Deletion as novel variants in VPS13B gene in Cohen syndrome: Case series
-
Li Kang


Abstract
Background
Cohen syndrome (OMIM No. # 216550) is a rare autosomal recessive disorder caused by homozygous mutation in the vacuolar protein sorting 13 homolog B (VPS13B) gene on chromosome 8q22.2. Clinical manifestations include hypermobile joints, microcephaly, intellectual disabilities, craniofacial and limb anomalies, and neutropenia. To date, more than 200 mutations of VPS13B have been reported in over 1,000 Cohen syndrome patients. This article reviews the clinical data of two cases of Cohen syndrome diagnosed by whole exome sequencing.
Results
Both children visited for psychomotor retardation. Gene detection showed a mutation in 8q22.2, NM_017890.4 Intron38 c.6940+1G > T and heterozygotic deletion of exon 3-19 of the VPS13B gene (Case 1), and a mutation in 8q22.2, NM_017890.4 Intron38 c.6940+1G > T and 8q22, NM_017890.4 Exon56 c10334_10335del in the VPS13B gene (Case 2). The variation was predicted to be pathogenic by related software, and they have not been reported.
Conclusion
Cohen syndrome should be considered in the differential diagnosis of any child with developmental retardation and neutropenia. The present study increases the mutation spectrum of the VPS13B gene and could be helpful in genetic diagnosis and genetic counseling in Cohen syndrome patients.
1 Introduction
Cohen syndrome (OMIM No. # 216550) is a rare autosomal recessive hereditary disease [1]. Cohen syndrome was first described by M. Michael Cohen Jr. in two affected siblings and one isolated case [2]. Mutation in the 13B vacuolar sorting protein gene, vacuolar protein sorting 13 homolog B (VPS13B) (8q22-8q23) gene, which is the only gene responsible for Cohen syndrome [3–5].
Cohen syndrome is a neurodevelopmental disease characterized by intellectual disabilities, short stature, hypotonia, microcephaly, musculoskeletal abnormalities, neutropenia, myopia, and facial abnormalities such as short philtrum and micrognathia [1,6–8]. The usual method of testing or screening for Cohen syndrome is whole exome sequencing [9]. To date, about 200 VPS13B mutations have been reported in nearly 1,000 cases of Cohen syndrome [10], of which about five cases have been genetically diagnosed in China [10,11]. Mutation includes missense/nonsense, regulatory, small deletions/insertions, and gross deletions/insertions, among which missense/nonsense variants are the most common [12]. The disease tends to be heterogeneous with a wide phenotypic variability. Distinct founder variants have been described in different areas.
In this study, we report the reviewed two novel pathogenic variants of VPS13B in patients from China and discuss the clinical and genetic variation characteristics of Cohen syndrome.
2 Case presentation
2.1 Case 1
A 2-year-old girl was admitted to the hospital because she showed psychomotor developmental delay. She had a speech delay and typical facial characteristics include microcephaly (head circumference 42 cm), hypertelorism, thick eyebrows, thick bushy hair, low hairline, and micrognathia (Figure 1a). The Autistic Behavior Checklist (ABC) score was 61. Developmental quotient (DQ) from five domains (adaptive, social, language, gross, and fine motor) was assessed using Gesell Developmental Schedules; they all showed moderate retardation (40 ≤ DQ ≤ 54) [13]. The laboratory examination results were as follows: the routine blood test showed that the white blood cell count was 8.74 × 109/L, the neutrophil count was 1.33 × 109/L, and the neutrophil percentage was 15.2%. High-throughput whole-exome sequencing: an ethylene diamine tetraacetic acid (ETDA) blood sample was taken, and next-generation sampling revealed a mutation in 8q22.2, NM_017890.4 Intron38 c.6940+1G > T in the VPS13B gene (Figure 2). This mutation was graded in relevant mutation interpretation guidelines by the American College of Medical Genetics (ACMG) as pathogenic. Heterozygotic deletion of exon 3–19 of the VPS13B gene was also revealed. Semi-quantitative PCR for specific gene deletion results: the copy number of case 1 (exon 3–19 of VPS13B gene) and her father was 1, heterozygous deletion. The mother’s copy number is 2, which is normal, and the deletion is from the father (Figure 3).

Characteristic facial features of case 1 (1a-left) and case 2 (1b-right).

VPS13B Intron38 c.6940+1G > T heterozygous mutation in case 1, carried by the mother, but not detected by the father.
