Order-disorder (OD) structures of Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11
-
Felix Eder

 ,
Berthold Stöger
und
Matthias Weil
,
Berthold Stöger
und
Matthias Weil

Abstract
Single crystals of the two alkali metal zinc oxidotellurates(IV), Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11, were obtained by reactions of mixtures of ZnO, TeO2, Rb2CO3 (molar ratios 2:3:6) and ZnO, TeO2, Na2CO3 (molar ratios 2:3:10), respectively, with small amounts of water as a mineralizer. Both compounds crystallize as order-disorder (OD) structures of layers and feature a high stacking fault probability. The crystal structure of Rb2Zn(TeO3)(CO3)·H2O is composed of layers extending parallel to (100). The structure is composed of two kinds of non-polar OD layers consisting of trigonal-pyramidal [TeO3]2−, tetrahedral [ZnO4]6−, Rb1+, and CO32−, H2O, Rb2+, respectively. Different centrings of the layer groups lead to an ambiguity in the stacking arrangement. The crystal structure of Na2Zn2Te4O11 is built from layers extending parallel to (001). Trigonal-pyramidal [TeO3]2− and bisphenoidal [TeO4]4− polyhedra form [Te4O11]6− groups, which are connected by longer Te–O-contacts to form 1∞[Te8O22]12− double chains oriented along either [100] or [010]. These chains form non-polar layers, which appear alternatingly in two orientations related by a fourfold rotoinversion. The Zn2+ and Na+ cations are located at the layer interface. The stacking ambiguity is due to different lattices of adjacent layers.
1 Introduction
The structural diversity of oxidotellurates(IV) can be attributed to the stereochemically active 5s 2 electron lone pair (ψ) at the TeIV atom [1]. Due to the space requirement of ψ, the resulting [ψTeO x ] (x = 3–5) coordination polyhedra [2] are one-sided and thus represent polar units, commonly with low point group symmetries. Such structural entities are promising building blocks in the search for new compounds with crystal structures lacking inversion symmetry. Hence, oxidotellurates(IV) are attractive candidates for crystal engineering intended for new materials having non-linear optical, ferro-, piezo- or pyroelectrical properties [3]. The space consumption of ψ can direct the dimensionality of the oxidotellurate(IV) units in the crystal structure, resulting in the formation of different structural elements like clusters, chains, layers, or channels [4]. An extensive overview about the structural richness of oxidotellurates(IV) and -(VI) has recently been published by Christy et al. [5].
For structural diversification of (transition) metal oxidotellurates(IV), simple secondary ions (Mn+; Xn−) or complex ions (XO n m−) with different sizes and/or point group symmetries can be incorporated. This concept proved successful by way of example for placement of Pb2+ into copper(II) oxidotellurates [6], or of tetrahedral oxidoanions like SO42− and SeO42− into various metal and transition metal oxidotellurates(IV) [7, 8]. With respect to the incorporation of secondary cations into (first row) transition metal oxidotellurates(IV), alkali metal cations are a good choice since they offer a different charge and a significantly larger ionic radius than (first row) transition metal cations and are easily soluble in water. The latter is of importance since the hydrothermal method is a common and easy to apply technique in the search for new oxidotellurates.
Based on the above concept, we were able to synthesize four new alkali metal copper(II) oxidotellurates(IV) [9]. For the current study, we focused on zinc oxidotellurates(IV) modified by the alkali metal cations Na+ or Rb+. Two types of sodium-zinc-oxidotellurates have already been structurally characterized. Na2Zn2(TeO3)3·xH2O (with Te in oxidation state +IV) crystallizes in the zemannite structure type [10] and consists of a three-dimensional framework of [Zn2Te3O9]2− with Na+ ions (and water molecules) occupying large channels in the direction of the hexagonal axis [11, 12]; Na2Zn2TeO6 (with Te in oxidation state +VI) is a good ion conductor [13]. However, rubidium-zinc-oxidotellurates have not been reported so far.
For crystal growth, the hydrothermal method was modified by a significant reduction of the water content. The applied synthetic conditions led to the formation of multi-phase products that contained the title compounds Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11.
The crystal structures of Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11 are both layered and show order-disorder (OD) behaviour. The results of a detailed OD analysis of the crystal structures and the diffraction pattern as well as refinement strategies are given in this communication.
2 Experimental
Only three drops of water (ca 0.1 g) were added to the solid educts instead of filling the reaction container to ½–¾ of its volume with water, which is the usual procedure. Hence, water acts here not as a classical solvent or reaction medium but rather functions as a mineralizer.
2.1 Rb2Zn(TeO3)(CO3)·H2O
The starting materials, 0.0417 g ZnO (0.51 mmol), 0.1225 g TeO2 (0.77 mmol) and 0.3464 g Rb2CO3 (1.50 mmol) (molar ratios 2:3:6), were manually mixed in a Teflon vessel with an inner volume of about 3 ml. Three drops of water (ca 0.1 g) were added on top of the mixture before placing the Teflon vessel inside a steel autoclave and heating at 483 K for one week. The autoclave was then cooled to room temperature within 4 h. The resulting product was a colourless solid. In the X-ray powder diffraction pattern of the bulk, Rb2Zn(TeO3)(CO3)·H2O was identified with a strong preferred orientation of the (100) plane. Rb2(CO3)·1.5H2O [14] and Rb4(HCO3)2(CO3)·H2O [15] were identified as minority products. Under the polarization microscope thin colourless plates of Rb2Zn(TeO3)(CO3)·H2O were manually isolated from the mixture.
2.2 Na2Zn2Te4O11
Single crystals of Na2Zn2Te4O11 were obtained following the same procedure described above for Rb2Zn(TeO3)(CO3)·H2O with the reactants 0.0767 g ZnO (0.94 mmol), 0.2254 g TeO2 (1.41 mmol) and 0.4998 g Na2CO3 (4.72 mmol) (molar ratios 2:3:10). After the reaction time, the shiny colourless product was washed with water in order to dissolve excess Na2CO3. In the X-ray powder pattern of the bulk, Na2Zn2Te4O11 was found as a side product besides ZnTeO3 [16] and Zn2Te3O8 [17] as the majority phases. Under the polarization microscope, small colourless blocks of Na2Zn2Te4O11 were manually isolated for single-crystal X-ray diffraction.
2.3 Single crystal diffraction
Intensity measurements were conducted on a Bruker KAPPA APEX II diffractometer equipped with a CCD detector by using graphite-monochromatized MoKα radiation. The optically preselected crystals were mounted on Kapton micro mounts with the help of perfluorinated oil and then measured at a temperature of 100 K.
2.4 Structure solution and refinement
After integration with SAINT [18] and absorption correction with SADABS [18] structure solution was performed with SHELXT [19]. For structure refinement of Rb2Zn(TeO3)(CO3)·H2O SHELXL [20] was used, for structure refinement of Na2Zn2Te4O11 JANA2006 [21] was employed.
The stacking disorder in both structures not only leads to diffuse scattering as observed on certain rods in the diffraction pattern, but in consequence also to the presence of so-called “phantom atoms” or “shadows” observable in difference-Fourier maps during the refinement. Their positions correspond to the atomic positions of the less preferred stacking possibility since the refined structure model corresponds to the most common stacking order.
