Hydrolytic depolymerization of major fibrous wastes
-
Arun Aneja


Abstract
Sustainability is one of the main issues of today’s research and development. One aspect of sustainability is also the efficient treatment of waste. Fibrous-based wastes have quite different reasons. They are in different stages of degradation and are more or less chemically complex or contain nonfibrous materials (such as zippers, buttons, membranes, etc.). There are, therefore, plenty of various combinations of mechanical and chemical methods for processing them. The main aim of this work was to describe a proven practical strategy for depolymerization of mixed fibrous wastes to their submicron particles and monomeric components. High-pressure neutral and acid hydrolysis kinetics of cotton, wool, and polyester (PET) fibers and their 50/50 blends representing common components of textile wastes were investigated. High-pressure water-neutral hydrolysis and acid (strong and weak) hydrolysis were used for both pure-component and bicomponent fiber blends. Hydrolysis was conducted in a newly designed high-pressure reactor. Parameters of mechanistic models for the kinetics of heterogeneous solid–liquid degradation were evaluated by nonlinear least squares regression. Model characteristics were basic parameters for efficient selection of suitable decomposition of basic types of individual and mixture fibrous waste including time factor. Hydrolysis agents and conditions of decomposition were selected as optimal based on our previous experimental findings and modeling. Two pure, untreated raw materials were used for the experiments on the decomposition of natural fibers: raw cotton yarn and wool fibers. Two multicomponent mixtures, cotton and PET (weight ratio, 50:50) and wool and PET (weight ratio, 50:50), prepared from the materials described above, were selected.
Nomenclature
- b
-
Solvent penetration rate (1/s)
- C
-
Integration constant (−)
- D
-
Penetration rate constant (with respect to surface geometry) (1/s2)
- d
-
Size of a soluble particle that is transferreds into solution (m)
- d 3
-
Soluble particle volume (m3)
- h
-
(Total) length of a fiber (m)
- k
-
Reaction rate constant (1/s)
- m V
-
Initial mass of the fiber sample (g)
- n(t)
-
Actual number of particles in a fiber (−)
- n[%]
-
Relative number of particles in athe fiber (%)
- n B(t)
-
Number of particles in the hydrolyzing bath in time t (−)
- n BE, m V
-
Total number of particles in a bath of the fully hydrolyzed material (−), Initial mass of the fiber sample [g]
- n S
-
Actual number of particles on the sample surface (−)
- n V
-
Initial number of particles in the fiber sample (−)
- Q S
-
Quantity scale parameter (−)
- R
-
Fiber aAverage radius of a fiber (assuming circularity of cross-section) (m)
- S
-
Surface of a fiber (m2)
- t
-
Time (s)
- t E
-
End of reaction time (s)
- V
-
Volume of a fiber (m3)
1 Introduction
Europe is the world’s greatest consumer and second-largest producer of textile materials (1), consuming 10 million tons of textiles annually. By 2025, the annual consumption of fiber in the world is expected to exceed 127 million tons, based on current economic and demographic trends (2).
Nowadays, just 15–20% of textiles are recycled, compared to 80% of steel, 65% of paper, and 30% of plastics (3). Approximately 1.5 million tons of post-consumer textiles are recycled, primarily by industrial and nonprofit organizations. The remaining 4.3 million tons are either dumped in landfills or burned in municipal garbage incinerators. This no longer aligns with society’s mandated worldwide sustainable recycling approach (1).
Reusing textiles is the most effective method of lowering carbon emissions. Numerous companies and merchants have expressed interest in assuming responsibility for their goods outside of the retail setting by creating take-back initiatives.
Downcycling textiles into fibrous items of lesser quality is not very useful because the resulting products are less comfortable, durable, and of poorer quality.
Upcycling textiles comprises the production of recycled fibers from monomers based on depolymerized wastes to obtain higher-quality fibrous products. It involves the correct identification and sorting of textile wastes according to their chemical composition (50% of textiles are made from fiber mixtures), which is complicated. The existing sorting and recycling technologies can be combined for a more effective recovery of monomers and conversion of them into recycled fibers or for using products of depolymerization (e.g., submicron particles) for another purpose (as nanofillers).
The quantity of textile waste collected for re-use and recycling has increased due to several factors:
Possible landfill restrictions or ban on textile waste for landfilling;
Increase in collection rates of textiles from governmental and retailer initiatives, textile producers, and in-store collection;
Increased demand from the public for a more sustainable society, which compels businesses to repurpose garbage as a feedstock;
Government regulation or modifications to current laws by offering rewards for reuse and recycling.
The basic strategies of post-consumer textiles processing are shown in Figure 1.

Major ways of textile waste processing.
This work proposes an upscaled textile recycling strategy to depolymerize fibrous wastes to their basic components (4).
This strategy is based on high-pressure hydrolysis under neutral or acidic conditions used alternatively for different fibers and their blends. Hydrolysis kinetics is described by a mechanistic model based on heterogeneous solid–liquid degradation. Model parameters are estimated from the experimental dependence of the number of particles produced by degradation n(t) as a function of time t by nonlinear least squares regression. The end of hydrolysis is characterized by a time of reaction t E.
This innovative approach broadens the scope of recycling textile waste and offers a higher value-added alternative to standard incineration.
2 Textile waste recycling
Both the chemical industry and textile industry are being pressed to consider alternative raw materials for the future to move toward a circular economy for at least three reasons:
The lack of resources necessitates the pursuit of novel approaches to maximize the utilization of existing ones.
The economy is noticeably strained by the cost of resources.
The requirement for sustainable materials to reduce the strain on existing resources, especially from products derived from petroleum.
The complicated treatment of post-consumer waste plastics, including fibrous wastes (Figure 2), is mainly due to several reasons:
Plastics that are difficult to sort (e.g., polyethylene [PE] versus polypropylene [PP]),
Multicomponent or composite plastics appear frequently (e.g., layers of different kinds of plastics), and
Different additives (such as flame retardants, plasticizers, or coloring agents) or foreign debris are present.
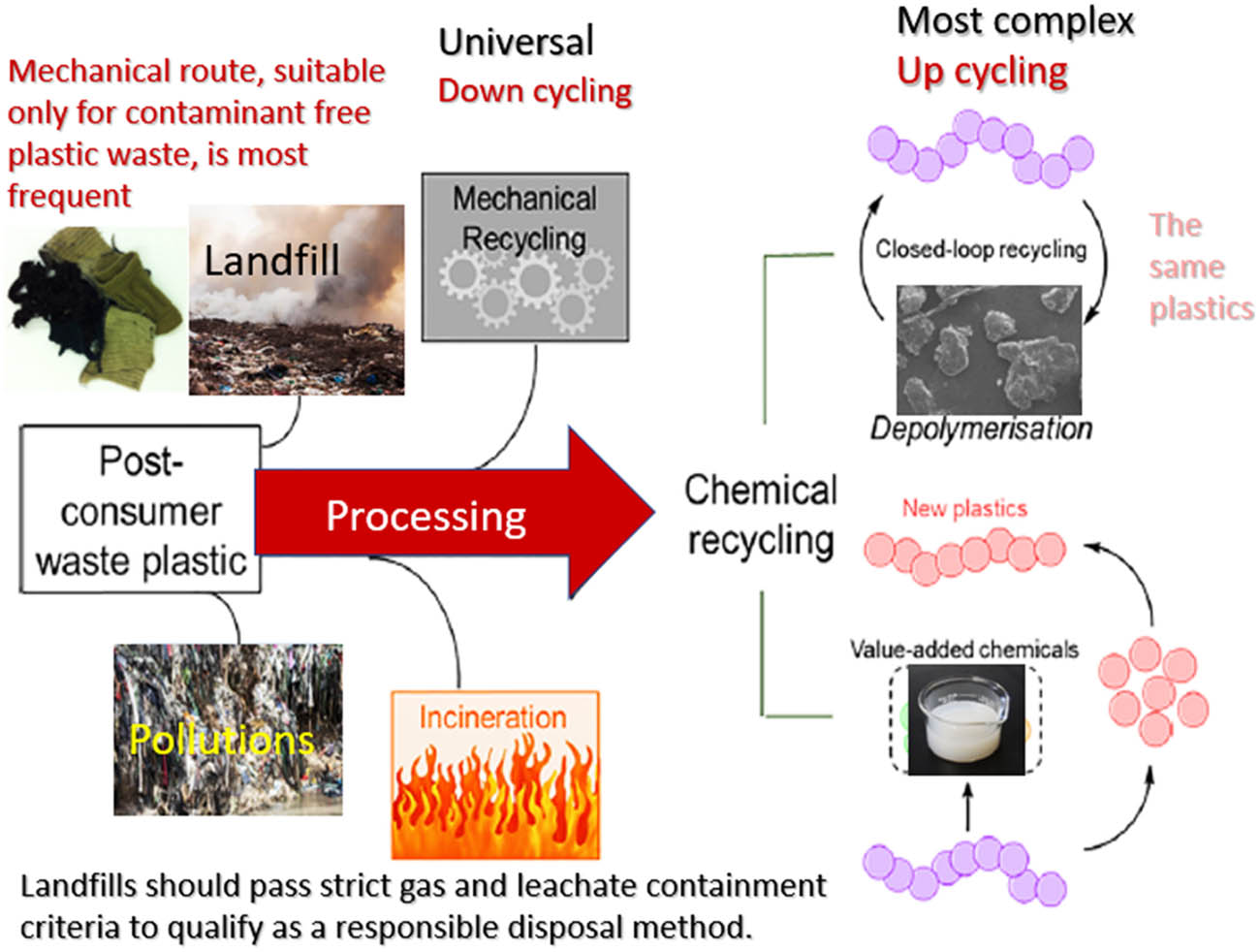
Different ways of processing plastic waste, including textiles.