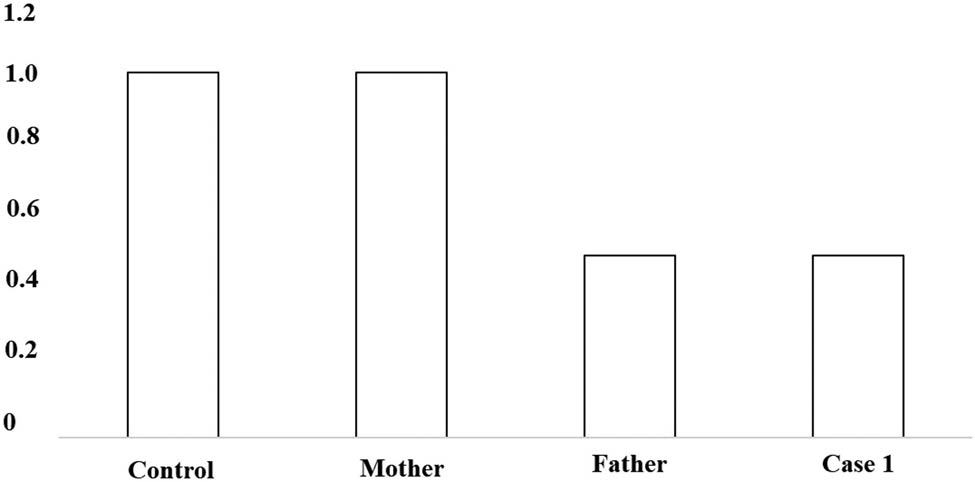
PCR verification: to the control, the ratio of copy number of VPS13B exon 3–19 in case 1 and her father was about 0.5, indicating that VPS13B had heterozygous deletion. To the control, the ratio of her mother’s copy number of VPS13B exon 3–19 was about 1, suggesting that her mother’s copy number was normal.
2.2 Case 2
A 1-year-old boy was admitted to the hospital because of developmental retardation, microcephaly (head circumference 43 cm), and micrognathia (Figure 1b). The modified Checklist for Autism in Toddlers (M-chat) score was 28. DQ from all five domains showed severe retardation (25 ≤ DQ ≤ 39) [13]. The laboratory examination results were as follows: the routine blood test showed that the white blood cell count was 7.60 × 109/L, the neutrophil count was 1.26 × 109/L, and the percentage of neutrophils was 16.5%. High-throughput whole-exome sequencing: an ETDA blood sample was taken, and using next-generation sampling, the results came out revealing a mutation in 8q22, NM_017890.4 Intron38 c.6940+1G > T (Figure 4) and 8q22, NM_017890.4 Exon56 c10334_10335del in the VPS13B gene (Figure 5). These two mutations were graded in relevant mutation interpretation guidelines by the ACMG as pathogenic.

VPS13B Intron38 c.6940+1G > T heterozygous mutation in case 2, carried by the mother, but not detected by the father.
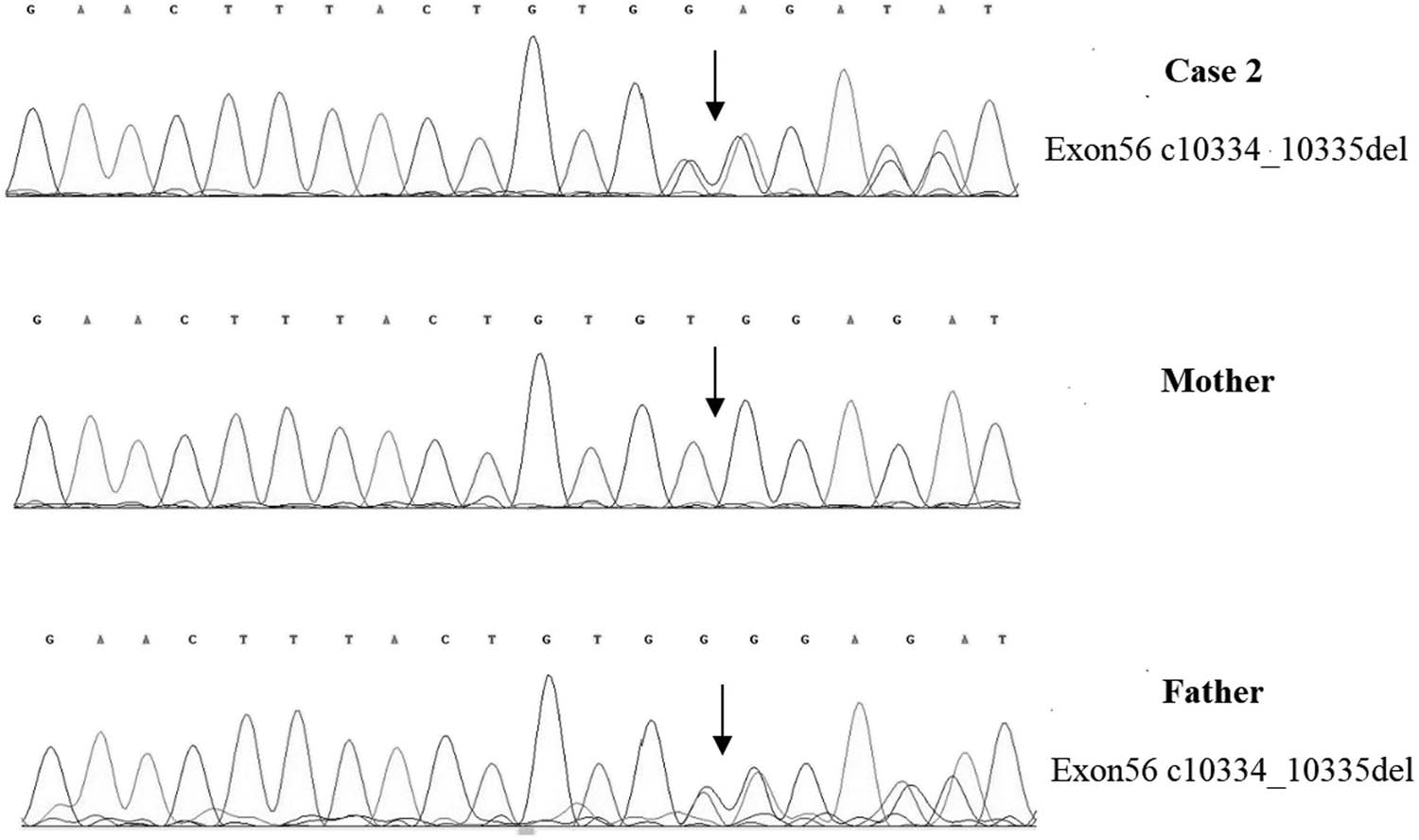
VPS13B Exon56 c10334_10335del heterozygous deletion in case 2, carried by the father, but not detected by the mother.
The clinical manifestations, relevant check results, and high-throughput whole-exome sequencing of the two children are summarized in Table 1. The pathogenicity of the mutations evaluated according to ACMG guidelines is detailed in Table 2.
Detailed clinical manifestations, relevant check results, and high-throughput whole-exome sequencing
| Case number | 1 | 2 |
|---|---|---|
| Gender (girl/boy) | Girl | Boy |
| Age (years) | 2 | 1 |
| Similar affected family member | No | No |
| Anthropometry | ||
| Height (cm) | 90 | 78 |
| Weight (kg) | 11.6 | 10.6 |
| Head circumference (cm) | 42 | 43 |
| Pregnancy history | Gravida 1 para 1 | Gravida 1 para 1 |
| Onset | About 1 year old | About 7 months |
| Clinical manifestations | Hypertelorism | Micrognathia |
