A β-ketoiminato palladium(II) complex for palladium deposition
-
Andrea Preuß


Abstract
The ¦-ketoiminato complex [Pd(OAc)L] (3) can be synthesized by the reaction of bis(benzoylacetone)diethylenetriamine (1, = LH) with [Pd(OAc)2] (2). The structure of 3 in the solid state has been determined by single X-ray diffraction analysis. Complex 3 crystallizes as a dimer (32), which is formed by hydrogen bonds between NH and OOAc functionalities of two adjacent ligands. Each of the Pd atoms is complexed by one ON2 donor unit of the polydentate ligand L− and an acetate group. Pd–Pd interactions and hydrogen bond formation between a NH and the C=O acetate moiety lead to a [4 + 2] coordination at Pd. The non-coordinated part of L exists in its ¦-keto-enamine form. The thermal decomposition behavior of 32 was studied by TG (thermogravimetry) and TG-MS showing that 32 decomposes between 200 and 500°C independent of the applied atmosphere. Under oxygen PdO is produced, while under argon Pd is formed as confirmed by PXRD measurements. Complex 32 was applied as a spin-coating precursor (conc. 0.1 mol L−1, volume 1.5 mL, 3000 rpm, deposition time 6 min, heating rate 50 K min−1, holding time 60 min (Ar) and 120 min (air) at T = 800°C). The as-obtained samples are characterized by granulated particles of Pd/PdO on the substrate surface. EDX (energy-dispersive X-ray spectroscopy) and XPS (X-ray photoelectron spectroscopy) measurements confirmed the formation of Pd (Ar) or PdO (O2) with up to 12 mol% C impurity.
1 Introduction
Recently, palladium-based materials attracted -attention, for example, in the field of microtechnology and can be found, for example, as integrant in gas sensors [1], [2], as metal contacts in field-effect transistors [3], or as parts in hydrogen permeable selective membranes [4]. Furthermore, palladium(II) complexes [5], [6], [7], [8], [9], palladium nanoparticles [10], [11], palladium films [12], or palladium composite materials [13] can be used as active components in homogeneous and heterogeneous catalysis, i.e. C,C cross-coupling [14] and C–H -activation reactions [15].
To produce thin films of palladium different deposition methods such as sputtering [2], electro-deposition [16], immersion deposition [17], spin-coating [18], [19], CVD (chemical vapor deposition) [20], [21], [22] and ALD (atomic layer deposition) [23], [24], [25] can be applied. For the deposition of Pd or PdO, the most commonly used gas-phase precursors are η3-allyl- and β-diketonato palladium complexes [21], [26]. For example, [Pd(η3-C3H7)2] [27] and [Pd(η3-2-Me-C3H4)2] [27] produce Pd films of high quality containing only 1.0 wt% carbon impurity without addition of any reactive gas [28]. However, these precursors feature thermal instability during evaporation and storage and are highly sensitive against air and moisture [26]. In contrast, bis(β-diketonato) palladium(II) complexes like [Pd(acac)2] (acac=acetylacetonate) are thermally stable but show low volatility and high decomposition temperatures [26]. The combination of allyl and β-diketonate groups within one complex resulted in a higher volatility and a higher thermal stability of the respective palladium complexes, allowing their use as CVD precursors for Pd layer formation [29].
Also β-ketoiminato palladium(II) complexes like [Pd(CF3C(O)CHC(CF3)NnBu)2] [25], [30], [Pd(CH3C(NH)CHC(O)CH3)2] [31], [32], [Pd(CH3(O)CCHCN(CH3)(CH2)3)2)] [33], [Pd(2-NC5H4)NHNCH-1-(O)-4-OMe-Ph)(PPh3)] [19] and palladium(II) allyl(β-ketoiminato) complexes [34], e.g. [Pd(ƞ3-2-Me-C3H4)(MeC(O)CHCMeNPh)] were synthesized with regard to their lower oxygen content as compared to β-diketonate species. This variant results in a decreased substrate oxidation upon metal film formation [31], [32]. In addition, these complexes show an improved stability towards moisture and air [35].
This prompted us to synthesize the β-ketoiminato palladium(II) complex [Pd(OAc)L] (L=PhC(O)CHC(Me)NCH2CH2NHCH2CH2NH(Me)C=CHC(O)Ph) (3), to study its thermal decomposition behavior and its use as spin-coating precursor for the formation of Pd and PdO deposits.
2 Results and discussion
For the synthesis of the β-ketoiminato palladium(II) complex [Pd(OAc)L] (3), bis(benzoylacetone)diethylenetriamine (1, =LH) [36] was reacted with [Pd(OAc)2] (2) in a 1:1 molar ratio in acetonitrile at ambient temperature for 16 h (eq. (1)). Even, when a two- or even three-fold excess of 2 was used for the reaction only 3 was formed. After work-up, complex 3 could be isolated as a yellow and air stable solid in a yield of 77% (Section 4). The new -organopalladium compound 3 is soluble in most common polar organic solvents, showing only reduced solubility in non-polar solvents, such as toluene or hexane.

Complex 3 was characterized by elemental analysis, IR and NMR (1H, 13C{1H}) spectroscopy and ESI-TOF mass spectrometry. TG (thermogravimetry), TG-MS (thermogravimetry-mass spectrometry) and vapor pressure studies were carried out to gain more information on the thermal decomposition behavior and volatility of 3. The molecular structure of 3 in the solid state was determined by single crystal X-ray -diffraction (see below).
The 1H and 13C{1H} NMR spectra of 3 are characterized by sets of signals for the various organic groups present in the complex molecule (Section 4). Very characteristic is the proton resonance signal of the Pd–NH unit appearing at 11.51 ppm, while the non-coordinated NH entity of L cannot be unequivocally assigned since it overlaps with the signals of the phenyl protons (Section 4). This observation strengthens the appearance of the β-keto-enamine tautomer of the non-complexed part of L in 3.
Characteristic for 3 in the IR spectrum is the presence of C=C, C=N and C–O stretching vibrations in the region of 1500–1600 cm−1, which is unique for this type of Pd complexes (Fig. SI1, Supporting information available online) [37], [38], [39], [40], [41], [42]. These vibrations are shifted to lower wavenumbers as compared to 1 (1, 1605 cm−1; 3, 1561 cm−1), confirming the coordination of L to Pd(II) [41], [42]. The NH stretching vibrations are observed between 3020 and 3340 cm−1. The weak band observed at 3339 cm−1 indicates the β-keto-enamine tautomer of the non-coordinated site of L [37], [40], [41], [43], [44], [45], while the bands at 3074, 3058 and 3018 cm−1 are characteristic for intra- and inter-molecular hydrogen bond formation [40]. An exact assignment of the binding motif of the Pd-bonded acetate ligand was not possible, due to a band overlap with the C=C vibrations (Section 4).
2.1 Solid-state structure
The molecular structure of 3 in the solid state was determined by single-crystal X-ray diffraction analysis. The Ortep plot of 3 is depicted in Fig. 1 and selected bond lengths (Å), angles (°), and torsion angles (°) are given in Table 1. Suitable crystals were obtained by crystallization of 3 from a saturated dichloromethane solution at ambient temperature.

