An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of 17β-estradiol in human serum and plasma
-
Myriam Ott
 ,
Andrea Geistanger
,
Andrea Geistanger


Abstract
Objectives
A new candidate isotope dilution-liquid chromatography-tandem mass spectrometry (ID-LC-MS/MS)-based reference measurement procedure (RMP) has been developed for the accurate and precise quantification of 17β-estradiol (E2) in human serum and plasma covering a measurement range from 0.400 to 5,000 pg/mL (1.47–18,357 pmol/L). To address this broad range, two separate methods were created: a high sensitivity (HS) method for concentrations between 0.400 and 5.00 pg/mL (1.47–18.4 pmol/L) and a standard range (SR) method for concentrations between 5.00 and 5,000 pg/mL (18.4–18,357 pmol/L).
Methods
As the primary reference material, E2 (CRM 6004-a) from the National Metrology Institute of Japan was used to ensure traceability to the international system (SI). A two-dimensional heart-cut LC approach was utilized for LC-MS/MS analysis, employing a supported liquid extraction sample preparation protocol for the SR method and a liquid-liquid extraction protocol followed by derivatization for the HS method. Assay validation was conducted following current guidelines. Selectivity and specificity were assessed using spiked serum samples, while potential matrix effects were evaluated through a post-column infusion experiment and comparison of standard line slopes. Precision, accuracy, and trueness were determined using an extensive 5-day protocol. Standard measurement uncertainty was evaluated according to the Guide to the Expression of Uncertainty in Measurement (GUM), with three individual sample preparations performed on at least two different days. Equivalence with higher-order RMPs was demonstrated through participation in the CDC Steroid Hormones Standardization (HoSt) program.
Results
The RMP enabled the quantification of E2 within the range of 0.400–5,000 pg/mL (1.47–18,357 pmol/L), demonstrating no interference from structurally related compounds and no evidence of matrix effects. The relative mean bias of the SR method ranged from −2.4 to 1.9 % across all levels, including secondary reference materials and spiked samples, whereas the HS method exhibited a mean bias ranging from −3.0 to 2.9 %. Expanded measurement uncertainties (k=2) for target value assignment ranged from 1.7 to 4.4 % for the SR method and were found to be ≤6.1 % for the HS method. The method’s transferability was demonstrated in a comparison study at a second laboratory. Additionally, the candidate RMP exhibited excellent correlation and equivalence to JCTLM-listed RMPs through the CDC HoSt program.
Conclusions
In summary, the ID-LC-MS/MS-based RMP accurately quantifies E2. Its robust performance makes it suitable for standardizing routine assays and measuring individual patient samples, ensuring traceability.
Introduction
Primarily produced in the ovaries, 17β-estradiol (E2) is the key pre-menopausal estrogen steroid hormone regulating the female reproductive system and secondary sexual characteristics [1], [2], [3], [4]. Though mainly found in female gonads, it is also produced in lower concentrations in male gonads, where it influences spermatogenesis [5], [6], [7], and in both sexes affecting neurological, musculoskeletal, and vascular functions [8], [9], [10]. In pre-menopausal women, E2 concentrations are high (200–400 pg/mL [734–1,469 pmol/L]) and decrease post-menopause to levels typical in men (5–20 pg/mL [18.4–73.4 pmol/L]) [11]. E2 is used to treat menopausal symptoms [12] but elevated levels can increase the risk of cardiovascular diseases [13] and various cancers [14], [15], [16]. While measurements of E2 are useful to detect possible pathologic conditions such as female tumors, more common applications of clinical testing include assessment of fertility [17], hormone dysregulation [18] and menopausal status [19].
High variability in E2 levels among individuals is a common issue in clinical settings, especially when measuring very low levels (<20 pg/mL [73.4 pmol/L]) [20], [21], [22], [23], [24], [25], which is often necessary for monitoring patients on aromatase inhibitor therapy [20] and diagnosing precocious puberty [26]. Additionally, E2 concentrations cover multiple orders of magnitude (approximately 1–3,000 pg/mL [3.67–11,014 pmol/L]), posing significant challenges to achieving high accuracy. This is particularly important for monitoring in vitro fertilization outcomes, which rely on accurate estrogen measurements to guide optimal therapy adjustments [27].
Routine analyses typically include immunoassays, which can be constrained by insufficient standardization and sensitivity, making detection inadequate for certain populations [25], 28]. Issues such as competing binding, cross-reactivity, and matrix effects further limit accuracy and specificity [29]. Isotope dilution (ID) gas chromatography-mass spectrometry (GS-MS) or liquid chromatography (LC)-MS-based methods have overcome many technical issues, improving sensitivity, specificity, and accuracy. However, various discrepancies between different ID-GS-MS or ID-LC-MS methods have been reported [24], 25], leading to an urgent need for standardization of highly sensitive and reliable methods. In addition, results from a study conducted by the Centers for Disease Control and Prevention (CDC) [30] identified calibration bias as the main cause of inter-assay variability and suggested that standardizing calibration with individual patient samples could significantly reduce this variability [24].
In this study, we introduce a novel candidate reference measurement procedure (RMP) based on ID-LC-MS/MS for the quantification of estradiol in human serum, utilizing a 2D heart-cut liquid chromatography (LC) approach for enhanced specificity. A broad measurement range of 0.400–5,000 pg/mL (1.47–18,357 pmol/L) was achieved by developing two distinct protocols: one for the high sensitivity (HS) range of 0.400–5.00 pg/mL (1.47–18.4 pmol/L) and another for the standard range (SR) of 5.00–5,000 pg/mL (18.4–18,357 pmol/L), exceeding the measurement ranges of all JCTLM-listed RMPs [31], 32]. In particular, the high sensitivity with the LLMI of 0.400 pg/mL (1.47 pmol/L) is crucial for the quantification of very low concentrations of estradiol in men, postmenopausal women and women under aromatase therapy treatment [20], 25], 28].
Since National Metrology Institutes (NMIs) are required to initiate traceability chains and assume a leading role in this process, the estradiol material CRM 6004-a from the National Measurement Institute of Japan (NMIJ) was employed as the primary reference material to ensure traceability to the International System of Units (SI). In scenarios where NMIs are unable to fulfil this role, alternative approaches must be considered. We also present a quantitative nuclear magnetic resonance (qNMR) methodology for the calculation of the mass fraction of E2 and establish SI traceability to both mass (kg) and amount of substance (mole) by utilizing qNMR internal standards (ISTDs) that are traceable to the National Institute of Standards and Technology (NIST) PS1 (primary standard qNMR) or NIST 350b [33], 34].
The analytical performance specifications (APS) [35] for the RMP were derived from the biological variability (BV) parameters reported in the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) [36] database and were utilized to assess precision and accuracy. For an E2 RMP, the desirable specifications include an imprecision of ≤3.8 %, a bias of ±2.0 %, a maximum allowable measurement uncertainty (MAu) of ≤2.5 %, and a total error of ≤8.1 % [36].
Equivalence with higher-order RMPs, C12RMP4R and NRMeth10 [37], 38], was demonstrated by participating in the CDC Steroid Hormones Standardization program (HoSt), which was established to assess the analytical accuracy and reliability of testosterone and estradiol tests performed in clinical research and public health laboratories [39], 40].
Materials and methods
A detailed operation procedure on the test procedure methodology can be found in the Supplementary Material 1.
Chemicals and reagents
As primary reference material, E2 (CAS 50-28-2) from the National Metrology Institute of Japan (NMIJ, CRM 6004-a) purchased from LGC (Wesel, Germany) was used. ISTD solutions in acetonitrile, 17β-estradiol-2,3,4–13C3 (13C3-E2, CAS 1261254-48-1, Supelco™) and 17β-estradiol-D5 (D5-E2, CAS 221093-45-4, Supelco™) were both obtained from Merck (Darmstadt, Germany). Three levels of E2 in human serum (BCR-576, BCR-577 and BCR-578) were sourced from the Joint Research Centre (Brussels, Belgium).
17α-Estradiol (CAS 57-91-0), estrone (CAS 53-16-7), ethyl acetate (CAS 141-78-6), n-heptane (CAS 142-82-5), dansyl chloride (CAS 605-65-2), and ammonium fluoride (CAS 12125-01-8) were obtained from Merck. ULC/MS grade acetonitrile (CAS 75-05-8), methanol (CAS 67-56-1) and formic acid (CAS 64-18-6) were purchased from Biosolve (Valkenwaard, Netherlands).
Steroid-free human serum was used as calibrator matrix and was purchased from Roche (Penzberg, Germany) or Golden West Diagnostics (Temecula, CA, USA). As an alternative calibrator matrix, a surrogate matrix composed of 1 % bovine serum albumin (Albumin BPLA 1 from Roche Penzberg, Germany) in phosphate-buffered saline (PBS, from VWR, Ismaning, Germany) was used.
Mixed gender native plasma pools (Li-heparin [CUST-BB-31072020-1], dipotassium (K2)-ethylene diamine tetraacetic acid (EDTA) [CUST-BB-17022020-4E], and tripotassium (K3) EDTA [CUST-BB-17022020-4D]) were obtained from Biotrend (Colonge, Germany). The native human serum pool consisted of at least five samples. According to the Declaration of Helsinki, all native serum and plasma patient samples were anonymized samples.
qNMR for determining the absolute purity of the standard materials
qNMR experiments were performed on a Jeol 600 MHz NMR equipped with a Helium-cryoprobe. Single-pulse-1HNMR was utilized for the quantitation (aromatic proton meta to phenolic group; δ=7.10 ppm; 1H; dimethyl terephthalate as qNMR ISTD; CD3CN as solvent) with an inter-scan delay of 70 s (Supplementary Material 2). The mass fraction value of our in-house method and that of the NMIJ 6004-a matches exactly, both being 98.4 %. Further details relevant to this manuscript are available in the Supplementary Material 2.
Additionally, we also performed quantum chemical calculations to ascertain the conformer distribution of estradiol in solution because the Boltzmann average of these structures constitute the NMR spectrum. Computational calculations to determine the most reactive chemical bond towards deprotonation were also performed to find the optimal resonance for quantitation. Finally DFT single-point calculations were performed at the wB97X-V/def2TZVP level of theory and all the information is presented in the Supplementary Material 2.
Matrix based calibrator and quality control (QC) preparation
Two independent primary stock solutions were prepared by weighing 5 mg E2 (NMIJ, CRM 6004-a) in a disposable weighing boat on an ultra-microbalance (XP6U/M, Mettler Toledo, readability of 0.0001 mg) and dissolving it in a 50 mL volumetric flask containing acetonitrile. The purity specified in the certificate of the reference material CRM 6004-a (98.4 % ± 0.3 %) was incorporated to yield the final stock solution concentrations. Based on these stock solutions four working and eight spike solutions were prepared in 50 % methanol (v + v) which were used for the generation of steroid-free matrix-based calibrator levels (1 + 49 v + v) with concentrations from 5.00 to 5,000 pg/mL (18.4 to 18,357 pmol/L) for the standard range (SR) method (See Supplementary Material 1, Section 3.2.3). Based on the same calibrator preparation concept the calibrator levels for the high sensitivity (HS) method were prepared with individual sample weighings and preparation of working and spike solutions. Final matrix based calibrator levels ranged from 0.400 to 5.00 pg/mL (1.47–18.4 pmol/L) (See Supplementary Material 1, Section 4.2.3).
A third independent weighing was used for the production of quality control samples (QCs) for both measuring ranges, respectively. To ensure accuracy and enable long-term monitoring, two spiked QC levels (15.0 pg/mL and 3,500 pg/mL [55.1 pmol/L and 12,850 pmol/L]), three levels of a certified secondary reference material (BCR-576 31.1 pg/mL [114 pmol/L], BCR-577 188 pg/mL [690 pmol/L], and BCR-578 365 pg/mL [1,340 pmol/L]), and one native patient sample (∼112 pg/mL [411 pmol/L]) were used (See Supplementary Material 1, Section 3.2.4). As there were no certified reference materials available for the concentration range of the HS method the certified material BCR-576 was diluted with steroid-free serum to a final concentration of 3.11 pg/mL (11.4 pmol/L) and used as a control sample. In addition, two spiked QC levels (steroid-free serum) with concentrations of 1.00 pg/mL (3.67 pmol/L) and 4.00 pg/mL (14.7 pmol/L) were prepared (See Supplementary Material 1, Section 4.2.4).
Sample preparation
Sample preparation was optimized and validated for serum matrix and plasma (lithium-heparin, K2-EDTA, K3-EDTA) matrices. To prepare the samples using the SR method a total volume of 450 µL of sample was pipetted into a screw cap vial. Then, 50 µL of the ISTD spiking solution (13C3-E2, 1.00 ng/mL [3,671 pmol/L] in 50 % methanol) was added, and the sample incubated for 30 min at room temperature. The sample was then diluted with deionized water and subjected to a supported liquid extraction (Novum SLE 6cc, Phenomenex) using ethyl acetate. The obtained extract was evaporated to dryness in a N2-evaporator, and the residue was reconstituted in 100 µL 100 mM Na2CO3–NaHCO3 buffer. After a final filtration step, the purified sample is ready for measurement.
