Design and synthesis of various 1,3,4-oxadiazoles as AChE and LOX enzyme inhibitors
-
Javed Iqbal
 ,
Ali Imran Mallhi
,
Ali Imran Mallhi

Abstract
N-Substituted-2-propanamide analogues of 1,3,4-oxadiazole have been synthesized using a multi-step synthetic protocol to explore new therapeutic anti-enzymatic agents. Herein, we have merged sulfonyl, piperidine, oxadiazole and amide into a single unit to synthesize a library of unique compounds, 8a–n. The molecular structures of all synthesized compounds were verified by 13C-NMR, 1H-NMR, HRMS and IR spectroscopy. Furthermore, the compounds were screened for their inhibition potential against acetylcholinesterase (AChE), urease and lipoxygenase (LOX) enzymes. A considerable inhibition potential was observed for three compounds against LOX with quercetin as a reference standard, two compounds against urease with thiourea as a reference standard and two compounds against AChE with eserine as a reference standard. Through molecular docking investigations, we were able to correlate the overall impact and inhibition criteria by the structure–activity relationship via the interactions between synthesized compounds and active sites of enzymes. Pharmacodynamics, pharmacokinetics and in vivo studies may be investigated further for the most active compounds to substantiate them as potential anti-enzymatic medications.
1 Introduction
The decrease in bioactivity potential of currently available medications has recognized the requirement of simulating solutions. The need of the hour is the availability of new active multifunctional drug candidates with a provocative response for the dangerous diseases that are present now. New active multifunctional drug candidates may help to solve the serious issues of current medications’ ineffectiveness. In this context, the heterocyclic analogues have been found to be active. These have been highlighted in the literature as bioactive agents against a number of enzymes and pathogens [1]. In terms of pharmaceutical research and drug development programs, the heterocyclic 1,3,4-oxadiazole and its analogues have drawn a lot of attention. This heterocyclic moiety has been employed as a medication, anticancer [2], antitumor [3], anticonvulsant [4], antibacterial [5,6], anti-HIV [7], anti-parasitic [8], anti-fungal [9,10], antioxidant [11], anti-diabetic [12] and anti-inflammatory [13] properties are just a few of its numerous applications. It has been observed that the 1,3,4-oxadiazole moiety has the capacity to bind ideally to the active site of an enzyme through its oxygen [14].
The enzymes known as lipoxygenase (LOX) are essential for the metabolism of arachidonic acid and for the production of a variety of lipids with biological activity. These have a significant impact on cancer, bronchial asthma, autoimmune disorders and inflammation. In the current study, we have established the structure–activity relationship (SAR) by docking interaction for the potential against LOX enzymes, as presented in Figure 3(a–d) [15]. Urease, also known as urea amido hydrolase, plays a role in pathogenic processes in both humans and animals. It contributes to peptic ulcers, kidney stones, hepatic encephalopathy, urolithiasis, urinary catheter incrustation and pyelonephritis [16]. Acetylcholine is terminated by the acetylcholinesterase (AChE) enzyme, a serine hydrolase, at cholinergic synapses, which in turn leads to cholinergic brain synapses and neuromuscular connections [17]. The development of novel therapeutic compounds like 1,3,4-oxadiazole analogues is thus imperative to treat many enzyme-generated diseases including inflammation, bronchial asthma, etc.
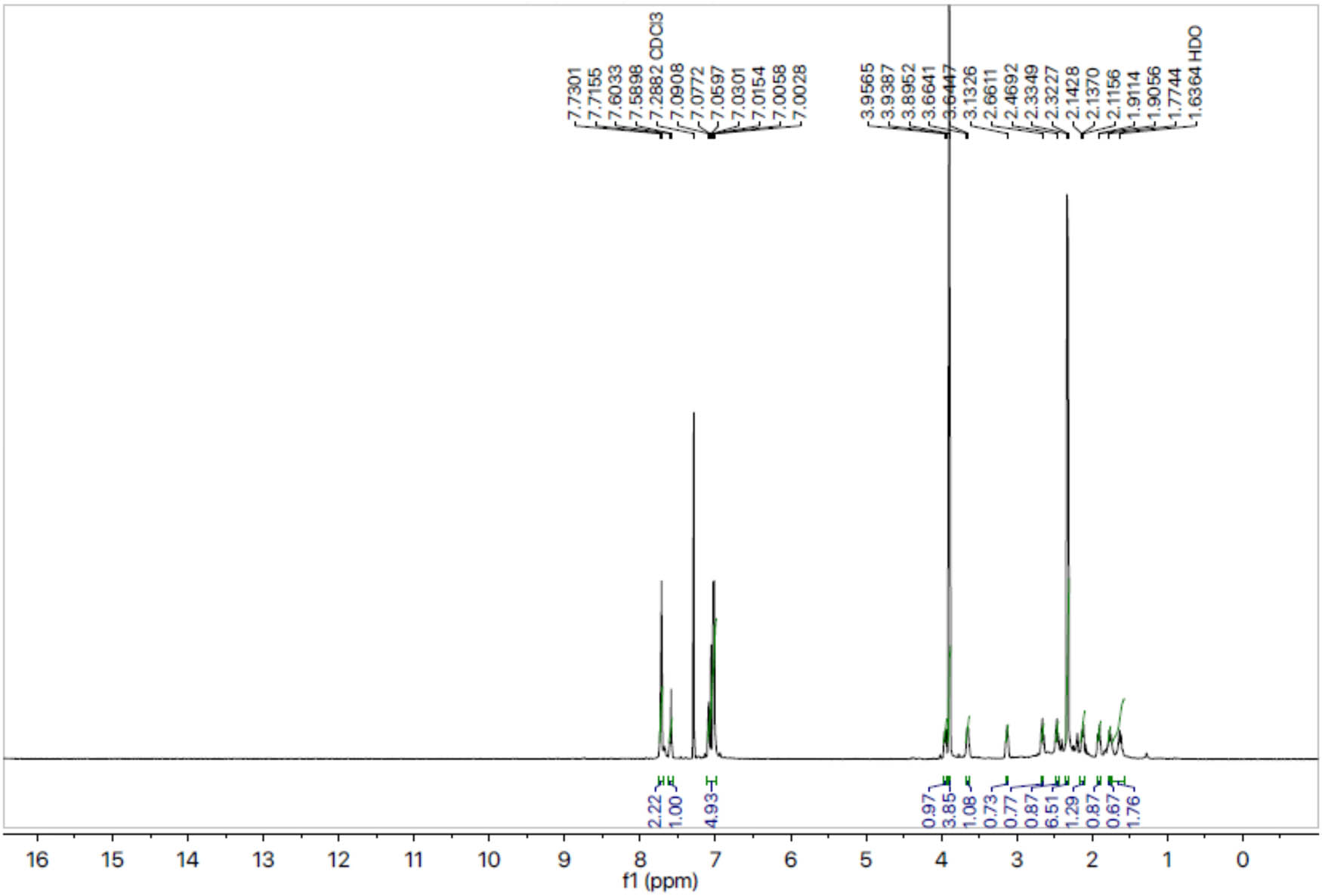
1H-NMR spectrum of compound 8a.
The biological activity of 1,3,4-oxadiazole analogues can be affected, both qualitatively and quantitatively, by slight structural modifications. The current research intends to synthesize N-substituted analogues of 1,3,4-oxadiazoles in search of new bioactive compounds with exceptional medicinal actions to cure disorders related to LOX, urease and AChE enzymes.
2 Results and discussion
2.1 Chemistry
The objectives of this study include the synthesis of N-substituted propanamide analogues (8a–n) of the heterocyclic 1,3,4-oxadiazole moiety and to evaluate their AChE and LOX inhibition potential along with SAR established by docking studies. We synthesized parent compound 5 using an intuitive three-step reaction. The initial step was the reaction of 4-methoxyphenylsulfonyl chloride (1) and ethyl piperidine-3-carboxylate (2) in an aqueous solution at pH 9–10 to obtain the corresponding ester (3), which was then heated with hydrazine hydrate to obtain carbohydrazide, 4. The cyclization of 4 was conducted with CS2 in a basic medium to yield compound 5 in the third step. Different amines (6a–n) were treated with 2-bromopropionyl bromide to generate a library of N-alkyl/aralkyl/aryl-2-proponamides (7a–n). Target compounds were synthesized by reacting 7a–n, with parent compound 5 in the presence of LiH using dimethylformamide as a solvent. After stirring for 12–14 h at room temperature, the reaction mixture was quenched by adding chilled distilled water, filtered and then washed with cold distilled water to yield target compounds 8a–n. Utilizing spectroscopic information from the 13C-NMR, 1H-NMR, HRMS and IR, the structures of newly synthesized compounds were established. All compounds, 8a–n, were tested against AChE, urease and LOX enzymes.
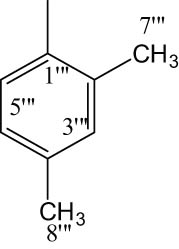
Compound 8a was obtained as an off-white solid. Its molecular formula was assigned C25H30N4O5S2 after analyzing 1H-NMR data and counting the number of protons present in it. The presence of the oxadiazole ring, benzene ring and amide linkage was confirmed by distinctive absorption bands that were shown by the IR spectrum at 3,351 (N–H), 3,077 (Ar C–H), 1,672 (C═N), 1,669 (C═O), 1,596 (Ar C═C), 1,433 (S═O), 1,079 (C–N) and 609 (C–S). The 1H-NMR spectrum (Figure 1) of compound 8a showed two doublets at 7.72 (d, J = 8.8 Hz, 2H, H-6″ and H-2″) and 7.02 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), which indicated the presence of a benzene ring attached to the sulfonyl group. A singlet at 3.89 (s, 3H, CH3-7″) proved that the benzene ring has a methoxy group linked to it. A doublet at 1.42 (d, J = 6.8 Hz, 3H, CH3-3″″) and a quartet at 3.63 (q, J = 6.8 Hz, 2H, CH2-2″″) indicated the presence of the propanamide group. Multiplet signals appearing at 3.95–3.93 (m, 1H, He-2′), 3.66–3.64 (m, 1H, Ha-2′), 3.13–3.12 (m, 1H, H-3′), 2.66–2.64 (m, 1H, He-6′), 2.46–2.36 (m, 1H, Ha-6′), 2.14–2.11 (m, 1H, Ha-5′), 1.91–1.90 (m, 1H, Ha-4′), 1.77–1.75 (m, 1H, He-5′) and 1.63–1.61 (m, 1H, He-4′) confirmed the presence of nine protons of the piperidine ring. The presence of two methyl groups attached to the benzene ring, which are further connected to the nitrogen of the amide functionality, was confirmed by two singlets which appeared at 2.33 (s, 3H, CH3-8‴) and 2.32 (s, 3H, CH3-7‴). The three protons of the benzene ring, which are additionally linked to the nitrogen of the amide functionality, were assigned to two doublets at 7.59 (d, J = 8.1 Hz, 1H, H-6‴) and 7.08 (d, J = 8.2 Hz, 1H, H-5‴); and a singlet at 7.05 (s, 1H, H-3‴). The 13C-NMR spectra (Figure 2) depicted that the most downfield signal appeared at 163.15 ppm for the quaternary carbon of the carbonyl group. Two quaternary carbons present in the oxadiazole ring resonated at 160.21 (C-5) and 160.89 (C-2). The quaternary carbons belonging to the 4-methoxy benzene sulfonyl moiety exhibited signals at 159.93 (C-4″) and 132.57 (C-1″) while the other four methane carbons of this group resonated at 129.73 (C-2″ and C-6″) and 114.32 (C-3″ and C-5″). The signals from three quaternary carbons, which are part of the benzene ring that is connected to the nitrogen of an amide functionality, have been identified at positions 127.65 (C-1‴), 131.49 (C-2‴) and 134.54 (C-4‴). The carbon signals for the two connected methyl groups could be observed at 20.77 (C-8‴) and 17.73 (C-7‴). Five carbon atoms comprising the core of the piperidine ring resonated at 48.45 Hz (C-2′), 46.26 Hz (C-6′), 27.08 Hz (C-3′), 30.92 Hz (C-4′) and 23.71 Hz (C-5′). The signal for the carbon of the methylene group resonated at 33.55 (C-2″″). The signal at 55.64 (C-7″) was assigned to the carbon of the methoxy group. Three signals at 127.60 (C-3‴), 120.01 (C-5‴) and 114.70 (C-6‴) confirmed the presence of three methane carbons of the benzene ring linked to the nitrogen of the amide ring. Therefore, the name of compound 8a was proposed as N-(2,4-dimethylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8a). The rest of the compounds were established in the same way.
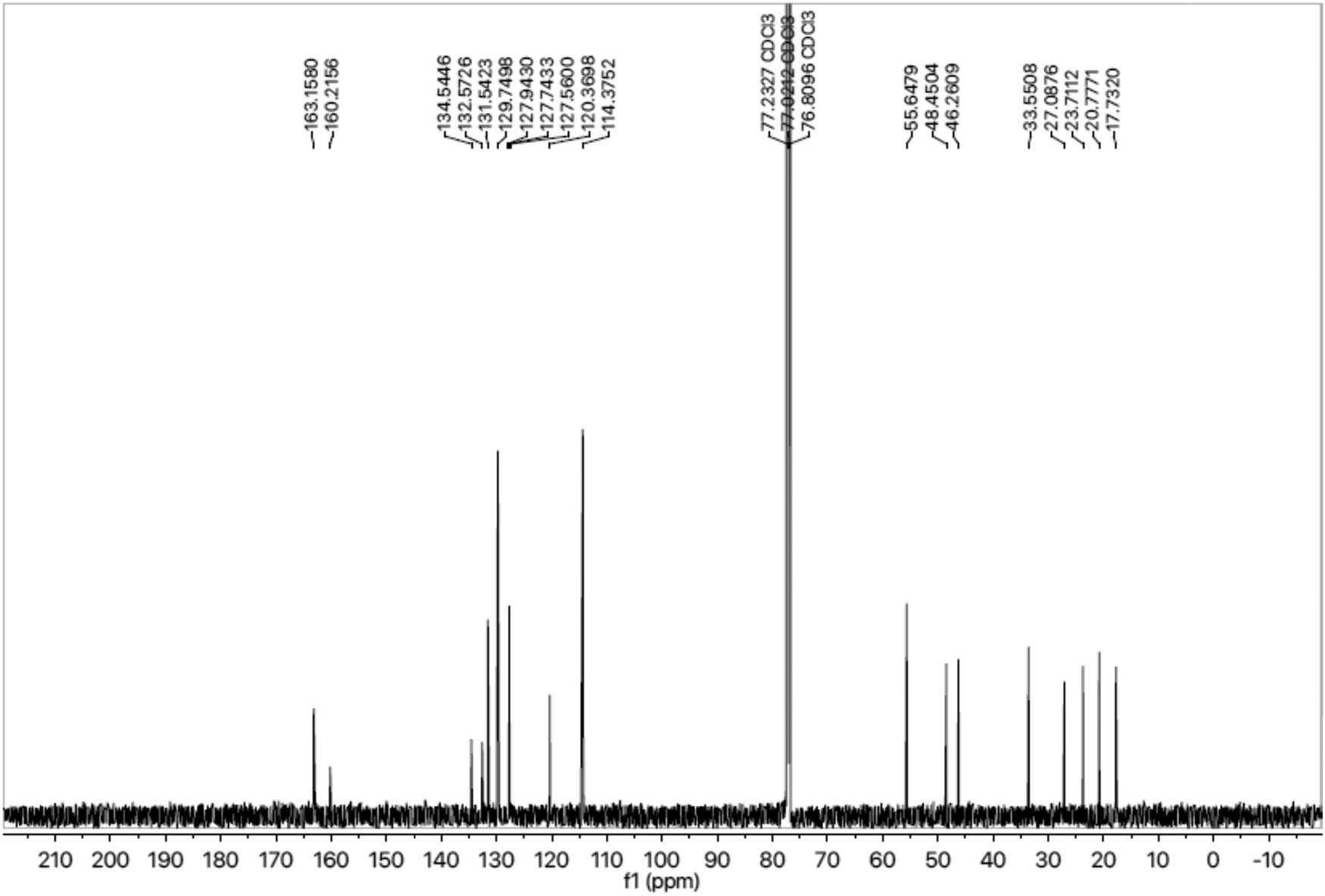
13C-NMR spectrum of compound 8a.
