NR4A1 inhibits the epithelial–mesenchymal transition of hepatic stellate cells: Involvement of TGF-β–Smad2/3/4–ZEB signaling
-
Qian Huang
 ,
Jingying Xu
,
Jingying Xu

Abstract
This study aimed to examine whether nuclear receptor 4a1 (NR4A1) is involved in inhibiting hepatic stellate cell (HSC) activation and liver fibrosis through the epithelial–mesenchymal transition (EMT). HSC-T6 cells were divided into the control group, the acetaldehyde (200 μM, an EMT activator) group, and the NR4A1 activation group (Cytosporone B; 1 μM). The expression levels of the epithelial marker E-cadherin, the mesenchymal markers fibronectin (FN), vimentin, smooth muscle alpha-actin (α-SMA), and fibroblast-specific protein 1 (FSP-1), and the components of the transforming growth factor (TGF)-β pathway were detected by real-time polymerase chain reaction and western blotting. Compared with the control group, E-cadherin in the acetaldehyde group was downregulated, whereas FN, FSP-1, vimentin, α-SMA, and COL1A1/COL1A2 were upregulated (P < 0.05). Compared with the acetaldehyde group, NR4A1 agonist upregulated E-cadherin and downregulated FN, FSP-1, vimentin, α-SMA, and COL1A1/COL1A2 (P < 0.05). After acetaldehyde stimulation, TGF-β, Smad2/3/4, and zinc finger E-box-binding homeobox (ZEB) were upregulated, while Smad7 mRNA levels were downregulated (all P < 0.05). Compared with acetaldehyde alone, NR4A1 agonist increased Smad7 mRNA levels and reduced TGF-β, Smad2/3/4, and ZEB mRNA levels (all P < 0.05). NR4A1 activation suppresses acetaldehyde-induced EMT, as shown by epithelial and mesenchymal marker expression. The inhibition of the TGF-β–Smad2/3/4–ZEB signaling during HSC activation might be involved.
1 Introduction
Liver fibrosis is a wound-healing response to chronic liver injury caused by viral hepatitis, alcohol, metabolic diseases, autoimmune conditions, and cholestatic liver diseases. The most common etiologies are alcoholic liver disease, non-alcoholic fatty liver disease, chronic viral hepatitis, genetic conditions (α − 1 antitrypsin deficiency, hereditary hemochromatosis, and Wilson disease), and autoimmune diseases (primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis), and drugs. Sustained liver damage leads to fibrosis, liver failure, and even hepatocellular carcinoma [1,2]. In the United States, the prevalence of chronic liver diseases was 1.8% in 2017, with 12.8 deaths per 100,000 population [3,4].
The epithelial–mesenchymal transition (EMT) is a transition of epithelial cells to a mesenchymal state, which is a reversible process [5]. The EMT plays an essential role in tissue development, wound healing, fibrosis, and cancer progression [6,7,8]. Liver fibrosis is caused by excess extracellular matrix (ECM) production by myofibroblasts [9]. Activation of hepatic stellate cells (HSCs) is a key event in the formation of liver fibrosis since it is the major source of myofibroblasts [10]. Several studies indicated that HSCs undergo EMT during their activation [7,11,12,13] to participate in liver fibrosis [14,15,16]. The main pathways involved in the EMT in liver fibrosis are the Hedgehog signaling pathway, transforming growth factor (TGF)-β signaling pathway, Notch signaling, and extracellular signal-regulated kinase (ERK) signaling pathway [7,17]. Excessive Hedgehog activation after liver injury participates in EMT and liver fibrosis [7,17]. TGF-β signaling is involved in ECM and collagen production by HSCs [7,17]. The ERK pathway plays a crucial role in cell growth and differentiation and represses EMT [17]. The Notch pathway is involved in cell differentiation [7]. Importantly, inhibition of the EMT of HSCs can suppress the activation of the HSCs, thereby alleviating the progression of hepatic fibrosis [11,18,19]. Hence, inhibition of the EMT is a promising strategy for reversing fibrosis.
Nuclear receptor 4a1 (NR4A1, also known as Nur77, TR3, or NGFIB) is a member of the NR4A family of nuclear orphan receptors. NR4A1 plays diverse and important regulatory roles in glucose and lipid metabolism and inflammatory responses [20,21]. NR4A1 is involved in the EMT in tumor metastasis and migration [22,23,24]. The loss of NR4A1 inhibits TGF-β-induced EMT and metastasis [22]. Furthermore, Palumbo-Zerr et al. [25] demonstrated that NR4A1 inhibits TGF-β signaling and can suppress experimental lung and liver fibrosis.
Additional recent studies have also demonstrated that NR4A1 inhibits TGF-β signaling [26,27,28]. Therefore, NR4A1 might represent a promising antifibrotic target, but since the EMT mechanisms associated with liver fibrosis and those associated with cancer might be different, it remains unclear whether NR4A1 inhibits HSC activation and liver fibrosis by modulating the EMT. This study aimed to examine whether NR4A1 is involved in inhibiting HSC activation and liver fibrosis through the EMT.
2 Materials and methods
2.1 Cells
The HSC-T6 cells were purchased from Shanghai Tongpai Technology Co., Ltd. (Shanghai, China). After synchronization, the HSC-T6 cells were divided into three groups: the control group (HSC-T6 cells plus an equal volume of medium), acetaldehyde (MACKLIN, Shanghai, China; purity: ≥99%), the treatment group (HSC-T6 cells plus acetaldehyde to a final concentration of 200 μM), and the NR4A1 activation group (HSC-T6 cells plus acetaldehyde to a final concentration of 200 μM and Cytosporone B [Csn-B; Santa Cruz Biotechnology, Inc., Dallas, TX, USA; purity: ≥98%] to a final concentration of 1 μM).