Post-consumer textile waste, which includes frequently used garment fragments, is frequently disposed of in landfills or burned (Figure 1). Textile waste has a calorific value of about 14,500 kJ·kg−1, which is similar to brown coal, having 15,000–20,000 kJ·kg−1 (5). The major limitation is that both landfilling and incineration have a huge negative impact on the environment and produce some toxic gases.
Based on the intricacy of its processing, textile waste can be divided into four categories (6):
Wastes from textile production, which are the easiest to process. They are mostly waste from one type of fiber only, without much pollution. The generation of these wastes is actually continuous during the processing of textiles, and their quantity and geometry can be easily determined.
Used apparel suitable for pre-sorting or disposal as municipal garbage. The primary issues with pre-sorted wastes are the inclusion of non-fibrous materials (buttons, zippers, plastic, metal components, etc.) and the combination of various fibers. Weathering or microbial action can partially decompose these wastes, which are frequently highly contaminated.
Carpets, drapes, and other used household textiles frequently have a high percentage of non-fibrous materials in some of their layers. These wastes are typically produced over a longer period of time.
Waste from composites and industrial textiles (such as building textiles) that have a fiber component along with a sizable amount of additional polymeric or non-polymeric components that are typically mixed in layers. These wastes are produced over a medium-to-long period of time.
Historically, yarns, non-woven textiles, and industrial cotton wool have all been made from some pre-consumer textile waste (6). Secondary textile raw materials are shredded on machines to create fibers that are added to primary fibers during the yarn-making process. It is necessary to measure the fibers’ length when processing fiber waste, especially in relation to their spinability (7).
Textile waste, when recycled, is a large source of natural and polymer materials for chemical, textile, and other industries. This is because of the following reasons:
Recycling natural fibers (wool and cotton) is a major alternative that can create some chemicals (bio-based materials) without the use of petroleum products and prepare solid submicron particles as potentially more environmentally friendly fillers.
Recycling synthetic fibers allows for the reduction of the consumption of polymeric raw materials (monomers) produced from petroleum and optimizes environmental balance and production costs.
Intimate blends of various types of fibers and mixed-color fabrics are considered limiting factors in textile recycling since they negatively affect the sorting processes and decrease the quality of recycled materials. Zahmani (3) has investigated the environmental performance of different recycling options, presenting a good overview of the current state of the art for textile recycling techniques.
Different mechanical techniques exist for recycling textile waste (8). The applicability of each method depends on the quality of waste.
Used textiles can be turned into yarns using a few mechanical procedures. Their characteristics depend on the caliber of the textile waste used. The only use for yarns made from recycled textiles with poor physical qualities is the production of synthetic technical textiles, like geotextiles or woven filtration systems. These yarns are primarily made of mixed-color fibers with different lengths.
Chemical recycling is an alternative to mechanical recycling for synthetic fibers (9). It is possible to use chemical methods for fibers like polyester (PET), nylon, or PP. According to these processes, fiber molecules are broken down, and the feedstock is repolymerized. Chemical processes produce not only monomeric substances but also oligomers and fine fibrils. Because all kinds of fibers are semicrystalline, they are often products of chemical recycling nanofibrillar solid particles based on crystalline arranged macromolecules with extreme durability during chemical treatment (10).
So far, the main fibers consumed in textile and garment production are PET, cotton, and their blends. Consequently, they are most predominant in textile multicomponent waste.
PET fibers are the most widely used synthetic polymer in the textile industry, with an annual production of 39 million tons.
PET waste recycling can be carried out in many ways (11,12,13,14,15,16,17,18,19,20,21,22). The main principle is the cleavage of ester bonds by attacking nucleophilic agents with labile hydrogen atoms (as water glycols) (20,21), leading to monomers terephthalic acid (TPA) and ethylene glycol (EG) (Figure 3).
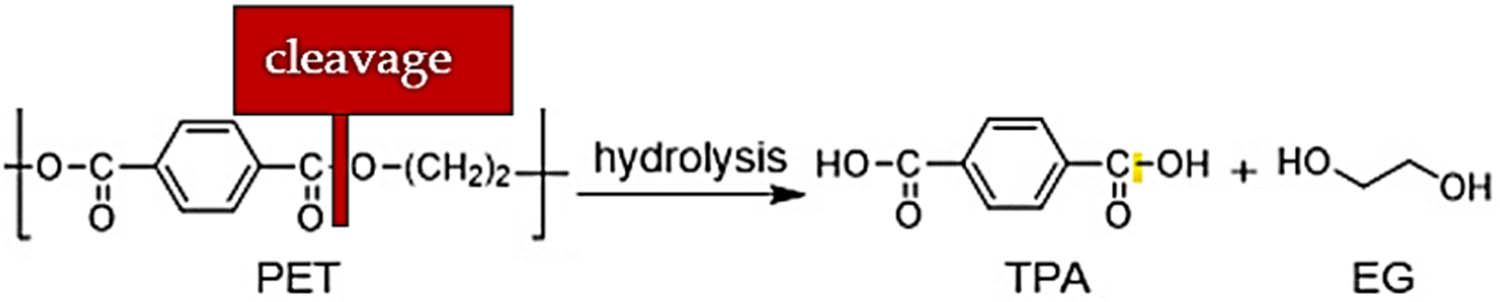
Depolymerization of PET by cleavage of esteric bonds.
Among the methods of polymer recycling, chemical recycling, applied above, is mostly for postconsumer condensation polymers, e.g., PET, which are very vulnerable to solvolytic chain cleavage and are of great interest (11,23,24,25,26). PET chemical recycling has several benefits, including the availability of a broad range of degrading (depolymerizing) agents and a wide variety of products, such as monomers for polymer and resin syntheses and other additives for polymeric materials (14,20,27). The final products of PET degradation are mainly TPA and EG (28). Often, the products of disintegration are monomers (soluble in water) as well as submillimeter or nanometer solid particles (Figure 4).

Gas NH3 selective disintegration of PET fibers to particles of sub-millimeter dimensions.
Cellulose is the most widely used natural polymer. The degradation and recycling of cellulose are well known in the paper sector, though they are not widely used in the textile sector.
Cellulose can be chemically recycled by using concentrated inorganic or organic acid at room temperature. One of the simplest ways is to prepare nanocellulose fibrils, which is shown in Figure 5. The scanning electron microscopy (SEM) image of microfibrils is shown in Figure 6.
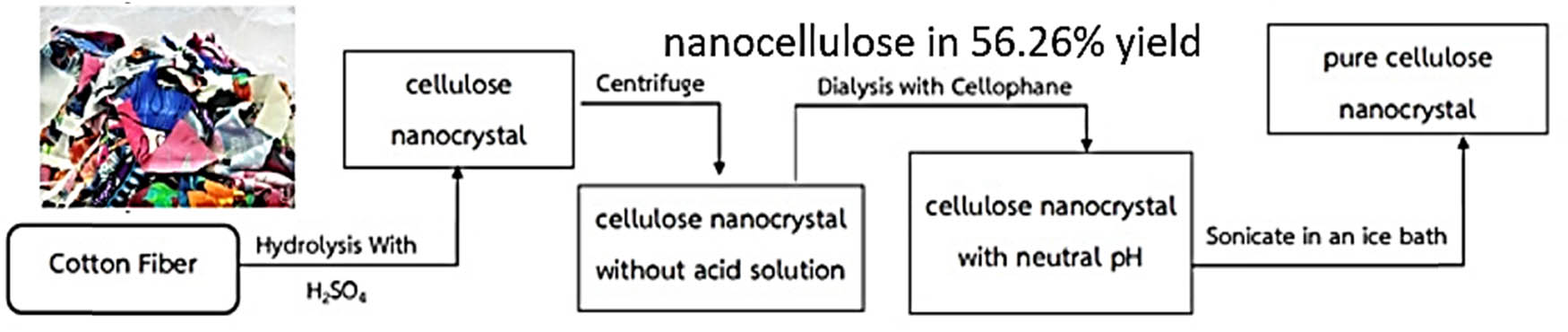
Preparation of nanocellulose from cotton-based wastes.

SEM image of cellulose fibrils after hydrolysis by 1:1 HCl for 2 h at 80°C.
The industry process of forming regenerated cellulosic fibers from wood using organic solvents is well known and widely used (29,30).
It is reported that yields of the reduced sugar from 64.3% to 73.9% were obtained by cotton cellulose hydrolysis using concentrated sulfuric acid at room temperature and fermentative hydrogen production (10). Also, the potential of textile cotton waste for energy production has been suggested. However, the main disadvantage of these concepts is ecological. This is primarily due to the use of very highly concentrated acids which decompose the cellulose. Further, cellulose can be hydrolyzed/decomposed at low acid concentrations (0.07–0.1 wt%) of sulfuric acid above 200°C with a yield of up to 90% (27). This ecologically acceptable method is energetically unacceptable due to high-temperature reaction requirements.
Researchers are trying to find green solvents for the dissolution and regeneration of cellulose, such as ionic liquids (31), and for the liquefaction of cellulose (32), such as acidified EG (33,34).
Kosan et al. (35) examined the characteristics of cellulose solutions in a variety of dissolving liquids, including ionic liquids with a range of cations and anions and N-methyl morpholine-N-oxide. Selective recovery of cellulose material from its blend by using ionic liquids is described by Wendler et al. (36).
The separation of PET from multicomponent textile waste presents a unique opportunity to produce valuable raw materials (monomers) that are otherwise produced from non-renewable resources (37).
3 Model of hydrolysis kinetics
The theory developed has already been discussed in detail in the article (4). It consists of pure and multi-component fiber blend degradation (15,21,38,39,40,41). Furthermore, it assumes the depolymerization process for two different rate-controlling steps:
Water transport in the cracks created in solid fibrous particles
Surface reaction as the rate-controlling mechanism.
