Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
-
Kamal M. El-Shaieb
 ,
Asmaa H. Mohamed
,
Asmaa H. Mohamed

Abstract
The reactivity of 4-amino-5-hydrazineyl-4H-1,2,4-triazole-3-thiol (1) towards several carbonyl compounds was investigated. We have found that on treatment of 1 with isatoic anhydride (2), 1,8-naphthalic anhydride (4), diphenic anhydride (6), pyromellitic dianhydride (8), and tetrabromophthalic anhydride (10), the reaction proceeds to give triazolotetrazine- and triazolophthalazine derivatives in good yields using simple experimental procedures. The structure of the synthesized compounds was confirmed using different spectroscopic data (1H NMR, 13C NMR, mass spectrometry, and elemental analysis). The mechanism of the obtained products was also discussed.
1 Introduction
In the last two decades, the chemistry of 1,2,4-triazole and their fused heterocyclic derivatives have received considerable attention owing to their synthetic and effective biological importance. Also, sulfur-containing triazolo-heterocycles are very important due to their practical applications. Among these heterocycles, the mercapto-substituted and thiono-substituted 1,2,4-triazoles were well studied [1], [2]. Triazolothiones possess various biological activities including anticancer [3], [4], antiviral [5], anti-inflammatory [6], antiproliferative [7], antifungal [8], antidepressant [9], antioxidant [10], antagonistic [11], antiulcer [12], antimicrobial [13], [14], and antimycotic activities [15]. Hypoglycemic activities have been observed as well [16]. Owing to their broad biological activity 1,2,4-triazole derivatives represent an attractive target for the elaboration of solid-phase synthesis methodology and the production of combinatorial libraries.
In addition, some imines derived from triazole were reported to possess antimicrobial [17], antianxiety, antidepressant [18], and plant growth regulatory activity [19].
[1,2,4]triazolo[4,3-b][1,2,4,5]tetrazines are molecules of wide interest and importance due to their use in synthetic chemistry [20] and as key scaffolds in energetic materials [21]. Some derivatives of these bicyclic compounds also possess a wide spectrum of biological activities with antihemostatic [22], antimicrobial [23], and antitumor properties [24], as inhibitors of a Bcl-2 protein [25], and as antibacterial serine-threonine protein kinases [26].
Considering the above reports and in continuation of our development of new and simple methods for the synthesis of polyfunctionally substituted heterocyclic compounds [27], [28], [29], [30], herein we describe the synthesis of novel compounds by treatment of 4-amino-5-hydrazineyl-4H-[1,2,4]triazole-3-thiol with different anhydrides and aldehydes.
2 Results and discussion
2.1 Reaction of 1 with isatoic anhydride (2)
The present study uses 4-amino-5-hydrazineyl-4H-1,2,4-triazole-3-thiol (1) as a target to get different biodynamic nitrogen heterocycles, such as [1,2,4,5]tetrazine and [1,2]diazepine fused with the 1,2,4-triazole ring by heating 1 with some selected anhydrides under reflux conditions. Refluxing equimolar amounts of 1 with isatoic anhydride (2) in glacial acetic acid (GAA) afforded the corresponding 6-(2-aminophenyl)-[1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine-3-thiol (3) in good yield as shown in Scheme 1.

Treatment of compound 1 with some anhydrides.
The structure of the newly synthesized compound 3 was confirmed mainly by NMR spectroscopy and ascertained by elemental analyses. For example, the 1H NMR spectrum of compound 3 showed a singlet at δ=14.09 ppm characteristic for the SH group in addition to a broad singlet attributed to the NH2 group at δ=6.55 ppm, while the aromatic protons are resonating in the range of δ=7.23–8.44 ppm. Moreover, the disappearance of the hydrazine protons of compound 1 confirmed the suggested structure. The 13C NMR spectrum of 3 showed six peaks at δ=118.49, 121.58, 124.83, 130.46, 114.73, and 141.22 ppm, which are assigned to aromatic carbon atoms, while peaks at δ=152.07, 165.43, 168.16 ppm are characteristic for the carbon atoms of the C=N units.
A plausible mechanism for the formation of product 3 is proposed in Scheme 2. With regard to the reaction mechanism, we propose that the reaction begins with a nucleophilic attack of the –NH2 of the hydazinyl group of 1 at the carbonyl carbon atom (C4) of 12 to give the intermediate 13. This intermediate loses a proton as well as a molecule of CO2 under the reaction conditions forming adduct 14 which in turn cyclizes to 15 by nucleophilic attack of the >N–NH2 group at the carbonyl carbon atom. Compound 15 suffers from dehydration and dehydrogenation to give the final product 3 under these reaction conditions (Scheme 2).

A rational pathway for the formation of compound 3.
2.2 Reaction of 1 with 1,8-naphthalic anhydride (4)
Surprisingly, reaction of compound 1 with 1,8-naphthalic anhydride (4) afforded 12-mercapto-7H,9H-[1,2,4]triazolo[3′,4′:3,4][1,2,4,5]tetrazino[6,1-a]benzo[de]isoquinolin-7-one (5) in excellent yield. The 1H NMR spectrum of 5 showed a singlet at δ=13.20 ppm attributed to the SH, as well as a singlet at δ=10.96 assigned to the NH group. The presence of a resonance at δ=165.56 ppm in the 13C NMR spectrum is attributed to carbonyl carbon atom while the other three peaks at δ=160.07, 149.49, and 144.03 ppm are assigned to 3 C=N.
Scheme 3 outlines a rational pathway for the formation of the product 5. We suggest that initial nucleophilic attack occurs by the most nucleophilic site of 1 (–NH–NH2) on one of the carbonyl groups of 4 affording the intermediate 16 which rearranged to 17. Adduct 17 suffers from another nucleophilic addition of >N–NH2 to the carbon atom of the carboxyl group with concomitant loss of a water molecule to form the cycloadduct 18. Then the lone pair of CO–NH attacks at the carbonyl group to produce the intermediate 19. The final product 5 is formed by dehydration of 19 under the reaction conditions.