2.2 Quantitative real-time PCR
Total RNA was extracted from the cells using TRIzol (MiniBEST Universal RNA Extraction Kit; Takara Bio, Otsu, Japan). Gene expression was measured by the quantitative real-time polymerase chain reaction (qRT-PCR) using the SYBR Green Real-time PCR Master Mix (Takara, Otsu, Japan) performed under standard conditions with an ABI 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). All primers were from Takara. The primer sequences are listed in Table 1.
Primer sequences for real-time PCR
| Gene | Primer sequences (5′–3″) | Product size (bp) | Designed T m (°C) |
|---|---|---|---|
| FSP-1 | Forward: ATGTAATAGTGTCCACCTTCC | 181 | 54.71 |
| Reverse: ACTTCATTGTCCCTGTTGCT | 57.34 | ||
| a-SMA | Forward: GGAGAAGCCCAGCCAGTCGC | 115 | 65.58 |
| Reverse: CCCGCCTTACAGAGCCCGGA | 66.26 | ||
| E-cadherin | Forward: TGTTGATAGCGTGCCCTTTG | 100 | 58.84 |
| Reverse: GTTCCGATTGCTTGCCTTTT | 57.56 | ||
| FN | Forward: GGATCCCCTCCCAGAGAAGT | 188 | 60.03 |
| Reverse: GGGTGTGGAAGGGTAACCAG | 59.96 | ||
| COL1A2 | Forward: AGGGTGTTCAAGGTGGCAAA | 166 | 60.03 |
| Reverse: CCACGTTCTCCTCTTGGACC | 60.04 | ||
| COL1A1 | Forward: AAAACCACCAAGACCTCCCG | 141 | 60.18 |
| Reverse: GGTGGGAGGGAACCAGATTG | 60.03 | ||
| Vimentin | Forward: ACCGCTTCGCCAACTACATC | 138 | 60.74 |
| Reverse: GCAACTCCCTCATCTCCTCCT | 60.97 | ||
| GADPH | Forward: TCTCTGCTCCTCCCTGTTCT | 95 | 59.59 |
| Reverse: ATCCGTTCACACCGACCTTC | 60.04 | ||
| Smad2 | Forward: GCCGCCCGAAGGGTAGAT | 164 | 61.55 |
| Reverse: TTCTGTTCTCCACCACCTGC | 59.89 | ||
| Smad3 | Forward: ATACGGATGTTCAAGTGTTCG | 242 | 56.38 |
| Reverse: ACTGGGTCCTCTTTGGTTTT | 56.85 | ||
| Smad4 | Forward: ATCCACCAAGTAATCGCGCA | 252 | 60.11 |
| Reverse: AGGTGGTAGTGCTGTTATGGTG | 60.03 | ||
| Smad7 | Forward: GTGGCATACTGGGAGGAGAA | 309 | 58.80 |
| Reverse: GATGGAGAAACCAGGGAACA | 57.12 | ||
| ZEB | Forward: CCAAAGCAACAGGGAGAGTTAC | 397 | 59.19 |
| Reverse: CTTGTCTTTCATCCTGGTTTCC | 57.23 | ||
| TGF-β | Forward: GAGGCGGTGCTCGCTTTGTA | 211 | 60.00 |
| Reverse: GCACTGCTTCCCGAATGTCTG | 57.14 |
2.2.1 Genomic DNA removal from total RNA
The genomic DNA was removed using the following components: 5× gDNA Eraser buffer 1 (2 μL), gDNA Eraser (1 μL), and total RNA (2 μL). The reaction conditions were as follows: 42°C, 2 min; 4°C, 5 min.
2.2.2 Calculation of the gene expression
First, we calculated the average Ct value of the sample, and then, the ∆Ct of the target gene in the sample. The relative value of the target gene in the sample to the internal reference gene was calculated. Afterward, the ∆Ct of a certain gene was calculated relative to the reference sample group and multiple relationships.
2.2.3 Reference gene selection
GAPDH has been used as a stable reference gene selected as a housekeeping conserved gene according to previous studies [29,30,31,32,33,34].
2.3 Western blot
Protein lysate was obtained from the cultured cells using the RIPA lysis buffer (Solarbio Corporation, Beijing, China). The protein concentration was measured with a bicinchoninic acid assay. Proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. The membrane was first incubated with rabbit anti-rat primary antibodies, such as E-cadherin (1:2,000; no. 20874-1-AP), fibronectin (FN; 1:3,000; no. 15613-1-AP), vimentin (1:2,000; no. 10366-1-AP), Smad2 (1:2,000; no. 12570-1-AP), Smad4 (1:2,000; no. 51144-1-AP), zinc-finger-enhancer-binding protein 1 (ZEB; 1:2,000; no. 21544-1-AP) (all from Proteintech Group Inc., Chicago, IL, USA), Smad3 (1:2,000, no. AF6362; Affinity BIO, Scoresby, Australia), Smad7 (1:2,000; no. AF5147; Affinity BIO), and β-actin (1:5,000; no. Ab8226; Abcam, Cambridge, UK), and then incubated with goat anti-rabbit horseradish peroxidase-conjugated-labeled secondary antibodies (1:5,000, # A0208, Beyotime Institute of Biotechnology, Haimen, China) for 1 h at room temperature. The blots were detected using enhanced chemiluminescence.
2.4 Statistical analysis
The data are presented as mean ± standard deviation and were analyzed using a one-way analysis of variance with Fisher’s least significant difference post hoc test. P-values <0.05 were considered statistically significant (*P < 0.05 and **P < 0.01).