The analysis used here does not consider the contribution of film diffusion of water. The governing kinetic equation for water transport (diffusion) in the solid phase as the rate-controlling step was fully derived in our previous paper (4). The main assumption is that the starting circular fiber has a length h [m] that is much greater than its radius R, h ≫ 2R. Ideally, this fiber can be divided into very small cubic particles (product of hydrolysis) with an edge size of d [m]. The number of particles n(t) (−) at time t (s) in the fiber is expressed by the final relation (full derivation is in (4)).
where
In fact, the term D (1/s2) is the penetration rate constant (with respect to surface geometry). In this case, quantity k (1/s) is the reaction rate constant, while quantity b (1/s) is the solvent penetration rate constant.
Because h and d are independent of temperature, activation energy E
A can be calculated from estimated reaction rate constants at various temperatures using the simple Arrhenius relation
For characterization of the overall effectivity of these processes, the total time of full depolymerization, i.e., end-of-reaction time t E (s), is calculated using Eq. 2
For two-component mixtures, Eq. 1 is valid for each component, and then the total molar amount is
4 Materials
Experiments were conducted by neutral and acid hydrolysis of pure and blended fibers for these cases, simulating common fibrous wastes. Hydrolysis agents and conditions of hydrolysis were selected to be optimal based on our previous experimental findings.
Case 1: Neutral hydrolysis of PET
Case 2: Acid (H3PO4) hydrolysis of pure cotton
Case 3: Acid (HCl) hydrolysis of the PET–cotton 50/50 blend
Case 4: Acid (H3PO4) hydrolysis of pure wool
Case 5: Acid (H3PO4) hydrolysis of the PET–cotton 50/50 blend
Case 6: Acid (H3PO4) hydrolysis of the PET–wool 50/50 blend
Two pure untreated raw materials were used for the experiments on the decomposition of natural fibers: raw cotton yarn and wool fibers. Two multicomponent mixtures, cotton/PET (weight ratio, 50:50) and wool/PET (weight ratio, 50:50), prepared from the materials described above, were selected.
For the degradation reaction of pure cotton, we used a crude unbleached cotton yarn. It was cut into segments of 1–2 cm in length and further treated. The crude carded sheep wool, which was used for the decomposition of pure wool and the preparation of a mixture of wool/PET, was also cut into segments of 1–2 cm in length. The mixture of cotton and PET was in the form of non-colored fabrics with a raw material composition of 50% cotton and 50% PET. Before the degradation reaction, it was cut into pieces of 0.5–1 cm2. The prepared samples were dried for 12 h in an oven at a temperature of 105°C and then stored in a desiccator until the experiment was carried out.
PET multifilament yarn (obtained from Julon Co) with fully drawn yarn-polyester of yarn count 78/36 was used for neutral hydrolysis reactions. Samples of PET yarn were cut into segments of 1–2 cm in length, conditioned for 24 h, or stored in a desiccator until the experiments were carried out. Two pure materials – 100% cotton yarn and wool fibers – as well as two 50:50 mixtures of these single materials with PET were used in acid hydrolysis experiments. A 50 wt% PET and 50 wt% wool mixture was made by weighing 0.5 g of PET and 0.5 g of wool fibers. A 50:50 combination of PET and cotton cloth was utilized to separate the two materials. A batch glass reactor fitted with a magnetic stirrer was used to hydrolyze pure and blended natural fibers using acid.
HCl and H3PO4 were used in acid hydrolysis at two different concentrations. Twenty milliliters of acid and 1 g of material were used in the reactions. All batch procedures were conducted with the solid:liquid ratio kept at 1:20. The temperature range of 55–120°C was used, and several agitation speeds were employed.
The initial stage of the procedure was using acid hydrolysis to remove natural fibers (wool and cotton) from mixtures. To determine the best way to remove natural fibers and to optimize the amount of data gathered, optimal conditions from the Box–Behnken experimental design were used. The PET fabric was neutrally hydrolyzed in a high-pressure reactor at 250°C and 35–38 bar pressure in the second stage. In keeping with minimum water use, the procedure was tested at two distinct weight (PET/water) ratios (1:8 and 1:10). By dissolving in sodium hydroxide solution, precipitating in hydrochloric acid, and washing with water, the reaction components were purified and then analyzed on a PerkinElmer Spectrum GX Fourier transform infrared (FT-IR) spectrometer (4).
4.1 Chemicals
Chemicals such as hydrochloric acid (HCl), with concentrations of 37% and 26%; phosphoric acid (H3PO4), with concentrations of 85% and 50%; and sodium hydroxide (NaOH), with concentrations of 10% were used. All aqueous solutions were prepared with deionized water (IKA Company, Germany). All chemicals were provided by Sigma-Aldrich.
5 Methods
5.1 Analytical methods
IR analysis was used to determine the structure of organic and inorganic substances or the presence of different functional groups. SEM analysis was used to gain insight into the surface structure of partially degraded cotton fibers, and gravimetry was used to study the reactions of degradation of natural fibers and the neutral hydrolysis of PET.
The FT-IR spectrophotometer PerkinElmer Spectrum GX for Chemistry, Dyes, and Polymers was used. IR spectra were recorded in the mid-IR range, each representing an average of 16 measurements. The relative number of particles n (%) in the fiber was evaluated from the analysis of IR spectra of the hydrolyzing bath based on the assumption that IR signal intensity is directly related to the number of particles in the hydrolyzing bath n B(t) in time t by using the relation:
where n BE is the total number of particles in a bath of fully hydrolyzed material (at the end of hydrolysis).
5.2 High-pressure reactor for hydrolysis
In order to carry out the neutral PET hydrolysis, a newly designed and constructed reactor (Figure 7) was used, which would allow the reaction to be carried out at elevated temperatures with increasing autogenous pressure.

High-pressure reactor for subcritical neutral hydrolysis in water (2).
Since terephthalic acid was formed during the reaction (the aqueous solution had a pH between 3 and 4), all parts that were in direct contact with the reaction mixture were made of stainless steel. In addition, all the elements were suitable for use under extreme conditions (40 bar and 250°C). The equipment was designed in such a way that it would be possible to depolymerize large quantities of material (15 g and more). Thus, a reaction vessel with a volume of 1 L was used. The heating of the reaction mixture was made possible by a special heater inserted into the wall of the reaction vessel. A temperature-sensitive sensor was added, which allowed monitoring and control of the temperature in the reaction system. The cooling coil, however, allowed the circulation of water and thus rapidly cooled the reaction mixture and the entire system at the end of the reaction. Pressure monitoring in the reaction vessel was enabled via a pressure gauge. For personal protection, a safety valve and a venting valve were added.
6 Results and discussion
The six experimental cases were analyzed for various fibers and their blends in neutral or acidic solvents. The solvents and conditions of hydrolysis were selected from previous findings as optimal. The kinetic and geometric parameters of Eq. 1 were estimated by the nonlinear least squares method (minimizing the sum of squared deviations of experimental points from the model curve) using the software package Darwin (Trilobyte Ltd., Czech Republic) (4). The basic parameters of the degradation reaction model for all cases are summarized in Table 1. The 95% confidence interval bands for the estimated curve are shown in Figures 8–13 as red lines. The activation energy plot was used to estimate the activation energy of the process (42).
Calculated model parameters of degradation reaction
| Case | Temperature T (°C) | Acid concentration c (%) | Rate parameter D (s−2) | Integration parameter C (−) | End time t E (min) |
|---|---|---|---|---|---|
| Case 1: neutral hydrolysis of PET | 250 | 0 | 2.69 × 10−5 | 1.04 | 197 |
| Case 2: acid (H3PO4) hydrolysis of pure cotton | 120 | 50 | 6.74 × 10−5 | 0.99 | 121 |
| 95 | 50 | 8.97 × 10−6 | 0.99 | 333 | |
| 75 | 50 | 3.84 × 10−6 | 1 | 509 | |
| Case 3: acid (HCl) hydrolysis of PET–cotton blend | 55 | 37 | 0.000134 | 0.97 | 85 |
| 70 | 37 | 0.000314 | 0.99 | 56 | |
| 55 | 26 | 3.71 × 10−5 | 0.99 | 163 | |
| 70 | 26 | 6.45 × 10−5 | 0.98 | 123 | |
| Case 4: acid (H3PO4) hydrolysis of wool | 120 | 50 | 2.63 × 10−5 | 1 | 195 |
| 95 | 50 | 1.13 × 10−6 | 1 | 939 | |
| Case 5: acid (H3PO4) hydrolysis of PET–cotton blend | 120 | 85 | 0.00127 | 0.98 | 28 |
| 80 | 85 | 0.000473 | 0.98 | 46 | |
| Case 6: acid (H3PO4) hydrolysis of PET–wool blend | 120 | 85 | 2.69 × 10−5 | 0.98 | 191 |
| 95 | 85 | 1.37 × 10−5 | 1 | 270 | |
| 120 | 50 | 1.38 × 10−5 | 0.99 | 268 | |
| 95 | 50 | 2.08 × 10−6 | 1 | 693 |
Case 1: Neutral hydrolysis of PET
A comparison of the solid phase penetrant transport model (water) with experimental data reveals good agreement.
Figure 8 shows the experimental data (black circles) and the kinetic model curve estimated by the nonlinear least squares model (black line). The model parameters are shown in Table 1 (first block). The 95% confidence interval band for the model curve is also shown (red lines).
