSC; LSC |
| 14 | 32,202 | 32,223 | P | 37 | −3 | 7.81 × 10−8 | IGS | LSC; LSC |
| 15 | 46,984 | 46,987 | P | 37 | −3 | 7.81 × 10−8 | IGS | LSC; LSC |
| 16 | 127,224 | 127,224 | R | 34 | −2 | 1.20 × 10−7 | ycf1; ycf1 | SSC; SSC |
| 17 | 8222 | 45,499 | P | 33 | −2 | 4.53 × 10−7 | trnS-GCU; trnS-GGA | LSC; LSC |
| 18 | 12,664 | 13,097 | P | 33 | −3 | 1.40 × 10−5 | atpF; atpF | LSC; LSC |
| 19 | 115,799 | 115,799 | R | 33 | −2 | 4.53 × 10−7 | IGS | SSC; SSC |
| 20 | 69,877 | 69,893 | F | 32 | −2 | 1.70 × 10−6 | IGS | LSC; LSC |
| 21 | 8220 | 35,994 | F | 32 | −3 | 5.10 × 10−5 | trnS-GCU; trnS-UGA | LSC; LSC |
| 22 | 35,997 | 45,499 | P | 32 | −3 | 5.10 × 10−5 | trnS-UGA; trnS-GGA | LSC; LSC |
| 23 | 9970 | 36,975 | F | 31 | −3 | 1.85 × 10−4 | trnT-CGU; trnG-UCC | LSC; LSC |
| 24 | 114,879 | 114,879 | P | 31 | −3 | 1.85 × 10−4 | IGS | SSC; SSC |
| 25 | 46,996 | 46,996 | P | 30 | −2 | 2.39 × 10−5 | IGS | LSC; LSC |
| 26 | 90,668 | 90,689 | F | 30 | −2 | 2.39 × 10−5 | ycf2; ycf2 | IRb; IRb |
| 27 | 90,668 | 151,623 | P | 30 | −2 | 2.39 × 10−5 | ycf2; ycf2 | IRb; IRa |
| 28 | 90,689 | 151,644 | P | 30 | −2 | 2.39 × 10−5 | ycf2; ycf2 | IRb; IRa |
| 29 | 88,000 | 88,023 | F | 30 | −3 | 6.68 × 10−4 | IGS; ycf2 | IRb; IRb |
| 30 | 88,000 | 154,289 | P | 30 | −3 | 6.68 × 10−4 | IGS; ycf2 | IRb; IRa |
| 31 | 88,023 | 154,312 | P | 30 | −3 | 6.68 × 10−4 | ycf2; IGS | IRb; IRa |
| 32 | 90,644 | 90,686 | F | 30 | −3 | 6.68 × 10−4 | ycf2; ycf2 | IRb; IRb |
| 33 | 90,644 | 151,626 | P | 30 | −3 | 6.68 × 10−4 | ycf2; ycf2 | IRb; IRa |
| 34 | 90,686 | 151,668 | P | 30 | −3 | 6.68 × 10−4 | ycf2; ycf2 | IRb; IRa |
| 35 | 151,623 | 151,665 | F | 30 | −3 | 6.68 × 10−4 | ycf2; ycf2 | IRa; IRa |
| 36 | 154,289 | 154,312 | F | 30 | −3 | 6.68 × 10−4 | ycf2; IGS | IRa; IRa |
Tandem repeat sequences were known as SSRs or microsatellites, usually consisting of 1–6 nucleotide repeat units. The majority of SSRs were mono- and tri-nucleotide repeats in A. setaceus cp genome, which had the number of 155 and 79 times, respectively. The mononucleotide repeats were almost A/T repeats (96.15%), and 76.92% of the dinucleotide repeats were AT/TA repeats. SSRs in cp genome of A. setaceus also preferred to use A/T bases, which was in line with previous studies on A. officinalis, that is, SSR markers in plant cps were rich in A/T repeats [25,31]. And 13 di-nucleotide, 12 tetra-nucleotide, and only one hexa-nucleotide SSRs were detected (Figure 4). The length of repeated sequences was found to range from 8 to 16 bp, similar with the lengths reported in other angiosperm plants [32]. Therefore, the high variation in SSRs in the A. setaceus cp genome is of great value for the development of molecular marker studies.
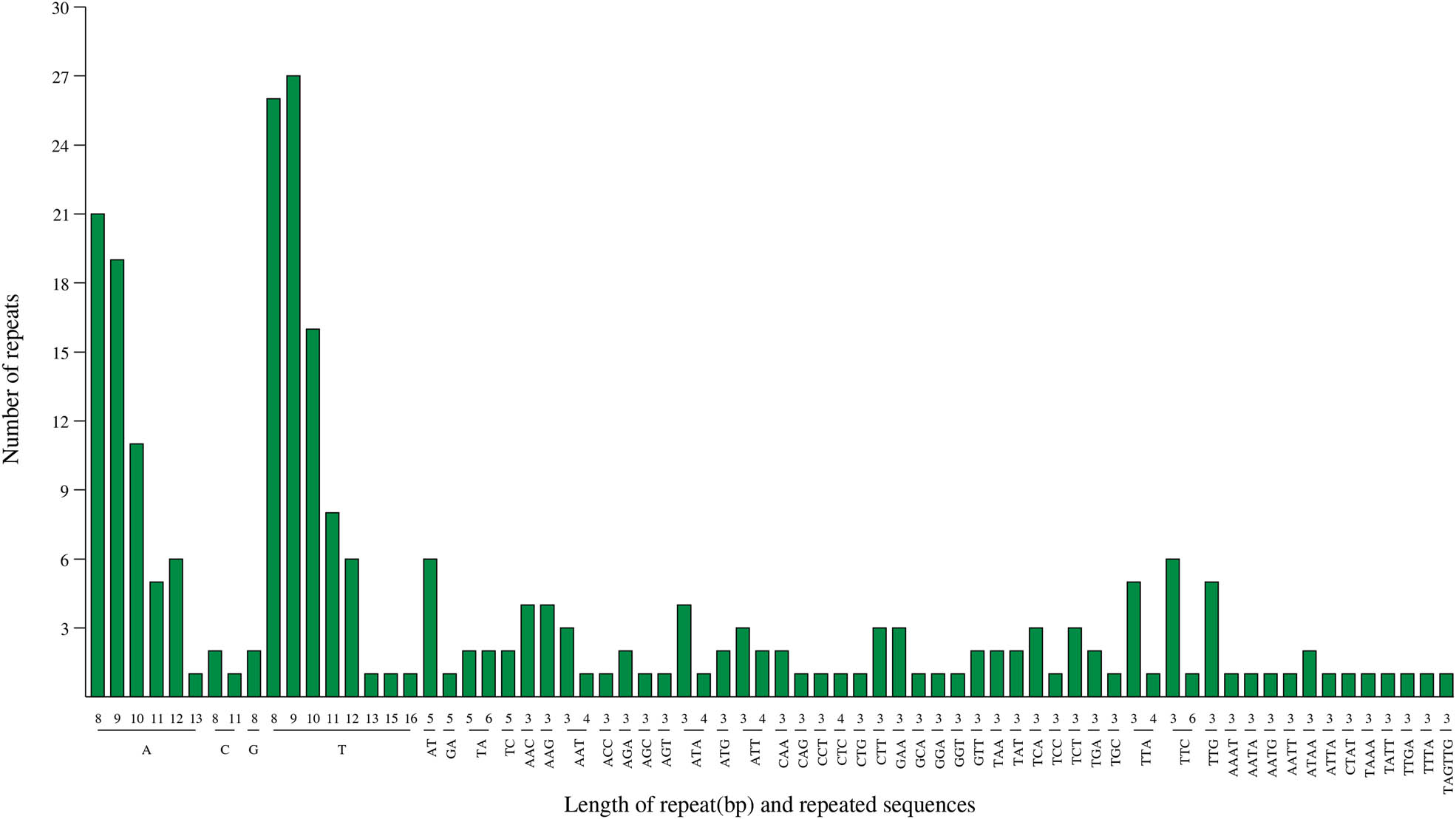
The type and number of SSR motif in the A. setaceus cp genome.
3.5 Non-synonymous/synonymous substitution value analysis
To further study the selection pressure on cp genes of A. setaceus and other Asparagus species in the process of evolution, the K a/K s values of protein-coding genes in A. setaceus vs A. officinalis, A. setaceus vs Asparagus schoberioides, A. setaceus vs Asparagus filicinus, and A. setaceus vs Asparagus racemasus were calculated by Dnasp software individually [19] (Figure 5). In total, 80 protein-coding genes were analyzed. The K a/K s average value was 0.1962, 0.2413, 0.1836, and 0.2547, respectively, and most of the genes had K a/K s < 1, which showed that the cp genes of the Asparagus species had been strongly purified and selected in the long-term evolution process.