3 Results
3.1 NR4A1 regulates the expression of EMT-related genes to prevent EMT in HSC-T6 cells
To explore the regulatory effects of NR4A1 on the EMT of HSC-T6 cells, we first used acetaldehyde to stimulate HSC-T6 cells and measured the changes in the expression of EMT-related genes. Compared with the control group, the mRNA levels of E-cadherin in the acetaldehyde group were significantly downregulated, whereas those of FN, fibroblast-specific protein 1 (FSP-1), and vimentin were significantly upregulated. In addition, the mRNA levels of HSC activation markers, including smooth muscle alpha-actin (α-SMA) and COL1A1/COL1A2, were significantly upregulated in the acetaldehyde group compared with the control group. When the NR4A1 agonist (Csn-B) was used with acetaldehyde, compared with the acetaldehyde group, the mRNA levels of E-cadherin in the NR4A1 activation group were significantly upregulated, while those of FN, FSP-1, vimentin, α-SMA, and collagen genes (COL1A1/COL1A2) were significantly downregulated (Figure 1). Similar changes were observed at the protein level (Figure 2). These results indicated that the acetaldehyde model induces EMT in HSC-T6 cells and that NR4A1 is involved in regulating the expression of EMT-related genes, probably preventing EMT in the cells.
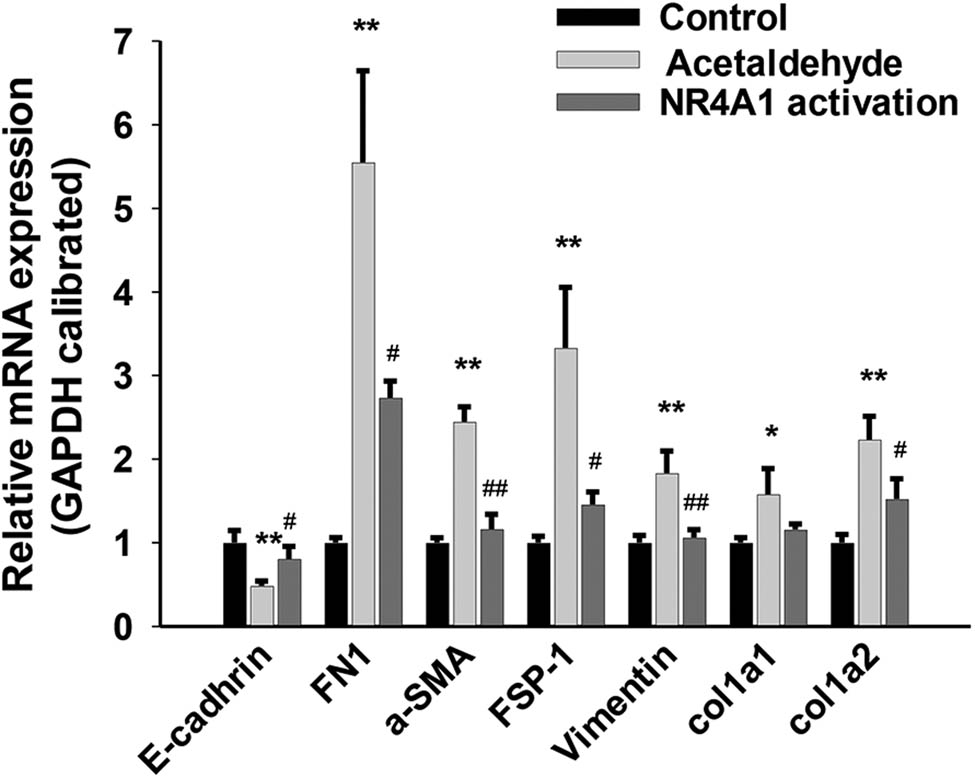
Effects of NR4A1 on EMT-related genes in HSC-T6 cells. The mRNA levels of E-cadherin in the acetaldehyde group were significantly downregulated, whereas FN, FSP-1, vimentin, α-SMA, and COL1A1/COL1A2 were significantly upregulated. The mRNA levels of E-cadherin in the NR4A1 activation group were significantly upregulated, while FN, FSP-1, vimentin, α-SMA, and COL1A1/COL1A2 were significantly downregulated. The mRNAs were analyzed by qRT-PCR analysis. *P < 0.05, **P < 0.01 vs control, # P < 0.05, ## P < 0.01 vs acetaldehyde. n = 3/group.