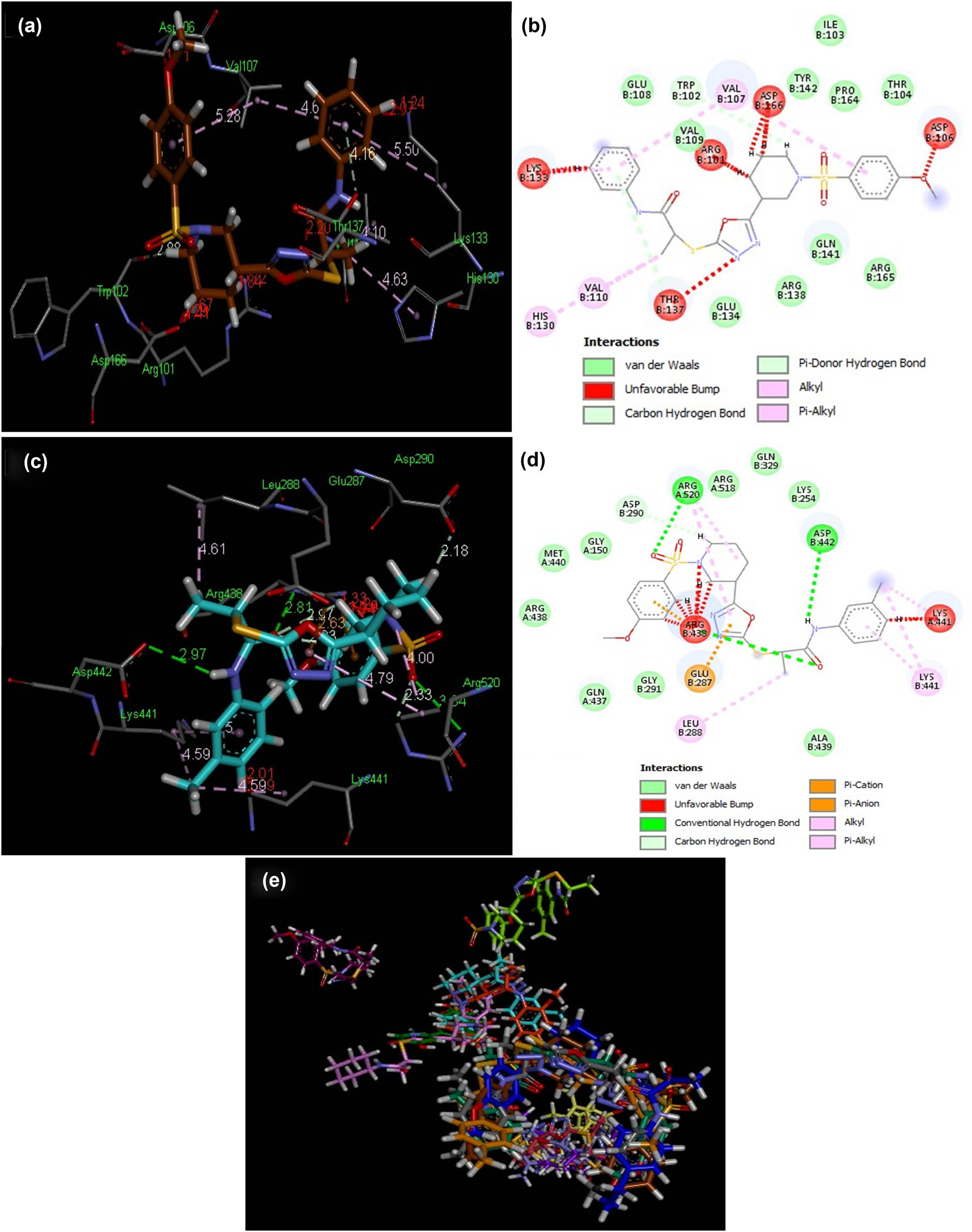
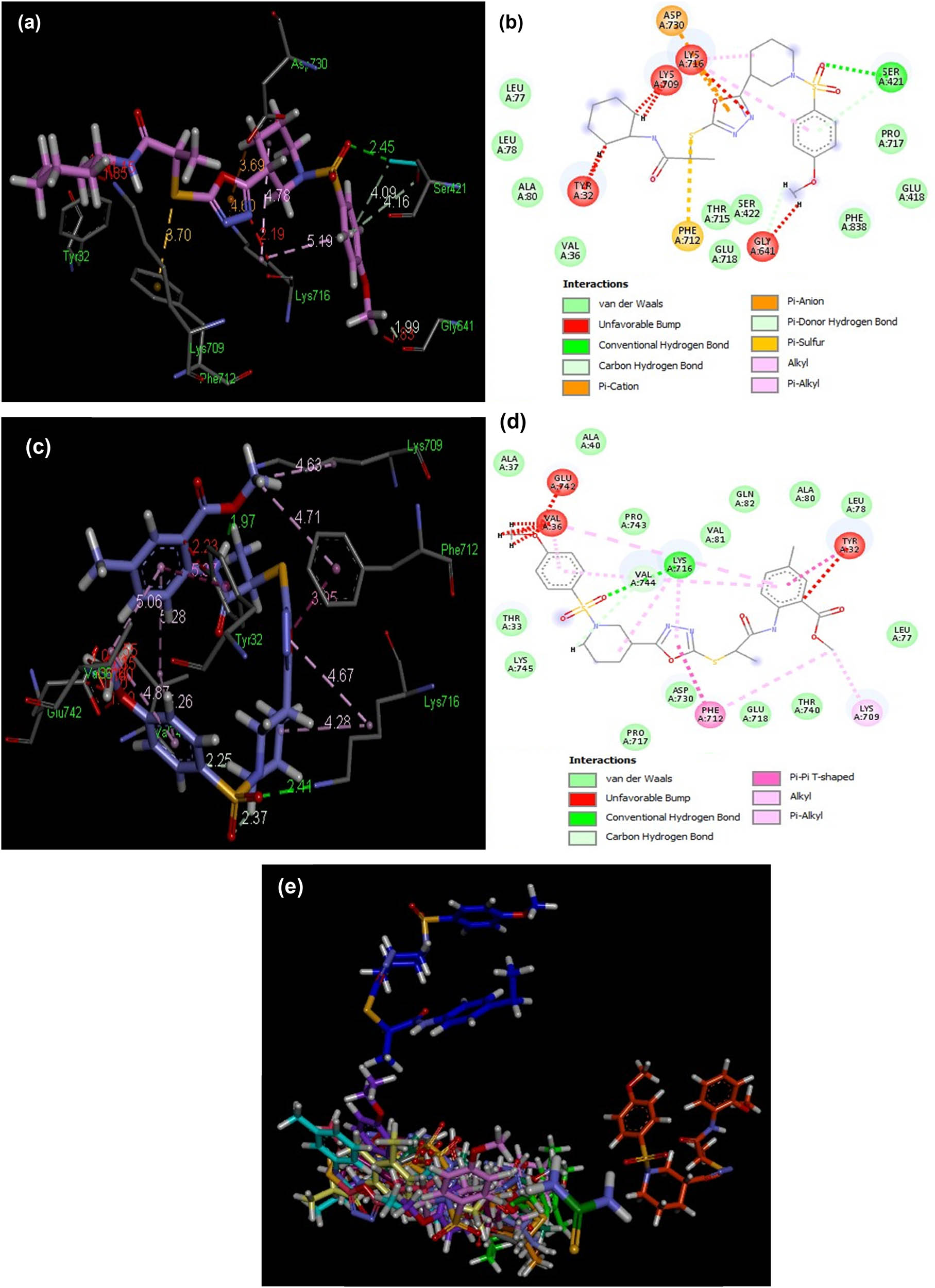
3D and 2D illustrations of docked ligands 8f (a and b) and 8h (c and d) within a hotspot of the LOX protein (PDB ID: 3v99). (e) Overlap of bound conformations of compounds 8 (a–n) showing overlap in two clusters in the hotspot 8a (gray), 8b (yellow), 8c (pink), 8d (magenta), 8e (parrot green), 8f (brown), 8g (sea green), 8h (sky blue), 8i (mustard), 8j (purple), 8k (blue), 8l (baby pink), 8m (orange), 8n (mauve) and quercetin (green).
2.2 Biological activity
2.2.1 LOX inhibition activity
The inhibition potential of LOX of all the synthesized compounds was studied in terms of percentage inhibition and IC50 values, and results are reported in Tables 1 and 2. Results were compared with the standard drug, quercetin. Among the 14 synthesized molecules, 6 compounds remained active against the LOX enzyme. Four compounds, 8f (bearing phenyl moiety), 8h (bearing 3-methylphenyl moiety), 8i (bearing 4-methylphenyl moiety) and 8m (bearing 2-methoxyphenyl moiety), showed significant inhibition potential, while 8j (bearing 4-ethoxyphenyl moiety) and 8n (bearing 2-methoxy-4-methylphenyl moiety)exhibited relatively low inhibition potential. The compounds bearing dimethylphenyl, 2-methylphenyl, 2-ethyl-6-methylphenyl and cyclohexyl moieties remained inactive. Compound 8f bearing a phenyl moiety remained more active as compared to 8l bearing a cyclohexyl moiety. Among mono-methyl substituted compounds, 8g–i, the compound 8h bearing a meta-substituted methylphenyl moiety remained more active as compared to 8i bearing a para-substituted methylphenyl moiety. The para-substitution of the phenyl ring in compound 8i by methyl rendered it more active as compared to 8j bearing a 4-ethoxyphenyl group. The ortho-substitution of the phenyl ring in compound 8m by methoxy rendered it more active as compared to 8g bearing a 4-methylphenyl group. Overall, compounds 8h, 8f and 8i exhibited good activity having inhibition values of 82.51, 81.32 and 78.42%, respectively.
Enzyme inhibition potential of compounds in terms of IC50 values
| Compound | IC50 (µM) | ||
|---|---|---|---|
| LOX | Urease | AChE | |
| 8a | — | — | 132.51 ± 0.52 |
| 8b | — | — | 92.34 ± 0.47 |
| 8c | — | — | — |
| 8d | — | — | |
| 8e | — | — | 63.72 ± 0.39 |
| 8f | 23.57 ± 0.48 | — | 102.46 ± 0.45 |
| 8g | — | — | >500 |
| 8h | 21.28 ± 0.42 | — | >500 |
| 8i | 40.51 ± 0.53 | — | — |
| 8j | 141.23 ± 0.59 | — | >500 |
| 8k | — | — | — |
| 8l | — | 75.7 ± 1.36 | >500 |
| 8m | 95.41 ± 0.23 | — | — |
| 8n | 215.32 ± 0.31 | 74.5 ± 1.25 | — |
| Quercetin | 2.43 ± 0.35 | ||
| Thiourea | 21.25 ± 0.35 | ||
| Eserine | 0.19 ± 0.05 | ||
Quercetin, Thiourea and Eserine are standard used against LOX, Urease and AChE respectively.
Enzyme inhibition potential of compounds in terms of % inhibition
| Compound | Inhibition (%) | ||
|---|---|---|---|
| LOX at 0.25 mM | Urease at 0.25 mM | AChE at 0.5 mM | |
| 8a | 45.23 ± 0.51 | 17.58 ± 1.35 | 71.49 ± 0.85 |
| 8b | 38.45 ± 0.62 | 32.36 ± 1.29 | 78.14 ± 0.91 |
| 8c | 42.52 ± 0.37 | 19.45 ± 1.52 | 34.78 ± 0.87 |
| 8d | 35.43 ± 0.52 | 37.48 ± 1.43 | 32.52 ± 0.63 |
| 8e | 39.54 ± 0.58 | 36.45 ± 1.57 | 84.23 ± 0.85 |
| 8f | 81.32 ± 0.76 | 12.63 ± 1.34 | 72.42 ± 0.72 |
| 8g | 45.26 ± 0.45 | 25.17 ± 1.58 | 29.85 ± 0.59 |
| 8h | 82.51 ± 0.68 | 23.46 ± 1.35 | 51.21 ± 0.65 |
| 8i | 78.42 ± 0.81 | 8.34 ± 1.27 | 34.13 ± 0.88 |
| 8j | 68.31 ± 0.75 | 34.53 ± 1.45 | 52.63 ± 0.48 |
| 8k | 42.23 ± 0.53 | 41.56 ± 1.59 | 42.61 ± 0.56 |
| 8l | 46.54 ± 0.46 | 91.24 ± 1.62 | 51.74 ± 0.52 |
| 8m | 68.35 ± 0.54 | 42.75 ± 1.58 | 43.62 ± 0.37 |
| 8n | 56.42 ± 0.59 | 91.25 ± 1.47 | 36.29 ± 0.64 |
| Quercetin | 89.25 ± 0.62 | ||
| Thiourea | 98.21 ± 0.78 | ||
| Eserine | 91.46 ± 1.25 | ||
Quercetin, Thiourea and Eserine are standard used against LOX, Urease and AChE respectively.
2.2.2 Urease inhibition potential
A study on the inhibition of urease by all the newly synthesized compounds, 8a–n, was carried out by utilizing thiourea as the reference, the results in terms of percentage inhibition and IC50 values are presented in Tables 1 and 2. The whole library of synthesized compounds was found active and showed variable urease inhibition potentials with IC50 values high enough relevant to the reference standard. Among the tested compounds, 8a–n, compound 8n was the most active urease inhibitor with an inhibition of 91.25 ± 1.47% and an IC50 value of 74.5 ± 1.25 as compared to the reference standard, thiourea, with an inhibition of 98.21 ± 0.78 % and an IC50 value of 21.25 ± 0.35. Compound 8n could be a better urease inhibitor and might be a new anti-urease drug in the drug discovery program after further evaluations.
2.2.3 AChE inhibition potential
The IC50 and percent inhibition of acetyl cholinesterase were determined and are presented in Tables 2 and 3. Eserine served as the benchmark. The whole library of synthesized compounds, 8a–n, showed variable inhibition potentials against the AChE enzyme. Four compounds, 8e, 8b, 8f and 8a, were found to be the most active with % inhibition values of 84.23 ± 0.85, 78.14 ± 0.91, 72.42 ± 0.72 and 71.49 ± 0.85, respectively. Similarly, 8e was the most active compound among the synthesized series of compounds with an IC50 value of 63.72 ± 0.39. The better potential possessed by compound 8e might be due to the presence of two methyl groups at positions 3 and 5 (Table 3).
