Analysis of genetic characteristics of 436 children with dysplasia and detailed analysis of rare karyotype
-
Zong-Yu Miao


Abstract
Chromosomal abnormality is one of the important causes of dysplasia in children. However, due to regional and ethnic differences, the reported rates of chromosomal abnormalities in patients with dysplasia vary greatly. Moreover, the clinical manifestations in children with rare chromosomal diseases were heterogeneous. So, we retrospectively analyzed the karyotype results of 436 children with dysplasia and conducted a detailed analysis of rare chromosomal diseases. The results showed that chromosomal abnormalities were present in 181 of 436 cases. Intellectual disability, dysmorphology, congenital malformations, the disorder of sexual development, and short stature were the main five clinical symptoms in children with chromosomal abnormalities. Moreover, 136 cases of Trisomy 21 (Tri21) were detected, of which 130 were standard Tri21, 5 were robertsonian Tri21, and 1 was chimera type. In addition, 16 cases of rare abnormal karyotype, including complex Tri21, complex Turner syndrome, 4p-syndrome, 18q-syndrome, and 5p-syndrome, were also detected. In summary, chromosome abnormality is one of the important causes of dysplasia in children. Furthermore, prenatal screening and diagnosis could play a great significance in preventing dysplasia in children. In addition, the retrospective analysis of rare cases is valuable for clinical diagnosis and risk assessment of recurrence.
1 Introduction
Dysplasia in children is increasingly becoming a prominent public health and social problem. Dysplasia in children affects children’s health and quality of life and the healthy and sustainable development of society. So, reducing congenital disabilities has great social significance. Therefore, it is necessary to delve into the causes of dysplasia in children.
Congenital heart disease (CHD) is one of the most common dysplasias in children. The incidence of CHD in neonates is 8–9/1,000, and nearly 1.35 million CHD neonates are born every year in the world [1]. Chromosomal causes of CHD include chromosome aneuploidies, such as Trisomy 21 (Tri21). Chromosomal aneuploidies represent 12.5% of CHD causes [2]. In addition, Tri21 syndrome, a common chromosomal disorder, is the most common cause of severe mental retardation, special facial features, and abnormal bone development in children.
Disorder of sexual development (DSD) and short stature are also the most common dysplasia symptoms in children. Sex chromosome abnormality is one of the most common causes of DSD and short stature. Common sex chromosomal disorders include Klinefelter syndrome (1/1,000–1/2,000 in male newborns), Turner syndrome (TS, approximately 1/5,000 in newborn girls), XYY syndrome (1/900 in males), and XXX syndrome (1/1,000 in newborn girls). Patients with sex chromosome diseases are often accompanied by gonadal dysplasia, secondary sexual signs dysplasia, fertility disorders, mild mental abnormalities, mental deficiencies, and other symptoms. In addition, children with TS often have short stature. Children with severe short stature are vulnerable to diverse developmental, social, and educational problems [3]. Therefore, the abnormality of the sex chromosome is one of the important causes of dysplasia in children.
There is a significant evidence in the literature that the chromosome partial monomer or trisomy can lead to severe dysplasia in children. The clinical examination of the patients with structurally abnormal chromosome 15 revealed a series of clinical symptoms, such as intellectual disability, CHD, severe growth retardation, hypertelorism, microcephalus, dysmorphology, and multiple sclerosis hyperpigmented or café-au-lait spots, short stature, and clinodactyly [4,5,6,7]. The common clinical features of the patients with partial trisomy of chromosome 8 included developmental delay, intellectual disability, mental retardation, severe hypotonia, hypospadias, skeletal anomalies, renal dysplasia, attention-deficit hyperactivity disorder, atypical facial appearance, and congenital hypoplasia of the tongue [8,9,10,11,12,13]. The short arm monosomy of chromosome 9 may present developmental delay, hypotonia, trigonocephaly, psychomotor developmental delay, learning difficulties, trigonocephaly, facial dysmorphia, and genital abnormalities [14,15].
Thus, the chromosomal abnormality is one of the important causes of dysplasia in children, which seriously endangers children’s physical and mental health and brings heavy spiritual and economic burdens to families and society. However, due to regional and ethnic differences, the reported rates of chromosomal abnormalities in patients with dysplasia vary greatly. So, we retrospectively analyzed the karyotype results of 436 children with dysplasia and conducted a detailed analysis of rare cases to clarify the chromosomal abnormality rate and distribution of abnormal chromosomes in local children with dysplasia and provide a theoretical basis for clinical diagnosis and prenatal diagnosis, and assess the risk of recurrence.
2 Patients and methods
2.1 Patients
Four hundred and thirty-six cases (359 male and 77 female) in children with clinically diagnosed dysplasia in the affiliated Yantai Yu Huang Ding Hospital of Qingdao University Medical College from January 1, 2012 to October 31, 2019, were recruited. The age group of the participants ranged from 1 day to 16 years. Patients with dysplasia caused by autoimmune, chemo- or radio-therapy, infectious, and iatrogenic injury were excluded. The clinical data of confirmed cases were retrospectively collected and analyzed.
-
Informed consent: Informed consent has been obtained from all individuals included in this study.
-
Ethical approval: The research related to human use has been complied with all the relevant national regulations, institutional policies, and in accordance with the tenets of the Helsinki Declaration and has been approved by the Ethical Committee of Yantai Yu Huang Ding Hospital.
2.2 G banding
Standard chromosomal analyses with G banding were performed on routinely cultured peripheral blood lymphocytes [16]. Thirty metaphases per patient were counted, and a minimum of five metaphases were analyzed. For the chimeric case, at least 100 metaphases were counted. Chromosome polymorphisms, such as pericentric inversion of chromosome 9, centromeric heterochromatin variants, and satellite variants, were classified as normal. The karyotypic reports were based on the International System for Human Cytogenetic Nomenclature.
3 Results
Chromosomal abnormalities were detected in 181 of 436 cases. The abnormal rate was 41.51%. Among them were 153 cases of autosomal abnormality (84.53%, 153/181, Table 1) and 28 cases of a sex chromosome abnormality (15.47%, 28/181, Table 2). The main clinical symptoms of 181 children with chromosomal abnormalities were intellectual disability (80.66%, 146/181), dysmorphology (71.82%, 130/181), congenital malformations (22.10%, 40/181), DSD (12.15%, 22/181), and short stature (8.29%, 15/181).
Distribution of autosomal abnormalities in children with dysplasia
| Classification | Chromosome karyotypes | Number of cases | Constituent ratio (%) | Abnormality rate (%) |
|---|---|---|---|---|
| Numerical abnormality | 132 | 86.27 | 30.27 | |
| 47,XX,+21 | 40 | 26.14 | 9.17 | |
| 47,XY,+21 | 86 | 56.21 | 19.72 | |
| 47,XX,+21[12]/46,XX[177] | 1 | 0.65 | 0.23 | |
| 47,XY,+21,inv(9)(p11q12) | 1 | 0.65 | 0.23 | |
| 47,XY,+21,13pstkstk | 1 | 0.65 | 0.23 | |
| 47,XX,+21,inv(9)(p11q13) | 1 | 0.65 | 0.23 | |
| 47,X,Yqs,+21 | 1 | 0.65 | 0.23 | |
| 47,XX,+mar[74]/46,XX[26] | 1 | 0.65 | 0.23 | |
| Structural abnormality | 21 | 13.73 | 4.82 | |
| 46,XX,der(13;14)(q10;q10),+21 | 1 | 0.65 | 0.23 | |
| 46,XY,der(14;21)(q10;q10),+21 | 2 | 1.31 | 0.46 | |
| 46,XX,der(21;21)(q10;q10), | 1 | 0.65 | 0.23 | |
| t(1;12)(q43;p12.1),inv(15)(q13q24) | ||||
| 46,XX,der(21;21)(q10;q10) | 1 | 0.65 | 0.23 | |
| 46,XY,del(4)(q33) | 1 | 0.65 | 0.23 | |
| 46,XY,del(5)(p14) | 1 | 0.65 | 0.23 | |
| 46,XY,del(5)(p14.3) | 1 | 0.65 | 0.23 | |
| 46,XX,del(13)(q31) | 1 | 0.65 | 0.23 | |
| 46,XX,del(8)(p23.1) | 1 | 0.65 | 0.23 | |
| 46,XX,add(16)(q24) | 1 | 0.65 | 0.23 | |
| 46,XY,r(9)(p24q34) | 1 | 0.65 | 0.23 | |
| 46,XY,der(9)t(2;9)(p25;p22)mat | 1 | 0.65 | 0.23 | |
| 46,XY,der(9)t(7;9)(p15;p22)pat | 1 | 0.65 | 0.23 | |
| 45,XX,der(15;21)(q10;q10)mat,del(18)(q21) | 1 | 0.65 | 0.23 | |
| 46,XX,der(3)del(3)(p21.3p23)t(2;3) | ||||
| (q11.2;p23) | 1 | 0.65 | 0.23 | |
| 46,XX,t(9;15)(p24;q13) | 1 | 0.65 | 0.23 | |
| 46,XX,t(9;16)(q13;q22) | 1 | 0.65 | 0.23 | |
| 46,XX,t(2;8)(q13;q23) | 1 | 0.65 | 0.23 | |
| 46,XX,t(1;3)(p22.1;q27) | 1 | 0.65 | 0.23 | |
| 46,XX,inv(12)(p12.2q15) | 1 | 0.65 | 0.23 | |
| Combination | 153 | 100 | 35.09 |
Distribution of sexual chromosomal abnormalities in children with dysplasia
| Classification | Chromosome karyotypes | Number of cases | Constituent ratio (%) | Abnormality rate (%) |
|---|---|---|---|---|
| Numerical abnormality | 10 | 35.71 | 2.29 | |
| 45,X | 7 | 25.00 | 1.61 | |
| 45,X[73]/47,XXX[27] | 1 | 3.57 | 0.23 | |
| 45,X[78]/47,XXX[22] | 1 | 3.57 | 0.23 | |
| 45,X[90]/46,XX[6] | 1 | 3.57 | 0.23 | |
| Structural abnormality | 16 | 57.15 | 3.67 | |
| 46,X,i(X)(q10) | 5 | 17.87 | 1.15 | |
| 45,X[91]/46,X,i(X)(q10)[9] | 1 | 3.57 | 0.23 | |
| 46,X,i(X)(q10)[89]/45,X[11] | 1 | 3.57 | 0.23 | |
| 45,X[61]/46,X,i(X)(p11.3)[39] | 1 | 3.57 | 0.23 | |
| 46,X,idic(Y)(q11.22)[94]/45,X[6] | 1 | 3.57 | 0.23 | |
| 45,X,inv(9)(p11q13)[72]/46,X,dic(Y)(q11.23),inv(9)(p11q13)[10]/46,XY, inv(9)(p11q13)[4] | 1 | 3.57 | 0.23 | |
| 46,X, idic(Y)(q11.23)[44]/45,X[56] | 1 | 3.57 | 0.23 | |
| 45,X[120]/46,X,r(X)(p22.2q22.2)[12]/ | 1 | 3.57 | 0.23 | |
| 46,X,rdup(X)(p22.2q22.2)[4] | ||||
| 45,X[80]/46,X,idic(X)(p11.3)[20] | 1 | 3.57 | 0.23 | |
| 46,X,idic(X)(p11.2)[92]/45,X[9]/ | 1 | 3.57 | 0.23 | |
| 47,X,idic(X)(p11.2),idic(X)(p11.2)[5] | ||||
| 46,X,idic(X)(p11.2)[90]/45,X[10] | 1 | 3.57 | 0.23 | |
| 46,X,idic(X;X)(q21.3;q11.1) | 1 | 3.57 | 0.23 | |
| 46,XX (male) | 2 | 7.14 | 0.46 | |
| Sex reversal combination | 28 | 100 | 6.42 |
One hundred and thirty-six cases of Tri21 were detected (31.19%, 136/436), including 89 male and 47 female, with a ratio of 1.89:1. Among them, 130 cases were standard Tri21 (95.59%, 130/136), 5 cases were Robertsonian Tri21 (3.79%, 5/132) and 1 case was a chimeric type (0.74%, 1/136).