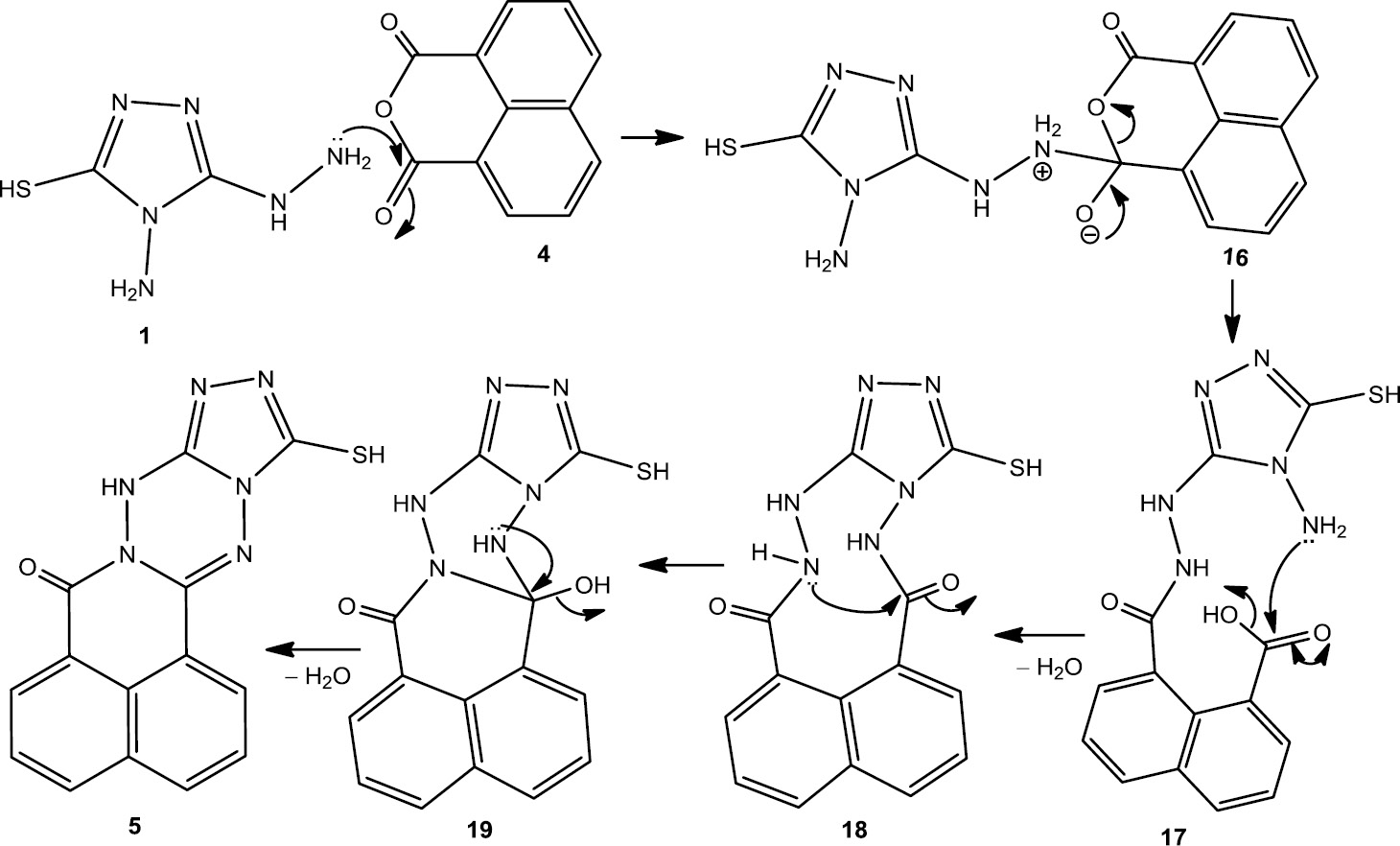
A rational pathway for the formation of compound 5.
2.3 Reaction of 1 with pyromellitic dianhydride (10)
Extending our strategy to refluxing compound 1 with pyromellitic dianhydride (10) in a molar ratio of 2:1 compound 11 (2,7-bis(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)-2,3,7,8-tetrahydropyridazino[4,5-g]phthalazine-1,4,6,9-tetraone) is formed in excellent yield. The molecular formula of compound 11 was elucidated by mass spectrometry and elemental analysis as C14H10N12O4S2. The 1H NMR spectrum of compound 11 shows a singlet at δ=13.20 ppm for the two SH groups, whereas the two protons of the NH groups appear as a singlet at δ=10.00 ppm in the 1H NMR spectrum. The two N–NH2 protons appear as a singlet at δ=5.55 ppm. The 13C NMR spectrum of compound 11 shows four carbonyl carbon atoms at δ=165.98 and 163.48 ppm in addition to four C=N carbon signals resonating at δ=150.03 ppm.
2.4 Reaction of 1 with 4-formyl[2.2]paracyclophane (20)
In the present study, new Schiff’s base derivatives were obtained by the condensation of compound 1 with some aldehydes, such as 4-formyl[2.2]paracyclophane (20) and piperonal (23). We have found that the product of the reaction between 1 and 20 depends of the molar ratios of both reactants. (E)-4-amino-5-(2-[2.2]paracyclophanylidene-hydrazineyl)-4H-1,2,4-triazole-3-thiol (21) is obtained on refluxing equal molar ratios of 1 and 20 in EtOH. In contrast, on using a two-fold molar quantity of 20, 4-((E)-([2.2]paracyclophanylidene)amino)-5-((E)-2-([2.2]paracyclophanylidene)-hydrazineyl)-4H-1,2,4-triazole-3-thiol (22) was produced (Scheme 4). The structure of the product is supported by elemental and spectral analyses. For example, compound 21 exhibits in the 1H NMR four characteristic singlets at δ=13.00, 10.45, 8.31, and 5.42 ppm assignable to SH, NH, HC=N, and NH2 protons, respectively, and multiplets at δ=2.81–3.55 ppm characteristic for the methylene protons. The aromatic protons resonate at δ=6.30–6.88 ppm. The 13C NMR spectrum of 21 shows 15 peaks at δ=130.42, 131.12, 131.65, 132.79, 133.02, 133.15, 134.38, 135.14, 138.61, 138.93, 138.98, 139.72, 144.16, 149.81, 164.40 ppm, which are assigned to aromatic carbons, while peaks at δ=33.25, 34.26, 34.43, 34.72 ppm are characteristic for the methylene carbon atoms. Moreover, the mass spectrum of 21 exhibits a molecular ion peak at m/z=364 (30%). The structure assigned to 22 is fully supported by spectral data and the mass spectrum (See section 4).