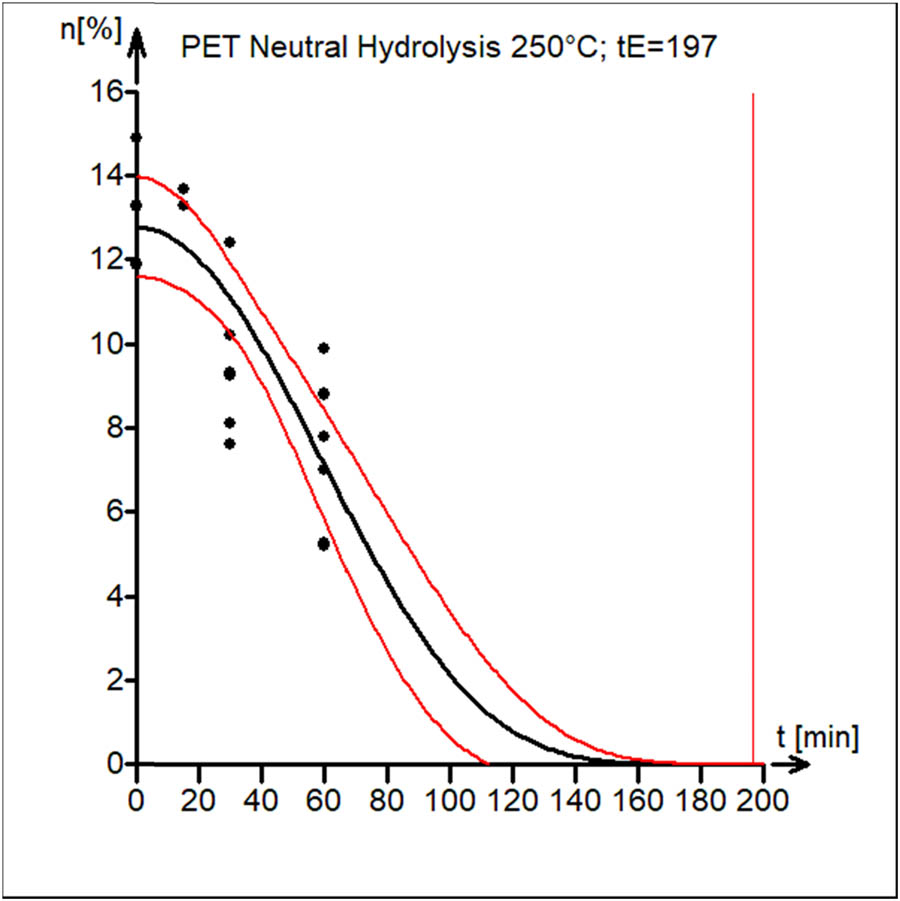
Kinetics of PET neutral hydrolysis with end-time of reaction t E = 197 min.
Because this reaction was carried out at only one temperature, activation energy was not evaluated. The form of the degradation reaction curve is a sigmoidal shape with a slow decline initially. This is consistent with the rate of water transport in the cracks created by solid PET particles as the controlling step. The availability of many active sites for the reaction to proceed is the cause of the rapid decline. Toward the end of the process, there are fewer sites available for hydrolytic reactions, which results in a lower rate.
Case 2: Acid (H3PO4) hydrolysis of cotton
A study of acid hydrolysis (50% H3PO4) of cotton fibers was conducted at three different temperatures, T = 75°C, 95°C, and 120°C, with constant stirring and approx. 30 times (mass) excess of the solvent. The reactants were stirred at 200 rpm. The degradation of cotton increased linearly with time. The rate of increase was higher at 120°C versus lower temperature. The activation energy of the acid hydrolysis reaction of cotton was found to be 72.9 kJ·mol−1. The experimental data (black circles) and kinetic model curve estimated by nonlinear least squares (black line) are shown in Figure 9a. Model parameters are shown in Table 1 (second block). The 95% confidence interval band for the model curve is shown as well (red lines).
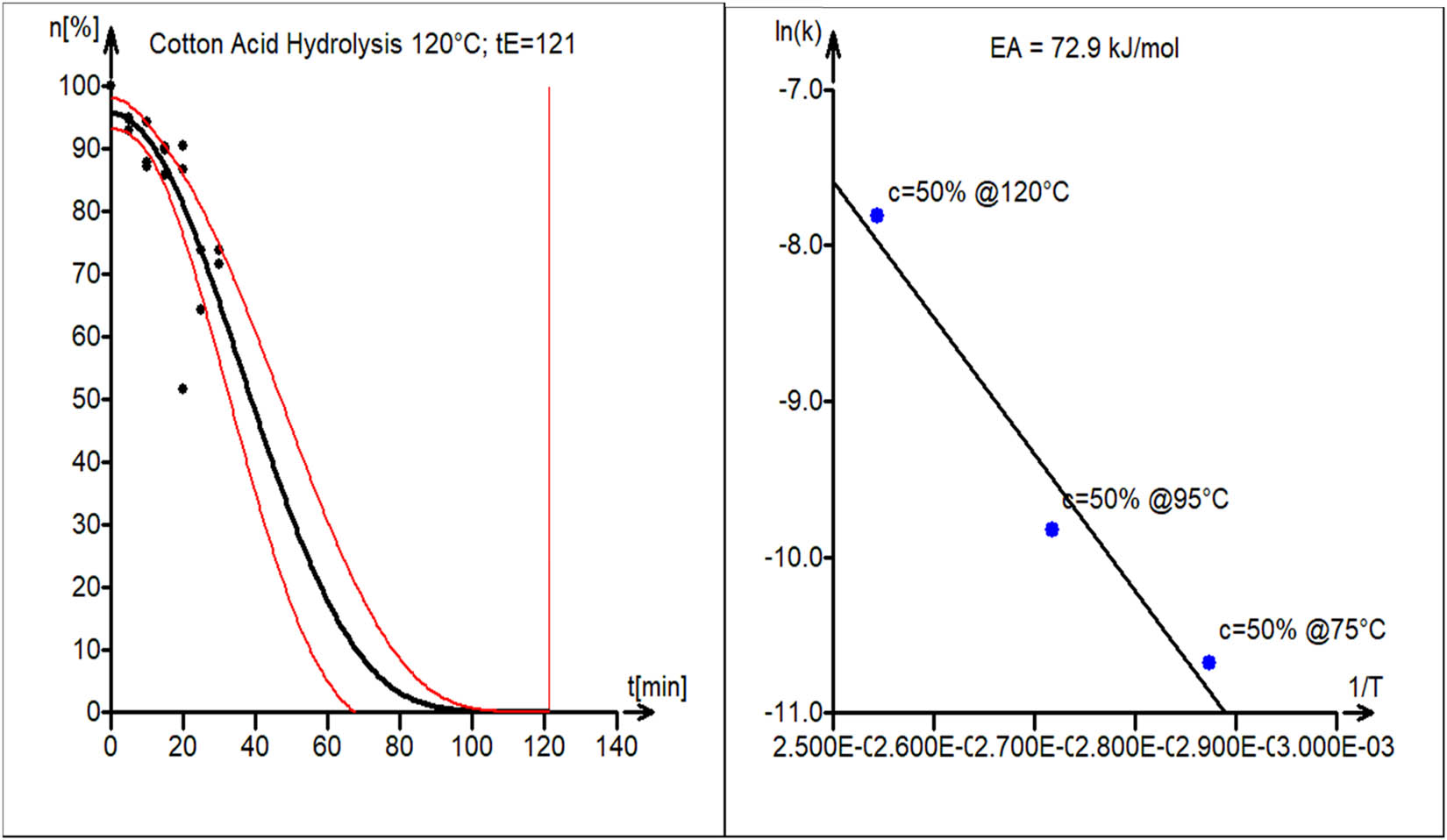
(a) Experimental kinetic data fitted with the theoretical model for T = 120°C. The estimated end-of-reaction time is t E = 121 min. (b) Estimation of activation energy for cotton acid hydrolysis, E A = 100.2 kJ·mol−1.
The activation energy plot (logarithm of estimated rate constants vs. reciprocal value of hydrolysis temperature), which is displayed in Figure 9b, was used to estimate the activation energy.
Case 3: Acid (HCl) hydrolysis of the PET–cotton 50/50 blend
The acid (HCl) hydrolysis of PET/cotton blend was conducted at 55°C and 70°C. HCl was selected to study the impact of a strong inorganic acid as compared to weak phosphoric acid. The activation energy of the acid hydrolysis reaction of the PET–cotton blend was found to be 43.7 kJ·mol−1. The experimental data (black circles) and kinetic model curve estimated by nonlinear least squares (black line) are shown in Figure 10a. Model parameters are shown in Table 1 (third block). The 95% confidence interval band for the model curve is shown as well (red lines).