During structure refinement of Rb2Zn(TeO3)(CO3)·H2O, phantom atoms were easily found for the Rb2 site, but could also be localized for the C1, O4, O5, O6 and O7 sites, which correspond to the carbonate ion and crystal water molecule, respectively. Except for Rb1, the anisotropic displacement parameters (ADPs) for the main atom and the shadow atom were constrained to the same value. The occupancy ratio of the main atoms and the shadow atoms refined to 86.12:13.88(17). It was not possible to localize the hydrogen atoms of the water molecule (O7) during refinement.
In the crystal structure of Na2Zn2Te4O11, only four oxygen positions are responsible for the different layer arrangement possibilities and are the only sites showing additional phantom atoms. This results in the characteristic reflections and the corresponding diffuse scattering being of rather low intensities. The relative occupancies of main and shadow atoms from the layer with the minor stacking possibility were refined to a ratio of 0.714/0.286(4). Instead of refining the structure as disordered, the phantom atoms were also suppressed by assigning distinct scale factors to the family reflections and the characteristic reflections in the JANA2006 software [21] resulting in a ratio between the scale factors of family reflections and characteristic reflections of 2.37.
Details of structure and refinement data for the two crystal structures are collated in Table 1. Further details of the crystal structure investigations may be obtained from the joint CCDC/FIZ Karlsruhe online deposition service: https://www.ccdc.cam.ac.uk/structures/ by quoting the deposition numbers specified at the end of Table 1.
Data collection and refinement details.
| Formula | Rb2Zn(TeO3)(CO3)·H2O | Na2Zn2Te4O11 |
|---|---|---|
| Formula weight | 489.95 | 863.14 |
| Measurement T (K) | 100 | |
| Radiation; λ (Å) | MoKα; 0.71073 | |
| Diffractometer | Bruker KAPPA APEX II CCD | |
| Cryst. dimensions (mm) | 0.14×0.09×0.04 | 0.14×0.10×0.06 |
| Cryst. colour | Colourless | Colourless |
| Cryst. shape | Plate | Block |
| Space group, No. | I2/c (15) |
|
| Formula units Z | 8 | 16 |
| a (Å) | 29.5915(12) | 15.2949(3) |
| b (Å) | 5.8670(3) | |
| c (Å) | 10.3140(5) | 18.7783(7) |
| β (Å) | 104.736(2) | |
| V (Å3) | 1731.75(14) | 4392.9(2) |
| μ (mm−1) | 17.3 | 14.9 |
| X-ray density (g⋅cm−3) | 3.76 | 5.22 |
| θmin–θmax (°) | 3.970–36.685 | 1.72–40.26 |
| h | −49–49 | −18–27 |
| k | −9–9 | −18–27 |
| l | −17–16 | −34–34 |
| Measured refl. | 24151 | 50971 |
| Independent refl. | 4275 | 6909 |
| Observed refl. (I > 2σ(I)) | 3306 | 4506 |
| R i | 0.0411 | 0.0211 |
| Abs. Tmin; Tmax | 0.542; 0.747 | 0.0589; 0.1174 |
| Refined parameters | 134 | 168 |
| Flack parameter | – | 0.03(2) |
| R1 (F2 > 2σ(F2)) | 0.0292 | 0.0255 |
| wR2(F2 all) | 0.0634 | 0.0472 |
| GOF | 1.046 | 1.072 |
| CSD-code | 2168667 | 2168668 |
3 Results and discussion
The formation of OD structures [22] is a quite common phenomenon in oxidotellurates(IV), as for example observed in Ca6Te5O15(NO3)2 [4] or KCa3Te5O12Cl3 [23]. This is in part due to the structure-directing properties of the lone pairs of the TeIV atoms leading to the formation of layers. Moreover, often the symmetry of the Te–O network is reduced with respect to the ions in the inner space, for example owing to formation of superstructures. In fact, the crucial aspect of an OD structure is that parts of the structure feature locally higher symmetry than the overall structure. In an OD interpretation, the structure is decomposed into OD layers reflecting this local symmetry. These layers do not necessarily correspond to layers in the crystal-chemical sense. An introduction to OD theory was given by Ferraris et al. [24].
Bond valence sums [25] were calculated based on the values of Brese and O’Keeffe [26] using the corresponding feature implemented in the PLATON software package [27]. Furthermore, for the Te atoms the revised parameters by Mills & Christy [28] under consideration of all oxygen contacts within a distance of 3.5 Å were employed as well.
3.1 Rb2Zn(TeO3)(CO3)·H2O
3.1.1 OD layers
The crystal structure of Rb2Zn(TeO3)(CO3)·H2O is built up of an alternation of two kinds of crystal-chemical layers, A1 and A2, extending parallel to (100) (Figure 1). The A1 layers ([TeO3]2−, Rb1+, Zn2+) possess higher (pseudo)-symmetry than the A2 (Rb2+, CO32−, H2O) layers, which leads to an ambiguity in the arrangement of the layers as described below. The sequence number of the layer is indicated in the subscript: … A1 n A2n+1A1n+2A2n+3….
![Figure 1:
The crystal structure of Rb2Zn(TeO3)(CO3)·H2O viewed down [010]. Layer names are indicated to the right, alternative stacking arrangements are omitted. Radii for all atoms are fixed at an arbitrary value.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_001.jpg)
The crystal structure of Rb2Zn(TeO3)(CO3)·H2O viewed down [010]. Layer names are indicated to the right, alternative stacking arrangements are omitted. Radii for all atoms are fixed at an arbitrary value.
The geometric parameters of the following description are derived from a model ignoring the minor stacking arrangement (“phantom atoms”). The Te1 atom is coordinated by three oxygen atoms with almost the same distance (1.860(2)–1.872(2) Å; Table 2). The resulting [TeO3]2− polyhedron (Figure 2) has the commonly observed trigonal-pyramidal shape. The BVS was calculated to 4.04 valence units (v.u.) and 3.94 v.u. using the revised parameters [28]. The [TeO3]2− units are isolated from each other but are connected with three [ZnO4]6− units by corner-sharing.
Selected interatomic distances d in the crystal structure of Rb2Zn(TeO3)(CO3)·H2O referring to the structure model consisting only of the preferred MDO2 stacking.
| Atoms | d (Å) | Atoms | d (Å) |
|---|---|---|---|
| Te1–O1 | 1.860(2) | Rb2–O7 | 2.894(4) |
| Te1–O2 | 1.868(2) | Rb2–O5iv | 2.946(5) |
| Te1–O3 | 1.871(2) | Rb2–O4i | 2.961(5) |
| Rb1–O3 | 2.960(2) | Rb2–O5 | 2.971(4) |
| Rb1–O2i | 2.977(2) | Rb2–O5v | 2.982(4) |
| Rb1–O1ii | 2.985(2) | Rb2–O5i | 3.063(4) |
| Rb1–O3iii | 3.089(2) | Zn1–O2 | 1.945(4) |
| Rb1–O2iii | 3.111(2) | Zn1–O1vi | 1.959(3) |
| Rb1–O2 | 3.118(2) | Zn1–O3ii | 1.967(3) |
| Rb1–O3ii | 3.150(2) | Zn1–O4 | 1.973(5) |
| Rb1–O1iii | 3.222(2) | C1–O4 | 1.309(5) |
| Rb1–O1i | 3.232(2) | C1–O5 | 1.226(5) |
| Rb1–O6 | 3.240(3) | C1–O6 | 1.21(3) |
-
Symmetry codes: (i) x, −1+y, z; (ii) x, 1−y, 1/2+z; (iii) 1−x, 1−y, 1−z; (iv) 1/2−x, 1/2−y, 1−z; (v) 1/2−x, −1/2+y, 3/2−z; (vi) x, 2−y, 1/2+z.

