Preparation of novel acyl pyrazoles and triazoles by means of oxidative functionalization reactions
-
Geoffrey P. Wadey


Abstract
Novel acyl pyrazoles and acyl triazoles have been prepared by means of the oxidative amidation of aldehydes in the presence of the requisite azole. Yields range from modest to good in both cases, and some limitations of the substrate scope have been discovered. Acyl pyrazoles were prepared by treatment of a mixture of aldehyde and pyrazole with an oxoammonium salt bearing the nitrate anion. In the case of acyl triazoles, the oxidative functionalization was performed using sodium persulfate as a terminal oxidant in the presence of a catalytic quantity of a nitroxide.
1 Introduction
The amide functional group is found not only in biological systems but also in pharmaceuticals, agrochemicals, and polymers [1,2,3]. In addition, amides serve as versatile starting materials for conversion to other classes of molecule including imines, aldehydes, amides, nitriles, and oximes [4,5]. As a result, simple and effective routes to amides are highly sought after both in academic and industrial settings. One of the most used routes to amides involves the coupling of amines with carboxylic acids, but this requires pre-functionalization steps and can be inefficient [6,7]. Metal-catalyzed routes are also available but they can be troublesome due to the elevated temperatures that are needed to facilitate the reaction, as well as requiring expensive transition-metal complexes [8]. The oxidative amidation of aldehydes represents an alternative approach to amides [9,10,11]. The aldehyde component serves as the carbonyl-containing progenitor of the amide and it is functionalized in an oxidative fashion with an amine [12,13]. From a cost and practicality standpoint, this route is particularly attractive; a wide range of aldehydes and amines being commercially available.
Within the class of amides, N-acyl pyrazoles are of interest not only on their own right [14,15,16,17], but also because they can participate readily in transamidation reactions and other functional-group interconversions [18]. Our research group has focused some significant effort on the preparation of N-acyl pyrazoles using an oxidative amidation approach and we have developed a number of methodologies centered around the use of oxoammonium salts and their corresponding nitroxide analogs as reagents and catalysts (Figure 1) [18,19,20,21,22]. The archetypal oxoammonium salt is 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (AcNH-TEMPO+ BF4 −, 1) [23,24,25], but when using this as a reagent to prepare N-acyl pyrazoles by the oxidative amidation of aldehydes with pyrazole, a super-stoichiometric quantity of the salt is required because the spent oxidant, hydroxylammonium salt 2, undergoes a comproportionation reaction with 1 to generate nitroxide 3 [23,26]. As a result, a sacrificial equivalent of 1 is required to achieve complete oxidation of the substrate. The analog of 1 bearing the nitrate anion (4) turns out not only to be effective for the transformation but can be used in substoichiometric amounts [22]. In addition, the reaction can be performed in the absence of an added base, making the approach more attractive than that using 1. We have previously shown the applicability of the methodology to the oxidative functionalization of some benzaldehydes, including those that are potentially challenging to use. We wanted to investigate this route further, and in so doing explore new chemical space and also expand the scope for the preparation of acyl triazoles. We report our results here.
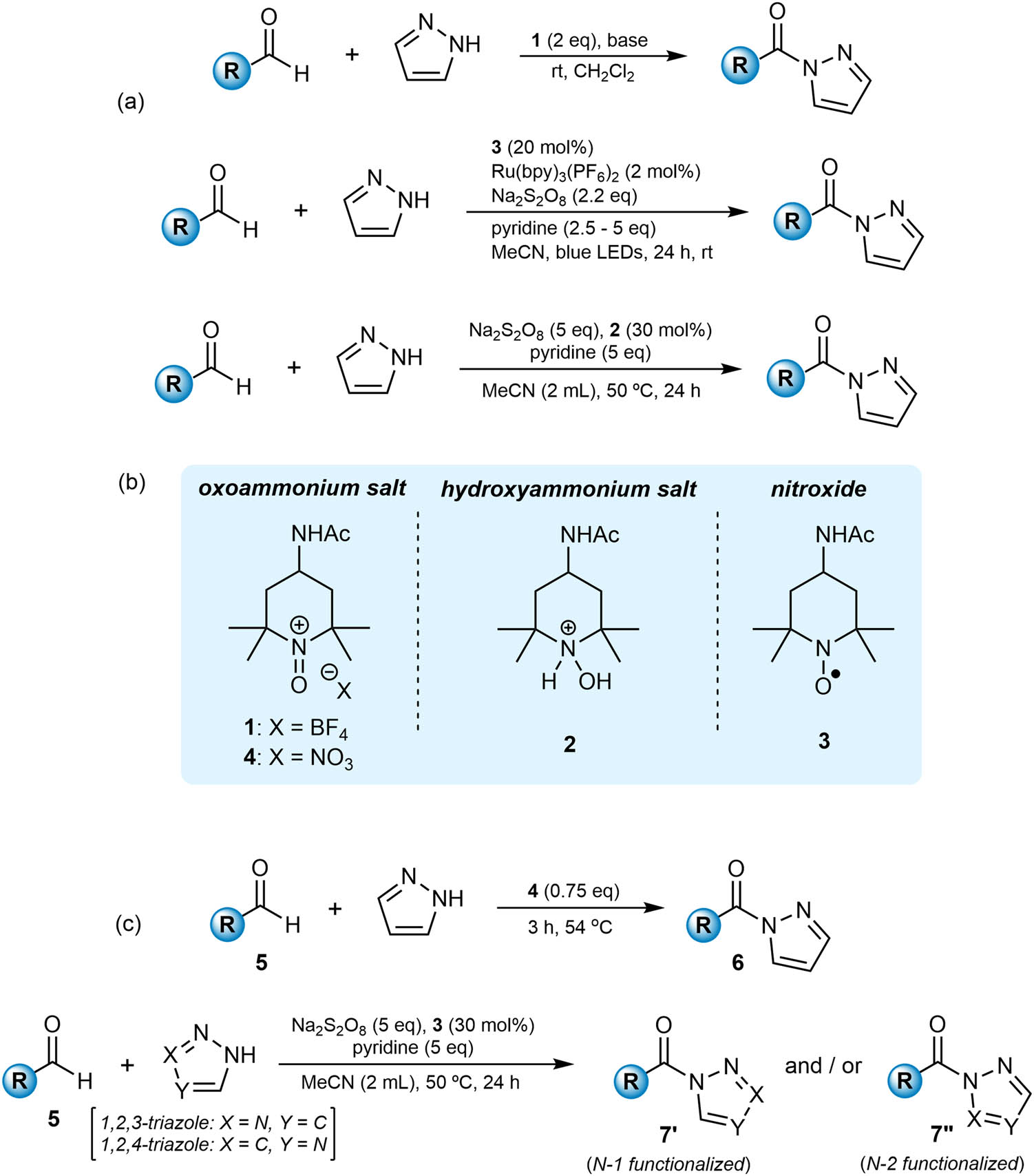
(a) Previous approaches to the oxidative amidation of aldehydes with pyrazole to prepare acyl pyrazoles. (b) Oxoammonium salts 1 and 4, and their hydroxylammonium and nitroxide analogs 2, and 3. (c) Methodologies presented here.