Selected bond lengths and angles (Å, °) of 32.a,b
| 32 (1) | 32 (2) | 32 (1) | 32 (2) | ||
|---|---|---|---|---|---|
| Pd1–O1 | 2.044(4) | 2.039(4) | N1–Pd1–O1 | 173.72(17) | 172.43(18) |
| Pd1–O3 | 1.989(4) | 1.978(4) | O3–Pd1–N2 | 178.82(17) | 178.40(18) |
| Pd1–N1 | 1.962(5) | 1.963(5) | O1–Pd1–O3 | 87.91(17) | 87.87(18) |
| Pd1–N2 | 2.060(5) | 2.048(5) | O3–Pd1–N1 | 94.9(2) | 94.4(2) |
| Pd1–Pd2 | 3.1412(7) | N1–Pd1–N2 | 84.5(2) | 85.0(2) | |
| C25–O1 | 1.278(7) | 1.276(7) | N2–Pd1–O1 | 92.84(19) | 92.9(2) |
| C25–O2 | 1.220(8) | 1.231(7) | |||
| O3–C7 | 1.293(8) | 1.295(8) | |||
| C7–C8 | 1.379(9) | 1.387(10) | MeCO2LON2Pd | 78.2(5) | 83.0(3) |
| C8–C9 | 1.420(10) | 1.412(10) | O4–C18–C16–N3 | 6.1(7) | 3.2(6) |
| C9–N1 | 1.300(9) | 1.292(9) | O3–C7–C9–N1 | 6.2(5) | 6.2(5) |
| N1–C11 | 1.488(8) | 1.492(9) | |||
| N3–C14 | 1.452(9) | 1.462(9) | |||
| N3–C16 | 1.335(9) | 1.340(9) | Short contacts within the sum of the van-der-Waals radii | ||
| C16–C17 | 1.393(9) | 1.384(11) | Pd1LO2 (3.15 Å) | 3.125(7) | |
| C17–C18 | 1.403(10) | 1.399(12) | Pd2LO6 (3.15 Å) | 3.106(6) | |
| C18–O4 | 1.270(8) | 1.254(9) | O1–Pd1LO2 | 45.21(15) | 45.61(14) |
a(1) and (2) refer to the individual mononuclear units in 32. bThe values of both molecules are shown, with regard to an equivalent position.
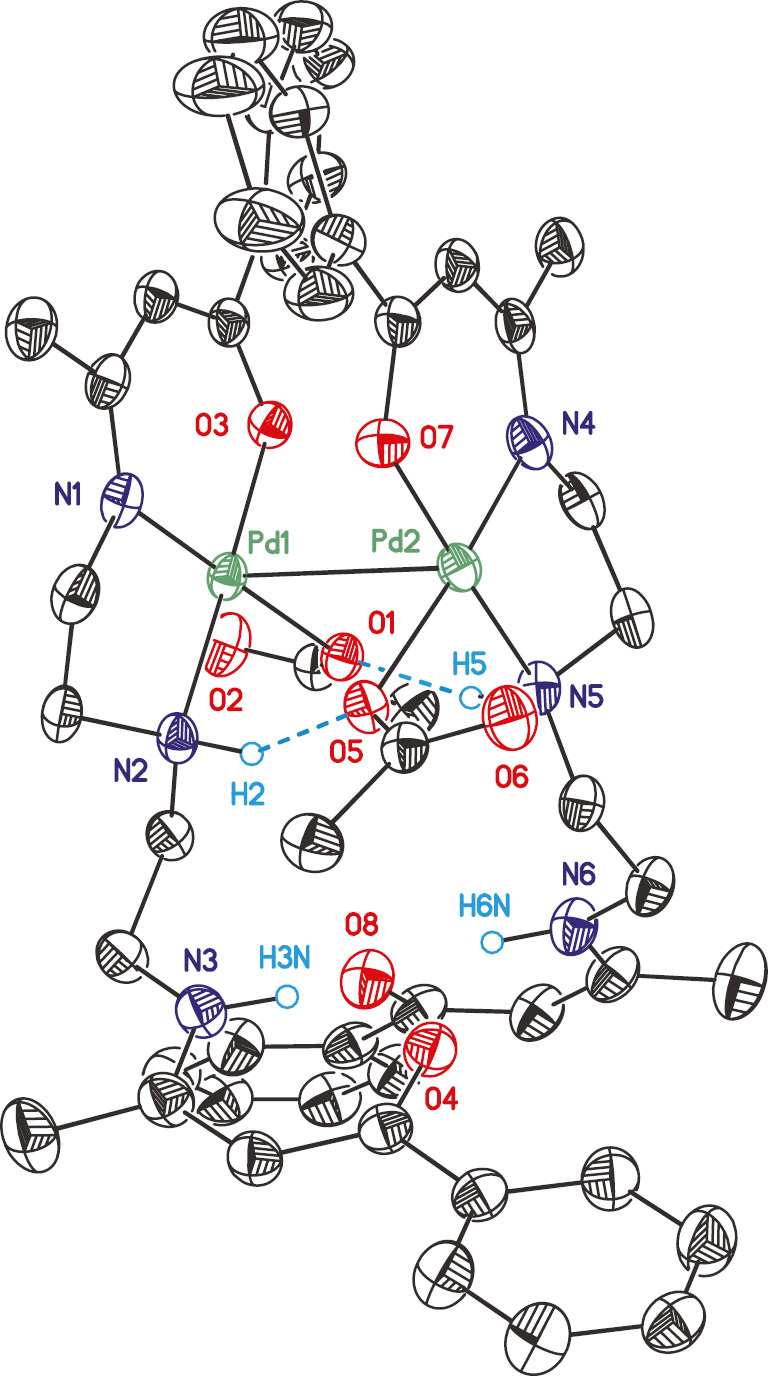
Complex 3 crystallizes in the triclinic space group P1̅ as a dimeric species (32) (Figs. 2–4, Tables 2 and 3). The dimer 32 is formed by hydrogen bonds between the NH functionalities and the acetate ligands (N2LO5 2.838(8) Å, N5LO1 2.882(8) Å) (Table 1, Fig. 2) of the neighboring mononuclear unit resulting in a Pd–Pd distance of 3.1412(7) Å (sum of the van-der-Waals radii: 3.26 Å [46]). Dimeric structures similar to 32 have been reported for [Pd(OPh)2(pyrrolidine)2] [47], [PdCl{2-(3-C12H8-2-O)-6-CH=N(Ar)C5H3N}] (Ar=2,4,6-Me3C6H2, 2,6-iPr2C6H3, 2,4,6-iPr3C6H2) [48], [49] and K[{hydroxyimio-acetate(1-)-O,N}{oxyimino-acetato(2-)-O,N}]palladate(II) [50] with comparable Pd–Pd distances.

Ball and stick view along the b axis. Hydrogen atoms are omitted for clarity (Pd: pale green; O: red; N: blue).
![Fig. 4: Ortep (50% probability level) plot of the core fragment of 32 with the atom numbering scheme of the heteroatoms showing the [4+2] coordination at Pd. All carbon bonded hydrogen atoms, parts of the ligand and the solvent molecules are omitted for clarity. Structure parameters are summarized in Tables 1–3.](/document/doi/10.1515/znb-2019-0172/asset/graphic/j_znb-2019-0172_fig_004.jpg)
Ortep (50% probability level) plot of the core fragment of 32 with the atom numbering scheme of the heteroatoms showing the [4+2] coordination at Pd. All carbon bonded hydrogen atoms, parts of the ligand and the solvent molecules are omitted for clarity. Structure parameters are summarized in Tables 1–3.
