Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
-
Hai-Wei Kuai
 ,
Ding-Yun Jiang
,
Ding-Yun Jiang

Abstract
1,3,5-Tri(1H-imidazol-4-yl)benzene (H3L) reacts with Ni(ClO4)2 · 6H2O under hydrothermal conditions at T = 100°C to yield a new complex: [Ni(H2L)2] · 5.5H2O (1). The product is characterized by single crystal and powder X-ray diffraction, infrared spectroscopy, and elemental and thermogravimetric analyses. Complex 1 crystallizes in the monoclinic space group C2/c and exhibits a binodal (3,6)-connected 2D kgd network structure with (43)2(46.66.83) topology.
1 Introduction
In the past decades, coordination polymers have attracted increasing attention due to their diverse structures as well as potential applications in many fields such as heterogeneous catalysis, ion-recognition, nonlinear optics, chemical absorption, electronic conductivity, and magnetism [1], [2], [3]. Therefore, many coordination polymers were prepared and reported [4], [5], [6]. The strategies for the assembly of coordination architectures involve the deliberate design of organic building blocks and the employment of suitable metal centers [7], [8]. The functional properties of complexes are thus largely dependent on the choice of the metal centers, the bridging ligands, and their architectures [9], and thus, the assembly of coordination compounds with variable structures seems to be important for the exploration of crystalline materials. Extensive research was carried out to manage influential factors such as acidic or basic media for the reaction system, solvent, and temperature as well as the ratio of metal-to-ligand for structural diversity [10], [11], [12]. Among the above-mentioned influential factors, the nature of the organic ligand was documented as crucial for the formation of supramolecular coordination compounds [13]. It is proven that N- or O-donor multidentate ligands, such as imidazolyl or carboxylate containing ligands are excellent candidates for building blocks for the construction of desirable frameworks [14].
Based on the above consideration, we have previously carried out research on N- and/or O-donor ligands such as 5-(1H-benzotriazol-1-ylmethyl)isophthalic acid, 5-(benzimidazol-1-ylmethyl)isophthalic acid, and 3,5-bis(2-pyridylmethyl)aminobenzoic acid [15]. In order to further explore the correlation between external reaction conditions and the structure of the resultant complexes, we selected the rigid tripodal ligand 1,3,5-tri(1H-imidazol-4-yl)benzene (H3L, Scheme 1) as an organic building block. The H3L is a multidentate N-donor ligand and may be expected to assemble complexes with porous frameworks in the presence of other anion ligands such as SCN− and N3− [16]. Moreover, this kind of ligand contains the 1H-imidazol-4-yl group, which maybe deprotonated in basic condition to give an imidazolate for adaptive building units, as demonstrated by zeolitic imidazolate framework materials [17]. We report herein the synthesis and structural characterization of [Ni(H2L)2]·5.5H2O (1).