Different alkyl/aralkyl/aryl groups
| Compound | R groups | Compound | R groups | Compound | R groups |
|---|---|---|---|---|---|
| 8a |
|
8f |
|
8k |
|
| 8b |
|
8g |
|
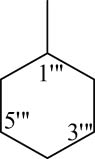
8l |
|
| 8c |
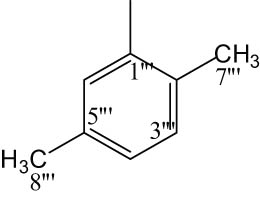
|
8h |
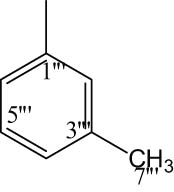
|
8m |
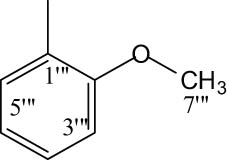
|
| 8d |
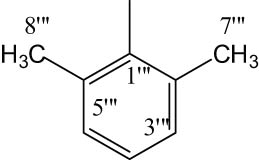
|
8i |
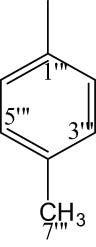
|
8n |
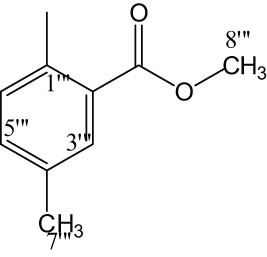
|
| 8e |
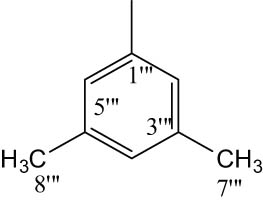
|
8j |
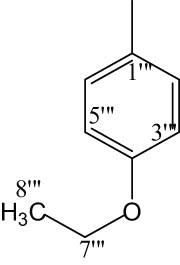
|
2.2.4 Molecular docking study
To obtain qualitative estimation and to identify the molecular basis of the calculated biological activities (IC50), the docked complexes of compounds 8(a–n) were examined. The binding contacts of new compounds 8(a–n) and the positive control (quercetin) with LOX 5-LOX (PDB ID 3V99) revealed that compounds 8f and 8h showed significant interaction patterns (Figure 3). Compound 8h exhibited the most potent interaction with LOX 5-LOX (PDB ID 3V99) with a score of 7,182 and an atomic contact energy (ACE) of –365.80 kcal/mol (Table 4). Compound 8h showed hydrogen bonds between NH of Arg520:one of the oxygen of the sulfur dioxide of compound 8h with an average distance of 3.04 Å, another hydrogen bond was observed between oxygen of the carboxylic acid of Asp442 and NH of the compound 8h (2.97 Å), and the third hydrogen bond was depicted between NH of Arg438 and the oxygen of the carbonyl directly attached with NH of compound 8h (2.81 Å). Compound 8h exhibited hydrophobic interaction potential with pocket amino acids Arg438, Met440, Gly150, Arg518, Gln329, Lys254, Ala439, Gly291, and Gln437 and also illustrated a pi–anion contact potential with Glu287 and a pi–cation interaction potential with Arg438 (Figure 4). Interestingly, ligands 8f exhibited no hydrogen bond interaction with the 5-LOX (PDB ID 3V99) receptor but instead found considerable geometric fit of these compounds in the receptor, and therefore scoring in PatchDock being based on shape complementarity principles it gave a score of 6,998 and an ACE value of –286.57 kcal/mol with 5-LOX (PDB ID 3V99). Compound 8f showed a hydrophobic interaction potential with hotspot amino acid residues Glu108, Val109, Tyr142, Ile103, Pro164, Thr104, Arg165, Gln141, Arg138 and Glu134.
Docking results for the highest-ranked biologically active ligands (LOX)
| Compound No. | Score | ACE (kcal/mol) | Amino acids show hydrogen bond contacts | Distance (Å) | Amino acids show hydrophobic contacts | Amino acids show pi–cation and pi–anion contacts |
|---|---|---|---|---|---|---|
| 8a | 6,846 | −256.42 | Val110 | 2.47 | Arg138, Glu108, His130, Tyr142, Asp166, Ile167, Thr104 | — |
| 8b | 5,824 | −155.25 | — | — | Ala439, Arg457, Val361, Phe286, Glu287, Arg246, Leu244, Lys441, Thr266, Arg370, Ser447, Thr444 | — |
| 8c | 5,916 | −134.83 | — | — | Thr366, Ala439, Glu287, Ilu283, Leu244, Phe286, Ala453 | Arg246, Arg370 |
| 8d | 5,758 | −226.65 | — | — | Asp156, Pro331, Leu305, Gly332, Asn328, Asn335, Asp290, Arg518, Gln329, Glu146, Met145, Asp333 | — |
| 8e | 5,956 | −201.29 | — | — | Val361, Thr366, Ala456, Arg457, Arg370, Phe286 | Arg246 |
| 8f | 6,998 | −286.57 | — | — | Glu108, Val109, Tyr142, Ile103, Pro164, Thr104, Arg165, Gln141, Arg138, Glu134 | — |
| 8g | 6,990 | −197.82 | Lys133, Val110, Arg101 | 2.75, 3.23, 3.05 | Trp102, Ile167, Thr104, Asp166, Val109, Tyr142, Glu134, Thr137, Glu108, His130 | — |
| 8h | 7,182 | −365.80 | Arg520, Asp442, Arg438 | 3.04, 2.97, 2.81 | Arg438, Met440, Gly150, Arg518, Gln329, Lys254, Ala439, Gly291, Gln437 | Arg438, Glu287 |
| 8i | 6,646 | −210.26 | Asp106, Arg165 | 2.82, 3.31 | Gly105, Trp102, Val109, Val110, Lys133, Glu134, Thr137, Arg138, Gln141 | Asp166 |
| 8j | 6,228 | −260.15 | — | — | Ala256, Val284, Phe286, Gln549, Tyr470, Ser447, Arg370, Asp442, Ile365, Ile283 | — |
| 8k | 6,720 | −205.20 | Thr104, Arg101 | 2.91, 3.20 | Leu111, Arg165, Glu134, Lys133, Val109, Thr137, Arg138, Tyr142, Gln141, Pro164, Trp102 | Glu108 |
| 8l | 6,986 | −178.42 | Gln434 | 3.25 | Met440, Gln437, Glu287, Lys254, Asp290, Arg518, Ile292, Gln329, Ile292, Gln434, Gly291, Gly430 | Arg438 |
| 8m | 6,068 | −69.93 | Ser522 | 3.01 | Arg438, Gln437, Gly150, Met440, Asp290, Arg518, Ala439, Leu288, Gly291, Gln437 | Glu287 |
| 8n | 6,120 | −215.17 | Intramolecular H-bonding | 1.97 | Ile245, Thr444, Leu289, Ile283, Asp285, Ile365, Asp442, Arg370, Ala453, Ala456 | — |
| Quercetin Std. | 4,352 | −70.76 | Gln434 | 2.46 | Arg438, Lys441, Arg518, Asp290, Gly150, Ile292, Gln437 | Arg438 |
No. = specific code assigned to the ligand; ACE = atomic contact energy calculated by PatchDock (kcal/mol); IC50 = experimental calculation of the inhibitory constant (µM); Distance = the hydrogen bond length calculated from the docked pose using Ligand interaction tool of PatchDock.
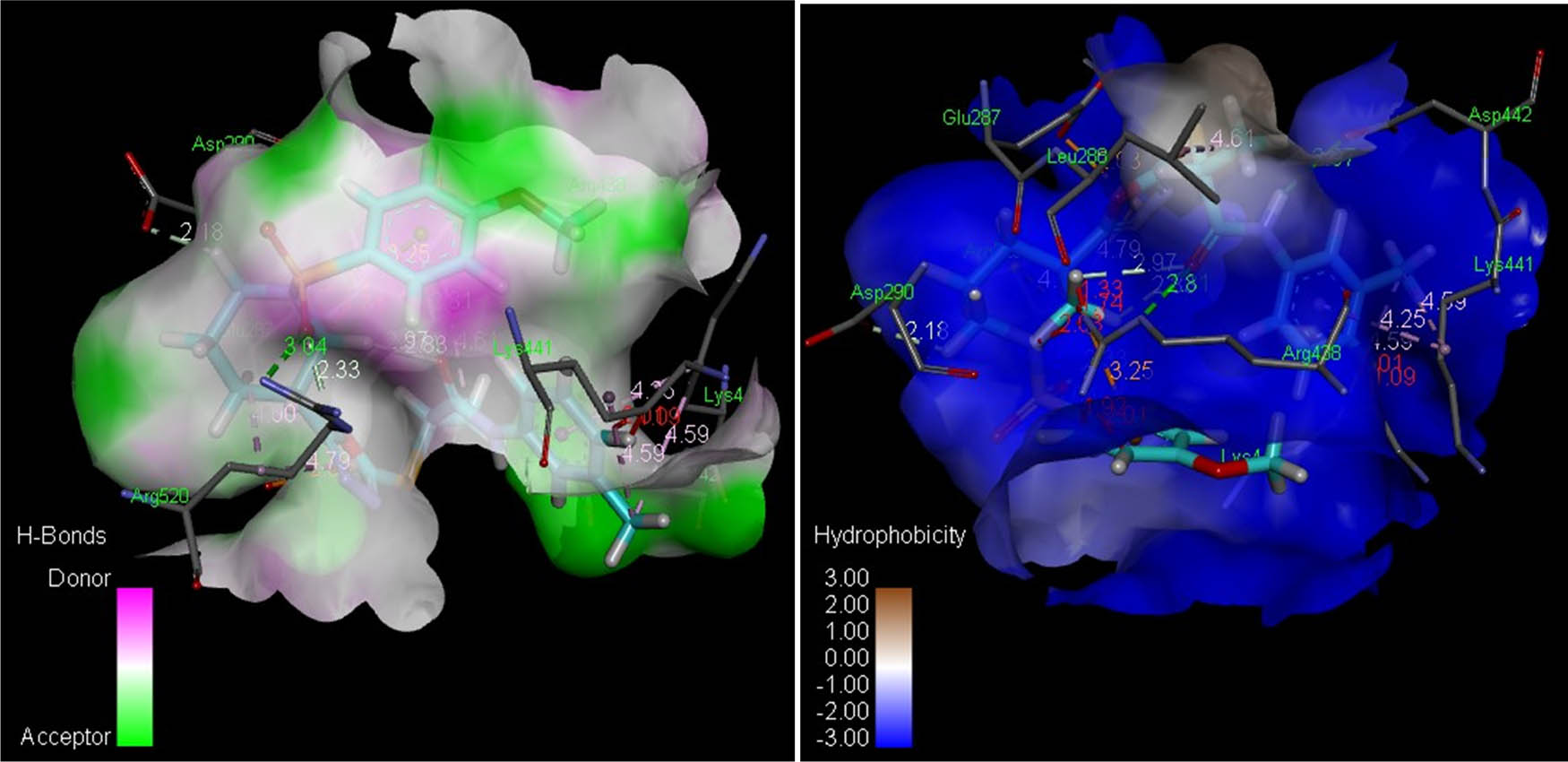
Analyses of H-bonds (left) and hydrophobic interactions (right) of compound 8h. The blue color in the hydrophobic contacts (right) depicts favorable structural elements (atoms and torsions) contributing to the entire binding energy within the LOX protein binding pocket (PDB ID: 3v99); the pink one is unfavorable, and the white one is neutral.
The docked complexes of Jack bean urease (JBU, PDB ID 4h9m) with compounds 8(a–n) and standard thiourea showed that compounds 8l and 8n depicted important interaction sequences as shown in Figure 5. Compound 8n showed the most potent contact with JBU (PDB ID 4h9m) with a score of 6,846 and an ACE of –368.49 kcal/mol (Table 5). A hydrogen bond was observed between the amino group of Lys716 and one of the oxygen of the sulfur dioxide of compound 8n (2.41 Å). Similarly, an intramolecular hydrogen bonding was observed between the oxygen of the methyl ester group and NH flanked between phenyl and carbonyl groups of compound 8n with an average distance of 1.97 Å. Compound 8n also showed hydrophobic contact potential with pocket amino acids Ala37, Ala40, Pro743, Val81, Gln82, Ala80, Leu78, Leu77, Thr740, Glu718, Asp730, Pro717, Lys745 and Thr33 and also illustrated a pi–pi T-shaped contact potential with Phe712 (Figure 6). Compound 8l has shown good interaction with JBU (PDB ID 4h9m) with a score of 6,806 and an ACE of –351.11 kcal/mol. A hydrogen bond was observed between Ser421 and one of the oxygen of the sulfur dioxide of compound 8l with an average distance of 2.45 Å. Compound 8l exhibited hydrophobic contact potential with pocket amino acids Pro717, Glu418, Phe838, Ser422, Thr715, Glu718, Val36, Ala80, Leu78 and Leu77.
3D and 2D representations of docked ligands 8l (a and b) and 8n (c and d) within a binding pocket of the JBU protein (PDB ID: 4h9m). (e) Overlap of bound conformations of compounds 8(a–n) illustrating overlap in two clusters in the binding pocket 8a (gray), 8b (yellow), 8c (pink), 8d (magenta), 8e (parrot green), 8f (brown), 8g (sea green), 8h (sky blue), 8i (mustard), 8j (purple), 8k (blue), 8l (baby pink), 8m (orange), 8n (mauve) and thiourea (green).
Docking results for the highest-ranked biologically active ligands (urease)
| Compound No. | Score | ACE (kcal/mol) | Amino acids show hydrogen bond contacts | Distance (Å) | Amino acids show hydrophobic contacts | Amino acids show pi–pi T-shaped contacts |
|---|---|---|---|---|---|---|
| 8a | 5,974 | −161.28 | Lys709 | 2.89 | Lys745, Tyr837, Ala37, Glu34, Tyr32, Val36, Phe712, Glu718, Thr740, Asp730, Ala80, Gln82 | — |
| 8b | 6,018 | −230.02 | — | — | Leu13, Gly12, Lys745, Met746, Glu746, Glu742, Phe838, Phe712, Asp730 | — |
| 8c | 5,666 | −235.75 | — | — | Met746, Thr33, Glu742, Lys709 | Phe712 |
| 8d | 5,724 | −262.97 | Val744 | 3.33 | Lys709, Lys745, Pro743, Met746 | Phe712 |
| 8e | 5,444 | −154.12 | Gln82 | 3.10 | Val81, Glu742, Val36, Asp730, Lys716, Pro717, Glu718, Leu77, Leu78, Thr740 | — |
| 8f | 6,722 | −149.78 | Lys709, Tyr32, Thr33, Lys716 | 2.58, 2.59, 3.34, 2.94 | Ala37, Leu13, Ala40, Glu718, Val36, Ala80, Gln82, Val81, Thr740, Asp730, Lys745, Pro743 | — |
| 8g | 6,140 | −152.51 | Thr33 | 2.83 | Leu13, Pro743, Tyr837, Phe838, Val36, Glu718, Ala80, Val81, Gln82, Pro741, Thr740, Asp730, Lys745, Ala37 | — |
| 8h | 5,474 | −314.18 | — | — | Glu742, Leu19, Gly12, Thr33, Met746, Asp730 | — |
| 8i | 6,730 | −157.34 | Thr33 | 3.26 | Thr740, Pro741, Val81, Gln82, Tyr32, Phe712, Val36, Leu13, Ala40, Ala16, Ala37, Tyr837, Lys745, Asp730, Lys709, Glu718 | — |
| 8j | 5,802 | −266.54 | — | — | Phe712, Lys745, Glu742, Phe840, Leu13, Ala16, Ala37, Thr33 | — |
| 8k | 5,904 | −212.23 | — | — | Asn836, Leu71, Leu839, Leu833, Arg23, Glu34, Asn31 | — |
| 8l | 6,806 | −351.11 | Ser421 | 2.45 | Pro717, Glu418, Phe838, Ser422, Thr715, Glu718, Val36, Ala80, Leu78, Leu77 | — |
| 8m | 5,518 | −302.84 | Ser711 | 2.65 | Pro134, Ile133, Gly135, Lys709, Leu397, Glu395, Glu136, Gly396, Lys277, Gly284, Pro285, Leu77, His76 | — |
| 8n | 6,846 | −368.49 | Lys716, Intramolecular H-bonding | 2.41, 1.97 | Ala37, Ala40, Pro743, Val81, Gln82, Ala80, Leu78, Leu77, Thr740, Glu718, Asp730, Pro717, Lys745, Thr33 | Phe712 |
| Thiourea Std. | 1,672 | −151.81 | Leu491 | 3.49 | Tyr544, Thr441, His519, His492, Leu489, Cys443, Ile518, Asn517 | — |
No. = specific code assigned to the ligand; ACE = atomic contact energy calculated by PatchDock (kcal/mol); IC50 = experimental calculation of the inhibitory constant (µM); Distance = hydrogen bond length calculated from the docked pose using Ligand interaction tool of PatchDock.