2 Results and discussion
2.1 Preparation of novel acyl pyrazoles
To start our preparative chemistry, we decided to prepare a number of N-acyl pyrazoles with various functionality on the aldehyde precursor. In each case, a mixture of 1 eq. of the aldehyde, 5, and 1.1 eq. of pyrazole was treated with 0.75 eq. of oxoammonium salt 4 and the reaction mixture was heated at 54°C for 3 h (Figure 2). Following this, the products were isolated by extraction, with no need for chromatographic purification. We focused initially on benzaldehydes bearing electron donating groups. In most cases, the N-acyl pyrazole was isolated in modest yield (6a–6c). An exception was when 4-(dimethylamino)benzaldehyde was used, in which case no reaction was observed (6d). We broadened the scope by next using 2-napthaldehyde as the aldehyde component. The desired N-acyl pyrazole, 6e, was obtained in 58% yield. Heterocyclic aldehydes could be used as reagents, a case in point being 1-methylindole-3-carboxaldehyde where the N-acyl pyrazole, 6f, could be prepared in 34% yield. However, when using 2-pyridinecarbaldehyde, no reaction was observed (6g). To complete our screen, we moved beyond benzaldehydes as substrates and instead chose (4-isobutylphenyl)acetaldehyde as a starting material, the corresponding N-acyl pyrazole, 6h, being obtained in 64% yield.
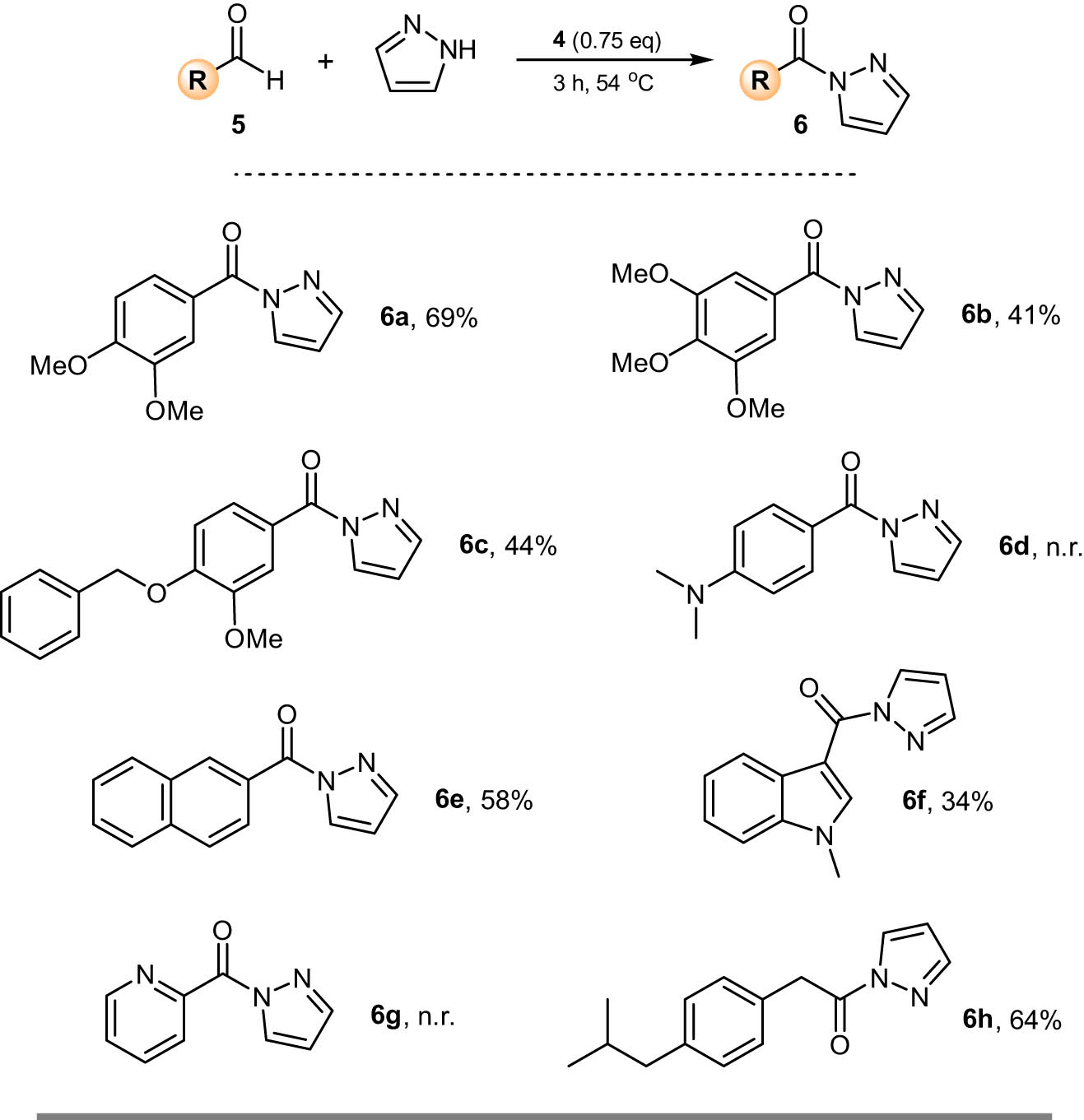
Oxidative amidation of aldehydes with pyrazole to prepare acyl pyrazoles. Reaction was carried out in a sealed vial using 5 (1 mmol, 1 eq.). Isolated yield after purification.