Twenty-eight cases of sex chromosome abnormalities were detected, among which 20 cases were TS (7 cases were 45,X, 13 cases were 45,X mosaic), 5 cases were 46,X,i(X)(q10), 2 cases were sexual reversal, and 1 case was 46,X,idic(X;X)(q21.3;q11.1).
Sixteen cases of rare abnormal karyotype were detected, including complex Tri21 (Figure 1), complex TS (Figures 2–4), partial monomer and partial trisomy (Figures 5), 4p-syndrome (Figure 6), 18q-syndrome (Figure 7), and 5p-syndrome.
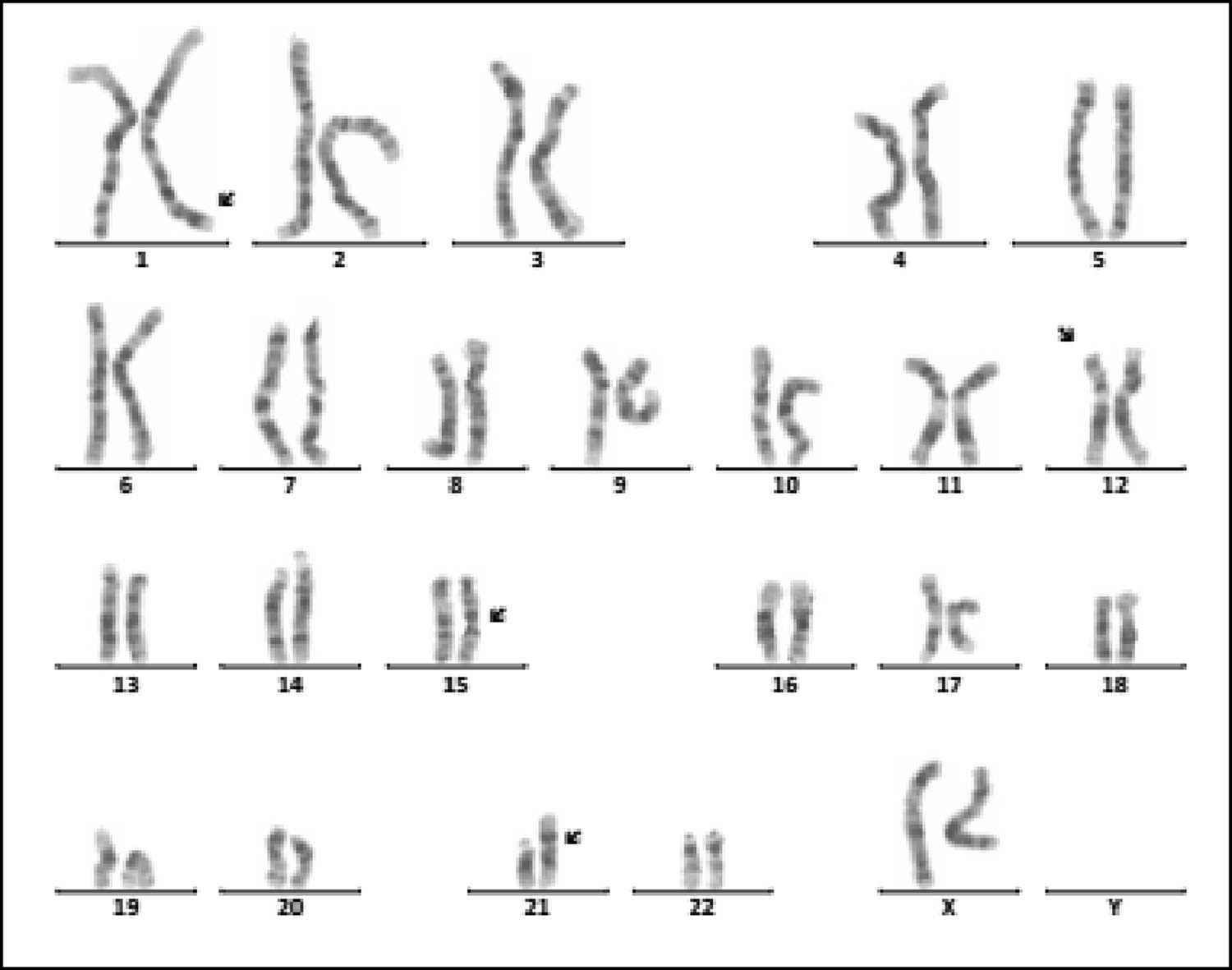
Karyotype of 46,XX, der(21;21)(q10;q10),t(1;12)(q43;p12.1),inv(15)(q13q24). The arrows show abnormal chromosomes.
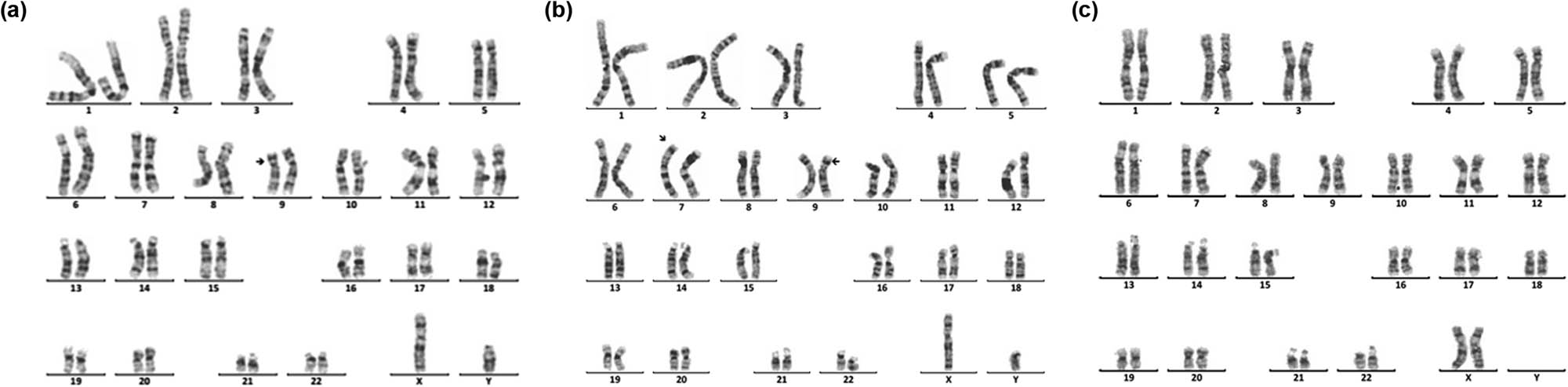
![Figure 2
Karyotype of 45,X,inv(9)(p11q13)[72]/46,X,dic(Y)(q11.23),inv(9)(p11q13)[10]/46,XY,inv(9)(p11 q13)[4]. The arrows show abnormal chromosomes. (a) 45,X,inv(9)(p11q13). (b) 46,X,dic(Y)(q11.23), inv(9)(p11q13). (c) 46,XY,inv(9) (p11q13).](/document/doi/10.1515/biol-2022-0046/asset/graphic/j_biol-2022-0046_fig_002.jpg)
Karyotype of 45,X,inv(9)(p11q13)[72]/46,X,dic(Y)(q11.23),inv(9)(p11q13)[10]/46,XY,inv(9)(p11 q13)[4]. The arrows show abnormal chromosomes. (a) 45,X,inv(9)(p11q13). (b) 46,X,dic(Y)(q11.23), inv(9)(p11q13). (c) 46,XY,inv(9) (p11q13).
![Figure 3
Karyotype of 45,X[120]/46,X,r(X)(p22.2q22.2)[12]/46,X,rdup(X)(p22.2q22.2)[4]. The arrows show abnormal chromosomes. (a) 45,X. (b) 46,X,r(X)(p22.2q22.2). (c) 46,X,rdup(X)(p22.2q22.2).](/document/doi/10.1515/biol-2022-0046/asset/graphic/j_biol-2022-0046_fig_003.jpg)
Karyotype of 45,X[120]/46,X,r(X)(p22.2q22.2)[12]/46,X,rdup(X)(p22.2q22.2)[4]. The arrows show abnormal chromosomes. (a) 45,X. (b) 46,X,r(X)(p22.2q22.2). (c) 46,X,rdup(X)(p22.2q22.2).
![Figure 4
Karyotype of 46,X,idic(X)(p11.2)[92]/45,X[9]/47,X,idic(X)(p11.2),idic(X)(p11.2)[5]. The arrows show abnormal chromosomes. (a) 45,X. (b) 46,X,idic(X)(p11.2). (c) 47,X,idic(X)(p11.2),idic(X)(p11.2).](/document/doi/10.1515/biol-2022-0046/asset/graphic/j_biol-2022-0046_fig_004.jpg)
Karyotype of 46,X,idic(X)(p11.2)[92]/45,X[9]/47,X,idic(X)(p11.2),idic(X)(p11.2)[5]. The arrows show abnormal chromosomes. (a) 45,X. (b) 46,X,idic(X)(p11.2). (c) 47,X,idic(X)(p11.2),idic(X)(p11.2).