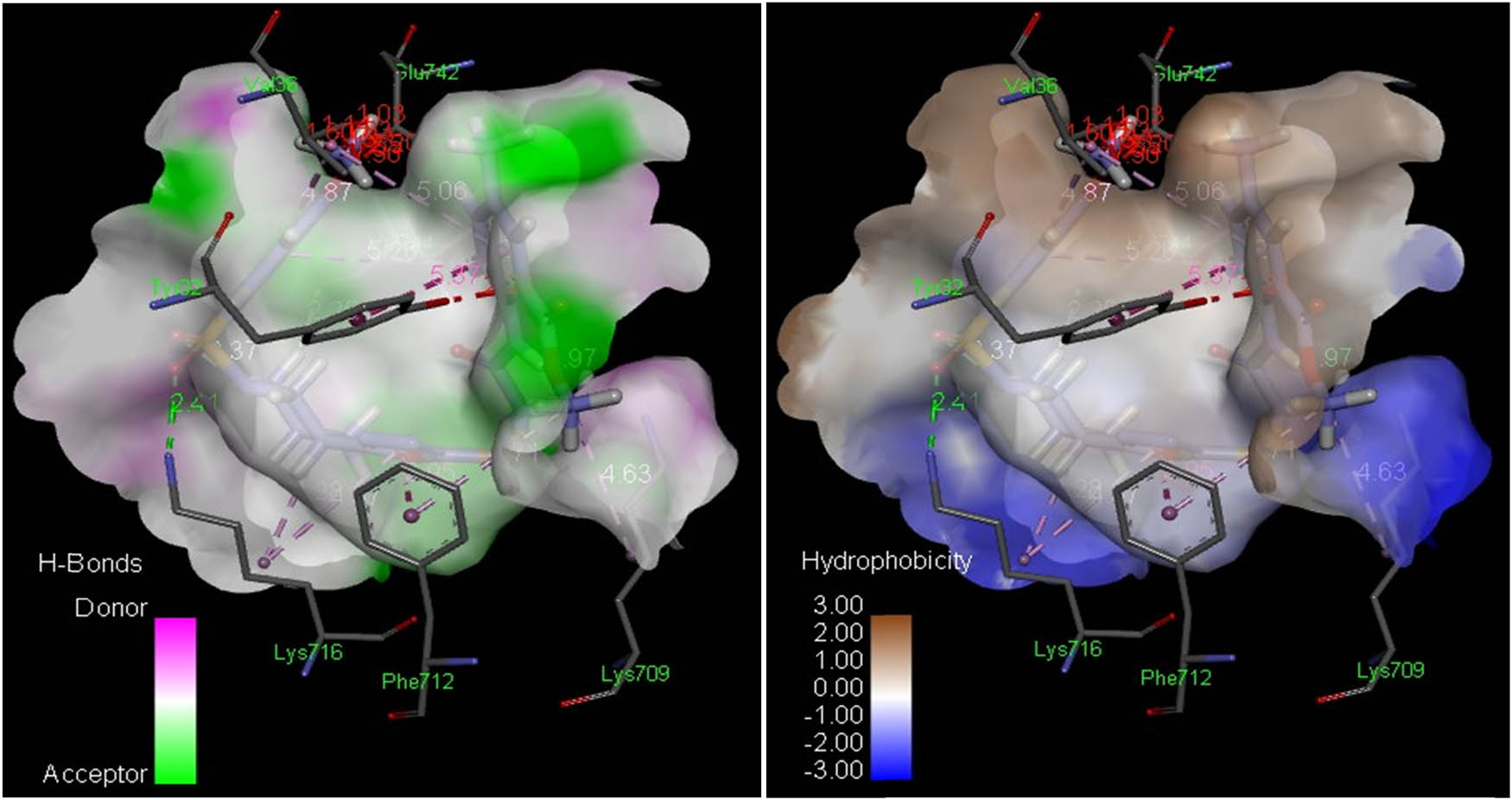
Analyses of H-bonds (left) and hydrophobic interactions (right) of compound 8n. The blue color in the hydrophobic contacts (right) exhibits favorable structural elements (atoms and torsions) contributing to the whole binding energy within the JBU protein binding pocket (PDB ID: 4h9m); the pink one is unfavorable, and the white one is neutral.
In the initial evaluation of the docked complexes of AChE (PDB ID 4m0e) with compounds 8(a–n) and eserine standard, it was predicted that compounds 8b and 8e exhibited excellent binding interactions (Figure 7). Compound 8e possessed the most potent interaction with AChE with a score of 6,340 and an ACE value of –325.34 kcal/mol. A hydrogen bond was observed between the hydroxyl group of Thr238 and sulfur was directly attached to the 1,3,4-oxadiazole moiety (2.87 Å). Compound 8e showed hydrophobic interaction potential with pocket amino acids Leu536, Trp532, Pro410, Gly234, Val303, Arg296, His405, Val370 and Val367 s as shown in Figure 8. Compound 8b has shown good interaction with AChE (PDB ID 4m0e) with a score of 6,122 and an ACE value of –317.71 kcal/mol. Interestingly, ligand 8b depicted no hydrogen bond contact with the AChE (PDB ID 4m0e) receptor but instead found considerable geometric fit of these ligands in the receptor, and hence scoring in PatchDock was based on shape complementarity principles (Table 6). Compound 8b showed hydrophobic contact potential with pocket amino acid residues Cys409, Gln413, Asn533, Gly234, Trp532, Leu536, Thr311, Pro312, Trp236, Ala314, Ala237, Val303, Val239 and Pro368. Compound 8b has shown potential for pi–pi T-shaped interaction with His405.
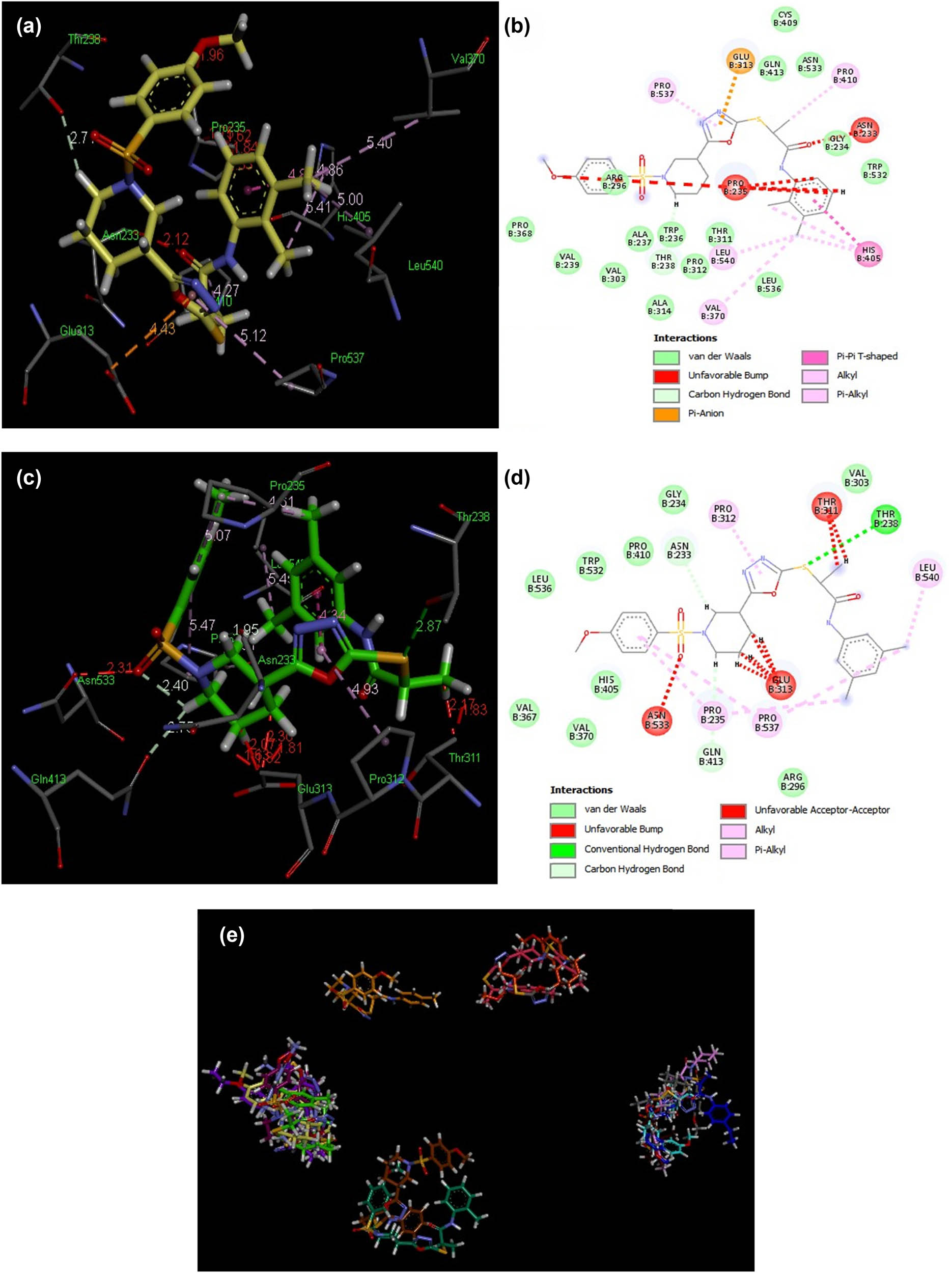
3D and 2D exhibitions of docked ligands 8b (a and b) and 8e (c and d) within a hotspot of the AChE protein (PDB ID: 4m0e). (e) Overlap of bound conformations of compounds 8(a–n) depicting overlap in two clusters in the hotspot 8a (gray), 8b (yellow), 8c (pink), 8d (magenta), 8e (parrot green), 8f (brown), 8g (sea green), 8h (sky blue), 8i (mustard), 8j (purple), 8k (blue), 8l (baby pink), 8m (orange), 8n (mauve) and eserine (green).
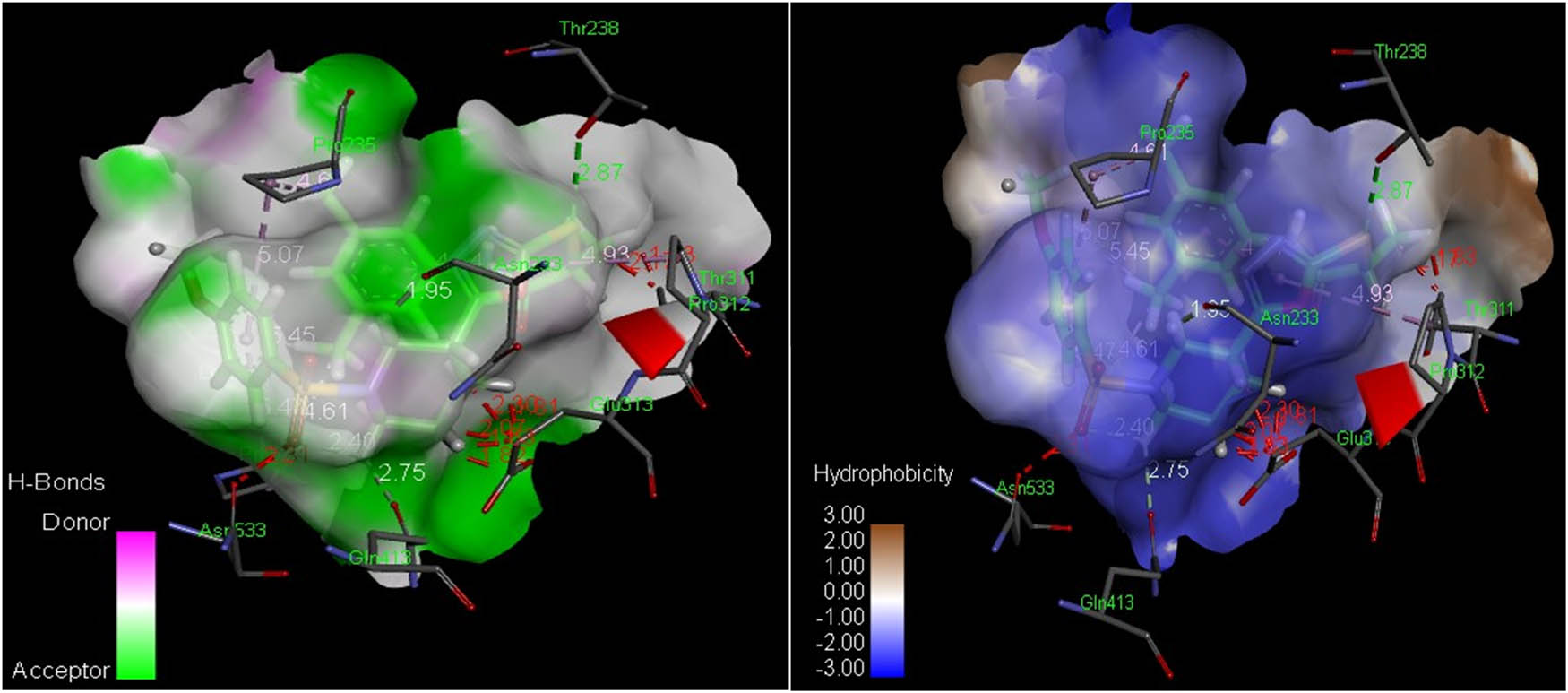
Analyses of H-bonds (left) and hydrophobic interactions (right) of compound 8e. The blue color in the hydrophobic contacts (right) shows favorable structural elements (atoms and torsions) contributing to the overall binding energy within the AChE protein hotspot (PDB ID: 4m0e); the pink one is unfavorable, and the white one is neutral.