2.2 Preparation of novel acyl triazoles
To expand the study, we next varied the azole coupling partner, we turned our attention to the preparation of triazoles. The N-acyl triazole products could have interesting biological properties based on previous literature [27,28,29]. Unlike the oxidative amidation with pyrazole, when preparing 1,2,3-triazoles or 1,2,4-triazoles, there is the potential for the formation of regioisomers; the acyl group being situated on either the 1-, 2-, or 4-position on the triazole ring. We initially employed the same synthetic approach as we had used for the preparation of acyl pyrazoles. While we did observe good product conversion, not all the aldehyde starting material was consumed, even after prolonged reaction times. This poses a challenge when it comes to product isolation. Separation of the acyl triazole from unreacted aldehyde is not a trivial process. To circumvent this, we decided to employ a different methodology. We have shown previously that acyl pyrazoles can be prepared using nitroxide 3 as a catalyst and sodium persulfate as a terminal oxidant [20]. The reaction is performed at 50°C for 24 h, using pyridine as a base. The reason for this is that the initial step of the reaction involves a thermally-induced homolytic cleavage of sodium persulfate to generate two equivalents of the sulfate radical anion (
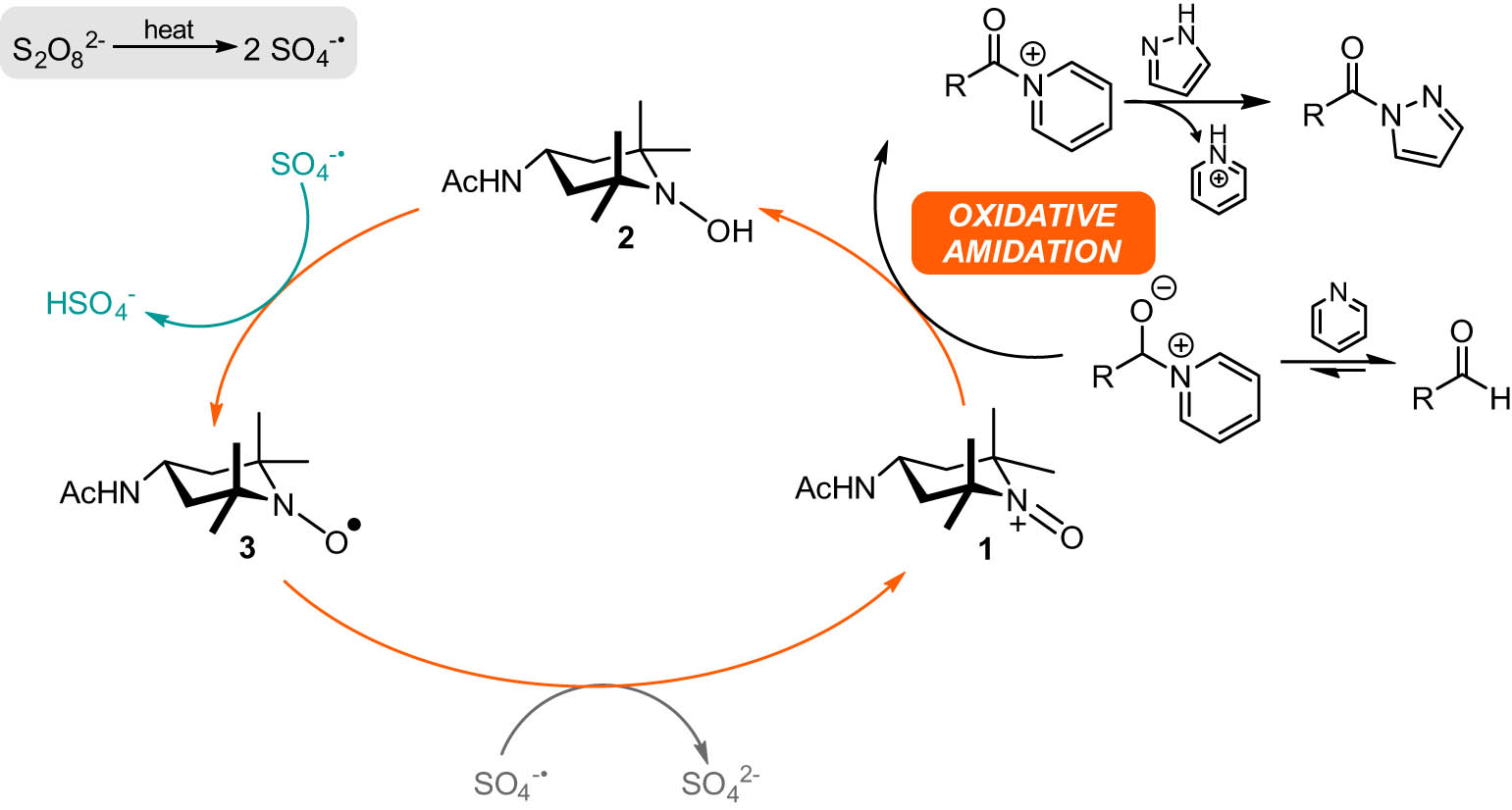
Catalytic cycle for oxidative amidation.