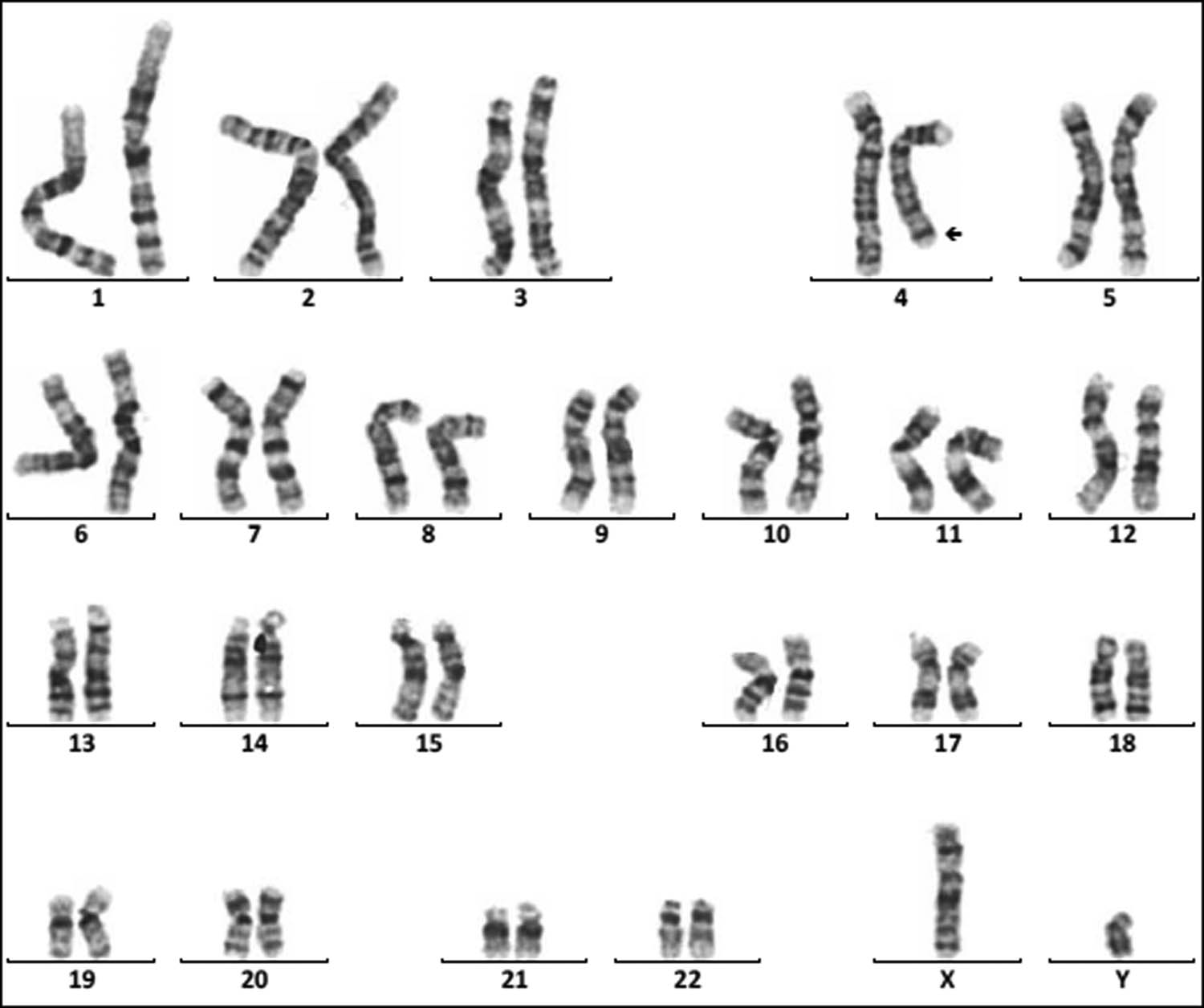
Karyotype of 46,XY,der(9)t(7;9)(p15;p22)pat. The arrows show abnormal chromosomes. (a) The karyotype of the patient. (b) The karyotype of the patient’s father. (c) The karyotype of the patient’s mother.
Karyotype of 46,XY,del(4)(q33). The arrows show abnormal chromosomes.
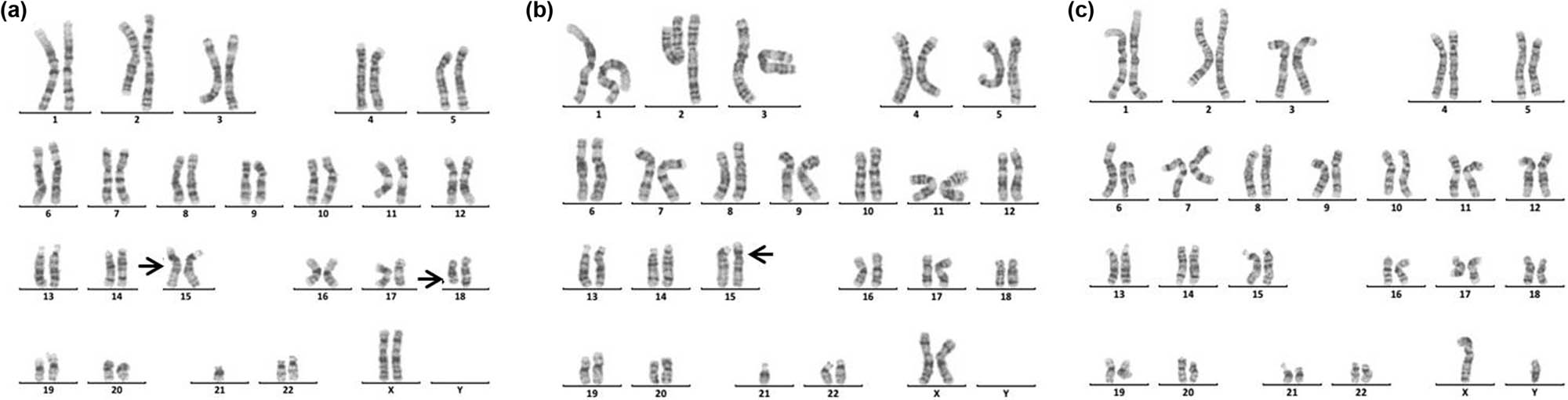
Karyotype of 45,XX,der(15;21)(q10;q10)mat,del(18)(q21). The arrows show abnormal chromosomes. (a) The karyotype of the patient. (b) The karyotype of the patient’s mother. (c) The karyotype of the patient’s father.
4 Discussion
Chromosomal diseases are often associated with growth retardation, mental retardation, and multiple malformations of organs, which are congenital diseases caused by chromosome number or structural abnormalities. In our study, 181 cases with abnormal karyotype were detected, and the abnormal rate was 41.51% (181/436), which indicated that chromosomal abnormalities were one of the important causes in children with dysplasia.
Intellectual disability (80.66%, 146/181) and dysmorphology (71.82%, 130/181) are the main clinical symptoms of 181 children with chromosomal abnormalities. As known, Tri21 is the most common chromosomal disorder, which implicated with intellectual disability and dysmorphology. The prevalence of Tri21 ranges from 1/700 to 1/2,000 in different ethnic populations investigated [17]. The cause of Tri21 is related to a variety of factors, such as increased reproductive age in women (>35 years old), degeneration of ovarian function, and genetic susceptibility [18,19]. In our study, 136 cases of Tri21 were detected (31.19%, 136/436), including 89 male and 47 female (1.89:1, 89/47). Among them, 130 cases were standard Tri21 (95.59%, 130/136), 5 cases were Robertson translocation Tri21 (3.79%, 5/132), and 1 case was a chimerical type (0.74%, 1/136), which is consistent with previous studies [20,21,22]. In 136 cases of Tri21, about two-thirds of pregnant women did not receive maternal serum screening for DS, and 16 pregnant women’s results of maternal serum screening for DS were high risk, but they did not receive a prenatal diagnosis. Therefore, enhancing people’s awareness of the importance of prenatal screening and genetic counseling in eugenics may be an important means of reducing birth in children with Tri21. In addition, one case with 46,XX,der(21;21)(q10;q10),t(1;12)(q43;p12.1), inv(15)(q13q24) (Figure 1), which is a rare complex Tri21 karyotype, was detected. So, this case is especially helpful to supplement the karyotype diversity of patients with Tri21.
SDS and short stature are the two other major clinical symptoms of 181 children with chromosomal abnormalities. As known, chromosomal abnormalities are one of the important causes of gonadal dysgenesis and physical retardation. In this study, 28 cases of sex chromosome abnormalities were detected, among which 20 cases were TS, and 5 cases were 46,X,i(X)(q10). The main manifestations of TS patients were immature uterus or no uterus, streak ovary or no ovary, primary amenorrhea, breast dysplasia, and short stature, and the main clinical symptoms of the patients with 46,X,i(X)(q10) were short stature and primary amenorrhea. These results suggested the necessity of two intact X chromosomes in the normal growth and development of a female. A single X chromosome in females randomly becomes inactive, but not all genes on which are transcriptionally silenced. Still, 15–20% of genes on inactive X chromosome remain operative and escape from X inactivation [23]. Moreover, normal female development is supported by a double dose of several specific genes on the X chromosome, such as the short stature homeobox (SHOX) gene, which is located at the Xp22, determines the height [24]. So, the haploinsufficiency of SHOX may be the main cause of short stature in patients. Dysmorphology and short stature caused by an X chromosome abnormality are more sensitive to estrogen before 12 years old. So, these kinds of patients need to receive treatment in the early stage. However, in this study, the average age of the patient with TS was11.94, greater than 6.6, which was reported by Massa et al. [25]. Consequently, the growth and development of children should be highly concerned during the preschool period. In addition, another three rare complex karyotypes were detected. One karyotype was 45,X,inv(9)(p11q13)[72]/46,X,dic(Y)(q11.23),inv(9)(p11q13)[10]/46,XY,inv(9)(p11q13)[4] (Figure 2), the other was 45,X[120]/46,X,r(X)(p22.2q22.2)[12]/46,X, rdup(X)(p22.2q22.2)[4] (Figure 3), and the third was 46,X,idic(X)(p11.2)[92]/45,X [9]/47,X,idic(X)(p11.2), idic(X)(p11.2)[5] (Figure 4).
Chromosome monomer or trisomy is one of the important causes of congenital malformation in children. Monomer 9p is a rare condition accompanied by trigonocephaly, facial dysmorphism, and developmental delay [26,27]. In this study, two cases with monosomy 9p were described. The karyotype of one patient was 46,XY,der(9)t(2;9)(p25;p22)mat, which presented with small ears, low ear position, small jaw, wide breast distance, short chest, long fingers and toes, inability to extend the second joint of the middle finger of both hands, special clenched fist posture (index finger pressed on the middle finger, little finger pressed on the ring finger) [28]. The karyotype of another patient was 46,XX,der(9)t(7;9)(p15;p22)pat (Figure 5). Studies have shown that patients with der(9)t(7;9) are mostly characterized by developmentally delayed psychomotor retardation and generalized developmental deficits (Table 3). In this study, the patient presents as developmentally delayed and psychomotor retardation, according to the previous report.