To achieve the required sensitivity, it was necessary to optimize the sample preparation protocol using derivatization for the HS method. The process involved pipetting 900 µL of the sample into a screw cap vial and adding 100 µL of the ISTD spiking solution (D5-E2, 25.0 pg/mL [91.8 pmol/L] in 50 % methanol) followed by an incubation step. For sample clean-up, a liquid-liquid-extraction (LLE) was performed using a mixture of heptane and ethyl acetate (9 + 1, v + v), and the resulting organic layer was evaporated to dryness in a N2-evaporator. The residue was then reconstituted in 100 µL 100 mM Na2CO3–NaHCO3 buffer. For derivatization, 100 µL of dansyl chloride in acetonitrile (1 mg/mL) was added to the sample, which was then vortexed and incubated at 65 °C for 3 min. After this step, the sample is ready for measurement and can be stored at 8 °C until analysis.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
To adjust for the differences in sample preparation and the requirement for a derivatization step in the HS method, two distinct LC-MS/MS methods were optimized. Both, the SR method and the HS method required a heart-cut two-dimensional (2D) LC-MS/MS analysis. This analysis was performed on an Agilent 1290 Infinity LC system coupled to an AB Sciex QTrap 6500 triple quad mass spectrometer. For the heart-cut approach, the analyte transfer between columns was accomplished by connecting them using a 100 µL sample loop. For specific details regarding the 2D LC-MS/MS analysis, including the gradient, source, and multi-reaction monitoring (MRM) parameters, please refer to Supplementary Material 1.
In the case of the SR method, the first dimension employed an Acquity BEH C18 column from Waters (2.1 × 50 mm, 1.7 µm, Milford, MA, USA), while the second dimension utilized a Raptor Biphenyl column from Restek (2.1 × 100 mm, 2.7 µm, Centre County, PA, USA). In the first dimension, chromatographic separation was carried out at a temperature of 50 °C, with a flow rate of 0.300 mL/min, while in the second dimension, the flow rate was set at 0.500 mL/min. Water (A1) and acetonitrile (B1) were used as mobile phases in the first dimension, and 0.2 mM NH4F in water (A2) and methanol (B2) in the second dimension. The retention time of E2 and 13C3-E2 was 7.6 min. Electrospray ionization (ESI) was performed in negative mode, and MRM was employed using the following transitions: E2 m/z 271.2>145.0 (quantitation ion (QI)) and m/z 271.2>182.9 (confirmation ion (CI)), 13C3-E2 m/z 274.2>147.9 (ISTD QI) and m/z 274.2>186.0 (ISTD CI). The total run time per sample was 9 min.
Correction note: Correction added November 6, 2025 after online publication May 26, 2025: The numbers 4 and 7 in 147.9 were mistakenly swapped, resulting in an incorrect value. The last sentence should correctly read:
Electrospray ionization (ESI) was performed in negative mode, and MRM was employed using the following transitions:E2 m/z 271.2>145.0 (quantitation ion (QI)) and m/z 271.2>182.9 (confirmation ion (CI)), 13C3-E2 m/z 274.2>147.9 (ISTD QI) and m/z 274.2>186.0 (ISTD CI).
In the HS method, the first dimension utilized an Acquity BEH C8 column from Waters (2.1 × 50 mm, 1.7 µm), while the second dimension employed a Raptor Biphenyl column from Restek (2.1 × 100 mm, 2.7 µm). Chromatographic separation was performed at a temperature of 50 °C in the first dimension, with a flow rate of 0.300 mL/min. In the second dimension, the flow rate was set at 0.500 mL/min. In the first dimension, the mobile phases used were water (A1) and acetonitrile (B1) both containing 0.1 % formic acid. In the second dimension, the mobile phases consisted of water (A2) and a mixture of methanol and acetonitrile (7 + 3, v/v) (B2) containing 0.1 % formic acid, respectively. The retention time of dansyl-E2 and dansyl-E2-d5 was 5.8 min. ESI was performed in positive mode. MRM was performed using the following transitions: dansyl-E2 m/z 506.1>171.1 (QI) and m/z 506.1>156.1 (CI), D5-dansyl-E2 m/z 511.1>171.1 (ISTD QI) and m/z 511.1>156.1 (CI). The total run time per sample was 9 min.
Structure of analytical series, calibration and data analysis
Prior to the start of any analytical series, a system suitability test was performed, which had to be passed. This included checking the instrument sensitivity, the chromatographic retention time, and potential analyte carry-over after injection of high concentrated samples. Further details can be found in the Supplementary Material 1.
For both the SR and HS method, an individual set of calibrator and quality control samples (QCs) was prepared for each measurement sequence. In each sequence, the calibrators and QCs were injected twice – once at the beginning and once at the end of a sequence. The peak area ratio (AR) of the analyte QI to ISTD QI was used for quantitation. The calibration functions used both injections of all calibration levels without forcing the functions through the origin. For the SR method a quadratic model was used in the form AR=a*cA2 + b*cA + c (cA=analyte concentration) and 1/cA weighting, while for the HS method, a linear model was used in the form AR=a*cA + b 1/cA weighting. Further information regarding the software utilized and the specific parameters for peak integration can be found the Supplementary Material 1.
Method validation
The method was validated as previously described in detail by Taibon et al. [41] and according to the Clinical & Laboratory Standard Institute Guidelines C62A: “Liquid Chromatography-Mass Spectrometry Methods” [42], the International Conference on Harmonization guidance document “Harmonized Tripartite Guideline Validation of Analytical Procedures: Text and Methodology Q2 (R1)” [43] and the GUM [44].
Selectivity/specificity
To identify potential interfering steroids (17α-estradiol, estrone, 1,3,5(10)-estratrien-16β, 17β-epoxy-3-ol, 5(10)-estren-3,17-dione, 4,16-androstadien-3β-ol, 5,16-androstadien-3β-ol, 16,(5α)-androsten-3-one, 2,(5α)-androsten-17-one, 3-deoxydehydroepiandrosterone, and 5a-dihydrotestosterone) in the first dimension of the SR method, individual standards at concentrations of 1,000 ng/mL were separately injected. Measurements were conducted using both ESI(+) and ESI(−) modes on an UPLC-QToF instrument (Synapt G2-Si with an Acquity UPLC, Waters Inc.). Column materials, HPLC gradient, and eluents were used as specified for the first dimension of the LC method parameters. As a single modification, ESI-additives have been added to the fluidics after HPLC separation and directly before the ESI-Probe, using a syringe pump (5 μL/mL continuous injection in “combined” fluidics mode with the HPLC-flow of 0.3 mL/min; Additive in ESI(+): 6.1 % Formic acid in deionized H2O (v/v); Additive in ESI(−): 6.3 mM Ammonium fluoride in deionized H2O). This ensured optimal electrospray ionization conditions in both positive and negative ion modes while maintaining the eluents from the first dimension of separation, which are free of ESI additives. ESI-MS settings and data acquisition were performed with the following parameters: capillary voltage at 3.0 kV, source temperature at 120 °C, desolvation temperature at 500 °C, mass range from m/z 10 to m/z 1,000, and full-scan centroid mode. Data analysis was conducted using MassLynx V4.2 SCN983 (Waters Inc.). The chromatographic separation of E2 and potential interferents was then assessed to ensure that any interferences were baseline-separated. Interferents that were not separated in the first dimension were analysed using the 2D-LC setup, and their chromatographic separation was evaluated.
Dansyl chloride was used as a derivatizing reagent for the high-sensitivity method. This reagent is commonly applied in fluorescence detection and mass spectrometry (MS) analysis of phenols and amines. Given the chemical structure of the potential interferents, 17α-estradiol, estrone, and 1,3,5(10)-estratrien-16β, 17β-epoxy-3-ol were selected for interference testing in the high-sensitivity method. Individual standards at concentrations of 225 pg/mL were injected separately, and measurements were performed using the 2D-LC setup. Chromatographic separation was assessed in both the first and second dimensions.
Steroid-free human serum spiked with the isotope labeled ISTD (with the specified concentration used for the SR and HS protocol) and was analyzed to determine the amount of residual E2 in the ISTD. The amount of unlabeled analyte in the ISTD must not exceed 20 % of the amount of the lower limit of the measuring interval (LLMI), corresponding to the concentration level of the lowest calibrator.
In addition, both the steroid-free human serum and the native serum pool were evaluated to identify potential interferences that may occur at the expected analyte retention time.
Matrix effects
For qualitative evaluation of possible signal enhancement or suppression effects, a post-column infusion experiment was performed. To achieve a consistent MS signal, a 50 % methanol (aq) (v + v) solution containing E2 and 13C3-E2 (c=150 ng/mL [550,701 pmol/L]) for the SR and D5-E2 (c=0.250 ng/mL [918 pmol/L]) for the HS method was infused into the post-column-flow (flow rate 10 μL/min). The SR was evaluated using different samples, such as a neat solution (50 % methanol v + v), the surrogate matrix, the steroid-free human serum, the native human serum pool, K2-EDTA plasma, K3-EDTA plasma, and lithium-heparin plasma. On the other hand, the HS method was tested using the neat solution, surrogate matrix, steroid-free human serum, and a native matrix pool diluted with steroid-free human serum to decrease the endogenous E2 concentration.
In addition, to quantitatively assess potential matrix effects, calibrator levels were spiked in the same matrices except the K2/3EDTA plasma matrices for the SR method and native matrix pool for the HS method. Calibrator levels were prepared once and injected twice. The slopes (x-term) of the standard lines were compared and confidence intervals (CIs) determined. Matrix effects were quantitatively assessed according to the formula: ME%=B/A × 100 (B=area ratio in matrix, A=area ratio in neat solution) based on Botelho et al. [37].
Precision, trueness and accuracy
To determine the precision of the method, a five-day validation experiment was performed, as previously described by Taibon et al. [41]. For data analysis, an internal statistical program (Biowarp), based on the VCA Roche Open Source software package in R, was utilized. An ANOVA-based variance component analysis (VCA) performed to estimate the total variability of the method (type A uncertainty) [45].
The experiment involved the analysis of seven samples, which covered the entire calibration range of the SR method. These samples consisted of three levels of certified secondary reference materials (BCR-576 [31.1 pg/mL [144 pmol/L]], BCR-577 [188 pg/mL [690 pmol/L]], and BCR-578 [365 pg/mL [1,340 pmol/L]]). Additionally, three levels (5.00 pg/mL [18.4 pmol/L], 15.0 pg/mL [55.1 pmol/L], and 3,500 pg/mL [12,850 pmol/L]) were spiked in steroid-free serum and one level of a native patient serum sample (approx. 30 pg/mL [110 pmol/L]) was included. For the HS method, three levels were used, with two samples spiked into steroid-free serum at concentrations of 1.00 pg/mL (3.67 pmol/L) and 5.00 pg/mL (18.4 pmol/L), and one sample prepared by diluting secondary reference material (BCR-576) with steroid-free serum to achieve a final concentration of 3.11 pg/mL (11.4 pmol/L). All samples were prepared six-fold at each day (three-fold in two separate measurement sequences named as part A and part B) and injected twice, resulting in n=12 measurements per day and n=60 measurements over five days. For each part A and B an independent calibration curve was generated preparing individual calibrator levels.
Trueness and accuracy for both methods was assed using the same samples as for the precision experiment with the exception of the native sample. Additionally, the accuracy of the SR method was evaluated by fortifying the lithium heparin plasma matrix with E2 concentrations of 15.0 pg/mL (55.1 pmol/L) and 3,500 pg/mL (12,850 pmol/L). To assess the dilution integrity of both methods, a highly concentrated sample at a concentration of 6,000 pg/mL (22,028 pmol/L) for the SR method and 5.50 pg/mL (20.2 pmol/L) for the HS method was used. Therefore, samples were diluted 1 + 1 with steroid-free human serum. A total of six sample preparations were performed, with each sample prepared in triplicate for both part A and part B on a single day. Accuracy was evaluated as closeness of agreement between the test result and the accepted reference value, whereas trueness was evaluated as closeness of agreement between the average value obtained from a series of test results and an accepted reference value.
Linearity
To evaluate linearity of both methods (0.400 pg/mL [1.47 pmol/L] to 5.00 pg/mL [18.4 pmol/L] HS and 5.00 pg/mL [18.4 pmol/L] to 5,000 pg/mL [18,357 pmol/L] SR), expanded (±20 %) calibrator sets in steroid-free serum were prepared in triplicate including individual weighings. Thus, the concentrations ranged from 4.00 pg/mL (14.7 pmol/L) to 6,000 pg/mL (22,028 pmol/L) for the SR range method and from 0.320 pg/mL (1.17 pmol/L) to 6.00 pg/mL (22.0 pmol/L) for the HS method. All samples of each calibrator set were prepared once and injected twice. Calibration curves with linear and quadratic fit (1/cA weighting) were created and the calibrations equation parameters were evaluated.
Additionally, the linearity of the method was validated by examining the recovery of serially diluted samples (n=1) using the preferred regression model for calculation. Measurement results have to show a linear dependency. Therefore, for the SR method 11 samples were prepared by serial dilution of a high concentrated sample (approximately 5,000 pg/mL [18,357 pmol/L]) with steroid free-serum following specific ratios (10 + 0, 9 + 1, 8 + 2, 7 + 3, etc.). To show linearity across both measuring ranges, a sample with an approximate concentration of 16.0 pg/mL (58.7 pmol/L) was prepared the same way, however samples >5.00 pg/mL (>18.4 pmol/L) were measured using the SR method and samples ≤5.0 pg/mL (≤18.4 pmol/L) were measured using the HS method.
Lower limit of the measuring interval (LLMI)
The LLMI was set to the concentration of the lowest calibrator level. Precision and accuracy at concentration of 5.00 pg/mL (18.4 pmol/L) for the SR method, as well at 0.400 pg/mL (1.47 pmol/L) for the HS method, were determined by six-fold preparation.