Selected hydrogen bond parameters (Å, °) of 32.
| D–HLA | D–H | DLA | D–HLA |
|---|---|---|---|
| N3–H3NLO4 | 0.95(3) | 2.642(9) | 142(5) |
| N6–H6NLO8 | 0.95(5) | 2.637(10) | 149(9) |
| N2–H2LO5 | 0.99(7) | 2.838(8) | 151(6) |
| N5–H5LO1 | 0.99(11) | 2.882(8) | 159(8) |
Dihedral angles around the Pd–Pd bond (Å) of 32.a
| X–Pd–Pd–Y | X–Pd–Pd–X | ||
|---|---|---|---|
| O1LN5 | 13.1(2) | O1LO5 | 106.07(18) |
| N2LO5 | 13.07(19) | N2LN5 | 80.0(2) |
| N1LO7 | 16.2(2) | N1LN4 | 110.7(2) |
| O3LN4 | 15.7(2) | O3LO7 | 78.80(17) |
aX refers to chemically equivalent atoms. Y refers to atoms on top of each other based on a top view.
Each half of 32 shows a square-planar coordination sphere at Pd, whereby L acts as a tri-dendate ligand interacting via one ß-ketoiminato unit and one NH functionality for Pd(II). Together with a mono-dentately bonded acetate a distorted square-planar geometry is set up. Although the exo-C=O moiety of the acetate is -positioned within a distance defined by the sum of the van-der-Waals radii of O and Pd (3.125(7)/3.106(6) Å; Σ=3.15 Å, Fig. 4, Table 1), the O1–Pd1–O2 angles of ~45° strongly deviate from that expected for an octahedral coordination domain. Together with the Pd–Pd interaction, which is also close to the maximum of the van-der-Waals radii, a [4+2] coordination of both Pd atoms in 32 should be considered.
The non-complexed part of L is directed towards the perpendicularly orientated second β-ketoiminato unit in order to avoid steric interactions (Fig. 2). Within the dimer 32, the two ligands L are stacked with opposite directions in a C2 fashion. Thus, the top view shows an eclipsed conformation of all Pd–X bonds (X=N, O) (Fig. 4, Table 2). The thus formed space between the two phenyl groups is occupied by a molecule of hexane, which is present as a guest solvent. The latter was refined disordered over two sets of sites (0.51:0.49). The packing motif of hexane results in a chain-type structure along [010], which is located on the midpoint of the crystallographic c axis. Hence, the 32 dimer is also stacked along [010], whereby adjacent units are orientated in opposite directions (Fig. 3). The non-complexed part of L shields the hexane channel.
According to NMR and IR spectroscopic investigations (vide supra), the non-complexed part of L in 32 crystallizes in the β-keto-enamine form, which is supported by the respective residual electron density allowing for the positioning of all NH hydrogen atoms.
The NH–C=C bond distances of the non-complexed building block of L is shortened from 1.420(10)/1.412(10) Å in a LNLCLCHL moiety (N1/N4) to 1.393(10)/1.384(11) (N3/N6) Å [51] and the C–N bond lengths are increased from 1.300(9)/1.292(9) (N1/N4) to 1.335(9)/1.340(9) Å (N3/N6) [51].
The PdN2O planes in 32 form an angle of 3.8(3)° and are thus almost co-planar (Fig. 4). The Pd–N and Pd–O distances are in agreement with those in other Pd complexes, i.e. in [Pd(OAc){2-(3-C12H8-2-O)-6-(CH=NAr)C5H3N}] (Ar=2,6-iPr2C6H3, 2,4,6-Me3C6H2, 2,4,6-iPr3C6H2) [47], [48], [49]. The Pd–Pd interaction between both fragments in 32 results in a minor shift of 0.019(3)/0.025(3) Å for the Pd atoms out of the N2O planes of the chelating part of L towards each other. The O atom of the Pd-bonded acetate ligand is placed below these planes by 0.241(9)/0.311(9) Å, which is the result of hydrogen bonding with the NH functionality of the -non-coordinated part of L. In order to minimize steric interactions, the acetate CO2 plane is directed away from the core fragment PdN2O by 78.2(5)/83.0(3)° (Table 1, Fig. 4).
2.2 Thermal behavior
To determine the thermal decomposition behavior of 32 (Fig. 5), thermogravimetric (TG) measurements were carried out both in an atmosphere of argon (gas flow of 20 mL min−1) and oxygen (gas flow of 20 mL min−1) in the temperature range of 40–800°C. It has found that 32 decomposes with similar onset temperatures of the first decomposition steps independent of the applied gas (Fig. 5). Additional decomposition steps at >300°C are observed when oxygen was used as reactive gas. The decomposition of 32 under argon produces Pd ([ICDD 00-046-1043]) (Fig. 6, left), while in presence of oxygen PdO ([ICDD 00-041-1107]) is formed as evidenced by PXRD (powder X-ray diffractometry) studies (Fig. 6, right).

TG traces of 32 under an atmosphere of argon (solid line) and oxygen (dashed line) (gas flow, 20 mL min−1, heating rate 10 K min−1).

PXRD patterns of the residues of 32 left in TG experiments under an atmosphere of argon (left) and oxygen (right) showing the characteristic reflections of crystalline Pd and PdO, respectively.
TG-MS (thermogravimetry – mass-spectrometry) studies were carried out on 32 to gain a deeper insight into the decomposition behavior of this complex. The results including the TG trace, its first derivative and the ion current curves of the mass-to-charge ratios (m/z) are depicted in Fig. 7 with assignments given in the caption. It was found that at the beginning of the heating a weight loss of ca. 3% occurred between T=50 and 150°C. Within this step -fragments of m/z=12 (C+), 14 (CH2+N+), 15 (CH3+), 26 (C2H2+, CN+), 27 (C2H3+, HCN+), 42 (C2H4N+, C2H2O+), 43 (C2H5N+, C2H3O+), 56 (C3H6N+) and 57 (C3H7N+, C3H5O+) could be detected indicating C–C and C–N bond cleavages. The next decomposition step with an overall weight loss of 35.8% was observed from an onset of 183°C displaying the fragments m/z=14, 15, 26, 27, 30 and 57. Above 190°C, fragments such as m/z=12 (C+), 14 (CH2+N+), 15 (CH3+), 26 (C2H2+, CN+), 27 (C2H3+, HCN+), 30 (CH4N+, CH2O+), 42 (C2H4N+, C2H2O+), 43 (C2H5N+, C2H3O+), 50 (C4H2+), 51 (C4H3+), 56 (C3H6N+) and 57 (C3H7N+, C3H5O+) were detected for a weight loss of 30.9%. A mass loss of ca. 5% perceived at a temperature >350°C confirming the release of volatile components from the residue. The remaining residue of 25.7% is higher than the calculated one for the formation of Pd (18.2%) signifying the presence of impurities.

TG trace and the 1st derivative (top) and TG-MS traces (bottom) of 32 (atmosphere of argon, gas flow 20 mL min−1; heating rate 5 K min−1; (m/z=12 (C+), 14 (CH2+N+), 15 (CH3+), 26 (C2H2+, CN+), 27 (C2H3+, HCN+), 30 (CH4N+, CH2O+), 42 (C2H4N+, C2H2O+), 43 (C2H5N+, C2H3O+), 50 (C4H2+), 51 (C4H3+), 56 (C3H6N+) and 57 (C3H7N+, C3H5O+).