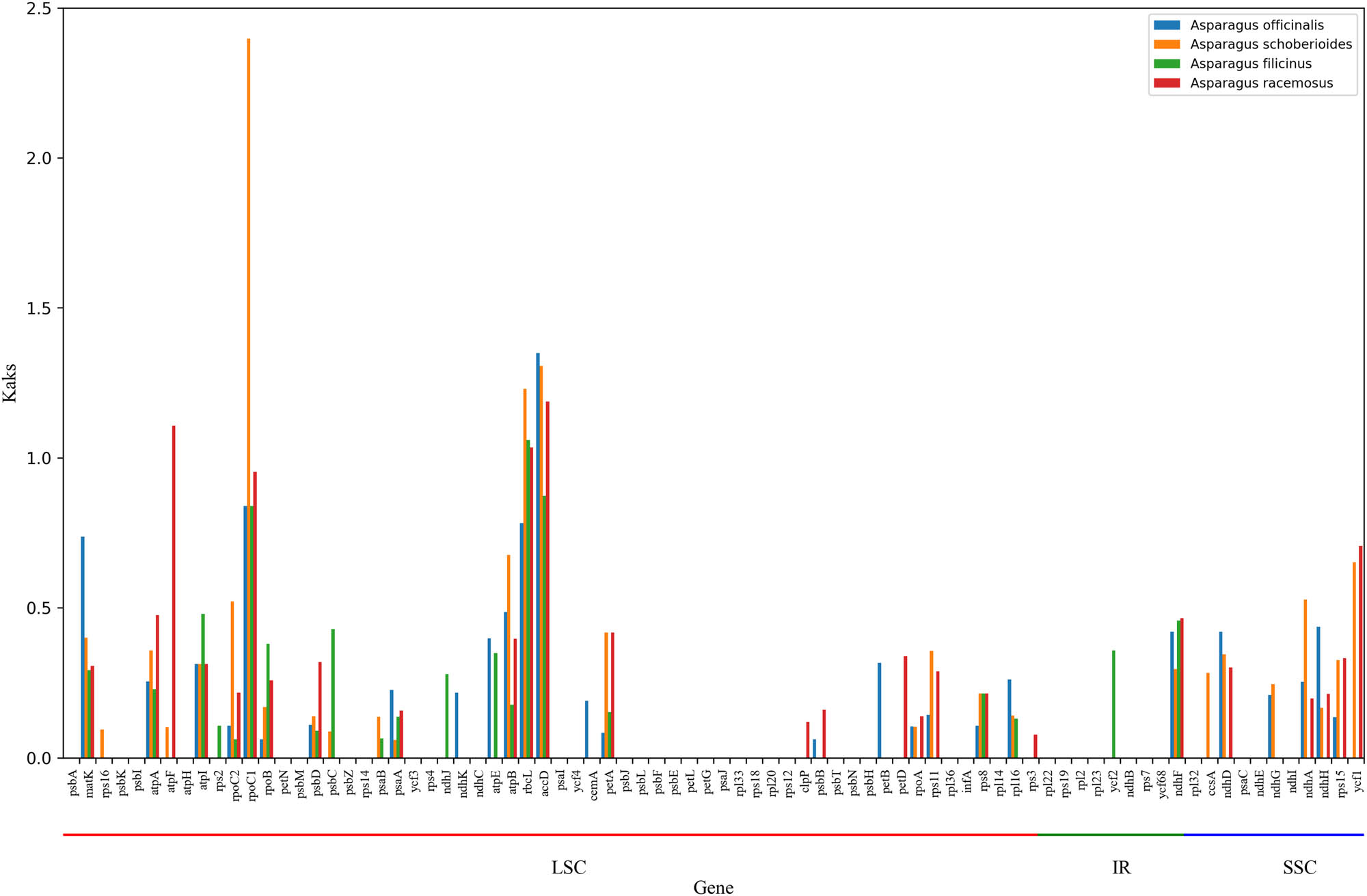
The K a/K s ratio of 80 protein-coding genes of the A. setaceus genome and four closely related species in the genus Asparagus.
3.6 IRScope analysis
The study showed that there were four boundaries in the cp genome of the Asparagus species, namely containing LSC region-inverted region b (LSC-IRb), inverted region b-SSC region (IRb-SSC), SSC region-inverted region a (SSC-IRa), and inverted region a-LSC region (IRa-LSC). The cp genome structure of the five selected Asparagus plants was relatively conservative (Figure 6). It was found that the boundaries between these species were consistent, and the difference was the length of genes from the boundary.
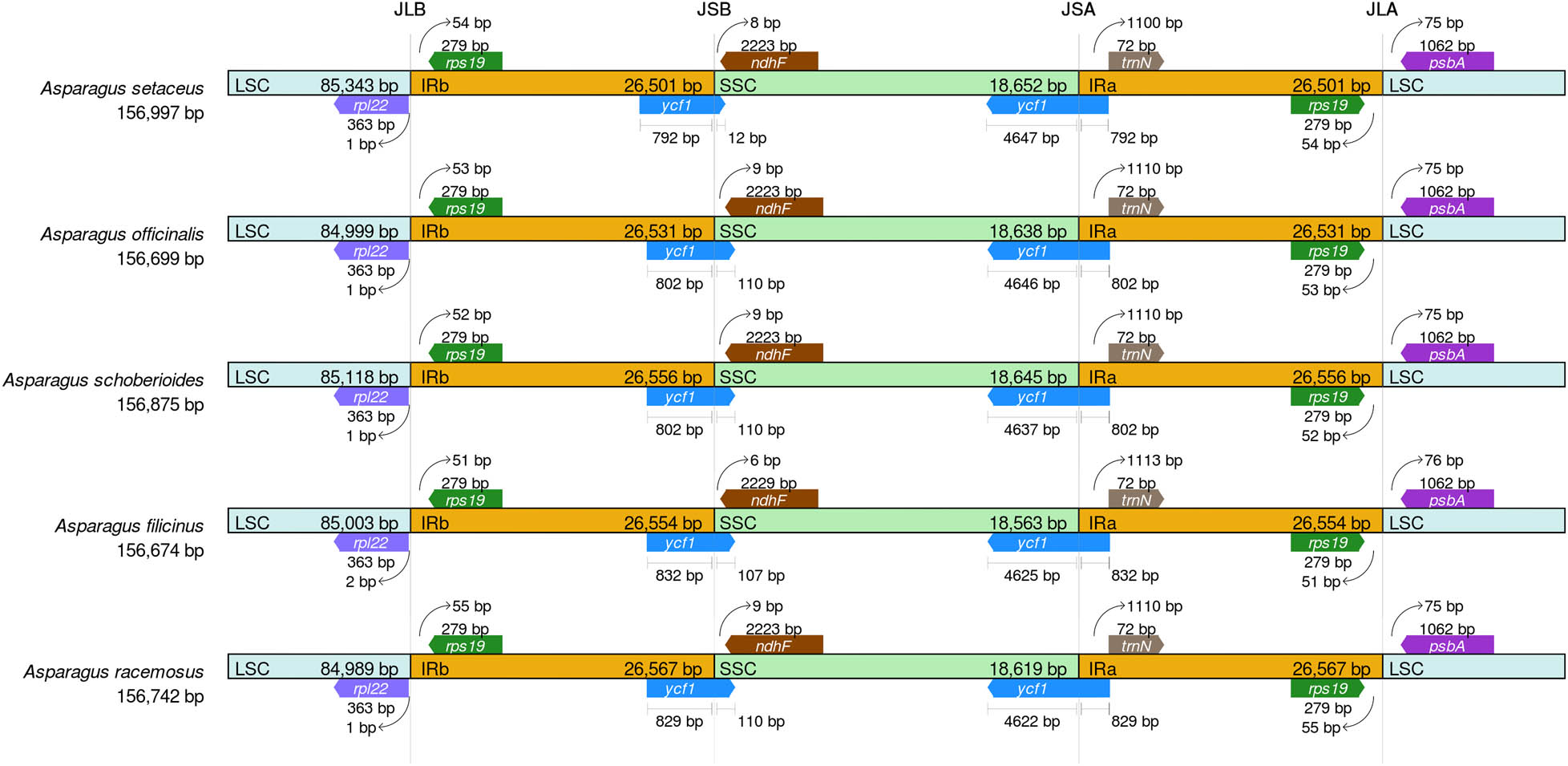
Comparative analysis of LSC, SSC, and IR regional boundaries of five Asparagus species.
3.7 Genome comparative analysis
The five known cp genome sequences in the genus Asparagus were compared. The result indicated that species with the largest genome was A. officinalis and that with the smallest was A. setacus. The gene order and content in the cp genome were used to analyze its difference with the online program mVISTA (https://genome.lbl.gov/vista/vista/bout.html). The gene order and contents of the Asparagus plants were found to be similar with those of other members in the genus Asparagus (Figure 7). It can be seen that all Asparagus species had conserved cp genomes, their coding regions were more conserved than their noncoding regions, and their IR regions were more conserved than their LSC and SSC regions.