| Thick eyebrows | Developmental retardation | |
| Thick bushy hair | ||
| Low hairline | ||
| Micrognathia | ||
| Developmental retardation | ||
| Radiological reports | ||
| Cranial magnetic functional evaluation | A widened brain gap | Cerebellar dysplasia |
| ECG | Sinus rhythm, right deviation of electrical axis, QR type of AVR, and no abnormality of ST segment | Normal |
| Autism assessment | ABC score was 61 | M-chat score was 28 |
| Gesell scale | Severe developmental retardation in five energy regions | Severe developmental retardation in five energy regions |
| Laboratory investigations | ||
| The percentage of neutrophils (%) | 25.0 | 20.5 |
| Variant | ||
| High-throughput whole-exome sequencing | 8q22.2, NM_017890.4 Intron38 c.6940+1G > T in the VPS13B gene; p.?. And heterozygotic deletion of exon 3–19 of VPS13B gene | 8q22, NM_017890.4 Intron38 c.6940+1G > T (Figure 3) and 8q22, NM_017890.4 Exon56 c10334_10335del in the VPS13B gene |
Pathogenicity evaluation
| ACMG evidence | |
|---|---|
| Case 1 | |
| c.6940+1G > T | PVS1: this variant affects the GT donor site of intron 38 and has been proved to cause exon 38 skip at mRNA level (PMID: 33959574) |
| PM3_strong: the variant was observed in trans with other pathogenic variants in several cases (PMID: 33959574, 33025479, 30138938, and in the current report) | |
| PS3: functional analysis has been conducted by Guiyu Lou et al. and Liangshan Li et al. (PMID: 33959574, 33025479) | |
| E3–19 del | PVS1: this variant causes loss of a long segment of the whole gene |
| PM2: this variant is very rare in common population | |
| PM3: the variant was observed in trans with other pathogenic variants in our case | |
| Case 2 | |
| c.6940+1G > T | Same as above |
| c.10334_10335del | PVS1: this variant causes frame shift and truncation of the 3′ terminal 7 exons |
| PM2: this variant is very rare in common population and has not yet been collected in gnomAD database | |
| PM3: the variant was observed in trans with other pathogenic variants in our case | |
-
Ethical approval: The research related to human use has been complied with all the relevant national regulations, institutional policies and in accordance with the tenets of the Helsinki Declaration, and has been approved by the authors’ institutional review board or equivalent committee.