Coordination polyhedra of the Te, Zn and Rb atoms in the structure of Rb2Zn(TeO3)(CO3)·H2O. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 2.
The Zn1 atoms are coordinated by four oxygen atoms forming a slightly distorted tetrahedron (Figure 2). The BVS of Zn1 is 2.00 v.u.. The [ZnO4]6− units are isolated from each other but share three of their corners with [TeO3]2− groups and the fourth corner (O4) with the carbonate group. In this way, 2∞[ZnTeO3] layers are formed extending parallel to (100). One side of this layer is connected to a layer of Rb1 atoms, the other side with the carbonate groups. This leads to two 2∞[ZnTeO3] layer pairs with a Rb1 layer situated in-between – the A1 layer (Figure 3a).
![Figure 3:
The (a) A1 and (b) A2 layers of Rb2Zn(TeO3)(CO3)·H2O projected on the layer plane (100). The (pseudo) symmetry operations are indicated using the standard graphical symbols [29]. Atomic radii are as in Figure 1.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_003.jpg)
The Rb1 position is coordinated by ten oxygen atoms with distances ranging from 2.961(2) to 3.240(4) Å. The BVS of Rb1 was calculated to 1.06 v.u.. Rb2 is surrounded by six oxygen atoms with bond lengths in the range of 2.894(4)–3.063(4) Å and three additional oxygen atoms at distances of 3.510(4)–3.596(4) Å resulting in a [6+3] coordination. The BVS amounts to 0.90 v.u. considering the six closer oxygen atoms only. This value increases to 0.99 v.u. when the three more distant oxygen contacts are also taken into account.
C1, O4, O5 and O6 form the carbonate ion (Figure 3b). O4 is part of the 2∞[ZnTeO3] layer and is positioned at a farther distance from C1 than O5 and O6, both of which do not have many other close bonding partners in their vicinity. The O7 position corresponds to the crystal water molecule, as the BVS of O7 amounts to only 0.19 v.u..
3.1.2 OD groupoid family
The structure belongs to a category IV OD family [22] of two kinds of non-polar layers. The OD groupoid layer symbol reads as
| A 1 | A 2 |
| A(1)2/m1 | P(1)21/c1 |
| [r, 0] | |
according to the notation of Grell and Dornberger-Schiff [30]. The first line in the symbol indicates the layer names, the second line the (idealized) symmetry of the layers. Accordingly, the A1 layer (Figure 3a) possesses idealized A(1)2/m1 and the A2 layer (Figure 3b) P(1)21/c1 symmetry. In the OD notation, capital Bravais symbols indicate the three-dimensionality of the layers (as opposed to the translation lattice) and parentheses give the direction missing translation.
The third line of the symbol gives one possible relative position of the two adjacent layers, viz. the origins of A1 and A2 are connected by the vector rb+a0 (Figure 3). The variable r is used instead of a fixed value, because OD groupoid families abstract from metric parameters in analogy to space group types. In this particular OD groupoid family (monoclinic point group with monoclinic axis parallel to the layer plane) no second parameter is needed. The translational component in the [001] direction is fixed for all stacking possibilities and reflected by the inclination of the a0 vector [31].
In Rb2Zn(TeO3)(CO3)·H2O the parameter r adopts the value 0, which is expressed by the symbol
| A 1 | A 2 |
| A(1)2/m1 | P(1)21/c1 |
| [0, 0] | |
and has consequences on the possible arrangements of the layers, as shown in the next section.
3.1.3 NFZ relationship
The NFZ relationship determines the number of stacking possibilities of a layer contact given a particular set of metric parameters. It reads as Z = N/F = [ n
:
n
: 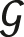 n
∩
n
∩  n+1].
n+1].  n
is the group of operations of the A
n
layer that do not invert the orientation of the layer with respect to the stacking direction. For A1 layers
n
is the group of operations of the A
n
layer that do not invert the orientation of the layer with respect to the stacking direction. For A1 layers  n
= A(1)m1 and for A2 layers
n
= A(1)m1 and for A2 layers  n
= P(1)c1.
n
= P(1)c1.  n
∩
n
∩  n+1 is the subgroup of
n+1 is the subgroup of  n
of those operations that also apply to the adjacent layer.
n
of those operations that also apply to the adjacent layer.
Since r = 0, the c glide reflection planes of the A1 and A2 layers overlap. Therefore, in all cases,  n
∩
n
∩  n+1 = P(1)c1. [
n+1 = P(1)c1. [ n
:
n
:  n
∩
n
∩  n+1] is the index of
n+1] is the index of  n
∩
n
∩  n+1 in
n+1 in  n
. Ultimately, we obtain for A1
n
→ A2n+1 contacts Z = [A(1)m1:P(1)c1] = 4/2 = 2 and for A2
n
→ A1n+1 contacts Z = [P(1)c1:P(1)c1] = 2/2 = 1. In other words, given an A1 layer, the adjacent A2 layers can be placed in two possible ways whereas for an A2 layer there is only one way of placing the A1 layer.
n
. Ultimately, we obtain for A1
n
→ A2n+1 contacts Z = [A(1)m1:P(1)c1] = 4/2 = 2 and for A2
n
→ A1n+1 contacts Z = [P(1)c1:P(1)c1] = 2/2 = 1. In other words, given an A1 layer, the adjacent A2 layers can be placed in two possible ways whereas for an A2 layer there is only one way of placing the A1 layer.
The two possibilities of placing the A2 layers are given by the coset decomposition of P(1)c1 in A(1)m1. In particular, they are related by the A-centering (translation along b/2+c/2) of the A1 layers. These two possibilities are denominated as A2+ and A2−.
3.1.4 Polytypes
The crucial point of the NFZ relationship is that the pairs of generated layers are geometrically equivalent. Thus, an infinity of polytypes, with arbitrary distributions of the A2+ and A2− positions can in principle be constructed, which are all locally equivalent. If interatomic interactions over more than one layer-width are neglected, these polytypes are also energetically equivalent.
However, experience shows that the stacking in ordered polytypes are often particularly simple. This can be explained by weak interactions over more than one layer width, which favor one of the geometrically non-equivalent triples of adjacent layers. Polytypes that cannot be decomposed into even simpler polytypes (containing only a subset of the triples, quadruples, and generally n-tuples) are said to be of a maximum degree of order (MDO) [32, 33].
In Rb2Zn(TeO3)(CO3)·H2O there are two MDO polytypes:

Schematic representations of the local symmetry of (a) the MDO1, (b) the MDO2 polytypes, and (c) the family structure. Triangles are red on one and blue on the other side or grey if they are symmetric by reflection at the drawing plane. A translational component of b/2 is represented by darker colors. Symmetry elements valid for the whole structure are drawn in red. The grey backdrop indicates the unit cell.
They play a special role in OD theory, because all polytypes can be decomposed into fragments of MDO polytypes.