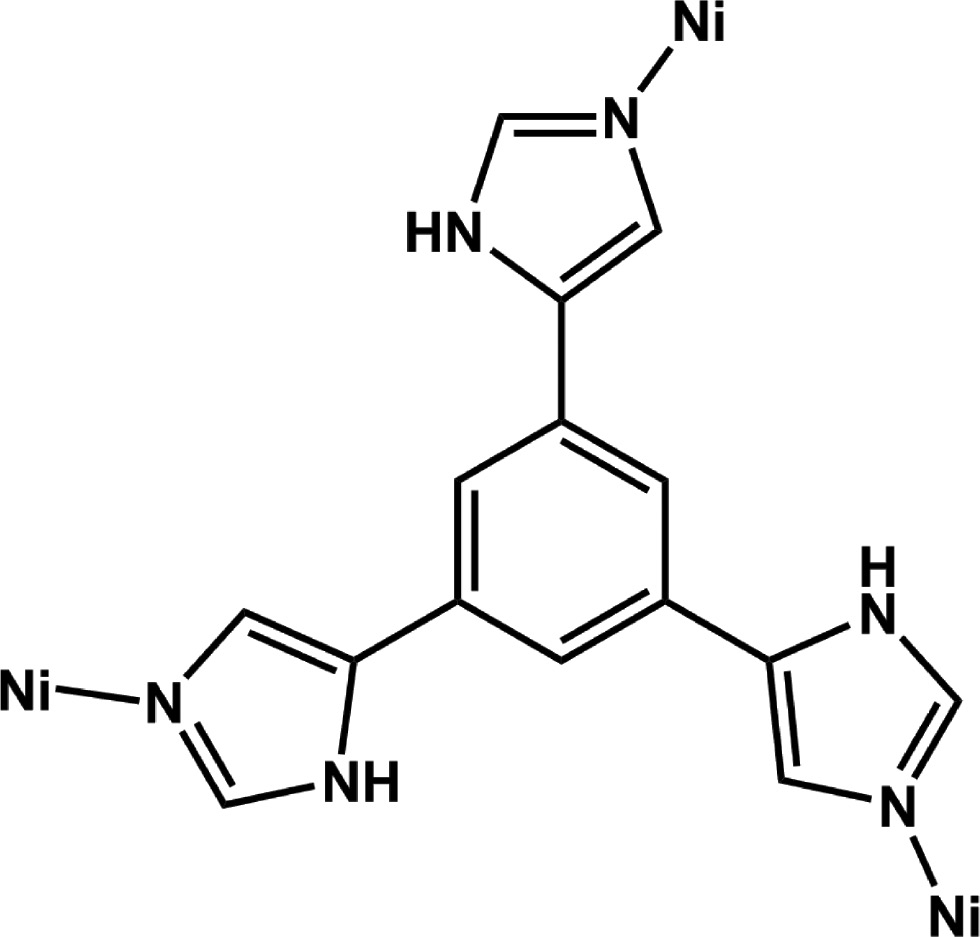
Coordination mode of H3L appearing in complex 1.
2 Results and discussion
2.1 Preparation
Ni(ClO4)2·6H2O reacts with 1,3,5-tri(1H-imidazol-4-yl)benzene(H3L) in the presence of ammonium hydroxide under hydrothermal conditions at T=100°C to yield the complex [Ni(H2L)2]·5.5H2O (1) which is stable in air.
2.2 Structural description of [Ni(H2L)2]·5.5H2O (1)
Single crystal X-ray structural analysis revealed that 1 crystallizes in the monoclinic space group C2/c (Table 1), exhibiting a 2D network structure. As shown in Fig. 1a with the atom numbering scheme, the asymmetric unit of 1 contains one half of a Ni2+ cation (located on an inversion center), and one H2L− ligand as counter anion. The H3L was deprotonated to give the H2L− anion, within which each H atom in the moiety of –NH– has the occupancy of 0.667. Each Ni2+ cation is six-coordinated with a slightly distorted octahedral coordination geometry with a (NiN6) donor set. The Ni–N bond lengths are in the range from 2.126(3)Å to 2.137(3)Å; the bond angles N–Ni–N from 88.87(11) to 180°. The average bond length around Ni2+ is 2.132Å (Table 2). Each H2L− links three Ni2+ cations (Scheme 1), and each Ni2+ is bound by six H2L−. This kind of connectivity repeats infinitely to yield a 2D network structure (Fig. 1b). Topology can be used to further analyze the structure of 1; each Ni2+ is bound by six H2L− and thus treated as a six-connected node; each H2L− ligand links three Ni2+ cations as a three-connected node. So, the structure of 1 can be simplified as a binodal (3,6)-connected 2D kgd network (Fig. 1c). The Point (Schläfli) symbol is (43)2(46.66.83) [18]. Adjacent layers are superposed and connected by hydrogen bonds to show a 2D+2D→3D stacking structure (Fig. 1d).