Clinical symptoms of the patient with der(9)t(7;9)
| Karyotype | Abnormal phenotype | Authors |
|---|---|---|
| 46, XY, der(9) t(7;9)(p15;p22) pat | Seizure, developmentally delayed, delayed myelination, and widened brain extracellular space | Zhong et al. [29] |
| 46, XY,t(7;9)(p22;q22)mat | Hypotrophic, full and wavy hair; a prominent forehead (middle facial part); microcephaly; low-set abnormal ears; hypertelorism; narrow, short eye slits; antimongoloid eye slant; broad, flat nasal bridge; bulbous nasal tip; microretrognathia; high palate; macrostomia; short neck; hollow stomach; short upper and lower extremities; bilateral clinodactyly of second and fifth fingers; thumbs and first toes are positioned far from other fingers (sandal gap); hypoplasia of toes nails; single transverse palmar crease; hypoplastic aortic arch; and hypoplastic lungs | Manvelyan et al. [30] |
| 46, XY,der(9)t(7;9)(p21.2;p23.5) | Bilateral choanal atresia, growth delay, marked psychomotor retardation, hydronephrosis, muscular hypotonia | Back et al. [31] |
| 46,XY, der(9),t(7;9)(q31.1;p23)pat | Generalized mild dysmorphic, heart failure, and hydrocephalus, sex reversal | Crocker et al. [32] |
| 46,XX,der(9)t(7;9)(p15.3;p24) | Psychomotor retardation, upward slant of palpebral fissures, and dolichomesophalangy | Teebi et al. [33] |
| 46,XX,der(9)t(7;9) (q35;q22.2) | Hypoplasia of the cerebellar vermis, dilated foramen Magendii, and dilatation of the cisterna magna | von Kaisenberg et al. [34] |
| 46, XX, der(9)t(7;9)(p15; p24) | Generalized developmental deficits, a high and large forehead, hypertelorism, and broad nasal bridge, hypothyroidism, obesity, cerebral palsy, severe mental retardation | Kozma et al. [35] |
| 46,XX,der(9)t(7;9)(p15;p22)pat | Developmental retardation and mental retardation | Present case |
Chromosome 4q deletion syndrome (4q-syndrome) is a rare condition, with an estimated incidence of 1 in 100,000 and the death rate was about 28% [36,37]. Although the clinical symptoms of patients with 4q-syndrome are complex and diverse (Table 4), through statistical analysis of the clinical symptoms of 101 patients, Strehle et al. found that craniofacial (99%), digital (88%), skeletal (54%), and cardiac (50%) were the most common anomalies [37]. Keeling et al. have reported that the critical region involved in the 4q terminal deletion syndrome may be 4q33 [43]. In this study, we reported a 16-month-old boy with 4q33-qter deletion (Figure 6). The patient presents with cleft palate, micrognathia, sydney line of the left hand, and developmental delay. Our results further support the idea that cleft palate-related genes might be located at 4q33 [43]. In addition, although most patients with 4q-syndrome have de novo deletions, familial cases have been reported, suggesting a high risk of recurrence of 4q-syndrome [47,48]. So, for these patients, prenatal diagnosis is necessary.
Clinical symptoms of the patient with 4q-syndrome
| Karyotype | Abnormal phenotype | Authors |
|---|---|---|
| Deletion of the segment 4q22.1-q23 | Slight developmental delay, mild dysmorphic features | Strehle et al. [38] |
| Deletion of segment 4q28.3-q31.23 | Growth failure, developmental delay, ventricular septum defect in the subaortic region, patent foramen ovale and patent ductus arteriosus, vascular malformation of the lung, dysgenesis of the corpus callosum and craniofacial dysmorphism | Duga et al. [39] |
| Deletion of segment 4q31-qter | Craniofacial dysmorphism, skeletal anomalies, ocular findings, and cardiac defect | Sandal et al. [40] |
| Deletion of segment q31.2-q35.2 | Craniofacial hypoplasia of left side of face, ipsilateral ptosis, erythroderma, and bilateral thumb anomalies | Kuldeep et al. [41] |
| Deletion of segment 4q31.21-q31.23 | Pseudohypoaldosteronism | Pritchard et al. [42] |
| Deletion of segment 4q31.3-qter | Complex CHD | Strehle et al. [38] |
| Deletion of segment 4q32-q34 | Mild developmental delay; a left ulnar ray defect with absent ulna and associated metacarpals, carpals, and phalanges; and a right ulnar nerve hypoplasia | Keeling et al. [43] |
| Deletion of segment 4q32.3-q34.3 | Congenital heart defects | Xu et al. [44] |
| Deletion of segment 4q33-qter | Mildly dysmorphic, heart failure, and hypercalcaemia | Strehle et al. [45] |
| Deletion of segment 4q33-qter | Cleft palate, micrognathia, sydney palm of left hand, and developmental delay | Present case |
| Deletion of segment 4q34.2-qter | CHD, submucosal cleft palate, hypernasal speech, learning difficulties, and right fifth finger anomaly manifestations | Tsai et al. [46] |
| Deletion of segment 4q34.3-qter | Asymptomatic cor triatriatum sinister | Marcì et al. [47] |
Deletion of the long arm of chromosome 18 (18q-) is relatively common among cytogenetic abnormality, which occurs incidentally in approximately 1 in 40,000 live births [49]. 18q-syndrome is characterized by a wide range of phenotypic abnormalities related to the size of the deletion and the position of breakpoints. The common clinical features of the 18q-syndrome are growth retardation, mental retardation, microcephaly, facial dysmorphisms, ear atresia, abnormal bone development, CHD, cerebral white matter abnormalities, and immature myelin formation [50,51,52]. In this study, the patient with a rare karyotype of 45,XX,der(15;21)(q10;q10)mat, del(18)(q21) (Figure 7) presented with mental retardation, unclear diction, facial dysmorphisms, abnormal bone development, the little finger end bending, poor balance ability, and unsteady walking. In addition, the patient also suffered from type I diabetes and Hashimoto’s thyroiditis, which were rarely reported. Therefore, this case is especially helpful in supplementing the phenotypic diversity of patients with 18q.
5 Conclusion
Chromosome abnormality is one of the important causes of dysplasia in children, and prenatal screening and diagnosis could play a great significance in preventing dysplasia in children. In addition, the retrospective analysis of the rare case is valuable for clinical diagnosis and risk assessment of recurrence.
Acknowledgements
We thank the patient and their family members for their participation in this study.
-
Funding information: This research was supported by Science and Technology Plan of Yantai (2021YD012).
-
Author contributions: H.Y.S. designed the experiments, Z.Y.M. and S.F.C. carried them out. H.W. and X.Y.L. were responsible for the summary and statistics of clinical data.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
[1] Wu XL, Li R, Fu F, Pan M, Han J, Yang X, et al. Chromosome microarray analysis in the investigation in children with congenital heart disease. BMC Pediatr. 2017;17:117.10.1186/s12887-017-0863-3Search in Google Scholar PubMed PubMed Central
[2] Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, et al. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol. 2011;32:1147–57.10.1007/s00246-011-0034-5Search in Google Scholar PubMed
[3] Huang Z, Sun Y, Fan Y, Wang L, Liu H, Gong Z, et al. Genetic evaluation of 114 Chinese short stature children in the next generation era: a single center study. Cell Physiol Biochem. 2018;49:295–305.10.1159/000492879Search in Google Scholar PubMed
[4] Britto IS, Regina Silva Herbest S, Tedesco GD, Drummond CL, Bussamra LC, Araujo Júnior E, et al. Prenatal diagnosis of a fetus with ring chromosomal 15 by two-and three-dimensional ultrasonography. Case Rep Obstet Gynecol. 2014;2014:495702.10.1155/2014/495702Search in Google Scholar PubMed PubMed Central
[5] Szabó A, Czakó M, Hadzsiev K, Duga B, Bánfai Z, Komlósi K, et al. Small supernumerary marker chromosome 15 and a ring chromosome 15 associated with a 15q26.3 deletion excluding the IGF1R gene. Am J Med Genet A. 2018;176:443–9.10.1002/ajmg.a.38566Search in Google Scholar PubMed
[6] Paz-Y-Miño C, Guevara-Aguirre J, Paz-Y-Miño A, Velarde F, Armendáriz-Castillo I, Yumiceba V, et al. Ring chromosome 15-cytogenetics and mapping arrays: a case report and review of the literature. J Med Case Rep. 2018;16(12):340.10.1186/s13256-018-1879-5Search in Google Scholar PubMed PubMed Central
[7] Ribeiro Dias Barroso C, Silveira Gomes L, Abrantes Silvestre V, Yamada Utagawa C. Cutis tricolor parvimaculata in ring chromosome 15 syndrome: a case report. Pediatr Dermatol. 2018;35:e204–5.10.1111/pde.13497Search in Google Scholar PubMed
[8] Chen CP, Lin SP, Chern SR, Wu PS, Chen YN, Chen SW, et al. Molecular cytogenetic characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 8 or r(8)(: p11.22 → q11.21:) in an 18-year-old female with short stature, obesity, attention deficit hyperactivity disorder, and intellectual disability. Taiwan J Obstet Gynecol. 2016;55:856–60.10.1016/j.tjog.2016.08.003Search in Google Scholar PubMed
[9] Chen CP, Lin SP, Lin YH, Chern SR, Wu PS, Chen YN, et al. Molecular cytogenetic characterization of mosaicism for a small Supernumerary marker chromosome derived from chromosome 8 or r(8)(: p12 → q13.1:) associated with phenotypicabnormalities. Taiwan J Obstet Gynecol. 2016;55:852–5.10.1016/j.tjog.2016.08.002Search in Google Scholar PubMed
[10] Farcas S, Erdelean D, Szekely FA, Navolan D, Andreescu N, Cioca A. A rare case of partial trisomy 8q24.12-q24.3 and partial monosomy of 8q24.3: prenatal diagnosis and clinical findings. Taiwan J Obstet Gynecol. 2019;58:36–9.10.1016/j.tjog.2018.11.005Search in Google Scholar PubMed
[11] Chen CP, Chen M, Ko TM, Ma GC, Tsai FJ, Tsai MS, et al. Prenatal diagnosis and molecular cytogenetic characterization of a small supernumerary marker chromosome derived from chromosome 8. Taiwan J Obstet Gynecol. 2010;49:500–5.10.1016/S1028-4559(10)60104-0Search in Google Scholar
[12] Chen CP, Chang SD, Su YN, Chen M, Chern SR, Su JW, et al. Rapid positive confirmation of mosaicism for a small supernumerary marker chromosome as r(8) by interphase fluorescence in situ hybridization, quantitative fluorescent polymerase chain reaction, and array comparative genomic hybridization on uncultured amniocytes in a pregnancy with fetal pyelectasis. Taiwan J Obstet Gynecol. 2012;51:405–10.10.1016/j.tjog.2012.07.016Search in Google Scholar PubMed