The family structure of an OD family is a fictitious polytype, which corresponds to an equal overlay of all stacking possibilities. The family structure in Rb2Zn(TeO3)(CO3)·H2O has A2/m symmetry with a = 2a0 [Figure 4c]. The A(1)2/m1 symmetry of the A1 layers is valid for the whole structure and the symmetry of the A2 layers is increased to A(1)2/m1 by ‘multiplying in’ the A-centering of the A1 layers. Such an overlay of two A2 layers is called A2±.
3.1.5 Diffraction
The diffraction pattern of Rb2Zn(TeO3)(CO3)·H2O (Figure 5) is composed of distinct rods with sharp reflections and rods with more diffuse reflections, as it is characteristic for OD structures with translationally equivalent layers [34]. The sharp reflection peaks are located on rods k+l even and correspond to the diffraction pattern of the A(1)2/m1 family structure. All polytypes, ordered or disordered, contribute equally to these family reflections. The peaks resulting from diffuse reflections on the k+l odd rods are called characteristic reflections, because they are characteristic for an individual polytype. In the crystal under investigation, the characteristic reflections correspond to the MDO2 polytype. Thus A2+A1A2− MDO2 triples are more common than A2+A1A2+ (or the equivalent A2−A1A2−) MDO1 triples. However, the diffuseness of the characteristic reflections means that there is also a tangible number of the latter, i.e., a high stacking fault probability.

(h2l)∗ plane of Rb2Zn(TeO3)(CO3)·H2O. Reciprocal lattice vectors are given with respect to the smallest common sublattice.
The occupancy (occ) ratio of the two orientations of the A2 layers refined to 86.12:13.88(17), which would correspond to 2occ−1 ≈ 72% of MDO2 and 28% MDO1 (note that refining a pure MDO1 crystal in the MDO2 setting would result in a 1:1 overlay of the A2 layers). However, such quantitative interpretations are treacherous, since random stackings are not equivalent to an overlay of MDO polytypes. Moreover, the broad peaks lead to systematic errors in the intensity evaluation, the ‘Ďurovič effect’ [35]. A proper evaluation of the stacking probabilities would require the modelling of the diffuse scattering.
3.2 Na2Zn2Te4O11
3.2.1 OD-description
Na2Zn2Te4O11 can be considered as a category I OD structure built of one kind of non-polar layers A n , where n is a sequential number. The layers are built of a 2∞[Te4O11]6− network and adopt (idealized) orthorhombic P212(2) symmetry (Figure 6a). In principle, the Na and Zn atoms form a crystal-chemically separate layer (Figure 6b). However, since they are located on (or in very close vicinity to) sites that fulfill the symmetry of both adjacent 2∞[Te4O11]6− layers, they can be considered from an OD point of view as being located at the interface between the OD layers.

Layers parallel to (001) in the crystal structure of Na2Zn2Te4O11 centred at z = 0 (a) and z = ⅛ (b). Te atoms are green, O atoms red, Zn atoms blue and Na atoms yellow. Radii for all atoms are fixed at an arbitrary value of 0.2 Å.
3.2.2 Crystal chemistry
For the four unique Te sites, two types of [TeO x ] polyhedra are present in the structure of Na2Zn2Te4O11. Te1 and Te3 are coordinated by four oxygen atoms (Table 3) in a bisphenoidal shape, which can be derived from a trigonal bipyramid with the 5s 2 lone pair (ψ) occupying an equatorial position. Te2 and Te4 are surrounded by three oxygen atoms and form a trigonal pyramid (Figure 7). The BVS of the Te atoms are 3.95 (Te1), 4.11 (Te2), 4.06 (Te3) and 4.07 (Te4) v.u.. Using the revised parameters [28], BVS of 4.03 (Te1), 4.04 (Te2), 4.06 (Te3) and 3.99 (Te4) v.u. are obtained.
Selected interatomic distances d in the crystal structure of Na2Zn2Te4O11 referring to the structure model consisting only of the preferred stacking order.
| Atoms | d (Å) | Atoms | d (Å) |
|---|---|---|---|
| Te1–O3 | 1.883(5) | Zn4–O2 | 1.958(6) |
| Te1–O2 | 1.887(6) | Zn4–O2xi | 1.958(6) |
| Te1–O1 | 1.985(2) | Zn4–O2vii | 1.958(6) |
| Te1–O4i | 2.290(5) | Zn4–O2xii | 1.958(6) |
| Te2–O4 | 1.831(5) | Na1–O10v | 2.380(5) |
| Te2–O5ii | 1.869(5) | Na1–O4 | 2.407(5) |
| Te2–O6iii | 1.881(5) | Na1–O9 | 2.473(7) |
| Te3–O9 | 1.878(5) | Na1–O3v | 2.478(7) |
| Te3–O8 | 1.883(5) | Na1–O5viii | 2.509(7) |
| Te3–O7iv | 1.969(2) | Na1–O2vi | 2.558(7) |
| Te3–O10v | 2.272(5) | Na1–O1 | 2.819(4) |
| Te4–O10 | 1.843(5) | Na2–O12 | 2.287(6) |
| Te4–O12 | 1.866(5) | Na2–O8 | 2.399(7) |
| Te4–O11 | 1.887(5) | Na2–O6iv | 2.426(6) |
| Zn1–O9 | 1.925(5) | Na2–O11iv | 2.528(7) |
| Zn1–O6 | 1.945(5) | Na2–O7iv | 2.729(4) |
| Zn1–O12 | 1.977(5) | Na2–O4i | 2.819(5) |
| Zn1–O3vi | 1.984(5) | Na2–O10 | 2.962(5) |
| Zn2–O8 | 1.963(6) | Te1–O5viii | 2.734(5) |
| Zn2–O8vii | 1.963(6) | Te1–O12 | 3.024(5) |
| Zn2–O5vi | 2.007(6) | Te2–O2xiii | 2.781(6) |
| Zn2–O5viii | 2.007(6) | Te2–O9 | 2.996(5) |
| Zn3–O11ix | 1.970(6) | Te3–O11x | 2.803(5) |
| Zn3–O11iii | 1.970(6) | Te3–O6iii | 3.110(5) |
| Zn3–O11v | 1.970(6) | Te4–O8xiv | 2.808(5) |
| Zn3–O11x | 1.970(6) | Te4–O3 | 3.058(5) |
-
Symmetry codes: (i) 1−y, 3/2−x, 1/4+z; (ii) y, 1−x, 1−z; (iii) 1/2−x, y, 3/4−z; (iv) 1−y, 1+x, 1−z; (v) 3/2−y, 1−x, −1/4+z; (vi) x, 3/2−y, 5/4−z; (vii) 1−x, 2−y, z; (viii) 1−x, 1/2+y, 5/4−z; (ix) −1/2+y, 1+x, −1/4+z; (x) 1/2+x, 2−y, 3/4−z; (xi) −1/2+y, 3/2−x, 3/2−z; (xii) 3/2−y, 1/2+x, 3/2−z; (xiii) −1/2+y, x, −1/4+z; (xiv) −1+y, 1/2+x, 1/4+z.
![Figure 7:
[TeO3]2− and [TeO4]4− coordination polyhedra in the crystal structure of Na2Zn2Te4O11. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 3.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_007.jpg)
[TeO3]2− and [TeO4]4− coordination polyhedra in the crystal structure of Na2Zn2Te4O11. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 3.