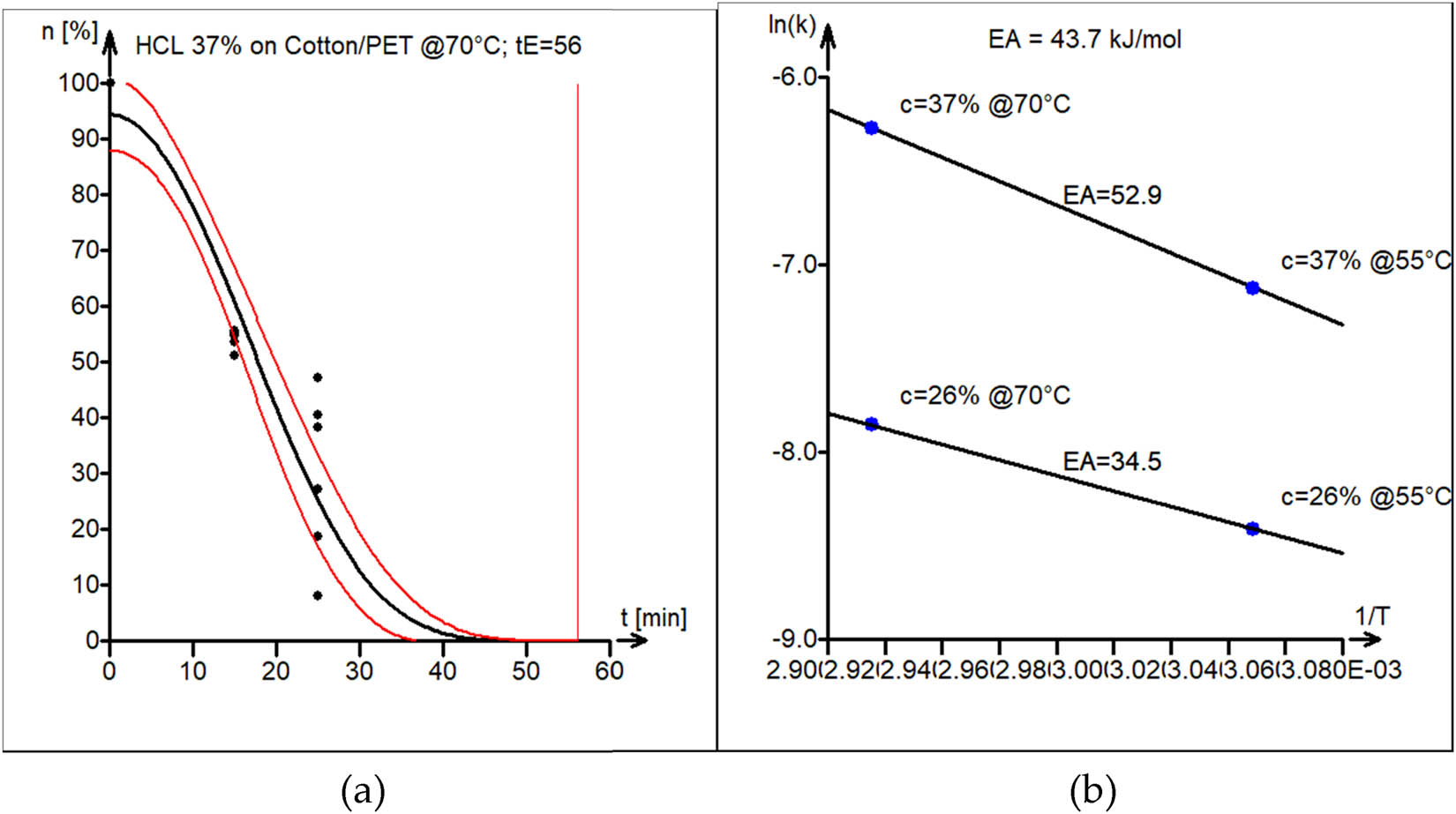
(a) Model versus experimental data of PET/cotton acid hydrolysis in 37% HCl at 70°C. The vertical line marks the estimated end-of-reaction time t E = 56 min. (b) Estimation of the activation energy of acid hydrolysis of PET/cotton, with average E A = 44 kJ·mol−1 from activation energies for different acid concentrations.
The activation energy plot, which is displayed in Figure 10b, was used to estimate the activation energy. The parameters are strongly dependent on the concentration of acid – 26% versus 37%.
Case 4: Acid (H3PO4) hydrolysis of wool
The acid (H3PO4) hydrolysis of wool was conducted at 95°C and 120°C with constant stirring at 200 rpm. The activation energy of hydrolysis was estimated to be 151.4 kJ·mol−1.
The experimental data (black circles) and kinetic model curve estimated by nonlinear least squares (black line) are shown in Figure 11a. Model parameters are shown in Table 1 (fourth block). The 95% confidence interval band for the model curve is also shown (red lines).
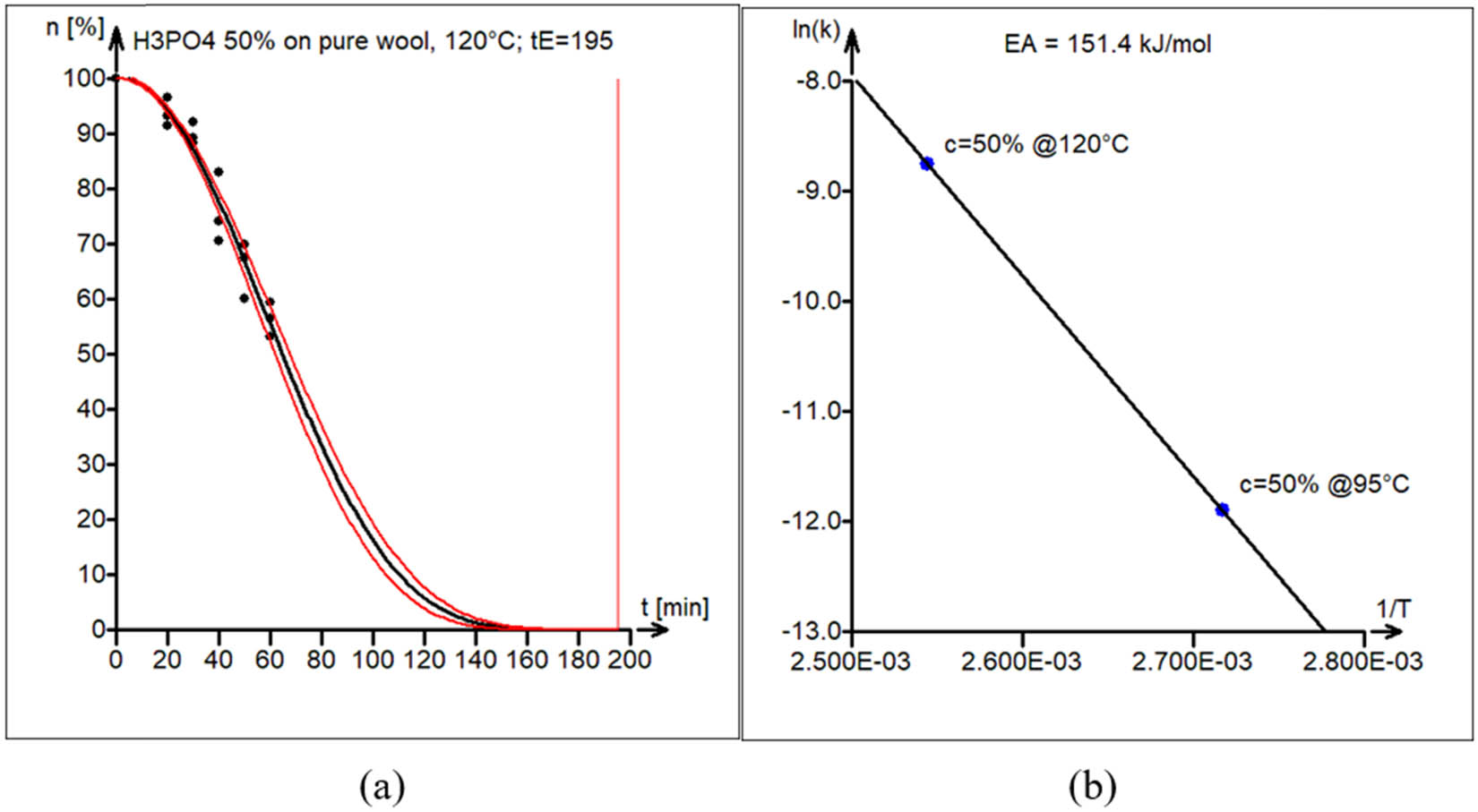
(a) Model versus experimental data of wool acid hydrolysis in 50% H3PO4 at 120°C. The vertical line marks the estimated end-of-reaction time t E = 195 min. (b) Estimation of the activation energy of acid hydrolysis of wool E A = 151 kJ·mol−1.
The activation energy plot, which is displayed in Figure 11b, was used to estimate the activation energy.
Case 5: Acid (H3PO4) hydrolysis of the PET–cotton 50/50 blend
The acid (H3PO4) hydrolysis of PET–cotton blend was conducted at 80°C and 120°C. The activation energy of the acid hydrolysis reaction of the PET–cotton blend was found to be 28.5 kJ·mol−1.
The experimental data (black circles) and kinetic model curve estimated by nonlinear least squares (black line) are shown in Figure 12a. Model parameters are shown in Table 1 (fifth block). The 95% confidence interval band for the model curve is shown as well (red lines).