Crystal structure data for complex 1 (R(F)=Σ||Fo|–|Fc||/Σ|Fo|).
| 1 | |
|---|---|
| Formula | C30H33N12O5.5Ni |
| Mr | 708.39 |
| Crystal size, mm3 | 0.20×0.12×0.10 |
| Crystal system | Monoclinic |
| Space group | C2/c |
| a, Å | 11.7114(17) |
| b, Å | 20.319(3) |
| c, Å | 14.159(2) |
| β, ° | 97.810(2) |
| V, Å3 | 3338.0(9) |
| Z | 4 |
| Dcalcd., g cm−3 | 1.41 |
| μ(MoKα), cm−1 | 0.6 |
| F(000), e | 1476 |
| hkl range | ±13, –24→+21, ±16 |
| θ range, ° | 2.00–25.00 |
| Refl. measured | 8238 |
| Refl. unique/Rint | 2847/0.0543 |
| Param. refined | 242 |
| R(F)a/wR(F2)b (all refls.) | 0.0809/0.2108 |
| GoF (F2)c | 1.095 |
| Δρfin (max/min), e Å−3 | 0.88/−0.80 |
aR(F)=Σ||Fo|–|Fc||/Σ|Fo|. bwR(F2)=[Σw(Fo2–Fc2)2/Σw(Fo2)2]1/2; w=[σ2(Fo2)+(AP)2+BP]−1, where P=(Max(Fo2, 0)+2Fc2)/3. cGoF= S=[Σw(Fo2–Fc2)2/(nobs–nparam)]1/2.
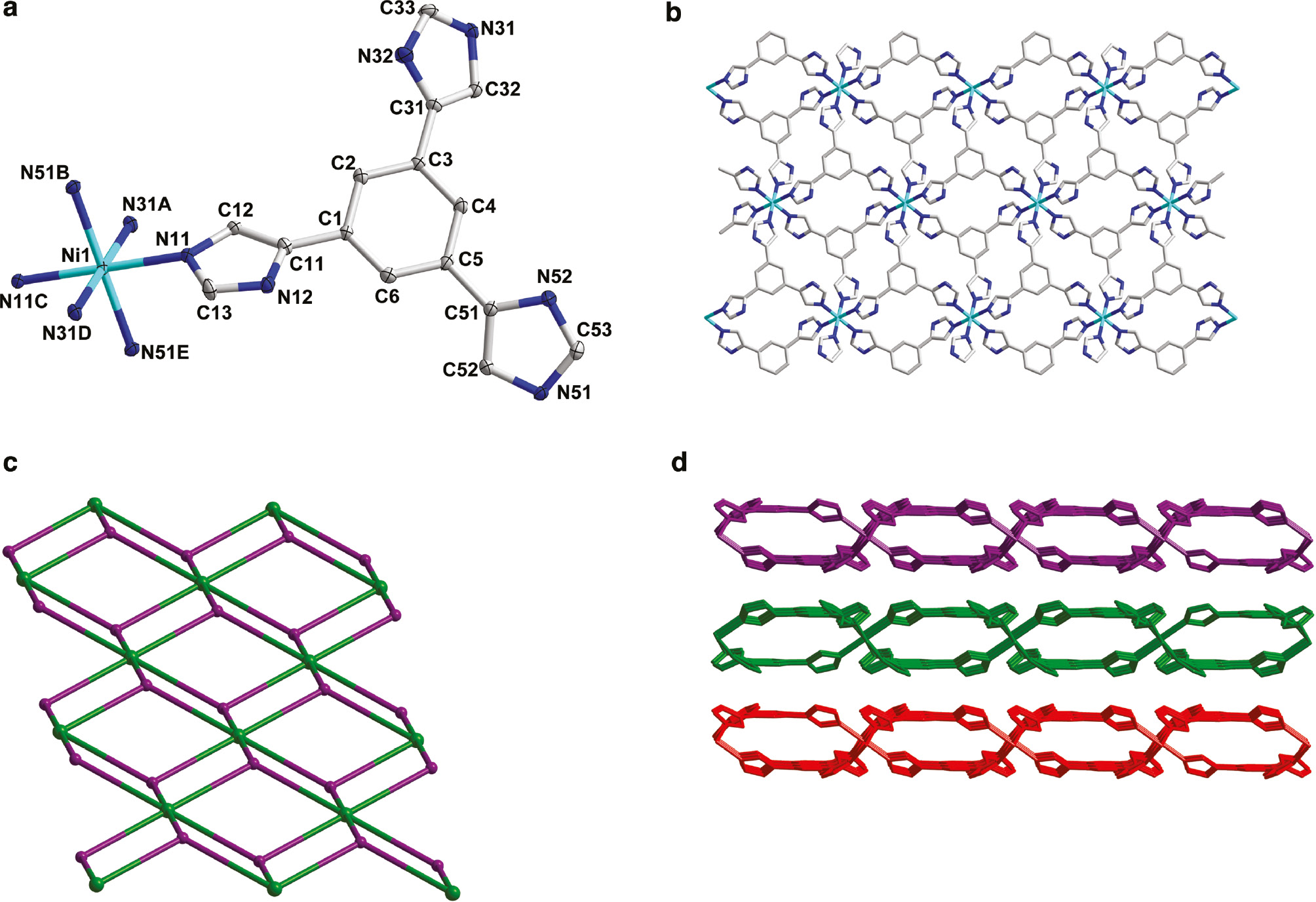
(a) The coordination environment of Ni(II) cations in complex 1 with the ellipsoids drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Symmetry operations: A–1/2–x, 3/2–y, –z; B–1/2+x, –1/2+y, z; C–x, 1–y, –z; D1/2+x, –1/2+y, z; E1/2–x, 3/2–y, –z. (b) View of the 2D structure of 1. (c) Schematic illustration of the topology of 1. (d) View of 2D+2D network-stacking structure in 1.