Sequence alignment of five cp genome in the genus Asparagus by mVISTA, with the annotation of A. setaceus as the reference.
3.8 Phylogenetic relationship reconstruction
The cp genome contains abundant information, and its structure, size, and gene composition are relatively constant, which has been widely utilized in phylogenetic analysis and species identification [33]. The cp genome can be used to resolve the deeper branches within species. To straighten out the phylogenetic positions of A. setaceus within the genus Asparagus, the ML method of phylogenetic analysis was performed based on the complete cp genome dataset from 24 plant taxa, with A. chinense used as the out-group. The ML tree had similar phylogenetic topologies, and most nodal support values were high. The higher was the branch’s credibility, the more consistent was the guiding value of the evolutionary analysis for the relationship [1]. Furthermore, the phylogenetic tree suggested that A. setaceus formed a single group, Asparagus cochinchinensis and Asparagus densiflorus were grouped into another group, and they were sister groups with a support rate of 100% (Figure 7). This was similar to Norup’s research result [34]. It was speculated that A. setaceus belonged to the subgenus Asparagopsis derived from the African origin, which had a certain genetic distance from other sub-genus Asparagus group in Asia (Figure 8).
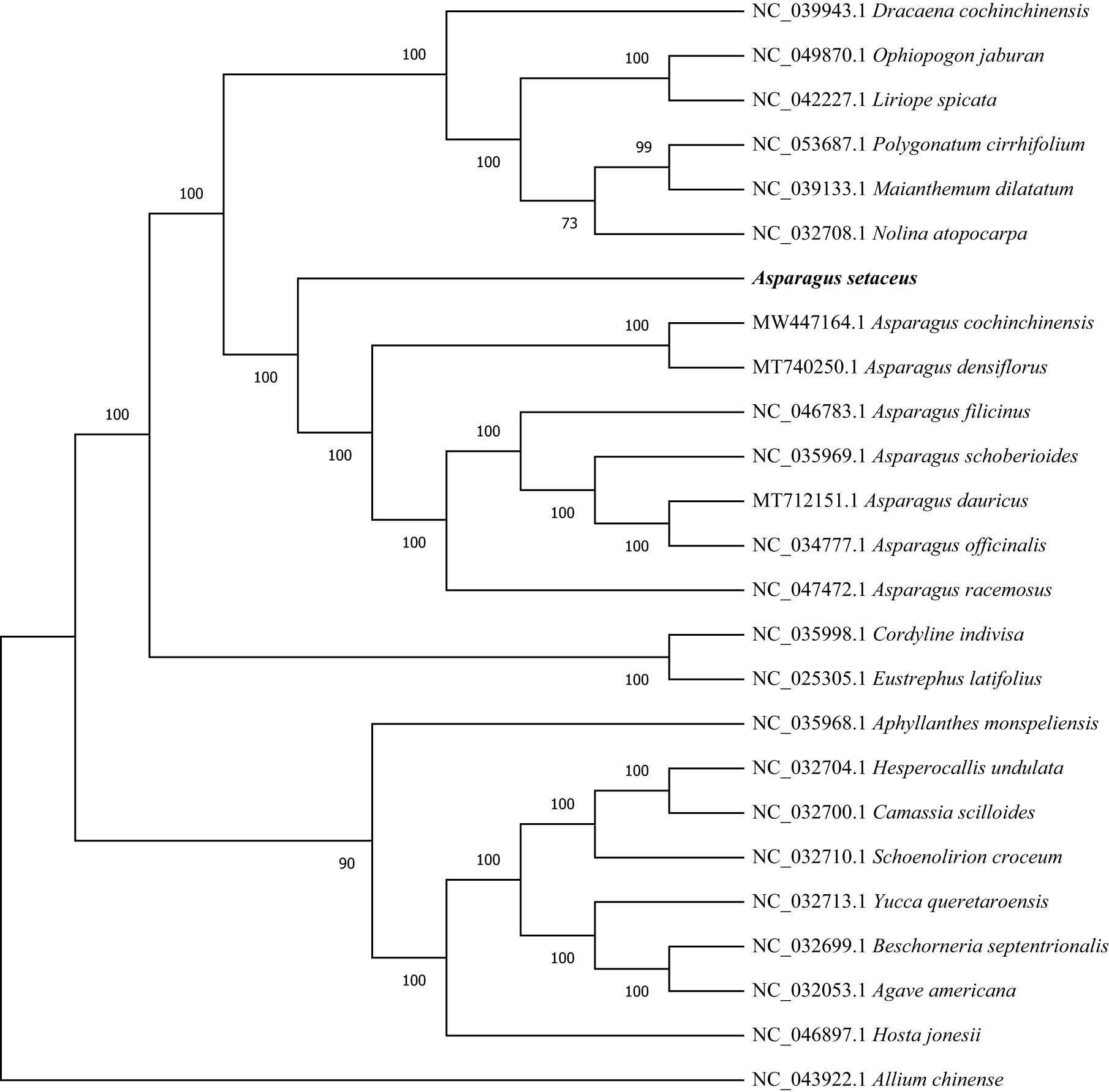
Phylogenetic tree was built based on the entire cp genomes from 25 species using RAxML. with GTRGAMMA model. And A. chinense was set as the out-group. Bootstrap values were displayed at the nodes.
4 Discussion
There are generally two traditional methods for obtaining plant cp genome. One is to isolate cp organelles from plant tissues, then extract cp DNA, and obtain plant cp genome with the first- or second-generation sequencing technology. But it is difficult to isolate whole cps and obtain high-quality cp DNA. The other method is to extract the whole plant genome DNA and then use the conserved region of cp genome to design primers with the first-generation sequencing method and finally splice the plant cp genome. The disadvantage of this method is that it is difficult to obtain complete cp genome sequence [32]. Along with the development of the new-generation sequencing technology, especially the second- and third-generation sequencing technology, and the extensive use of a large number of Bioinformatics software, the whole genome DNA of plants can be extracted for high-throughput sequencing, and the cp reads of the samples are extracted and assembled to obtain the cp genome of plants. This method does not require the separation of cp DNA, reduces the labor intensity, and improves the success rate of the experiment [35]. The Illumina HiSeq second-generation and PacBio Sequel third-generation sequencing platforms have high flux, and this method can effectively obtain the cp genome under the premise of containing cp sequences from related species [36]. Therefore, Illumina HiSeq sequencing platform was used to re-sequence the whole genome of A. setaceus and the cp genome of A. setaceus was assembled with related species by the software Canu and SOAPdenovo2 in this study, which provided a successful example for cp genome sequencing and assembly annotation of other species.
In the genus Asparagus, it belongs to a group of commonly used Chinese medicinal materials. Many medicinal plants are under great pressure of artificial selection in the long-term selection process, resulting in the similarity of many plants in this group, which is difficult to distinguish and identify [5,7]. Therefore, the study of cp genome is of great value to the genetic research of this genus. To detect the differences between the cp genomes of the genus Asparagus, four published species (A. filicinus, A. schoberioides, A. officinalis, and Asparagus racemosus) were downloaded from GenBank for comparison. The results showed that there was little difference in the length of cp genome between A. setaceus and its related species, with the length between 156,674 and 157,119 bp, and the type and number of genes were roughly the same, which proved that the cp genome was highly conserved. The length difference of cp genome in Asparagus plants mainly occurred in LSC region, which may be caused by the insertion and deletion of gene spacer, which was in line with the cp genome of most angiosperms [37].
On the basis of obtaining the structure and composition in A. setaceus cp genome, this study analyzed its codon preference, repeat sequence, SSR characteristics, boundary differences, and polymorphism sites, which provided a data basis for the study of cp genome in this genus. Phylogenetic analysis showed that A. setaceus was closely related to A. cochinchinensis and A. densiflorus. Due to the close genetic relationship of Asparagus plants, inter-specific hybridization within the genus was easy, and the intermediate type and transitional type were quite common, so the systematic classification was difficult [38]. The use of cp genome can provide a reference for the classification of plants in the genus, but the number of published cp genomes in the genus Asparagus is still very limited (https://www.ncbi.nlm.nih.gov/genome/browse#!/Organelles/Asparagus); the relevant research only stays in the comparative analysis of different species cp genomes. Therefore, it is necessary to obtain more cp genomes of this genus to better solve the phylogenetic problem of the genus Asparagus in Asparagaceae.