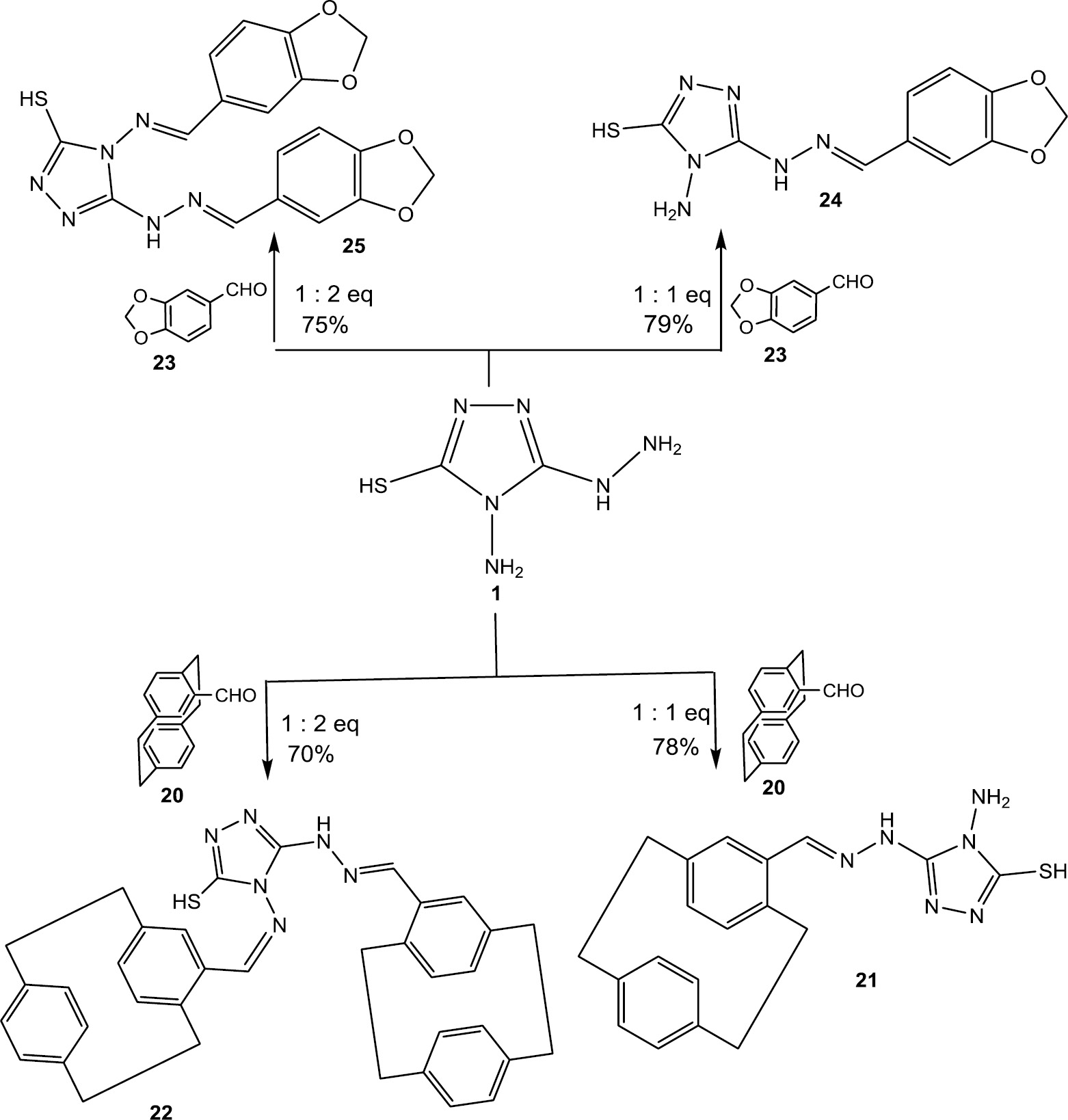
Synthesis of Schiff’s base derivatives 21, 22, 24, and 25.
2.5 Reaction of 1 with piperonal (23)
The reaction of 1 with piperonal (23) was found to proceed relatively smoothly to give (E)-4-amino-5-(2-(benzo[d][1,3]dioxol-5-ylmethylene)hydrazineyl)-4H-1,2,4-triazole-3-thiol (24) and 4-((E)-(benzo[d][1,3]dioxol-5-ylmethylene)amino)-5-((E)-2-(benzo[d][1,3]dioxol-5-ylmethylene) hydrazineyl)-4H-1,2,4-triazole-3-thiol (25) depending on the molar ratio of 1–23 (Scheme 4). The structure of compounds 24 and 25 were assigned by 1H and 13C NMR spectroscopy and mass spectrometry, in addition to elemental analyses.
2.6 The proclivity of compound 1 towards other electrophilic reagents such as terephthalaldehyde (26), o-phthalaldehyde (28), and ninhydrin (30)
Condensation of 1,2,4-triazol 1 with terephthalaldehyde (26) in the presence of a catalytic amount of p-TSA in boiling ethanol afforded 5,5′-((2E,2′E)-2,2′-(1,4-phenylenebis(methaneylylidene))bis(hydrazin-1-yl-2-ylidene))bis(4-amino-4H-1,2,4-triazole-3-thiol) (27).