Selected bond lengths (Å) and angles (°) for complex 1.
| [Ni(H2L)2]·5.5H2O (1) | |||
|---|---|---|---|
| Ni(1)–N(11) | 2.134(3) | Ni(1)–N(51)B | 2.126(3) |
| Ni(1)–N(31)D | 2.136(3) | ||
| N(11)–Ni(1)–N(11)C | 180 | N(11)–Ni(1)–N(51)B | 91.11(10) |
| N(11)–Ni(1)–N(31)D | 90.44(10) | N(11)–Ni(1)–N(31)A | 89.57(10) |
| N(11)–Ni(1)–N(51)E | 88.89(10) | N(11)C–Ni(1)–N(31)A | 90.44(10) |
| N(31)D–Ni(1)–N(51)B | 90.65(11) | N(31)A–Ni(1)–N(51)B | 89.35(11) |
| N(31)D–Ni(1)–N(31)A | 180 | N(51)B–Ni(1)–N(51)E | 180 |
Symmetry transformations used to generate equivalent atoms: for 1: A–1/2–x, 3/2–y, –z; B–1/2+x, –1/2+y, z; C–x, 1–y, –z; D1/2+x, –1/2+y, z; E1/2–x, 3/2–y, –z.
2.3 Powder X-ray diffractionand thermogravimetric analysis measurements of complex 1
The phase purity of 1 could be proven by powder X-ray diffraction (PXRD) measurement. As shown in Fig. 2, the PXRD pattern of the as-synthesized sample is consistent with the simulated one.