5 Conclusion
Through the methods of second-generation and third-generation sequencing platform, combined with the homology sequence alignment of related species and the use of cp splicing software, the whole cp genome sequence can be obtained. This program establishes a reference for the report of cp genome in other species. In this research, a typical quadripartite structure was exhibited in A. setaceus cp genome with 158,076 bp, including 89 protein-coding, 38 tRNA, and 8 rRNA genes. Contrast with the previous A. setaceus cp genome in NCBI, we had detected 7 SNPs and 16 Indels, which were mostly distributed in noncoding areas. In addition, 260 SSRs and 36 repeat sequences marked out in the cp genome could be utilized for species identification. Furthermore, A/T ending bias was detected and C-to-U transitions were found for the identified RNA editing sites in this cp genome. It was also seen that the cp genome had similarity with the sequenced species in genome size, gene composition, and genetic organization in the genus Asparagus. By the phylogenetic reconstruction of the whole cp genome, it was shown that A. setaceus was closely related with A. cochinchinensis in the genus. Therefore, the reported cp genome provided information for sequence variation, genomic comparison, and phylogenetic relationship studies in Asparagaceae.
-
Funding information: This work was supported by the Natural Science Foundation of China (32060078), the Natural Science Foundation of Jiangxi (20171BAB214024 and 20202BABL203044), the Special Program of Science and Technology Cooperation of Jiangxi Provincial Department of Science and Technology (20212BDH81022), the Science and Technology Program of Jiangxi Provincial Department of Education (GJJ202619), and Nanchang Normal University “11531” project.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih. gov/under the accession number MT712152.1. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA752952, SRR15371399, and SAMN20668211, respectively.
References
[1] Daniell H, Lin CS, Yu M, Chang WJ. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016;17(1):134. 10. 1186/s13059-016-1004-2.Search in Google Scholar
[2] Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. P Natl Acad Sci. 2007;104(49):19369–74.10.1073/pnas.0709121104Search in Google Scholar PubMed PubMed Central
[3] Cui YX, Chen XL, Nie LP, Sun W, Hu HY, Lin YL, et al. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int J Mol Sci. 2019;20(16):4040. 10.3390/ijms20164040.Search in Google Scholar PubMed PubMed Central
[4] Li JR, Li SF, Wang J, Dong R, Zhu HW, Li N, et al. Characterization of the complete chloroplast genome of Asparagus setaceus. Mitochondrial DNA B. 2019;4(2):2639–40. 10.1080/23802359.2019.1643798.Search in Google Scholar PubMed PubMed Central
[5] Li SF, Wang J, Dong R, Zhu HW, Nan LN, Li N, et al. Chromosome-level genome assembly, annotation and evolutionary analysis of the ornamental plant Asparagus setaceus. Hortic Res. 2020;7:48. 10.1038/s41438-020-0271-y.Search in Google Scholar PubMed PubMed Central
[6] McGaw LJ, Eloff JN. Ethnoveterinary use of southern African plants and scientific evaluation of their medicinal properties. J Ethnopharmacol. 2008;119:559–74.10.1016/j.jep.2008.06.013Search in Google Scholar PubMed
[7] Bansal RK, Menzies SA, Broadhurst PG. Screening of Asparagus species for resistance to Stemphylium leaf spot. NZ J Agric Res. 1986;29:539–45.10.1080/00288233.1986.10423507Search in Google Scholar
[8] Li JL, Wang S, Yu J, Wang L, Zhou SL. A modified CTAB protocol for plant DNA extraction. Chin Bull Bot. 2013;48:72–8.10.3724/SP.J.1259.2013.00072Search in Google Scholar
[9] Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. 10.1101/gr.215087.116.Search in Google Scholar PubMed PubMed Central
[10] Hu J, Fan JP, Sun ZY, Liu SL. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36(7):2253–5. 10. 1093/bioinformatics/btz891.Search in Google Scholar
[11] Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2015;4(1):s13742-015-0069-2. 10.1186/s13742-015-0069-2.Search in Google Scholar PubMed PubMed Central
[12] Qu XJ, Moore MJ, Li DZ, Yi TS. PGA: a software package for rapid, accurate and flexible batch annotation of plastomes. Plant Methods. 2019;15(1):1–12.10.1186/s13007-019-0435-7Search in Google Scholar PubMed PubMed Central
[13] Matthew K, Richard M, Amy W, Steven SH, Matthew C, Shane S, et al. Geneious basic: an integrated and extend-able desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.10.1093/bioinformatics/bts199Search in Google Scholar PubMed PubMed Central
[14] Greiner S, Lehwark P, Bock R. Organellar Genome DRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47:59–64.10.1093/nar/gkz238Search in Google Scholar PubMed PubMed Central
[15] Delcher AL, Salzberg SL, Phillippy AM. Using MUMmer to identify similar regions in large sequence sets. Current Prot Bioinform. 2003;1:10.10.1002/0471250953.bi1003s00Search in Google Scholar PubMed
[16] Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106:411–22. 10.1007/s00122-002-1031-0.Search in Google Scholar PubMed
[17] Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22):4633–42.10.1093/nar/29.22.4633Search in Google Scholar PubMed PubMed Central
[18] Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20(4):1160–6.10.1093/bib/bbx108Search in Google Scholar PubMed PubMed Central
[19] Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–302.10.1093/molbev/msx248Search in Google Scholar PubMed
[20] Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3.10.1093/bioinformatics/btp348Search in Google Scholar PubMed PubMed Central
[21] Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.10.1093/bioinformatics/btu033Search in Google Scholar PubMed PubMed Central
[22] Qian J, Song JY, Gao HH, Zhu YJ, Xu J, Pang XH, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE. 2013;8(2):e57607.10.1371/journal.pone.0057607Search in Google Scholar PubMed PubMed Central
[23] Asaf S, Waqas M, Khan AL, Khan MA, Kang SM, Imran QM, et al. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front Plant Sci. 2017;8:304.10.3389/fpls.2017.00304Search in Google Scholar PubMed PubMed Central
[24] Shirasawa K, Asamizu E, Sato S, Nakamura Y, Tabata S, Sasamoto S, et al. An interspecific linkage map of SSR and intronic polymorphism markers in tomato. Theor Appl Genet. 2010;121(4):731–9. 10.1007/s00122-010-1344-3.Search in Google Scholar PubMed PubMed Central
[25] Sheng WT, Chai XW, Rao YS, Tu XT, Du SG. The complete chloroplast genome sequence of Asparagus (Asparagus officinalis L.) and its phylogenetic position within Asparagales. J Plant Breed Genet. 2017;5(3):121–8.Search in Google Scholar
[26] Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23. 10.1016/0378-1119 (90)90491-9.Search in Google Scholar
[27] Mower JP. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009;37(suppl_2):W253–9.10.1093/nar/gkp337Search in Google Scholar PubMed PubMed Central
[28] Cui G, Wang C, Wei X, Wang H, Wang X, Zhu X, et al. Complete chloroplast genome of Hordeum brevisubulatum: Genome organization, synonymous codon usage, phylogenetic relationships, and comparative structure analysis. PLoS ONE. 2021;16(12):e0261196. 10.1371/journal.pone.0261196.Search in Google Scholar PubMed PubMed Central
[29] Cavalier-Smith T. Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr Biol. 2002;12(2):62–4.10.1016/S0960-9822(01)00675-3Search in Google Scholar
[30] Nie X, Lv S, Zhang Y, Du X, Wang L, Biradar SS, et al. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE. 2012;7(5):e36869.10.1371/journal.pone.0036869Search in Google Scholar PubMed PubMed Central
[31] Kuang DY, Wu H, Wang YL, Gao LM, Zhang SZ, Lu L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 2011;54(8):663–73.10.1139/g11-026Search in Google Scholar PubMed
[32] Liu FX, Movahedi A, Yang WG, Xu L, Xie JG, Zhang Y. The complete chloroplast genome and characteristics analysis of Callistemon rigidus R.Br. Mol Biol Rep. 2020;47:5013–24. 10.1007/s11033-020-05567-4.Search in Google Scholar PubMed
[33] Carbonell-Caballero J, Alonso R, Ibanez V, Terol J, Talon M, Dopazo J. A Phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol and Evol. 2015;32(8):2015–35. 10.1093/molbev/msv082.Search in Google Scholar PubMed PubMed Central
[34] Norup MF, Petersen G, Burrows S, Bouchenak-Khelladi J, Leebens-Mack J, Pires JC, et al. Evolution of Asparagus L. (Asparagaceae): Out-of-South-Africa and multiple origins of sexual dimorphism. Mol Phylogenet Evol. 2015;92:25–44.10.1016/j.ympev.2015.06.002Search in Google Scholar PubMed
[35] Yan C, Du JC, Cao L, Li Y, Hou XL. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene. 2019;699:24–36. 10.1016/j.gene.2019.02.075.Search in Google Scholar PubMed
[36] Huang SN, Ge XJ, Cano A, Salazar BGM, Deng YF. Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): molecular structure, phylogenetic relationships, and adaptive evolution. Peer J. 2020;8:e8450. 10.7717/peerj.8450. PMID: 32071806.Search in Google Scholar PubMed PubMed Central
[37] Daniell H, Jin SX, Zhu XG, Gitzendanner MA, Soltis DE, Soltis PS. Green giant-a tiny chloroplast genome with mighty power to produce high-value proteins: history and phylogeny. Plant Biotechnol J. 2021;19(8):430–47. 10.1111/pbi.13556.Search in Google Scholar PubMed PubMed Central
[38] Kubota S, Konno I, Kanno A. Molecular phylogeny of the genus Asparagus (Asparagaceae) explains interspecific crossability between the garden asparagus (A. officinalis) and other Asparagus species. Theor Appl Genet. 2012;124:345–54. 10.1007/s00122-011-1709-2.Search in Google Scholar PubMed
© 2022 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Biomedical Sciences
- Effects of direct oral anticoagulants dabigatran and rivaroxaban on the blood coagulation function in rabbits
- The mother of all battles: Viruses vs humans. Can humans avoid extinction in 50–100 years?