Condensation of 1 with o-phthalaldehyde (28) under the same reaction conditions furnished 10H,12H-[1,2,4]triazolo[3′,4′:3,4][1,2,4,5]tetrazino[6,1-a]isoindole-3-thiol (29) as shown in Scheme 5. The structures of the products 27 and 29 are supported by elemental and spectral analyses.

Reaction of 1 with 26, o-phthalaldehyde (28), and ninhydrin (30).
A rational pathway for the formation of compound 29 is that the amino group of (–NH–NH2) attacks at one of the carbonyl carbon atoms by its lone pair of electrons leading to the formation of the intermediate 32, which yields the azomethine 33 after losing a molecule of water. The azomethine nitrogen atom attacks by its lone pair of electrons on the carbon atom of the second carbonyl group to give the cycloadduct 34 which undergoes rearrangement to 35 which in turn becomes susceptible for attack by the amino group of >N–NH2 of 1 to form the adduct 36 which dehydrates under the reaction conditions to give the final product 29 (Scheme 6).

Proposed mechanism for the formation of compound 29.
The reactivity of 1 towards ninhydrin 30 has been also studied. The product of this reaction was found to be 3-amino-10b-hydroxy-2-mercapto-3,10b-dihydro-6H- indeno[1,2-e][1,2,4]triazolo[5,1-c][1,2,4]triazin-6-one (31) as is shown in Scheme 5. The structural assignment of the product is made on the basis of the NMR spectra and is supported by its mass spectrum. The 1H NMR of 31 displayed broad singlet signals at δ=5.70, 12.31, 13.72, and 13.51 ppm assigned to NH2, 2 OH, and 2 SH groups, respectively. Furthermore, the presence of a multiplet in the region of δ=7.70–8.00 ppm is due to eight aromatic protons, which proves the existence of another isomer for the molecule due to the presence of a chiral carbon atom. Moreover, the quaternary carbon atom of 31 is resonating in the 13C NMR spectrum at δ=92.00 ppm. While, the carbonyl carbon atom and 3 C=N carbon atoms of 31 are resonating at δ=169.90, 160.53, 158.95, 154.40 ppm, respectively. The mass spectrum exhibits the molecular ion peak at m/z=288.
A suggested mechanism for formation of product 31 is shown in Scheme 7. We postulate that the electron pair of the amino group attacks the most electrophilic center in 30 (C2) leading to the formation of adduct 37. This adduct loses a molecule of H2O to give the intermediate 38. The final product 31 is formed upon cyclization by nucleophilic attack of the electron pair of the triazole nitrogen atom (one of the pyridine-like nitrogen atoms) on one of the carbonyl carbon atom.

A rational pathway for the formation of compound 31.
3 Summary
This study reports facile syntheses of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives in good yields by the reaction of 4-amino-5-hydrazineyl- 4H-1,2,4-triazole-3-thiol with some selected anhydrides, aldehydes, and ketones. The novel compounds are characterized by spectral data and elemental analyses.
4 Experimental section
All reagents were purchased from Fluka and were used without further purification. Melting points were measured in capillary tubes using a Büchi 530 melting point apparatus and are uncorrected. Infrared (IR) spectra were measured using a Bruker Tensor 27 instrument. 1H NMR (300 or 400 MHz) and 13C NMR (75 or 101 MHz) spectra were recorded in DMSO-d6 using BrukerAvance II-300 and Avance DRX-400 spectrometers with tetramethylsilane as the internal standard. Mass spectra measurements (EI, 70 eV) were performed using a Finnigan MAT 8430 spectrometer.
4.1 General procedure for the synthesis of compounds 3, 5, 7, and 9
A mixture of 4-amino-5-hydrazineyl-4H-1,2,4-triazole-3-thiol (1) (2 mmol) and anhydrides 2, 4, 6, 8, and 10 (2 mmol) in GAA (10 mL) was heated under reflux conditions for 2-4 h. A precipitate begins to form. After completion of the reaction (monitoring by thin layer chromatography, TLC), the precipitate was collected by filtration and washed with hot ethanol.
4.1.1 6-(2-Aminophenyl)-[1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine-3-thiol (3)
Pink crystal, yield 72%, m. p. 210°C–212°C. –IR (film): ν=3320, 1620, 1612 cm−1. –1H NMR (400 MHz, DMSO-d6: δ=6.55 (brs, 2H, NH2), 6.90–6.94 (t, J=7.2 Hz, 1H, ArH), 7.23–7.26 (t, J=7.2 Hz, 1H, ArH), 7.97–7.99 (d, J=7.2 Hz, 1H, ArH), 8.42–8.44 (d, J=8 Hz, 1H, ArH), 14.09 (brs, 1H, SH). –13CNMR (150 MHz, DMSO-d6: δ=118.49, 121.58, 124.83, 130.46, 114.73, 141.22, 152.07, 165.43, 168.16. –MS (EI, 70 eV): m/z (%)=245 [M]+ (20), 230 (30), 188 (40), 146 (50), 131 (15), 90 (22). –C9H7N7S (245.26): calcd. C 44.07, H 2.88, N 39.98; found C 44.23, H 2.92, N 40.13.
4.1.2 12-Mercapto-7H,9H-[1,2,4]triazolo[3′,4′:3,4][1,2,4,5]tetrazino[6,1-a]benzo[de]isoquinolin-7-one (5)
Pale yellow crystal, yield 77%, m. p. 260°C–262°C. –IR (film): ν=3221, 1656, 1638, 1618 cm−1. –1H NMR (400 MHz, DMSO-d6: δ=7.68 (s, 1H, CH), 7.80–7.90 (m, 2H, ArH), 8.06–8.06 (d, J=8 Hz, 2H, ArH), 8.19 (s, 1H, ArH), 10.78 (s, 1H, NH), 13.18 (brs, 1H, SH). –13CNMR (150 MHz, DMSO-d6: δ=127.27, 127.38, 129.64, 130.41, 135.772, 142.07, 144.03, 149.49, 165.56, 169.14. –MS (EI, 70 eV): m/z (%)=308 [M]+ (22), 197 (10), 153 (5), 125 (8), 86 (6), 62 (4). –C14H8N6OS (308.32): calcd. C 54.54, H 2.62, N 27.26; found C 54.72, H 2.67, N 27.44.