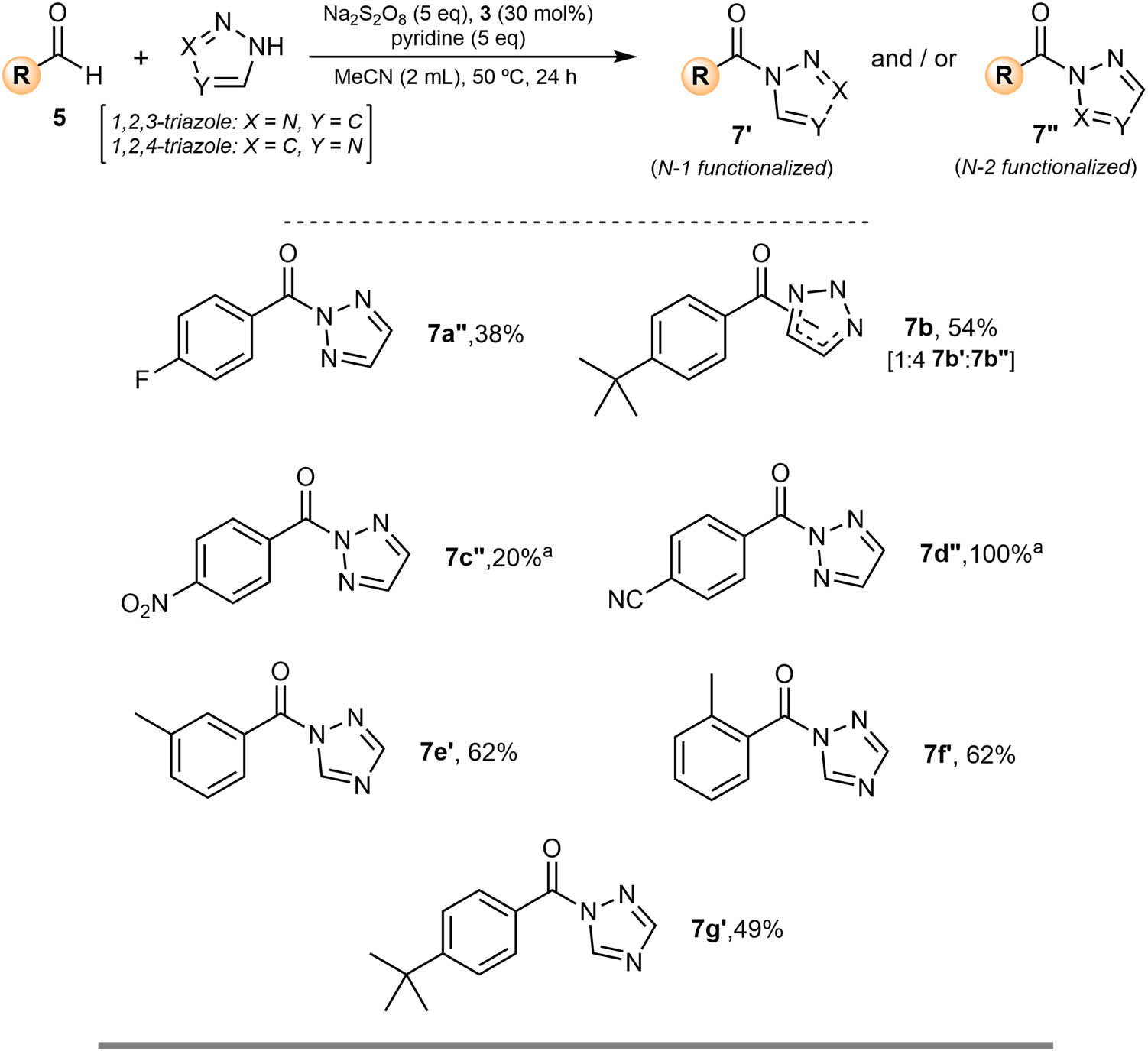
Oxidative amidation of aldehydes with triazole to prepare acyl triazoles. Reaction performed in a sealed vial using 5 (1 mmol, 1 eq.). Isolated yield after purification, unless noted otherwise. aProduct conversion determined by GC-MS.
3 Conclusion
In summary, we have prepared a range of novel acyl azoles by means of the oxidative amidation of aldehydes. Acyl pyrazoles were prepared by treatment of a mixture of aldehyde and pyrazole with an oxoammonium salt bearing the nitrate anion. In the case of acyl triazoles, a different approach was taken. The oxidative functionalization was performed using sodium persulfate as a terminal oxidant in the presence of a catalytic quantity of a nitroxide. Yields ranged from modest to good in both cases, and some limitations of the substrate scope were discovered.
4 Experimental section
4.1 General considerations
NMR spectra (1H, 13C, and 19F) were recorded at 300 K on a Brüker Avance Ultra Shield 300 MHz, Bruker DRX-400 400 MHz, or Brüker Avance 500 MHz spectrometer. 1H-NMR spectra were referenced to residual chloroform (7.26 ppm) in CDCl3. 13C-NMR spectra were referenced to CDCl3 (77.16 ppm). 19F-NMR spectra were referenced to hexafluorobenzene (−161.64 ppm) [30]. Reactions were monitored by an Agilent Technologies 7820A Gas Chromatograph attached to a 5975 Mass Spectrometer, 19F-NMR, and/or by TLC on silica gel plates (60 Å porosity, 250 μm thickness). TLC analysis was performed using a solution of 8:2 hexanes:ethyl acetate and visualized with UV light.
Deuterated chloroform (CDCl3) was purchased from Cambridge Isotope Laboratories. 4-acetamido-TEMPO (ACT, 3) and 4-Acetamido-2,2,6,6-tetramethylpiperidin-1-oxoammonium nitrate (4) were prepared using previously reported protocols [31,32]. All aldehydes and heterocycles used were purchased from Oakwood Chemicals, Sigma-Aldrich, or Alfa Aesar and distilled before use if required.
4.2 Procedure for the preparation of acyl pyrazoles from aldehydes
To a 14 mL capacity vial equipped with a stir bar was added the aldehyde (5, 1 mmol, 1 eq.), 4-acetamido-2,2,6,6-tetramethylpiperidin-1-oxoammonium nitrate (4, 206 mg, 0.75 eq.), and pyrazole (75 mg, 1.1 eq.). The vial was closed tightly and the contents heated in an oil bath at 50°C. Upon completion of the heating step, the vial and its contents were allowed to cool to room temperature and then the product mixture was transferred to a 250 mL separatory funnel using acetonitrile (1 mL) and the vial was washed with hexanes (50 mL) and this was also added to the separatory funnel. Deionized water (50 mL) was added to the separatory funnel and the aqueous layer then was washed twice with hexanes (2 × 25 mL). The organic washes were combined and washed with 2 M hydrochloric acid (15 mL) and saturated aqueous sodium bicarbonate (20 mL). The organic layer was dried over sodium sulfate, and the solvent removed in-vacuo to afford the product 6.