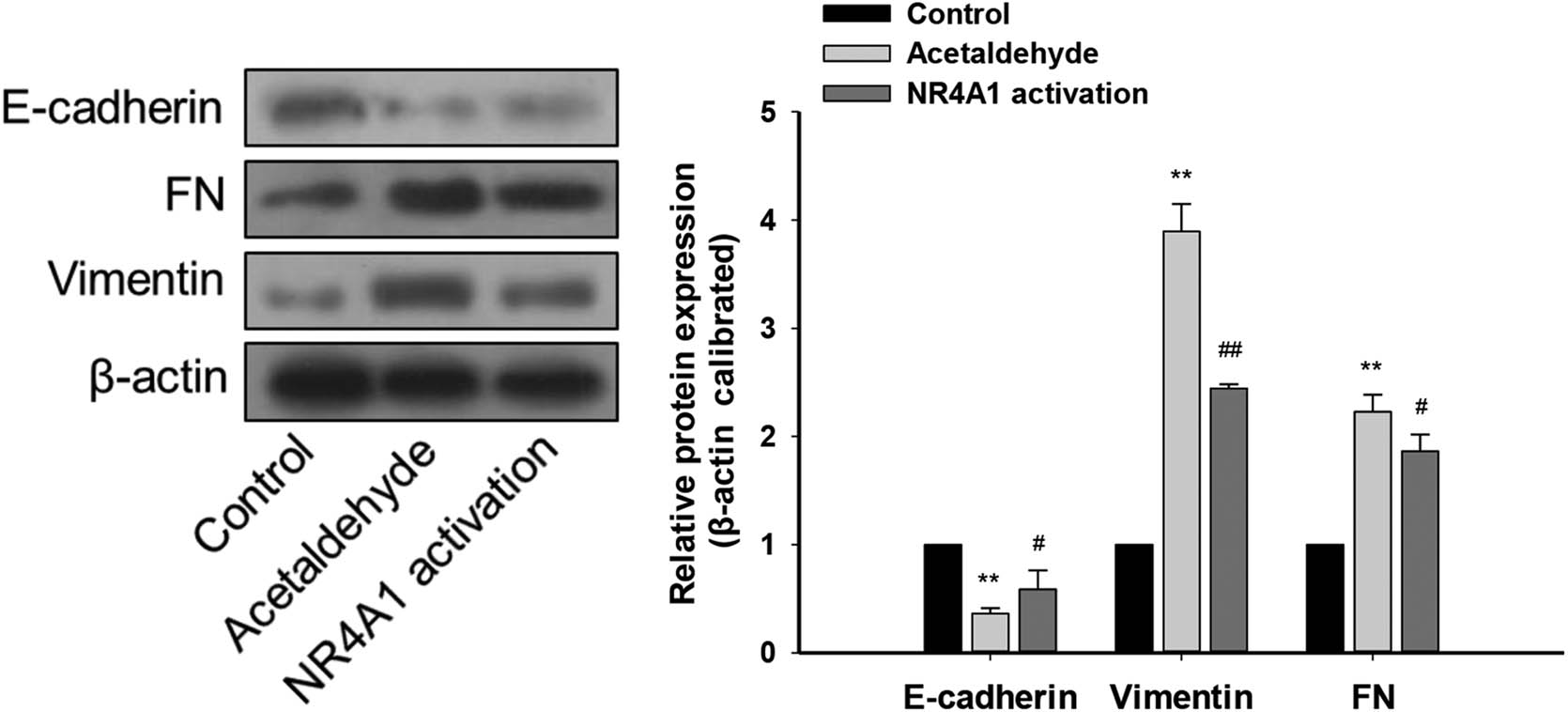
Effects of NR4A1 on EMT-related proteins in HSC-T6 cells. Protein levels of E-cadherin (epithelial marker) in the acetaldehyde group were downregulated, while those of FN and vimentin (mesenchymal markers) were upregulated. The protein levels of E-cadherin were upregulated in the NR4A1 activation group, while those of FN and vimentin were downregulated. The proteins were analyzed by western blotting. β-Actin was used as an internal control. **P < 0.01 vs control, # P < 0.05, ## P < 0.01 vs acetaldehyde. n = 3/group.
3.2 NR4A1 possibly inhibits EMT through the TGF-β–Smad–ZEB signaling pathway
The TGF-β–Smad–ZEB signaling pathway is involved in the EMT [6]. Therefore, we explored whether NR4A1 activation inhibits EMT by regulating the TGF-β–Smad–ZEB signaling pathway. After acetaldehyde was used to stimulate HSC-T6 cells, TGF-β, Smad2/3/4, and ZEB mRNA levels were significantly upregulated, while Smad7 mRNA levels were significantly downregulated. Compared to these levels following acetaldehyde alone, acetaldehyde combined with an NR4A1 agonist (Csn-B) resulted in increased Smad7 mRNA levels and reduced TGF-β, Smad2/3/4, and ZEB mRNA levels (Figure 3). Similar changes were also observed at the protein level (Figure 4). Collectively, these results suggest that during acetaldehyde-induced HSC activation and EMT, NR4A1 activation suppresses the EMT, at least in part, by inhibiting the TGF-β–Smad–ZEB signaling pathway.