4.1.3 2′-(3-Mercapto-5,8-dihydro[1,2,4]triazolo[4,3-b][1,2,4,5]tetrazin-6-yl)[1,1′-biphenyl]-2-carboxylic acid (7)
Yellow crystal, yield 77%, m. p. 198°C–200°C. –IR (film): ν=3313–3201, 1710, 1643, 1612 cm−1. –1H NMR (400 MHz, DMSO-d6: δ=7.11–7.27 (m, 3H, ArH), 7.40–7.50 (m, 3H, ArH), 7.59–7.61 (d, J=7.2 Hz, 1H, ArH,), 7.81–7.83 (d, J=7.6 Hz, 1H, ArH), 8.74 (s, 1H, NH), 9.79 (s, 1H, NH), 10.92 (s, 1H, OH), 12.93 (s, 1H, SH). –13CNMR (150 MHz, DMSO-d6: δ=125.78, 127.40, 128.19, 128.68, 129.60, 129.37, 129.85, 130.62, 130.79, 131.18, 133.32, 152.10, 165.41, 168.83, 169.35. –C16H12N6O2S (352.37): calcd. C 54.54, H 3.43, N 23.85; found C 54.72, H 3.46, N 24.00.
4.1.4 2-(4-Amino-5-mercapto-4H-1,2,4-triazol-3-yl)-5,6,7,8-tetrabromo-2,3-dihydrophthalazine-1,4-dione (9)
Pale yellow crystal, yield 80%, m. p. 300°C–302°C. –1H NMR (300 MHz, DMSO-d6: δ=5.65 (brs, 2H, NH2), 9.89 (s, 1H, NH), 12.90 (s, 1H, SH). –13C NMR (75 MHz, DMSO-d6: δ=121.43; 108.34, 128.52, 137.67, 149.92, 161.30, 165.95. –MS (EI, 70 eV): m/z (%)=594 (20), 592 (20), 590 (30), 460 (40), 418 (20), 391 (20), 340 (18), 314 (20), 254 (18), 231 (32), 150 (33), 131 (34), 93 (18), 43 (100). –C10H4Br4N6O2S (591.86): calcd. C 20.29, H 0.68, N 14.20; found C 20.48, H 0.7, N 14.36.
4.2 Synthesis of 2,7-bis(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)-2,3,7,8-tetrahydropyridazino-[4,5-g]phthalazine-1,4,6,9-tetraone (11)
A mixture of 1 (2 mmol) and pyromellitic dianhydride (10) (1 mmol) in GAA (10 mL) was heated under reflux for 4 h. A precipitate begins to form. After completion of the reaction (monitoring by TLC), the precipitate was collected by filtration and washed with hot ethanol. Pale yellow crystals, yield 65%, m. p. 354°C–356°C. –1H NMR (300 MHz, DMSO-d6: δ=5.50 (brs, 4H, 2NH2), 8.10 (s, 2H, ArH), 9.98 (s, 2H, 2NH), 12.90 (s, 2H, 2SH). –13C NMR (75 MHz, DMSO-d6: δ=119.25, 134.90, 150.03, 163.46, 165.98. –MS (EI, 70 eV): m/z (%)=474 [M]+ (40), 216 (100), 173 (88), 145 (30), 131 (42), 102 (54), 74 (42). –C14H10N12O4S2 (474.44): calcd. C 35.44, H 2.12, N 14.20; found C 35.61, H 2.15, N 14.36.
4.3 General procedure for the synthesis of compounds 21 and 24
A mixture of compound 1 (1 mmol) was mixed and warmed with a solution of an equimolar amount of 4-formyl[2.2]paracyclophane (20) (1 mmol) and/or piperonal (23) (1 mmol) in dry EtOH (15 mL) in the presence of a catalytic amount of p-TSA. The mixture was refluxed for 2 h. A pale yellow precipitate was formed. The precipitate was collected, washed, and recrystallized from DMF-EtOH.
4.3.1 (E)-4-amino-5-(2-[2.2]paracyclophanylidene-hydrazineyl)-4H-1,2,4-triazole-3-thiol (21)
Pale yellow crystal, yield 78%, m. p. 238°C–240°C. –1H NMR (300 MHz, DMSO-d6: δ=2.81–3.55 (m, 8H, 4CH2), 5.45 (brs, 2H, NH2), 6.30–6.63 (m, 6H, ArH), 6.75 (s, 1H, ArH), 8.45 (s, 1H, NH), 10.55 (s, 1H, HC=N), 12.92 (s, 1H, SH). –13C NMR (75 MHz, DMSO-d6: δ=33.25, 34.26, 34.43, 34.72, 130.42, 131.12, 131.65, 132.79, 133.02, 133.15, 134.38, 135.14, 138.61, 138.93, 138.98, 139.72, 144.16, 149.81, 164.40. –MS (EI, 70 eV): m/z (%)=364 [M]+ (32), 233 (20), 130 (98), 104 (100). –C19H20N6S (364.47): calcd. C 62.61, H 5.53, N 23.06; found C 62.81, H 5.57, N 23.23.
4.3.2 4-Amino-5-(2-(benzo[d][1,3]dioxol-5-yl-methylene)hydrazinyl)-4H-1,2,4-triazole-3-thiol (24)
Pale yellow crystal, yield 79%, m. p. 213°C–214°C. –1H NMR (300 MHz, DMSO-d6: δ=5.48 (brs, 2H, NH2), 6.01 (s, 2H, CH2), 6.80–7.10 (m, 2H, ArH), 7.22 (s, 1H, ArH), 8.25 (brs, 1H, NH), 10.49 (brs, 1H, HC=N), 12.98 (s, 1H, SH). –13C NMR (75 MHz, DMSO-d6: δ=101.34 (CH2), 104.52, 108.34, 122.22, 129.12, 143.78, 147.78, 148.33, 149.62, 164.29. –MS (EI, 70 eV): m/z (%)=278 [M]+ (78), 148 (44), 134 (60), 121 (50), 76 (49), 65 (100). –C10H10N6O2S (278.29): calcd. C 43.16, H 3.62, N 30.20; found C 43.35, H 3.65, N 30.39.