Uncertainty of measurements
Standard measurement uncertainty was evaluated following the GUM guidelines [44]. The overall standard uncertainty in a single measurement was determined by considering two main factors: the standard uncertainty in calibrator preparation (unccal) and the calculated standard uncertainty in the precision experiment (uncprec). To ensure accurate estimation of the error, the calibrator level with the highest standard uncertainty (unccal) was selected for each sample concentration level when combining uncertainties. Reference or target values were established by averaging multiple sample preparations conducted on at least two different days. The standard measurement uncertainty was obtained by combining the standard uncertainty in calibrator preparation (unccal) with the standard uncertainty (standard deviation) of the mean of the measurement results (uncmean). The resulting expanded measurement uncertainty was then multiplied by a coverage factor of k=2 for a confidence level of 95 %, assuming a normal distribution. Detailed information can be found in Supplementary Material 3.
Equivalence to JCTLM listed reference measurement procedures (RMPs)
To establish equivalence with the JCTLM-listed Reference Measurement Procedures (RMPs), the CDC Hormones Standardization Program’s Phase 1 Panel was utilized. This panel, provided by the Centers for Disease Control and Prevention (CDC), includes 40 non-pooled human serum samples with assigned target values determined using either the Ghent University’s ID-GC-MS reference method [38] or the CDC ID-LC-MS/MS reference method [37]. Since these samples only encompass the concentration range of the SR method, an additional set of 10 single donor human serum samples were measured using the HS method. These samples had assigned reference values provided by the CDC and covered concentrations ranging from 1.10 to 4.70 pg/mL (4.04 to 17.3 pmol/L). The purpose of this measurement was to assess the equivalence of the HS method to the JCTLM RMPs for concentrations below 5.00 pg/mL (18.4 pmol/L). To prove the robustness of the candidate RMP and enable ongoing method comparison and bias assessment in accordance with the CLSI EP09C (formerly CLSI EP09-A2) guideline [39], 40], regular participation in the Phase 2 CDC HoST program was carried out from Q2 2023 to Q1 2024.
Equivalence of results between independent laboratories
To assess the agreement of the RMP between two independent laboratories (Laboratory 1: Roche Diagnostics GmbH, Penzberg, Germany; Laboratory 2: Department of Pediatrics and Adolescent Medicine, University Hospital Erlangen, Erlangen, Germany), a method comparison study was performed on n=94 native residual, anonymized patient samples. In addition, a three-day precision experiment was performed at Laboratory 2. Spiked samples were provided by Laboratory 1. Both laboratories prepared their own calibrator levels using the primary reference material.
For SR measurements adaptations to the sample preparation protocol were made in the second laboratory in order to use the 96-well plate format. All steps before the sample extraction were done in one single 96-well deep well plate from polypropylene. In brief, 400 µL sample and 40 µL ISTD were incubated before adding 360 µL water. The sample was mixed and equally distributed onto two 96-well plates (Phenomenex Novum SLE) for extraction. The eluate of both parts was collected in one set of glass inserts (Hirschmann, Eberstadt, Germany). After evaporation, the samples were reconstituted, mixed, sealed and measured.
A second method comparison study (n=51) was carried out by Laboratory 1 and Laboratory 3 (Institute of Laboratory Medicine, Clinical Chemistry, and Molecular Diagnostics, University of Leipzig Medical Center, Germany) to compare a high-throughput, in-house developed LC-MS/MS routine assay to the candidate RMP performed at Laboratory 1. The routine assay utilized commercially available calibration material and employed online solid phase extraction for sample preparation [46].
Results
Traceability to SI units
The availability and use of the JTCLM listed primary reference material NMIJ-6004a from NMIJ enables the metrological traceability to the SI unit of mass (kilogram), the most important parameter for an RMP.
Selectivity
To assess the chromatographic separation of E2 and known interferences in the SR method (Figure 1) and HS method (Figure 2), neat solutions were prepared and injected individually. All interferences were effectively separated from E2 in the first chromatographic dimension and were not transferred to the second dimension. The compound 1,3,5(10)-Estratriene-16β,17β-epoxy-3-ol contained an unidentified impurity probably due to insufficient purification. This impurity was not fully separated from E2 in the first dimension of the HS method, leading to its transfer to the second dimension, where it was successfully separated. However, it has to be considered that the concentration used for the interference testing was about 45 times higher than the highest calibrator level concentration.

Chromatographic separation of E2 and potential interferents in the first dimension of the SR method.
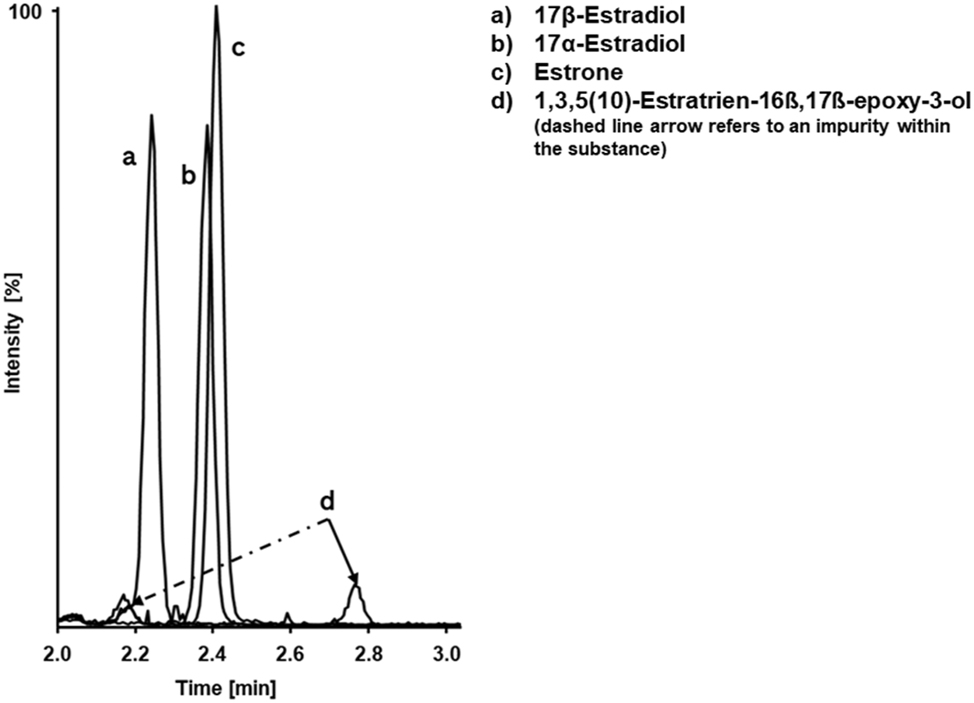
Chromatographic separation of E2 and potential interferents in the first dimension of the HS method.
For ISTD spiked steroid-free serum samples, in both methods, no peaks were observed in the analyte SRM traces. This indicates the absence of residual E2 in the used ISTDs. Additionally, the steroid-free serum and native serum samples did not show any potential interferences at the expected analyte retention times.
Matrix effects specificity
A post-column infusion experiment was performed to evaluate ion suppression or enhancement effects across various matrices for both methods. Neither method showed significant ion suppression or enhancement at the analyte and ISTD retention times. Quantitative evaluation of matrix effects was conducted by comparison of the standard line slopes of different matrices. Due to a pipetting error, calibrator level four had to be excluded for the steroid-free human serum matrix in the SR method. As a result, the following slopes (x-term) and CIs were obtained: 0.0087 (95 % CI 0.0086 to 0.0089) for the neat solution, 0.0086 (95 % CI 0.0085 to 0.0087) for the surrogate matrix, 0.0087 (95 % CI 0.0085 to 0.0089) for the steroid-free human serum, 0.0087 (95 % CI 0.0085 to 0.0090) for the native human serum pool, and 0.0086 (95 % CI 0.0084 to 0.0088) for lithium-heparin plasma. For the HS method slopes and CIs were found to be 0.37 (95 % CI 0.35 to 0.38) for the neat solution, 0.40 (95 % CI 0.36 to 0.44) for the surrogate matrix, 0.40 (95 % CI 0.39 to 0.41) for the steroid-free human serum. Based on the comparable slopes and overlapping estimated CIs for both methods, it can be concluded that there is no matrix effect present.
The evaluation of sample matrix effects, showed no effect for the SR method and HS method with matrix effects ranging from 96 to 102 % with the exception of the steroid-free human serum for HS method indicated a slight ion enhancement resulting in a mean ME% of 107 %. However, the comparison of standard line slopes along with our evaluations on the accuracy and precision of the HS method showed no significant influence from this effect, thereby proving the method to be matrix independent.
Precision, trueness and accuracy
To evaluate the overall variability of the RMP, a validation experiment was conducted over multiple days. The intermediate precision of the SR method ranged from 1.3 to 3.1 %, and the repeatability consistently remained ≤2.9 % across different concentration levels, except for the lowest level of 5.00 pg/mL (18.4 pmol/L), where the intermediate precision and repeatability were found to be 8.7 % and 7.8 %, respectively. Due to technical issues, two measurements for BCR-576 and one for BCR-577 were excluded (Table 1). When employing the HS method for levels ≤5.00 pg/mL (≤18.4 pmol/L), the ANOVA analysis revealed an intermediate precision ≤5.9 % and a repeatability ≤5.6 % (Table 2).
Standard range method – precision performance parameters for E2 quantification using the cRMP (n=60 measurements).
| Variance source | CV, % | ||||||
|---|---|---|---|---|---|---|---|
| Level 1 5.00 pg/mL (18.4 pmol/L) |
Level 2 15.0 pg/mL (55.1 pmol/L) |
Level 3 3,500 pg/mL (12,850 pmol/L) |
BCR-576a
31.1 pg/mL (114 pmol/L) |
BCR-577a
188 pg/mL (690 pmol/L) |
BCR-578 365 pg/mL (1,340 pmol/L) |
Patient sample 32.6 pg/mL (120 pmol/L) |
|
| Intermediate | 8.7 | 3.1 | 1.3 | 2.6 | 1.8 | 1.9 | 2.3 |
| Between-day | 2.9 | 0.0 | 0.6 | 1.7 | 0.5 | 0.7 | 0.5 |
| Between-calibration | 2.5 | 0.9 | 0.4 | 0.3 | 1.2 | 1.2 | 1.1 |
| Repeatability | 7.8 | 2.9 | 1.1 | 1.9 | 1.2 | 1.4 | 2.0 |
| Between-preparation | 6.6 | 1.5 | 0.8 | 0.5 | 0.0 | 0.6 | 0.0 |
| Between-injection | 4.2 | 2.5 | 0.8 | 1.8 | 1.2 | 1.2 | 2.0 |
-
CV, coefficient of variation. Conversion factor pg/mL to pmol/L: 3.67. aBCR-576 (n=58) and BCR-577 (n=59). The coefficients of variation for repeatability and intermediate precision, which were determined from the individual variances, are printed in bold.
High sensitivity method – precision performance parameters for E2 quantification using the cRMP (n=60 measurements).
| Variance source | CV, % | ||
|---|---|---|---|
| Level 1 1.00 pg/mL (3.67 pmol/L) |
Level 2 5.00 pg/mL (18.4 pmol/L) |
BCR-576 diluted 3.11 pg/mL (11.4 pmol/L) |
|
| Intermediate | 5.8 | 5.9 | 5.6 |
| Between-day | 1.7 | 1.0 | 1.3 |
| Between-calibration | 0.0 | 3.1 | 1.8 |
| Repeatability | 5.6 | 4.9 | 5.1 |
| Between-preparation | 2.1 | 0.0 | 0.0 |
| Between-injection | 5.2 | 4.9 | 5.1 |
-
CV, coefficient of variation. Conversion factor pg/mL to pmol/L: 3.67. The coefficients of variation for repeatability and intermediate precision, which were determined from the individual variances, are printed in bold.
The accuracy and trueness were assessed by preparing six samples at each concentration level in a single day (Tables 3 and 4). The relative mean bias for the SR method ranged from −2.4 to 1.9 % for all levels including secondary reference material as well as self-spiked steroid-free human serum samples. The two evaluated levels for the lithium-heparin plasma matrix were found with a relative mean bias of −0.8 % and −0.4 %, respectively. In addition one sample >ULMI (approximately 6,000 pg/mL [22,028 pmol/L]) was diluted 1 + 1 (v + v) with steroid-free human serum and prepared six-fold resulting in a relative mean bias of 0.0 %. For the HS method the mean bias of all three levels ranged from −3.0 to 2.9 %. A sample with a concentration >ULMI (approximately 5.50 pg/mL [20.2 pmol/L]) was diluted 1 + 1 (v + v) with steroid-free serum and prepared six-fold resulting in a relative mean bias of 2.4 %.
Standard range method – bias and 95 % CI of spiked steroid-free serum samples, secondary reference material samples, lithium-heparin plasma samples and one diluted sample. The mean bias and corresponding confidence intervals were calculated using the individual sample biases of n=6 preparations.
| Concentration | Steroid-free serum samples and secondary reference material | Concentration | Plasma | ||
|---|---|---|---|---|---|
| Mean bias, % | 95 % CI, % | Mean bias, % | 95 % CI, % | ||
|
Level 1
5.00 pg/mL (18.4 pmol/L) |
−2.0 | −4.5 to 0.5 |
Level 1
59.8 pg/mL (220 pmol/L) |
−0.8 | −1.9 to 0.3 |
|
Level 2
15.0 pg/mL (55.1 pmol/L) |
0.3 | −2.2 to 2.8 |
Level 2
3,580 pg/mL (13,143 pmol/L) |
−0.4 | −1.2 to 0.4 |
|
Level 3
3,500 pg/mL (12,850 pmol/L) |
1.5 | 0.3 to 2.6 | |||
|
BCR-576
31.1 pg/mL (114 pmol/L) |
1.9 | 0.3 to 3.5 | |||
|
BCR-577
188 pg/mL(690 pmol/L) |
−2.4 | −2.8 to −1.9 | |||
|
BCR-578
365 pg/mL (1,340 pmol/L) |
−1.4 | −2.6 to −0.1 | |||
|
Dilution 1
6,000 pg/mL (22,028 pmol/L) |
0.0 | −1.3 to 1.3 | |||
-
CI, confidence interval. Conversion factor pg/mL to pmol/L: 3.67.