-
Informed consent: Informed consent has been obtained from all individuals included in this study.
3 Discussion
We reported two novel compound heterozygous variants in the VPS13B gene inherited from non-inbred parents. Although this variant (cases 1 and 2) 8q22, NM_017890.4 Intron38 c.6940+1G > T in the VPS13B gene had been detected in patients with Cohen syndrome [7], the deletion of exon 3–19 and Exon56 c10334_10335del of VPS13B gene could constitute complex heterozygous mutations that cause Cohen syndrome with mutation in Intron38 c.6940+1G > T. The deletion range of these two fragments, which has not been reported in the literature, is a newly discovered variation that extends the spectrum of genes associated with Cohen syndrome.
The most significant manifestations of the cases reported in this article are developmental retardation and neutropenia. This result is consistent with the clinical features reported in other literature [14,15]. The possible mechanism of VPS13B causing neutropenia is related to increased neutrophil apoptosis and decreased SerpinB1 expression, which is a critical component of neutrophil survival [15]. In addition, in Cohen syndrome neutrophils, the SERPINB1 gene, which encodes a critical component of neutrophil survival, is significantly reduced, leading to excessive apoptosis of neutrophils, which is a possible cause of neutropenia [16]. The probable cause of developmental retardation is that VPS13B gene mutation leads to the reduction of coding protein which is a Golgi-enhanced scaffold protein that helps to maintain the structure and function of the Golgi complex. It has been reported that VPS13B regulates functional neuronal networks [7].
Cohen syndrome is a clinically heterogeneous disease with extensive and variable clinical manifestations, such as myopia, retinal dystrophy, and heart disease [14,15]. Unlike other cases, our patient did not present any ophthalmologic or cardiologic abnormality [17]. Therefore, there are some difficulties in the early diagnosis of the disease. And for clinical, there is no unified diagnostic standard for Cohen syndrome. The diagnosis can be determined mainly based on clinical manifestations, including obesity, hypotonia, mental deficiency, and facial, oral, ocular and limb anomalies: leukopenia, especially neutropenia. The pathogenic variation of the VPS13B gene was detected by molecular genetics [1,6,15]. According to previous research reports, there is no clear correlation between severity of the disorder and variant [1,12]. Further research is needed on the relationship between genotype and phenotype of Cohen syndrome.
Management of Cohen syndrome includes regular monitoring, rehabilitation, and genetic counseling. Ophthalmology should regularly evaluate visual acuity, refractive errors, and retinal dystrophy. Routine blood tests are performed to assess neutropenia. More frequent monitoring may be required for individuals with low absolute neutrophil count or recurring infection. Growth and weight gain also should be monitored. Treatment for patients with Cohen syndrome is symptomatic, including early physical, occupational, and speech therapy to address developmental retardation, hypotonia, joint hyperactivity, and granulocyte colony-stimulating factor to improve neutropenia [15].
4 Conclusion
We found two novel compound heterozygous variants in the VPS13B gene that can induce Cohen syndrome. In clinical diagnosis, if the children had special facial features, joint hyperactivity, neutropenia, and multi-system involvement, great attention should be paid to the possibility of genetic diseases. Therefore, genetic testing is significant for early diagnosis.
Acknowledgements
Special thanks to the Tianjin Children Hospital for supporting us in this project. Published with written consent of the patient.
-
Funding information: This research was funded by Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-040A).