4.4 General procedure for the synthesis of compounds 22 and 25
Compound 1 (1 mmol) and 4-formyl[2.2]paracyclophane (20) (2 mmol) or piperonal (23) (2 mmol) in presence of a catalytic amount of p-TSA was heated in absolute ethanol (20 mL). A pale yellow precipitate was formed after 30 min. The reaction was continued for 2 h. After completion of the reaction (TLC analysis), the precipitate was filtered off, washed, dried and recrystallized from DMF-EtOH.
4.4.1 4-((E)-([2.2]paracyclophanylidene)amino)-5-((E)-2-(paracyclophanylidene)hydrazineyl)-4H-1,2,4-triazole-3-thiol (22)
Pale yellow crystal, yield 70%, m. p. 280°C–282°C. –1H NMR (400 MHz, DMSO-d6: δ=3.03–3.63 (m, 16H, 8CH2), 6.28–6.81 (m, 8H, ArH), 7.05 (s, 2H, ArH), 7.32–7.36 (m, 4H, ArH), 8.32 (s, 1H, HC=N), 10.52 (s, 1H, HC=N), 13.40 (s, 1H, SH). –MS (EI, 70 eV): m/z (%)=582 [M]+ (33), 375 (20), 348 (46). –C36H34N6S (582.76): calcd. C 74.20, H 5.88, N 14.42; found C 74.02, H 5.83, N 14.30.
4.4.2 4-((E)-(Benzo[d][1,3]dioxol-5-ylmethylene)amino)-5-((E)-2-(benzo[d][1,3]dioxol-5-yl-methylene)hydrazineyl)-4H-1,2,4-triazole-3-thiol (25)
Pale yellow crystal, yield 75%, m. p. 300°C–302°C. –1H NMR (400 MHz, DMSO-d6: δ=6.07 (s, 2H, CH2), 6.16 (s, 2H, CH2), 6.95–6.97 (d, J=8 Hz, 1H, ArH), 7.07–7.09 (d, J=8 Hz, 2H, ArH), 7.20 (s, 1H, ArH), 7.38–7.40 (d, J=8 Hz, 1H, ArH), 7.71 (s, 1H, ArH), 8.26 (brs, 1H, NH), 10.05 (s, 1H, HC=N), 10.51 (s, 1H, HC=N), 13.25 (s, 1H, SH). –13C NMR (100 MHz, DMSO-d6: δ=101.90, 102.46, 105.16, 106.56, 108.89, 108.97, 122.88, 127.10, 127.20, 129.41, 145.26, 148.41, 148.67, 149.04, 151.53, 159.79, 161.19. –MS (EI, 70 eV): m/z (%)=410 [M]+ (22), 409 [M]−1 (100). –C18H14N6O4S (410.41): calcd. C 52.68, H 3.44, N 20.48; found C 52.88, H 3.48, N 20.65.
4.5 Synthesis of 5,5′-(((1E,1′E)-1,4-phenylenebis(methaneylylidene))bis(hydrazin-1-yl-2-ylidene))bis(4-amino-4H-1,2,4-triazole-3-thiol) (27)
Compound 1 (2 mmol) and terephthalaldehyde (26) (1 mmol) in presence of a catalytic amount of p-TSA was heated in absolute ethanol (20 mL). Yellow precipitate was formed after 30 min. The reaction was continued for 2 h. After completion of the reaction (TLC analysis), the precipitate was filtered off, washed, dried and recrystallized from DMF-EtOH. Yellow powder, yield 75%, m. p. 342°C–344°C. –1H NMR (400 MHz, DMSO-d6: δ=5.45 (brs, 2H, NH2), 6.64 (s, 4H, ArH), 8.24 (s, 2H, 2NH), 10.71 (s, 2H, 2HC=N), 12.98 (brs, 2H, 2SH). –13C NMR (100 MHz, DMSO-d6: δ=126.34, 133.14, 135.29, 149.50, 164.44. –MS (EI, 70 eV): m/z (%)=390 [M]+ (30), 244 (28), 128 (98), 116 (97), 85 (100), 75 (72). –C12H14N12S2 (390.45): calcd. C 36.91, H 3.61, N 43.05; found C 37.11, H 3.64, N 43.23.
4.6 Synthesis of 10H,12H-[1,2,4]triazolo[3′,4′:3,4][1,2,4,5]tetrazino[6,1-a]isoindole-3-thiol (29)
Equimolar amounts of compound 1 and o-phthalaldehyde (28) was heated in absolute ethanol (20 mL) in presence of p-TSA as a catalyst under reflux conditions for 2 h (the reaction was followed by TLC). The resulting precipitate that was formed after cooling was filtered off, dried, and recrystallized from ethanol. Gray powder, yield 60%, m. p. 278°C–280°C. –1H NMR (400 MHz, DMSO-d6: δ=4.58 (s, 2H, CH2), 7.10–7.22 (m, 2H, ArH), 7.47–7.49 (d, J=8 Hz, 1H, ArH), 7.60–7.62 (d, J=8 Hz, 1H, ArH), 8.13 (s, 1H, NH), 13.59 (s, 1H, SH). –13C NMR (100 MHz, DMSO-d6: δ=54.98, 121.58, 124.83, 128.37, 129.60, 130.46, 131.65, 141.22, 152.07, 165.43. –MS (EI, 70 eV): m/z (%)=246 [M]+2 (8), 245 [M]+1 (12), 244 [M]+ (12), 230 (27), 186 (5), 130 (26), 116 (20), 102 (18), 76 (12), 59 (10). –C10H8N6S (244.28): calcd. C 49.17, H 3.30, N 34.40; found C 49.34, H 3.33, N 34.57.