High sensitivity method – bias and 95 % CI of spiked steroid-free serum samples, one diluted secondary reference material sample and one diluted sample. The mean bias and corresponding confidence intervals were calculated using the individual sample biases of n=6 preparations.
| Concentration | Steroid-free serum samples and diluted secondary reference material | |
|---|---|---|
| Mean bias, % | 95 % CI, % | |
|
Level 1
1.00 pg/mL (3.67 pmol/L) |
−0.8 | −4.6 to 3.0 |
|
Level 2
5.00 pg/mL (18.4 pmol/L) |
2.9 | 1.2 to 4.6 |
|
BCR-576 diluted
3.11 pg/mL (11.4 pmol/L) |
−3.0 | −6.3 to 0.3 |
|
Dilution 1
5.52 pg/mL (20.3 pmol/L) |
2.4 | −0.6 to 5.4 |
-
CI, confidence interval. Conversion factor pg/mL to pmol/L: 3.67.
Overall these results are in a very good agreement with the RMP performance specifications defined based on the biological variation as well as the suggested performance criteria for total estradiol based on Vesper et al. [24].
Linearity
Linearity was evaluated over a concentration range of 0.320–6,000 pg/mL (1.17–22,028 pmol/L), which was divided into the two separate measurement ranges (SR and HS method). Both methods were assessed using linear regression, employing a linear model and a quadratic model.
All prepared calibrator sets demonstrated correlations coefficients of ≥0.999 for the SR method and ≥0.996 for the HS method, indicating a good fit for both models. Considering the smaller residuals, the linear regression model with a quadratic fit (AR=a*cA2 + b*cA + c) and a 1/cA weighting was selected for calibration of the SR method. For the HS method the residuals of the linear and quadratic model were found to be similar, wherefore the linear model with a 1/cA weighting was chosen for calibration.
In addition, the linearity of both measuring ranges was proven through the measurement of serially diluted samples, resulting in correlation coefficients of ≥0.999. The average recovery for the SR method was determined to be 98 %, while the average recovery for samples bridging the SR and HS method was found to be 94 %.
Lower limit of the measuring interval (LLMI)
The LLMI was determined for both LC-MS/MS methods by six-fold preparation of the lowest calibration level (Figures 3 and 4). For the SR method, a relative deviation of −5.4 % and a CV of 2.8 % was determined at 4.94 pg/mL (18.1 pmol/L). For the HS method, the LLMI was determined at a concentration of 0.400 pg/mL (1.47 pmol/L), with a relative deviation of 1.5 % and a CV of 9.9 %. Based on the criteria established performance criteria [24], 37], the RMP allows for an imprecision of 5.7 % and a bias of ±0.8 pg/mL (±2.94 pmol/L) for samples with concentrations ≤20 pg/mL (≤73.4 pmol/L). As a result, these findings fall within the anticipated range of values.
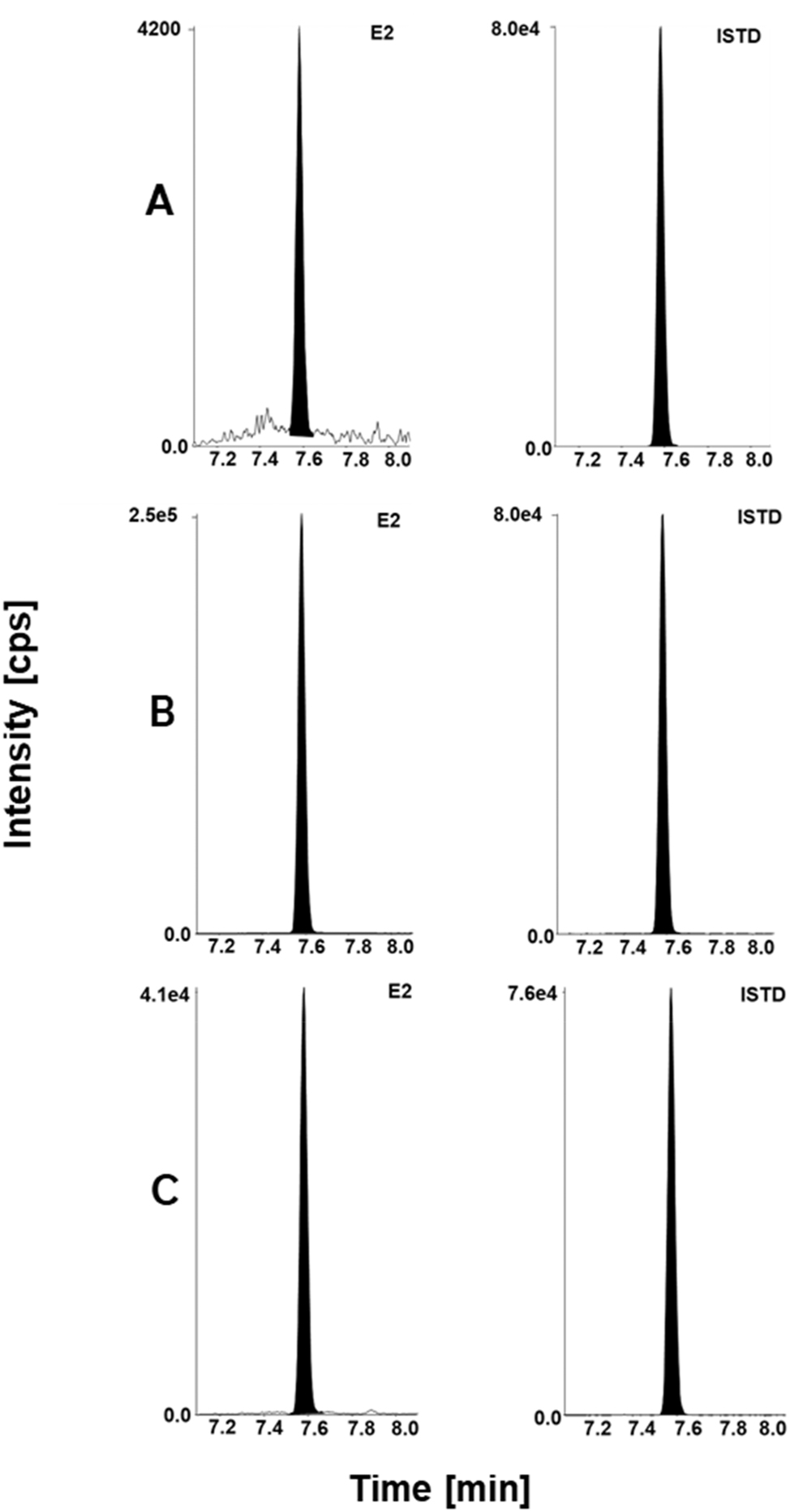
Estradiol LC-MS/MS chromatographic readouts. For SR method: (A) calibrator level 1 with a concentration of 5.00 pg/mL spiked in steroid-free serum matrix, analyte (left) and ISTD (right). (B) Secondary reference material BCR-578 (lyophilized native human serum) with a concentration of 365 pg/mL, analyte (left) and ISTD (right). (C) Fortified Li-heparin patient sample with a concentration of 59.8 pg/mL, analyte (left) and ISTD (right). SR, standard range method; HS, high sensitivity method; ISTD, internal standard.
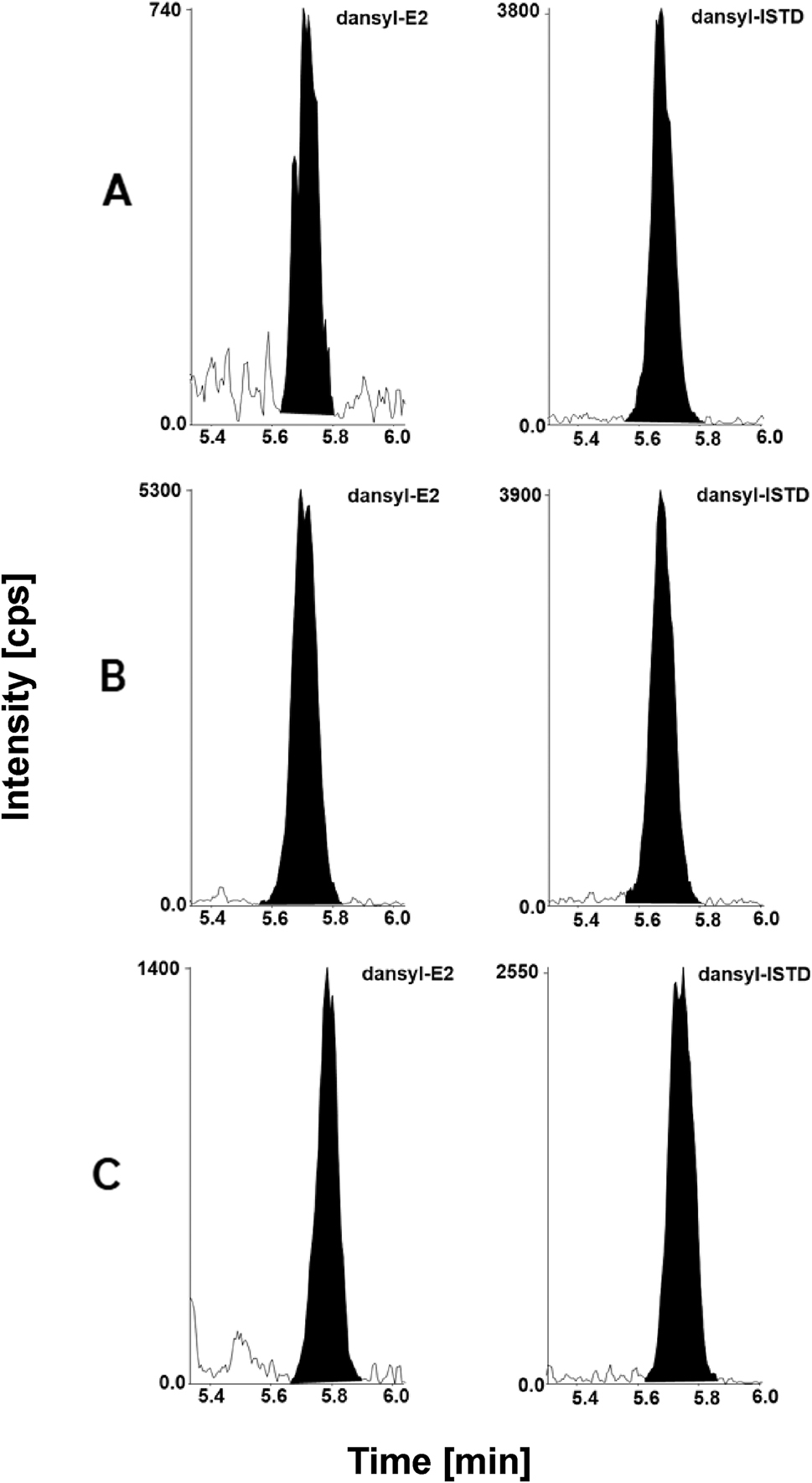
Estradiol LC-MS/MS chromatographic readouts. For HS method: (A) calibrator level 1 with a concentration of 0.400 pg/mL spiked in steroid-free serum matrix, analyte (left) and ISTD (right). (B) Quality control sample with a concentration of 4.02 pg/mL in steroid-free serum analyte (left) and ISTD (right). (C) Native serum sample with a concentration of 1.02 pg/mL, analyte (left) and ISTD (right). SR, standard range method; HS, high sensitivity method; ISTD, internal standard.
Uncertainty of measurements
The overall standard uncertainty for a single measurement for the SR method varied between 1.5 % and 3.2 %, with the exception of the lowest level, where it was found to be 8.8 %, as presented in Table 5. By multiplying the overall standard uncertainty by a coverage factor of k=2 (corresponding to a confidence level of 95 % assuming a normal distribution), the corresponding expanded uncertainty ranged from 3.0 to 6.3 %, and was determined to be 17.5 % for the lowest level. These uncertainties can be further reduced by performing multiple measurements. Therefore, for each level multiple sample preparations and measurements were performed on two different days, and the arithmetic mean (n=6) was calculated. As shown in Table 6, the overall standard uncertainties were reduced to 0.8–2.2 %, resulting in expanded uncertainties ranging from 1.7 to 4.4 %.