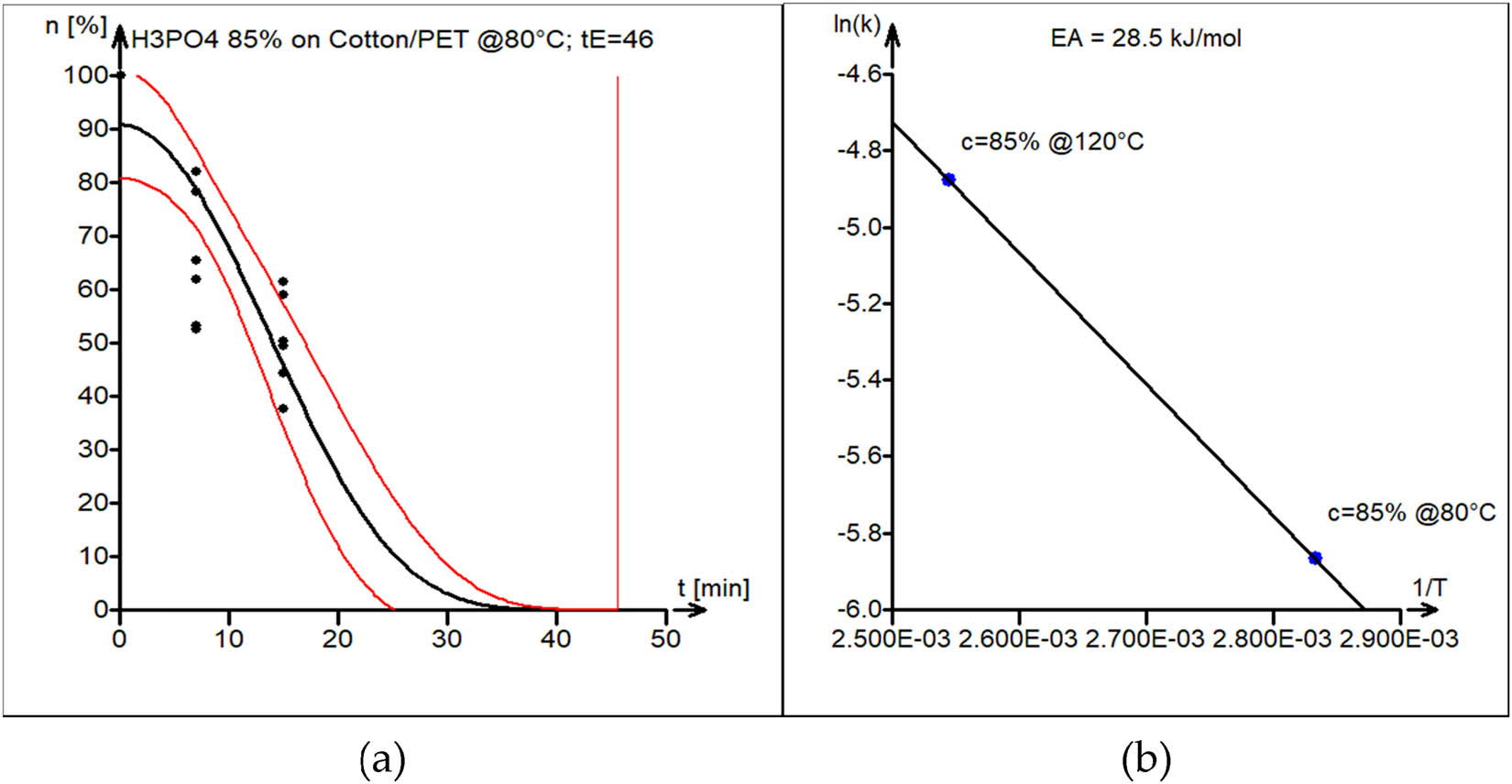
(a) Model versus experimental data of PET/cotton blend acid hydrolysis in 50% H3PO4 at 80°C. The vertical line marks the estimated end-of-reaction time t E = 46 min. (b) Estimation of the activation energy of acid hydrolysis of PET/cotton blend, with estimated E A = 28.5 kJ·mol−1.
The activation energy plot, which is displayed in Figure 12b, was used to estimate the activation energy.
Case 6: Acid Hydrolysis of the PET–wool 50/50 blend
The acid (H3PO4) hydrolysis of the PET/wool blend was conducted at 95°C and 120°C. The activation energy of the acid hydrolysis reaction of the PET–cotton blend was found to be 91 kJ·mol−1.
The experimental data (black circles) and kinetic model curve estimated by nonlinear least squares (black line) are shown in Figure 13a. Model parameters are shown in Table 1 (sixth block). The 95% confidence interval band for the model curve is shown as well (red lines).
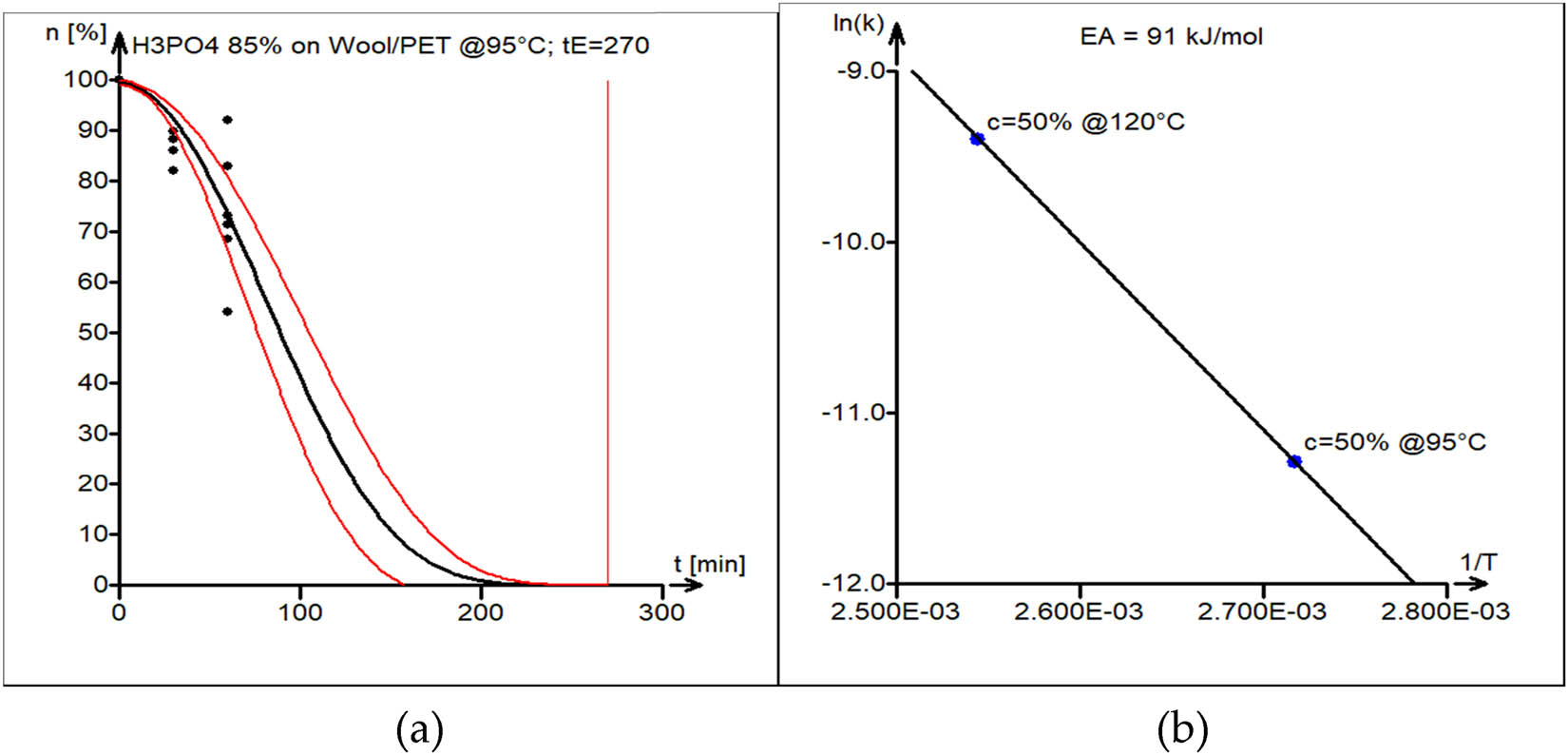
(a) Model versus experimental data of PET/wool acid hydrolysis in 50% H3PO4 at 120°C. The vertical line marks the estimated end-of-reaction time t E = 270 min. (b) Estimation of the activation energy of acid hydrolysis of PET/wool blend, with estimated E A = 91 kJ·mol−1.
The activation energy plot, which is displayed in Figure 13b, was used to estimate the activation energy.
It was, therefore, possible to use this proposed model with sufficient precision for all tested cases (different fibers and mixtures). From Table 1, it is evident that the rate parameters D of this model are varied for different fibers in a huge range because they are composed of kinetic and geometrical terms and are too complex to be compared with the rate constant of a chemical reaction. This parameter can be used only to compare the differences between the kinetic behavior of different fiber hydrolysis. The integration parameter C is nearly constant for all investigated systems. This is due to the meaning of this parameter
Activation energies are in the range of subcritical water hydrolysis of PET/cotton blended fabrics (62.2–87.8 kJ·mol−1) (39) and are much less than the activation energies of uncatalyzed degradation of PET by using the polarizable continuum model (200.8–213.4 kJ·mol−1) (43). Carta et al. found that the activation energy was 101.3 kJ·mol−1 using HNO3 for hydrolysis and 88.7 kJ·mol−1 using H2SO4, respectively (44). The activation energy for the neutral solid-state heterogeneous hydrolysis (94.5 + 2 kJ·mol−1) was obtained by Golike and Lazoski (45). All these values were obtained under different experimental conditions using different types of models and from different forms of materials, and therefore are not directly comparable.
7 Conclusions
Waste recycling is a direction and process of progress rather than an issue of reaching a goal. For all living species, we must constantly improve and secure our surroundings. This is only possible by taking a holistic approach to waste problem-solving that considers social, environmental, and economic factors. The direct and indirect effects of waste treatment on the environment must be considered, in addition to the local economic benefits, in order to try on a longer time horizon. It is necessary to balance not just ecology at the expense of economy, but the entire system of human demands entangled with the environment, in order to accomplish this. Linear systems, in which wastes are the last step, must give way to closed-loop and sustainable systems, in which wastes are the primary inputs for new products.
Here, a proposed new model for the kinetics of heterogeneous solid–liquid interaction of natural and synthetic fibers with water (neutral hydrolysis) and acid (strong and weak) reactions was successfully used for typical fibers and their mixtures. This model was successfully used for describing experimental data of all fibers and their mixtures using nonlinear least squares criteria with great precision. The activation energy of hydrolysis for different fibers and reactants is also typical for individual cases. For neutral hydrolysis, a special high-pressure reactor was developed. Selected process conditions were in accordance with the concept of less pollution caused by catalysts or hazardous chemicals.
For all cases (neutral hydrolysis of PET or pure component natural fiber acid hydrolysis), the model using solid phase penetrant transport as the predominant mechanism is acceptable. The model gave a good fit with the experimental data. The kinetics of the reaction of cotton/phosphoric acid hydrolysis resulted in an activation energy of 72.9 kJ·mol−1. In the case of acid hydrolysis (HCl) of the multi-component blend of PET and cotton, the activation energy was found to be 43.7 kJ·mol−1. The end-of-reaction time t E was proposed as a good overall characteristic of the hydrolysis process efficiency.
In the range of used stirrer speeds (80 and 150 rpm, or 100 and 200 rpm, respectively), no significant impact on hydrolysis was found. A possible reason may be that the difference between the stirrer speed is not significant for the used rate of fiber hydrolysis.