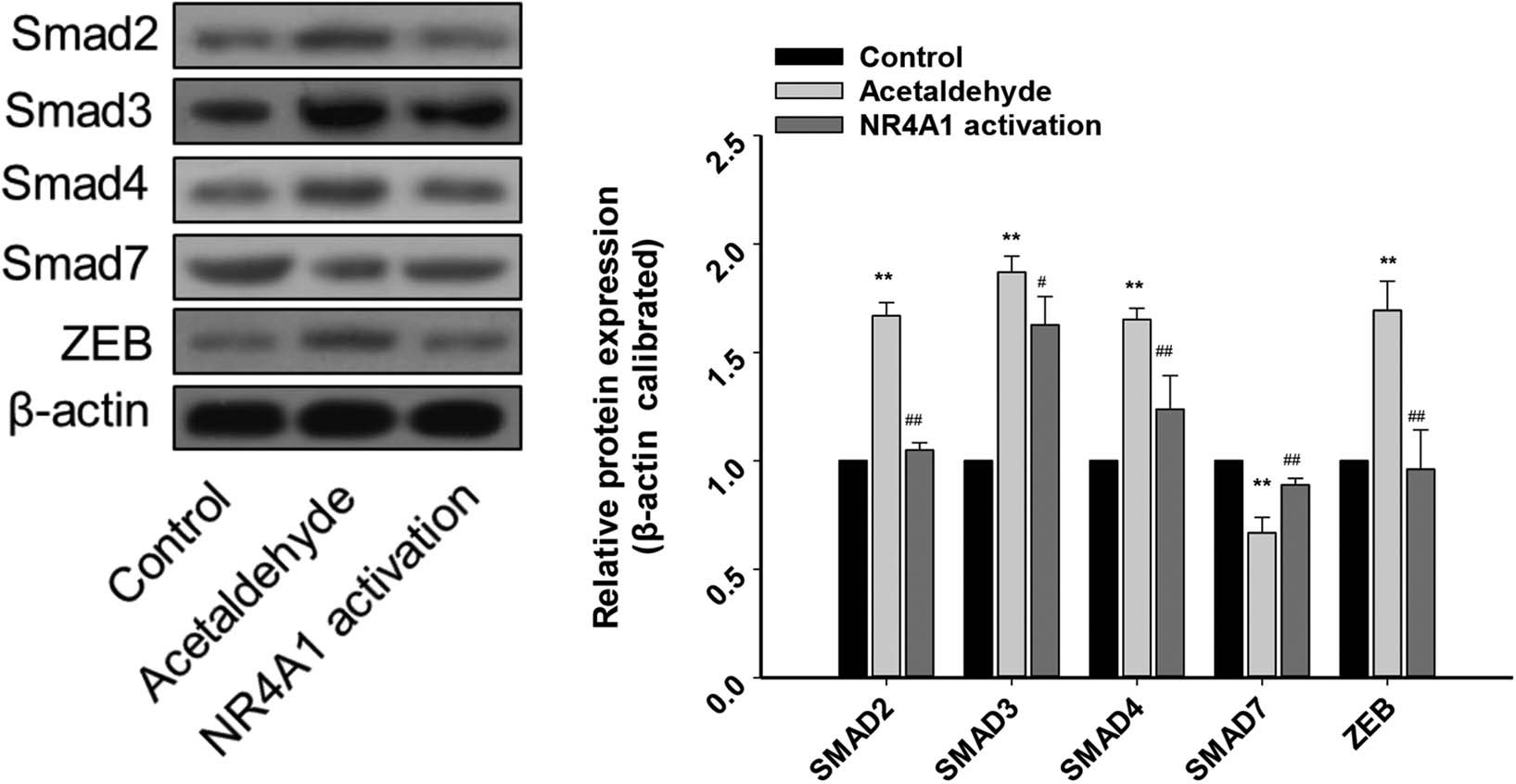
Effects of NR4A1 on the protein levels of Smad2/3/4, Smad 7, and ZEB. Protein levels of Smad2/3/4 and ZEB in the acetaldehyde group were upregulated, while that of Smad 7 was downregulated. The expression of these proteins was reversed in the NR4A1 activation group. The proteins were analyzed by western blotting. β-Actin was used as an internal control. *P < 0.05, **P < 0.01 vs control, # P < 0.05, ## P < 0.01 vs acetaldehyde. n = 3/group.
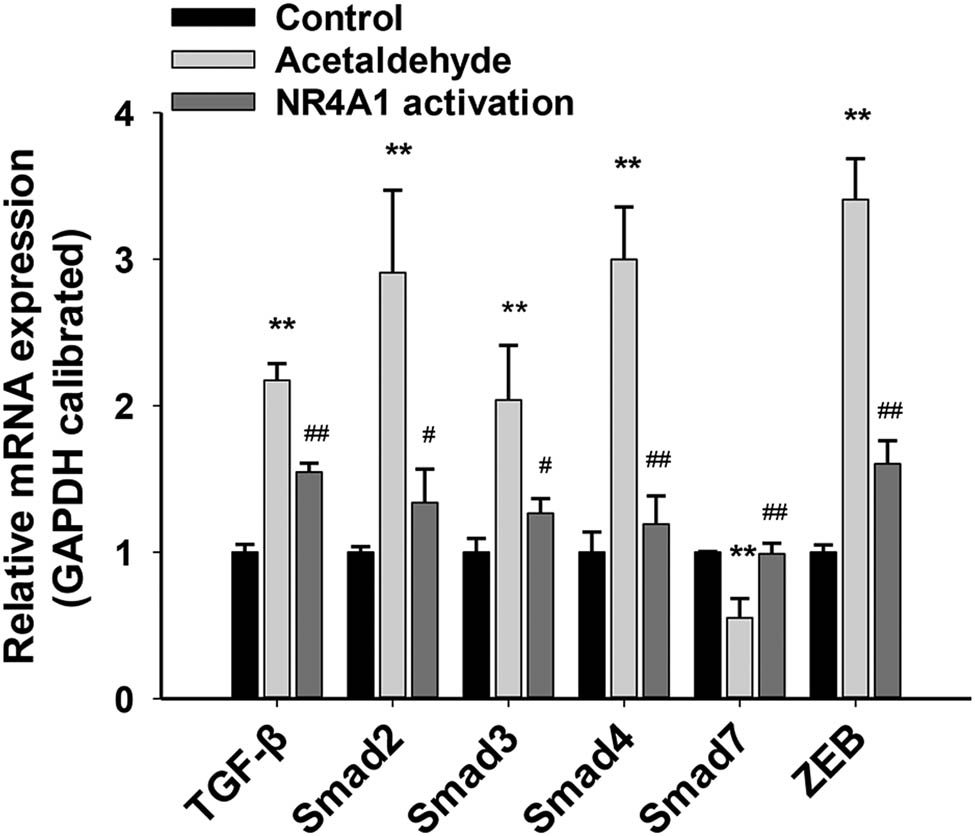
mRNA levels of the components of the TGF-β–Smad–ZEB signal pathway in HSC-T6 cells. The mRNA levels of TGF-β, Smad2/3/4, and ZEB were significantly upregulated, while Smad7 mRNA levels were significantly downregulated. The mRNA levels of Smad7 in the NR4A1 activation group were significantly upregulated, while those of TGF-β, Smad2/3/4, and ZEB were significantly downregulated. *P < 0.05, **P < 0.01 vs control, # P < 0.05, ## P < 0.01 vs acetaldehyde. n = 3/group.