Docking results for the highest-ranked biologically active ligands (AChE)
| Compound No. | Score | ACE (kcal/mol) | Amino acids show hydrogen bond contacts | Distance (Å) | Amino acids show hydrophobic contacts | Amino acids show pi–pi T-shaped contacts |
|---|---|---|---|---|---|---|
| 8a | 6,074 | −73.72 | Arg463 | 2.47 | Phe137, Asp134, Tyr449, Glu81, Ser438, Leu437 | Tyr465 |
| 8b | 6,122 | −317.71 | — | — | Cys409, Gln413, Asn533, Gly234, Trp532, Leu536, Thr311, Pro312, Trp236, Ala314, Ala237, Val303, Val239, Pro368 | His405 |
| 8c | 5,244 | −225.43 | Arg247, Thr238 | 3.31, 2.97 | Val239, Trp236, Leu536, Trp532, Asn533, His405, Arg296, Pro368, Val370, Asn233, Pro312, Ala237, Gly240, Glu243 | — |
| 8d | 5,280 | −256.71 | Thr238 | 3.09 | Val239,Val303, Ala237, Thr311, Pro232, Gly234, Asn233, Trp532, Val370, Asn533, Gln413, Pro410, Cys409, Gln369 | His405 |
| 8e | 6,340 | −325.34 | Thr238 | 2.87 | Leu536, Trp532, Pro410, Gly234, Val303, Arg296, His405, Val370, Val367 | — |
| 8f | 5,902 | −50.79 | Lys538, Gln413 | 3.10, 3.39 | Pro537, Trp385, Arg356, Val389, Leu360, Glu376, Gly416, Ala412, Tyr508, Ala505, Gln508, Gly506 | — |
| 8g | 5,896 | −141.90 | Arg417, Arg534 | 2.44, 3.36 | Leu360, Trp385, Gly506 Gln508, Tyr508, Gly416, Ala412, Lys538 | — |
| 8h | 5,870 | −117.56 | Tyr465 | 3.34 | Ser438, Leu437, Gly82, Tyr449, Asp134, Phe137, Asp460, Glu452, Asp131 | — |
| 8i | 5,870 | −117.56 | Arg365, Ala505 | 2.73, 1.96 | Gly506, Gln508, Gly416, Ala412, Gln413, Arg417, Lys538, Leu360 | — |
| 8j | 5,868 | −404.73 | Glu313 | 3.79 | Val370, Leu540, Leu536, Pro537, Asn533, Gln413, Trp236, Val300, Arg247, Val239, Thr311, Pro312, Gly234 | — |
| 8k | 5,840 | −128.51 | Arg463, Tyr465, Thr436 | 3.17, 2.22, 2.93 | Trp439, Leu437, Tyr449, Leu130, Phe80, Gly79, Glu84 | — |
| 8l | 5,700 | −26.50 | Tyr465 | 2.45 | Leu22, Ile457, Asp131, Glu452, Tyr449, Ser438, Asp460, Phe137, Asp134 | — |
| 8m | 5,608 | −254.87 | Arg296 | 3.35 | Pro312, Val239, Trp236, Arg247, Gly234, Gln369, Pro410, Leu536, His405, Pro537, Trp532, Pro368, Gly240, Thr311 | — |
| 8n | 5,758 | −336.76 | Intramolecular H-bonding | 1.97 | Pro312, Gly234, Pro410, Pro537, Arg296, Gln369, Pro368, Val370, Leu536, Cys409, Asn533, Gln413 | — |
| Eserine Std. | 4,342 | −85.74 | Glu81, Glu81 | 2.69, 2.77 | Asp131, Val132, Ile457, Tyr449, Leu437, Ser438 | — |
No. = specific code assigned to the ligand; ACE = atomic contact energy calculated by PatchDock (kcal/mol); IC50 = experimental calculation of the inhibitory constant (µM); Distance = hydrogen bond length calculated from the docked pose using Ligand interaction tool of PatchDock.
Our docking experiments have shown that compounds 8f and 8h against 5-LOX (PDB ID 3V99) revealed significant interaction patterns and compounds 8l and 8n depicted important contact against JBU (PDB ID 4h9m), while compounds 8b and 8e for AChE (PDB ID 4m0e) could possibly show inhibitory potential against AChE. This is consistent with the IC50 values of the ligands 8(a–n).
3 Conclusion
In an attempt to identify new therapeutic agents for drug formulation research, we formulated a library of novel compounds with piperidine and 1,3,4-oxadiazole moieties. The structures of the synthesized compounds have been revealed by spectroscopic data. The ability of all compounds to inhibit the enzymes AChE, urease and LOX was examined. A number of compounds demonstrated excellent enzyme inhibition capability against the above enzymes. The SAR relationship of active anti-enzymatic compounds has been established by docking studies. New bioactive compounds may be developed as a result of the modest modifications to the primary molecular structure. To improve the results of drug discovery and drug formulation programs, new derivatives could be synthesized with slight changes to the substituents.
4 Experimental
The chemicals required to synthesize the targeted molecules were provided by Sigma Aldrich and Alfa Aesar. The potassium bromide approach was utilized to record IR spectra using a Jasco-320A spectrometer. The Nuclear magnetic resonance spectra for carbon and protons were obtained using a Bruker spectrometer operating at 150 and 600 MHz, respectively. DMSO/CDCl3 was employed as a solvent. To verify the reaction accuracy and purity of freshly synthesized compounds, thin-layer chromatography (TLC) was carried out on aluminum plates (pre-coated with silica gel) using the solvent system of n-hexane and ethyl acetate. A UV lamp (254 nm) and iodine solution were utilized to visualize the TLC plates. Uncorrected melting points of the newly synthesized compounds were recorded using the Griffin and George apparatus.
4.1 Synthesis of ethyl 1-[(4-methoxyphenyl)sulfonyl]piperidin-3-carboxylate (3)
Ethyl piperidin-3-carboxylate (2, 0.05 mol) was dispersed in 50 mL distilled water. Aqueous Na2CO3 (10 mL) was added with stirring. Equimolar 4-methoxyphenylsulfonyl chloride (1) was added dropwise with stirring. The pH of the reaction was maintained at 9–10 by adding 5% aqueous Na2CO3. The reaction progress was monitored by TLC. The reaction mixture was neutralized with dilute HCl on completion. A white solid was collected by filtration. The product was washed with water, and dried to yield compound 3.
4.2 Synthesis of 1-[(4-methoxyphenyl)sulfonyl]piperidin-3-carbohydrazide (4)
Equimolar amounts of compound 3 and hydrazine hydrate were refluxed for 2–4 h in methanol. TLC was used to confirm the formation of product 4. The extra solvent present in the reaction mixture was evaporated by slow heating, followed by cooling. The formed product was filtered and dried at room temperature for further utilization.
4.3 Synthesis of 5-[1-(4-methoxyphenyl)sulfonyl)piperidin-3-yl]-1,3,4-oxadiazol-2-thiol (5)
Compound 4 was refluxed with equimolar aq. KOH for half an hour in the presence of MeOH used as a solvent. It was followed by the addition of equimolar CS2 at room temperature. The whole mixture was refluxed for a further 6h, and the reaction progress was monitored by TLC. At completion, cold distilled water was added to the reaction mixture and pH was adjusted to 6 by adding dilute HCl dropwise. Product 5 was obtained as a fine precipitate, which was filtered, washed and dried at room temperature for further use.
4.4 General procedure for the synthesis of N-alkyl/aralkyl/aryl-2-proponamides (7a–n)
Equimolar amounts of 2-bromopropionyl bromide with various alkyl/aralkyl/aryl amines (6a–n) were stirred at room temperature in an aqueous basic media. The pH was maintained at 9–10 with 20% sodium carbonate solution. The reaction progress was monitored by TLC. Compounds, 7a–n, were obtained in the form of precipitates through filtration, washing and drying at room temperature.
4.5 Synthesis of N-alkyl/aralkyl/aryl-2-proponamide derivatives of 1,3,4-oxadiazole (8a–n)
Compound 5 (0.03 mol) was dissolved in 20 mL dimethyl formamide followed by the addition of equimolar LiH. The mixture was stirred for 30 min. An equimolar amount of various electrophiles, 7a–n, were added and set to stir for a further 12–14 h at room temperature. Reaction progress was monitored by TLC. At completion, cold distilled water was added to obtain the title compounds, 8a–n, which were further filtered or extracted according to the nature of the product.
4.6 Biological activity assays
4.6.1 LOX inhibition assay
The inhibition potential of compounds against the 15-LOX enzyme was performed using the UV-based protocol as reported earlier [18]. A mixture of test compound, buffer solution (pH 6.8) and 15-LOX was made in a 96-well plate. After pre-incubation for 5 min, the absorbance was measured at 234 nm. Then, a 25 μL solution of the linoleic acid substrate was added to initiate the reaction and contents were incubated for 10 min. After incubation, the absorbance was measured at 234 nm. All experiments were performed in triplicate. The IC50 values of active compounds were calculated as given for the AChE inhibition assay.
4.6.2 Urease inhibition assay
The reported protocol [19] was adopted to evaluate the anti-urease activity of the whole series of synthesized compounds, 8a–n. The enzyme and sample solution were mixed in the presence of a phosphate buffer solution. The absorbance was measured at 625 nm using a Synergy HT (BioTek, USA) microplate reader before and after the addition of urea stock solution. Reagents for phenol hypochlorite were also included. IC50 values and percentage inhibition were calculated by the method given for the AChE inhibition assay.
4.6.3 AChE inhibition assay
Screening of compounds against AChE was carried out as mentioned earlier [17], wherein eserine was used as a standard. The chemistry of the reaction is as follows:
The 100 μL reaction mixture contained 60 μL of phosphate buffer (pH 7.7), test solutions and the enzyme AChE. After 5 min pre-incubation at 37°C, the absorbance was measured at 405 nm. The reaction was started by the addition of 0.5 mM of 10 μL acetylthiocholine (for AChE) and 10 μL of 0.5 mM DTNB. After incubation for 20–25 min at 37°C, the contents were read at 405 nm by using a 96-well plate reader (Synergy HT, BioTek, USA). The inhibition (%) was calculated by the following formula:
IC50 values of the active compounds were computed using Ez-Fit software (Perrella Scientific Inc. Amherst, USA).
4.6.4 Molecular docking
The crystal structures of human AChE complexed with dihydrotanshinone, JBU complexed with acetohydroxamic acid and LOX complexed with arachidonic acid were retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank with PDB ID 4m0e, resolution 2.00 Å, 4h9m, resolution 1.52 Å and 3v99, resolution 2.25 Å, respectively. The coordinate files were subjected to Discovery Studio 4.5 Visualizer for pre-docking receptor preparation by removing water molecules and adding hydrogen atoms. Compounds 8(a–n) were docked with AChE (4m0e), JBU (4h9m) and LOX (3v99) by PatchDock. PatchDockis a molecular docking tool, targeted to find docking transformations that produce good molecular shape complementarity based on shape complementarity principles [20]. The input files comprise the receptor protein and ligand in PDB format. PatchDock server affords multiple solutions and “solution 1” was selected as it is surrounded by the most significant residues as hotspots for docking analyses assigned in crystal structures of AChE (4m0e), JBU (4h9m) and LOX (3v99) receptors [21,22,23,24]. The docked structures 8(a–n) were observed using Discovery Studio 4.5 Visualizer. The binding affinities of the docked ligands 8(a–n) were assessed as scores and the ACE of the docked complexes. The hydrogen bonding and hydrophobic contacts of each ligand 8(a–n) were evaluated within the hotspot of receptor proteins. The conformation of compounds 8(a–n), which demonstrated the maximum biological activities, is shown in Table 3 and Figures 1 and 2 with their favorable contacts in the binding pockets.
4.6.5 Statistical analysis
Statistical analysis was performed using Microsoft Excel 2010 by making all the calculations in triplicate. Results were presented as mean ± SEM. The IC50 values are calculated using EZ-Fit Enzyme Kinetics Software (Perrella Scientific Inc. Amherst, USA).
4.6.6 Spectral data
4.6.6.1 N-(2,4-Dimethylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8a)
Off-white solid; yield: 81%; m.p.: 155–157°C; HRMS: [M]•+ 530.6618 (calcd for C25H30N4O5S2; 530.6629); Anal. Calcd for C25H30N4O5S2: C, 56.58; H, 5.70; N, 10.56; O, 15.08; S, 12.08; found C, 56.52; H, 5.64; N, 10.50; O, 15.03; S, 12.02; IR (KBr, υ max, cm−1): 3,351 (N–H), 3,077 (ArC–H), 1,672 (C═N), 1,669 (C═O), 1,596 (Ar C═C), 1,433 (S═O), 1,079 (C–N), 609 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.72 (d, J = 8.8 Hz, 2H, H-6″ and H-2″), 7.59 (d, J = 8.1 Hz, 1H, H-6‴), 7.08 (d, J = 8.2 Hz, 1H, H-5‴), 7.05 (s, 1H, H-3‴), 7.02 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 3.95–3.93 (m, 1H, He-2′), 3.89 (s, 3H, H-7″), 3.66–3.64 (m, 1H, Ha-2′), 3.63 (q, J = 6.8 Hz, 2H, H-2″″), 3.13 (br.s, 1H, H-3′), 2.66 (br.s, 1H, He-6′), 2.46 (br.s, 1H, Ha-6′), 2.33 (s, 3H, H-8‴), 2.32 (s, 3H, H-7‴), 2.14–2.11 (m, 1H, He-5′), 1.91–1.90 (m, 1H, He-4′), 1.77 (br.s, 1H, Ha-5′), 1.63 (br.s, 1H, Ha-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz,CDCl3, δ/ppm): 163.15 (C-1″″, amidic), 160.21 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.93 (C-4″, aromatic), 134.54 (C-4‴, aromatic), 132.57 (C-1″, aromatic), 131.49 (C-2‴, aromatic), 129.73 (C-2″ and C-6″, aromatic), 127.65 (C-1‴, aromatic), 127.60 (C-3‴, aromatic), 120.01 (C-5‴, aromatic), 114.70 (C-6‴, aromatic), 114.32 (C-3″ and C-5″, aromatic), 55.64 (C-7″, methoxy), 48.45 (C-2′, aliphatic,heterocyclic), 46.26 (C-6′, aliphatic, heterocyclic), 33.55 (C-2″″, amidic), 30.92 (C-4′, aliphatic, heterocyclic), 27.08 (C-3′, aliphatic, heterocyclic), 23.71 (C-5′, aliphatic, heterocyclic), 20.77 (C-8‴, aliphatic), 18.42 (C-3″″, amidic), 17.73 (C-7‴, aliphatic).
4.6.6.2 N-(2,3-Dimethylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8b)
Light pink solid; yield: 83%; m.p.: 162–164°C; HRMS: [M]•+ 530.6618 (calcd for C25H30N4O5S2; 530.6629); Anal. Calcd for C25H30N4O5S2:C, 56.58; H, 5.70; N, 10.56; O, 15.08; S, 12.08; found C, 56.52; H, 5.64; N, 10.50; O, 15.03; S, 12.02; IR (KBr, υ max, cm−1): 3,359 (N–H), 3,074 (C–H stretching of aromatic ring), 1,674 (C═N stretching), 1,667 (C═O), 1,593 (Ar C═C), 1,439 (S═O), 1,072 (C–N), 611 (C–S); 1H-NMR (600 MHz, CDCl3,δ/ppm): 7.72 (d, J = 8.8 Hz, 2H, H-6″ and H-2″), 7.53 (d, J = 8.0 Hz, 1H, H-6‴), 7.17 (t, J = 8.2 Hz, 1H, H-5‴), 7.03 (d, J = 7.7 Hz, 1H, H-4‴), 7.02 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 3.89 (s, 3H, H-7″), 3.96–3.94 (m, 1H, He-2′), 3.67–3.65 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.14–3.11 (m, 1H, H-3′), 2.67–2.63 (m, 1H, He-6′), 2.48–2.44 (m, 1H, Ha-6′), 2.35 (s, 3H, H-8‴), 2.26 (s, 3H, H-7‴), 2.14–2.13 (m, 1H, Ha-5′), 1.93–1.89 (m, 1H, Ha-4′), 1.79–1.72 (m, 1H, He-5′), 1.65–1.59 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.15 (C-1″″), 160.38 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 160.31 (C-4″, aromatic), 137.87 (C-1‴, aromatic), 135.08 (C-3‴, aromatic), 132.57 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 127.58 (C-2‴, aromatic), 126.83 (C-4‴, aromatic), 126.38 (C-5‴, aromatic), 118.75 (C-6‴, aromatic), 114.37 (C-3″ and C-5″, aromatic), 55.64 (C-7″, aromatic), 48.45 (C-2′, aliphatic, heterocyclic), 46.25 (C-6′, aliphatic, heterocyclic), 33.56 (C-2″″, amidic), 30.92 (C-4′, aliphatic, heterocyclic), 27.12 (C-3′, aliphatic, heterocyclic), 23.72 (C-5′, aliphatic, heterocyclic), 20.63 (C-8‴, aliphatic), 18.42 (C-3″″, amidic), 13.65 (C-7‴, aliphatic).