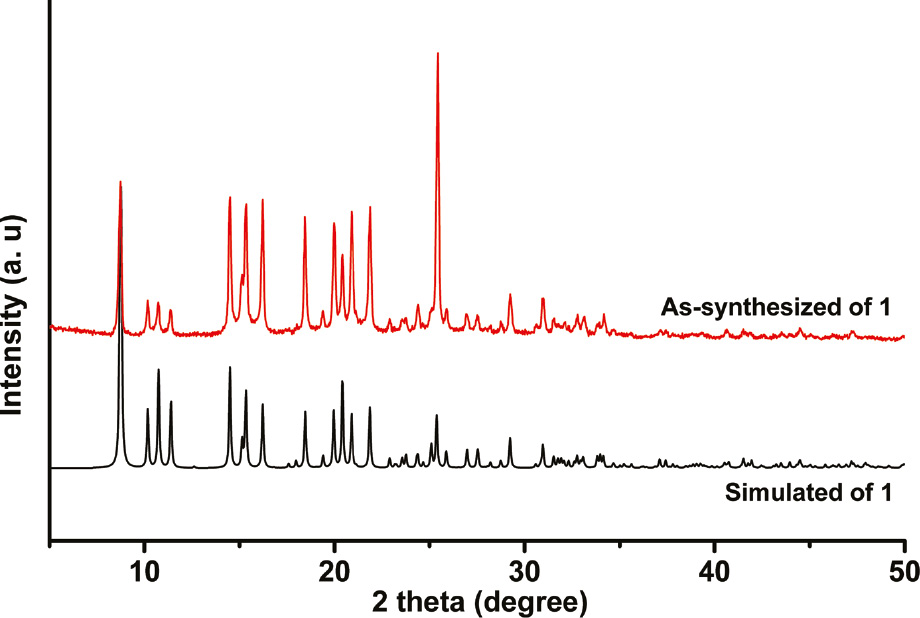
The experimental and simulated powder X-ray diffraction pattern of complex 1.
Thermogravimetric analysis (TGA) was carried out for complex 1, and the result is shown in Fig. 3. A continuous weight loss (13.7%) in the temperature range of 92°C–195°C, corresponding to the release of lattice water (calcd 13.98%), and the decomposition of the residue can be observed starting near 400°C.
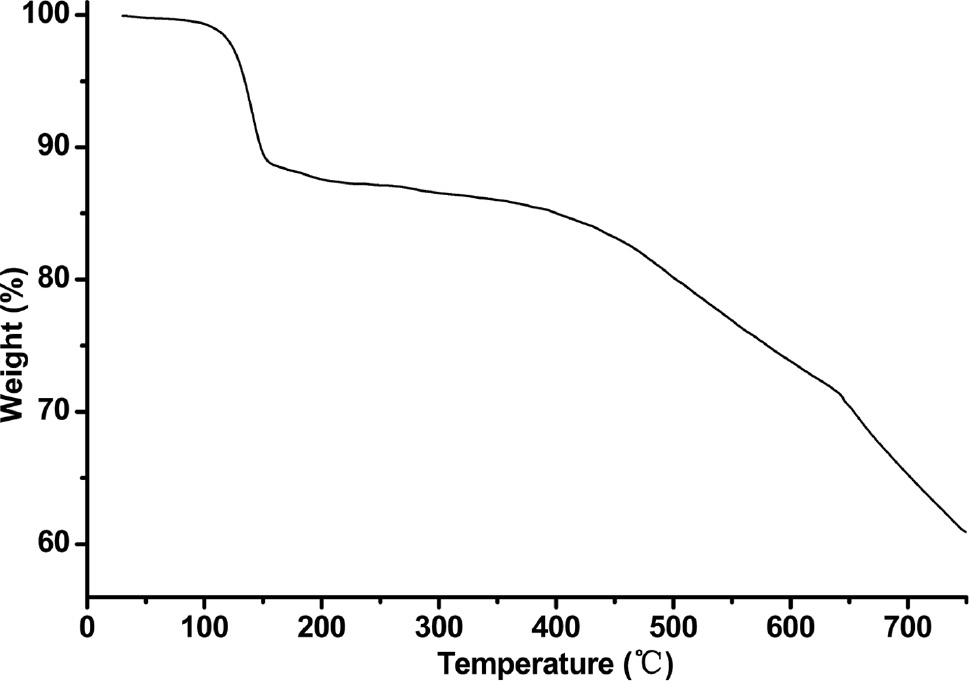
Thermogravimetric analysis curve of complex 1.