The [TeO3]2− and [TeO4]4− units are connected to each other by a common corner forming linear [Te4O11]6− units. Such [Te4O11]6− units have already been described several times [5], usually in compounds with the composition MIII2Te4O11. Using the notation introduced by Christy et al. [5], the Te–O units are denoted as (Δ–◊–◊–Δ). Additionally, each Te atom has another oxygen contact at distances between 2.734(5) and 2.808(5) Å. Through these contacts, each [Te4O11]6− unit is linked to two neighbouring units resulting in 1∞[Te8O22]12− (double)chains oriented either parallel to [100] or [010].
Each of the four Zn atoms is surrounded by four oxygen atoms between 1.925(5) and 2.007(6) Å (Table 3) forming an isolated and distorted [ZnO4]6− tetrahedron (Figure 8); the O–Zn–O angles are in a range of 101.6(2)°–126.8(2)°. The BVS of the Zn atoms are 2.02 (Zn1), 1.89 (Zn2), 1.94 (Zn3) and 2.00 (Zn4) v.u.. The Na atoms are coordinated by seven oxygen atoms at distances of 2.287(6)–2.962(5) Å (Figure 9). The [NaO7]13− polyhedra are connected to each other by the longest contacts to form chains oriented parallel to [001]. The BVS of the Na sites are 1.07 (Na1) and 0.99 (Na2) v.u..
![Figure 8:
[ZnO4]6− tetrahedra in the crystal structure of Na2Zn2Te4O11. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 3.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_008.jpg)
[ZnO4]6− tetrahedra in the crystal structure of Na2Zn2Te4O11. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 3.
![Figure 9:
[NaO7]13− coordination polyhedra in the crystal structure of Na2Zn2Te4O11. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 3.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_009.jpg)
[NaO7]13− coordination polyhedra in the crystal structure of Na2Zn2Te4O11. Displacement ellipsoids are drawn at the 90% probability level. Symmetry codes refer to Table 3.
3.2.3 OD groupoid family
As it is the case for Rb2Zn(TeO3)(CO3)·H2O, a crucial factor of the OD description is the presence of different translation lattices of adjacent layers. In the former, the differing lattices were due to the different Bravais classes of the layers group. In the conventional setting, the basis vectors of the layer lattices are, however, identical. In contrast, adjacent layers in Na2Zn2Te4O11 possess different basis vectors when expressed in the conventional setting.
The smallest common sublattice[1] of the lattices of all A n layers in Na2Zn2Te4O11 is a rectangular lattice spanned by (a, b) with a = b = |a| = |b| = 15.2949(3) Å. The vector c0 is perpendicular to (001) and has the length of one layer width (c0 = |c0| = 4.6946(2) Å).
The translation lattices of the A2n (even) layers are spanned by (a, b/2) and of the A2n+1 (odd) layers by (a/2, b), i.e., there are additional centering vectors at (0, ½, 0) T and (½, 0, 0) T , respectively, which will be expressed by the Bravais symbols X and X′. It follows that the orientations of the even and odd A n layers are related by a rotation of ±90° about c0. Owing to the orthorhombic layer group, and therefore a rectangular translation lattice, this can only be realized if a = b and γ = 90°. In particular, in the conventional (here primitive) setting of the P212(2) layer group, the cell parameters must fulfill a = 2b. This represents a non-crystallographic layer lattice restriction, which may appear in OD structures of layers of non-equal lattices [36].
The OD groupoid family symbols for OD structures of layers of one kind list the layer symmetry and one possible set of σ-POs relating adjacent layers [22]. This notation has not been worked out for cases such as Na2Zn2Te4O11, where layers possess different lattices. However, the five placed symbols for tetragonal OD groupoid families can be adapted for this case. We suggest the following symbol
| [100] | [010] | [001] | [110] |
|
|
| X | 21 | 2 | (2) | ||
|
|
n 2, r−s | n 2, r−s+½ | |||
| n 2, r+s−½ | n 2, r−s+1 |
The first line is usually not part of the symbol but given here for convenience. It indicates the directions of the operations in the lines below. The second line gives the operations of a single layer (λ-POs), including the Bravais centering (X stands for the (0, ½, 0)
T
centering vector, see above). In the third line, the partial operations mapping a layer on an adjacent layer (σ-POs) are listed. The point group of the OD groupoid family (the group generated by the linear parts of all POs) is
As for the previously discussed case of Rb2Zn(TeO3)(CO3)·H2O, the metric parameters (r, s) adopt special values. Here, r = s = 0, which is due to the Na and Zn atoms that are located at the layer interfaces. This means that the axes of the 2[001] operations of adjacent layers have to overlap. As it can be seen in Figure 10, this is the case for half of the 2[001] axes. Again, this can be represented by the symbol
| [100] | [010] | [001] | [110] |
|
|
| X | 21 | 2 | (2) | ||
|
|
c 2 | n 2, ½ | |||
| n 2, −½ | n 2, 1 |
![Figure 10:
The A2n (X) and A2n+1 (X′) layers of Na2Zn2Te4O11. The (pseudo)-symmetry operations are indicated using the standard graphical symbols [29]. Additional translational symmetry is indicated by the dashed unit cells. Na and Zn atoms were omitted, as they are half-assigned to two adjacent layers.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_010.jpg)
The A2n (X) and A2n+1 (X′) layers of Na2Zn2Te4O11. The (pseudo)-symmetry operations are indicated using the standard graphical symbols [29]. Additional translational symmetry is indicated by the dashed unit cells. Na and Zn atoms were omitted, as they are half-assigned to two adjacent layers.
3.2.4 NFZ relationship
The NFZ relationship again is used to derive the stacking possibilities in the case of Na2Zn2Te4O11. For even and odd A
n
layers  n
= X11(2) and
n
= X11(2) and  n
= X′11(2), respectively. In both cases
n
= X′11(2), respectively. In both cases  n
∩
n
∩  n+1 = P11(2) because half of the 2[001] axes of adjacent layers coincide (Figure 10). Thus, given the position of an even A
n
layer, there are Z = [X11(2):P11(2)] = 2 possibilities of placing the adjacent layers. The ambiguity arises owing to the different translation lattices. The origin of the adjacent (odd) layers may be located at nc0 or b/2+nc0, which corresponds to the X-centering vector. Z = 2 can also be obtained from the before-mentioned fact that only half of the 2[001] axes are shared by adjacent layers.
n+1 = P11(2) because half of the 2[001] axes of adjacent layers coincide (Figure 10). Thus, given the position of an even A
n
layer, there are Z = [X11(2):P11(2)] = 2 possibilities of placing the adjacent layers. The ambiguity arises owing to the different translation lattices. The origin of the adjacent (odd) layers may be located at nc0 or b/2+nc0, which corresponds to the X-centering vector. Z = 2 can also be obtained from the before-mentioned fact that only half of the 2[001] axes are shared by adjacent layers.
The same argument can be made for odd A n layers: Z = [X′11(2):P11(2)] = 2 and the origin of the adjacent (even) layers can be located at nc0 or a/2+nc0. The two possible locations of the A n layers will be designated as A n + and A n −, respectively.