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
[1] Rodrigues JM, Fernandes HD, Caruthers C, Braddock SR, Knutsen AP. Cohen Syndrome: review of the literature. Cureus. 2018;10(9):e3330.10.7759/cureus.3330Search in Google Scholar PubMed PubMed Central
[2] Cohen MM Jr, Hall BD, Smith DW, Graham CB, Lampert KJ. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J Pediatr. 1973;83(2):280–4.10.1016/S0022-3476(73)80493-7Search in Google Scholar PubMed
[3] Rejeb I, Jilani H, Elaribi Y, Hizem S, Hila L, Zillahrdt JL, et al. First case report of Cohen syndrome in the Tunisian population caused by VPS13B mutations. BMC Med Genet. 2017;18(1):134.10.1186/s12881-017-0493-5Search in Google Scholar PubMed PubMed Central
[4] Karimzadeh MR, Omidi F, Sahebalzamani A, Saeidi K. A novel VPS13B mutation identified by whole-exome sequencing in Iranian patients with Cohen syndrome. J Mol Neurosci. 2021;71(12):2566–74.10.1007/s12031-021-01852-4Search in Google Scholar PubMed
[5] Douzgou S, Petersen MB. Clinical variability of genetic isolates of Cohen syndrome. Clin Genet. 2011;79(6):501–6.10.1111/j.1399-0004.2011.01669.xSearch in Google Scholar PubMed
[6] Zhao S, Luo Z, Xiao Z, Li L, Zhao R, Yang Y, et al. Case report: two novel VPS13B mutations in a Chinese family with Cohen syndrome and hyperlinear palms. BMC Med Genet. 2019;20(1):187.10.1186/s12881-019-0920-xSearch in Google Scholar PubMed PubMed Central
[7] Yang C, Hou M, Li Y, Sun D, Guo Y, Liu P, et al. Gene analysis: a rare gene disease of intellectual deficiency-Cohen syndrome. Int J Dev Neurosci. 2018;68:83–8.10.1016/j.ijdevneu.2018.05.004Search in Google Scholar PubMed
[8] Kivitie-Kallio S, Norio R. Cohen syndrome: essential features, natural history, and heterogeneity. Am J Med Genet. 2001;102(2):125–35.10.1002/1096-8628(20010801)102:2<125::AID-AJMG1439>3.0.CO;2-0Search in Google Scholar
[9] Ghzawi A, Hirbawi H, Negida A, Abu-Farsakh H. A case of a Jordanian male twin with Cohen’s syndrome, with genetic analysis and muscle biopsy; case report. Ann Med Surg (Lond). 2021;71:103014.10.1016/j.amsu.2021.103014Search in Google Scholar
[10] Hu X, Huang T, Liu Y, Zhang L, Zhu L, Peng X, et al. Identification of a novel VPS13B mutation in a Chinese patient with Cohen syndrome by whole-exome sequencing. Pharmgenomics Pers Med. 2021;14:1583–9.10.2147/PGPM.S327252Search in Google Scholar
[11] Zhou JL, HUang HL, Wen FY. Severe congenital neutropenia: a case report and literature review. Lin Chuang Er Ke Za Zhi. 2020;38(1):61–4(Chinese).Search in Google Scholar
[12] Koehler K, Schuelke M, Hell AK, Schittkowski M, Huebner A, Brockmann K. A novel homozygous nonsense mutation of VPS13B associated with previously unreported features of Cohen syndrome. Am J Med Genet A. 2020;182(3):570–5.10.1002/ajmg.a.61435Search in Google Scholar PubMed
[13] Li Z, Pan L, Chen Y, Meng D, Liu Y, Li L, et al. The value of prenatal magnetic resonance imaging and postnatal follow-up using Gesell Developmental Schedules score for mild-to-moderate simple bilateral fetal ventriculomegaly. J Matern Fetal Neonatal Med. 2022;35(25):6229–35.10.1080/14767058.2021.1910657Search in Google Scholar PubMed
[14] Li L, Bu X, Ji Y, Tan P, Liu S. A novel homozygous VPS13B splice-site mutation causing the skipping of exon 38 in a Chinese family with Cohen syndrome. Front Pediatr. 2021;9(9):651621.10.3389/fped.2021.651621Search in Google Scholar PubMed PubMed Central
[15] Momtazmanesh S, Rayzan E, Shahkarami S, Rohlfs M, Klein C, Rezaei N. A novel VPS13B mutation in Cohen syndrome: a case report and review of literature. BMC Med Genet. 2020;21(1):140.10.1186/s12881-020-01075-1Search in Google Scholar PubMed PubMed Central
[16] Duplomb LRJ, Jego G. Serpin B1 defect and increased apoptosis of neutrophils in Cohen syndrome neutropenia. J Mol Med. 2019;97:633–45.10.1007/s00109-019-01754-4Search in Google Scholar PubMed