3 Experimental section
All commercially available chemicals were of reagent grade and used as received without further purification. The H3L ligand was synthesized via the experimental procedure reported in the literature [17]. Elemental analysis of C, H, and N were taken using a Perkin-Elmer 240°C elemental analyzer. Infrared (IR) spectra was recorded using a Bruker Vector22 FT-IR spectrophotometer by using KBr pellets. The PXRD patterns were measured using a Shimadzu XRD-6000 X-ray diffractometer with CuKα (λ=1.5418 Å) radiation at room temperature. TGA was performed using a simultaneous SDT 2960 thermal analyzer under nitrogen atmosphere with a heating rate of 10 K min−1.
3.1 Preparation of [Ni(H2L)2]·5.5H2O (1)
The reaction mixture of H3L (0.1 mmol, 27.6 mg), Ni(ClO4)2·6H2O (73.1 mg, 0.2 mmol), and aqueous ammonia (25%, 2 mL) in 10 mL H2O was sealed in a 16 mL Teflon-lined stainless steel container and heated at 100°C for 3 days. After cooling to room temperature, green block crystals of 1 were collected by filtration and washed with water and ethanol several times (yield 40% based on the H3L). –C30H33N12O5.5Ni (708.39): calcd C 50.87, H 4.70, N 23.73; found C 50.59, H 4.42, N 23.44%.–IR (KBr pellet, cm−1): ν=3466 (m), 1617 (s), 1571 (s), 1495 (s), 1448 (s), 1382 (s), 1318 (s), 1271 (s), 1125 (s), 1089 (s), 992 (m), 949 (m), 870 (m), 831 (s), 759 (m), 626 (s).
Caution: The perchlorate salt must be handled with care for the danger of potential explosion.
3.2 X-ray structure determination
The crystallographic data collection for complex 1 was carried out using a Bruker Smart ApexII CCD area-detector diffractometer using graphite-monochromatized MoKα radiation (λ=0.71073 Å) at T=200 K. The diffraction data were integrated by using the program Saint [19], which was also used for the intensity corrections for Lorentz and polarization effects. Semi-empirical absorption corrections were applied using the program Sadabs [20]. The structure of 1 was solved by direct methods, and all nonhydrogen atoms were refined anisotropically on F2 by full-matrix least-squares techniques using the Shelxl-97 crystallographic software package [21], [22], [23]. In 1, all hydrogen atoms at C atoms were generated geometrically, while the ones at N12, N32, and N52 were found at reasonable positions in the difference Fourier maps and located there. The H atoms at the interstitial(lattice) water molecule could not be located and thus were excluded from the refinement. The details of crystal parameters, data collection, and refinement are summarized in Table 1; selected bond lengths and angles are listed in Table 2.
CCDC 1538208 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
Funding source: Natural Science Foundation for Universities in Jiangsu Province
Award Identifier / Grant number: 16KJB150005
Funding source: Huaiyin Institute of Technology
Award Identifier / Grant number: 15HGZ006
Award Identifier / Grant number: 491713325
Funding statement: The authors gratefully acknowledge Natural Science Foundation for Universities in Jiangsu Province (16KJB150005) and Huaiyin Institute of Technology (15HGZ006 and 491713325) for financial support of this work.