[13] Shao HY, Miao ZY, Liu XY, Hou XF, Wu H. Molecular cytogenetic characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 8 associated with congenital hypoplasia of the tongue and review of the literature. Taiwan J Obstet Gynecol. 2020;59:323–6.10.1016/j.tjog.2020.01.025Search in Google Scholar PubMed
[14] Spazzapan P, Arnaud E, Baujat G, Nizon M, Malan V, Brunelle F, et al. Clinical and neuroradiological features of the 9p deletion syndrome. Childs Nerv Syst. 2016;32:327–35.10.1007/s00381-015-2957-2Search in Google Scholar PubMed
[15] Hou QF, Wu D, Chu Y, Liao SX. Clinical findings and molecular cytogenetic study of de novo pure chromosome 9p deletion: Pre- and postnatal diagnosis. Taiwan J Obstet Gynecol. 2016;55:867–70.10.1016/j.tjog.2016.11.001Search in Google Scholar PubMed
[16] Tawn EJ, Curwen GB, Jonas P, Riddell AE, Hodgson L. Chromosome aberrations determined by sFISH and G-banding in lymphocytes from workers with internal deposits of plutonium. Int J Radiat Biol. 2016;92:312–20.10.3109/09553002.2016.1152414Search in Google Scholar PubMed PubMed Central
[17] Wu H, Miao ZY, Hou XF, Liu XY, Shao HY. Prenatal diagnosis of low-level mosaicism for trisomy 21 with rare karyotype detected by noninvasive prenatal testing. Taiwan J Obstet Gynecol. 2017;56:703–5.10.1016/j.tjog.2017.08.027Search in Google Scholar PubMed
[18] Yamamot T, Shimojima K, Nishizawa T, Matsuo M, Ito M, Imai K. Clinical manifestations of the deletion of Down syndrome critical region including DYRKlA and KCNJ6. Am J Med Genet A. 2011;155A:113–9.10.1002/ajmg.a.33735Search in Google Scholar PubMed
[19] Grebe C, Klingebiel TM, Graus P, Toischer K, Didié M, Jacobshagen C, et al. Enhanced expression of DYRKlA in cardiomyocytes inhibits acute NFAT activation but does not prevent hypertrophy in vivo. Cardiovase Res. 2011;90:521–8.10.1093/cvr/cvr023Search in Google Scholar PubMed
[20] Zhao W, Chen F, Wu M, Jiang S, Wu B, Luo H, et al. Postnatal identification of trisomy 21: an overview of 7,133 postnatal trisomy 21 cases identified in a diagnostic reference laboratory in China. PLoS One. 2015;10:e0133151.10.1371/journal.pone.0133151Search in Google Scholar PubMed PubMed Central
[21] Balkan M, Akbas H, Isi H, Oral D, Turkyilmaz A, Kalkanli S, et al. Cytogenetic analysis of 4216 patients referred for suspected chromosomal abnormalities in Southeast Turkey. Genet Mol Res. 2010;9:1094–103.10.4238/vol9-2gmr827Search in Google Scholar PubMed
[22] Mandava S, Koppaka N, Bhatia V, Das BR. Cytogenetic analysis of 1572 cases of down syndrome: a report of double aneuploidy and novel findings 47,XY,t(14;21)(q13;q22.3)mat, + 21 and 45,XX,t(14;21)in an Indian population. Genet Test Mol Biomarkers. 2010;14:499–504.10.1089/gtmb.2009.0167Search in Google Scholar PubMed
[23] Carrel L, Cottle AA, Goglin KC, Willard HF. A first-generation X inactivation profile of the human X chromosome. Proc Natl Acad Sci USA. 1999;96:14440–4.10.1073/pnas.96.25.14440Search in Google Scholar PubMed PubMed Central
[24] Seo GH, Kang E, Cho JH, Lee BH, Choi JH, Kim GH, et al. Turner syndrome presented with tall stature due to overdosage of the SHOX gene. Ann Pediatr Endocrinol Metab. 2015;20:110–3.10.6065/apem.2015.20.2.110Search in Google Scholar PubMed PubMed Central
[25] Massa G, Verlinde F, De Schepper J, Thomas M, Bourguignon JP, Craen M, et al. Trends in age at diagnosis of turner syndrome. Arch Dis Child. 2005;90:267–8.10.1136/adc.2004.049817Search in Google Scholar PubMed PubMed Central
[26] León-Carlos NY, García-Delgado C, Morales-Jiménez AB, Serrano-Bello C, Cervantes A, Morán Barroso VF. Monosomy 9p24 in two non-related patients as result of a translocation (2;9). Arch Argent Pediatr. 2018;116:e603–8.Search in Google Scholar
[27] Spazzapan P, Arnaud E, Baujat G, Nizon M, Malan V, Brunelle F, et al. Clinical and neuroradiological features of the 9p deletion syndrome. Childs Nerv Syst. 2016;32:327–35.10.1007/s00381-015-2957-2Search in Google Scholar PubMed
[28] Miao ZY, Shao HY, Wu H. One case of 46,XY,der(9)t(2;9)(p25;p22)mat. Chin J Med Genet. 2016;1:47.Search in Google Scholar
[29] Zhong M, Dong Y, Li M, Yao. H. Infantile spasms in a boy with an abnormal karyotype (46, XY, der(9)t(7;9)(p15;p22)pat). Neurol India. 2014;62:189–91.10.4103/0028-3886.132393Search in Google Scholar PubMed
[30] Manvelyan M, Simonyan I, Hovhannisyan G, Aroutiounian R, Hamid AB, Liehr T. A new case of a complex small supernumerary marker chromosome: a der(9)t(7;9)(p22;q22) due to a maternal balanced rearrangement. J Pediatr Genet. 2015;4:199–200.10.1055/s-0035-1565270Search in Google Scholar PubMed PubMed Central
[31] Back E, Jung C, Zeitler S, Schempp W. De novo duplication of 7pter-- > p21.2 and deletion of 9pter-- > p23.5: clinical and cytogenetic diagnosis. Clin Genet. 1997;51:56–60.10.1111/j.1399-0004.1997.tb02416.xSearch in Google Scholar
[32] Crocker M, Coghill SB, Cortinho. R. An unbalanced autosomal translocation (7;9) associated with feminization. Clin Genet. 1988;34:70–3.10.1111/j.1399-0004.1988.tb02618.xSearch in Google Scholar PubMed
[33] Teebi AS, Gibson L, McGrath J, Meyn MS, Breg WR, Yang-Feng TL. Molecular and cytogenetic characterization of 9p- abnormalities. Am J Med Genet. 1993;46:288–92.10.1002/ajmg.1320460310Search in Google Scholar
[34] von Kaisenberg CS, Caliebe A, Krams M, Hackelöer BJ, Jonat W. Absence of 9q22-9qter in trisomy 9 does not prevent a Dandy-Walker phenotype. Am J Med Genet. 2000;95:425–8.10.1002/1096-8628(20001218)95:5<425::AID-AJMG3>3.0.CO;2-DSearch in Google Scholar
[35] Kozma C, Haddad BR, Meck JM. Trisomy 7p resulting from 7p15;9p24 translocation: report of a new case and review of associated medical complications. Am J Med Genet. 2000;91:286–90.10.1002/(SICI)1096-8628(20000410)91:4<286::AID-AJMG9>3.0.CO;2-2Search in Google Scholar
[36] Strehle EM, Yu L, Rosenfeld JA, Donkervoort S, Zhou Y, Chen TJ, et al. Genotype-phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet A. 2012;158A(9):2139–51.10.1002/ajmg.a.35502Search in Google Scholar
[37] Strehle EM, Bantock HM. The phenotype of patients with 4q-syndrome. Genet Couns. 2003;14(2):195–205.Search in Google Scholar
[38] Strehle EM, Gruszfeld D, Schenk D, Mehta SG, Simonic I, Huang T. The spectrum of 4q- syndrome illustrated by a case series. Gene. 2012;506(2):387–91.10.1016/j.gene.2012.06.087Search in Google Scholar
[39] Duga B, Czako M, Komlosi K, Hadzsiev K, Torok K, Sumegi K, et al. Deletion of 4q28.3-31.23 in the background of multiple malformations with pulmonary hypertension. Mol Cytogenet. 2014;7:36.10.1186/1755-8166-7-36Search in Google Scholar
[40] Sandal G, Ormeci AR, Oztas S. De novo terminal 4q deletion syndrome with new ocular findings in Turkish twins: case report. Genet Couns. 2013;24(2):217–22.Search in Google Scholar
[41] Kuldeep CM, Khare AK, Garg A, Mittal A, Gupta L. Terminal 4q deletion syndrome. Indian J Dermatol. 2012;57(3):222–4.10.4103/0019-5154.96203Search in Google Scholar
[42] Pritchard AB, Ritter A, Kearney HM, Izumi. K. Interstitial 4q deletion syndrome including NR3C2 causing pseudohypoaldosteronism. Mol Syndromol. 2020;10(6):327–31.10.1159/000505279Search in Google Scholar
[43] Keeling SL, Lee-Jones L, Thompson P. Interstitial deletion 4q32-34 with ulnar deficiency: 4q33 may be the critical region in 4q terminal deletion syndrome. Am J Med Genet. 2001;99(2):94–8.10.1002/1096-8628(2000)9999:999<00::AID-AJMG1134>3.0.CO;2-DSearch in Google Scholar
[44] Xu W, Ahmad A, Dagenais S, Iyer RK, Innis JW. Chromosome 4q deletion syndrome: narrowing the cardiovascular critical region to 4q32.2-q34.3. Am J Med Genet A. 2012;158A(3):635–40.10.1002/ajmg.a.34425Search in Google Scholar
[45] Strehle EM, Ahmed OA, Hameed M, Russell A. The 4q-syndrome. Couns. 2001;12(4):327–39.Search in Google Scholar
[46] Tsai CH, Van Dyke DL, Feldman GL. Child with velocardiofacial syndrome and del (4)(q34.2): another critical region associated with a velocardiofacial syndrome-like phenotype. Am J Med Genet. 1999;82(4):336–39.10.1002/(SICI)1096-8628(19990212)82:4<336::AID-AJMG11>3.0.CO;2-ISearch in Google Scholar
[47] Marcì M, Guarina A, Castiglione MC, Sanfilippo N. Different cardiac anomalies in mother and son with 4q-syndrome. Genet. 2015;2015:932651.10.1155/2015/932651Search in Google Scholar
[48] Lall M, Puri R, Saviour P, Verma I. A familial deletion 4q syndrome: an outcome of a paracentric inversion. Indian J Hum Genet. 2012;18(2):238–40.10.4103/0971-6866.100780Search in Google Scholar
[49] Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, et al. Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet. 1999;85(5):455–62.10.1002/(SICI)1096-8628(19990827)85:5<455::AID-AJMG5>3.0.CO;2-ZSearch in Google Scholar
[50] Mark PR, Radlinski BC, Core N, Fryer A, Kirk EP, Haldeman-Englert CR. Narrowing the critical region for congenital vertical talus in patients with interstitial 18q deletions. Am J Med Genet A. 2013;161A(5):1117–21.10.1002/ajmg.a.35791Search in Google Scholar
[51] Perry BP, Sebold C, Hasi M, Heard P, Carter E, Hill A, et al. Sensorineural hearing loss in people with deletions of 18q: hearing in 18q-. Otol Neurotol. 2014;35(5):782–86.10.1097/MAO.0000000000000363Search in Google Scholar
[52] Tassano E, Severino M, Rosina S, Papa R, Tortora D, Gimelli G, et al. Interstitial de novo 18q22.3q23 deletion: clinical, neuroradiological and molecular characterization f a new case and review of the literature. Mol Cytogenet. 2016;9:78.10.1186/s13039-016-0285-1Search in Google Scholar
© 2022 Zong-Yu Miao et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Biomedical Sciences
- Effects of direct oral anticoagulants dabigatran and rivaroxaban on the blood coagulation function in rabbits
- The mother of all battles: Viruses vs humans. Can humans avoid extinction in 50–100 years?