3.2.5 Polytypes
There are two kinds of triples A n An+1An+2 of consecutive layers: with the A n and An+2 possessing the same (e.g., A+A?A+) or different origins when projected on (001) (e.g., A+A?A−). The two MDO polytypes are each formed by only one kind of triple:
MDO1: … A+A+A+A+ …,
MDO2: … A+A+A−A− …,
All other polytypes can be decomposed into fragments of MDO1 and MDO2. In the family structure, even and odd n layers are an equal superposition of two layers related by translation along a/2 and b/2, respectively. Such a layer, which can be written as A±, has P22(2) symmetry with the lattice basis (a/2, b/2). Adjacent layers are related by
3.2.6 Family structure
First measurements on smaller crystals of Na2Zn2Te4O11 did not immediately reveal the actual stacking sequence as only strong reflections of the family structure were observed. The asymmetric unit of the family structure (
The oxygen sites, which are responsible for the various layer orientations, are only half occupied in this structure model since the different arrangements are all projected into a smaller unit cell. The O4 site, which corresponds to O1 and O7 in the main structure, has a very short distance of 0.714(11) Å to its own symmetry-equivalent and can therefore only be half occupied. The same can be said for the O3 site (corresponding to the O4 and O10 sites), which has a distance of 2.822(12) Å to its own symmetry-equivalent.
3.2.7 Diffraction
As has been discussed for Rb2Zn(TeO3)(CO3)·H2O, OD structures with translationally equivalent layers have characteristic diffraction patterns, where on rows with family reflections only these reflections are observed. No streaking or characteristic reflections are expected if the layers are perfectly translationally equivalent. In the case of Na2Zn2Te4O11, even and odd n layers are not translationally equivalent. However, since the Fourier transform is linear, the same argument can be applied when considering the two substructures of the even- and odd A n layers only.
When indexing with (a∗, b∗, c 0 ∗) on rods h and k even, only sharp reflections are observed at l = n/2, n ∈ ℤ, corresponding to the family structure (c = 2c0) (Figure 11a). The characteristic reflections originate from one of the two substructures (A n with n = even and n = odd, respectively). The even A n layers contribute to diffraction intensities of the odd-h rods and the odd A n layers to the odd-k rods.

(a) (4kl)∗ plane of Na2Zn2Te4O11, (b) (3kl)∗ plane of Na2Zn2Te4O11. Reciprocal lattice vectors are given with respect to the smallest common sublattice.
As listed in Table 4, sharp diffraction spots would be expected for MDO1 at all h and k values, with l = n/2 (n ∈ ℤ) as the only restriction. This is not the case as only the family reflections are present on the h, k = even rods with no intensity in between. In the characteristic h or k = odd planes (Figure 11b) there are also reflections at l = n/4, which are in contradiction with a basis vector 2c0.
Stacking order and symmetry of MDO-polytypes, sublattices and family structure of Na2Zn2Te4O11.
| Stacking order | a (Å) | b (Å) | c (Å) | Space group | |
|---|---|---|---|---|---|
| MDO1 | … A+A+A+A+ … | 15.2949(3) | b = a | 9.3892(4) |
|
| MDO2 | … A+A+A−A− … | 15.2949(3) | b = a | 18.7783(7) |
|
| A2n (n ∈ ℤ) | … A+__A−__ … | 15.2949(3) | 7.6474(3) | 18.7783(7) | B222 |
| A2n+1 (n ∈ ℤ) | … A+__A−__ … | 7.6474(3) | 15.2949(3) | 18.7783(7) | A222 |
| Family structure | … A±A± … | 7.6474(3) | b = a | 9.3892(4) |
|
For the MDO2 stacking order, reflections are expected at h+k+4l = 2n, n ∈ ℤ. All observed reflections can be explained by the unit cell of the MDO2 polytype, making it the preferred stacking order (Figure 12).
![Figure 12:
The preferred (MDO2) stacking arrangement of Na2Zn2Te4O11 in a projection along [010]. Te atoms are green, O atoms red, Zn atoms blue and Na atoms yellow. Atomic radii were fixed to an arbitrary value of 0.2 Å for clarity.](/document/doi/10.1515/zkri-2022-0030/asset/graphic/j_zkri-2022-0030_fig_012.jpg)
The preferred (MDO2) stacking arrangement of Na2Zn2Te4O11 in a projection along [010]. Te atoms are green, O atoms red, Zn atoms blue and Na atoms yellow. Atomic radii were fixed to an arbitrary value of 0.2 Å for clarity.
However, the group of reflections at h, k = odd, l = n/2 (n ∈ ℤ) (with respect to a∗, b∗, c 0 ∗), is systematically absent. Referring to (a∗, b∗, c∗) of the MDO2 polytype (Table 4; c = 4c 0 ) this means that in the l = even planes all characteristic reflections are absent (only the family reflections remain), while in the l = odd planes the characteristic reflections are present. Again, this can be explained by the individual presence of the two substructures of the even- and odd A n layers. The A2n (n ∈ ℤ) substructure only allows the k = even reflections since its b is half of the MDO2 unit cell. The B-centring of the substructure unit cell results in the condition h+l = 2n (n ∈ ℤ). In the l = even planes, where the absences appear, this results in the condition h = 2n (n ∈ ℤ) since l is already even. Therefore, only the reflections of the family structure (h, k, l = even) are present for the A2n sublattice in the l = 2n (n ∈ ℤ) planes. The same reasoning is valid for the A2n+1 sublattice, for which a summary is given in Table 5.
Reflection conditions of the two substructures in the stacking of Na2Zn2Te4O11. h, k, l refer to (a*, b*, c*) of the MDO2 polytype.
| h | k | l | A2n (n ∈ ℤ) | A2n+1 (n ∈ ℤ) | |
|---|---|---|---|---|---|
| Even | Even | Even | Present | Present | Family structure |
| Even | Odd | Odd | – | Present | Characteristic |
| Odd | Even | Odd | Present | – | Characteristic |
| Odd | Odd | Even | – | – | Non-space group systematic absences |
In the actual diffraction pattern, the characteristic h or k odd reflections are very weak (Figure 11b) since the deviation from the family structure is only due to the ordering of the O1, O4, O7 and O10 atoms. Distinct maxima are located at the positions expected for MDO2, which means that the MDO2 triples are preferred over MDO1 triples. As in the case of Rb2Zn(TeO3)(CO3)·H2O, the reflection peaks are very broad and located on streaks of continuous diffuse scattering.
As discussed before, a refinement with different scale factors for characteristic and family reflections in JANA2006 [21] resulted in a ratio of 2.37. If P is the probability of an MDO2 triple appearing and Q the probability of MDO1, then the remaining intensity ratio should be
The fact that there is a preferred stacking order can be explained by the influence of the Na atoms. The Zn atoms are located exactly or almost exactly between the layers on sites with higher symmetry and do not have any bonds towards the “critical” O1/O7 and O4/O10 sites which create the differences between the A n + and A n − layers. In fact, the Zn atoms form bonds to all other oxygen sites except these four.
The Na1 and Na2 positions are located farther away from the idealized high-symmetric sites. Each Na site forms bonds towards both O4 and O10 and to one of O1 and O7. The deviation from the idealized position is the largest in the z-direction. The Na-atoms located in the gap between two 1∞[Te8O22]12− chains are pulled closer towards the layer while the ones directly above and below the chains are pushed away and located closer to the next layer. This is responsible for the fact that two subsequent layers A n and An+1 can either be positioned straight above each other or be translated by a/2 or b/2 but not translated by a/4+b/4. Furthermore, Na2 also has a dislocation of 0.115 Å from the idealized high-symmetric position within the plane. These small dislocations of the Na sites lead to a desymmetrization of the ideal OD-symmetry [37] and therefore are the presumable cause for the existence of the preference for the MDO2 stacking over MDO1.