References
[1] M. Hardy, N. Struch, F. Topić, G. Schnakenburg, K. Rissanen, A. Lützen, Inorg. Chem.2018, 57, 3507.10.1021/acs.inorgchem.7b02516Suche in Google Scholar PubMed
[2] A. Rossin, M. Peruzzini, Chem. Rev.2016, 116, 8848.10.1021/acs.chemrev.6b00043Suche in Google Scholar PubMed
[3] X. H. Zhu, X. C. Cheng, D. H. Li, Y. H. Qian, Z. Naturforsch.2018, 73b, 423.10.1515/znb-2018-0071Suche in Google Scholar
[4] J. Li, S. Chen, L. Y. Jiang, Y. S. Li, B. Li, Cryst. Growth Des.2018, 18, 1931.10.1021/acs.cgd.7b01701Suche in Google Scholar
[5] X. H. Zhu, D. Y. Jiang, X. C. Cheng, D. H. Li, W. G. Du, Z. Naturforsch.2019, 74b, 409.10.1515/znb-2018-0270Suche in Google Scholar
[6] B.L. Chen, S.C. Xiang, G.D. Qian, Acc. Chem. Res.2010, 43, 1115.10.1021/ar100023ySuche in Google Scholar PubMed
[7] L. Luo, G. C. Lv, P. Wang, Q. Liu, K. Chen, W.Y. Sun, Cryst. Eng. Commun.2013, 15, 9537.10.1039/c3ce41056kSuche in Google Scholar
[8] L. Luo, K. Chen, Q. Liu, Y. Lu, T. A. Okamura, G.C. Lv, Y. Zhao, W. Y. Sun, Cryst. Growth Des.2013, 13, 2312.10.1021/cg301815wSuche in Google Scholar
[9] H. W. Kuai, T. Hu, D. Y. Jiang, Y. H. Qian, Z. Naturforsch.2019, 74b, 415.10.1515/znb-2018-0284Suche in Google Scholar
[10] H. W. Kuai, X. C. Cheng, X. H. Zhu, Polyhedron2013, 50, 390.10.1016/j.poly.2012.10.044Suche in Google Scholar
[11] H. W. Kuai, T. Hu, D. Y. Jiang, X. H. Zhu, Z. Naturforsch.2018, 73b, 407.10.1515/znb-2018-0034Suche in Google Scholar
[12] H. W. Kuai, X. C. Cheng, L. D. Feng, X. H. Zhu, Z. Anorg. Allg. Chem.2011, 637, 1560.10.1002/zaac.201100160Suche in Google Scholar
[13] P. Wang, L. Luo, J. Fan, G.C. Lv, Y. Song, W.Y. Sun, Micropor. Mesopor. Mater.2013, 175, 116.10.1016/j.micromeso.2013.03.021Suche in Google Scholar
[14] S. Hazra, S. Bhattacharya, M. K. Singh, L. Carrella, E. Rentschler, T. Weyhermüller, G. Rajaraman, S. Mohanta, Inorg. Chem.2013, 52, 12881.10.1021/ic400345wSuche in Google Scholar PubMed
[15] Z. H. Chen, Y. Zhao, S. S. Chen, P. Wang, W.Y. Sun, J. Solid State Chem.2013, 202, 215.10.1016/j.jssc.2013.03.042Suche in Google Scholar
[16] X. H. Zhu, D. Y. Jiang, T. Hu, D. H. Li, Z. Naturforsch.2018, 73b, 265.10.1515/znb-2017-0199Suche in Google Scholar
[17] S. S. Chen, M. Chen, S. Takamizawa, M. S. Chen, Z. Su, W. Y. Sun, Chem. Commun.2011, 47, 752.10.1039/C0CC04085ASuche in Google Scholar PubMed
[18] V. A. Blatov, IUCr CompCommun Newsletter2006, 7, 4.Suche in Google Scholar
[19] Saint, Program for Data Extraction and Reduction, Bruker AXS, Inc., Madison, WI (USA) 2001.Suche in Google Scholar
[20] G. M. Sheldrick, Sadabs, Program for Bruker Area Detector Absorption Correction, University of Göttingen, Göttingen (Germany) 1997.Suche in Google Scholar
[21] G. M. Sheldrick, Shelxs/l-97, Programs for the Determination of Crystal Structure, University of Göttingen, Göttingen (Germany) 1997.Suche in Google Scholar
[22] G. M. Sheldrick, Acta Crystallogr.1990, A46, 467.10.1107/S0108767390000277Suche in Google Scholar
[23] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112.10.1107/S0108767307043930Suche in Google Scholar PubMed
©2019 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Electron densities of two cyclononapeptides from invariom application
- Crystal structures, Hirshfeld surface analysis and Pixel energy calculations of three trifluoromethylquinoline derivatives: further analyses of fluorine close contacts in trifluoromethylated derivatives
- Synthesis and antifungal activities of 3-substituted phthalide derivatives
- Unexpected isolation of a cyclohexenone derivative