Acknowledgments
This work was supported by the research project “Hybrid materials for hierarchical structures” (HyHi, Reg. No. CZ.02.1.01/0.0/0.0/16_019/0000843) granted by the Ministry of Education, Youth and Sports of the Czech Republic and the European Union – European Structural and Investment Funds in the frames of operational programme research, development.
-
Funding information: This research was funded by the Department of Material Engineering, Faculty of Textile Engineering, Technical University of Liberec, Czech Republic.
-
Author contributions: Arun Aneja: conceptualization, methodology, formal analysis, resources; Karel Kupka: formal analysis, data curation, visualization, validation; Jiří Militký: writing – original draft, resources, supervision, investigation, project administration, funding acquisition; Mohanapriya Venkataraman: writing – review and editing, resources, methodology, visualization, validation.
-
Conflict of interest: The authors state no conflict of interest.
-
Data availability statement: All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
(1) Juanga-Labayen JP, Labayen IV, Yuan Q. A review on textile recycling practices and challenges. Textiles. 2022;2:174–88. 10.3390/textiles2010010.Search in Google Scholar
(2) Gschwandtner C. Outlook on global fiber demand and supply 2030. vol. 97, Lenzinger Berichte; 2022. p. 11–9.Search in Google Scholar
(3) Zahmani B. Carbon footprint and energy use of textile recycling techniques [MSc. Thesis]. Goteborg, Sweden: Chalmers University of Technology; 2012.Search in Google Scholar
(4) Aneja A, Kupka K, Militký J, Venkataraman M. Kinetics of hydrolytic depolymerization of textile waste containing polyester. Fibers. 2024;12:82. 10.3390/fib12100082.Search in Google Scholar
(5) Wang Y. Fiber and textile waste utilization. Waste Biomass Valorization. 2010;1:135–43. 10.1007/s12649-009-9005-y.Search in Google Scholar
(6) Hanuš J, Dvořáková E. Possibilities of using cryogenic technologies to recycle textiles and shoes. [Report TUL]. Liberec: 2008 (in Czech).Search in Google Scholar
(7) Křemenáková D, Krupincová G, Militký J. Properties of rotor yarns from reprocessed cotton fibers. Proceedings of Beltwide Cotton Conference. Nashville: 2008 Jan.Search in Google Scholar
(8) Roosen M, Mys N, Kusenberg M, Billen P, Dumoulin A, Dewulf J, et al. Detailed analysis of the composition of selected plastic packaging waste products and its implications for mechanical and thermochemical recycling. Environ Sci Technol. 2020;54:13282–93. 10.1021/acs.est.0c03371.Search in Google Scholar PubMed
(9) Coates GW, Getzler YD. Chemical recycling to monomer for an ideal, circular polymer economy. Nat Rev Mater. 2020;5:501–516.1. 10.1038/s41578-020-0190-4.Search in Google Scholar
(10) Veit D. Fibers: history, production, properties, market. Cham, Switzerland: Springer International Publishing; 2022. ISBN 978-3-031-15308.10.1007/978-3-031-15309-9Search in Google Scholar
(11) Awaja FD. Recycling of PET. Eur Polym J. 2005;41:1453–77. 10.1016/j.eurpolymj.2005.02.005.Search in Google Scholar
(12) Campanelli JM, Kamal D, Cooper A. A kinetic study of the hydrolytic degradation of polyethylene terephthalate at high temperatures. J Appl Polym Sci. 1993;48:443–51. 10.1002/app.1993.070480309.Search in Google Scholar
(13) Chen JC, Ou Y, Hu C. Depolymerization of poly(ethylene terephthalate) resin under pressure. J Appl Polym Sci. 1991;42:1501–7. 10.1002/app.1991.070420603.Search in Google Scholar
(14) Damayanti W. Strategic possibility routes of recycled PET. Polymer. 2021;13:1475–512. 10.3390/polym13091475.Search in Google Scholar PubMed PubMed Central
(15) de Carvalho GM, Muniz EC, Rubira AF. Hydrolysis of post-consume poly(ethylene terephthalate) with sulfuric acid and product characterization by WAXD, 13C NMR and DSC. Polym Degrad Stab. 2006;91:1326–32. 10.1016/j.polymdegradstab.2005.08.005.Search in Google Scholar
(16) Goje AS, Thakur SA, Diware VR, Patil SA, Dalwale PS, Mishra S. Hydrolytic depolymerization of poly(ethylene terephthalate) waste at high temperatures under autogenous pressure. Polym-Plast Technol. 2004;43(4):1093–113. 10.1081/PPT-200030031.Search in Google Scholar
(17) Hosseini SS, Taheri S, Zadhoush A, Mehrabani‐Zeinabad A. Hydrolytic degradation of poly(ethylene terephthalate). J Appl Polym Sci. 2007;103:2304–9. 10.1002/app.24142.Search in Google Scholar
(18) Launay AF, Thominette J. Hydrolysis of poly(ethylene terephthalate): a kinetic study. Polym Degrad Stab. 1994;46:319–24. 10.1016/0141-3910(94)90148-1.Search in Google Scholar
(19) Noritake A, Hori M, Shigematsu M, Tanahashi M. Recycling of polyethylene terephthalate using high-pressure steam treatment. Polym J. 2008;40:498–502. 10.1295/polymj.PJ2007237.Search in Google Scholar
(20) Paszun D, Spychaj T, Nowaczek N. Chemical recycling of Poly-(ethylene terephthalate) – PET. Ekoplast. 1993;1:25. (in Polish).Search in Google Scholar
(21) Rudakova T, Moiseyev Y, Pal’vanov V, Zaikov G. Kinetics and mechanism of breakdown of PET in dilute sulphuric acid solutions. J Chem Phys. 1974; U.S.S.R. Academy of Sciences: 1572–81.10.1016/0032-3950(74)90424-9Search in Google Scholar
(22) George N, Kurian T. Recent developments in the chemical recycling of postconsumer poly(ethylene terephthalate) waste. Ind Eng Chem Res. 2014;53:14185–98. 10.1021/ie501995m.Search in Google Scholar
(23) Carta D, Cao G, D'Angeli C. Chemical recycling of poly(ethylene terephthalate) (PET) by hydrolysis and glycolysis. Environ Sci Pollut Res. 2003;10(6):390–4. 10.1065/espr2001.12.104.8.Search in Google Scholar PubMed
(24) Allen NS, Edge M, Mohammadian M, Jones K. Hydrolytic degradation of poly(ethylene terephthalate): importance of chain scission versus crystallinity. Eur Polym J. 1991;27(12):1373–8. 10.1016/0014-3057(91)90237-I.Search in Google Scholar
(25) Sadrmohaghegh C, Scott G, Setudeh E. Recycling mixed plastics. Polym-Plast Technol. 1985;24:149–85. 10.1080/03602558508070064.Search in Google Scholar
(26) Park SO, Kim SH. Poly (ethylene terephthalate) recycling for high value-added textiles. Fash Text. 2014;1(1):1. 10.1186/s40691-014-0001-x.Search in Google Scholar
(27) Chandrashekhar B, Mishra SM, Sharma K, Dubey S. Bio-ethanol production from textile cotton waste via dilute acid hydrolysis and fermentation by saccharomyces cerevisiae. J Ecobiotechnol. 2011;3(4):6–9.Search in Google Scholar
(28) Tustin GC, Pell Jr TM, Jenkins DA, Jernigan MT. Process for the recovery of terephthalic acid and ethylene glycol from poly(ethylene terephthalate). US Patent 5,413,681; 1995.Search in Google Scholar
(29) Lin JH, Chang YH, Hsu YH. Degradation of cotton cellulose treated with hydrochloric acid either in water or in ethanol. Food Hydrocoll. 2009;23:1548–53. 10.1016/j.foodhyd.2008.10.005.Search in Google Scholar
(30) Gholamzad E, Karimi K, Masoomi M. Effective conversion of waste polyester–cotton textile to ethanol and recovery of polyester by alkaline pretreatment. J Chem Eng. 2014;253:40–5. 10.1016/j.cej.2014.04.109.Search in Google Scholar
(31) Kim JS. Cellulose hydrolysis under extremely low sulfuric acid and high-temperature conditions. Appl Biochem Biotechnol. 2001;91–93:331–40. 10.1007/978-1-4612-0217-2_28.Search in Google Scholar
(32) Chu C-Y, Wu SY, Tsai CY, Lin CY. Kinetics of cotton cellulose hydrolysis using concentrated acid and fermentative hydrogen production from hydrolysate. Int J Hydrogen Energy. 2011;36(14):8743–50. 10.1016/j.ijhydene.2010.07.072.Search in Google Scholar
(33) Lee YY. Dilute-acid hydrolysis of lignocellulosic biomass. Adv Biochem Eng/Biotechnol. 1999;65:93–116. 10.1007/3-540-49194-5_5.Search in Google Scholar
(34) Jasiukaitytė E, Kunaver M, Strlič M. Cellulose liquefaction in acidified ethylene glycol. Cellulose. 2009;16:393–405. 10.1007/s10570-009-9288-y.Search in Google Scholar
(35) Kosan K, Schwikal F, Meister F. Effects of pre-treatment and dissolution conditions for improved solution and processing properties of cellulose in ionic liquids. Lenzinger Ber. 2012;90:76–84.Search in Google Scholar
(36) Wendler F, Kosan B, Krieg M, Meister F. Possibilities for the physical modification of cellulose shapes using ionic liquids. Macrom Symp. 2009;280:112–22. 10.1002/masy.200950613.Search in Google Scholar
(37) Šerod JB, Ivanusic BA, Aneja AP, Majcen le Marechal A, Voncina B, Volmajer Valh J, et al. Multicomponent textile blend recycling. Proceedings of AUTEX Conference. Croatia: 2012; p. 6.Search in Google Scholar
(38) Wang Y, Wan J, Ma Y, Huang M. Hydrolysis kinetics characteristic of recycled fiber in subcritical water. Bioresour Technol. 2012;105:152–9. 10.1016/j.biortech.2011.11.072.Search in Google Scholar PubMed
(39) Zhang L. Kinetics of hydrolysis of poly(ethylene terephthalate) wastes catalyzed by dual functional phase transfer catalyst: A mechanism of chain-end scission. Eur Polym J. 2014;60:1–5. 10.1016/j.eurpolymj.2014.08.007.Search in Google Scholar
(40) Kao C, Wan B, Cheng W. Kinetics of hydrolytic depolymerization of melt poly(ethyleneterephthalate). J Am Chem Soc. 1998;37:1228–34. 10.1021/ie970543q.Search in Google Scholar
(41) Yang C, Xing X, Li Z, Zhang S. A comprehensive review on water diffusion in polymers focusing on the polymer–metal interface combination. Polymer. 2020;12:138. 10.3390/polym12010138.Search in Google Scholar PubMed PubMed Central