4 Discussion
Activation of HSCs is a key event in liver fibrosis [10]. HSCs undergo EMT during activation [7,11,12,13]. NR4A1 inhibits TGF-β signaling, which can suppress experimental lung and liver fibrosis [25], but whether NR4A1 inhibits HSC activation and liver fibrosis through the EMT is unknown. Therefore, this study aimed to examine whether NR4A1 is involved in inhibiting HSC activation and liver fibrosis through the EMT. The results suggest that NR4A1 activation suppresses acetaldehyde-induced EMT in HSCs, as shown by increased epithelial and decreased mesenchymal marker expression. The inhibition of the TGF-β–Smad2/3/4–ZEB signaling during HSC activation might be involved.
Liver fibrosis is a wound-healing response to various liver damage forms, such as hepatitis, alcohol, drugs, metabolic diseases, biliary injury, and toxins. Importantly, HSC activation is a key event in liver fibrosis [10], and wounding and repair are dynamic processes that include matrix synthesis, deposition, and degradation [35]. HSC activation undergoes EMT-like changes and participates in fibrogenesis [11,12]. Consistent with these previous studies, the present study indicated that the acetaldehyde-induced activation of HSCs upregulated the expression levels of FN, FSP-1, vimentin, and α-SMA (all myofibroblastic markers) while downregulating those of E-cadherin (an epithelial marker). Hence, these findings confirm that HSC activation involves the EMT.
Several previous studies have shown that NR4A1 can promote tumor cells’ EMT [22,23,24]. Still, it remains unclear whether the role of NR4A1 in EMT in cancer is the same as in EMT in liver fibrosis and HSC activation. Csn-B is an NR4A1 agonist that enhances the transcriptional activity of NR4A1. The present study showed that Csn-B upregulated the expression levels of epithelial markers and decreased the expression levels of mesenchymal markers in acetaldehyde-induced EMT in HSCs, compared to these levels in the presence of acetaldehyde alone. These findings suggest that NR4A1 activation suppressed the EMT during acetaldehyde-induced activation of HSCs. It is contrary to the findings in tumor cells, which is not surprising since the pathogeneses and cellular characteristics of tumors and fibrosis exhibit different characteristics. In the present study, the mechanism of NR4A1-mediated inhibition of the EMT of HSCs was explored. Palumbo-Zerr et al. [25] found that NR4A1 is an endogenous inhibitor of TGF-β signaling and inhibits skin, lung, liver, and kidney fibrosis. TGF-β signaling has also been identified as one of the predominant inducers of the EMT [36,37], and the inhibition of TβRI activity can block TGFβ‑induced EMT. TGF-β binds to TβR receptors to form a complex, activating Smad2/Smad3 to interact with Smad4 to form trimeric Smad complexes. The TGFβ/Smad signaling pathway activates the expression of EMT transcription factors and initiates the EMT [6,38]. Smad complexes interact with ZEB1 and ZEB2 to mediate TGFβ‑regulated gene expression. ZEB is one of the key transcription factors of the EMT, and its functions are finely regulated at the transcriptional, translational, and post-translational levels. ZEB expression is activated early during the EMT and plays a central role in developing both fibrosis and cancer. The TGF–Smad–ZEB pathway is involved in the EMT [6]. In the present study, we found that acetaldehyde-mediated stimulation of HSCs significantly upregulated TGF-β, Smad2/3/4, and ZEB levels and significantly downregulated Smad7 levels. Furthermore, NR4A1 activation in the presence of acetaldehyde resulted in higher Smad7 levels and lower TGF-β, Smad 2/3/4, and ZEB expression levels than acetaldehyde treatment alone. Previous studies confirmed that Smad7 inhibits TGF-β signaling [39]. The present study illustrates that NR4A1 can suppress EMT by inhibiting TGF-β–Smad2/3/4–ZEB signaling. NR4A1 might negatively regulate TGF-β signaling, at least in part, by promoting SMAD7 expression.
NR4A1 activates TGF signaling and promotes EMT in tumor cells [22,23], whereas the present study in HSCs differs from those in tumor cells. Interestingly, NR4A1 exhibits both tumor-suppressive and pro-oncogenic effects in cancer development [40,41,42]. NR4A1 translocation from the nucleus to the cytoplasm in colon cancer cells may initiate apoptotic cascades [43]. In contrast, NR4A1 exhibits anti-apoptotic effects when it is not exported from the nucleus [41,44]. TGF-mediated induction of the EMT is dependent on the nuclear export of NRA41 in breast cancer cells, and NR4A1 antagonists inhibit the nuclear export of NR4A1 and thereby block the TGF-induced EMT [23]. NR4A1 phosphorylation decreases the transcriptional activity of NR4A1, and pNR4A1 is strongly associated with hepatic/lung fibrosis and is mainly located in the cytoplasm, whereas pan-NR4A1 localizes in both nuclear and cytoplasmic compartments [25]. Collectively, these studies suggest that the effects of NR4A1 depend on its subcellular localization and the cell type in which it is signaling.
This study has limitations. The subcellular localization and expression of NRA4A1 were not examined, which could be an added benefit for this study. Thus, this should be tested in future experiments. In addition, the pathways involved in EMT and fibrosis were examined superficially. Only an agonist of NRA4A1 was used, and future studies should also use an antagonist. In addition, agonists/antagonists and silencing/overexpression of TGF-β and other proteins involved in that pathway should be used to determine the contribution of the TGF-β pathway in the EMT in liver fibrosis. Nonetheless, this study only used GAPDH as a reference gene due to the fact that several studies used GAPDH as an internal reference [29–34]. However, it is recommended to use at least two reference genes to obtain more reliable results, and therefore, other reference genes will be considered for future studies.