4.6.6.3 N-(2,5-Dimethylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8c)
Light pink solid; yield: 79%; m.p.: 154–156°C; HRMS: [M]•+ 530.6618 (calcd for C25H30N4O5S2; 530.6629); Anal. Calcd for C25H30N4O5S2:C, 56.58; H, 5.70; N, 10.56; O, 15.08; S, 12.08; found C, 56.52; H, 5.64; N, 10.50; O, 15.03; S, 12.02; IR (KBr, υ max, cm−1): 3,357 (N–H), 3,068 (C–H stretching of aromatic ring), 1,671 (C═N stretching), 1,665 (C═O), 1,588 (Ar C═C), 1,437 (S═O), 1,069 (C–N), 602 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.72 (d, J = 8.8 Hz, 2H, H-6″ and H-2″), 7.64 (s, 1H, H-6‴), 7.11 (t, J = 7.7 Hz, 1H, H-3‴), 7.02 (d, J = 8.8 Hz, 2H, H-3″ and H-5″), 6.90 (d, J = 7.6 Hz, 1H, H-4‴), 3.89 (s, 3H, H-7″), 3.96–3.94 (m, 1H, He-2′), 3.66–3.65 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.16–3.12 (m, 1H, H-3′), 2.69–2.66 (m, 1H, He-6′), 2.50–2.47 (m, 1H, Ha-6′), 2.37 (s, 3H, H-8‴), 2.31 (s, 3H, H-7‴), 2.15–2.12 (m, 1H, Ha-5′), 1.95–1.91 (m, 1H, Ha-4′), 1.80–1.74 (m, 1H, He-5′), 1.67–1.63 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.16 (C-1″″, amidic), 160.29 (C-5, heterocyclic), 160.88 (C-2, heterocyclic), 159.93 (C-4″, aromatic), 137.23 (C-1‴, aromatic), 134.97 (C-5‴, aromatic), 130.58 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 127.58 (C-2‴, aromatic), 125.22 (C-3‴, aromatic), 124.01 (C-4‴, aromatic), 120.22 (C-6‴, aromatic), 114.37 (C-3″ and C-5″, aromatic), 55.64 (C-7″, methoxy), 48.48 (C-2′, aliphatic, heterocyclic), 46.27 (C-6′, aliphatic, heterocyclic), 33.53 (C-2″″, amidic), 30.90 (C-4′, aliphatic, heterocyclic), 27.07 (C-3′, aliphatic, heterocyclic), 23.71 (C-5′, aliphatic, heterocyclic), 21.24 (C-8‴, aliphatic), 18.44 (C-3″″, amidic), 17.28 (C-7‴, aliphatic).
4.6.6.4 N-(2,6-Dimethylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8d)
Off-white solid; yield: 88%; m.p.: 110–112°C; HRMS: [M]•+ 530.6618 (Calcd for C25H30N4O5S2; 530.6629); Anal. Calcd for C25H30N4O5S2: C, 56.58; H, 5.70; N, 10.56; O, 15.08; S, 12.08; found C, 56.52; H, 5.64; N, 10.50; O, 15.03; S, 12.02; IR (KBr, υ max, cm−1): 3,353 (N–H), 3,077 (C–H stretching of aromatic ring), 1,676 (C═N stretching), 1,661 (C═O), 1,590 (Ar C═C), 1,433 (S═O), 1,075 (C–N), 608 (C–S); 1H-NMR (600 MHz, CDCl3,δ/ppm): 7.69 (d, J = 8.4 Hz, 2H, H-2″ and H-6″), 7.19 (d, J = 8.8 Hz, 2H, H-3‴ and H-5‴), 7.02 (d, J = 8.8 Hz, 2H, H-3″ and H-5″), 7.17 (d, J = 8.8 Hz, 1H, H-4‴), 3.89 (s, 3H, H-7″), 3.97–3.94 (m, 1H, He-2′), 3.67–3.65 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.15–3.12 (m, 1H, H-3′), 2.69–2.67 (m, 1H, He-6′), 2.50–2.46 (m, 1H, Ha-6′), 2.12 (s, 6H, H-8‴ and H-7‴), 2.15–2.12 (m, 1H, Ha-5′), 1.96–1.92 (m, 1H, Ha-4′), 1.81–1.74 (m, 1H, He-5′), 1.67–1.62 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz,CDCl3, δ/ppm): 163.21 (C-1″″, amidic), 160.29 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.93 (C-4″, aromatic), 135.30 (C-1‴, aromatic), 132.43 (C-1″, aromatic), 129.82 (C-2‴, aromatic), 129.73 (C-2″ and C-6″, aromatic), 128.87 (C-3‴ and C-5‴, aromatic), 128.38 (C-6‴, aromatic), 127.49 (C-4‴, aromatic), 114.36 (C-3″ and C-5″, aromatic), 55.64 (C-7″, methoxy), 47.98 (C-2′, aliphatic, heterocyclic), 46.19 (C-6′, aliphatic, heterocyclic), 33.49 (C-2″″), 30.90 (C-4′, aliphatic, heterocyclic), 26.87 (C-3′, aliphatic, heterocyclic), 23.48 (C-5′, aliphatic, heterocyclic), 18.40 (C-3″″, amidic), 17.78 (C-7‴ and C-8‴, aliphatic).
4.6.6.5 N-(3,5-Dimethylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8e)
Off-white solid; yield: 86%; m.p.: 176–178°C; HRMS: [M]•+ 530.6618 (calcd for C25H30N4O5S2; 530.6629); Anal. Calcd for C25H30N4O5S2:C, 56.58; H, 5.70; N, 10.56; O, 15.08; S, 12.08; found C, 56.52; H, 5.64; N, 10.50; O, 15.03; S, 12.02; IR (KBr, υ max, cm−1): 3,360 (N–H), 3,079 (C–H stretching of aromatic ring), 1,667 (C═N stretching), 1,666 (C═O), 1,589 (Ar C═C), 1,430 (S═O), 1,074 (C–N), 614 (C–S); 1H-NMR (600 MHz, CDCl3,δ/ppm): 7.71 (d, J = 8.8 Hz, 2H, H-6″ and H-2″), 7.10 (s, 2H, H-2‴ and H-6‴), 7.01 (d, J = 8.8 Hz, 2H, H-3″ and H-5″), 6.75 (s, 1H, H-4‴), 3.88 (s, 3H, H-7″), 3.95–3.90 (m, 1H, He-2′), 3.65–3.63 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.16–3.12 (m, 1H, H-3′), 2.70–2.66 (m, 1H, He-6′), 2.51–2.44 (m, 1H, Ha-6′), 2.34 (s, 6H, H-7‴, H-8‴), 2.23–2.12 (m, 1H, Ha-5′), 1.94–1.91 (m, 1H, Ha-4′), 1.80–1.74 (m, 1H, He-5′), 1.68–1.64 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.15 (C-1″″, amidic), 160.21 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.72 (C-4″, aromatic), 139.21 (C-1‴, aromatic), 137.06 (C-3‴, aromatic), 132.43 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 127.55 (C-5‴, aromatic), 125.31 (C-4‴, aromatic), 115.74 (C-2‴ and C-6‴, aromatic), 114.37 (C-3″ and C-5″, aromatic), 55.63 (C-7″, methoxy), 48.54 (C-2′, aliphatic, heterocyclic), 46.28 (C-6′, aliphatic, heterocyclic), 33.48 (C-2″″), 30.74 (C-4′, aliphatic, heterocyclic), 27.08 (C-3′, aliphatic, heterocyclic), 23.71 (C-5′, aliphatic, heterocyclic) 18.42 (C-3″″), 21.42 (C-7‴ and C-8‴).
4.6.6.6 2-((5-(1-((4-Methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)-N-phenylpropanamide (8f)
Off-white solid; yield: 86%; m.p.: 189–191°C; HRMS: [M]•+ 502.6124 (calcd for C23H36N4O5S2; 530.6631); Anal. Calcd for C23H36N4O5S2:C, 54.96; H, 5.21; N, 11.15; O, 15.92; S, 12.76; found C, 54.91; H, 5.18; N, 11.11; O, 15.89; S, 12.71; IR (KBr, υ max, cm−1): 3,354 (N–H), 3,067 (C–H stretching of aromatic ring), 1,680 (C═N stretching), 1,658 (C═O), 1,599 (Ar C═C), 1,434 (S═O), 1,079 (C–N), 606 (C–S); 1H-NMR (600 MHz, CDCl3,δ/ppm): 7.71 (d, J = 8.7 Hz, 2H, H-6″ and H-2″), 7.49 (d, J = 8.0 Hz, 2H, H-2‴ and H-6‴), 7.37 (t, J = 7.9 Hz, 2H, H-3‴ and H-3‴), 7.10 (t,J = 7.3 Hz, 1H, H-4‴), 7.01 (d, J = 8.7 Hz, 2H, H-3″ and H-5″), 3.88 (s, 3H, H-7″), 3.96–3.95 (m, 1H, He-2′), 3.66–3.64 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.16–3.13 (m, 1H, H-3′), 2.70–2.66 (m, 1H, He-6′), 2.50–2.47 (m, 1H, Ha-6′), 2.19–2.12 (m, 1H, Ha-5′), 1.92–1.90 (m, 1H, Ha-4′), 1.77–1.73 (m, 1H, He-5′), 1.68–1.62 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.16 (C-1″″, amidic), 160.34 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.98 (C-4″, aromatic), 137.53 (C-1‴, aromatic), 132.43 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 129.38 (C-3‴ and C-5‴, aromatic), 123.22 (C-4‴,aromatic), 117.76 (C-2‴ and C-6‴, aromatic), 114.38 (C-3″ and C-5″, aromatic), 55.64 (C-7″, methoxy), 48.57 (C-2′, aliphatic, heterocyclic), 46.29 (C-6′, aliphatic, heterocyclic), 33.52 (C-2″″, amidic), 30.72 (C-4′, aliphatic, heterocyclic), 27.12 (C-3′, aliphatic, heterocyclic), 23.73 (C-5′, aliphatic, heterocyclic), 18.42 (C-3″″, amidic).
4.6.6.7 2-((5-(1-((4-Methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)-N-(o-tolyl)propanamide (8g)
Light brown solid; yield: 89%; m.p.: 104–106°C; HRMS: [M]•+ 516.6338 (calcd for C24H28N4O5S2; 516.6347); Anal. Calcd for C24H28N4O5S2:C, 55.80; H, 5.46; N, 10.84; O, 15.48; S, 12.41; found C, 55.78; H, 5.43; N, 10.80; O, 15.43; S, 12.38; IR (KBr, υ max, cm−1): 3,353 (N–H), 3,076 (C–H stretching of aromatic ring), 1,677 (C═N stretching), 1,665 (C═O), 1,593 (Ar C═C), 1,429 (S═O), 1,078 (C–N), 615 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.82 (d, J = 8.0 Hz, 1H, H-6‴), 7.71 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.27 (t, J = 7.4 Hz, 1H, H-5‴), 7.22 (d, J = 7.4 Hz, 1H, H-3‴), 7.06 (t, 8.5 Hz, 1H, H-4‴), 7.01 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 6.75 (s, 1H, H-4‴), 3.88 (s, 3H, H-7″), 3.98–3.96 (m, 1H, He-2′), 3.68–3.66 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.15–3.12 (m, 1H, H-3′), 2.66–2.63 (m, 1H, He-6′), 2.47–2.36 (m, 1H, Ha-6′), 2.15–2.12 (m, 1H, Ha-5′), 2.11 (s, 3H, H-7‴), 1.93–1.89 (m, 1H, Ha-4′), 1.77–1.75 (m, 1H, He-5′), 1.63–1.61 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.14 (C-1″″, amidic), 160.48 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 160.40 (C-4″, aromatic),), 135.74 (C-1‴, aromatic),), 130.71 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic),), 127.59 (C-2‴, aromatic),), 127.26 (C-3‴, aromatic),), 126.77 (C-4‴, aromatic),), 123.97 (C-5‴, aromatic),), 119.24 (C-6‴, aromatic),), 114.36 (C-3″ and C-5″, aromatic),), 55.63 (C-7″, methoxy), 48.56 (C-2′, aliphatic, heterocyclic), 46.27 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 33.56 (C-2″″), 27.18 (C-3′, aliphatic, heterocyclic), 23.79 (C-5′, aliphatic, heterocyclic), 18.42 (C-3″″, amidic), 17.74 (C-7‴, aliphatic).
4.6.6.8 2-((5-(1-((4-Methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)-N-(m-tolyl)propanamide (8h)
Off-white solid; yield: 85%; m.p.: 146–148°C; HRMS: [M]•+ 516.6338 (calcd for C24H28N4O5S2; 516.6347; Anal. Calcd for C24H28N4O5S2:C, 55.80; H, 5.46; N, 10.84; O, 15.48; S, 12.41; found C, 55.78; H, 5.43; N, 10.80; O, 15.43; S, 12.38; IR (KBr, υ max, cm−1): 3,355 (N–H), 3,066 (C–H stretching of aromatic ring), 1,681 (C═N stretching), 1,659 (C═O), 1,586 (Ar C═C), 1,429 (S═O), 1,067 (C–N), 601 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.82 (d, J = 8.0 Hz, 1H, H-6‴), 7.71 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.27 (t, J = 7.4 Hz, 1H, H-5‴), 7.22 (d, J = 7.4 Hz, 1H, H-3‴), 7.06 (t, 8.5 Hz, 1H, H-4‴) 7.01 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 6.75 (s, 1H, H-4‴), 3.88 (s, 3H, H-7″), 3.98–3.96 (m, 1H, He-2′), 3.68–3.66 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.15–3.12 (m, 1H, H-3′), 2.66–2.63 (m, 1H, He-6′), 2.47–2.36 (m, 1H, Ha-6′), 2.15–2.12 (m, 1H, Ha-5′), 2.11 (s, 3H, H-7‴), 1.93–1.89 (m, 1H, Ha-4′), 1.77–1.75 (m, 1H, He-5′), 1.63–1.61 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.17 (C-1″″, amidic), 160.16 (C-5, heterocyclic), 160.89 (C-2, heterocyclic),159.37 (C-4″, aromatic), 139.53 (C-3‴, aromatic), 136.80 (C-1‴, aromatic), 132.11 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 129.25 (C-5‴, aromatic), 124.71 (C-4‴, aromatic), 118.92 (C-2‴, aromatic), 115.31 (C-6‴, aromatic), 114.38 (C-3″ and C-5″, aromatic), 55.64 (C-7″, methoxy), 48.45 (C-2′, aliphatic, heterocyclic), 46.26 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 33.47 (C-2″″, amidic), 27.01 (C-3′, aliphatic, heterocyclic), 23.63 (C-5′, aliphatic, heterocyclic), 21.52 (C-7‴, aliphatic), 18.42 (C-3″″, amidic).