- Knockdown of G1P3 inhibits cell proliferation and enhances the cytotoxicity of dexamethasone in acute lymphoblastic leukemia
- LINC00665 regulates hepatocellular carcinoma by modulating mRNA via the m6A enzyme
- Association study of CLDN14 variations in patients with kidney stones
- Concanavalin A-induced autoimmune hepatitis model in mice: Mechanisms and future outlook
- Regulation of miR-30b in cancer development, apoptosis, and drug resistance
- Informatic analysis of the pulmonary microecology in non-cystic fibrosis bronchiectasis at three different stages
- Swimming attenuates tumor growth in CT-26 tumor-bearing mice and suppresses angiogenesis by mediating the HIF-1α/VEGFA pathway
- Characterization of intestinal microbiota and serum metabolites in patients with mild hepatic encephalopathy
- Functional conservation and divergence in plant-specific GRF gene family revealed by sequences and expression analysis
- Application of the FLP/LoxP-FRT recombination system to switch the eGFP expression in a model prokaryote
- Biomedical evaluation of antioxidant properties of lamb meat enriched with iodine and selenium
- Intravenous infusion of the exosomes derived from human umbilical cord mesenchymal stem cells enhance neurological recovery after traumatic brain injury via suppressing the NF-κB pathway
- Effect of dietary pattern on pregnant women with gestational diabetes mellitus and its clinical significance
- Potential regulatory mechanism of TNF-α/TNFR1/ANXA1 in glioma cells and its role in glioma cell proliferation
- Effect of the genetic mutant G71R in uridine diphosphate-glucuronosyltransferase 1A1 on the conjugation of bilirubin
- Quercetin inhibits cytotoxicity of PC12 cells induced by amyloid-beta 25–35 via stimulating estrogen receptor α, activating ERK1/2, and inhibiting apoptosis
- Nutrition intervention in the management of novel coronavirus pneumonia patients
- circ-CFH promotes the development of HCC by regulating cell proliferation, apoptosis, migration, invasion, and glycolysis through the miR-377-3p/RNF38 axis
- Bmi-1 directly upregulates glucose transporter 1 in human gastric adenocarcinoma
- Lacunar infarction aggravates the cognitive deficit in the elderly with white matter lesion
- Hydroxysafflor yellow A improved retinopathy via Nrf2/HO-1 pathway in rats
- Comparison of axon extension: PTFE versus PLA formed by a 3D printer
- Elevated IL-35 level and iTr35 subset increase the bacterial burden and lung lesions in Mycobacterium tuberculosis-infected mice
- A case report of CAT gene and HNF1β gene variations in a patient with early-onset diabetes
- Study on the mechanism of inhibiting patulin production by fengycin
- SOX4 promotes high-glucose-induced inflammation and angiogenesis of retinal endothelial cells by activating NF-κB signaling pathway
- Relationship between blood clots and COVID-19 vaccines: A literature review
- Analysis of genetic characteristics of 436 children with dysplasia and detailed analysis of rare karyotype
- Bioinformatics network analyses of growth differentiation factor 11
- NR4A1 inhibits the epithelial–mesenchymal transition of hepatic stellate cells: Involvement of TGF-β–Smad2/3/4–ZEB signaling
- Expression of Zeb1 in the differentiation of mouse embryonic stem cell
- Study on the genetic damage caused by cadmium sulfide quantum dots in human lymphocytes
- Association between single-nucleotide polymorphisms of NKX2.5 and congenital heart disease in Chinese population: A meta-analysis
- Assessment of the anesthetic effect of modified pentothal sodium solution on Sprague-Dawley rats
- Genetic susceptibility to high myopia in Han Chinese population
- Potential biomarkers and molecular mechanisms in preeclampsia progression
- Silencing circular RNA-friend leukemia virus integration 1 restrained malignancy of CC cells and oxaliplatin resistance by disturbing dyskeratosis congenita 1
- Endostar plus pembrolizumab combined with a platinum-based dual chemotherapy regime for advanced pulmonary large-cell neuroendocrine carcinoma as a first-line treatment: A case report
- The significance of PAK4 in signaling and clinicopathology: A review
- Sorafenib inhibits ovarian cancer cell proliferation and mobility and induces radiosensitivity by targeting the tumor cell epithelial–mesenchymal transition
- Characterization of rabbit polyclonal antibody against camel recombinant nanobodies
- Active legumain promotes invasion and migration of neuroblastoma by regulating epithelial-mesenchymal transition
- Effect of cell receptors in the pathogenesis of osteoarthritis: Current insights
- MT-12 inhibits the proliferation of bladder cells in vitro and in vivo by enhancing autophagy through mitochondrial dysfunction
- Study of hsa_circRNA_000121 and hsa_circRNA_004183 in papillary thyroid microcarcinoma
- BuyangHuanwu Decoction attenuates cerebral vasospasm caused by subarachnoid hemorrhage in rats via PI3K/AKT/eNOS axis
- Effects of the interaction of Notch and TLR4 pathways on inflammation and heart function in septic heart
- Monosodium iodoacetate-induced subchondral bone microstructure and inflammatory changes in an animal model of osteoarthritis
- A rare presentation of type II Abernethy malformation and nephrotic syndrome: Case report and review
- Rapid death due to pulmonary epithelioid haemangioendothelioma in several weeks: A case report
- Hepatoprotective role of peroxisome proliferator-activated receptor-α in non-cancerous hepatic tissues following transcatheter arterial embolization
- Correlation between peripheral blood lymphocyte subpopulations and primary systemic lupus erythematosus
- A novel SLC8A1-ALK fusion in lung adenocarcinoma confers sensitivity to alectinib: A case report
- β-Hydroxybutyrate upregulates FGF21 expression through inhibition of histone deacetylases in hepatocytes
- Identification of metabolic genes for the prediction of prognosis and tumor microenvironment infiltration in early-stage non-small cell lung cancer
- BTBD10 inhibits glioma tumorigenesis by downregulating cyclin D1 and p-Akt
- Mucormycosis co-infection in COVID-19 patients: An update
- Metagenomic next-generation sequencing in diagnosing Pneumocystis jirovecii pneumonia: A case report
- Long non-coding RNA HOXB-AS1 is a prognostic marker and promotes hepatocellular carcinoma cells’ proliferation and invasion
- Preparation and evaluation of LA-PEG-SPION, a targeted MRI contrast agent for liver cancer
- Proteomic analysis of the liver regulating lipid metabolism in Chaohu ducks using two-dimensional electrophoresis
- Nasopharyngeal tuberculosis: A case report
- Characterization and evaluation of anti-Salmonella enteritidis activity of indigenous probiotic lactobacilli in mice
- Aberrant pulmonary immune response of obese mice to periodontal infection
- Bacteriospermia – A formidable player in male subfertility
- In silico and in vivo analysis of TIPE1 expression in diffuse large B cell lymphoma
- Effects of KCa channels on biological behavior of trophoblasts
- Interleukin-17A influences the vulnerability rather than the size of established atherosclerotic plaques in apolipoprotein E-deficient mice
- Multiple organ failure and death caused by Staphylococcus aureus hip infection: A case report
- Prognostic signature related to the immune environment of oral squamous cell carcinoma
- Primary and metastatic squamous cell carcinoma of the thyroid gland: Two case reports
- Neuroprotective effects of crocin and crocin-loaded niosomes against the paraquat-induced oxidative brain damage in rats
- Role of MMP-2 and CD147 in kidney fibrosis
- Geometric basis of action potential of skeletal muscle cells and neurons
- Babesia microti-induced fulminant sepsis in an immunocompromised host: A case report and the case-specific literature review
- Role of cerebellar cortex in associative learning and memory in guinea pigs
- Application of metagenomic next-generation sequencing technique for diagnosing a specific case of necrotizing meningoencephalitis caused by human herpesvirus 2
- Case report: Quadruple primary malignant neoplasms including esophageal, ureteral, and lung in an elderly male
- Long non-coding RNA NEAT1 promotes angiogenesis in hepatoma carcinoma via the miR-125a-5p/VEGF pathway
- Osteogenic differentiation of periodontal membrane stem cells in inflammatory environments
- Knockdown of SHMT2 enhances the sensitivity of gastric cancer cells to radiotherapy through the Wnt/β-catenin pathway
- Continuous renal replacement therapy combined with double filtration plasmapheresis in the treatment of severe lupus complicated by serious bacterial infections in children: A case report
- Simultaneous triple primary malignancies, including bladder cancer, lymphoma, and lung cancer, in an elderly male: A case report
- Preclinical immunogenicity assessment of a cell-based inactivated whole-virion H5N1 influenza vaccine
- One case of iodine-125 therapy – A new minimally invasive treatment of intrahepatic cholangiocarcinoma
- S1P promotes corneal trigeminal neuron differentiation and corneal nerve repair via upregulating nerve growth factor expression in a mouse model