- Knockdown of G1P3 inhibits cell proliferation and enhances the cytotoxicity of dexamethasone in acute lymphoblastic leukemia
- LINC00665 regulates hepatocellular carcinoma by modulating mRNA via the m6A enzyme
- Association study of CLDN14 variations in patients with kidney stones
- Concanavalin A-induced autoimmune hepatitis model in mice: Mechanisms and future outlook
- Regulation of miR-30b in cancer development, apoptosis, and drug resistance
- Informatic analysis of the pulmonary microecology in non-cystic fibrosis bronchiectasis at three different stages
- Swimming attenuates tumor growth in CT-26 tumor-bearing mice and suppresses angiogenesis by mediating the HIF-1α/VEGFA pathway
- Characterization of intestinal microbiota and serum metabolites in patients with mild hepatic encephalopathy
- Functional conservation and divergence in plant-specific GRF gene family revealed by sequences and expression analysis
- Application of the FLP/LoxP-FRT recombination system to switch the eGFP expression in a model prokaryote
- Biomedical evaluation of antioxidant properties of lamb meat enriched with iodine and selenium
- Intravenous infusion of the exosomes derived from human umbilical cord mesenchymal stem cells enhance neurological recovery after traumatic brain injury via suppressing the NF-κB pathway
- Effect of dietary pattern on pregnant women with gestational diabetes mellitus and its clinical significance
- Potential regulatory mechanism of TNF-α/TNFR1/ANXA1 in glioma cells and its role in glioma cell proliferation
- Effect of the genetic mutant G71R in uridine diphosphate-glucuronosyltransferase 1A1 on the conjugation of bilirubin
- Quercetin inhibits cytotoxicity of PC12 cells induced by amyloid-beta 25–35 via stimulating estrogen receptor α, activating ERK1/2, and inhibiting apoptosis
- Nutrition intervention in the management of novel coronavirus pneumonia patients
- circ-CFH promotes the development of HCC by regulating cell proliferation, apoptosis, migration, invasion, and glycolysis through the miR-377-3p/RNF38 axis
- Bmi-1 directly upregulates glucose transporter 1 in human gastric adenocarcinoma
- Lacunar infarction aggravates the cognitive deficit in the elderly with white matter lesion
- Hydroxysafflor yellow A improved retinopathy via Nrf2/HO-1 pathway in rats
- Comparison of axon extension: PTFE versus PLA formed by a 3D printer
- Elevated IL-35 level and iTr35 subset increase the bacterial burden and lung lesions in Mycobacterium tuberculosis-infected mice
- A case report of CAT gene and HNF1β gene variations in a patient with early-onset diabetes
- Study on the mechanism of inhibiting patulin production by fengycin
- SOX4 promotes high-glucose-induced inflammation and angiogenesis of retinal endothelial cells by activating NF-κB signaling pathway
- Relationship between blood clots and COVID-19 vaccines: A literature review
- Analysis of genetic characteristics of 436 children with dysplasia and detailed analysis of rare karyotype
- Bioinformatics network analyses of growth differentiation factor 11
- NR4A1 inhibits the epithelial–mesenchymal transition of hepatic stellate cells: Involvement of TGF-β–Smad2/3/4–ZEB signaling
- Expression of Zeb1 in the differentiation of mouse embryonic stem cell
- Study on the genetic damage caused by cadmium sulfide quantum dots in human lymphocytes
- Association between single-nucleotide polymorphisms of NKX2.5 and congenital heart disease in Chinese population: A meta-analysis
- Assessment of the anesthetic effect of modified pentothal sodium solution on Sprague-Dawley rats
- Genetic susceptibility to high myopia in Han Chinese population
- Potential biomarkers and molecular mechanisms in preeclampsia progression
- Silencing circular RNA-friend leukemia virus integration 1 restrained malignancy of CC cells and oxaliplatin resistance by disturbing dyskeratosis congenita 1
- Endostar plus pembrolizumab combined with a platinum-based dual chemotherapy regime for advanced pulmonary large-cell neuroendocrine carcinoma as a first-line treatment: A case report
- The significance of PAK4 in signaling and clinicopathology: A review
- Sorafenib inhibits ovarian cancer cell proliferation and mobility and induces radiosensitivity by targeting the tumor cell epithelial–mesenchymal transition
- Characterization of rabbit polyclonal antibody against camel recombinant nanobodies
- Active legumain promotes invasion and migration of neuroblastoma by regulating epithelial-mesenchymal transition
- Effect of cell receptors in the pathogenesis of osteoarthritis: Current insights
- MT-12 inhibits the proliferation of bladder cells in vitro and in vivo by enhancing autophagy through mitochondrial dysfunction
- Study of hsa_circRNA_000121 and hsa_circRNA_004183 in papillary thyroid microcarcinoma
- BuyangHuanwu Decoction attenuates cerebral vasospasm caused by subarachnoid hemorrhage in rats via PI3K/AKT/eNOS axis
- Effects of the interaction of Notch and TLR4 pathways on inflammation and heart function in septic heart
- Monosodium iodoacetate-induced subchondral bone microstructure and inflammatory changes in an animal model of osteoarthritis
- A rare presentation of type II Abernethy malformation and nephrotic syndrome: Case report and review
- Rapid death due to pulmonary epithelioid haemangioendothelioma in several weeks: A case report
- Hepatoprotective role of peroxisome proliferator-activated receptor-α in non-cancerous hepatic tissues following transcatheter arterial embolization
- Correlation between peripheral blood lymphocyte subpopulations and primary systemic lupus erythematosus
- A novel SLC8A1-ALK fusion in lung adenocarcinoma confers sensitivity to alectinib: A case report
- β-Hydroxybutyrate upregulates FGF21 expression through inhibition of histone deacetylases in hepatocytes
- Identification of metabolic genes for the prediction of prognosis and tumor microenvironment infiltration in early-stage non-small cell lung cancer
- BTBD10 inhibits glioma tumorigenesis by downregulating cyclin D1 and p-Akt
- Mucormycosis co-infection in COVID-19 patients: An update
- Metagenomic next-generation sequencing in diagnosing Pneumocystis jirovecii pneumonia: A case report
- Long non-coding RNA HOXB-AS1 is a prognostic marker and promotes hepatocellular carcinoma cells’ proliferation and invasion
- Preparation and evaluation of LA-PEG-SPION, a targeted MRI contrast agent for liver cancer
- Proteomic analysis of the liver regulating lipid metabolism in Chaohu ducks using two-dimensional electrophoresis
- Nasopharyngeal tuberculosis: A case report
- Characterization and evaluation of anti-Salmonella enteritidis activity of indigenous probiotic lactobacilli in mice
- Aberrant pulmonary immune response of obese mice to periodontal infection
- Bacteriospermia – A formidable player in male subfertility
- In silico and in vivo analysis of TIPE1 expression in diffuse large B cell lymphoma
- Effects of KCa channels on biological behavior of trophoblasts
- Interleukin-17A influences the vulnerability rather than the size of established atherosclerotic plaques in apolipoprotein E-deficient mice
- Multiple organ failure and death caused by Staphylococcus aureus hip infection: A case report
- Prognostic signature related to the immune environment of oral squamous cell carcinoma
- Primary and metastatic squamous cell carcinoma of the thyroid gland: Two case reports
- Neuroprotective effects of crocin and crocin-loaded niosomes against the paraquat-induced oxidative brain damage in rats
- Role of MMP-2 and CD147 in kidney fibrosis
- Geometric basis of action potential of skeletal muscle cells and neurons
- Babesia microti-induced fulminant sepsis in an immunocompromised host: A case report and the case-specific literature review
- Role of cerebellar cortex in associative learning and memory in guinea pigs
- Application of metagenomic next-generation sequencing technique for diagnosing a specific case of necrotizing meningoencephalitis caused by human herpesvirus 2
- Case report: Quadruple primary malignant neoplasms including esophageal, ureteral, and lung in an elderly male
- Long non-coding RNA NEAT1 promotes angiogenesis in hepatoma carcinoma via the miR-125a-5p/VEGF pathway
- Osteogenic differentiation of periodontal membrane stem cells in inflammatory environments
- Knockdown of SHMT2 enhances the sensitivity of gastric cancer cells to radiotherapy through the Wnt/β-catenin pathway
- Continuous renal replacement therapy combined with double filtration plasmapheresis in the treatment of severe lupus complicated by serious bacterial infections in children: A case report
- Simultaneous triple primary malignancies, including bladder cancer, lymphoma, and lung cancer, in an elderly male: A case report
- Preclinical immunogenicity assessment of a cell-based inactivated whole-virion H5N1 influenza vaccine
- One case of iodine-125 therapy – A new minimally invasive treatment of intrahepatic cholangiocarcinoma
- S1P promotes corneal trigeminal neuron differentiation and corneal nerve repair via upregulating nerve growth factor expression in a mouse model
- Early cancer detection by a targeted methylation assay of circulating tumor DNA in plasma
- Calcifying nanoparticles initiate the calcification process of mesenchymal stem cells in vitro through the activation of the TGF-β1/Smad signaling pathway and promote the decay of echinococcosis
- Evaluation of prognostic markers in patients infected with SARS-CoV-2
- N6-Methyladenosine-related alternative splicing events play a role in bladder cancer
- Characterization of the structural, oxidative, and immunological features of testis tissue from Zucker diabetic fatty rats
- Effects of glucose and osmotic pressure on the proliferation and cell cycle of human chorionic trophoblast cells
- Investigation of genotype diversity of 7,804 norovirus sequences in humans and animals of China
- Characteristics and karyotype analysis of a patient with turner syndrome complicated with multiple-site tumors: A case report
- Aggravated renal fibrosis is positively associated with the activation of HMGB1-TLR2/4 signaling in STZ-induced diabetic mice
- Distribution characteristics of SARS-CoV-2 IgM/IgG in false-positive results detected by chemiluminescent immunoassay
- SRPX2 attenuated oxygen–glucose deprivation and reperfusion-induced injury in cardiomyocytes via alleviating endoplasmic reticulum stress-induced apoptosis through targeting PI3K/Akt/mTOR axis
- Aquaporin-8 overexpression is involved in vascular structure and function changes in placentas of gestational diabetes mellitus patients
- Relationship between CRP gene polymorphisms and ischemic stroke risk: A systematic review and meta-analysis
- Effects of growth hormone on lipid metabolism and sexual development in pubertal obese male rats
- Cloning and identification of the CTLA-4IgV gene and functional application of vaccine in Xinjiang sheep
- Antitumor activity of RUNX3: Upregulation of E-cadherin and downregulation of the epithelial–mesenchymal transition in clear-cell renal cell carcinoma
- PHF8 promotes osteogenic differentiation of BMSCs in old rat with osteoporosis by regulating Wnt/β-catenin pathway
- A review of the current state of the computer-aided diagnosis (CAD) systems for breast cancer diagnosis
- Bilateral dacryoadenitis in adult-onset Still’s disease: A case report
- A novel association between Bmi-1 protein expression and the SUVmax obtained by 18F-FDG PET/CT in patients with gastric adenocarcinoma
- The role of erythrocytes and erythroid progenitor cells in tumors
- Relationship between platelet activation markers and spontaneous abortion: A meta-analysis
- Abnormal methylation caused by folic acid deficiency in neural tube defects
- Silencing TLR4 using an ultrasound-targeted microbubble destruction-based shRNA system reduces ischemia-induced seizures in hyperglycemic rats
- Plant Sciences
- Seasonal succession of bacterial communities in cultured Caulerpa lentillifera detected by high-throughput sequencing
- Cloning and prokaryotic expression of WRKY48 from Caragana intermedia
- Novel Brassica hybrids with different resistance to Leptosphaeria maculans reveal unbalanced rDNA signal patterns
- Application of exogenous auxin and gibberellin regulates the bolting of lettuce (Lactuca sativa L.)
- Phytoremediation of pollutants from wastewater: A concise review
- Genome-wide identification and characterization of NBS-encoding genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.)