[17] Taban M, Memoracion-Peralta DS, Wang H, Al-Gazali LI, Traboulsi EI. Cohen syndrome: report of nine cases and review of the literature, with emphasis on ophthalmic features. J Am Assoc Pediatr Ophthalmol Strabismus. 2007;11:431–7.10.1016/j.jaapos.2007.01.118Search in Google Scholar PubMed
© 2023 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- HIF-1α participates in secondary brain injury through regulating neuroinflammation
- Omega-3 polyunsaturated fatty acids alleviate early brain injury after traumatic brain injury by inhibiting neuroinflammation and necroptosis
- The correlation between non-arteritic anterior ischemic optic neuropathy and cerebral infarction
- Enriched environment can reverse chronic sleep deprivation-induced damage to cellular plasticity in the dentate gyrus of the hippocampus
- Middle cerebral artery dynamic cerebral autoregulation is impaired by infarctions in the anterior but not the posterior cerebral artery territory in patients with mild strokes
- Leptin ameliorates Aβ1-42-induced Alzheimer’s disease by suppressing inflammation via activating p-Akt signaling pathway
- TIPE2 knockdown exacerbates isoflurane-induced postoperative cognitive impairment in mice by inducing activation of STAT3 and NF-κB signaling pathways
- Does the patellar tendon reflex affect the postural stability in stroke patients with blocked vision?
- Inactivation of CACNA1H induces cell apoptosis by initiating endoplasmic reticulum stress in glioma
- miR-101-3p improves neuronal morphology and attenuates neuronal apoptosis in ischemic stroke in young mice by downregulating HDAC9
- A custom-made weight-drop impactor to produce consistent spinal cord injury outcomes in a rat model
- Arterial spin labeling for moyamoya angiopathy: A preoperative and postoperative evaluation method
- Thyroid hormone levels paradox in acute ischemic stroke
- Geniposide protected against cerebral ischemic injury through the anti-inflammatory effect via the NF-κB signaling pathway
- The clinical characteristics of acute cerebral infarction patients with thalassemia in a tropic area in China
- Comprehensive behavioral study of C57BL/6.KOR-ApoEshl mice
- Incomplete circle of Willis as a risk factor for intraoperative ischemic events during carotid endarterectomies performed under regional anesthesia – A prospective case-series
- HOTAIRM1 knockdown reduces MPP+-induced oxidative stress injury of SH-SY5Y cells by activating the Nrf2/HO-1 pathway
- Esmolol inhibits cognitive impairment and neuronal inflammation in mice with sepsis-induced brain injury
- EHMT2 affects microglia polarization and aggravates neuronal damage and inflammatory response via regulating HMOX1
- Hematoma evacuation based on active strategies versus conservative treatment in the management of moderate basal ganglia hemorrhage: A retrospective study
- Knockdown of circEXOC6 inhibits cell progression and glycolysis by sponging miR-433-3p and mediating FZD6 in glioma
- CircYIPF6 regulates glioma cell proliferation, apoptosis, and glycolysis through targeting miR-760 to modulate PTBP1 expression
- Relationship between serum HIF-1α and VEGF levels and prognosis in patients with acute cerebral infarction combined with cerebral-cardiac syndrome
- The promoting effect of modified Dioscorea pills on vascular remodeling in chronic cerebral hypoperfusion via the Ang/Tie signaling pathway
- Effects of enriched environment on the expression of β-amyloid and transport-related proteins LRP1 and RAGE in chronic sleep-deprived mice
- An interventional study of baicalin on neuronal pentraxin-1, neuronal pentraxin-2, and C-reactive protein in Alzheimer’s disease rat model
- PD98059 protects SH-SY5Y cells against oxidative stress in oxygen–glucose deprivation/reperfusion
- TPVB and general anesthesia affects postoperative functional recovery in elderly patients with thoracoscopic pulmonary resections based on ERAS pathway
- Brain functional connectivity and network characteristics changes after vagus nerve stimulation in patients with refractory epilepsy
- Association between RS3763040 polymorphism of the AQP4 and idiopathic intracranial hypertension in a Spanish Caucasian population