- Preparation and structure of 4-(dimethylamino)thiopivalophenone – intermolecular interactions in the crystal
- A new binuclear NiII complex with tetrafluorophthalate and 2,2′-bipyridine ligands: synthesis, crystal structure and magnetic properties
- Two mononuclear zinc(II) complexes constructed by two types of phenoxyacetic acid ligands: syntheses, crystal structures and fluorescence properties
- Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
- Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
- Crystal structure and photocatalytic degradation properties of a new two-dimensional zinc coordination polymer based on 4,4ʹ-oxy-bis(benzoic acid)
- Intermetallics of the types REPd3X2 and REPt3X2 (RE=La–Nd, Sm, Gd, Tb; X=In, Sn) with substructures featuring tin and In atoms in distorted square-planar coordination
- A 119Sn Mössbauer-spectroscopic characterization of the diamagnetic birefringence material Sn2B5O9Cl
- Synthesis, crystal structure and photoluminescence of the salts Cation+ [M(caffeine)Cl]− with Cation+=NnBu4+, AsPh4+ and M==Zn(II), Pt(II)
- Synthesis and characterization of two bifunctional pyrazole-phosphonic acid ligands
- A β-ketoiminato palladium(II) complex for palladium deposition
- Orthoamide und Iminiumsalze, XCVIa. Push-pull-substituierte 1,3,5-Hexatriene aus Orthoamiden von Alkincarbonsäuren und Birckenbach-analogen Acetophenonen
- Orthoamide und Iminiumsalze, IIICa. Weitere Ergebnisse bei der Umsetzung von Orthoamiden der Alkincarbonsäuren mit CH2- und CH2/NH-aciden Verbindungen
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Electron densities of two cyclononapeptides from invariom application
- Crystal structures, Hirshfeld surface analysis and Pixel energy calculations of three trifluoromethylquinoline derivatives: further analyses of fluorine close contacts in trifluoromethylated derivatives
- Synthesis and antifungal activities of 3-substituted phthalide derivatives
- Unexpected isolation of a cyclohexenone derivative
- Preparation and structure of 4-(dimethylamino)thiopivalophenone – intermolecular interactions in the crystal
- A new binuclear NiII complex with tetrafluorophthalate and 2,2′-bipyridine ligands: synthesis, crystal structure and magnetic properties
- Two mononuclear zinc(II) complexes constructed by two types of phenoxyacetic acid ligands: syntheses, crystal structures and fluorescence properties
- Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
- Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
- Crystal structure and photocatalytic degradation properties of a new two-dimensional zinc coordination polymer based on 4,4ʹ-oxy-bis(benzoic acid)
- Intermetallics of the types REPd3X2 and REPt3X2 (RE=La–Nd, Sm, Gd, Tb; X=In, Sn) with substructures featuring tin and In atoms in distorted square-planar coordination
- A 119Sn Mössbauer-spectroscopic characterization of the diamagnetic birefringence material Sn2B5O9Cl
- Synthesis, crystal structure and photoluminescence of the salts Cation+ [M(caffeine)Cl]− with Cation+=NnBu4+, AsPh4+ and M==Zn(II), Pt(II)
- Synthesis and characterization of two bifunctional pyrazole-phosphonic acid ligands
- A β-ketoiminato palladium(II) complex for palladium deposition
- Orthoamide und Iminiumsalze, XCVIa. Push-pull-substituierte 1,3,5-Hexatriene aus Orthoamiden von Alkincarbonsäuren und Birckenbach-analogen Acetophenonen
- Orthoamide und Iminiumsalze, IIICa. Weitere Ergebnisse bei der Umsetzung von Orthoamiden der Alkincarbonsäuren mit CH2- und CH2/NH-aciden Verbindungen