- Early cancer detection by a targeted methylation assay of circulating tumor DNA in plasma
- Calcifying nanoparticles initiate the calcification process of mesenchymal stem cells in vitro through the activation of the TGF-β1/Smad signaling pathway and promote the decay of echinococcosis
- Evaluation of prognostic markers in patients infected with SARS-CoV-2
- N6-Methyladenosine-related alternative splicing events play a role in bladder cancer
- Characterization of the structural, oxidative, and immunological features of testis tissue from Zucker diabetic fatty rats
- Effects of glucose and osmotic pressure on the proliferation and cell cycle of human chorionic trophoblast cells
- Investigation of genotype diversity of 7,804 norovirus sequences in humans and animals of China
- Characteristics and karyotype analysis of a patient with turner syndrome complicated with multiple-site tumors: A case report
- Aggravated renal fibrosis is positively associated with the activation of HMGB1-TLR2/4 signaling in STZ-induced diabetic mice
- Distribution characteristics of SARS-CoV-2 IgM/IgG in false-positive results detected by chemiluminescent immunoassay
- SRPX2 attenuated oxygen–glucose deprivation and reperfusion-induced injury in cardiomyocytes via alleviating endoplasmic reticulum stress-induced apoptosis through targeting PI3K/Akt/mTOR axis
- Aquaporin-8 overexpression is involved in vascular structure and function changes in placentas of gestational diabetes mellitus patients
- Relationship between CRP gene polymorphisms and ischemic stroke risk: A systematic review and meta-analysis
- Effects of growth hormone on lipid metabolism and sexual development in pubertal obese male rats
- Cloning and identification of the CTLA-4IgV gene and functional application of vaccine in Xinjiang sheep
- Antitumor activity of RUNX3: Upregulation of E-cadherin and downregulation of the epithelial–mesenchymal transition in clear-cell renal cell carcinoma
- PHF8 promotes osteogenic differentiation of BMSCs in old rat with osteoporosis by regulating Wnt/β-catenin pathway
- A review of the current state of the computer-aided diagnosis (CAD) systems for breast cancer diagnosis
- Bilateral dacryoadenitis in adult-onset Still’s disease: A case report
- A novel association between Bmi-1 protein expression and the SUVmax obtained by 18F-FDG PET/CT in patients with gastric adenocarcinoma
- The role of erythrocytes and erythroid progenitor cells in tumors
- Relationship between platelet activation markers and spontaneous abortion: A meta-analysis
- Abnormal methylation caused by folic acid deficiency in neural tube defects
- Silencing TLR4 using an ultrasound-targeted microbubble destruction-based shRNA system reduces ischemia-induced seizures in hyperglycemic rats
- Plant Sciences
- Seasonal succession of bacterial communities in cultured Caulerpa lentillifera detected by high-throughput sequencing
- Cloning and prokaryotic expression of WRKY48 from Caragana intermedia
- Novel Brassica hybrids with different resistance to Leptosphaeria maculans reveal unbalanced rDNA signal patterns
- Application of exogenous auxin and gibberellin regulates the bolting of lettuce (Lactuca sativa L.)
- Phytoremediation of pollutants from wastewater: A concise review
- Genome-wide identification and characterization of NBS-encoding genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.)
- Alleviative effects of magnetic Fe3O4 nanoparticles on the physiological toxicity of 3-nitrophenol to rice (Oryza sativa L.) seedlings
- Selection and functional identification of Dof genes expressed in response to nitrogen in Populus simonii × Populus nigra
- Study on pecan seed germination influenced by seed endocarp
- Identification of active compounds in Ophiopogonis Radix from different geographical origins by UPLC-Q/TOF-MS combined with GC-MS approaches
- The entire chloroplast genome sequence of Asparagus cochinchinensis and genetic comparison to Asparagus species
- Genome-wide identification of MAPK family genes and their response to abiotic stresses in tea plant (Camellia sinensis)
- Selection and validation of reference genes for RT-qPCR analysis of different organs at various development stages in Caragana intermedia
- Cloning and expression analysis of SERK1 gene in Diospyros lotus
- Integrated metabolomic and transcriptomic profiling revealed coping mechanisms of the edible and medicinal homologous plant Plantago asiatica L. cadmium resistance
- A missense variant in NCF1 is associated with susceptibility to unexplained recurrent spontaneous abortion
- Assessment of drought tolerance indices in faba bean genotypes under different irrigation regimes
- The entire chloroplast genome sequence of Asparagus setaceus (Kunth) Jessop: Genome structure, gene composition, and phylogenetic analysis in Asparagaceae
- Food Science
- Dietary food additive monosodium glutamate with or without high-lipid diet induces spleen anomaly: A mechanistic approach on rat model
- Binge eating disorder during COVID-19
- Potential of honey against the onset of autoimmune diabetes and its associated nephropathy, pancreatitis, and retinopathy in type 1 diabetic animal model
- FTO gene expression in diet-induced obesity is downregulated by Solanum fruit supplementation
- Physical activity enhances fecal lactobacilli in rats chronically drinking sweetened cola beverage
- Supercritical CO2 extraction, chemical composition, and antioxidant effects of Coreopsis tinctoria Nutt. oleoresin
- Functional constituents of plant-based foods boost immunity against acute and chronic disorders
- Effect of selenium and methods of protein extraction on the proteomic profile of Saccharomyces yeast
- Microbial diversity of milk ghee in southern Gansu and its effect on the formation of ghee flavor compounds
- Ecology and Environmental Sciences
- Effects of heavy metals on bacterial community surrounding Bijiashan mining area located in northwest China
- Microorganism community composition analysis coupling with 15N tracer experiments reveals the nitrification rate and N2O emissions in low pH soils in Southern China
- Genetic diversity and population structure of Cinnamomum balansae Lecomte inferred by microsatellites
- Preliminary screening of microplastic contamination in different marine fish species of Taif market, Saudi Arabia
- Plant volatile organic compounds attractive to Lygus pratensis
- Effects of organic materials on soil bacterial community structure in long-term continuous cropping of tomato in greenhouse
- Effects of soil treated fungicide fluopimomide on tomato (Solanum lycopersicum L.) disease control and plant growth
- Prevalence of Yersinia pestis among rodents captured in a semi-arid tropical ecosystem of south-western Zimbabwe
- Effects of irrigation and nitrogen fertilization on mitigating salt-induced Na+ toxicity and sustaining sea rice growth
- Bioengineering and Biotechnology
- Poly-l-lysine-caused cell adhesion induces pyroptosis in THP-1 monocytes
- Development of alkaline phosphatase-scFv and its use for one-step enzyme-linked immunosorbent assay for His-tagged protein detection
- Development and validation of a predictive model for immune-related genes in patients with tongue squamous cell carcinoma
- Agriculture
- Effects of chemical-based fertilizer replacement with biochar-based fertilizer on albic soil nutrient content and maize yield
- Genome-wide identification and expression analysis of CPP-like gene family in Triticum aestivum L. under different hormone and stress conditions
- Agronomic and economic performance of mung bean (Vigna radiata L.) varieties in response to rates of blended NPS fertilizer in Kindo Koysha district, Southern Ethiopia
- Influence of furrow irrigation regime on the yield and water consumption indicators of winter wheat based on a multi-level fuzzy comprehensive evaluation
- Discovery of exercise-related genes and pathway analysis based on comparative genomes of Mongolian originated Abaga and Wushen horse
- Lessons from integrated seasonal forecast-crop modelling in Africa: A systematic review
- Evolution trend of soil fertility in tobacco-planting area of Chenzhou, Hunan Province, China
- Animal Sciences
- Morphological and molecular characterization of Tatera indica Hardwicke 1807 (Rodentia: Muridae) from Pothwar, Pakistan
- Research on meat quality of Qianhua Mutton Merino sheep and Small-tail Han sheep
- SI: A Scientific Memoir
- Suggestions on leading an academic research laboratory group
- My scientific genealogy and the Toronto ACDC Laboratory, 1988–2022
- Erratum
- Erratum to “Changes of immune cells in patients with hepatocellular carcinoma treated by radiofrequency ablation and hepatectomy, a pilot study”
- Erratum to “A two-microRNA signature predicts the progression of male thyroid cancer”
- Retraction
- Retraction of “Lidocaine has antitumor effect on hepatocellular carcinoma via the circ_DYNC1H1/miR-520a-3p/USP14 axis”
Articles in the same Issue
- Biomedical Sciences
- Effects of direct oral anticoagulants dabigatran and rivaroxaban on the blood coagulation function in rabbits
- The mother of all battles: Viruses vs humans. Can humans avoid extinction in 50–100 years?