- Effects of γ-oryzanol on motor function in a spinal cord injury model
- Electroacupuncture inhibits the expression of HMGB1/RAGE and alleviates injury to the primary motor cortex in rats with cerebral ischemia
- Effects of edaravone dexborneol on neurological function and serum inflammatory factor levels in patients with acute anterior circulation large vessel occlusion stroke
- CST3 alleviates bilirubin-induced neurocytes’ damage by promoting autophagy
- Excessive MALAT1 promotes the immunologic process of neuromyelitis optica spectrum disorder by upregulating BAFF expression
- Evaluation of cholinergic enzymes and selected biochemical parameters in the serum of patients with a diagnosis of acute subarachnoid hemorrhage
- 7-Day National Institutes of Health Stroke Scale as a surrogate marker predicting ischemic stroke patients’ outcome following endovascular therapy
- Cdk5 activation promotes Cos-7 cells transition towards neuronal-like cells
- 10.1515/tnsci-2022-0313
- PPARα agonist fenofibrate prevents postoperative cognitive dysfunction by enhancing fatty acid oxidation in mice
- Predicting functional outcome in acute ischemic stroke patients after endovascular treatment by machine learning
- EGCG promotes the sensory function recovery in rats after dorsal root crush injury by upregulating KAT6A and inhibiting pyroptosis
- Preoperatively administered single dose of dexketoprofen decreases pain intensity on the first 5 days after craniotomy: A single-centre placebo-controlled, randomized trial
- Myeloarchitectonic maps of the human cerebral cortex registered to surface and sections of a standard atlas brain
- The BET inhibitor apabetalone decreases neuroendothelial proinflammatory activation in vitro and in a mouse model of systemic inflammation
- Carthamin yellow attenuates brain injury in a neonatal rat model of ischemic–hypoxic encephalopathy by inhibiting neuronal ferroptosis in the hippocampus
- Functional connectivity in ADHD children doing Go/No-Go tasks: An fMRI systematic review and meta-analysis
- Review Articles
- Human prion diseases and the prion protein – what is the current state of knowledge?
- Nanopharmacology as a new approach to treat neuroinflammatory disorders
- Case Report
- Deletion as novel variants in VPS13B gene in Cohen syndrome: Case series
- Commentary
- Translation of surface electromyography to clinical and motor rehabilitation applications: The need for new clinical figures
- Revealing key role of T cells in neurodegenerative diseases, with potential to develop new targeted therapies
- Retraction
- Retraction of “Eriodictyol corrects functional recovery and myelin loss in SCI rats”
- Special Issue “Advances in multimedia-based emerging technologies...”
- Evaluation of the improvement of walking ability in patients with spinal cord injury using lower limb rehabilitation robots based on data science
Articles in the same Issue
- Research Articles
- HIF-1α participates in secondary brain injury through regulating neuroinflammation
- Omega-3 polyunsaturated fatty acids alleviate early brain injury after traumatic brain injury by inhibiting neuroinflammation and necroptosis
- The correlation between non-arteritic anterior ischemic optic neuropathy and cerebral infarction
- Enriched environment can reverse chronic sleep deprivation-induced damage to cellular plasticity in the dentate gyrus of the hippocampus
- Middle cerebral artery dynamic cerebral autoregulation is impaired by infarctions in the anterior but not the posterior cerebral artery territory in patients with mild strokes
- Leptin ameliorates Aβ1-42-induced Alzheimer’s disease by suppressing inflammation via activating p-Akt signaling pathway
- TIPE2 knockdown exacerbates isoflurane-induced postoperative cognitive impairment in mice by inducing activation of STAT3 and NF-κB signaling pathways
- Does the patellar tendon reflex affect the postural stability in stroke patients with blocked vision?