Standard range method – exemplary overview of measurement uncertainty for E2 quantification with the candidate RMP in serum samples for single measurements.
| Level | |||||||
|---|---|---|---|---|---|---|---|
| Level 1 5.00 pg/mL (18.4 pmol/L) |
Level 2 15.0 pg/mL (55.1 pmol/L) |
Level 3 3,500 pg/mL (12,850 pmol/L) |
BCR-576a
31.1 pg/mL (114 pmol/L) |
BCR-577a
188 pg/mL (690 pmol/L) |
BCR-578 365 pg/mL (1,340 pmol/L) |
Patient 32.6 pg/mL (120 pmol/L) |
|
|
Type B uncertainty
Calibrator preparation, CV (%) |
0.9 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| Characterization of reference material | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 |
| Preparation of | |||||||
| Stock solution | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 |
| Working solution | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 |
| Spike solution | 0.73 | 0.61 | 0.60 | 0.61 | 0.62 | 0.62 | 0.61 |
| Matrix based calibrator | 0.89 | 0.80 | 0.79 | 0.80 | 0.80 | 0.80 | 0.80 |
|
Type A uncertainty
intermediate precision, CV (%) |
8.7 | 3.1 | 1.3 | 2.6 | 1.8 | 1.9 | 2.3 |
| Measurement uncertainty (k=1), CV (%) | 8.8 | 3.2 | 1.5 | 2.7 | 2.0 | 2.1 | 2.4 |
| Expanded measurement uncertainty (k=2), CV (%) | 17.5 | 6.3 | 3.0 | 5.4 | 3.9 | 4.1 | 4.8 |
-
CV, coefficient of variation. Conversion factor pg/mL to pmol/L: 3.67. aBCR-576 (n=58) and BCR-577 (n=59). The measurement uncertainty of the whole approach for a single measurement estimated as a combination of the uncertainty of calibrator preparation (type B uncertainty) and uncertainty of the precision experiment (type A uncertainty) are given in bold.
Standard range method – exemplary overview of measurement uncertainty for E2 target value assignment (n=6) with the candidate RMP in serum samples.
| Level | |||||||
|---|---|---|---|---|---|---|---|
| Level 1 5.00 pg/mL (18.4 pmol/L) |
Level 2 15.0 pg/mL (55.1 pmol/L) |
Level 3 3,500 pg/mL (12,850 pmol/L) |
BCR-576a
31.1 pg/mL (114 pmol/L) |
BCR-577a
188 pg/mL (690 pmol/L) |
BCR-578 365 pg/mL (1,340 pmol/L) |
Patient sample 32.6 pg/mL (120 pmol/L) |
|
|
Type B uncertainty
Calibrator preparation, CV (%) |
0.9 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| Characterization of reference material | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 |
| Preparation of | |||||||
| Stock solution | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 |
| Working solution | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 |
| Spike solution | 0.73 | 0.61 | 0.60 | 0.61 | 0.62 | 0.62 | 0.61 |
| Matrix based calibrator | 0.89 | 0.80 | 0.79 | 0.80 | 0.80 | 0.80 | 0.80 |
|
Type A uncertainty
intermediate precision, CV (%) |
2.0 | 1.1 | 0.3 | 0.6 | 0.3 | 0.5 | 1.7 |
| Measurement uncertainty (k=1), CV (%) | 2.2 | 1.4 | 0.8 | 1.0 | 0.9 | 1.0 | 1.9 |
| Expanded measurement uncertainty (k=2), CV (%) | 4.5 | 2.8 | 1.7 | 1.9 | 1.7 | 1.9 | 3.7 |
-
CV, coefficient of variation. Conversion factor pg/mL to pmol/L: 3.67. aBCR-576 (n=58) and BCR-577 (n=59). The measurement uncertainty of the whole approach for a target value assignment estimated as a combination of the uncertainty of calibrator preparation (type B uncertainty) and uncertainty of the precision experiment (type A uncertainty) are given in bold.
For the HS method standard uncertainty for a single measurement ranged from 5.9 to 6.2 %, as shown in Table 7. By multiplying the standard uncertainty by a coverage factor of k=2 the expanded uncertainty ranged from 11.7 to 12.4 %. To assign target values, samples were prepared on a minimum of two different days, and the arithmetic mean (n=6) was determined. The standard uncertainties were found to be ≤3.1 % with corresponding expanded uncertainties ≤6.1 % (Table 8).
High sensitivity method – exemplary overview of measurement uncertainty for E2 quantification with the candidate RMP in serum samples for single measurements.
| Level | |||
|---|---|---|---|
| Level 1 1.00 pg/mL (3.67 pmol/L) |
Level 2 5.00 pg/mL (18.4 pmol/L) |
BCR-576 diluted 3.11 pg/mL (11.4 pmol/L) |
|
|
Type B uncertainty
Calibrator preparation, CV (%) |
2.2 | 1.8 | 1.7 |
| Characterization of reference material | 0.15 | 0.15 | 0.15 |
| Preparation of | |||
| Stock solution | 0.16 | 0.16 | 0.16 |
| Working solution | 1.51 | 1.51 | 1.51 |
| Spike solution | 2.13 | 1.71 | 1.65 |
| Matrix based calibrator | 2.19 | 1.79 | 1.73 |
|
Type A uncertainty
intermediate precision, CV (%) |
5.8 | 5.9 | 5.6 |
| Measurement uncertainty (k=1), CV (%) | 6.2 | 6.1 | 5.9 |
| Expanded measurement uncertainty (k=2), CV (%) | 12.4 | 12.3 | 11.7 |
-
CV, coefficient of variation. Conversion factor pg/mL to pmol/L: 3.67. The measurement uncertainty of the whole approach for a single measurement estimated as a combination of the uncertainty of calibrator preparation (type B uncertainty) and uncertainty of the precision experiment (type A uncertainty) are given in bold.
High sensitivity method – exemplary overview of measurement uncertainty for E2 target value assignment (n=6) with the candidate RMP in serum samples.
| Level | |||
|---|---|---|---|
| Level 1 1.00 pg/mL (3.67 pmol/L) |
Level 2 5.00 pg/mL (18.4 pmol/L) |
BCR-576 diluted 3.11 pg/mL (11.4 pmol/L) |
|
|
Type B uncertainty
Calibrator preparation, CV (%) |
2.2 | 1.8 | 1.7 |
| Characterization of reference material | 0.15 | 0.15 | 0.15 |
| Preparation of | |||
| Stock solution | 0.16 | 0.16 | 0.16 |
| Working solution | 1.51 | 1.51 | 1.51 |
| Spike solution | 2.13 | 1.71 | 1.65 |
| Matrix based calibrator | 2.19 | 1.79 | 1.73 |
|
Type A uncertainty
intermediate precision, CV (%) |
2.0 | 2.5 | 2.5 |
| Measurement uncertainty (k=1), CV (%) | 3.0 | 3.1 | 3.1 |
| Expanded measurement uncertainty (k=2), CV (%) | 5.9 | 6.1 | 6.1 |
-
CV, coefficient of variation. Conversion factor pg/mL to pmol/L: 3.67. The measurement uncertainty of the whole approach for a target value assignment estimated as a combination of the uncertainty of calibrator preparation (type B uncertainty) and uncertainty of the precision experiment (type A uncertainty) are given in bold.
Equivalence to JCTLM listed reference measurement procedures (RMPs)
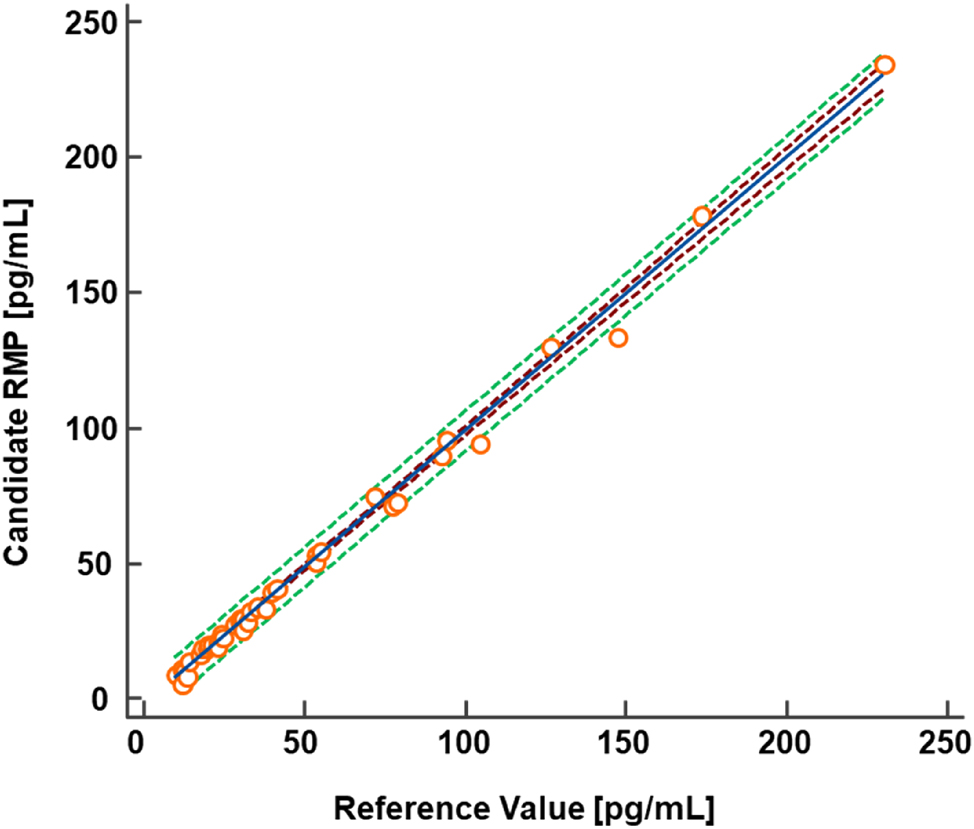
To show equivalence between the established candidate RMP and two of the JCTLM listed RMPs from the University of Ghent [38] and CDC [37] the CDC HoSt Phase 1 panel, which consisted of 40 individual human serum samples, was analyzed with n=1 preparations. For samples with concentrations ≤5.00 pg/mL (≤18.4 pmol/L), the HS method was used (n=4 samples), while samples with concentrations >5.00 pg/mL (>18.4 pmol/L) were measured using the SR method (n=36 samples). Passing-Bablok regression showed an excellent correlation with a slope of 0.99 (95 % CI 0.97 to 1.01) and an intercept of 0.12 (95 % CI −0.51 to 0.59) (Figure 5A). The Bland-Altman plot showed a mean bias of −0.8 % (95 % CI −2.9 to 1.3) and the 2S interval of the relative difference was 12.8 % (Figure 5B). This implies a strong agreement between the measured concentrations using the candidate RMP and the reference values assigned with JCTLM listed RMPs [37], 38]. In addition, as the phase 1 panel covers mostly the SR method, 10 individual human serum samples (1.10–4.70 pg/mL [4.04–17.3 pmol/L]) were measured using the HS method. These measurements showed a maximum absolute bias ranging from −0.17 to 0.22 pg/mL compared to the reference values (Table 9).
![Figure 5:
Equivalence testing results between the cRMP and JCTLM-listed methods [37], 38] were analyzed using the CDC HoSt Phase 1 reference panel, which included 40 individual human serum samples ranging from 1.15 to 268 pg/mL (n=1 preparation). The Passing-Bablok regression (A) showed an excellent correlation, with a slope of 0.99 (95 % CI 0.97 to 1.01) and an intercept of 0.12 pg/mL (95 % CI −0.51 to 0.59 pg/mL). The Bland-Altman plot (B) revealed a mean bias of −0.8 % (95 % CI −2.9 to 1.3) and a 2S interval of the relative difference at 12.8 %. CI, confidence interval; RMP, reference measurement procedure; SD, standard deviation.](/document/doi/10.1515/cclm-2024-1255/asset/graphic/j_cclm-2024-1255_fig_005.jpg)
Equivalence testing results between the cRMP and JCTLM-listed methods [37], 38] were analyzed using the CDC HoSt Phase 1 reference panel, which included 40 individual human serum samples ranging from 1.15 to 268 pg/mL (n=1 preparation). The Passing-Bablok regression (A) showed an excellent correlation, with a slope of 0.99 (95 % CI 0.97 to 1.01) and an intercept of 0.12 pg/mL (95 % CI −0.51 to 0.59 pg/mL). The Bland-Altman plot (B) revealed a mean bias of −0.8 % (95 % CI −2.9 to 1.3) and a 2S interval of the relative difference at 12.8 %. CI, confidence interval; RMP, reference measurement procedure; SD, standard deviation.
High sensitivity method – calculated absolute bias [pg/mL] between the concentrations measured by the cRMP and provided reference values from CDC samples (concentrations <5.00 pg/mL [18.4 pmol/L]).
| Sample No. | Reference value, pg/mL | Absolute bias, pg/mL |
|---|---|---|
| 1 | 3.95 | 0.07 |
| 2 | 1.10 | 0.01 |
| 3 | 2.50 | 0.02 |
| 4 | 4.70 | 0.00 |
| 5 | 3.40 | −0.17 |
| 6 | 2.70 | 0.22 |
| 7 | 3.01 | 0.21 |
| 8 | 3.05 | 0.20 |
| 9 | 1.15 | −0.04 |
| 10 | 4.10 | 0.04 |
In order to demonstrate the robustness of the RMP candidate method, participation in the CDC HoSt Phase 2 program was carried out on a regular basis. Data evaluation was done based on the official report provided by CDC. Findings on measurement bias and imprecision were compared against the suggested performance criteria for E2 of ±12.5 % bias for samples >20 pg/mL (>73.4 pmol/L), ±2.5 pg/mL bias for samples ≤20 pg/mL (≤73.4 pmol/L), and ≤11.4 % for imprecision at all concentrations [24]. Linear regression analysis resulted in a slope of 1.01 (95 % CI 0.99 to 1.03) and an intercept of −1.82 (95 % CI −3.48 to −0.15 pg/mL) (Figure 6). Bias evaluation showed a relative mean bias of −3.4 % (95 % CI −5.4 to −1.4 %) for samples >20 pg/mL (>73.4 pmol/L) and an absolute mean bias of −1.25 (95 % CI −2.92 to 0.41) for samples <20 pg/mL (<73.4 pmol/L). The results demonstrate that the method is accurate and robust, and the results remain consistent throughout the year, between lots, and over the reported measurement range.