4 Conclusion and outlook
The crystal structures of Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11 are both OD-structures with high stacking fault probabilities. Both structures are remarkable as the OD character is caused by different translation lattices of adjacent layers. For Rb2Zn(TeO3)(CO3)·H2O, the different symmetries of two different crystal-chemical layers cause the stacking ambiguity. In Na2Zn2Te4O11, charge-balance leads to the occupationally modulated twofold superstructures and ultimately to the stacking ambiguity resulting in the OD behaviour.
As noted in the introduction, the stereo-chemically active 5s 2 electron lone pair frequently leads to modular layer structures and thus is the main reason for the occurrence of numerous OD structures in this family of compounds. The modification of the hydrothermal synthesis by a significant reduction of the water content might also encourage the formation of structures with a layered set-up. Under the chosen synthetic conditions with water as a mineralizer instead as a solvent, the ions are less mobile than in a classical hydrothermal set-up. Therefore, further solid-state reaction experiments of transition metal oxidotellurates with water as a mineralizer seem to be a promising path to find even more modular structures featuring OD character. This already proved to be the case for the stacking-disordered structure of K4Sn3Te8O24, K2Cu3Te4O12, which consists of rods being disordered relative to each other, as well as the structures of K2Mn2Te3O9·H2O and K2Cd2Te3O9·H2O, which seem to show allotwinning. Details on these structures will follow in forthcoming publications.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: The authors acknowledge TU Wien Bibliothek for financial support through its Open Access Funding Program.
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
1. Galy, J., Meunier, G., Andersson, S., Åström, A. Stéréochimie des eléments comportant des paires non liées: Ge (II), As (III), Se (IV), Br (V), Sn (II), Sb (III), Te (IV), I (V), Xe (VI), Tl (I), Pb (II), et Bi (III) (oxydes, fluorures et oxyfluorures). J. Solid State Chem. 1975, 13, 142–159; https://doi.org/10.1016/0022-4596(75)90092-4.Suche in Google Scholar
2. Zemann, J. Zur Stereochemie des Te(IV) gegenüber Sauerstoff. Monatsh. Chem. 1971, 102, 1209–1216; https://doi.org/10.1007/bf00917174.Suche in Google Scholar
3. Ok, K. M., Chi, E. O., Halasyamani, P. S. Bulk characterization methods for non-centrosymmetric materials: second-harmonic generation, piezoelectricity, pyroelectricity, and ferroelectricity. Chem. Soc. Rev. 2006, 35, 710–717; https://doi.org/10.1039/b511119f.Suche in Google Scholar PubMed
4. Stöger, B., Weil, M. The calcium oxotellurate(IV) nitrates Ca5Te4O12(NO3)2(H2O)2 and Ca6Te5O15(NO3)2. Mineral. Petrol. 2013, 107, 253–263.10.1007/s00710-012-0259-xSuche in Google Scholar
5. Christy, A. G., Mills, S. J., Kampf, A. R. A review of the structural architecture of tellurium oxycompounds. Miner. Mag. 2016, 80, 415–545; https://doi.org/10.1180/minmag.2016.080.093.Suche in Google Scholar
6. Weil, M., Shirkhanlou, M., Stürzer, T. Phase formation studies of lead(II) copper(II) oxotellurates: the crystal structures of dimorphic PbCuTeO5, PbCuTe2O6, and [Pb2Cu2(Te4O11)](NO3)2. Z. Anorg. Allg. Chem. 2019, 645, 347–353; https://doi.org/10.1002/zaac.201800262.Suche in Google Scholar
7. Weil, M., Shirkhanlou, M. Incorporation of sulfate or selenate groups into oxotellurates(IV): I. Calcium, cadmium, and strontium compounds. Z. Anorg. Allg. Chem. 2017, 643, 330–339; https://doi.org/10.1002/zaac.201600406.Suche in Google Scholar
8. Weil, M., Shirkhanlou, M. Incorporation of sulfate or selenate groups into oxotellurates(IV): II. Compounds with divalent lead. Z. Anorg. Allg. Chem. 2017, 643, 757–765; https://doi.org/10.1002/zaac.201700016.Suche in Google Scholar
9. Eder, F., Weil, M. The alkali metal copper(II) oxidotellurates(IV) Li2Cu2Te3O9, Li2Cu3Te4O12, Rb2Cu3Te6O16 and Cs2Cu3Te6O16 – four new structure types. Z. Anorg. Allg. Chem. 2022. https://doi.org/10.1002/zaac.202200089.10.1002/zaac.202200089Suche in Google Scholar
10. Cametti, G., Churakov, S., Armbruster, T. Reinvestigation of the zemannite structure and its dehydration behavior: a single-crystal X-ray and atomistic simulation study. Eur. J. Mineral 2017, 29, 53–61; https://doi.org/10.1127/ejm/2017/0029-2587.Suche in Google Scholar
11. Matzat, E. Die Kristallstruktur eines unbenannten zeolithartigen Telluritminerals, {(Zn, Fe)2[TeO3]3}NaxH2 − x·yH2O. Tschermaks Mineral. Petrogr. Mitt. 1967, 12, 108–117; https://doi.org/10.1007/bf01127687.Suche in Google Scholar
12. Miletich, R. The synthetic microporous tellurites Na2[Me 2(TeO3)3]·3H2O (Me=Zn, Co): crystal structure, de- and rehydration, and ion exchange properties. Monatsh. Chem. 1995, 126, 417–430; https://doi.org/10.1007/bf00813204.Suche in Google Scholar
13. Evstigneeva, M. A., Nalbandyan, V. B., Petrenko, A. A., Medvedev, B. S., Kataev, A. A. A new family of fast sodium ion conductors: Na2M2TeO6 (M = Ni, Co, Zn, Mg). Chem. Mater. 2011, 23, 1174–1181; https://doi.org/10.1021/cm102629g.Suche in Google Scholar
14. Cirpus, V., Wittrock, J., Adam, A. Carbonat-Hydrate der schweren Alkalimetalle: Darstellung und Struktur von Rb2CO3·1.5 H2O und Cs2CO3·3 H2O. Z. Anorg. Allg. Chem. 2001, 627, 533–538. https://doi.org/10.1002/1521-3749(200103)627:3<533::aid-zaac533>3.0.co;2-a.10.1002/1521-3749(200103)627:3<533::AID-ZAAC533>3.0.CO;2-ASuche in Google Scholar
15. Cirpus, V., Adam, A. Die ersten Hydrogencarbonate mit einer trimeren [H2(CO3)3]4−-Baugruppe: Zur Darstellung und Kristallstruktur von Rb4H2(CO3)3·H2O und K4H2(CO3)3·1,5 H2O. Z. Anorg. Allg. Chem. 1995, 621, 1197–1204; https://doi.org/10.1002/zaac.19956210715.Suche in Google Scholar
16. Hanke, K. Zinktellurit: Kristallstruktur und Beziehungen zu einigen Seleniten. Sci. Nat. 1967, 54, 199; https://doi.org/10.1007/bf00594516.Suche in Google Scholar
17. Hanke, K. Die Kristallstruktur von Zn2Te3O8. Sci. Nat. 1966, 55, 273; https://doi.org/10.1007/bf00621643.Suche in Google Scholar
18. Bruker APEX3 and SAINT; Bruker AXS Inc.: Madison, Wisconsin, USA, 2016.Suche in Google Scholar