- Inactivation of CACNA1H induces cell apoptosis by initiating endoplasmic reticulum stress in glioma
- miR-101-3p improves neuronal morphology and attenuates neuronal apoptosis in ischemic stroke in young mice by downregulating HDAC9
- A custom-made weight-drop impactor to produce consistent spinal cord injury outcomes in a rat model
- Arterial spin labeling for moyamoya angiopathy: A preoperative and postoperative evaluation method
- Thyroid hormone levels paradox in acute ischemic stroke
- Geniposide protected against cerebral ischemic injury through the anti-inflammatory effect via the NF-κB signaling pathway
- The clinical characteristics of acute cerebral infarction patients with thalassemia in a tropic area in China
- Comprehensive behavioral study of C57BL/6.KOR-ApoEshl mice
- Incomplete circle of Willis as a risk factor for intraoperative ischemic events during carotid endarterectomies performed under regional anesthesia – A prospective case-series
- HOTAIRM1 knockdown reduces MPP+-induced oxidative stress injury of SH-SY5Y cells by activating the Nrf2/HO-1 pathway
- Esmolol inhibits cognitive impairment and neuronal inflammation in mice with sepsis-induced brain injury
- EHMT2 affects microglia polarization and aggravates neuronal damage and inflammatory response via regulating HMOX1
- Hematoma evacuation based on active strategies versus conservative treatment in the management of moderate basal ganglia hemorrhage: A retrospective study
- Knockdown of circEXOC6 inhibits cell progression and glycolysis by sponging miR-433-3p and mediating FZD6 in glioma
- CircYIPF6 regulates glioma cell proliferation, apoptosis, and glycolysis through targeting miR-760 to modulate PTBP1 expression
- Relationship between serum HIF-1α and VEGF levels and prognosis in patients with acute cerebral infarction combined with cerebral-cardiac syndrome
- The promoting effect of modified Dioscorea pills on vascular remodeling in chronic cerebral hypoperfusion via the Ang/Tie signaling pathway
- Effects of enriched environment on the expression of β-amyloid and transport-related proteins LRP1 and RAGE in chronic sleep-deprived mice
- An interventional study of baicalin on neuronal pentraxin-1, neuronal pentraxin-2, and C-reactive protein in Alzheimer’s disease rat model
- PD98059 protects SH-SY5Y cells against oxidative stress in oxygen–glucose deprivation/reperfusion
- TPVB and general anesthesia affects postoperative functional recovery in elderly patients with thoracoscopic pulmonary resections based on ERAS pathway
- Brain functional connectivity and network characteristics changes after vagus nerve stimulation in patients with refractory epilepsy
- Association between RS3763040 polymorphism of the AQP4 and idiopathic intracranial hypertension in a Spanish Caucasian population
- Effects of γ-oryzanol on motor function in a spinal cord injury model
- Electroacupuncture inhibits the expression of HMGB1/RAGE and alleviates injury to the primary motor cortex in rats with cerebral ischemia
- Effects of edaravone dexborneol on neurological function and serum inflammatory factor levels in patients with acute anterior circulation large vessel occlusion stroke
- CST3 alleviates bilirubin-induced neurocytes’ damage by promoting autophagy
- Excessive MALAT1 promotes the immunologic process of neuromyelitis optica spectrum disorder by upregulating BAFF expression
- Evaluation of cholinergic enzymes and selected biochemical parameters in the serum of patients with a diagnosis of acute subarachnoid hemorrhage
- 7-Day National Institutes of Health Stroke Scale as a surrogate marker predicting ischemic stroke patients’ outcome following endovascular therapy
- Cdk5 activation promotes Cos-7 cells transition towards neuronal-like cells
- 10.1515/tnsci-2022-0313
- PPARα agonist fenofibrate prevents postoperative cognitive dysfunction by enhancing fatty acid oxidation in mice
- Predicting functional outcome in acute ischemic stroke patients after endovascular treatment by machine learning
- EGCG promotes the sensory function recovery in rats after dorsal root crush injury by upregulating KAT6A and inhibiting pyroptosis
- Preoperatively administered single dose of dexketoprofen decreases pain intensity on the first 5 days after craniotomy: A single-centre placebo-controlled, randomized trial
- Myeloarchitectonic maps of the human cerebral cortex registered to surface and sections of a standard atlas brain
- The BET inhibitor apabetalone decreases neuroendothelial proinflammatory activation in vitro and in a mouse model of systemic inflammation
- Carthamin yellow attenuates brain injury in a neonatal rat model of ischemic–hypoxic encephalopathy by inhibiting neuronal ferroptosis in the hippocampus
- Functional connectivity in ADHD children doing Go/No-Go tasks: An fMRI systematic review and meta-analysis
- Review Articles
- Human prion diseases and the prion protein – what is the current state of knowledge?
- Nanopharmacology as a new approach to treat neuroinflammatory disorders
- Case Report
- Deletion as novel variants in VPS13B gene in Cohen syndrome: Case series
- Commentary
- Translation of surface electromyography to clinical and motor rehabilitation applications: The need for new clinical figures
- Revealing key role of T cells in neurodegenerative diseases, with potential to develop new targeted therapies
- Retraction
- Retraction of “Eriodictyol corrects functional recovery and myelin loss in SCI rats”
- Special Issue “Advances in multimedia-based emerging technologies...”
- Evaluation of the improvement of walking ability in patients with spinal cord injury using lower limb rehabilitation robots based on data science