To demonstrate the robustness of the RMP candidate RMP, regular participation in the CDC HoST Phase 2 program was undertaken. Linear regression analysis resulted in a slope of 1.01 (95 % CI 0.99 to 1.03) and an intercept of −1.82 pg/mL (95 % CI −3.48 to −0.15 pg/mL). Bias evaluation showed a relative mean bias of −3.4 % (95 % CI −5.4 to −1.4 %) for samples >20 pg/mL and an absolute mean bias of −1.25 pg/mL (95 % CI −2.92 to 0.41 pg/mL). CI, confidence interval; RMP, reference measurement procedure; SD, standard deviation.
Equivalence of results between independent laboratories
The analysis of anonymized native patient samples (n=94) was conducted at Laboratories 1 and 2. Out of the total samples n=1 sample was identified as outlier based on the LORELIA outlier test and was not included in the analysis [47]. Passing-Bablok analysis revealed a good agreement between the two laboratories, resulting in a regression equation with a slope of 0.97 (95 % CI 0.97 to 0.98) and an intercept of −0.01 (95 % CI −0.13 to 0.21) (Figure 7A). Additionally, Pearson’s correlation coefficient was ≥0.999. Relative Bland-Altman analysis was conducted for samples greater than 20 pg/mL (73.4 pmol/L) (n=54), while absolute Bland-Altman analysis was performed for samples with a concentration of 20 pg/mL (73.4 pmol/L) or less (n=39). The relative Bland-Altman analysis revealed a mean bias of −1.2 % (95 % CI: −3.5 to 1.1 %), with the ±1.96*SD range of relative differences spanning from −17.4 to 15.0 % (lower limit 95 % CI: −21.3 to −13.6 %, upper limit 95 % CI: 11.2–18.9 %) (Figure 7B). The absolute Bland-Altman analysis indicated a mean bias of −0.4 pg/mL (95 % CI: −0.7 to −0.1 pg/mL), with the ±1.96*SD range of differences between −2.2 and 1.5 pg/mL (lower limit 95 % CI: −2.7 to −1.7 pg/mL, upper limit 95 % CI: 0.9–2.0 pg/mL) (Figure 7C).
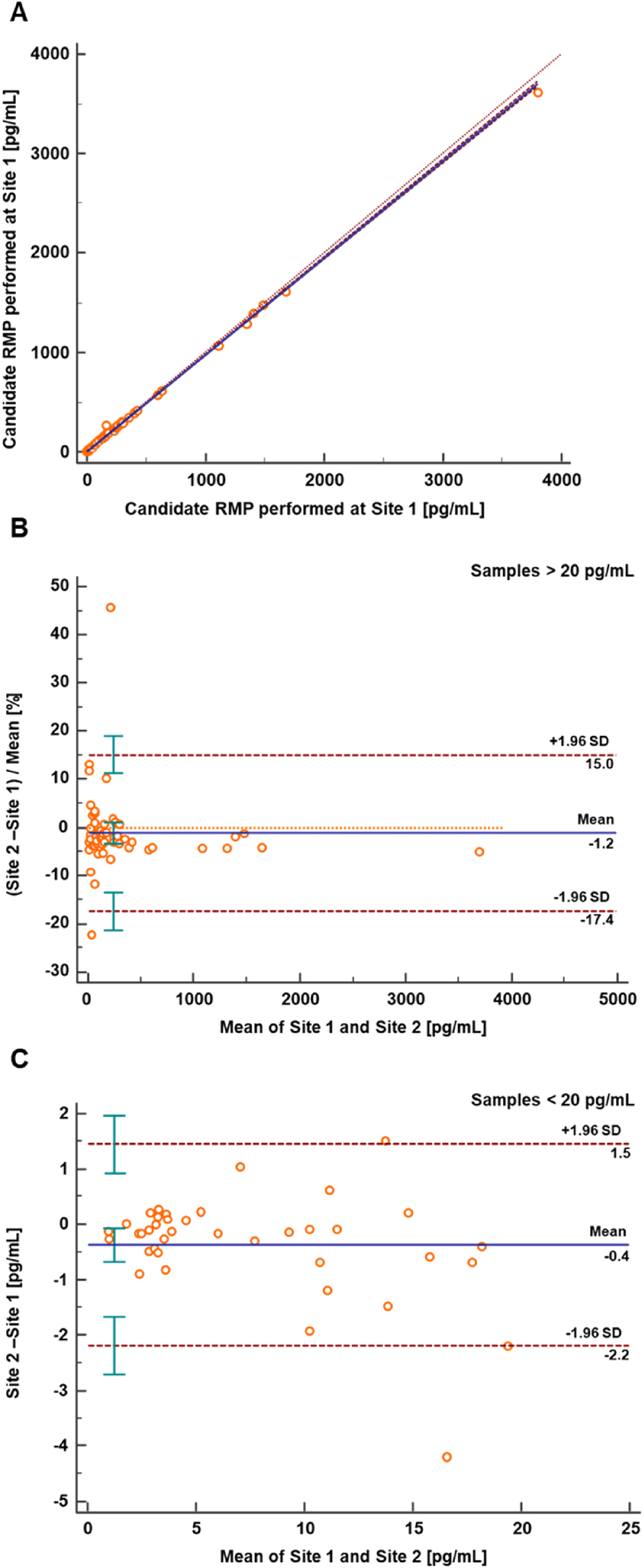
Method comparison study results between two independent laboratories. (A) Passing-Bablok regression plot with Pearson regression analysis for the RMP method comparison (n=93) between Laboratory 1 (Roche Diagnostics GmbH, Penzberg) and Laboratory 2 (Department of Pediatrics and Adolescent Medicine, University Hospital Erlangen). The regression equation had a slope of 0.97 (95 % CI 0.97 to 0.98) and an intercept of −0.01 (95 % CI −0.13 to 0.21), with a Pearson correlation value of ≥0.999. (B) Relative Bland-Altman plot for the RMP method comparison (n=54) between the two laboratories. The interlaboratory measurement bias was −1.2 % (95 % CI −3.5 to 1.1), with a ±1.96*SD range of relative differences from −17.4 to 15.0 % (lower limit 95 % CI −21.3 to −13.6, upper limit 95 % CI 11.2 to 18.9). (C) Absolute Bland-Altman analysis (n=39) showed a mean bias of −0.4 pg/mL (95 % CI −0.7 to −0.1), with a ±1.96*SD range of differences between −2.2 and 1.5 pg/mL (lower limit 95 % CI −2.7 to −1.7, upper limit 95 % CI 0.9 to 2.0). CI, confidence interval; RMP, reference measurement procedure; SD, standard deviation.
At Laboratory 2, the three-day precision experiment shows SR method intermediate precision ranging from 1.9 to 4.9 % and repeatability ranging from 1.3 to 3.9 % across all levels. For the HS method (Levels ≤5.00 pg/mL [≤18.4 pmol/L]), intermediate precision was ≤5.8 % and repeatability ≤5.0 %. All CVs were comparable to those at Laboratory 1.
In order to establish comparability with an in-house developed high-throughput LC-MS/MS based assay [46], an additional method comparison study was conducted (Figure 8). This assay used a commercially available kit for calibration. A total of n=51 native patient samples were analysed, with two samples exceeding the upper limit of the measuring interval in the in-house developed assay and one sample confirmed as outlier performing the LORELIA outlier test [47]. The Passing-Bablok regression analysis resulted in a regression equation with a slope of 1.11 (95 % CI 1.07 to 1.15) and an intercept of −3.4 (95 % CI −8.1 to −0.9) with r=0.998. Furthermore, the Bland-Altman analysis revealed a mean bias of 7.2 % (95 % CI 2.7–11.6 %) and a corresponding 2S interval ranging from −23.0 to 37.4 %.
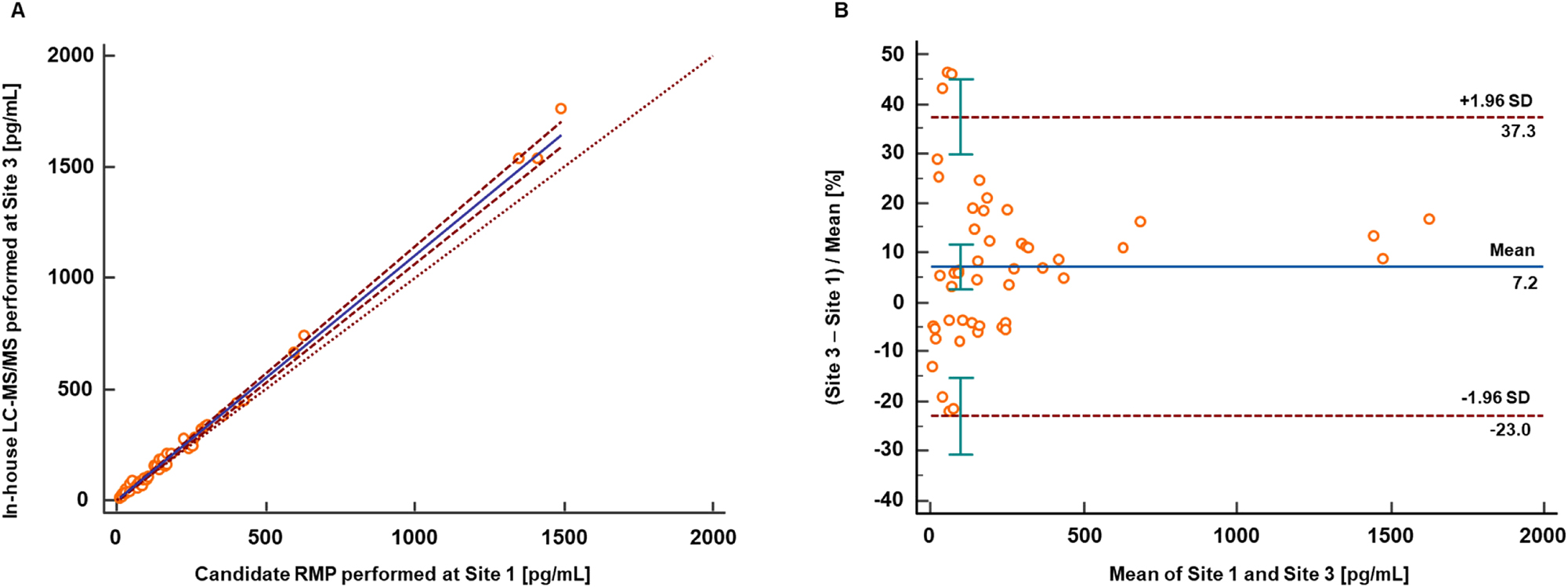
Results from the method comparison study performed between the candidate RMP (Site 1, Roche Diagnostics GmbH, Penzberg) and an in-house developed high-throughput LC-MS/MS based assay (Site 3, Institute of Laboratory Medicine, Clinical Chemistry, and Molecular Diagnostics, University of Leipzig Medical Center). (A) Passing-Bablok regression analysis resulted in a regression equation with a slope of 1.11 (95 % CI 1.07 to 1.15) and an intercept of −3.4 (95 % CI −8.1 to −0.9) with r=0.998. (B) Bland-Altman analysis revealed a mean bias of 7.2 % (95 % CI 2.7 to 11.6) and a corresponding 2S interval ranging from −23.0 to 37.4 %. CI, confidence interval; RMP, reference measurement procedure; SD, standard deviation.
Discussion
A new candidate ID-LC-MS/MS-based RMP has been developed for quantifying estradiol in human serum and plasma with high specificity, using a 2D heart-cut LC approach. The measurement range of 0.400–5,000 pg/mL (1.47–18,357 pmol/L) is broader than any JCTLM-listed RMPs (Table 10) and supports various clinical applications. This range was achieved by creating two methods for concentrations of 0.400–5.00 pg/mL (1.47–18.4 pmol/L) (HS) and 5.00–5,000 pg/mL (18.4–18,357 pmol/L) (SR). For the HS method, a derivatization step was added to the preparation protocol to 1the ionization process, increasing instrument sensitivity.