In summary, this study indicates that NR4A1 suppresses the EMT during acetaldehyde-induced HSC activation. NR4A1-mediated inhibition of the EMT of HSCs is involved in the suppression of TGF-β–SMAD2/3/4–ZEB signaling and increased SMAD7 expression, but confirmation is needed. Hence, the findings suggest that NR4A1 plays an important role during HSC activation and that NR4A1 might be a promising therapeutic target for treating liver fibrosis.
-
Funding information: This work was supported by a grant from the General Project of Hangzhou Health Science and Technology Program (No. 2015A31).
-
Conflict of interest: The authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during this study are available from the corresponding author on reasonable request.
References
[1] Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–69.10.1007/978-94-007-1042-9_4Search in Google Scholar
[2] Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. 2015;63(4):1038–9.10.1016/j.jhep.2015.03.039Search in Google Scholar PubMed
[3] Kochanek KD, Murphy SL, Xu J, Arias E. Deaths: final data for 2017. Natl Vital Stat Rep. 2019;68(9):1–77.Search in Google Scholar
[4] Murphy SL, Xu J, Kochanek KD, Arias E. Mortality in the United States, 2017. NCHS. Data Brief. 2018;328:1–8.Search in Google Scholar
[5] Dewidar B, Meyer C, Dooley S, Meindl B, Nadja. TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8:11.10.3390/cells8111419Search in Google Scholar PubMed PubMed Central
[6] Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96.10.1038/nrm3758Search in Google Scholar PubMed PubMed Central
[7] Chen Y, Fan Y, Guo D-Y, Xu B, Shi X-Y, Li J-T, et al. Study on the relationship between hepatic fibrosis and epithelial-mesenchymal transition in intrahepatic cells. Biomed Pharmacother. 2020;129:110413.10.1016/j.biopha.2020.110413Search in Google Scholar PubMed
[8] Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.10.1016/j.cell.2009.11.007Search in Google Scholar PubMed
[9] Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–94.10.1038/nri3623Search in Google Scholar PubMed
[10] Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–10.10.1172/JCI66369Search in Google Scholar PubMed PubMed Central
[11] Zheng J, Wang W, Yu F, Dong P, Chen B, Zhou M-T. MicroRNA-30a suppresses the activation of hepatic stellate cells by inhibiting epithelial-to-mesenchymal transition. Cell Physiol Biochem. 2018;46(1):82–92.10.1159/000488411Search in Google Scholar PubMed
[12] Yue HY, Yin C, Hou JL, Zeng X, Chen YX, Zhong W, et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut. 2010;59(2):236–46.10.1136/gut.2008.174904Search in Google Scholar PubMed
[13] Lim YS, Kim KA, Jung JO, Yoon JH, Suh KS, Kim CY, et al. Modulation of cytokeratin expression during in vitro cultivation of human hepatic stellate cells: evidence of transdifferentiation from epithelial to mesenchymal phenotype. Histochem Cell Biol. 2002;118(2):127–36.10.1007/s00418-002-0436-9Search in Google Scholar PubMed
[14] Nitta T, Kim JS, Mohuczy D, Behrns KE. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology. 2008;48(3):909–19.10.1002/hep.22397Search in Google Scholar PubMed PubMed Central
[15] Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135(2):642–59.10.1053/j.gastro.2008.04.038Search in Google Scholar PubMed
[16] Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46(1):48–57.10.1002/hep.21672Search in Google Scholar PubMed
[17] Yu K, Li Q, Shi G, Li N. Involvement of epithelial-mesenchymal transition in liver fibrosis. Saudi J Gastroenterol. 2018;24(1):5–11.10.4103/sjg.SJG_297_17Search in Google Scholar PubMed PubMed Central
[18] Dai W, Zhao J, Tang N, Zeng X, Wu K, Ye C, et al. MicroRNA-155 attenuates activation of hepatic stellate cell by simultaneously preventing EMT process and ERK1 signalling pathway. Liver Int. 2015;35(4):1234–43.10.1111/liv.12660Search in Google Scholar PubMed
[19] Yu F, Geng W, Dong P, Huang Z, Zheng J. LncRNA-MEG3 inhibits activation of hepatic stellate cells through SMO protein and miR-212. Cell Death Dis. 2018;9(10):1014.10.1038/s41419-018-1068-xSearch in Google Scholar PubMed PubMed Central
[20] Fassett MS, Jiang W, D’Alise AM, Mathis D, Benoist C. Nuclear receptor Nr4a1 modulates both regulatory T-cell (Treg) differentiation and clonal deletion. Proc Natl Acad Sci U S A. 2012;109(10):3891–6.10.1073/pnas.1200090109Search in Google Scholar PubMed PubMed Central
[21] Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol. 2011;12(8):778–85.10.1038/ni.2063Search in Google Scholar PubMed PubMed Central
[22] Zhou F, Drabsch Y, Dekker TJA, de Vinuesa AG, Li Y, Hawinkels LJAC, et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nature Commun. 2014;5:3388.10.1038/ncomms4388Search in Google Scholar PubMed
[23] Hedrick E, Safe S. Transforming growth factor β/NR4A1-inducible breast cancer cell migration and epithelial-to-mesenchymal transition Is p38α (mitogen-activated protein kinase 14) dependent. Mol Cell Biol. 2017;37:18.10.1128/MCB.00306-17Search in Google Scholar PubMed PubMed Central