4.2.1 (3,4-Dimethoxyphenyl)(1H-pyrazol-1-yl)methanone (6a)
Obtained as a pale-yellow solid (0.161 g, 69%), reaction run for 3 h. 1H-NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 2.9, 0.7 Hz, 1H), 7.97 (dd, J = 8.5, 2.1 Hz, 1H), 7.83–7.75 (m, 2H), 6.96 (d, J = 8.6 Hz, 1H), 6.54–6.48 (m, 1H), 3.97 (s, 3H), 3.95 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 165.48, 153.56, 148.69, 144.32, 130.85, 126.99, 123.64, 114.50, 110.22, 109.15, 56.25, 56.22. IR (neat, ν/cm−1): 1,671 (C═0). HRMS (ESI) m/z calculated for C12H13N2O [M + H]+ 226.0980, found 226.0979.
4.2.2 (3,4,5-Trimethoxyphenyl)(1H-pyrazol-1-yl)methanone (6b)
Obtained as a pale-yellow solid (0.108 g, 41%), reaction run for 1 h. 1H-NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.9, 0.8 Hz, 1H), 7.80 (dt, J = 1.5, 0.7 Hz, 1H), 7.49 (s, 2H), 6.54–6.49 (m, 1H), 3.93 (s, 3H), 3.91 (s, 6H). 13C-NMR (101 MHz, CDCl3) δ 165.57, 152.75, 144.54, 142.73, 130.88, 126.10, 109.65, 109.37, 61.04, 56.41. IR (neat, ν/cm−1): 1,685 (C = O). HRMS (ESI) m/z calculated for C13H15N2O4 [M + H]+ 263.1032, found 263.1033.
4.2.3 (4-(Benzyloxy)-3-methoxyphenyl)(1H-pyrazol-1-yl)methanone (6c)
Obtained as a yellow solid (0.137 g, 44%), reaction run for 3 h. 1H-NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.8, 0.7 Hz, 1H), 7.89 (dd, J = 8.5, 2.1 Hz, 1H), 7.79 (d, J = 2.1 Hz, 2H), 7.46–7.29 (m, 5H), 6.97 (d, J = 8.5 Hz, 1H), 6.53–6.48 (m, 1H), 5.26 (s, 2H), 3.96 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 165.42, 152.71, 149.13, 144.29, 136.38, 130.82, 128.83, 128.25, 127.31, 126.74, 123.86, 114.95, 112.29, 109.12, 70.96, 56.28. IR (neat, ν/cm−1): 1,685 (C═0). HRMS (ESI) m/z calculated for C12H13N2O [M + H]+ 309.1239, found 309.1237.
4.2.4 (Naphthalene-2-yl)(1H-pyrazol-1-yl)methanone (6e)
Obtained as a white solid (0.129 g, 58%), reaction run for 1 h. 1H-NMR (400 MHz, CDCl3) δ 8.77 (s, 1H), 8.50 (dd, J = 2.9, 0.7 Hz, 1H), 8.14 (dd, J = 8.7, 1.8 Hz, 1H), 8.02–7.88 (m, 3H), 7.85 (dd, J = 1.4, 0.7 Hz, 1H), 7.60 (dddd, J = 23.3, 8.1, 6.9, 1.4 Hz, 2H), 6.59–6.53 (m, 1H). 13C-NMR (101 MHz, CDCl3) δ 166.54, 144.68, 135.56, 133.84, 132.32, 130.71, 129.81, 128.83, 127.97, 127.87, 126.92, 126.89, 109.55. IR (neat, ν/cm−1): 1,690 (C═0). HRMS (ESI) m/z calculated for C14H11N2O [M + H]+ 223.0871, found 223.0869.
4.2.5 (1-Methylindol-3-yl)(1H-pyrazol-1-yl)methanone (6f)
Obtained as a red solid (0.077 g, 34%), reaction run for 1. 1H-NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 8.54–8.46 (m, 2H), 7.77 (dd, J = 1.5, 0.8 Hz, 1H), 7.44–7.32 (m, 3H), 6.47 (dd, J = 2.8, 1.5 Hz, 1H), 3.92 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 160.98, 143.23, 140.18, 136.95, 129.78, 128.97, 123.57, 122.95, 122.52, 109.92, 108.15, 106.20, 33.84. IR (neat, ν/cm−1): 1,650 (C═0). HRMS (ESI) m/z calculated for C13H12N3O [M + H]+ 226.0980, found 226.0979.
4.2.6 2-(4-Isobutylphenyl)-1-(1H-pyrazol-1-yl)propan-1-one (6h)
Obtained as a colorless oil (0.164 g, 64%), reaction run for 2 h. 1H-NMR (400 MHz, CDCl3) δ 8.24 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 0.8 Hz, 1H), 7.36 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 6.42–6.36 (m, 1H), 5.20 (q, J = 7.1 Hz, 1H), 2.44 (d, J = 7.2 Hz, 2H), 1.84 (hept, J = 6.8 Hz, 1H), 1.63 (d, J = 7.1 Hz, 3H), 0.90 (d, J = 6.6 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ 173.41, 143.94, 140.78, 137.21, 129.51, 128.77, 127.81, 109.82, 45.16, 42.51, 30.23, 22.50, 18.93. IR (neat, ν/cm−1): 1,729 (C═0). HRMS (ESI) m/z calculated for C16H21N2O [M + H]+ 257.1654, found 257.1654.
4.3 Procedure for the preparation of acyl triazoles from aldehydes
To a 14 mL capacity vial equipped with a stir bar were added the aldehyde (5, 1 mmol, 1 eq.), pyridine (404 µL, 5 eq.), acetonitrile (2 mL), 3 (63 mg, 0.30 eq.), triazole (83 mg, 1.2 eq.), and sodium persulfate (1.190 g, 5 eq.). The vial was closed tightly, and the contents heated in an oil bath at 50°C for 24 h. Upon completion of the heating step, the vial and its contents were allowed to cool to room temperature and then the product mixture was transferred to a 250 mL separatory funnel, and the vial was washed with deionized water (20 mL) and this was also added to the separatory funnel. The contents of the separatory funnel were then further diluted with deionized water (30 mL) and the aqueous layer then washed three times with hexanes (1 × 50 mL followed by 2 × 25 mL). The organic washes were combined and washed with 0.5 M hydrochloric acid (15 mL), saturated aqueous sodium bicarbonate (20 mL), and deionized water (15 mL). The organic layer was dried over sodium sulfate, and the solvent removed in-vacuo to afford the product 7.