2.3 Vapor pressure measurements
Vapor pressure measurements were carried out to prove if 32 is suited to be used as a CVD precursor. The methodology used is based on the mass loss of the samples as a function of increasing temperature at atmospheric pressure (Section 4) [52]. A TG system with a horizontal balance was applied to determine the weight loss in an isothermal phase at different temperatures as described previously [52]. To minimize the measurement errors and provide reliable experimental data, each study was carried out thrice.
For the measurements the temperature range of 130–190°C was chosen according to the conditions of the TG studies (Fig. 5). The results are depicted in Fig. 8. For comparison, also the vapor pressure of LH (1) is given [53]. The studies allowed the determination of the Antoine parameters, which are summarized in Table 4.

Vapor pressure traces of 1 and 32 in an atmosphere of nitrogen (40 mL min−1).
Linear regression parameter of the vapor pressure measurements of 1 [53] and 32 as well as their molar enthalpy of evaporation.
| Compd. | log p (bar)=A–B/Ta | ΔHvap (kJ mol−1) | ||
|---|---|---|---|---|
| A | B | R2 | ||
| 1 | 5.28 | 3020 | 0.997 | 57.8 |
| 32 | 5.41 | 3856 | 0.994 | 73.8 |
aA and B=Antoine parameters; T=absolute temperature; R2=coefficient of determination.
From Fig. 8 it can be seen that 32 is characterized by low volatility, with the highest vapor pressure of 1.2 mbar at 190°C. Palladium(II) complexes with bis(β-diketonato) or allyl-(β-diketonato) ligands show a higher volatility at lower temperatures (e.g. 2.22 mbar at 80°C for [Pd(accp)2] [54] (accp=2-acetylcyclopentanoate), 1.4 mbar at 60°C for [Pd(acac)2] [55] or 3.3 mbar at 100°C for [Pd(ƞ3-2-Me-C3H4)(acac)] [26]). In contrast, the copper(II) complex [Cu(μ-OAc)(L)(H2O)]2 reported previously [53] shows a higher vapor pressure than 32.
As a result of the low volatility of 32 this complex was applied only as a spin-coating precursor for the formation of Pd and PdO deposits.
2.4 Spin-coating experiments
The spin-coating process allows the deposition of metal or metal oxide layers by utilizing non-volatile precursors [56], [57]. Hence, complex 32 was applied in the formation of Pd and PdO deposits exploiting this technique. Silicon wafers covered with a native SiO2 layer were used as -substrates for the deposition studies (Section 4). The respective precursor solution was prepared by -dissolving 32 in acetonitrile (0.05 m solutions, Section 4). The spin coater was operated at 3000 rpm for 2 min and the -deposition procedure was repeated thrice. The respective deposition parameters are summarized in Table 5. The as-obtained deposits were then heated in a horizontal tube furnace (Pd, sample A) or in a muffle furnace (PdO, sample B) to 800°C for one (A) or 2 h (B) to leave granulated particles on the substrate surface.
Deposition parameters for Pd and PdO formation by using 32 as spin-coating precursor.
| Sample | t (min) | Conc. (mol L−1) | ΔT (°C) | Holding time (min) | Particle diameter (nm) | Volume (mL) |
|---|---|---|---|---|---|---|
| A | 6 | 0.05 | 800 | 60 | 20±9 | 1.5 |
| B | 6 | 0.05 | 800 | 120 | 130±28 | 1.5 |
2.5 Sample characterization
The morphology and the particle diameter of the as--deposited Pd clusters were studied by using SEM -(scanning electron microscopy) (Fig. 9, Figs. SI2 and SI3, Supporting Information). EDX (energy-dispersive X-ray spectroscopy) (Fig. SI4, Supporting Information) and XPS (X-ray photoelectron spectroscopy) were applied to determine the elemental composition of the respective samples (Figs. SI5 and SI6, Supporting Information).

SEM images of the Pd (sample A; top view, top left) and PdO (B, top view, bottom left and middle) residues and their cross-sectional images (A, top right; B, bottom right) deposited on silicon substrates covered with a native SiO2 layer.
Depending on the annealing conditions, different morphologies of the as-formed deposits were observed (Fig. 9). Sample A, when annealed in an atmosphere of argon, is characterized by spherical particles at the substrate surface with a narrow size distribution (20±9 nm) (Table 5) and with the particles agglomerated to small islands. In contrast, sample B shows aspherical particles spread all over the substrate surface (Fig. 9 and SI3, left) with a higher size distribution (130±28 nm) (Table 5), probably due to the longer decomposition time compared to sample A. Next to the particles, rod like structures were found consisting of agglomerated particles. Such structures are often associated with the formation of composite materials [54]. Comparable palladium nanoparticles (NPs) were received via a CVD process using precursors like [Pd(η3-C3H5)(η5-C5H5)] [13], [Pd(acac)2] [58], [Pd(hfac)2] [59], [60] (hfac=1,1,1,5,5,5-hexafluoro-2,4-pentanedionate), [Pd(η3-2-Me-C3H4)(acch)] or [Pd(η3-2-tBu-C3H4)(accp)] (acch=2-acetylcyclohexanoate; accp=2-acetylcyclopentanoate)) [54]. Roy and coworkers investigated the influence of the electro-less deposition of Pd NPs depending on the surface energy of the substrate [61]. Spherical-shaped particles were found at the substrate surface of Si(100) when a deposition time of 10 min was applied, while increased deposition times of 20 and 60 min led to the formation of aspherical NPs comparable to particles observed for sample B [61].
The chemical composition of the deposits was investigated by EDX spectroscopy, showing signals for Pd as well as carbon, oxygen and silicon (Fig. SI4).
Furthermore, surface sensitive ex situ XPS measurements were performed. All XPS investigations were carried out without and with sputtering of the deposits (Table SI1, see the ESI). The survey XPS spectra are depicted in Figures SI5 and SI6. Since XPS is a surface sensitive measurement method, contaminations on the surface may be overestimated. Therefore, the airborne hydrocarbon impurities and carbon surface contaminations of the precursor molecules on the surface were removed by argon sputtering (4 keV, for 60 min).
As expected, the elemental composition of the original and sputtered substrates of samples A and B differs (Table SI1, ESI), but for both deposits the palladium content increased up to approximately 1.6 at% and no carbon was detected after sputtering. Sample A exhibits a higher silicon content compared to sample B, but B also shows a significantly higher amount of oxygen after sputtering, indicating the presence of PdO and silicon oxide formation.