[24] To SKY, Zeng WJ, Zeng JZ, Wong AST. Hypoxia triggers a Nur77-β-catenin feed-forward loop to promote the invasive growth of colon cancer cells. Br J Cancer. 2014;110(4):935–45.10.1038/bjc.2013.816Search in Google Scholar PubMed PubMed Central
[25] Palumbo-Zerr K, Zerr P, Distler A, Fliehr J, Mancuso R, Huang J, et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-beta signaling and fibrosis. Nat Med. 2015;21(2):150–8.10.1038/nm.3777Search in Google Scholar PubMed
[26] Wu J, Sun H, Yang X, Sun X. Nur77 suppression facilitates androgen deprivation-induced cell invasion of prostate cancer cells mediated by TGF-β signaling. Clin Transl Oncol. 2018;20(10):1302–13.10.1007/s12094-018-1862-zSearch in Google Scholar PubMed
[27] Hiwatashi N, Mukudai S, Bing R, Branski RC. The effects of cytosporone-B, a novel antifibrotic agent, on vocal fold fibroblasts. Laryngoscope. 2018;128(12):E425–E8.10.1002/lary.27361Search in Google Scholar PubMed PubMed Central
[28] Hiwatashi N, Bing R, Kraja I, Branski RC. NR4A1 is an endogenous inhibitor of vocal fold fibrosis. Laryngoscope. 2017;127(9):E317–23.10.1002/lary.26678Search in Google Scholar PubMed PubMed Central
[29] Li Z, Li X, Zhang Q, Yuan L, Zhou X. Reference gene selection for transcriptional profiling in Cryptocercus punctulatus, an evolutionary link between Isoptera and Blattodea. Sci Rep. 2020;10(1):22169.10.1038/s41598-020-79030-6Search in Google Scholar PubMed PubMed Central
[30] Mehta A, Dobersch S, Dammann RH, Bellusci S, Ilinskaya ON, Braun T, et al. Validation of Tuba1a as appropriate internal control for normalization of gene expression analysis during mouse lung development. Int J Mol Sci. 2015;16(3):4492–511.10.3390/ijms16034492Search in Google Scholar PubMed PubMed Central
[31] Mu J, Chen L, Gu Y, Duan L, Han S, Li Y, et al. Genome-wide identification of internal reference genes for normalization of gene expression values during endosperm development in wheat. J Appl Genet. 2019;60(3–4):233–41.10.1007/s13353-019-00503-0Search in Google Scholar PubMed
[32] Adeola F. Normalization of gene expression by quantitative RT-PCR in human cell line: comparison of 12 endogenous reference genes. Ethiop J Health Sci. 2018;28(6):741–8.10.4314/ejhs.v28i6.9Search in Google Scholar PubMed PubMed Central
[33] Feria-Romero IA, Bribiesca-Cruz I, Coyoy-Salgado A, Segura-Uribe JJ, Bautista-Poblet G, Granados-Cervantes A, et al. Validation of housekeeping genes as an internal control for gene expression studies in the brain of ovariectomized rats treated with tibolone. Gene. 2021;769:145255.10.1016/j.gene.2020.145255Search in Google Scholar
[34] Nazari F, Parham A, Maleki AF. GAPDH, β-actin and β2-microglobulin, as three common reference genes, are not reliable for gene expression studies in equine adipose- and marrow-derived mesenchymal stem cells. J Anim Sci Technol. 2015;57:18.10.1186/s40781-015-0050-8Search in Google Scholar
[35] Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3:2.10.1016/S1542-3565(04)00445-8Search in Google Scholar
[36] Zhang J, Tian X-J, Zhang H, Teng Y, Li R, Bai F, et al. TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal. 2014;7(345):ra91.10.1126/scisignal.2005304Search in Google Scholar PubMed
[37] Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65(4):798–808.10.1016/j.jhep.2016.05.007Search in Google Scholar PubMed
[38] Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30.10.1038/nrm3434Search in Google Scholar PubMed PubMed Central
[39] Ganai AA, Husain M. Genistein attenuates D-GalN induced liver fibrosis/chronic liver damage in rats by blocking the TGF-β/Smad signaling pathways. Chem Biol Interact. 2017;261:80–5.10.1016/j.cbi.2016.11.022Search in Google Scholar PubMed
[40] Mullican SE, Zhang S, Konopleva M, Ruvolo V, Andreeff M, Milbrandt J, et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med. 2007;13(6):730–5.10.1038/nm1579Search in Google Scholar PubMed
[41] Lee S-O, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, et al. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res. 2010;70(17):6824–36.10.1158/0008-5472.CAN-10-1992Search in Google Scholar PubMed PubMed Central
[42] Ramirez-Herrick AM, Mullican SE, Sheehan AM, Conneely OM. Reduced NR4A gene dosage leads to mixed myelodysplastic/myeloproliferative neoplasms in mice. Blood. 2011;117(9):2681–90.10.1182/blood-2010-02-267906Search in Google Scholar PubMed PubMed Central
[43] Wilson AJ, Arango D, Mariadason JM, Heerdt BG, Augenlicht LH. TR3/Nur77 in colon cancer cell apoptosis. Cancer Res. 2003;63(17):5401–7.Search in Google Scholar
[44] Kolluri SK, Bruey-Sedano N, Cao X, Lin B, Lin F, Han Y-H, et al. Mitogenic effect of orphan receptor TR3 and its regulation by MEKK1 in lung cancer cells. Mol Cell Biol. 2003;23(23):8651–67.10.1128/MCB.23.23.8651-8667.2003Search in Google Scholar PubMed PubMed Central
© 2022 Qian Huang et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.