4.3.1 (4-Fluorophenyl)(2H-1,2,3-triazol-2-yl)methanone (7a″)
Obtained as white solid (0.072 g, 38%). 1H-NMR (400 MHz, CDCl3) δ 8.31–8.21 (m, 2H), 8.00 (s, 2H), 7.27–7.17 (m, 2H). 13C-NMR (101 MHz, CDCl3) δ 166.31 (d, J = 257.1 Hz), 162.29, 138.46, 134.96 (d, J = 9.3 Hz), 126.59, 115.86 (d, J = 22.2 Hz). 19F-NMR (376 MHz, CDCl3) δ −102.96 (ddd, J = 13.2, 8.3, 5.1 Hz). IR (neat, ν/cm−1): 1,717 (C═0). HRMS (ESI) m/z calculated for C9H7N3O [M + H]+ 192.0573, found 192.0569.
4.3.2 Mixture of (4-(tert-butyl)phenyl)(1H-1,2,3-triazol-2-yl)methanone (7b′) and (4-(tert-butyl)phenyl)(2H-1,2,3-triazol-1-yl)methanone (7b″)
Obtained as a yellow oil (0.124 g, 54%, 1:4 ratio of 7b′ to 7b″). 1H-NMR (400 MHz, CDCl3) 7b′: δ 8.48 (d, J = 1.4 Hz, 1H), 8.26–8.18 (m, 2H), 7.82 (d, J = 1.4 Hz, 1H), 7.61–7.58 (m, 2H), 1.38 (s, 9H); 7b″: δ 8.14–8.07 (m, 2H), 7.98 (s, 2H), 7.58–7.53 (m, 2H), 1.37 (s, 9H). 13C-NMR (101 MHz, CDCl3) δ 163.42, 158.79, 157.99, 157.55, 138.18, 133.60, 132.37, 131.99, 130.62, 130.19, 127.51, 125.79, 125.54, 35.46, 35.38, 31.21, 31.13. IR (neat, ν/cm−1): 1,719 (C═0). HRMS (ESI) m/z calculated for C13H16N3O [M + H]+ 230.1293, found 230.1293.
4.3.3 m-Tolyl(1H-1,2,4-triazol-1-yl)methanone (7e′)
Obtained as a white solid (0.083 g, 44%). 1H-NMR (400 MHz, CDCl3) δ 9.07 (s, 1H), 8.12 (s, 1H), 8.05–7.97 (m, 2H), 7.52–7.41 (m, 2H), 2.46 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 164.87, 153.42, 145.92, 138.62, 135.29, 132.15, 129.94, 129.01, 128.51, 21.45. IR (neat, ν/cm−1): 1,681 (C═0). HRMS (ESI) m/z calculated for C10H10N3O [M + H]+ 188.0824, found 188.0819.
4.3.4 o-Tolyl(1H-1,2,4-triazol-1-yl)methanone (7f′)
Obtained as a colorless oil (0.116 g, 62%). 1H-NMR (400 MHz, CDCl3) δ 9.00 (s, 1H), 8.07 (s, 1H), 7.58 (dd, J = 7.7, 1.4 Hz, 1H), 7.51 (td, J = 7.6, 1.5 Hz, 1H), 7.39–7.29 (m, 2H), 2.44 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 166.02, 153.69, 145.34, 138.81, 132.62, 131.48, 130.52, 130.06, 125.62, 20.15. IR (neat, ν/cm−1): 1,722 (C═0). HRMS (ESI) m/z calculated for C10H10N3O [M + H]+ 188.0824, found 188.0819.
4.3.5 (4-(Tert-butyl)phenyl)(1H-1,2,4-triazol-2-yl)methanone (7g″)
Obtained as a colorless oil (0.113 g, 49%). 1H-NMR (400 MHz, CDCl3) δ 9.06 (s, 1H), 8.17–8.12 (m, 2H), 8.09 (s, 1H), 7.56–7.50 (m, 2H), 1.33 (s, 9H). 13C-NMR (101 MHz, CDCl3) δ 164.32, 158.45, 153.24, 145.80, 131.78, 126.96, 125.60, 35.31, 30.98. IR (neat, ν/cm−1): 1,709 (C═0). HRMS (ESI) m/z calculated for C13H16N3O [M + H]+ 230.1293, found 230.1278.
-
Funding information: This research was funded by the University of Connecticut, Department of Chemistry, Charles Waring Fund (KED and GPW).