4.7 Synthesis of 3-amino-10b-hydroxy-2-mercapto-3,10b-dihydro-6H-indeno[1,2-e][1,2,4]triazolo[5,1-c][1,2,4]triazin-6-one (31)
A mixture of ninhydrin (30) (1 mmol) and compound 1 (1 mmol) in the presence of a catalytic amount of p-TSA in ethanol (20 mL) was heated under reflux conditions. A reddish-brown precipitate was formed and the reaction was continued for a further 2 h. The precipitate was filtered off and recrystallized from 1,4-dioxan. Reddish brown crystal, yield 72%, m. p. 220°C–222°C. –1H NMR (400 MHz, DMSO-d6: δ=5.70 (brs, 2H, NH2), 7.70–8.00 (m, 8H, ArH), 12.31 (s, 1H, OH), 12.72 (brs, 1H, OH), 13.51 (s, 2H, 2SH). –13C NMR (100 MHz, DMSO-d6: δ=92.80, 119.98, 121.10, 123.24, 124.16, 131.02, 148.20, 158.10, 169.80. –MS (EI, 70 eV): m/z (%)=288 [M]+ (10), 270 (20), 240 (10), 158 (30), 129 (40), 102 (28), 76 (32). –C11H8N6O2S (288.29): calcd. C 45.83, H 2.80, N 29.15; found C 45.98, H 2.82, N 29.30.
References
[1] J. H. Mansoory, S. S. Rajput, Int. J. Pharm. Pharm. Sci. 2015, 7, 20–32.Suche in Google Scholar
[2] S. Karakuş, U. Çoruh, B. Barlas-Durgun, M. Ezequiel, Ö. Suna, T. VázquezLópez, R. Sevim, Marmara Pharm. J.2010, 14, 84–90.10.12991/201014454Suche in Google Scholar
[3] Y. A. Al-Soud, M. N. Al-Dweri, N. A. Al-Masoudi, Farmaco2004, 59, 775–783.10.1016/j.farmac.2004.05.006Suche in Google Scholar PubMed
[4] H. Abdel-Gawad, H. A. Mohamed, K. M. Dawood, F. A. Badria, Chem. Pharm. Bull. 2010, 58, 1529–1531.10.1248/cpb.58.1529Suche in Google Scholar PubMed
[5] M. Abdel-Megeed, M. A. Hamdy, S. A. Gamal-Eldien, A. E. Mahmoud, Eur. J. Med. Chem. 2009, 44, 117–123.10.1016/j.ejmech.2008.03.017Suche in Google Scholar PubMed
[6] Z. Li, Z. Gu, K. Yin, R. Zhang, Q. Deng, J. Xiang, Eur. J. Med. Chem. 2009, 44, 4716–4720.10.1016/j.ejmech.2009.05.030Suche in Google Scholar PubMed
[7] D. Gupta, D. K. Jain, J. Adv. Pharm. Technol. Res. 2015, 6, 141–146.10.4103/2231-4040.161515Suche in Google Scholar PubMed PubMed Central
[8] D. Mares, C. Romagnoli, E. Andreotti, M. Manfrini, C. Beatr, J. Agric. Food Chem. 2004, 52, 2003–2009.10.1021/jf030695ySuche in Google Scholar PubMed
[9] J. M. Kane, M. W. Dudley, S. M. Sorensen, P. M. Francis, J. Med. Chem. 1988, 31, 1253–1258.10.1021/jm00401a031Suche in Google Scholar PubMed
[10] I. Khan, S. Ali, S. Hameed, N. H. Rama, M. T. Hussain, A. Wadood, R. Uddin, Z. Ul-Haq, A. Khan, S. Ali, M. I. Choudhary, Eur. J. Med. Chem. 2010, 45, 5200–5207.10.1016/j.ejmech.2010.08.034Suche in Google Scholar PubMed
[11] A. Manilal, B. Sabarathnam, G. S. Kiran, S. Sujith, C. Shakir, J. Selvin, Asian J. Med. Sci. 2010, 2, 195–200.Suche in Google Scholar
[12] T. Akhtar, S. Hameed, K. M. Khan, A. Khan, M. I. Choudhary, J. Enzyme Inhib. Med. Chem. 2010, 25, 572–576.10.3109/14756360903389864Suche in Google Scholar PubMed
[13] N. D. Heindel, J. R. Reid, J. Heterocycl. Chem.1980, 17, 1087–1088.10.1002/jhet.5570170547Suche in Google Scholar
[14] B. S. Holla, B. Kalluraya, K. R. Sridhar, E. Drake, L. M. Thomas, K. K. Bhandary, M. S. Levine, Euro. J. Med. Chem. 1994, 29, 301–308.10.1016/0223-5234(94)90100-7Suche in Google Scholar
[15] J. Haber, Cas. Lek. Cesk. 2001, 140, 596–604.Suche in Google Scholar
[16] M. Y. Mhasalkar, M. H. Shah, S. T. Nikam, J. Med. Chem. 1971, 14, 260–262.10.1021/jm00285a029Suche in Google Scholar PubMed
[17] S. Jubie, P. Sikdar, R. Kalirajan, B. Gowramma, S. Gomathy, S. Sankar, K. Elango, J. Pharm. Res.2010, 3, 511–513.Suche in Google Scholar
[18] S. Jubie, P. Sikdar, S. Antony, R. Kalirajan, B. Gowramma, S. Gomathy, K. Elango, Pak. J. Pharm. Sci.2011, 24, 109–112.Suche in Google Scholar
[19] X. X. Ye, Z. F. Chen, A. J. Zhang, L. X. Zhang, Molecules2007, 12, 1202–1209.10.3390/12061202Suche in Google Scholar PubMed PubMed Central
[20] I. N. Ganebnykh, S. G. Tolshchina, R. I. Ishmetova, N. K. Gnatenko, P. A. Slepukhin, G. L. Rusinov, V. N. Charushin, Eur. J. Org. Chem. 2011, 12, 2309–2318.10.1002/ejoc.201001590Suche in Google Scholar
[21] T. Wei, W. Zhu, J. Zhang, H. J. Xiao, J. Hazard. Mater. 2010, 179, 581–590.10.1016/j.jhazmat.2010.03.043Suche in Google Scholar PubMed
[22] R. R. Kamble, B. S. Sudha, D. B. Biradar, Indian J. Heterocycl. Chem. 2007, 17, 121–124.Suche in Google Scholar
[23] F. M. A. Altalbawy, E. S. S. Darwish, Asian J. Chem. 2011, 23, 2951–2955.Suche in Google Scholar
[24] F. Xu, C. Zheng, T. Zhang, D. Yuan, H. Chen, Y. Yang, T. Lin, Chinese Patent CN, 103012410, 2013.Suche in Google Scholar
[25] J. C. Reed, K. Yip, E. Sergienko, Y. Su, PCT Int. Appl. WO2008154207, 2008.Suche in Google Scholar
[26] O. B. Bekker, V. N. Danilenko, R. I. Ishmetova, D. A. Maslov, G. L. Rusinov, S. G. Tolshchina, V. N Charushin, Russian Patent RU2462466, 2012.Suche in Google Scholar
[27] A. H. Mohamed, Z. Naturforsch. 2018, 73b, 399–406.10.1515/znb-2018-0010Suche in Google Scholar
[28] K. M. El-Shaieb, J. Chem. Res. 2015, 39, 515–517.10.3184/174751915X14398000795121Suche in Google Scholar
[29] K. M. El-Shaieb, H. Hopf, P. G. Jones, Arkivoc2009, x, 146–160.10.3998/ark.5550190.0010.a15Suche in Google Scholar
[30] K. M. El-Shaieb, M. A. Ameen, F. F. Abdel-latif, A. H. Mohamed, Z. Naturforsch. 2013, 68b, 905–912.10.5560/znb.2013-3117Suche in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/znb-2019-0140).