4.6.6.9 2-((5-(1-((4-Methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)-N-(p-tolyl)propanamide (8i)
White solid; yield: 87%; m.p.: 142–144°C; HRMS: [M]•+ 516.6338 (Calcd for C24H28N4O5S2; 516.6347); Anal. Calcd for C24H28N4O5S2:C, 55.80; H, 5.46; N, 10.84; O, 15.48; S, 12.41; found C, 55.78; H, 5.43; N, 10.80; O, 15.43; S, 12.38; IR (KBr, υ max, cm−1): 3,351 (N–H), 3,076 (C–H stretching of aromatic ring), 1,672 (C═N stretching), 1,669 (C═O), 1,596 (Ar C═C), 1,438 (S═O), 1,066 (C–N), 612 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.82 (d, J = 8.0 Hz, 1H, H-6‴), 7.72 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.32 (s, 1H, H-2‴), 7.26 (d, (t, J = 8.7 Hz 1H, H-5‴), J = 8.8 Hz, 2H, H-4‴ and H-6‴), 7.01 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 6.95 3.89 (s, 3H, H-7″), 3.93–3.91 (m, 1H, He-2′), 3.63–3.61 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.18–3.14 (m, 1H, H-3′), 2.73–2.70 (m, 1H, He-6′), 2.54–2.50 (m, 1H, Ha-6′), 2.34 (s, 3H, H-7‴), 2.19–2.12 (m, 1H, Ha-5′), 1.94–1.91 (m, 1H, Ha-4′), 1.79–1.76 (m, 1H, He-5′), 1.69–1.66 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.22 (C-1″″, amidic), 160.16 (C-5, heterocyclic), 160.88 (C-2, heterocyclic), 159.37 (C-4″, aromatic), 134.69 (C-4‴, aromatic), 133.39 (C-1‴, aromatic), 131.11 (C-1″, aromatic), 130.09 (C-3‴ and C-5‴, aromatic), 129.75 (C-2″ and C-6″, aromatic), 127.48 (C-2‴, aromatic), 119.23 (C-6‴, aromatic), 114.42 (C-3″ and C-5″, aromatic), 55.67 (C-7″, methoxy), 48.27 (C-2′, aliphatic, heterocyclic), 46.23 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 33.45 (C-2″″), 26.85 (C-3′, aliphatic, heterocyclic), 23.46 (C-5′, aliphatic, heterocyclic), 20.84 (C-7‴, aliphatic), 18.42 (C-3″″, amidic).
4.6.6.10 N-(4-Ethoxyphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8j)
Light brown solid; yield: 87%; m.p.: 138–140°C; HRMS: [M]•+ 546.6672 (calcd for C25H30N4O5S2; 546.6681); Anal. Calcd for C25H30N4O5S2: C, 54.93; H, 5.53; N, 10.25; O, 17.56; S, 11.73; found C, 54.93; H, 5.51; N, 10.22; O, 17.51; S, 11.70; IR (KBr, υ max, cm−1): 3,348 (N–H), 3,074 (C–H stretching of aromatic ring), 1,681 (C═N stretching), 1,668 (C═O), 1,596 (Ar C═C), 1,427 (S═O), 1,064 (C–N), 607 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.71 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.37 (d, J = 8.9 Hz, 2H, H-2‴ and H-6‴), 7.01 (d, J = 8.5 Hz, 2H, H-3″ and H-5″), 6.90 (d, J = 8.9 Hz, 2H, H-3‴ and H-5‴), 4.03 (q, J = 7.0 Hz, 3H, H-7‴), 3.89 (s, 3H, H-7″), 3.92–3.90 (m, 1H, He-2′), 3.63–3.61 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.15–3.11 (m, 1H, H-3′), 2.70–2.66 (m, 1H, He-6′), 2.51–2.47 (m, 1H, Ha-6′), 2.13–2.10 (m, 1H, Ha-5′), 1.93–1.89 (m, 1H, Ha-4′), 1.79–1.73 (m, 1H, He-5′), 1.67–1.63 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″), 1.32 (t, J = 7.0 Hz, 3H, H-8‴); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.16 (C-1″″, amidic), 160.01 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.92 (C-4″, aromatic), 155.62 (C-4‴, aromatic), 130.07 (C-1‴, aromatic), 132.11 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 120.28 (C-2‴ and C-6‴, aromatic), 115.27 (C-3‴ and C-5‴, aromatic), 114.37 (C-3″ and C-5″, aromatic), 63.81 (C-7‴, aliphatic), 55.64 (C-7″, methoxy), 48.48 (C-2′, aliphatic, heterocyclic), 46.26 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 33.49 (C-2″″, amidic), 27.04 (C-3′, aliphatic, heterocyclic), 23.67 (C-5′, aliphatic, heterocyclic), 18.42 (C-3″″, amidic), 14.83 (C-8‴, aliphatic).
4.6.6.11 N-(2-Ethyl-6-methylphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8k)
Off-white solid; yield: 88%; m.p.: 134–136°C; HRMS: [M]•+ 544.6921 (calcd for C26H32N4O5S2; 544.6926); Anal. Calcd for C26H32N4O5S2:C, 57.33; H, 5.92; N, 10.29; O, 14.69; S, 11.77; found C, 57.29; H, 5.90; N, 10.21; O, 14.64; S, 11.73; IR (KBr, υ max, cm−1): 3,351 (N–H), 3,080 (C–H stretching of aromatic ring), 1,682 (C═N stretching), 1,669 (C═O), 1,587 (Ar C═C), 1,428 (S═O), 1,066 (C–N), 604 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.68 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.24 (t, J = 8.8 Hz, 1H, H-4‴), 7.20 (d, J = 8.6 Hz, 1H, H-5‴), 7.02 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 7.00 (d, J = 8.6 Hz, 1H, H-3‴), 3.89 (s, 3H, H-7″), 3.90 (3H, H-7‴), 3.92–3.90 (m, 1H, He-2′), 3.63–3.61 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.15–3.11 (m, 1H, H-3′), 2.70–2.66 (m, 1H, He-6′), 2.60 (q, J = 7.0 Hz, 3H, H-8‴), 2.51–2.47 (m, 1H, Ha-6′), 2.13–2.10 (m, 1H, Ha-5′), 1.93–1.89 (m, 1H, Ha-4′), 1.79–1.73 (m, 1H, He-5′), 1.67–1.63 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″), 1.25 (t, J = 7.0 Hz, 3H, H-9‴); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.16 (C-1″″, amidic), 160.01 (C-5, heterocyclic), 160.89 (C-2, heterocyclic),159.92 (C-4″, aromatic), 135.87 (C-1‴, aromatic), 132.11 (C-1″, aromatic), 130.23 (C-2‴, aromatic), 129.73 (C-2″ and C-6″, aromatic), 129.05 (C-6‴, aromatic), 127.22 (C-5‴, aromatic), 124.67 (C-4‴, aromatic), 121.32 (C-3‴, aromatic), 114.49 (C-3″ and C-5″, aromatic), 55.74 (C-7″, methoxy), 48.43 (C-2′, aliphatic, heterocyclic), 46.13 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 33.36 (C-2″″), 27.04 (C-3′, aliphatic, heterocyclic), 24.77 (C-8‴, aliphatic), 23.67 (C-5′, aliphatic, heterocyclic), 18.42 (C-3″″, amidic), 17.64 (C-7‴, aliphatic), 14.62 (C-9‴, aliphatic).
4.6.6.12 N-Cyclohexyl-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8l)
Off-white solid; yield: 82%; m.p.: 113–115°C; HRMS: [M]•+ 544.6923 (calcd for C23H32N4O5S2; 508.6528); Anal. Calcd for C23H32N4O5S2:C, 54.31; H, 6.34; N, 11.01; O, 15.73; S, 12.61; found C, 54.28; H, 6.30; N, 11.00; O, 15.71; S, 12.57; IR (KBr, υ max, cm−1): 3,349 (N–H), 3,066 (C–H stretching of aromatic ring), 1,670 (C═N stretching), 1,663 (C═O), 1,588 (Ar C═C), 1,430 (S═O), 1,074 (C–N), 606 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.72 (d, J = 8.8 Hz, 2H, H-2″, H-6″), 7.01 (d, J = 8.6 Hz, 2H, H-3″, H-5″), 3.92–3.90 (m, 1H, He-2′), 3.86 (s, 3H, H-7″), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.63–3.61 (m, 1H, Ha-2′), 3.54 (m, 1H, H-1‴), 3.15–3.11 (m, 1H, H-3′), 2.70–2.66 (m, 1H, He-6′), 2.51–2.47 (m, 1H, Ha-6′), 2.13–2.10 (m, 1H, Ha-5′), 1.93–1.89 (m, 1H, Ha-4′), 1.79–1.73 (m, 1H, He-5′), 1.67–1.63 (m, 1H, He-4′), 1.74–1.72 (m, 2H, He-2‴ and He-6‴), 1.51–1.50 (m, 1H, He-4‴), 1.49–1.48 (m, 2H, Ha-2‴ and Ha-6‴), 1.47–1.45 (m, 1H, Ha-4‴), 1.42 (d, J = 7.0 Hz, 1H, H-3″″), 1.21–1.18 (m, 2H, He-3‴ and He-5‴), 1.12–1.11 (m, 2H, Ha-3‴ and Ha-5‴); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.13 (C-1″″, amidic), 160.01 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.92 (C-4″, aromatic), 132.11 (C-1″, aromatic), 129.73 (C-2″ and C-6″, aromatic), 114.38 (C-3″ and C-5″, aromatic), 55.74 (C-7″, methoxy), 49.63 (C-1‴, aromatic), 48.43 (C-2′, aliphatic, heterocyclic), 46.13 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 33.36 (C-2″″, amidic), 32.68 (C-2‴ and C-6‴, aromatic), 27.04 (C-3′, aliphatic, heterocyclic), 25.44 (C-4‴, aromatic), 24.92 (C-3‴ and C-5‴, aromatic), 23.67 (C-5′, aliphatic, heterocyclic), 18.42 (C-3″″, amidic).
4.6.6.13 N-(2-Methoxyphenyl)-2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamide (8m)
Light pink solid; yield: 78%; m.p.: 108–110°C; HRMS: [M]•+ 532.6367 (calcd for C24H28N4O5S2; 532.6372); Anal. Calcd for C24H28N4O5S2:C, 54.12; H, 5.30; N, 10.52; O, 18.02; S, 12.04; found C, 54.07; H, 5.26; N, 10.49; O, 18.00; S, 12.01; IR (KBr, υ max, cm−1): 3,349 (N–H), 3,080 (C–H stretching of aromatic ring), 1,669 (C═N stretching), 1,658 (C═O), 1,587 (Ar C═C), 1,426 (S═O), 1,082 (C–N), 601 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 8.08 (d, J = 9.5 Hz, 1H, H-6‴) 7.72 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.24–7.22 (m, 2H, H-3‴ and H-4‴), 7.01 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 6.92 (t, J = 9.4 Hz, 1H, H-5‴), 3.94 (s, 3H, H-7‴), 3.88 (s, 3H, H-7″), 3.92–3.90 (m, 1H, He-2′), 3.71–3.69 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.17–3.13 (m, 1H, H-3′), 2.65–2.61 (m, 1H, He-6′), 2.46–2.42 (m, 1H, Ha-6′), 2.17–2.14 (m, 1H, Ha-5′), 1.94–1.90 (m, 1H, Ha-4′), 1.79–1.76 (m, 1H, He-5′), 1.65–1.61 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.13 (C-1″″, amidic), 160.32 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.68 (C-4″, aromatic), 147.16 (C-2‴, aromatic), 131.11 (C-1″, aromatic), 129.74 (C-2″ and C-6″, aromatic), 126.95 (C-4‴, aromatic), 122.83 (C-1‴, aromatic), 121.33 (C-5‴, aromatic), 117.21 (C-6‴, aromatic), 114.35 (C-3″ and C-5″, aromatic), 110.07 (C-3‴, aromatic), 55.63 (C-7″ and C-7‴, methoxy, aliphatic), 48.58 (C-2′, aliphatic, heterocyclic),), 46.28 (C-6′, aliphatic, heterocyclic),), 30.92 (C-4′, aliphatic, heterocyclic),), 33.53 (C-2″″, amidic), 27.20 (C-3′, aliphatic, heterocyclic),), 23.83 (C-5′, aliphatic, heterocyclic),), 18.42 (C-3″″, amidic).
4.6.6.14 Methyl 2-(2-((5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propanamido)-5-methylbenzoate (8n)
White solid; yield: 79%; m.p.: 180–182°C; HRMS: [M]•+ 574.6741 (calcd for C26H30N4O5S2; 574.6748); Anal. Calcd for C26H30N4O5S2:C, 54.34; H, 5.26; N, 9.75; O, 19.49; S, 11.16; found C, 54.30; H, 5.21; N, 9.72; O, 19.43; S, 11.11; IR (KBr, υ max, cm−1): 3,361 (N–H), 3,067 (C–H stretching of aromatic ring), 1,682 (C═N stretching), 1,669 (C═O), 1,587 (Ar C═C), 1,433 (S═O), 1,070 (C–N), 612 (C–S); 1H-NMR (600 MHz, CDCl3, δ/ppm): 7.84 (s, 1H, H-3‴) 7.72 (d, J = 8.9 Hz, 2H, H-6″ and H-2″), 7.42 (d, J = 8.9 Hz, 1H, H-5‴), 7.01 (d, J = 8.9 Hz, 2H, H-3″ and H-5″), 6.92 (d, J = 8.4 Hz, 1H, H-6‴), 3.98–3.96 (m, 1H, He-2′), 3.89 (s, 3H, H-7‴), 3.88 (s, 3H, H-7″), 3.87–3.78 (m, 1H, Ha-2′), 3.64 (q, J = 6.8 Hz, 2H, H-2″″), 3.17–3.13 (m, 1H, H-3′), 2.65–2.61 (m, 1H, He-6′), 2.46–2.42 (m, 1H, Ha-6′), 2.34 (s, 3H, H-8‴), 2.17–2.14 (m, 1H, Ha-5′), 1.94–1.90 (m, 1H, Ha-4′), 1.79–1.76 (m, 1H, He-5′), 1.65–1.61 (m, 1H, He-4′), 1.42 (d, J = 6.8 Hz, 3H, H-3″″); 13C-NMR (150 MHz, CDCl3, δ/ppm): 163.13 (C-1″″, amidic), 162.35 (C-8‴, aliphatic), 160.16 (C-5, heterocyclic), 160.89 (C-2, heterocyclic), 159.37 (C-4″, aromatic), 134.62 (C-1‴, aromatic), 132.11 (C-1″, aromatic), 129.63 (C-2″ and C-6″, aromatic), 129.20 (C-4‴, aromatic), 128.72 (C-6‴, aromatic), 128.61 (C-3‴, aromatic), 128.56 (C-5‴, aromatic), 127.23 (C-2‴, aromatic), 114.37 (C-3″ and C-5″, aromatic), 55.64 (C-7″, aromatic), 51.56 (C-8‴, aliphatic), 49.06 (C-2′, aliphatic, heterocyclic), 46.09 (C-6′, aliphatic, heterocyclic), 30.92 (C-4′, aliphatic, heterocyclic), 34.02 (C-2″″, amidic), 28.52 (C-3′, aliphatic, heterocyclic), 24.26 (C-5′, aliphatic, heterocyclic), 21.13 (C-7‴, aliphatic), 18.42 (C-3″″, amidic) (Scheme 1).