-
Author contributions: Geoffrey Wadey: undergraduate student, compound preparation, product isolation and characterization, and writing – review and editing. Katrina Doherty: undergraduate student, compound preparation, product isolation and characterization, and writing – review and editing. Arturo León Sandoval: graduate student, compound preparation, product isolation and characterization, undergraduate supervision, project design, and writing – review and editing. Nicholas Leadbeater: conceptualization, project administration, and writing – original draft, review, and editing.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
[1] Pattabiraman VR, Bode JW. Rethinking amide bond synthesis. Nature. 2011;480(7378):471–9.10.1038/nature10702Suche in Google Scholar PubMed
[2] Humphrey JM, Chamberlin AR. Chemical synthesis of natural product peptides: Coupling methods for the incorporation of noncoded amino acids into peptides. Chem Rev. 1997;97(6):2243–66.10.1021/cr950005sSuche in Google Scholar PubMed
[3] Dunetz JR, Magano J, Weisenburger GA. Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org Process Res Dev. 2016;20(2):140–77.10.1021/op500305sSuche in Google Scholar
[4] Szostak M, editor. Amide Bond Activation. Basel: MDPI; 2019.Suche in Google Scholar
[5] Czerwiński PJ, Furman B. Reductive functionalization of amides in synthesis and for modification of bioactive compounds. Front Chem. 2021;9:655849.10.3389/fchem.2021.655849Suche in Google Scholar PubMed PubMed Central
[6] Ojeda-Porras A, Gamba-Sánchez D. Recent developments in amide synthesis using nonactivated starting materials. J Org Chem. 2016;81(23):11548–55.10.1021/acs.joc.6b02358Suche in Google Scholar PubMed
[7] Smith MB. Compendium of organic synthetic methods. New York: Wiley; 2001.10.1002/9780471224471Suche in Google Scholar
[8] Allen CL, Williams JMJ. Metal-catalysed approaches to amide bond formation. Chem Soc Rev. 2011;40(7):3405–15.10.1039/c0cs00196aSuche in Google Scholar PubMed
[9] Lee W, Jeon HJ, Jung H, Kim D, Seo S, Chang S. Controlled relay process to access N-centered radicals for catalyst-free amidation of aldehydes under visible light. Chem. 2021;7(2):495–508.10.1016/j.chempr.2020.12.004Suche in Google Scholar
[10] Tan B, Toda N, Barbas CF. Organocatalytic amidation and esterification of aldehydes with activating reagents by a cross-coupling strategy. Angew Chemie Int Ed. 2012;51(50):12538–41.10.1002/anie.201205921Suche in Google Scholar PubMed
[11] Ekoue-Kovi K, Wolf C. One-pot oxidative esterification and amidation of aldehydes. Chem Eur J. 2008;14(21):6302–15.10.1002/chem.200800353Suche in Google Scholar PubMed
[12] Oldenhuis NJ, Whittaker AM, Dong VM. Greener methods for amide bond synthesis. In: Richardson PF, editor. Green chemistry in drug discovery: from academia to industry. New York, NY: Springer New York; 2022. p. 35–9610.1007/978-1-0716-1579-9_2Suche in Google Scholar
[13] Montalbetti CAGN, Falque V. Amide bond formation and peptide coupling. Tetrahedron. 2005;61(46):10827–52.10.1016/j.tet.2005.08.031Suche in Google Scholar
[14] Ferguson TEG, Reihill JA, Martin SL, Walker B. Novel inhibitors and activity-based probes targeting trypsin-like serine proteases. Front Chem. 2022;10:423.10.3389/fchem.2022.782608Suche in Google Scholar PubMed PubMed Central
[15] Otrubova K, Chatterjee S, Ghimire S, Cravatt BF, Boger DL. N-acyl pyrazoles: Effective and tunable inhibitors of serine hydrolases. Bioorg Med Chem. 2019;27(8):1693–703.10.1016/j.bmc.2019.03.020Suche in Google Scholar PubMed PubMed Central
[16] Tokumasu K, Yazaki R, Ohshima T. Direct catalytic chemoselective α-amination of acylpyrazoles: A concise route to unnatural α-amino acid derivatives. J Am Chem Soc. 2016;138(8):2664–9.10.1021/jacs.5b11773Suche in Google Scholar PubMed
[17] Ma J, Ding X, Hu Y, Huang Y, Gong L, Meggers E. Metal-templated chiral Brønsted base organocatalysis. Nat Commun. 2014;5(1):4531.10.1038/ncomms5531Suche in Google Scholar PubMed
[18] Ovian JM, Kelly CB, Pistritto VA, Leadbeater NE. Accessing N-acyl azoles via oxoammonium salt-mediated oxidative amidation. Org Lett. 2017;19(6):1286–9.10.1021/acs.orglett.7b00060Suche in Google Scholar PubMed
[19] Nandi J, Vaughan MZ, León Sandoval A, Paolillo JM, Leadbeater NE. Oxidative amidation of amines in tandem with transamidation: A route to amides using visible-light energy. J Org Chem. 2020;85(14):9219–29.10.1021/acs.joc.0c01222Suche in Google Scholar PubMed
[20] Politano F, León Sandoval A, Witko ML, Doherty KE, Schroeder CM, Leadbeater NE. Nitroxide-catalyzed oxidative amidation of aldehydes to yield N-acyl azoles using sodium persulfate. European J Org Chem. 2022;2022(4):e202101239.10.1002/ejoc.202101239Suche in Google Scholar
[21] Nandi J, Ovian JM, Kelly CB, Leadbeater NE. Oxidative functionalisation of alcohols and aldehydes via the merger of oxoammonium cations and photoredox catalysis. Org Biomol Chem. 2017;15(39):8295–301.10.1039/C7OB02243CSuche in Google Scholar
[22] León Sandoval A, Doherty KE, Wadey GP, Leadbeater NE. Solvent- and additive-free oxidative amidation of aldehydes using a recyclable oxoammonium salt. Org Biomol Chem. 2022;20(11):2249–54.10.1039/D2OB00307DSuche in Google Scholar
[23] Leadbeater NE, Bobbitt JM. TEMPO-derived oxoammonium salts as versatile oxidizing agents. Aldrichimica Acta. 2014;47(3):65–74.Suche in Google Scholar
[24] Bobbitt JM. Oxoammonium salts. 6.1 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium perchlorate: A Stable and convenient reagent for the oxidation of alcohols. Silica gel catalysis. J Org Chem. 1998;63(25):9367–74.10.1021/jo981322cSuche in Google Scholar
[25] Bobbitt JM, Merbouh N, Inokuchi T, Ma L-J. 4-Acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium Tetrafluoroborate. Encyclopedia of Reagents for Organic Synthesis. New York: John Wiley & Sons, Ltd; 2013.10.1002/047084289X.rn00346.pub3Suche in Google Scholar
[26] Bobbitt JM, Bartelson AL, Bailey WF, Hamlin TA, Kelly CB. Oxoammonium salt oxidations of alcohols in the presence of pyridine bases. J Org Chem. 2014;79(3):1055–67.10.1021/jo402519mSuche in Google Scholar PubMed