- Knockdown of G1P3 inhibits cell proliferation and enhances the cytotoxicity of dexamethasone in acute lymphoblastic leukemia
- LINC00665 regulates hepatocellular carcinoma by modulating mRNA via the m6A enzyme
- Association study of CLDN14 variations in patients with kidney stones
- Concanavalin A-induced autoimmune hepatitis model in mice: Mechanisms and future outlook
- Regulation of miR-30b in cancer development, apoptosis, and drug resistance
- Informatic analysis of the pulmonary microecology in non-cystic fibrosis bronchiectasis at three different stages
- Swimming attenuates tumor growth in CT-26 tumor-bearing mice and suppresses angiogenesis by mediating the HIF-1α/VEGFA pathway
- Characterization of intestinal microbiota and serum metabolites in patients with mild hepatic encephalopathy
- Functional conservation and divergence in plant-specific GRF gene family revealed by sequences and expression analysis
- Application of the FLP/LoxP-FRT recombination system to switch the eGFP expression in a model prokaryote
- Biomedical evaluation of antioxidant properties of lamb meat enriched with iodine and selenium
- Intravenous infusion of the exosomes derived from human umbilical cord mesenchymal stem cells enhance neurological recovery after traumatic brain injury via suppressing the NF-κB pathway
- Effect of dietary pattern on pregnant women with gestational diabetes mellitus and its clinical significance
- Potential regulatory mechanism of TNF-α/TNFR1/ANXA1 in glioma cells and its role in glioma cell proliferation
- Effect of the genetic mutant G71R in uridine diphosphate-glucuronosyltransferase 1A1 on the conjugation of bilirubin
- Quercetin inhibits cytotoxicity of PC12 cells induced by amyloid-beta 25–35 via stimulating estrogen receptor α, activating ERK1/2, and inhibiting apoptosis
- Nutrition intervention in the management of novel coronavirus pneumonia patients
- circ-CFH promotes the development of HCC by regulating cell proliferation, apoptosis, migration, invasion, and glycolysis through the miR-377-3p/RNF38 axis
- Bmi-1 directly upregulates glucose transporter 1 in human gastric adenocarcinoma
- Lacunar infarction aggravates the cognitive deficit in the elderly with white matter lesion
- Hydroxysafflor yellow A improved retinopathy via Nrf2/HO-1 pathway in rats
- Comparison of axon extension: PTFE versus PLA formed by a 3D printer
- Elevated IL-35 level and iTr35 subset increase the bacterial burden and lung lesions in Mycobacterium tuberculosis-infected mice
- A case report of CAT gene and HNF1β gene variations in a patient with early-onset diabetes
- Study on the mechanism of inhibiting patulin production by fengycin
- SOX4 promotes high-glucose-induced inflammation and angiogenesis of retinal endothelial cells by activating NF-κB signaling pathway
- Relationship between blood clots and COVID-19 vaccines: A literature review
- Analysis of genetic characteristics of 436 children with dysplasia and detailed analysis of rare karyotype
- Bioinformatics network analyses of growth differentiation factor 11
- NR4A1 inhibits the epithelial–mesenchymal transition of hepatic stellate cells: Involvement of TGF-β–Smad2/3/4–ZEB signaling
- Expression of Zeb1 in the differentiation of mouse embryonic stem cell
- Study on the genetic damage caused by cadmium sulfide quantum dots in human lymphocytes
- Association between single-nucleotide polymorphisms of NKX2.5 and congenital heart disease in Chinese population: A meta-analysis
- Assessment of the anesthetic effect of modified pentothal sodium solution on Sprague-Dawley rats
- Genetic susceptibility to high myopia in Han Chinese population
- Potential biomarkers and molecular mechanisms in preeclampsia progression
- Silencing circular RNA-friend leukemia virus integration 1 restrained malignancy of CC cells and oxaliplatin resistance by disturbing dyskeratosis congenita 1
- Endostar plus pembrolizumab combined with a platinum-based dual chemotherapy regime for advanced pulmonary large-cell neuroendocrine carcinoma as a first-line treatment: A case report
- The significance of PAK4 in signaling and clinicopathology: A review
- Sorafenib inhibits ovarian cancer cell proliferation and mobility and induces radiosensitivity by targeting the tumor cell epithelial–mesenchymal transition
- Characterization of rabbit polyclonal antibody against camel recombinant nanobodies
- Active legumain promotes invasion and migration of neuroblastoma by regulating epithelial-mesenchymal transition
- Effect of cell receptors in the pathogenesis of osteoarthritis: Current insights
- MT-12 inhibits the proliferation of bladder cells in vitro and in vivo by enhancing autophagy through mitochondrial dysfunction
- Study of hsa_circRNA_000121 and hsa_circRNA_004183 in papillary thyroid microcarcinoma
- BuyangHuanwu Decoction attenuates cerebral vasospasm caused by subarachnoid hemorrhage in rats via PI3K/AKT/eNOS axis
- Effects of the interaction of Notch and TLR4 pathways on inflammation and heart function in septic heart
- Monosodium iodoacetate-induced subchondral bone microstructure and inflammatory changes in an animal model of osteoarthritis
- A rare presentation of type II Abernethy malformation and nephrotic syndrome: Case report and review
- Rapid death due to pulmonary epithelioid haemangioendothelioma in several weeks: A case report
- Hepatoprotective role of peroxisome proliferator-activated receptor-α in non-cancerous hepatic tissues following transcatheter arterial embolization
- Correlation between peripheral blood lymphocyte subpopulations and primary systemic lupus erythematosus
- A novel SLC8A1-ALK fusion in lung adenocarcinoma confers sensitivity to alectinib: A case report
- β-Hydroxybutyrate upregulates FGF21 expression through inhibition of histone deacetylases in hepatocytes
- Identification of metabolic genes for the prediction of prognosis and tumor microenvironment infiltration in early-stage non-small cell lung cancer
- BTBD10 inhibits glioma tumorigenesis by downregulating cyclin D1 and p-Akt
- Mucormycosis co-infection in COVID-19 patients: An update
- Metagenomic next-generation sequencing in diagnosing Pneumocystis jirovecii pneumonia: A case report
- Long non-coding RNA HOXB-AS1 is a prognostic marker and promotes hepatocellular carcinoma cells’ proliferation and invasion
- Preparation and evaluation of LA-PEG-SPION, a targeted MRI contrast agent for liver cancer
- Proteomic analysis of the liver regulating lipid metabolism in Chaohu ducks using two-dimensional electrophoresis
- Nasopharyngeal tuberculosis: A case report
- Characterization and evaluation of anti-Salmonella enteritidis activity of indigenous probiotic lactobacilli in mice
- Aberrant pulmonary immune response of obese mice to periodontal infection
- Bacteriospermia – A formidable player in male subfertility
- In silico and in vivo analysis of TIPE1 expression in diffuse large B cell lymphoma
- Effects of KCa channels on biological behavior of trophoblasts
- Interleukin-17A influences the vulnerability rather than the size of established atherosclerotic plaques in apolipoprotein E-deficient mice
- Multiple organ failure and death caused by Staphylococcus aureus hip infection: A case report
- Prognostic signature related to the immune environment of oral squamous cell carcinoma
- Primary and metastatic squamous cell carcinoma of the thyroid gland: Two case reports
- Neuroprotective effects of crocin and crocin-loaded niosomes against the paraquat-induced oxidative brain damage in rats
- Role of MMP-2 and CD147 in kidney fibrosis
- Geometric basis of action potential of skeletal muscle cells and neurons
- Babesia microti-induced fulminant sepsis in an immunocompromised host: A case report and the case-specific literature review
- Role of cerebellar cortex in associative learning and memory in guinea pigs
- Application of metagenomic next-generation sequencing technique for diagnosing a specific case of necrotizing meningoencephalitis caused by human herpesvirus 2
- Case report: Quadruple primary malignant neoplasms including esophageal, ureteral, and lung in an elderly male
- Long non-coding RNA NEAT1 promotes angiogenesis in hepatoma carcinoma via the miR-125a-5p/VEGF pathway
- Osteogenic differentiation of periodontal membrane stem cells in inflammatory environments
- Knockdown of SHMT2 enhances the sensitivity of gastric cancer cells to radiotherapy through the Wnt/β-catenin pathway
- Continuous renal replacement therapy combined with double filtration plasmapheresis in the treatment of severe lupus complicated by serious bacterial infections in children: A case report
- Simultaneous triple primary malignancies, including bladder cancer, lymphoma, and lung cancer, in an elderly male: A case report
- Preclinical immunogenicity assessment of a cell-based inactivated whole-virion H5N1 influenza vaccine
- One case of iodine-125 therapy – A new minimally invasive treatment of intrahepatic cholangiocarcinoma