Summary of established RMPs and the new proposed RMP.
| RMP | JCTLM database | Measurement technique | Sample preparation | Sample size | LLMI | ULMI | Expected uncertainty |
|---|---|---|---|---|---|---|---|
| Thienpont et al. [38] | NRMeth 10 University of Ghent RMP for estradiol |
ID-GC-MS | N/A | Serum from 15 men (11.9–30.8 pg/mL) and 25 women (2.5–285 pg/mL) | 0.015 nmol/L | 30 nmol/L | 1.5–3% |
| Nelson et al. [49] | NRMeth40_Estradiol Mayo RMP for 17β-estradiol in blood plasma |
LC-MS/MS | LLE with dichloromethane and derivatization | 0.5 mL | 0.037 nmol/L | 7.4 nmol/L | N/A |
| Zhang et al. [50] | C17RMP7 GPHCM RMP for serum 17β-estradiol |
ID-LC-MS | A liquid-liquid extraction procedure | 0.5 mL of serum was used for high E2 concentration sample value assignments (>100 pg/mL), and 1.0 mL of serum was used for low-concentration samples (≤100 pg/mL) | 0.018 nmol/L | 13.889 nmol/L | 1.9–6.1 % |
| Botelho et al. [37] | C12 RMP4R CDC ID LC-MS/MS RMP for total 17β-estradiol |
ID LC-MS/MS | 2 LLE steps with ethyl acetate (x2) | 0.5–5 mL | 0.00261 nmol/L | 3.67 nmol/L | 4–4.8 % |
| Siekmann et al. [51] | NRMeth 61 DGKC RMO for serum 17β-estradiol |
ID/GC/MS | LLE with dichloromethane; chromatography purification; derivatization | 2 mL | 0.1 nmol/L | 5 nmol/L | 1–4% |
| Tai et al. [52] | C3RMMP9 NIST LC/MC/MS method for 17β-estradiol |
ID/LC/MS/MS | SPE, chromatography purification; derivatization | 3–5 mL | 25 ng/L (0.092 nmol/L) | 360 ng/L (1.32 nmol/L) | 3 % |
| This method | ID-2D-LC-MS/MS | SLE with ethyl acetate for the SR method; LLE with heptane/ethyl acetate for HS, followed by derivatization | Standard range method 0.5 mL, high sensitivity method 1.0 mL | 0.400 pg/mL (0.00147 nmol/L) | 5,000 pg/mL(18.4 nmol/L) | 1.7–4.4 % |
Traceability to SI units (kg) was achieved using either primary reference materials or materials characterized by qNMR. Furthermore, the analytical performance specifications (APS) for the SR RMP including imprecision, bias, and maximal allowable expanded measurement uncertainty (MAu) performing target value assignment were successfully met. For the high sensitivity method, the MAu for target value assignment was ≤3.1 %, which was higher than the desired specification of ≤2.5 %. The calculation of the desired MAu employed a preferred factor of 0.5. However, the EFLM database suggests using a factor of 0.75 corresponding to the minimal requirements. Therefore, the MAu is estimated to be ≤3.8 %, proving that the high sensitivity method is suitable for its intended use [36].
The equivalence of the candidate RMP to the JCTLM-listed RMPs [33], 34] was demonstrated through participation in the CDC HoSt program resulting in an an excellent agreement and correlation. Consistent participation in Phase 2 of the CDC HoST program validated the robustness of the method comparison and bias assessment, in accordance with the CLSI EP09C guideline. Furthermore, the HS method demonstrated strong alignment with the reference values established using JCTLM-listed methods.
The method’s transferability was effectively demonstrated through a comparison study at the Department of Pediatrics and Adolescent Medicine, University Hospital Erlangen. The resulting data revealed very good agreement between the two laboratories for both measuring ranges. The SLE sample preparation protocol, originally employed in the first laboratory, was successfully adapted to an automated 96-well plate format in the second laboratory. This adaptation was validated by precision experiments, which produced consistent inter-laboratory CV values.
The method comparison study between the RMP and the in-house LC-MS/MS-based assay exhibited a strong correlation, despite a noted positive deviation. These findings are in line with the broader context of estradiol measurements. The proficiency test for hormone assays, organized by the Reference Institute for Bioanalytics (RfB), permits a maximum deviation of 35 % from the target value, as per the guidelines of the German Medical Association (RiliBÄK). According to the RfB database [48], this year’s overall inter-laboratory variability for LC-MS/MS estradiol measurements ranges from 16.2 to 35.1 % CV. This indicates that the variability between laboratories is greater than the positive deviation observed in our method comparison study.
Conclusions
In summary, the presented ID-LC-MS/MS-based RMP is suitable for the accurate quantification of 17β-estradiol in a working range from 0.400 to 5,000 pg/mL (1.47 to 18,357 pmol/L) and the performance of the method enables its use for the evaluation and standardization of routine assays and the measurement of individual patient samples with ensured traceability.
Acknowledgments
We would like to express our sincere gratitude to Manuel Seitz and Martin Rempt for their technical support and the execution of the interference testing. We would also like to thank Aline Hoffmeister, Monika Kriner, and Michael Dedio for their support in selecting and providing samples. Editorial support, under the direction of the authors, was provided by Graziella Greco and Jade Drummond of inScience Communications, Springer Healthcare Ltd, UK, and was funded by Roche Diagnostics GmbH. (Penzberg, Germany).
-
Research ethics: All procedures were in accordance with the Helsinki Declaration. All samples used were exclusively anonymized samples.
-
Informed consent: Not applicable.
-
Author contributions: All authors have contributed to the manuscript conception and design; acquisition, or analysis and interpretation of data; drafting or revision; and final approval of the published article.
-
Use of Large Language Models, AI and Machine Learning Tools: Roche Chat, Roche’s artificial intelligence (AI) Technology, was used to improve the language of the manuscript.
-
Conflict of interest: Judith Taibon, Christian Geletneky, Neeraj Singh, Myriam Ott and Andrea Geistanger are all employees of Roche Diagnostics GmbH. Tobias Santner was an employee of Roche Diagnostics GmbH at the time the study was conducted. Friederike Bauland is an employee of Chrestos Concept GmbH & Co. KG, Essen, Germany. Manfred Rauh and Daniel Köppl are employees of the Department of Pediatrics and Adolescent Medicine, University Hospital Erlangen. Uta Ceglarek and Alexander Gaudl are employees of the Institute of Laboratory Medicine, Clinical Chemistry and Molecular Diagnostics, University of Leipzig Medical Center. Roche employees holding Roche non-voting equity securities (Genussscheine): Judith Taibon, Christian Geletneky, Andrea Geistanger, Myriam Ott.
-
Research funding: This research was funded by Roche Diagnostics GmbH. Manfred Rauh and Uta Ceglarek with team are funded cooperation partners of Roche Diagnostics GmbH.
-
Data availability: The raw data can be obtained on request from the corresponding author.
References
1. Estradiol. StatPearls [Internet]. Treasure Island (FL). https://www.ncbi.nlm.nih.gov/books/NBK549797/ [Accessed 8 Jul 2024].Suche in Google Scholar
2. Albrecht, ED, Pepe, GJ. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. Int J Dev Biol 2010;54:397–408. https://doi.org/10.1387/ijdb.082758ea.Suche in Google Scholar PubMed PubMed Central
3. Wang, L, Moenter, SM. Differential roles of hypothalamic AVPV and arcuate kisspeptin neurons in estradiol feedback regulation of female reproduction. Neuroendocrinology 2020;110:172–84. https://doi.org/10.1159/000503006.Suche in Google Scholar PubMed PubMed Central
4. Parisi, F, Fenizia, C, Introini, A, Zavatta, A, Scaccabarozzi, C, Biasin, M, et al.. The pathophysiological role of estrogens in the initial stages of pregnancy: molecular mechanisms and clinical implications for pregnancy outcome from the periconceptional period to end of the first trimester. Hum Reprod Update 2023;29:699–720. https://doi.org/10.1093/humupd/dmad016.Suche in Google Scholar PubMed PubMed Central
5. Hess, RA, Bunick, D, Lee, KH, Bahr, J, Taylor, JA, Korach, KS, et al.. A role for oestrogens in the male reproductive system. Nature 1997;390:509–12. https://doi.org/10.1038/37352.Suche in Google Scholar PubMed PubMed Central
6. Wibowo, E, Schellhammer, P, Wassersug, RJ. Role of estrogen in normal male function: clinical implications for patients with prostate cancer on androgen deprivation therapy. J Urol 2011;185:17–23. https://doi.org/10.1016/j.juro.2010.08.094.Suche in Google Scholar PubMed
7. Schulster, M, Bernie, AM, Ramasamy, R. The role of estradiol in male reproductive function. Asian J Androl 2016;18:435–40. https://doi.org/10.4103/1008-682x.173932.Suche in Google Scholar
8. Luine, VN. Estradiol and cognitive function: past, present and future. Horm Behav 2014;66:602–18. https://doi.org/10.1016/j.yhbeh.2014.08.011.Suche in Google Scholar PubMed PubMed Central
9. Yoest, KE, Cummings, JA, Becker, JB. Estradiol, dopamine and motivation. Cent Nerv Syst Agents Med Chem 2014;14:83–9. https://doi.org/10.2174/1871524914666141226103135.Suche in Google Scholar PubMed PubMed Central
10. Barakat, R, Oakley, O, Kim, H, Jin, J, Ko, CJ. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep 2016;49:488–96. https://doi.org/10.5483/bmbrep.2016.49.9.141.Suche in Google Scholar PubMed PubMed Central
11. O’Connell, MB. Pharmacokinetic and pharmacologic variation between different estrogen products. J Clin Pharmacol 1995;35:18s–24s. https://doi.org/10.1002/j.1552-4604.1995.tb04143.x.Suche in Google Scholar PubMed
12. Langer, RD, Hodis, HN, Lobo, RA, Allison, MA. Hormone replacement therapy - where are we now? Climacteric 2021;24:3–10. https://doi.org/10.1080/13697137.2020.1851183.Suche in Google Scholar PubMed
13. Shufelt, CL, Manson, JE. Menopausal hormone therapy and cardiovascular disease: the role of formulation, dose, and route of delivery. J Clin Endocrinol Metab 2021;106:1245–54. https://doi.org/10.1210/clinem/dgab042.Suche in Google Scholar PubMed PubMed Central
14. Brinton, LA, Felix, AS. Menopausal hormone therapy and risk of endometrial cancer. J Steroid Biochem Mol Biol 2014;142:83–9. https://doi.org/10.1016/j.jsbmb.2013.05.001.Suche in Google Scholar PubMed PubMed Central
15. Johansson, Å, Schmitz, D, Höglund, J, Hadizadeh, F, Karlsson, T, Ek, WE. Investigating the effect of estradiol levels on the risk of breast, endometrial, and ovarian cancer. J Endocr Soc 2022;6:bvac100. https://doi.org/10.1210/jendso/bvac100.Suche in Google Scholar PubMed PubMed Central
16. Key, TJ, Appleby, PN, Reeves, GK, Travis, RC, Alberg, AJ, Barricarte, A, et al.. Sex hormones and risk of breast cancer in premenopausal women: a collaborative reanalysis of individual participant data from seven prospective studies. Lancet Oncol 2013;14:1009–19. https://doi.org/10.1016/S1470-2045(13)70301-2.Suche in Google Scholar PubMed PubMed Central
17. Usala, SJ, Trindade, AA. A novel fertility indicator equation using estradiol levels for assessment of phase of the menstrual cycle. Medicina 2020;56. https://doi.org/10.3390/medicina56110555.Suche in Google Scholar PubMed PubMed Central
18. Su, HI, Freeman, EW. Hormone changes associated with the menopausal transition. Minerva Ginecol 2009;61:483–9.Suche in Google Scholar
19. Shieh, A, Greendale, GA, Cauley, JA, Karvonen-Gutierrez, C, Crandall, CJ, Karlamangla, AS. Estradiol and follicle-stimulating hormone as predictors of onset of menopause transition-related bone loss in pre- and perimenopausal women. J Bone Miner Res 2019;34:2246–53. https://doi.org/10.1002/jbmr.3856.Suche in Google Scholar PubMed PubMed Central
20. Niravath, P, Bhat, R, Al-Ameri, M, AlRawi, A, Foreman, C, Trivedi, MV. Challenges of measuring accurate estradiol levels in aromatase inhibitor-treated postmenopausal breast cancer patients on vaginal estrogen therapy. Pharmacol Res Perspect 2017;5. https://doi.org/10.1002/prp2.330.Suche in Google Scholar PubMed PubMed Central
21. Hogervorst, E, Williams, J, Combrinck, M, David Smith, A. Measuring serum oestradiol in women with Alzheimer’s disease: the importance of the sensitivity of the assay method. Eur J Endocrinol 2003;148:67–72. https://doi.org/10.1530/eje.0.1480067.Suche in Google Scholar PubMed
22. Dowsett, M, Folkerd, E. Deficits in plasma oestradiol measurement in studies and management of breast cancer. Breast Cancer Res 2005;7:1–4. https://doi.org/10.1186/bcr960.Suche in Google Scholar PubMed PubMed Central
23. Toniolo, P, Lukanova, A. The challenge of measuring circulating estradiol at low concentrations. Breast Cancer Res 2005;7:45–7. https://doi.org/10.1186/bcr987.Suche in Google Scholar PubMed PubMed Central
24. Vesper, HW, Botelho, JC, Vidal, ML, Rahmani, Y, Thienpont, LM, Caudill, SP. High variability in serum estradiol measurements in men and women. Steroids 2014;82:7–13. https://doi.org/10.1016/j.steroids.2013.12.005.Suche in Google Scholar PubMed PubMed Central
25. Rosner, W, Hankinson, SE, Sluss, PM, Vesper, HW, Wierman, ME. Challenges to the measurement of estradiol: an endocrine society position statement. J Clin Endocrinol Metab 2013;98:1376–87. https://doi.org/10.1210/jc.2012-3780.Suche in Google Scholar PubMed PubMed Central
26. Qudsiya, Z, Gupta, V. Peripheral precocious puberty. StatPearls. Treasure Island (FL) ineligible companies; 2024. Disclosure: Vikas Gupta declares no relevant financial relationships with ineligible companies.Suche in Google Scholar
27. Garnett, E, Bruno-Gaston, J, Cao, J, Zarutskie, P, Devaraj, S. The importance of estradiol measurement in patients undergoing in vitro fertilization. Clin Chim Acta 2020;501:60–5. https://doi.org/10.1016/j.cca.2019.09.021.Suche in Google Scholar PubMed
28. Taieb, J, Benattar, C, Birr, AS, Lindenbaum, A. Limitations of steroid determination by direct immunoassay. Clin Chem 2002;48:583–5. https://doi.org/10.1093/clinchem/48.3.583.Suche in Google Scholar
29. Blair, IA. Analysis of estrogens in serum and plasma from postmenopausal women: past present, and future. Steroids 2010;75:297–306. https://doi.org/10.1016/j.steroids.2010.01.012.Suche in Google Scholar PubMed PubMed Central
30. Centers for disease control and prevention. https://www.cdc.gov/index.html [Accessed 11 Jul 2024].Suche in Google Scholar