- Alleviative effects of magnetic Fe3O4 nanoparticles on the physiological toxicity of 3-nitrophenol to rice (Oryza sativa L.) seedlings
- Selection and functional identification of Dof genes expressed in response to nitrogen in Populus simonii × Populus nigra
- Study on pecan seed germination influenced by seed endocarp
- Identification of active compounds in Ophiopogonis Radix from different geographical origins by UPLC-Q/TOF-MS combined with GC-MS approaches
- The entire chloroplast genome sequence of Asparagus cochinchinensis and genetic comparison to Asparagus species
- Genome-wide identification of MAPK family genes and their response to abiotic stresses in tea plant (Camellia sinensis)
- Selection and validation of reference genes for RT-qPCR analysis of different organs at various development stages in Caragana intermedia
- Cloning and expression analysis of SERK1 gene in Diospyros lotus
- Integrated metabolomic and transcriptomic profiling revealed coping mechanisms of the edible and medicinal homologous plant Plantago asiatica L. cadmium resistance
- A missense variant in NCF1 is associated with susceptibility to unexplained recurrent spontaneous abortion
- Assessment of drought tolerance indices in faba bean genotypes under different irrigation regimes
- The entire chloroplast genome sequence of Asparagus setaceus (Kunth) Jessop: Genome structure, gene composition, and phylogenetic analysis in Asparagaceae
- Food Science
- Dietary food additive monosodium glutamate with or without high-lipid diet induces spleen anomaly: A mechanistic approach on rat model
- Binge eating disorder during COVID-19
- Potential of honey against the onset of autoimmune diabetes and its associated nephropathy, pancreatitis, and retinopathy in type 1 diabetic animal model
- FTO gene expression in diet-induced obesity is downregulated by Solanum fruit supplementation
- Physical activity enhances fecal lactobacilli in rats chronically drinking sweetened cola beverage
- Supercritical CO2 extraction, chemical composition, and antioxidant effects of Coreopsis tinctoria Nutt. oleoresin
- Functional constituents of plant-based foods boost immunity against acute and chronic disorders
- Effect of selenium and methods of protein extraction on the proteomic profile of Saccharomyces yeast
- Microbial diversity of milk ghee in southern Gansu and its effect on the formation of ghee flavor compounds
- Ecology and Environmental Sciences
- Effects of heavy metals on bacterial community surrounding Bijiashan mining area located in northwest China
- Microorganism community composition analysis coupling with 15N tracer experiments reveals the nitrification rate and N2O emissions in low pH soils in Southern China
- Genetic diversity and population structure of Cinnamomum balansae Lecomte inferred by microsatellites
- Preliminary screening of microplastic contamination in different marine fish species of Taif market, Saudi Arabia
- Plant volatile organic compounds attractive to Lygus pratensis
- Effects of organic materials on soil bacterial community structure in long-term continuous cropping of tomato in greenhouse
- Effects of soil treated fungicide fluopimomide on tomato (Solanum lycopersicum L.) disease control and plant growth
- Prevalence of Yersinia pestis among rodents captured in a semi-arid tropical ecosystem of south-western Zimbabwe
- Effects of irrigation and nitrogen fertilization on mitigating salt-induced Na+ toxicity and sustaining sea rice growth
- Bioengineering and Biotechnology
- Poly-l-lysine-caused cell adhesion induces pyroptosis in THP-1 monocytes
- Development of alkaline phosphatase-scFv and its use for one-step enzyme-linked immunosorbent assay for His-tagged protein detection
- Development and validation of a predictive model for immune-related genes in patients with tongue squamous cell carcinoma
- Agriculture
- Effects of chemical-based fertilizer replacement with biochar-based fertilizer on albic soil nutrient content and maize yield
- Genome-wide identification and expression analysis of CPP-like gene family in Triticum aestivum L. under different hormone and stress conditions
- Agronomic and economic performance of mung bean (Vigna radiata L.) varieties in response to rates of blended NPS fertilizer in Kindo Koysha district, Southern Ethiopia
- Influence of furrow irrigation regime on the yield and water consumption indicators of winter wheat based on a multi-level fuzzy comprehensive evaluation
- Discovery of exercise-related genes and pathway analysis based on comparative genomes of Mongolian originated Abaga and Wushen horse
- Lessons from integrated seasonal forecast-crop modelling in Africa: A systematic review
- Evolution trend of soil fertility in tobacco-planting area of Chenzhou, Hunan Province, China
- Animal Sciences
- Morphological and molecular characterization of Tatera indica Hardwicke 1807 (Rodentia: Muridae) from Pothwar, Pakistan
- Research on meat quality of Qianhua Mutton Merino sheep and Small-tail Han sheep
- SI: A Scientific Memoir
- Suggestions on leading an academic research laboratory group
- My scientific genealogy and the Toronto ACDC Laboratory, 1988–2022
- Erratum
- Erratum to “Changes of immune cells in patients with hepatocellular carcinoma treated by radiofrequency ablation and hepatectomy, a pilot study”
- Erratum to “A two-microRNA signature predicts the progression of male thyroid cancer”
- Retraction
- Retraction of “Lidocaine has antitumor effect on hepatocellular carcinoma via the circ_DYNC1H1/miR-520a-3p/USP14 axis”
Articles in the same Issue
- Biomedical Sciences
- Effects of direct oral anticoagulants dabigatran and rivaroxaban on the blood coagulation function in rabbits
- The mother of all battles: Viruses vs humans. Can humans avoid extinction in 50–100 years?
- Knockdown of G1P3 inhibits cell proliferation and enhances the cytotoxicity of dexamethasone in acute lymphoblastic leukemia
- LINC00665 regulates hepatocellular carcinoma by modulating mRNA via the m6A enzyme
- Association study of CLDN14 variations in patients with kidney stones
- Concanavalin A-induced autoimmune hepatitis model in mice: Mechanisms and future outlook
- Regulation of miR-30b in cancer development, apoptosis, and drug resistance
- Informatic analysis of the pulmonary microecology in non-cystic fibrosis bronchiectasis at three different stages
- Swimming attenuates tumor growth in CT-26 tumor-bearing mice and suppresses angiogenesis by mediating the HIF-1α/VEGFA pathway
- Characterization of intestinal microbiota and serum metabolites in patients with mild hepatic encephalopathy
- Functional conservation and divergence in plant-specific GRF gene family revealed by sequences and expression analysis
- Application of the FLP/LoxP-FRT recombination system to switch the eGFP expression in a model prokaryote
- Biomedical evaluation of antioxidant properties of lamb meat enriched with iodine and selenium
- Intravenous infusion of the exosomes derived from human umbilical cord mesenchymal stem cells enhance neurological recovery after traumatic brain injury via suppressing the NF-κB pathway
- Effect of dietary pattern on pregnant women with gestational diabetes mellitus and its clinical significance
- Potential regulatory mechanism of TNF-α/TNFR1/ANXA1 in glioma cells and its role in glioma cell proliferation
- Effect of the genetic mutant G71R in uridine diphosphate-glucuronosyltransferase 1A1 on the conjugation of bilirubin
- Quercetin inhibits cytotoxicity of PC12 cells induced by amyloid-beta 25–35 via stimulating estrogen receptor α, activating ERK1/2, and inhibiting apoptosis
- Nutrition intervention in the management of novel coronavirus pneumonia patients
- circ-CFH promotes the development of HCC by regulating cell proliferation, apoptosis, migration, invasion, and glycolysis through the miR-377-3p/RNF38 axis
- Bmi-1 directly upregulates glucose transporter 1 in human gastric adenocarcinoma
- Lacunar infarction aggravates the cognitive deficit in the elderly with white matter lesion
- Hydroxysafflor yellow A improved retinopathy via Nrf2/HO-1 pathway in rats
- Comparison of axon extension: PTFE versus PLA formed by a 3D printer
- Elevated IL-35 level and iTr35 subset increase the bacterial burden and lung lesions in Mycobacterium tuberculosis-infected mice
- A case report of CAT gene and HNF1β gene variations in a patient with early-onset diabetes
- Study on the mechanism of inhibiting patulin production by fengycin
- SOX4 promotes high-glucose-induced inflammation and angiogenesis of retinal endothelial cells by activating NF-κB signaling pathway
- Relationship between blood clots and COVID-19 vaccines: A literature review
- Analysis of genetic characteristics of 436 children with dysplasia and detailed analysis of rare karyotype
- Bioinformatics network analyses of growth differentiation factor 11
- NR4A1 inhibits the epithelial–mesenchymal transition of hepatic stellate cells: Involvement of TGF-β–Smad2/3/4–ZEB signaling
- Expression of Zeb1 in the differentiation of mouse embryonic stem cell
- Study on the genetic damage caused by cadmium sulfide quantum dots in human lymphocytes
- Association between single-nucleotide polymorphisms of NKX2.5 and congenital heart disease in Chinese population: A meta-analysis
- Assessment of the anesthetic effect of modified pentothal sodium solution on Sprague-Dawley rats
- Genetic susceptibility to high myopia in Han Chinese population
- Potential biomarkers and molecular mechanisms in preeclampsia progression
- Silencing circular RNA-friend leukemia virus integration 1 restrained malignancy of CC cells and oxaliplatin resistance by disturbing dyskeratosis congenita 1
- Endostar plus pembrolizumab combined with a platinum-based dual chemotherapy regime for advanced pulmonary large-cell neuroendocrine carcinoma as a first-line treatment: A case report
- The significance of PAK4 in signaling and clinicopathology: A review
- Sorafenib inhibits ovarian cancer cell proliferation and mobility and induces radiosensitivity by targeting the tumor cell epithelial–mesenchymal transition
- Characterization of rabbit polyclonal antibody against camel recombinant nanobodies
- Active legumain promotes invasion and migration of neuroblastoma by regulating epithelial-mesenchymal transition
- Effect of cell receptors in the pathogenesis of osteoarthritis: Current insights
- MT-12 inhibits the proliferation of bladder cells in vitro and in vivo by enhancing autophagy through mitochondrial dysfunction
- Study of hsa_circRNA_000121 and hsa_circRNA_004183 in papillary thyroid microcarcinoma
- BuyangHuanwu Decoction attenuates cerebral vasospasm caused by subarachnoid hemorrhage in rats via PI3K/AKT/eNOS axis
- Effects of the interaction of Notch and TLR4 pathways on inflammation and heart function in septic heart
- Monosodium iodoacetate-induced subchondral bone microstructure and inflammatory changes in an animal model of osteoarthritis
- A rare presentation of type II Abernethy malformation and nephrotic syndrome: Case report and review
- Rapid death due to pulmonary epithelioid haemangioendothelioma in several weeks: A case report
- Hepatoprotective role of peroxisome proliferator-activated receptor-α in non-cancerous hepatic tissues following transcatheter arterial embolization
- Correlation between peripheral blood lymphocyte subpopulations and primary systemic lupus erythematosus
- A novel SLC8A1-ALK fusion in lung adenocarcinoma confers sensitivity to alectinib: A case report
- β-Hydroxybutyrate upregulates FGF21 expression through inhibition of histone deacetylases in hepatocytes
- Identification of metabolic genes for the prediction of prognosis and tumor microenvironment infiltration in early-stage non-small cell lung cancer
- BTBD10 inhibits glioma tumorigenesis by downregulating cyclin D1 and p-Akt
- Mucormycosis co-infection in COVID-19 patients: An update