(42) Timm EW, Hinshelwood CN. The activation energy of organic reactions. Part III. the kinetics of acid hydrolysis of esters. J Chem Soc. 1938;862–9. 10.1039/JR9380000862.Search in Google Scholar
(43) Yan Z, Lian J, Li M, Meng L, Zhang Y, Ge C, et al. Deeper insight into hydrolysis mechanisms of polyester/cotton blended fabrics for separation by explicit solvent models. Int J Biol Macromol. 2020;154:596–605. 10.1016/j.ijbiomac.2020.03.130.Search in Google Scholar PubMed
(44) Carta D, Cao G, D'Angeli C. Chemical recycling of poly(ethylene terephthalate) (pet) by hydrolysis and glycolysis. Environ Sci Pollut Res. 2003;10:390–4. 10.1065/espr2001.12.104.8.Search in Google Scholar PubMed
(45) Golike RC, Lazoski SW. Kinetics of hydrolysis of polyethylene terephthalate films. J Phys Chem. 1960;64:895–8. 10.1021/j100836a018.Search in Google Scholar
© 2025 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- Flow-induced fiber orientation in gas-powered projectile-assisted injection molded parts
- Research on thermal aging characteristics of silicone rubber composite materials for dry-type distribution transformers
- Kinetics of acryloyloxyethyl trimethyl ammonium chloride polymerization in aqueous solutions
- Influence of siloxane content on the material performance and functional properties of polydimethylsiloxane copolymers containing naphthalene moieties
- Enhancement effect of electron beam irradiation on acrylonitrile–butadiene–styrene (ABS) copolymers from waste electrical and electronic equipment by adding 1,3-PBO: A potential way for waste ABS reuse
- Model construction and property study of poly(ether-ether-ketone) by molecular dynamics simulation with meta-modeling methods
- Zinc–gallic acid–polylysine nanocomplexes with enhanced bactericidal activity for the treatment of bacterial keratitis
- Effect of pyrogallol compounds dosage on mechanical properties of epoxy coating
- Preparation of in situ polymerized polypyrrole-modified braided cord and its electrical conductivity investigation under varied mechanical conditions
- Hydrophobicity, UV resistance, and antioxidant properties of carnauba wax-reinforced CG bio-polymer film
- Janus nanofiber membrane films loading with bioactive calcium silicate for the promotion of burn wound healing
- Synthesis of migration-resistant antioxidant and its application in natural rubber composites
- Influence of the flow rate on the die swell for polymer micro coextrusion process
- Fatty acid filled polyaniline nanofibres with dual electrical conductivity and thermo-regulatory characteristics: Futuristic material for thermal energy storage
- Hydrolytic depolymerization of major fibrous wastes
- Performance of epoxy hexagonal boron nitrate underfill materials: Single and mixed systems
- Blend electrospinning of citronella or thyme oil-loaded polyurethane nanofibers and evaluating their release behaviors
- Efficiency of flexible shielding materials against gamma rays: Silicon rubber with different sizes of Bi2O3 and SnO
- A comprehensive approach for the production of carbon fibre-reinforced polylactic acid filaments with enhanced wear and mechanical behaviour
- Electret melt-blown nonwovens with charge stability for high-performance PM0.3 purification under extreme environmental conditions
- Study on the failure mechanism of suture CFRP T-joints under/after the low-velocity impact loading
- Experimental testing and finite element analysis of polyurethane adhesive joints under Mode I loading and degradation conditions
- Optimizing recycled PET 3D printing using Taguchi method for improved mechanical properties and dimensional precision
- Effect of stacking sequence of the hybrid composite armor on ballistic performance and damage mechanism
- Bending crack propagation and delamination damage behavior of orthogonal ply laminates under positive and negative loads
- Molecular dynamics simulation of thermodynamic properties of Al2O3-modified silicone rubber under silane coupling agent modification
- Precision injection molding method based on V/P switchover point optimization and pressure field balancing
- Heparin and zwitterion functionalized small-diameter vascular grafts for thrombogenesis prevention
- Metal-free N, S-co-doped carbon materials derived from calcined aromatic co-poly(urea-thiourea)s as efficient alkaline oxygen reduction catalysts
- Influence of stitching parameters on the tensile performance and failure mechanisms of CFRP T-joints
- Synthesis of PEGylated polypeptides bearing thioether pendants for injectable ROS-responsive hydrogels
- A radiation-resistant MWCNT/Nafion/MWCNT composite film for humidity perception
- Study on the crosslinking network and mechanical properties of Taraxacum kok-saghyz natural rubber
- Preparation of maleic anhydride-modified curcumin/PBAT composite film for food packaging
- Rapid Communication
- RAFT-mediated polymerization-induced self-assembly of poly(ionic liquid) block copolymers in a green solvent
- Corrigendum
- Corrigendum to “High-strength polyvinyl alcohol-based hydrogel by vermiculite and lignocellulosic nanofibrils for electronic sensing”
Articles in the same Issue
- Research Articles
- Flow-induced fiber orientation in gas-powered projectile-assisted injection molded parts
- Research on thermal aging characteristics of silicone rubber composite materials for dry-type distribution transformers
- Kinetics of acryloyloxyethyl trimethyl ammonium chloride polymerization in aqueous solutions
- Influence of siloxane content on the material performance and functional properties of polydimethylsiloxane copolymers containing naphthalene moieties
- Enhancement effect of electron beam irradiation on acrylonitrile–butadiene–styrene (ABS) copolymers from waste electrical and electronic equipment by adding 1,3-PBO: A potential way for waste ABS reuse
- Model construction and property study of poly(ether-ether-ketone) by molecular dynamics simulation with meta-modeling methods
- Zinc–gallic acid–polylysine nanocomplexes with enhanced bactericidal activity for the treatment of bacterial keratitis
- Effect of pyrogallol compounds dosage on mechanical properties of epoxy coating
- Preparation of in situ polymerized polypyrrole-modified braided cord and its electrical conductivity investigation under varied mechanical conditions
- Hydrophobicity, UV resistance, and antioxidant properties of carnauba wax-reinforced CG bio-polymer film
- Janus nanofiber membrane films loading with bioactive calcium silicate for the promotion of burn wound healing
- Synthesis of migration-resistant antioxidant and its application in natural rubber composites
- Influence of the flow rate on the die swell for polymer micro coextrusion process
- Fatty acid filled polyaniline nanofibres with dual electrical conductivity and thermo-regulatory characteristics: Futuristic material for thermal energy storage
- Hydrolytic depolymerization of major fibrous wastes
- Performance of epoxy hexagonal boron nitrate underfill materials: Single and mixed systems
- Blend electrospinning of citronella or thyme oil-loaded polyurethane nanofibers and evaluating their release behaviors
- Efficiency of flexible shielding materials against gamma rays: Silicon rubber with different sizes of Bi2O3 and SnO
- A comprehensive approach for the production of carbon fibre-reinforced polylactic acid filaments with enhanced wear and mechanical behaviour
- Electret melt-blown nonwovens with charge stability for high-performance PM0.3 purification under extreme environmental conditions
- Study on the failure mechanism of suture CFRP T-joints under/after the low-velocity impact loading
- Experimental testing and finite element analysis of polyurethane adhesive joints under Mode I loading and degradation conditions
- Optimizing recycled PET 3D printing using Taguchi method for improved mechanical properties and dimensional precision
- Effect of stacking sequence of the hybrid composite armor on ballistic performance and damage mechanism
- Bending crack propagation and delamination damage behavior of orthogonal ply laminates under positive and negative loads
- Molecular dynamics simulation of thermodynamic properties of Al2O3-modified silicone rubber under silane coupling agent modification
- Precision injection molding method based on V/P switchover point optimization and pressure field balancing
- Heparin and zwitterion functionalized small-diameter vascular grafts for thrombogenesis prevention
- Metal-free N, S-co-doped carbon materials derived from calcined aromatic co-poly(urea-thiourea)s as efficient alkaline oxygen reduction catalysts
- Influence of stitching parameters on the tensile performance and failure mechanisms of CFRP T-joints
- Synthesis of PEGylated polypeptides bearing thioether pendants for injectable ROS-responsive hydrogels
- A radiation-resistant MWCNT/Nafion/MWCNT composite film for humidity perception
- Study on the crosslinking network and mechanical properties of Taraxacum kok-saghyz natural rubber
- Preparation of maleic anhydride-modified curcumin/PBAT composite film for food packaging
- Rapid Communication
- RAFT-mediated polymerization-induced self-assembly of poly(ionic liquid) block copolymers in a green solvent
- Corrigendum
- Corrigendum to “High-strength polyvinyl alcohol-based hydrogel by vermiculite and lignocellulosic nanofibrils for electronic sensing”