3 Conclusion
As a potential spin-coating precursor for the deposition of Pd and PdO, complex [Pd(OAc)L] (3) (L =PhC(O)CHC(Me)NCH2CH2NHCH2CH2NH(Me)C=CHC(O)Ph) was synthesized by the reaction of bis(benzoylacetone)diethylene--triamine (1, =LH) with [Pd(OAc)2] (2), whereby only one of the two β-ketoiminato functionalities in L is coordinated to the Pd(II) ion. Complex 3 has been structurally characterized by single-crystal X-ray diffraction analysis. In the solid state a dimeric species (32) exists in which two molecules are connected via hydrogen bonds between the NH functionalities and the acetate ligands complemented by a Pd–Pd distance of 3.1412(7) Å. TG studies have indicated that the decomposition with the highest weight loss of 32 occurs between 200 and 500°C independent of the gas used (argon and oxygen). PXRD investigations of the as-obtained residues confirmed the formation of Pd in an inert gas atmosphere and of PdO when oxygen as reactive gas was applied. TG-MS measurements of 32 reveal a multi-step decomposition starting at 50°C. The detection of -fragments such as m/z=12, 14, 15, 26, 27, 30, 42, 43, 50, 51, 56 and 57 approve Pd–O, Pd–N, C–N and C–C bond cleavages. The vapor pressure of 32 was determined to 1.2 mbar at 190 °C, much lower than conventional palladium(II) complexes used for Pd CVD [26], [54], [55]. Hence, 32 was applied only as a spin-coating precursor for the deposition of Pd (sample A) and PdO (sample B). The as-deposited samples show separated particles on the substrate surface. The appearance of the particles depends on the annealing atmosphere (Ar or air). Deposit A treated in argon shows spherical particles with a size distribution of 20±9 nm, while sample B treated in air shows aspherical particles of size 130±28 nm. Similar -morphologies were obtained, when precursors like [Pd(η3-C3H5)(η5-C5H5)] [13], [Pd(acac)2] [58], [Pd(hfac)2] [59], [60] or [Pd(η3-2-Me-C3H4)(β-diketonate)] (β-diketonate=acch, accp) [54] (acac=acetylacetonate, hfac=1,1,1,5,5,5-hexafluoro-2,4-pentanedionate, acch=2-acetylcyclohexanoate, accp=2-acetylcyclopenta-noate) were used in CVD processes. XPS studies of samples A and B showed no carbon impurities after sputtering.
4 Experimental section
4.1 General procedure
All chemicals were purchased from commercial suppliers and were used without any further purification. Bis(benzoylacetone)diethylenetriamine (1) was synthesized according to a previously published method [36].
4.2 Instruments
NMR spectra (500.3 MHz for 1H, 125.7 MHz for 13C{1H}) were recorded using a Bruker Avance III 500 FT-NMR spectrometer at ambient temperature. Chemical shifts are reported in ppm downfield from tetramethylsilane with the solvent as reference signal (1H NMR, δ (CDCl3) 7.26 ppm; 13C{1H} NMR, δ (CDCl3) 77.16 ppm). The infrared spectrum was recorded with a Thermo Nicolet 200 FT-IR spectrometer. Elemental analysis was performed with a Thermo FLASHEA 1112 Series instrument. The high-resolution mass spectrum was recorded with a Bruker Daltonik micrOTOF-QII mass spectrometer performing in the ESI mode. The melting point was determined by using a Gallenkamp MFB 595 010 M melting point apparatus. Vapor pressure measurements were performed with a Mettler Toledo TGA/DSC1 1100 system with a UMX1 balance. The TG and TG-MS experiments were performed with a Mettler Toledo TGA/DSC1 1600 system with a MX1 balance coupled with a Pfeifer Vacuum MS Thermostar GSD 301 T2 mass spectrometer. PXRD measurements of the respective TG residues were done with a STOE-STADI P diffractometer equipped with a Ge(111) monochromator and CuKα radiation (λ=1.540598 Å, 40 kV, 40 mA). The layer formation experiments were carried out using the spin coater ws-650. The surface morphology and cross-sectional details were studied by field-emission scanning electron microscopy using a ZEISS Supra60 SEM. Energy-dispersive X-ray analysis using a Bruker Quantax 400 system attached to a SEM was applied to determine the chemical composition of the samples. The composition of the samples was investigated using a PREVAC XPS system. Monochromatic AlKα radiation (1486.6 eV) was provided by a VG Scienta MX 650 X-ray source and a monochromator. The energy distribution of the photoelectrons was measured using a VG Scienta EW3000 analyzer, which was operated at 1 eV step size and 1.0 s measurement time at each point for survey spectra. The Casa XPS 2.3.16 Pre-rel 1.4 software was used for the deconvolution of the XPS peaks. For the calculation of the atomic concentration, Scofield relative sensitivity factors (RSFs) were used. These RSFs were corrected for a monochromator-analyzer angle of 52.55°. For the escape depth correction in Casa XPS, a value of −0.75 was applied.
4.3 Synthesis of [Pd(OAc)L]2 (32)
Palladium(II) acetate (2) (0.224 g, 1 mmol) was dissolved in 50 mL of acetonitrile and 0.39 g (1 mmol) of bis(benzoylacetone)diethylenetriamine (1, =LH) was added in a single portion at ambient temperature. The reaction mixture was stirred at this temperature for 16 h. Afterwards, all volatiles were removed in vacuum (1 mbar). The residue was dissolved in 20 mL of dichloromethane and the solution added dropwise to 100 mL of hexane. The precipitate was collected by filtration. After recrystallization from dichloromethane at ambient temperature, complex 32 could be isolated in form of yellow needles. Yield: 0.426 g (0.077 mmol, 77% based on 1). M. p. 166°C (decomposition). –Analysis calcd for C52H62N6O8Pd2 (1110.26 g mol−1): C 56.17, H 5.62, N 7.56; found C 55.74, H 5.64, N 7.62. –IR data (KBr, cm−1): ν=3402 (w), 3191 (w), 3178 (w), 3095 (w), 3054 (w), 2965 (w), 2928 (w), 2877 (w), 1560 (s), 1508 (s), 1484 (m), 1458 (m), 1433 (m), 1407 (m), 1386 (m), 1347 (m), 1319 (w), 1292 (m), 1281 (w), 1260 (w), 1178 (w), 1179 (w), 1157 (w), 1143 (w), 1124 (w), 1064 (m), 1022 (w), 987 (w), 781 (m), 708 (s), 668 (m). –1H NMR (CDCl3): δ=2.08 (s, 6H, NCCH3), 2.09 (s, 6H, PdNCCH3), 2.16 (s, 6H, O2CCH3), 2.61–2.68 (m, 2H, CH2), 2.89–2.98 (m, 2H, CH2), 3.13–3.21 (m, 2H, CH2), 3.32–3.40 (m, 2H, CH2), 3.44–3.54 (m, 2H, CH2), 3.57–3.65 (m, 2H, CH2), 3.72–3.82 (m, 2H, CH2), 3.92–4.03 (m, 2H, CH2), 5.53 (s, 2H, CHCO), 5.71 (s, 2H, CHCOPd), 7.28–7.40 (m, 14H, mCH, pCH, C6H5; NH), 7.73–7.75 (m, 4H, oCH, C6H5), 7.80–7.85 (m, 4H, oCH, C6H5), 11.51 (t, 3JHH=5.6 Hz, 2H, NH) ppm. –13C NMR (CDCl3): δ=19.6 (s, O2CCH3), 21.1 (s, 3H, NCCH3), 24.3 (s, PdNCCH3), 41.7 (s, CH2), 51.5 (s, CH2), 54.3 (s, CH2), 64.0 (s, CH2), 93.1 (s, CHCOPd), 98.0 (s, CHCOH), 127.1 (s, oC, C6H5), 127.2 (s, oC, C6H5), 128.2 (s, mC, pC, C6H5), 128.4 (s, mCH, pCH, C6H5), 129.9 (s, mC, pC, C6H5), 130.8 (s, mC, pC, C6H5), 138.0 (s, iC, C6H5) 140.3 (s, iC, C6H5) 163.1 (s, NCCH3) 165.0 (s, PdNCCH3) 172.1 (s, CHCOH) 180.8 (s, CHCOPd) 188.5 (s, O2CCH3) ppm. –HRMS (ESI-TOF): m/z=496.1351 (calcd. 496.1220 for C24H28N3O2Pd], [M–OAc]+).