31. International Organization of Standardization (ISO). ISO 15193:2009(E): in vitro diagnostic medical devices−measurement of quantities in samples of biological origin−requirements for content and presentation of reference measurement procedures. Geneva, Switzerland: International Organization of Standardization. https://www.iso.org/standard/42021.html [Accessed 10 Jul 2024].Suche in Google Scholar
32. Joint committee on traceability in laboratory medicine (JCTLM)_Estradiol RMPs. https://www.jctlmdb.org/#/app/search/search-results?keywords=estradiol&code_type=rmp [Accessed 11 Jul 2024].Suche in Google Scholar
33. Nelson, MA, Waters, JF, Toman, B, Lang, BE, Rück, A, Breitruck, K, et al.. A new realization of SI for organic chemical measurement: NIST PS1 primary standard for quantitative NMR (benzoic acid). Anal Chem 2018;90:10510–17. https://doi.org/10.1021/acs.analchem.8b02575.Suche in Google Scholar PubMed PubMed Central
34. Westwood, S, Lippa, K, Shimuzu, Y, Lalerle, B, Saito, T, Duewer, D, et al.. Methods for the SI-traceable value assignment of the purity of organic compounds (IUPAC Technical Report). Pure Appl Chem 2023;95:1–77. https://doi.org/10.1515/pac-2020-0804.Suche in Google Scholar
35. Miller, WG. The role of analytical performance specifications in international guidelines and standards dealing with metrological traceability in laboratory medicine. Clin Chem Lab Med 2024;62:1462–9. https://doi.org/10.1515/cclm-2023-1201.Suche in Google Scholar PubMed
36. European Federation of Clinical Chemistry and Laboratory Medicine (EFLM). EFLM biological variation database – estradiol. https://biologicalvariation.eu/search?query=Estradiol [Accessed 10 Jul 2024].Suche in Google Scholar
37. Botelho, JC, Ribera, A, Cooper, HC, Vesper, HW. Evaluation of an isotope dilution HPLC tandem mass spectrometry candidate reference measurement procedure for total 17-β estradiol in human serum. Anal Chem 2016;88:11123–9. https://doi.org/10.1021/acs.analchem.6b03220.Suche in Google Scholar PubMed PubMed Central
38. Thienpont, LM, Verhaeghe, PG, Van Brussel, KA, De Leenheer, AP. Estradiol-17 beta quantified in serum by isotope dilution-gas chromatography-mass spectrometry: reversed-phase C18 high-performance liquid chromatography compared with immuno-affinity chromatography for sample pretreatment. Clin Chem 1988;34:2066–9. https://doi.org/10.1093/clinchem/34.10.2066.Suche in Google Scholar
39. Steroid hormones standardization programs. https://www.cdc.gov/clinical-standardization-programs/php/hormones/index.html [Accessed 11 July 2024].Suche in Google Scholar
40. Clinical Laboratory Standards Institute. CLSI document EP9-A2. Wayne, PA: Clinical Laboratory Standards Institute; 2002. https://clsi.org/standards/products/method-evaluation/documents/ep09/ [Accessed 11 Jul 2022].Suche in Google Scholar
41. Taibon, J, Santner, T, Singh, N, Ibrahim, SC, Babitzki, G, Köppl, D, et al.. An isotope dilution-liquid chromatography-tandem mass spectrometry (ID-LC-MS/MS)-based candidate reference measurement procedure (RMP) for the quantification of aldosterone in human serum and plasma. Clin Chem Lab Med 2023;61:1902–16. https://doi.org/10.1515/cclm-2022-0996.Suche in Google Scholar PubMed
42. Clinical and Laboratory Standards Institute (CLSI). C62-A. Liquid chromatography-mass spectrometry methods; approved guideline. In: CLSI document C62-A. Wayne, PA: Clinical and Laboratory Standard Institute; 2014.Suche in Google Scholar
43. ICH Harmonised Tripartite Guideline. Validation of analytical procedures: text and methodology Q2(R1). https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf [Accessed 4 Sep 2024].Suche in Google Scholar
44. Evaluation of measurement data – guide to the expression of uncertainty in measurement, JCGM 100:2008, GUM 1995 with minor corrections, JCGM, 2008. Available from: https://www.bipm.org/documents/20126/2071204/JCGM_100_2008_E.pdf/cb0ef43f-baa5-11cf-3f85-4dcd86f77bd6?version=1.10&t=1659082531978&download=true.Suche in Google Scholar
45. VCA. Variance component analysis. https://cran.r-project.org/web/packages/VCA/index.html [Accessed 2024].Suche in Google Scholar
46. Gaudl, A, Kratzsch, J, Bae, YJ, Kiess, W, Thiery, J, Ceglarek, U. Liquid chromatography quadrupole linear ion trap mass spectrometry for quantitative steroid hormone analysis in plasma, urine, saliva and hair. J Chromatogr A 2016;1464:64–71. https://doi.org/10.1016/j.chroma.2016.07.087.Suche in Google Scholar PubMed
47. Rauch, G, Geistanger, A, Timm, J. A new outlier identification test for method comparison studies based on robust regression. J Biopharm Stat 2011;21:151–69. https://doi.org/10.1080/10543401003650275.Suche in Google Scholar PubMed
48. RfB database – estradiol. https://www.rfb.bio/cgi/surveys?searchType=analyte&rv_type=all&analyte=Estradiol-17beta&year=2024 [Accessed 2 September 2024].Suche in Google Scholar
49. Nelson, RE, Grebe, SK, O’Kane, DJ, Singh, RJ. Liquid chromatography–tandem mass spectrometry assay for simultaneous measurement of estradiol and estrone in human plasma. Clin Chem 2004;50:373–84. https://doi.org/10.1373/clinchem.2003.025478.Suche in Google Scholar PubMed
50. Zhang, Q, Cai, Z, Liu, Y, Lin, H, Wang, Q, Yan, J, et al.. Comparison of bracketing calibration and classical calibration curve quantification methods in establishing a candidate reference measurement procedure for human serum 17β-estradiol by isotope dilution liquid chromatography tandem mass spectrometry. Microchem J 2020;152:104270. https://doi.org/10.1016/j.microc.2019.104270.Suche in Google Scholar
51. Siekmann, L, Siekmann, A, Breuer, H. Isotopic dilution mass spectrometry of oestriol and oestradiol-17β – an approach to definitive methods. Fresenius’ Z für Anal Chem 1978;290:122–3. https://doi.org/10.1007/bf00482284.Suche in Google Scholar
52. Tai, SS, Welch, MJ. Development and evaluation of a reference measurement procedure for the determination of estradiol-17beta in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal Chem 2005;77:6359–63. https://doi.org/10.1021/ac050837i.Suche in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/cclm-2024-1255).
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Editorial
- Keeping pace with patient safety by developing and qualifying higher-order reference measurement procedures for laboratory measurement standardization
- Review
- The role of AI in pre-analytical phase – use cases
- Opinion Paper
- Total laboratory automation: fit for its intended purposes?
- Guidelines and Recommendations
- EFLM checklist for the assessment of AI/ML studies in laboratory medicine: enhancing general medical AI frameworks for laboratory-specific applications
- Candidate Reference Measurement Procedures and Materials
- An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of cortisol in human serum and plasma
- Isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedures for the quantification of 24(R),25-dihydroxyvitamin D2 and 24(R),25-dihydroxyvitamin D3 in human serum and plasma
- An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of cortisone in human serum and plasma
- Candidate reference measurement procedure based on isotope dilution-two dimensional-liquid chromatography-tandem mass spectrometry for the quantification of androstenedione in human serum and plasma
- An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of 17β-estradiol in human serum and plasma
- Isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedures for the quantification of total and free phenytoin in human serum and plasma
- An isotope dilution-liquid chromatography-tandem mass spectrometry based candidate reference measurement procedure for the simultaneous quantification of 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 in human serum and plasma
- General Clinical Chemistry and Laboratory Medicine
- Quality assurance using patient split samples: recommendations for primary healthcare laboratories
- Age distorts the interpretation of FIB-4
- Not all anti-parietal cell antibody tests are equal for diagnosing pernicious anemia
- Impact of renal and hepatic function on dihydropyrimidine dehydrogenase phenotype assessed by enzyme activity in peripheral blood mononuclear cells and uracilemia
- Fecal leukocyte esterase levels predict endoscopic severity as an alternative biomarker in inflammatory bowel disease
- Cancer Diagnostics
- CA-125 glycovariant assays enhance diagnostic sensitivity in the detection of epithelial ovarian cancer
- Cardiovascular Diseases
- Defining the analytical characteristics of a novel high-sensitivity point-of-care troponin I assay in its intended clinical environment
- An automatic chemiluminescence immunoassay for a novel biomarker NT-IGFBP-4: analytical performance and clinical relevance in heart failure
- Analysis of total cholesterol results measured in the initial period of the Croatian screening program for familial hypercholesterolemia: a pilot study
- Diabetes
- Comparison of seven different enzymatic methods for serum glycated albumin in pregnant women: a multicenter study
- Infectious Diseases
- Comparative analysis of monocyte distribution width alterations in Escherichia coli sepsis: insights from in vivo and ex vivo models
- Proadrenomedullin for prediction of early and mid-term mortality in patients hospitalized for community-acquired pneumonia
- Annual Reviewer Acknowledgment
- Reviewer Acknowledgment
- Letters to the Editor
- Biological variation of serum Golgi protein 73 concentrations
- Are vitamins A and E results truly traceable and clinically useful? A practical and critical inquiry
- Tafasitamab interference in immunofixation electrophoresis
- Improvement in the turnaround time of PTH(1–84) as part of the intraoperative PTH monitoring for parathyroidectomy
- Rethinking the use of “one-way ANOVA” in CLSI EP15-A3 – a call for terminological precision and methodological clarity
- Toxic beauty: acute kidney injury triggered by hair-straightening treatment
- Congress Abstracts
- 57th National Congress of the Italian Society of Clinical Biochemistry and Clinical Molecular Biology (SIBioC – Laboratory Medicine)
Artikel in diesem Heft
- Frontmatter
- Editorial
- Keeping pace with patient safety by developing and qualifying higher-order reference measurement procedures for laboratory measurement standardization
- Review
- The role of AI in pre-analytical phase – use cases
- Opinion Paper
- Total laboratory automation: fit for its intended purposes?
- Guidelines and Recommendations
- EFLM checklist for the assessment of AI/ML studies in laboratory medicine: enhancing general medical AI frameworks for laboratory-specific applications
- Candidate Reference Measurement Procedures and Materials
- An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of cortisol in human serum and plasma
- Isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedures for the quantification of 24(R),25-dihydroxyvitamin D2 and 24(R),25-dihydroxyvitamin D3 in human serum and plasma
- An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of cortisone in human serum and plasma
- Candidate reference measurement procedure based on isotope dilution-two dimensional-liquid chromatography-tandem mass spectrometry for the quantification of androstenedione in human serum and plasma
- An isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedure for the quantification of 17β-estradiol in human serum and plasma
- Isotope dilution-liquid chromatography-tandem mass spectrometry-based candidate reference measurement procedures for the quantification of total and free phenytoin in human serum and plasma
- An isotope dilution-liquid chromatography-tandem mass spectrometry based candidate reference measurement procedure for the simultaneous quantification of 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 in human serum and plasma
- General Clinical Chemistry and Laboratory Medicine
- Quality assurance using patient split samples: recommendations for primary healthcare laboratories
- Age distorts the interpretation of FIB-4
- Not all anti-parietal cell antibody tests are equal for diagnosing pernicious anemia
- Impact of renal and hepatic function on dihydropyrimidine dehydrogenase phenotype assessed by enzyme activity in peripheral blood mononuclear cells and uracilemia
- Fecal leukocyte esterase levels predict endoscopic severity as an alternative biomarker in inflammatory bowel disease
- Cancer Diagnostics
- CA-125 glycovariant assays enhance diagnostic sensitivity in the detection of epithelial ovarian cancer
- Cardiovascular Diseases
- Defining the analytical characteristics of a novel high-sensitivity point-of-care troponin I assay in its intended clinical environment
- An automatic chemiluminescence immunoassay for a novel biomarker NT-IGFBP-4: analytical performance and clinical relevance in heart failure
- Analysis of total cholesterol results measured in the initial period of the Croatian screening program for familial hypercholesterolemia: a pilot study
- Diabetes
- Comparison of seven different enzymatic methods for serum glycated albumin in pregnant women: a multicenter study
- Infectious Diseases
- Comparative analysis of monocyte distribution width alterations in Escherichia coli sepsis: insights from in vivo and ex vivo models
- Proadrenomedullin for prediction of early and mid-term mortality in patients hospitalized for community-acquired pneumonia
- Annual Reviewer Acknowledgment
- Reviewer Acknowledgment
- Letters to the Editor
- Biological variation of serum Golgi protein 73 concentrations
- Are vitamins A and E results truly traceable and clinically useful? A practical and critical inquiry
- Tafasitamab interference in immunofixation electrophoresis
- Improvement in the turnaround time of PTH(1–84) as part of the intraoperative PTH monitoring for parathyroidectomy
- Rethinking the use of “one-way ANOVA” in CLSI EP15-A3 – a call for terminological precision and methodological clarity
- Toxic beauty: acute kidney injury triggered by hair-straightening treatment
- Congress Abstracts
- 57th National Congress of the Italian Society of Clinical Biochemistry and Clinical Molecular Biology (SIBioC – Laboratory Medicine)