- S1P promotes corneal trigeminal neuron differentiation and corneal nerve repair via upregulating nerve growth factor expression in a mouse model
- Early cancer detection by a targeted methylation assay of circulating tumor DNA in plasma
- Calcifying nanoparticles initiate the calcification process of mesenchymal stem cells in vitro through the activation of the TGF-β1/Smad signaling pathway and promote the decay of echinococcosis
- Evaluation of prognostic markers in patients infected with SARS-CoV-2
- N6-Methyladenosine-related alternative splicing events play a role in bladder cancer
- Characterization of the structural, oxidative, and immunological features of testis tissue from Zucker diabetic fatty rats
- Effects of glucose and osmotic pressure on the proliferation and cell cycle of human chorionic trophoblast cells
- Investigation of genotype diversity of 7,804 norovirus sequences in humans and animals of China
- Characteristics and karyotype analysis of a patient with turner syndrome complicated with multiple-site tumors: A case report
- Aggravated renal fibrosis is positively associated with the activation of HMGB1-TLR2/4 signaling in STZ-induced diabetic mice
- Distribution characteristics of SARS-CoV-2 IgM/IgG in false-positive results detected by chemiluminescent immunoassay
- SRPX2 attenuated oxygen–glucose deprivation and reperfusion-induced injury in cardiomyocytes via alleviating endoplasmic reticulum stress-induced apoptosis through targeting PI3K/Akt/mTOR axis
- Aquaporin-8 overexpression is involved in vascular structure and function changes in placentas of gestational diabetes mellitus patients
- Relationship between CRP gene polymorphisms and ischemic stroke risk: A systematic review and meta-analysis
- Effects of growth hormone on lipid metabolism and sexual development in pubertal obese male rats
- Cloning and identification of the CTLA-4IgV gene and functional application of vaccine in Xinjiang sheep
- Antitumor activity of RUNX3: Upregulation of E-cadherin and downregulation of the epithelial–mesenchymal transition in clear-cell renal cell carcinoma
- PHF8 promotes osteogenic differentiation of BMSCs in old rat with osteoporosis by regulating Wnt/β-catenin pathway
- A review of the current state of the computer-aided diagnosis (CAD) systems for breast cancer diagnosis
- Bilateral dacryoadenitis in adult-onset Still’s disease: A case report
- A novel association between Bmi-1 protein expression and the SUVmax obtained by 18F-FDG PET/CT in patients with gastric adenocarcinoma
- The role of erythrocytes and erythroid progenitor cells in tumors
- Relationship between platelet activation markers and spontaneous abortion: A meta-analysis
- Abnormal methylation caused by folic acid deficiency in neural tube defects
- Silencing TLR4 using an ultrasound-targeted microbubble destruction-based shRNA system reduces ischemia-induced seizures in hyperglycemic rats
- Plant Sciences
- Seasonal succession of bacterial communities in cultured Caulerpa lentillifera detected by high-throughput sequencing
- Cloning and prokaryotic expression of WRKY48 from Caragana intermedia
- Novel Brassica hybrids with different resistance to Leptosphaeria maculans reveal unbalanced rDNA signal patterns
- Application of exogenous auxin and gibberellin regulates the bolting of lettuce (Lactuca sativa L.)
- Phytoremediation of pollutants from wastewater: A concise review
- Genome-wide identification and characterization of NBS-encoding genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.)
- Alleviative effects of magnetic Fe3O4 nanoparticles on the physiological toxicity of 3-nitrophenol to rice (Oryza sativa L.) seedlings
- Selection and functional identification of Dof genes expressed in response to nitrogen in Populus simonii × Populus nigra
- Study on pecan seed germination influenced by seed endocarp
- Identification of active compounds in Ophiopogonis Radix from different geographical origins by UPLC-Q/TOF-MS combined with GC-MS approaches
- The entire chloroplast genome sequence of Asparagus cochinchinensis and genetic comparison to Asparagus species
- Genome-wide identification of MAPK family genes and their response to abiotic stresses in tea plant (Camellia sinensis)
- Selection and validation of reference genes for RT-qPCR analysis of different organs at various development stages in Caragana intermedia
- Cloning and expression analysis of SERK1 gene in Diospyros lotus
- Integrated metabolomic and transcriptomic profiling revealed coping mechanisms of the edible and medicinal homologous plant Plantago asiatica L. cadmium resistance
- A missense variant in NCF1 is associated with susceptibility to unexplained recurrent spontaneous abortion
- Assessment of drought tolerance indices in faba bean genotypes under different irrigation regimes
- The entire chloroplast genome sequence of Asparagus setaceus (Kunth) Jessop: Genome structure, gene composition, and phylogenetic analysis in Asparagaceae
- Food Science
- Dietary food additive monosodium glutamate with or without high-lipid diet induces spleen anomaly: A mechanistic approach on rat model
- Binge eating disorder during COVID-19
- Potential of honey against the onset of autoimmune diabetes and its associated nephropathy, pancreatitis, and retinopathy in type 1 diabetic animal model
- FTO gene expression in diet-induced obesity is downregulated by Solanum fruit supplementation
- Physical activity enhances fecal lactobacilli in rats chronically drinking sweetened cola beverage
- Supercritical CO2 extraction, chemical composition, and antioxidant effects of Coreopsis tinctoria Nutt. oleoresin
- Functional constituents of plant-based foods boost immunity against acute and chronic disorders
- Effect of selenium and methods of protein extraction on the proteomic profile of Saccharomyces yeast
- Microbial diversity of milk ghee in southern Gansu and its effect on the formation of ghee flavor compounds
- Ecology and Environmental Sciences
- Effects of heavy metals on bacterial community surrounding Bijiashan mining area located in northwest China
- Microorganism community composition analysis coupling with 15N tracer experiments reveals the nitrification rate and N2O emissions in low pH soils in Southern China
- Genetic diversity and population structure of Cinnamomum balansae Lecomte inferred by microsatellites
- Preliminary screening of microplastic contamination in different marine fish species of Taif market, Saudi Arabia
- Plant volatile organic compounds attractive to Lygus pratensis
- Effects of organic materials on soil bacterial community structure in long-term continuous cropping of tomato in greenhouse
- Effects of soil treated fungicide fluopimomide on tomato (Solanum lycopersicum L.) disease control and plant growth
- Prevalence of Yersinia pestis among rodents captured in a semi-arid tropical ecosystem of south-western Zimbabwe
- Effects of irrigation and nitrogen fertilization on mitigating salt-induced Na+ toxicity and sustaining sea rice growth
- Bioengineering and Biotechnology
- Poly-l-lysine-caused cell adhesion induces pyroptosis in THP-1 monocytes
- Development of alkaline phosphatase-scFv and its use for one-step enzyme-linked immunosorbent assay for His-tagged protein detection
- Development and validation of a predictive model for immune-related genes in patients with tongue squamous cell carcinoma
- Agriculture
- Effects of chemical-based fertilizer replacement with biochar-based fertilizer on albic soil nutrient content and maize yield
- Genome-wide identification and expression analysis of CPP-like gene family in Triticum aestivum L. under different hormone and stress conditions
- Agronomic and economic performance of mung bean (Vigna radiata L.) varieties in response to rates of blended NPS fertilizer in Kindo Koysha district, Southern Ethiopia
- Influence of furrow irrigation regime on the yield and water consumption indicators of winter wheat based on a multi-level fuzzy comprehensive evaluation
- Discovery of exercise-related genes and pathway analysis based on comparative genomes of Mongolian originated Abaga and Wushen horse
- Lessons from integrated seasonal forecast-crop modelling in Africa: A systematic review
- Evolution trend of soil fertility in tobacco-planting area of Chenzhou, Hunan Province, China
- Animal Sciences
- Morphological and molecular characterization of Tatera indica Hardwicke 1807 (Rodentia: Muridae) from Pothwar, Pakistan
- Research on meat quality of Qianhua Mutton Merino sheep and Small-tail Han sheep
- SI: A Scientific Memoir
- Suggestions on leading an academic research laboratory group
- My scientific genealogy and the Toronto ACDC Laboratory, 1988–2022
- Erratum
- Erratum to “Changes of immune cells in patients with hepatocellular carcinoma treated by radiofrequency ablation and hepatectomy, a pilot study”
- Erratum to “A two-microRNA signature predicts the progression of male thyroid cancer”
- Retraction
- Retraction of “Lidocaine has antitumor effect on hepatocellular carcinoma via the circ_DYNC1H1/miR-520a-3p/USP14 axis”