4.4 Single-crystal X-ray diffraction analysis of 32
Diffraction data was collected with an Oxford Gemini S diffractometer using graphite-monochromatized CuKα radiation (λ=1.54184 Å) at T=120 K with an oil-coated shock-cooled crystal (Table 6). The structure was solved by Direct Methods and refined by full-matrix least squares procedures on F2 [62], [63], [64]. All non-hydrogen atoms were refined anisotropically, and a riding model was employed in the refinement of the hydrogen atom positions. Graphics of the molecular structures were created by using Shelxtl and Ortep [65].
CCDC 1939768 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystallographic data for 32.
| Empiric formula | C58H76Pd2N6O8 |
| Mr | 1198.04 |
| Size, mm3 | 0.38×0.14×0.14 |
| Crystal system | Triclinic |
| Space group | P1̅ |
| a, Å | 14.9440(15) |
| b, Å | 15.4460(17) |
| c, Å | 15.4548(17) |
| α, ° | 62.757(11) |
| β, ° | 62.965(11) |
| γ, ° | 66.799(10) |
| V, Å3 | 2746.1(6) |
| Dcalcd g cm−3 | 1.45 |
| Z | 2 |
| μ(CuKα), mm−1 | 5.8 |
| F(000), e | 1244.0 |
| hkl range | ±17, ±18, ±18 |
| T, K | 120 |
| θ range, ° | 3.311–64.998 |
| Measured reflections | 15 160 |
| Independent reflections | 9179 |
| Rint | 0.0273 |
| R1/wR2, (I>2 σ(I)) | 0.0695/0.1916 |
| R(F)/wR(F2)a (all reflexions) | 0.0828/0.2035 |
| Δρfin (max/min), e Å−3 | 2.52/−1.54 |
| CCDC no. | 1939768 |
aR(F)=||Fo|–|Fc||/Σ|Fo|, wR(F2)=[Σw(Fo2–Fc2)2/Σw(Fo2)2]1/2, w=[σ2(Fo2)+(AP)2+BP]−1, where P=(Max(Fo2, 0)+2Fc2)/3.
4.5 Spin coating experiments
For the spin-coating experiments 2 mL of a 0.05 m solution of 32 (111 mg) in acetonitrile was prepared. The deposition experiments were repeated thrice. The respective substrates of size 20 mm×20 mm were cleaned with ethanol by bath sonication and were afterwards air dried at ambient temperature.
5 Supporting information
Further data including the IR spectrum of 32, SEM images, EDX and XPS spectra of the samples is given as supplementary material available online (DOI: 10.1515/znb-2019-0172).
Acknowledgement
This study has received funding from the European Social Fund (ESF). We thank Ute Stöß for carrying out elemental analysis, Cornelia Kowol for performing the SEM/EDX measurements and Natalia Rüffer for TG and vapor pressure measurements and acknowledge the help of Lutz Mertens, Marcus Weber and Prof. Dr. Michael Mehring in performing the PXRD measurements.
References
[1] E. Czerwosz, E. Kowalska, M. Kozłowski, J. Radomska, H. Wronka, M. Angiola, A. Martucci, W. Włodarski, Procedia Eng.2014, 87, 963–966.10.1016/j.proeng.2014.11.318Suche in Google Scholar
[2] J. H. Yoon, B. J. Kim, J. S. Kim, Mater. Chem. Phys.2012, 133, 987–991.10.1016/j.matchemphys.2012.02.002Suche in Google Scholar
[3] H. Zhong, Z. Zhang, B. Chen, H. Xu, D. Yu, L. Huang, L. Peng, Nano Res.2015, 8, 1669–1679.10.1007/s12274-014-0656-zSuche in Google Scholar
[4] K. Sato, M. Natsui, Y. Hasegawa, Mater. Trans.2014, 56, 473–478.10.2320/matertrans.MF201402Suche in Google Scholar
[5] C. Gäbler, M. Korb, D. Schaarschmidt, J. Matthäus Speck, A. Hildebrandt, H. Lang, Inorg. Chem. Commun.2015, 54, 96–99.10.1016/j.inoche.2015.02.018Suche in Google Scholar
[6] A. Jakob, B. Milde, P. Ecorchard, C. Schreiner, H. Lang, J. Organomet. Chem.2008, 693, 3821–3830.10.1016/j.jorganchem.2008.09.040Suche in Google Scholar
[7] J. Kühnert, M. Lamač, J. Demel, A. Nicolai, H. Lang, P. Štěpnička, J. Mol. Catal. A-Chem.2008, 285, 41–47.10.1016/j.molcata.2008.01.026Suche in Google Scholar
[8] M. Lohan, B. Milde, S. Heider, J. M. Speck, S. Krauße, D. Schaarschmidt, T. Rüffer, H. Lang, Organometallics2012, 31, 2310–2326.10.1021/om201220wSuche in Google Scholar
[9] B. Milde, M. Lohan, C. Schreiner, T. Rüffer, H. Lang, Eur. J. Inorg. Chem.2011, 2011, 5437–5449.10.1002/ejic.201100842Suche in Google Scholar
[10] C. Gäbler, J. Jeschke, G. Nurgazina, S. Dietrich, D. Schaarschmidt, C. Georgi, M. Schlesinger, M. Mehring, H. Lang, Catal. Lett.2013, 143, 317–323.10.1007/s10562-013-0967-9Suche in Google Scholar
[11] A. M. Trzeciak, A. W. Augustyniak, Coord. Chem. Rev.2019, 384, 1–20.10.1016/j.ccr.2019.01.008Suche in Google Scholar
[12] P. Serp, P. Kalck, R. Feurer, Chem. Rev.2002, 102, 3085–3128.10.1021/cr9903508Suche in Google Scholar
[13] M. Zhang, Y. Gao, C. Li, C. Liang, Chin. J. Catal.2015, 36, 588–594.10.1016/S1872-2067(14)60292-8Suche in Google Scholar
[14] N. T. S. Phan, M. Van Der Sluys, C. W. Jones, Adv. Synth. Catal.2006, 348, 609–679.10.1002/adsc.200505473Suche in Google Scholar
[15] X. Chen, K. M. Engle, D. H. Wang, Y. Jin-Quan, Angew. Chem. Int. Ed.2009, 48, 5094–5115.10.1002/anie.200806273Suche in Google Scholar PubMed PubMed Central
[16] S. Shrestha, E. J. Biddinger, Electrochim. Acta2015, 174, 254–263.10.1016/j.electacta.2015.05.164Suche in Google Scholar
[17] Y. Wang, W. Li, W. Wang, N. Mitsuzak, W. Bao, Z. Chen, Thin Solid Films2015, 586, 35–40.10.1016/j.tsf.2015.04.038Suche in Google Scholar
[18] A. M. Saleem, S. Shafiee, T. Krasia-Christoforou, I. Savva, G. Göransson, V. Desmaris, P. Enoksson, Sci. Technol. Adv. Mater.2015, 16, 015007.10.1088/1468-6996/16/1/015007Suche in Google Scholar PubMed PubMed Central
[19] K. B. Manjunatha, R. Dileep, G. Umesh, B. Ramachandra Bhat, Mater. Lett.2013, 105, 173–176.10.1016/j.matlet.2013.03.076Suche in Google Scholar
[20] H. Lang, D. Adner, C. Georgi, in PATAI’s Chemistry of Functional Groups, Metal Enolates (Ed.: I. Marek), Wiley, Chichester, 2016, pp. 1–28.10.1002/9780470682531.pat0901Suche in Google Scholar
[21] H. Lang, S. Dietrich, in Comprehensive Inorganic Chemistry II, From Elements to Applications, 2nd edition, Vol. 4 (Eds.: J. Reedijk, K. Poeppelmeier), Elsevier Ltd., Amsterdam, 2013, pp. 211–269.10.1016/B978-0-08-097774-4.00412-5Suche in Google Scholar