- Metagenomic next-generation sequencing in diagnosing Pneumocystis jirovecii pneumonia: A case report
- Long non-coding RNA HOXB-AS1 is a prognostic marker and promotes hepatocellular carcinoma cells’ proliferation and invasion
- Preparation and evaluation of LA-PEG-SPION, a targeted MRI contrast agent for liver cancer
- Proteomic analysis of the liver regulating lipid metabolism in Chaohu ducks using two-dimensional electrophoresis
- Nasopharyngeal tuberculosis: A case report
- Characterization and evaluation of anti-Salmonella enteritidis activity of indigenous probiotic lactobacilli in mice
- Aberrant pulmonary immune response of obese mice to periodontal infection
- Bacteriospermia – A formidable player in male subfertility
- In silico and in vivo analysis of TIPE1 expression in diffuse large B cell lymphoma
- Effects of KCa channels on biological behavior of trophoblasts
- Interleukin-17A influences the vulnerability rather than the size of established atherosclerotic plaques in apolipoprotein E-deficient mice
- Multiple organ failure and death caused by Staphylococcus aureus hip infection: A case report
- Prognostic signature related to the immune environment of oral squamous cell carcinoma
- Primary and metastatic squamous cell carcinoma of the thyroid gland: Two case reports
- Neuroprotective effects of crocin and crocin-loaded niosomes against the paraquat-induced oxidative brain damage in rats
- Role of MMP-2 and CD147 in kidney fibrosis
- Geometric basis of action potential of skeletal muscle cells and neurons
- Babesia microti-induced fulminant sepsis in an immunocompromised host: A case report and the case-specific literature review
- Role of cerebellar cortex in associative learning and memory in guinea pigs
- Application of metagenomic next-generation sequencing technique for diagnosing a specific case of necrotizing meningoencephalitis caused by human herpesvirus 2
- Case report: Quadruple primary malignant neoplasms including esophageal, ureteral, and lung in an elderly male
- Long non-coding RNA NEAT1 promotes angiogenesis in hepatoma carcinoma via the miR-125a-5p/VEGF pathway
- Osteogenic differentiation of periodontal membrane stem cells in inflammatory environments
- Knockdown of SHMT2 enhances the sensitivity of gastric cancer cells to radiotherapy through the Wnt/β-catenin pathway
- Continuous renal replacement therapy combined with double filtration plasmapheresis in the treatment of severe lupus complicated by serious bacterial infections in children: A case report
- Simultaneous triple primary malignancies, including bladder cancer, lymphoma, and lung cancer, in an elderly male: A case report
- Preclinical immunogenicity assessment of a cell-based inactivated whole-virion H5N1 influenza vaccine
- One case of iodine-125 therapy – A new minimally invasive treatment of intrahepatic cholangiocarcinoma
- S1P promotes corneal trigeminal neuron differentiation and corneal nerve repair via upregulating nerve growth factor expression in a mouse model
- Early cancer detection by a targeted methylation assay of circulating tumor DNA in plasma
- Calcifying nanoparticles initiate the calcification process of mesenchymal stem cells in vitro through the activation of the TGF-β1/Smad signaling pathway and promote the decay of echinococcosis
- Evaluation of prognostic markers in patients infected with SARS-CoV-2
- N6-Methyladenosine-related alternative splicing events play a role in bladder cancer
- Characterization of the structural, oxidative, and immunological features of testis tissue from Zucker diabetic fatty rats
- Effects of glucose and osmotic pressure on the proliferation and cell cycle of human chorionic trophoblast cells
- Investigation of genotype diversity of 7,804 norovirus sequences in humans and animals of China
- Characteristics and karyotype analysis of a patient with turner syndrome complicated with multiple-site tumors: A case report
- Aggravated renal fibrosis is positively associated with the activation of HMGB1-TLR2/4 signaling in STZ-induced diabetic mice
- Distribution characteristics of SARS-CoV-2 IgM/IgG in false-positive results detected by chemiluminescent immunoassay
- SRPX2 attenuated oxygen–glucose deprivation and reperfusion-induced injury in cardiomyocytes via alleviating endoplasmic reticulum stress-induced apoptosis through targeting PI3K/Akt/mTOR axis
- Aquaporin-8 overexpression is involved in vascular structure and function changes in placentas of gestational diabetes mellitus patients
- Relationship between CRP gene polymorphisms and ischemic stroke risk: A systematic review and meta-analysis
- Effects of growth hormone on lipid metabolism and sexual development in pubertal obese male rats
- Cloning and identification of the CTLA-4IgV gene and functional application of vaccine in Xinjiang sheep
- Antitumor activity of RUNX3: Upregulation of E-cadherin and downregulation of the epithelial–mesenchymal transition in clear-cell renal cell carcinoma
- PHF8 promotes osteogenic differentiation of BMSCs in old rat with osteoporosis by regulating Wnt/β-catenin pathway
- A review of the current state of the computer-aided diagnosis (CAD) systems for breast cancer diagnosis
- Bilateral dacryoadenitis in adult-onset Still’s disease: A case report
- A novel association between Bmi-1 protein expression and the SUVmax obtained by 18F-FDG PET/CT in patients with gastric adenocarcinoma
- The role of erythrocytes and erythroid progenitor cells in tumors
- Relationship between platelet activation markers and spontaneous abortion: A meta-analysis
- Abnormal methylation caused by folic acid deficiency in neural tube defects
- Silencing TLR4 using an ultrasound-targeted microbubble destruction-based shRNA system reduces ischemia-induced seizures in hyperglycemic rats
- Plant Sciences
- Seasonal succession of bacterial communities in cultured Caulerpa lentillifera detected by high-throughput sequencing
- Cloning and prokaryotic expression of WRKY48 from Caragana intermedia
- Novel Brassica hybrids with different resistance to Leptosphaeria maculans reveal unbalanced rDNA signal patterns
- Application of exogenous auxin and gibberellin regulates the bolting of lettuce (Lactuca sativa L.)
- Phytoremediation of pollutants from wastewater: A concise review
- Genome-wide identification and characterization of NBS-encoding genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.)
- Alleviative effects of magnetic Fe3O4 nanoparticles on the physiological toxicity of 3-nitrophenol to rice (Oryza sativa L.) seedlings
- Selection and functional identification of Dof genes expressed in response to nitrogen in Populus simonii × Populus nigra
- Study on pecan seed germination influenced by seed endocarp
- Identification of active compounds in Ophiopogonis Radix from different geographical origins by UPLC-Q/TOF-MS combined with GC-MS approaches
- The entire chloroplast genome sequence of Asparagus cochinchinensis and genetic comparison to Asparagus species
- Genome-wide identification of MAPK family genes and their response to abiotic stresses in tea plant (Camellia sinensis)
- Selection and validation of reference genes for RT-qPCR analysis of different organs at various development stages in Caragana intermedia
- Cloning and expression analysis of SERK1 gene in Diospyros lotus
- Integrated metabolomic and transcriptomic profiling revealed coping mechanisms of the edible and medicinal homologous plant Plantago asiatica L. cadmium resistance
- A missense variant in NCF1 is associated with susceptibility to unexplained recurrent spontaneous abortion
- Assessment of drought tolerance indices in faba bean genotypes under different irrigation regimes
- The entire chloroplast genome sequence of Asparagus setaceus (Kunth) Jessop: Genome structure, gene composition, and phylogenetic analysis in Asparagaceae
- Food Science
- Dietary food additive monosodium glutamate with or without high-lipid diet induces spleen anomaly: A mechanistic approach on rat model
- Binge eating disorder during COVID-19
- Potential of honey against the onset of autoimmune diabetes and its associated nephropathy, pancreatitis, and retinopathy in type 1 diabetic animal model
- FTO gene expression in diet-induced obesity is downregulated by Solanum fruit supplementation
- Physical activity enhances fecal lactobacilli in rats chronically drinking sweetened cola beverage
- Supercritical CO2 extraction, chemical composition, and antioxidant effects of Coreopsis tinctoria Nutt. oleoresin
- Functional constituents of plant-based foods boost immunity against acute and chronic disorders
- Effect of selenium and methods of protein extraction on the proteomic profile of Saccharomyces yeast
- Microbial diversity of milk ghee in southern Gansu and its effect on the formation of ghee flavor compounds
- Ecology and Environmental Sciences
- Effects of heavy metals on bacterial community surrounding Bijiashan mining area located in northwest China
- Microorganism community composition analysis coupling with 15N tracer experiments reveals the nitrification rate and N2O emissions in low pH soils in Southern China
- Genetic diversity and population structure of Cinnamomum balansae Lecomte inferred by microsatellites
- Preliminary screening of microplastic contamination in different marine fish species of Taif market, Saudi Arabia
- Plant volatile organic compounds attractive to Lygus pratensis
- Effects of organic materials on soil bacterial community structure in long-term continuous cropping of tomato in greenhouse
- Effects of soil treated fungicide fluopimomide on tomato (Solanum lycopersicum L.) disease control and plant growth
- Prevalence of Yersinia pestis among rodents captured in a semi-arid tropical ecosystem of south-western Zimbabwe
- Effects of irrigation and nitrogen fertilization on mitigating salt-induced Na+ toxicity and sustaining sea rice growth
- Bioengineering and Biotechnology
- Poly-l-lysine-caused cell adhesion induces pyroptosis in THP-1 monocytes
- Development of alkaline phosphatase-scFv and its use for one-step enzyme-linked immunosorbent assay for His-tagged protein detection
- Development and validation of a predictive model for immune-related genes in patients with tongue squamous cell carcinoma
- Agriculture
- Effects of chemical-based fertilizer replacement with biochar-based fertilizer on albic soil nutrient content and maize yield
- Genome-wide identification and expression analysis of CPP-like gene family in Triticum aestivum L. under different hormone and stress conditions
- Agronomic and economic performance of mung bean (Vigna radiata L.) varieties in response to rates of blended NPS fertilizer in Kindo Koysha district, Southern Ethiopia
- Influence of furrow irrigation regime on the yield and water consumption indicators of winter wheat based on a multi-level fuzzy comprehensive evaluation
- Discovery of exercise-related genes and pathway analysis based on comparative genomes of Mongolian originated Abaga and Wushen horse
- Lessons from integrated seasonal forecast-crop modelling in Africa: A systematic review
- Evolution trend of soil fertility in tobacco-planting area of Chenzhou, Hunan Province, China
- Animal Sciences
- Morphological and molecular characterization of Tatera indica Hardwicke 1807 (Rodentia: Muridae) from Pothwar, Pakistan
- Research on meat quality of Qianhua Mutton Merino sheep and Small-tail Han sheep
- SI: A Scientific Memoir
- Suggestions on leading an academic research laboratory group
- My scientific genealogy and the Toronto ACDC Laboratory, 1988–2022
- Erratum
- Erratum to “Changes of immune cells in patients with hepatocellular carcinoma treated by radiofrequency ablation and hepatectomy, a pilot study”
- Erratum to “A two-microRNA signature predicts the progression of male thyroid cancer”
- Retraction
- Retraction of “Lidocaine has antitumor effect on hepatocellular carcinoma via the circ_DYNC1H1/miR-520a-3p/USP14 axis”