19. Sheldrick, G. M. SHELXT – integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8; https://doi.org/10.1107/s2053273314026370.Suche in Google Scholar
20. Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8; https://doi.org/10.1107/s2053229614024218.Suche in Google Scholar
21. Petříček, V., Dušek, M., Palatinus, L. Crystallographic computing system JANA2006: general features. Z. Kristallogr. 2014, 229, 345–352.10.1515/zkri-2014-1737Suche in Google Scholar
22. Dornberger-Schiff, K., Grell-Niemann, H. On the theory of order-disorder (OD) structures. Acta Crystallogr. 1961, 14, 167–177; https://doi.org/10.1107/s0365110x61000607.Suche in Google Scholar
23. Larvor, C., Stöger, B., Weil, M. The allotwinning of KCa3Te5O12Cl3: an OD interpretation. Z. Kristallogr. 2018, 233, 849–859; https://doi.org/10.1515/zkri-2018-2071.Suche in Google Scholar
24. Ferraris, G., Makovicky, E., Merlino, S. Crystallography of Modular Materials. IUCr Monographs on Crystallography, Vol. 15; Oxford University Press: Oxford, 2008.10.1093/acprof:oso/9780199545698.001.0001Suche in Google Scholar
25. Brown, I. D. The Chemical Bond in Inorganic Chemistry: The Bond Valence Model; Oxford University Press: Oxford, 2002.Suche in Google Scholar
26. Brese, N. E., O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. 1991, B47, 192–197; https://doi.org/10.1107/s0108768190011041.Suche in Google Scholar
27. Spek, A. L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13; https://doi.org/10.1107/s0021889802022112.Suche in Google Scholar
28. Mills, S. J., Christy, A. G. Revised values of the bond-valence parameters for TeIV–O, TeVI–O and TeIV–Cl. Acta Crystallogr. 2013, B69, 145–149; https://doi.org/10.1107/s2052519213004272.Suche in Google Scholar PubMed
29. Hahn, T., Aroyo, M. I. Symbols of symmetry elements. In Space-Group Symmetry, Volume A of International Tables for Crystallography; IUCr: Chester, 2016; pp. 144–148, chapter 2.1.2.10.1107/97809553602060000114Suche in Google Scholar
30. Dornberger-Schiff, K., Grell, H. Symbols for OD groupoid families referring to OD structures (polytypes) consisting of more than one kind of layer. Acta Crystallogr. 1982, A38, 49–54.10.1107/S0567739482000096Suche in Google Scholar
31. Fichtner, K. On the description of symmetry of OD structure (II) The Parameters. Krist. Tech. 1979, 14, 1453–1461; https://doi.org/10.1002/crat.19790141207.Suche in Google Scholar
32. Dornberger-Schiff, K. Geometrical properties of MDO polytypes and procedures for their derivation. I. General concept and applications to polytype families consisting of OD layers all of the same kind. Acta Crystallogr. 1982, A38, 483–491; https://doi.org/10.1107/s0567739482001041.Suche in Google Scholar
33. Dornberger-Schiff, K., Grell, H. Geometrical properties of MDO polytypes and procedures for their derivation. II. OD families containing OD layers of M > 1 kinds and their MDO polytypes. Acta Crystallogr. 1982, A38, 491–498; https://doi.org/10.1107/s0567739482001053.Suche in Google Scholar
34. Jeffery, J. W. Unusual X-ray diffraction effects from a crystal of wollastonite. Acta Crystallogr. 1953, 6, 821–825; https://doi.org/10.1107/s0365110x53002428.Suche in Google Scholar
35. Nespolo, M., Ferraris, G. Effects of the stacking faults on the calculated electron density of mica polytypes—the Ďurovič effect. Eur. J. Mineral 2001, 13, 1035–1045; https://doi.org/10.1127/0935-1221/2001/0013-1035.Suche in Google Scholar
36. Stöger, B. Non-crystallographic layer lattice restrictions in order-disorder (OD) structures. Symmetry 2014, 6, 589–621; https://doi.org/10.3390/sym6030589.Suche in Google Scholar
37. Ďurovič, S. Desymmetrization of OD structures. Krist. Tech. 1979, 14, 1047–1053.10.1002/crat.19790140904Suche in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/zkri-2022-0030).
© 2022 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- In this issue
- Inorganic Crystal Structures (Original Paper)
- Crystal structure and magnetic properties of some compounds with GdNi2Ga3In type structure
- Partially disordered pyrochlore: time-temperature dependence of recrystallization and dehydration
- Lu37Ru16.4In4 – coloring and vacancy formation in a new structure type closely related to a 8 × 8 × 8 bcc superstructure
- BaFe0.875Re0.125O3−δ and BaFe0.75Ta0.25O3−δ as potential cathodes for solid-oxide fuel-cells: a structural study from neutron diffraction data
- Unbalanced racemates: solid state solutions containing enantiomeric pairs crystallizing in Sohncke space groups with (L:D) ratios other than (1:1) – illustrated with crystals of a Co(III) coordination compound
- Crystal structure and specific heat of calcium lanthanide oxyborates Ca4LnO(BO3)3
- Order-disorder (OD) structures of Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11
- Na2Cu+[Cu2+3O](AsO4)2Cl and Cu3[Cu3O]2(PO4)4Cl2: two new structure types based upon chains of oxocentered tetrahedra
- Organic and Metalorganic Crystal Structures (Original Paper)
- Two isomers Ba5Mg4C54O48H114 and Pb5Mg4C54O48H114
- Layered structures based on antimony tartrate dimers
Artikel in diesem Heft
- Frontmatter
- In this issue
- Inorganic Crystal Structures (Original Paper)
- Crystal structure and magnetic properties of some compounds with GdNi2Ga3In type structure
- Partially disordered pyrochlore: time-temperature dependence of recrystallization and dehydration
- Lu37Ru16.4In4 – coloring and vacancy formation in a new structure type closely related to a 8 × 8 × 8 bcc superstructure
- BaFe0.875Re0.125O3−δ and BaFe0.75Ta0.25O3−δ as potential cathodes for solid-oxide fuel-cells: a structural study from neutron diffraction data
- Unbalanced racemates: solid state solutions containing enantiomeric pairs crystallizing in Sohncke space groups with (L:D) ratios other than (1:1) – illustrated with crystals of a Co(III) coordination compound
- Crystal structure and specific heat of calcium lanthanide oxyborates Ca4LnO(BO3)3
- Order-disorder (OD) structures of Rb2Zn(TeO3)(CO3)·H2O and Na2Zn2Te4O11
- Na2Cu+[Cu2+3O](AsO4)2Cl and Cu3[Cu3O]2(PO4)4Cl2: two new structure types based upon chains of oxocentered tetrahedra
- Organic and Metalorganic Crystal Structures (Original Paper)
- Two isomers Ba5Mg4C54O48H114 and Pb5Mg4C54O48H114
- Layered structures based on antimony tartrate dimers