[22] H. Lang, R. Buschbeck, in PATAI’s Chemistry of Functional Groups (Ed.: Z. Rappoport), The Chemistry of Metal Enolates, (Ed.: J. Zabicky), Wiley, Chichester, 2009, pp. 929–1017.Suche in Google Scholar
[23] D. N. Goldstein, S. M. George, Appl. Phys. Lett.2009, 95, 143106.10.1063/1.3238558Suche in Google Scholar
[24] X. Zhou, Y. Zhang, Z. Dong, S. Liu, C. Zhang, B. Huang, K. Cao, B. Shan, R. Chen, Mater. Res. Soc. Symp. Proc.2013, 1548, 7–12.10.1557/opl.2013.847Suche in Google Scholar
[25] T. Aaltonen, M. Ritala, Y. L. Tung, Y. Chi, K. Arstila, K. Meinander, M. Leskelä, J. Mater. Res.2004, 19, 3353–3358.10.1557/JMR.2004.0426Suche in Google Scholar
[26] J.-C. Hierso, R. Feurer, P. Kalck, Coord. Chem. Rev.1998, 178–180, 1811–1834.10.1016/S0010-8545(98)00161-1Suche in Google Scholar
[27] B. Henc, P. W. Jolly, R. Salz, S. Stobbe, G. Wilke, R. Benn, R. Mynott, K. Seevogel, R. Goddard, C. Krüger, J. Organomet. Chem.1980, 191, 449–475.10.1016/S0022-328X(00)81073-6Suche in Google Scholar
[28] J. E. Gozum, D. M. Pollina, J. A. Jensen, G. S. Girolami, J. Am. Chem. Soc.1988, 110, 2688–2689.10.1021/ja00216a073Suche in Google Scholar
[29] Z. Yuan, R. J. Puddephatt, Adv. Mater.1996, 6, 51–54.10.1002/adma.19940060109Suche in Google Scholar
[30] Y. H. Liu, Y. C. Cheng, Y. L. Tung, Y. Chi, Y. L. Chen, C. S. Liu, S. M. Peng, G. H. Lee, J. Mater. Chem.2003, 13, 135–142.10.1039/B208535FSuche in Google Scholar
[31] G. I. Zharkova, P. A. Stabnikov, I. A. Baidina, A. I. Smolentsev, S. V. Tkachev, Polyhedron2009, 28, 2307–2312.10.1016/j.poly.2009.02.043Suche in Google Scholar
[32] G. I. Zharkova, S. V. Sysoev, P. A. Stabnikov, V. A. Logvinenko, I. K. Igumenov, J. Therm. Anal. Calorim.2011, 103, 381–385.10.1007/s10973-010-0949-8Suche in Google Scholar
[33] E. S. Vikulova, S. A. Cherkasov, N. S. Nikolaeva, A. I. Smolentsev, S. V. Sysoev, N. B. Morozova, J. Therm. Anal. Calorim.2019, 135, 2573–2582.10.1007/s10973-018-7371-zSuche in Google Scholar
[34] Y.-L. Tung, W.-C. Tseng, C.-Y. Lee, P.-F. Hsu, Y. Chi, S.-M. Peng, G.-H. Lee, Organometallics1999, 18, 864–869.10.1021/om980728cSuche in Google Scholar
[35] I. Giebelhaus, R. Müller, W. Tyrra, I. Pantenburg, T. Fischer, S. Mathur, Inorg. Chim. Acta2011, 372, 340–346.10.1016/j.ica.2011.02.052Suche in Google Scholar
[36] A. A. Ensafi, S. Meghdadi, A. R. Allafchian, IEEE Sensors J.2008, 8, 248–254.10.1109/JSEN.2007.913146Suche in Google Scholar
[37] J. Cisterna, V. Artigas, M. Fuentealba, P. Hamon, C. Manzur, V. Dorcet, J. R. Hamon, D. Carrillo, Inorg. Chim. Acta2017, 462, 266–280.10.1016/j.ica.2017.04.001Suche in Google Scholar
[38] J. Cisterna, V. Dorcet, C. Manzur, I. Ledoux-Rak, J.-R. Hamon, D. Carrillo, Inorg. Chim. Acta2015, 430, 82–90.10.1016/j.ica.2015.02.030Suche in Google Scholar
[39] M. Fuentealba, J. R. Hamon, D. Carrillo, C. Manzur, New J. Chem.2007, 31, 1815–1825.10.1039/b707934fSuche in Google Scholar
[40] M. Fuentealba, A. Trujillo, J.-R. Hamon, D. Carrillo, C. Manzur, J. Mol. Struct.2008, 881, 76–82.10.1016/j.molstruc.2007.08.030Suche in Google Scholar
[41] G. Ahumada, J. Oyarce, T. Roisnel, S. Kahlal, M. A. Del Valle, D. Carrillo, J. Y. Saillard, J. R. Hamon, C. Manzur, New J. Chem.2018, 42, 19294–19304.10.1039/C8NJ04923HSuche in Google Scholar
[42] G. Ahumada, M. Fuentealba, T. Roisnel, S. Kahlal, D. Carrillo, R. Cordova, J.-Y. Saillard, J.-R. Hamon, C. Manzur, Polyhedron2018, 151, 279–286.10.1016/j.poly.2018.05.048Suche in Google Scholar
[43] S. Celedon, M. Fuentealba, T. Roisnel, J. R. Hamon, D. Carrillo, C. Manzur, Inorg. Chim. Acta2012, 390, 184–189.10.1016/j.ica.2012.04.028Suche in Google Scholar
[44] M. Á. Gaona, F. Montilla, E. Álvarez, A. Galindo, Dalton Trans.2015, 44, 6516–6525.10.1039/C5DT00358JSuche in Google Scholar
[45] J. V Greenhill, Chem. Soc. Rev.1977, 6, 277–294.10.1039/cs9770600277Suche in Google Scholar
[46] A. Bondi, J. Phys. Chem.1964, 68, 441–451.10.1021/j100785a001Suche in Google Scholar
[47] P. L. Alsters, P. J. Baesjou, M. D. Janssen, H. Kooijman, A. Sicherer-Roetman, A. L. Spek, G. Van Kotten, Organometallics1992, 11, 4124–4135.10.1021/om00060a032Suche in Google Scholar
[48] O. Adeyi, W. B. Cross, G. Forrest, L. Godfrey, E. G. Hope, A. McLeod, A. Singh, K. Singh, G. A. Solan, Y. Wang, L. A. Wright, Dalton Trans.2013, 42, 7710–7723.10.1039/c3dt50176kSuche in Google Scholar PubMed
[49] W. B. Cross, E. G. Hope, G. Forrest, K. Singh, G. A. Solan, Polyhedron2013, 59, 124–132.10.1016/j.poly.2013.04.049Suche in Google Scholar
[50] N. I. Dodoff, M. Kubiak, J. Kuduk-Jaworska, A. Mastalarz, A. Kochel, V. Vassilieva, N. Vassilev, N. Trendafilova, I. Georgieva, M. Lalia-Kantouri, M. Apostolova, Chemija2009, 20, 208–217.Suche in Google Scholar
[51] Although in some cases the high value of the standard deviations does not allow for a comparison, a trend can be observed. The observations are supported by the above--mentioned spectroscopic results.Suche in Google Scholar
[52] A. Tuchscherer, C. Georgi, N. Roth, D. Schaarschmidt, T. Rüffer, T. Waechtler, S. E. Schulz, S. Oswald, T. Gessner, H. Lang, Eur. J. Inorg. Chem.2012, 30, 4867–4876.10.1002/ejic.201200601Suche in Google Scholar
[53] A. Preuß, M. Korb, T. Rüffer, J. Bankwitz, C. Georgi, A. Jakob, S. E. Schulz, H. Lang, Z. Anorg. Allg. Chem.2019, submitted.Suche in Google Scholar
[54] K. Assim, M. Melzer, M. Korb, T. Rüffer, A. Jakob, J. Noll, C. Georgi, S. E. Schulz, H. Lang, RSC Adv.2016, 6, 102557–102569.10.1039/C6RA22887ASuche in Google Scholar