©2019 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Electron densities of two cyclononapeptides from invariom application
- Crystal structures, Hirshfeld surface analysis and Pixel energy calculations of three trifluoromethylquinoline derivatives: further analyses of fluorine close contacts in trifluoromethylated derivatives
- Synthesis and antifungal activities of 3-substituted phthalide derivatives
- Unexpected isolation of a cyclohexenone derivative
- Preparation and structure of 4-(dimethylamino)thiopivalophenone – intermolecular interactions in the crystal
- A new binuclear NiII complex with tetrafluorophthalate and 2,2′-bipyridine ligands: synthesis, crystal structure and magnetic properties
- Two mononuclear zinc(II) complexes constructed by two types of phenoxyacetic acid ligands: syntheses, crystal structures and fluorescence properties
- Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
- Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
- Crystal structure and photocatalytic degradation properties of a new two-dimensional zinc coordination polymer based on 4,4ʹ-oxy-bis(benzoic acid)
- Intermetallics of the types REPd3X2 and REPt3X2 (RE=La–Nd, Sm, Gd, Tb; X=In, Sn) with substructures featuring tin and In atoms in distorted square-planar coordination
- A 119Sn Mössbauer-spectroscopic characterization of the diamagnetic birefringence material Sn2B5O9Cl
- Synthesis, crystal structure and photoluminescence of the salts Cation+ [M(caffeine)Cl]− with Cation+=NnBu4+, AsPh4+ and M==Zn(II), Pt(II)
- Synthesis and characterization of two bifunctional pyrazole-phosphonic acid ligands
- A β-ketoiminato palladium(II) complex for palladium deposition
- Orthoamide und Iminiumsalze, XCVIa. Push-pull-substituierte 1,3,5-Hexatriene aus Orthoamiden von Alkincarbonsäuren und Birckenbach-analogen Acetophenonen
- Orthoamide und Iminiumsalze, IIICa. Weitere Ergebnisse bei der Umsetzung von Orthoamiden der Alkincarbonsäuren mit CH2- und CH2/NH-aciden Verbindungen
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Electron densities of two cyclononapeptides from invariom application
- Crystal structures, Hirshfeld surface analysis and Pixel energy calculations of three trifluoromethylquinoline derivatives: further analyses of fluorine close contacts in trifluoromethylated derivatives
- Synthesis and antifungal activities of 3-substituted phthalide derivatives
- Unexpected isolation of a cyclohexenone derivative
- Preparation and structure of 4-(dimethylamino)thiopivalophenone – intermolecular interactions in the crystal
- A new binuclear NiII complex with tetrafluorophthalate and 2,2′-bipyridine ligands: synthesis, crystal structure and magnetic properties
- Two mononuclear zinc(II) complexes constructed by two types of phenoxyacetic acid ligands: syntheses, crystal structures and fluorescence properties
- Investigation of the reactivity of 4-amino-5-hydrazineyl-4H-1,2, 4-triazole-3-thiol towards some selected carbonyl compounds: synthesis of novel triazolotriazine-, triazolotetrazine-, and triazolopthalazine derivatives
- Synthesis and structural characterization of a Ni(II) coordination polymer with a tripodal 4-imidazolyl-functional ligand
- Crystal structure and photocatalytic degradation properties of a new two-dimensional zinc coordination polymer based on 4,4ʹ-oxy-bis(benzoic acid)
- Intermetallics of the types REPd3X2 and REPt3X2 (RE=La–Nd, Sm, Gd, Tb; X=In, Sn) with substructures featuring tin and In atoms in distorted square-planar coordination
- A 119Sn Mössbauer-spectroscopic characterization of the diamagnetic birefringence material Sn2B5O9Cl
- Synthesis, crystal structure and photoluminescence of the salts Cation+ [M(caffeine)Cl]− with Cation+=NnBu4+, AsPh4+ and M==Zn(II), Pt(II)
- Synthesis and characterization of two bifunctional pyrazole-phosphonic acid ligands
- A β-ketoiminato palladium(II) complex for palladium deposition
- Orthoamide und Iminiumsalze, XCVIa. Push-pull-substituierte 1,3,5-Hexatriene aus Orthoamiden von Alkincarbonsäuren und Birckenbach-analogen Acetophenonen
- Orthoamide und Iminiumsalze, IIICa. Weitere Ergebnisse bei der Umsetzung von Orthoamiden der Alkincarbonsäuren mit CH2- und CH2/NH-aciden Verbindungen