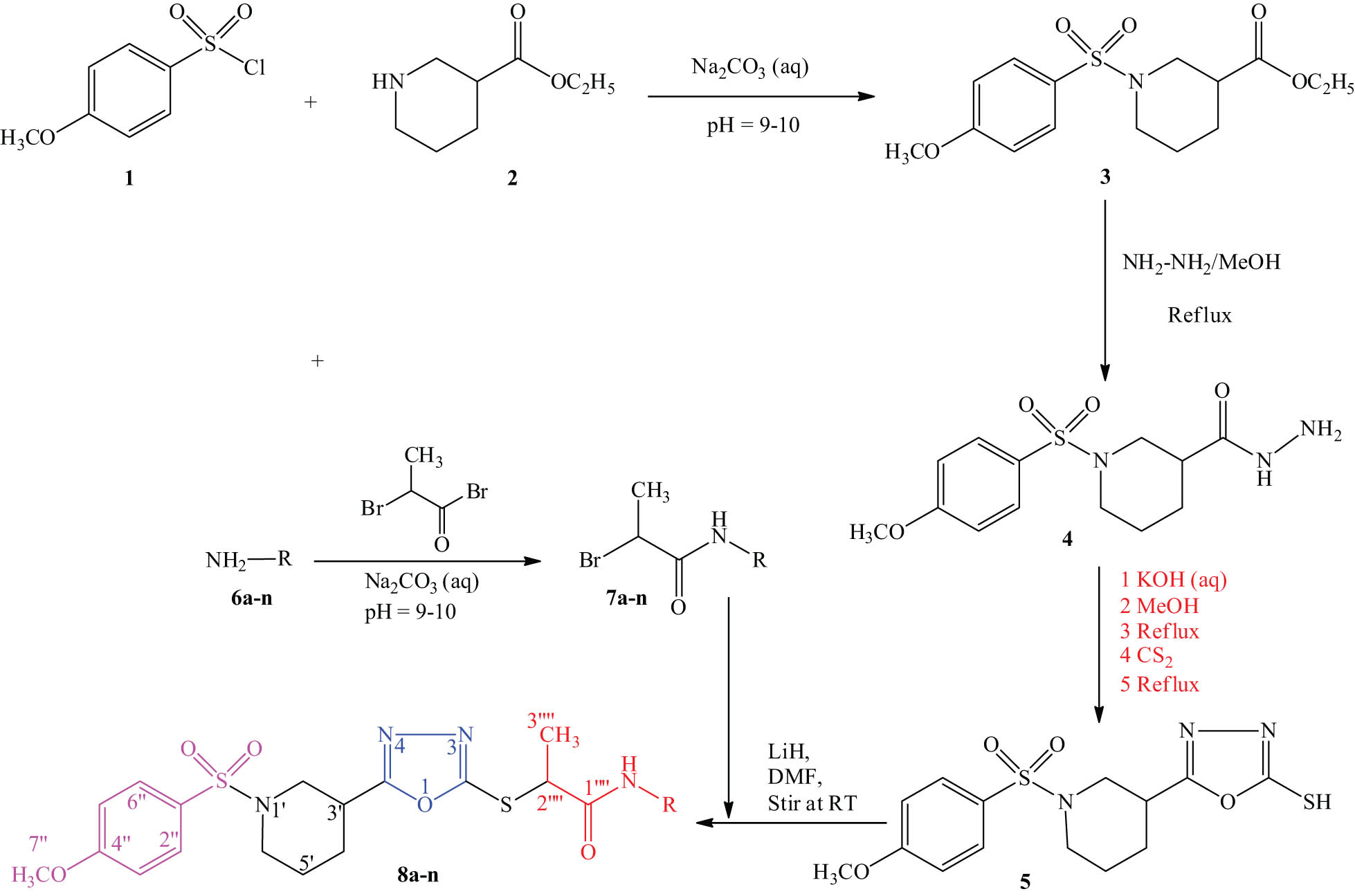
Synthesis of N-substituted propanamide derivatives of 5-(1-((4-methoxyphenyl)sulfonyl)piperidin-3-yl)-1,3,4-oxadiazol-2-thiol.
Acknowledgements
The authors express their gratitude to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R158), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
-
Funding information: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R158), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
-
Author contributions: J.I. reviewed, finalized and submitted the manuscript; A.I.M. conducted the molecular docking analysis; A.-ur-R. designed the research project; S.R. proofread the submitted manuscript; S.H.A.M. conducted the enzyme inhibition activities; F.Z. worked as coworker in the current project with J.I.; M.A.A. assisted in the laboratory work; M.I. contributed in the chemistry part of the manuscript; S.A.A.S. conducted the spectral analysis of the synthesized compounds; H.J. assisted in the laboratory work.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
[1] Iqbal J, Aziz-ur-Rehman, Abbasi MA, Siddiqui SZ, Khalid H, Laulloo SJ, et al. BSA Binding, molecular docking and in vitro biological screening of some new 1, 2, 4-triazole heterocycles bearing azinane nucleus. Pak J Pharm Sci. 2020;33:149–60.Search in Google Scholar
[2] Rashid M, Husain A, Mishra R. Synthesis of benzimidazoles bearing oxadiazole nucleus as anticancer agents. Eur J Med Chem. 2012;54:855–66.10.1016/j.ejmech.2012.04.027Search in Google Scholar PubMed
[3] Murty MSR, Penthala R, Nath LR, Anto RJ. Synthesis of salicylic acid-based 1,3,4-oxadiazole derivatives coupled with chiral oxazolidinones: novel hybrid heterocycles as antitumor agents. Lett Drug Des Discov. 2014;11:1133–42.10.2174/1570180811666140627004607Search in Google Scholar
[4] Tabatabai SA, Lashkari SB, Zarrindast MR, Gholibeikian M, Shafiee A. Design, synthesis and anticonvulsant activity of 2-(2-phenoxy) phenyl- 1,3,4-oxadiazole derivatives. Iran J Pharm Res. 2013;12(Supp l):105–11.Search in Google Scholar
[5] Deshmukh R, Jha AK, Thakur AS, Dewangan D. Synthesis and antibacterial activity of some 1,3,4-oxadiazole derivatives and their thione analogues. Int J Res Pharm Biomed Sci. 2011;2:215–9.Search in Google Scholar
[6] Maslat AO, Mahmud A, Hasan T, Mahmoud AT. Synthesis antibacterial, antifungal andgenotoxic activity of bis-1,3,4- oxadiazole derivatives. Pol J Pharmacol. 2002;54:55–9.Search in Google Scholar
[7] Hajimahdi Z, Zarghi A, Zabihollahi R, Aghasadeghi MR. Synthesis, biological evaluation, and molecular modeling studies of new 1,3,4-oxadiazole and 1,3,4-thiadiazole-substituted 4-oxo-4H-pyrido[1,2-a] pyrimidines as anti-HIV-1 agents. Med Chem Res. 2013;22:2467–75.10.1007/s00044-012-0241-5Search in Google Scholar
[8] Omar MT. Synthesis of new xanthenone derivatives of expected antibilharzial activity. Arch Pharm Res. 1997;20:602–9.10.1007/BF02975219Search in Google Scholar PubMed
[9] Hasan A, Thomas NF, Gapil S. Synthesis, characterization and antifungal evaluation of 5-substituted-4-amino-1,2,4triazole-3-thioesters. Molecules. 2011;16:1297–309.10.3390/molecules16021297Search in Google Scholar PubMed PubMed Central
[10] Holla BS, Poojary KN, Kalluraya B, Gowda PV. 5-Substituted-1,3,4-oxadiazoline-2-thiones. Indian J Heterocycl Chem. 1996;5:273–6.Search in Google Scholar
[11] Kerimov I, Ayhan-Kilcigil G, Ozdamar ED, Can-Eke B, Coban T, Ozbey S. Design and one-pot and microwave-assisted synthesis of 2-amino/5-aryl-1,3,4-oxadiazoles bearing a benzimidazole moiety as antioxidants. Arch Pharm. 2012;345:549–56.10.1002/ardp.201100440Search in Google Scholar PubMed
[12] Saitoh M, Kunitomo J, Kimura E, Hayase Y, Kobayashi H, Uchiyama N, et al. Design, synthesis and structure–activity relationships of 1, 3, 4-oxadiazole derivatives as novel inhibitors of glycogen synthase kinase-3b. Bioorg Med Chem. 2009;17:2017–29.10.1016/j.bmc.2009.01.019Search in Google Scholar PubMed
[13] Akhter M, Husain A, Azad B, Ajmal M. Aroylpropionic acid based 2,5-disubstituted-1,3,4-oxadiazoles: synthesis and their anti-inflammatory and analgesic activities. Eur J Med Chem. 2009;44:2372–8.10.1016/j.ejmech.2008.09.005Search in Google Scholar PubMed
[14] Zhang X, Qiu M, Sun J, Zhang Y, Yang Y, Wang X, et al. Synthesis, biological evaluation and molecular docking studies of 1,3,4-oxadiazole derivatives possessing 1,4-benzodioxan moiety as potential anticancer agents. Bioorg Med Chem. 2011;19:6518–24.10.1016/j.bmc.2011.08.013Search in Google Scholar PubMed
[15] Alitonou GA, Avlessi F, Sohounhloue DK, Agnaniet H, Bessiere JM, Menut C. Investigations on the essential oil of Cymbopogongiganteus from benin for its potential use as an anti inflammatory agent. Int J Aromather. 2006;16:37–41.10.1016/j.ijat.2006.01.001Search in Google Scholar
[16] Lodhi MA, Abbasi MA, Choudhary N, Ahmad VU. Kinetics studies on triacontanylpalmitate: a urease inhibitor. Nat Prod Res. 2007;21:721–5.10.1080/14786410600906913Search in Google Scholar PubMed
[17] Tougu V. Acetylcholinesterase: Mechanism of catalysis and inhibition. Curr Med Chem. 2001;1:155–70.10.2174/1568015013358536Search in Google Scholar
[18] Baylac S, Racine P. Inhibition of 5-lipoxygenase by essential oils and other natural fragrant extracts. Int J Aromather. 2003;13:138–42.10.1016/S0962-4562(03)00083-3Search in Google Scholar
[19] Mahernia S, Bagherzadeh K, Mojab F, Amanlou M. Urease inhibitory activities of some commonly consumed herbal medicines. Iran J Pharm Res. 2015;14:943–7.Search in Google Scholar
[20] Dina SD, Yuval I, Ruth N, Haim JW. Patchdock and symmdock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–7.10.1093/nar/gki481Search in Google Scholar PubMed PubMed Central
[21] Jan MS, Ahmad S, Hussain F, Ahmad A, Mahmood F, Rashid U, et al. Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2,5-dione derivatives as multitarget anti-inflammatory agents. Eur J Med Chem. 2020;186:111863.10.1016/j.ejmech.2019.111863Search in Google Scholar PubMed
[22] Gilbert NC, Rui Z, Neau DB, Waight MT, Bartlett SG, Boeglin WE, et al. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine 663. FASEB J. 2012;26(8):3222–9.10.1096/fj.12-205286Search in Google Scholar PubMed PubMed Central
[23] Munawar MA, Chattha FA, Kousar S, Munir J, Ismail T, Ashraf M, et al. Synthesis of novel triazoles and a tetrazole of escitalopram as cholinesterase inhibitors. Bioorg Med Chem. 2015;23:6014–24.10.1016/j.bmc.2015.06.051Search in Google Scholar PubMed
[24] Ullah A, Iftikhar F, Arfan M, Batool-Kazmi ST, Anjum MN, Haq IU, et al. Amino acid conjugated antimicrobial drugs: Synthesis, lipophilicity-activity relationship, antibacterial and urease inhibition activity. Eur J Med Chem. 2018;145:140–53.10.1016/j.ejmech.2017.12.089Search in Google Scholar PubMed
© 2023 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- Synthesis, characterization, and antibacterial activity of a new poly azo compound containing N-arylsuccinimid and dibenzobarrelene moieties
- Design, synthesis, and antiviral activities evaluation of novel quinazoline derivatives containing sulfonamide moiety
- Design, synthesis, and anticancer activity of novel 4,6-dimorpholinyl-1,3,5-triazine compounds
- Design, synthesis, biological evaluation, and bio-computational modeling of imidazo, thieno, pyrimidopyrimidine, pyrimidodiazepene, and motifs as antimicrobial agents
- Synthesis of a novel phosphate-containing ligand rhodium catalyst and exploration of its optimal reaction conditions and mechanism for the polymerization of phenylacetylene
- Design, synthesis, and antiproliferative activity of novel 1,2,4-triazole-chalcone compounds
- Synthesis of metal–organic nanofiber/rGO nanocomposite as the sensing element for electrochemical determination of hypoxanthine
- Design and synthesis of various 1,3,4-oxadiazoles as AChE and LOX enzyme inhibitors
- Bis(2-cyanoacetohydrazide) as precursors for synthesis of novel azoles/azines and their biological evaluation
- Synthesis, characterization, and biological target prediction of novel 1,3-dithiolo[4,5-b]quinoxaline and thiazolo[4,5-b]quinoxaline derivatives
- Sustainable conversion of carbon dioxide into novel 5-aryldiazenyl-1,2,4-triazol-3-ones using Fe3O4@SP-vanillin-TGA nanocomposite
- Erratum
- Erratum to “Design, synthesis and study of antibacterial and antitubercular activity of quinoline hydrazone hybrids”
- SI: Undergraduate Research in the Synthesis of Biologically Active Small Molecules and Their Applications
- Preparation of novel acyl pyrazoles and triazoles by means of oxidative functionalization reactions
- Synthesis and conformational analysis of N-BOC-protected-3,5-bis(arylidene)-4-piperidone EF-24 analogs as anti-cancer agents
- SI: Development of Heterocycles for Biomedical and Bioanalytical Applications
- Influence of octreotide on apoptosis and metabolome expression in lipopolysaccharide-induced A549 cells
- Crude extract of J1 fermentation promotes apoptosis of cervical cancer cells
Articles in the same Issue
- Research Articles
- Synthesis, characterization, and antibacterial activity of a new poly azo compound containing N-arylsuccinimid and dibenzobarrelene moieties
- Design, synthesis, and antiviral activities evaluation of novel quinazoline derivatives containing sulfonamide moiety
- Design, synthesis, and anticancer activity of novel 4,6-dimorpholinyl-1,3,5-triazine compounds
- Design, synthesis, biological evaluation, and bio-computational modeling of imidazo, thieno, pyrimidopyrimidine, pyrimidodiazepene, and motifs as antimicrobial agents
- Synthesis of a novel phosphate-containing ligand rhodium catalyst and exploration of its optimal reaction conditions and mechanism for the polymerization of phenylacetylene
- Design, synthesis, and antiproliferative activity of novel 1,2,4-triazole-chalcone compounds
- Synthesis of metal–organic nanofiber/rGO nanocomposite as the sensing element for electrochemical determination of hypoxanthine
- Design and synthesis of various 1,3,4-oxadiazoles as AChE and LOX enzyme inhibitors
- Bis(2-cyanoacetohydrazide) as precursors for synthesis of novel azoles/azines and their biological evaluation
- Synthesis, characterization, and biological target prediction of novel 1,3-dithiolo[4,5-b]quinoxaline and thiazolo[4,5-b]quinoxaline derivatives
- Sustainable conversion of carbon dioxide into novel 5-aryldiazenyl-1,2,4-triazol-3-ones using Fe3O4@SP-vanillin-TGA nanocomposite
- Erratum
- Erratum to “Design, synthesis and study of antibacterial and antitubercular activity of quinoline hydrazone hybrids”
- SI: Undergraduate Research in the Synthesis of Biologically Active Small Molecules and Their Applications
- Preparation of novel acyl pyrazoles and triazoles by means of oxidative functionalization reactions
- Synthesis and conformational analysis of N-BOC-protected-3,5-bis(arylidene)-4-piperidone EF-24 analogs as anti-cancer agents
- SI: Development of Heterocycles for Biomedical and Bioanalytical Applications
- Influence of octreotide on apoptosis and metabolome expression in lipopolysaccharide-induced A549 cells
- Crude extract of J1 fermentation promotes apoptosis of cervical cancer cells