[55] J. R. Vargas Garcia, T. Goto, Mater. Trans.2003, 44, 1717–1728.10.2320/matertrans.44.1717Suche in Google Scholar
[56] E. Pousaneh, A. Preuß, K. Assim, J. Noll, A. Jakob, T. Rüffer, H. Lang, J. Rare Earth2017, 35, 1248–1254.10.1016/j.jre.2017.08.002Suche in Google Scholar
[57] P. Frenzel, A. Preuß, J. Bankwitz, C. Georgi, F. Ganss, L. Mertens, S. E. Schulz, O. Hellwig, M. Mehring, H. Lang, RSC Adv.2019, 9, 10657–10669.10.1039/C9RA00585DSuche in Google Scholar PubMed PubMed Central
[58] F. E. Annanouch, Z. Haddi, M. Ling, F. Di Maggio, S. Vallejos, T. Vilic, Y. Zhu, T. Shujah, P. Umek, C. Bittencourt, C. Blackman, E. Llobet, ACS Appl. Mater. Interf.2016, 8, 10413–10421.10.1021/acsami.6b00773Suche in Google Scholar PubMed
[59] L. Wang, G. L. Griffin, J. Electrochem. Soc.2007, 154, D151–D155.10.1149/1.2430648Suche in Google Scholar
[60] B. V. Bhaskaran, M. J. Hampden-Smith, T. Kodas, Chem. Vap. Dep.1997, 3, 281–286.10.1002/cvde.19970030506Suche in Google Scholar
[61] A. Roy, S. S. Singha, S. Majumder, A. Singha, S. Banerjee, B. Satpati, ACS Appl. Nano Mater.2019, 2, 2503–2514.10.1021/acsanm.9b00420Suche in Google Scholar
[62] G. M. Sheldrick, Acta Crystallogr.1990, A46, 467–473.10.1107/S0108767390000277Suche in Google Scholar
[63] G. M. Sheldrick, Shelxl-97, Program for Crystal Structure Refinement, Universität Göttingen, Göttingen (Germany) 1997.Suche in Google Scholar
[64] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112–122.10.1107/S0108767307043930Suche in Google Scholar PubMed
[65] L. J. Farrugia, J. Appl. Crystallogr.2012, 45, 849–854.10.1107/S0021889812029111Suche in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/znb-2019-0172).
©2019 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Electron densities of two cyclononapeptides from invariom application
- Crystal structures, Hirshfeld surface analysis and Pixel energy calculations of three trifluoromethylquinoline derivatives: further analyses of fluorine close contacts in trifluoromethylated derivatives
- Synthesis and antifungal activities of 3-substituted phthalide derivatives
- Unexpected isolation of a cyclohexenone derivative
- Preparation and structure of 4-(dimethylamino)thiopivalophenone – intermolecular interactions in the crystal
- A new binuclear NiII complex with tetrafluorophthalate and 2,2′-bipyridine ligands: synthesis, crystal structure and magnetic properties
- Two mononuclear zinc(II) complexes constructed by two types of phenoxyacetic acid ligands: syntheses, crystal structures and fluorescence properties
- Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
- Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
- Crystal structure and photocatalytic degradation properties of a new two-dimensional zinc coordination polymer based on 4,4ʹ-oxy-bis(benzoic acid)
- Intermetallics of the types REPd3X2 and REPt3X2 (RE=La–Nd, Sm, Gd, Tb; X=In, Sn) with substructures featuring tin and In atoms in distorted square-planar coordination
- A 119Sn Mössbauer-spectroscopic characterization of the diamagnetic birefringence material Sn2B5O9Cl
- Synthesis, crystal structure and photoluminescence of the salts Cation+ [M(caffeine)Cl]− with Cation+=NnBu4+, AsPh4+ and M==Zn(II), Pt(II)
- Synthesis and characterization of two bifunctional pyrazole-phosphonic acid ligands
- A β-ketoiminato palladium(II) complex for palladium deposition
- Orthoamide und Iminiumsalze, XCVIa. Push-pull-substituierte 1,3,5-Hexatriene aus Orthoamiden von Alkincarbonsäuren und Birckenbach-analogen Acetophenonen
- Orthoamide und Iminiumsalze, IIICa. Weitere Ergebnisse bei der Umsetzung von Orthoamiden der Alkincarbonsäuren mit CH2- und CH2/NH-aciden Verbindungen
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Electron densities of two cyclononapeptides from invariom application
- Crystal structures, Hirshfeld surface analysis and Pixel energy calculations of three trifluoromethylquinoline derivatives: further analyses of fluorine close contacts in trifluoromethylated derivatives
- Synthesis and antifungal activities of 3-substituted phthalide derivatives
- Unexpected isolation of a cyclohexenone derivative
- Preparation and structure of 4-(dimethylamino)thiopivalophenone – intermolecular interactions in the crystal
- A new binuclear NiII complex with tetrafluorophthalate and 2,2′-bipyridine ligands: synthesis, crystal structure and magnetic properties
- Two mononuclear zinc(II) complexes constructed by two types of phenoxyacetic acid ligands: syntheses, crystal structures and fluorescence properties
- Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
- Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
- Crystal structure and photocatalytic degradation properties of a new two-dimensional zinc coordination polymer based on 4,4ʹ-oxy-bis(benzoic acid)
- Intermetallics of the types REPd3X2 and REPt3X2 (RE=La–Nd, Sm, Gd, Tb; X=In, Sn) with substructures featuring tin and In atoms in distorted square-planar coordination
- A 119Sn Mössbauer-spectroscopic characterization of the diamagnetic birefringence material Sn2B5O9Cl
- Synthesis, crystal structure and photoluminescence of the salts Cation+ [M(caffeine)Cl]− with Cation+=NnBu4+, AsPh4+ and M==Zn(II), Pt(II)
- Synthesis and characterization of two bifunctional pyrazole-phosphonic acid ligands
- A β-ketoiminato palladium(II) complex for palladium deposition
- Orthoamide und Iminiumsalze, XCVIa. Push-pull-substituierte 1,3,5-Hexatriene aus Orthoamiden von Alkincarbonsäuren und Birckenbach-analogen Acetophenonen
- Orthoamide und Iminiumsalze, IIICa. Weitere Ergebnisse bei der Umsetzung von Orthoamiden der Alkincarbonsäuren mit CH2- und CH2/NH-aciden Verbindungen