[27] Odin IS, Cao S, Hughes D, Zamaratskii EV, Zarubin YP, Purygin PP, et al. Synthesis of new N-acyl-1,2,3-triazole chalcones and determination of their antibacterial activity. Dokl Chem. 2020;492(2):89–92.10.1134/S0012500820360021Suche in Google Scholar
[28] Huang L, Liu M, Man S, Ma D, Feng D, Sun Z, et al. Design, synthesis and bio-evaluation of novel 2-aryl-4-(3,4,5-trimethoxy-benzoyl)-5-substituted-1,2,3-triazoles as the tubulin polymerization inhibitors. Eur J Med Chem. 2020;186:111846.10.1016/j.ejmech.2019.111846Suche in Google Scholar PubMed
[29] Brand S, Ko EJ, Viayna E, Thompson S, Spinks D, Thomas M, et al. Discovery and optimization of 5-Amino-1,2,3-triazole-4-carboxamide series against Trypanosoma cruzi. J Med Chem. 2017;60(17):7284–99.10.1021/acs.jmedchem.7b00463Suche in Google Scholar PubMed PubMed Central
[30] Rosenau CP, Jelier BJ, Gossert AD, Togni A. Exposing the origins of irreproducibility in fluorine NMR spectroscopy. Angew Chemie Int Ed. 2018;57(30):9528–33.10.1002/anie.201802620Suche in Google Scholar PubMed
[31] Mercadante MA, Kelly CB, Bobbitt JM, Tilley LJ, Leadbeater NE. Synthesis of 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate and 4-acetamido-(2,2,6,6-tetramethyl-piperidin-1-yl)oxyl and their use in oxidative reactions. Nat Protoc. 2013;8(4):666–76.10.1038/nprot.2013.028Suche in Google Scholar PubMed
[32] Miller SA, León Sandoval A, Leadbeater NE. Oxidation of alcohols using an oxoammonium salt bearing the nitrate anion. Tetrahedron Lett. 2020;61(6):151464.10.1016/j.tetlet.2019.151464Suche in Google Scholar
© 2023 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Research Articles
- Synthesis, characterization, and antibacterial activity of a new poly azo compound containing N-arylsuccinimid and dibenzobarrelene moieties
- Design, synthesis, and antiviral activities evaluation of novel quinazoline derivatives containing sulfonamide moiety
- Design, synthesis, and anticancer activity of novel 4,6-dimorpholinyl-1,3,5-triazine compounds
- Design, synthesis, biological evaluation, and bio-computational modeling of imidazo, thieno, pyrimidopyrimidine, pyrimidodiazepene, and motifs as antimicrobial agents
- Synthesis of a novel phosphate-containing ligand rhodium catalyst and exploration of its optimal reaction conditions and mechanism for the polymerization of phenylacetylene
- Design, synthesis, and antiproliferative activity of novel 1,2,4-triazole-chalcone compounds
- Synthesis of metal–organic nanofiber/rGO nanocomposite as the sensing element for electrochemical determination of hypoxanthine
- Design and synthesis of various 1,3,4-oxadiazoles as AChE and LOX enzyme inhibitors
- Bis(2-cyanoacetohydrazide) as precursors for synthesis of novel azoles/azines and their biological evaluation
- Synthesis, characterization, and biological target prediction of novel 1,3-dithiolo[4,5-b]quinoxaline and thiazolo[4,5-b]quinoxaline derivatives
- Sustainable conversion of carbon dioxide into novel 5-aryldiazenyl-1,2,4-triazol-3-ones using Fe3O4@SP-vanillin-TGA nanocomposite
- Erratum
- Erratum to “Design, synthesis and study of antibacterial and antitubercular activity of quinoline hydrazone hybrids”
- SI: Undergraduate Research in the Synthesis of Biologically Active Small Molecules and Their Applications
- Preparation of novel acyl pyrazoles and triazoles by means of oxidative functionalization reactions
- Synthesis and conformational analysis of N-BOC-protected-3,5-bis(arylidene)-4-piperidone EF-24 analogs as anti-cancer agents
- SI: Development of Heterocycles for Biomedical and Bioanalytical Applications
- Influence of octreotide on apoptosis and metabolome expression in lipopolysaccharide-induced A549 cells
- Crude extract of J1 fermentation promotes apoptosis of cervical cancer cells
Artikel in diesem Heft
- Research Articles
- Synthesis, characterization, and antibacterial activity of a new poly azo compound containing N-arylsuccinimid and dibenzobarrelene moieties
- Design, synthesis, and antiviral activities evaluation of novel quinazoline derivatives containing sulfonamide moiety
- Design, synthesis, and anticancer activity of novel 4,6-dimorpholinyl-1,3,5-triazine compounds
- Design, synthesis, biological evaluation, and bio-computational modeling of imidazo, thieno, pyrimidopyrimidine, pyrimidodiazepene, and motifs as antimicrobial agents
- Synthesis of a novel phosphate-containing ligand rhodium catalyst and exploration of its optimal reaction conditions and mechanism for the polymerization of phenylacetylene
- Design, synthesis, and antiproliferative activity of novel 1,2,4-triazole-chalcone compounds
- Synthesis of metal–organic nanofiber/rGO nanocomposite as the sensing element for electrochemical determination of hypoxanthine
- Design and synthesis of various 1,3,4-oxadiazoles as AChE and LOX enzyme inhibitors
- Bis(2-cyanoacetohydrazide) as precursors for synthesis of novel azoles/azines and their biological evaluation
- Synthesis, characterization, and biological target prediction of novel 1,3-dithiolo[4,5-b]quinoxaline and thiazolo[4,5-b]quinoxaline derivatives
- Sustainable conversion of carbon dioxide into novel 5-aryldiazenyl-1,2,4-triazol-3-ones using Fe3O4@SP-vanillin-TGA nanocomposite
- Erratum
- Erratum to “Design, synthesis and study of antibacterial and antitubercular activity of quinoline hydrazone hybrids”
- SI: Undergraduate Research in the Synthesis of Biologically Active Small Molecules and Their Applications
- Preparation of novel acyl pyrazoles and triazoles by means of oxidative functionalization reactions
- Synthesis and conformational analysis of N-BOC-protected-3,5-bis(arylidene)-4-piperidone EF-24 analogs as anti-cancer agents
- SI: Development of Heterocycles for Biomedical and Bioanalytical Applications
- Influence of octreotide on apoptosis and metabolome expression in lipopolysaccharide-induced A549 cells
- Crude extract of J1 fermentation promotes apoptosis of cervical cancer cells