
The role of poly(acrylic acid) in conventional glass polyalkenoate cements
-
Adel M.F. Alhalawani
 ,
Daniel Boyd
,
Daniel Boyd

Abstract
Glass polyalkenoate cements (GPCs) have been used in dentistry for over 40 years. These novel bioactive materials are the result of a reaction between a finely ground glass (base) and a polymer (acid), usually poly(acrylic acid) (PAA), in the presence of water. This article reviews the types of PAA used as reagents (including how they vary by molar mass, molecular weight, concentration, polydispersity and content) and the way that they control the properties of the conventional GPCs (CGPCs) formulated from them. The article also considers the effect of PAA on the clinical performance of CGPCs, including biocompatibility, rheological and mechanical properties, adhesion, ion release, acid erosion and clinical durability. The review has critically evaluated the literature and clarified the role that the polyacid component of CGPCs plays in setting and maturation. This review will lead to an improved understanding of the chemistry and properties of the PAA phase which will lead to further innovation in the glass-based cements field.
1 Introduction
Conventional glass polyalkenoate cements (CGPCs), commonly referred to as the glass ionomer cements, were developed in the early 1970s at the laboratory of the Government Chemist in London, England [1]. These adhesive materials have been subjected to continuous improvement and diversification [2]. CGPCs are acid-base cements typically formed by the reaction of an organic aqueous solution of polyalkenoic acid, mainly a copolymer of poly(acrylic acid) (PAA) (Figure 1) with an inorganic acid-degradable fluoro-alumino-silicate glass. The reaction between both components results in a composite cement material consisting of reacted and unreacted glass particles embedded in a polysalt matrix [2–4]. GPCs are used in dentistry due to a selection of clinical advantages as follows [2, 5–9]:
Single-step adhesion characteristics of both enamel and dentine.
Biocompatible and considered bioactive.
High-dimensional stability.
Harden and form a cement and exhibit good resistance to cohesive failure, upon aging.
Satisfactory aesthetics and have negligible shrinkage upon setting.
![Figure 1: General structure of the repeating unit for PAA (reprinted with permission from [2]).](/document/doi/10.1515/polyeng-2015-0079/asset/graphic/j_polyeng-2015-0079_fig_001.jpg)
General structure of the repeating unit for PAA (reprinted with permission from [2]).
These features have made them attractive candidates for expanded applications in hard tissue repair. However, whilst their use in orthopedic applications has long been mooted, their non-dental clinical use has been limited to ear, nose and throat applications [10–13]. This is due to certain outages in their properties, including the following [2, 14]:
Clinical literature supporting defective osteoneogenesis and fatal encephalopathy arising from aluminum ions (Al3+) release in vivo.
Relatively poor mechanical properties vs. conventional acrylic bone cements.
Sensitivity to ambient room conditions, such as temperature and humidity, during mixing.
Susceptibility to both water desiccation and contamination during cement maturation.
The majority of research into CGPCs has focused on changing the glass composition because it is capable of controlling both setting chemistry, strength [15, 16] and ion release. However, the polyacid phase also plays a major role in controlling rheological (working and setting times) and mechanical properties, chemical adhesion to substrates, ion release and durability [17]. These characteristics can be influenced by altering the molar mass [18, 19], concentration [20], molecular weight [21, 22] and polydispersity index (PDI) [23] of the polyacid alongside other factors such as the powder: liquid ratio [24–26], and the addition of surface conditioners and chelating agents [8].
Moshaverinia et al. [7] have reviewed the literature describing the trends of modifying the polymer phase in GPCs. They focused on the advantages and disadvantages of various PAA-based polymers currently used in GPC systems (including new acrylic acid copolymers, amino acid containing polyelectrolytes, N-vinylpyrrolidone and N-vinylcaprolactam modified terpolymers). This review, however, focuses on the chemistry and properties of PAA and their subsequent effect on the early performance, characteristics and clinical applicability of CGPCs. The aim of this review is to complement existing works by evaluating three important questions:
How does the chemistry of PAA influence setting and maturation reactions of CGPCs?
To what extent can the different properties of PAA (molar mass, molecular weight, concentration, PDI and content and incorporation of chelating agents) affect the mechanical and rheological properties of CGPCs?
How does the PAA phase of CGPCs influence their clinical performance?
Following this brief introduction to CGPCs, Section 2 aims to answer the first research question by providing a historical background of PAA and the effect of its chemistry on the setting and maturation reactions of CGPCs. The main objective of Section 3 is to answer the second research question by providing a detailed evaluation as to how different properties of the PAA component may influence the characteristics of CGPCs, particularly mechanical and rheological properties. Section 4 provides a critical evaluation of the role of PAA in the clinical performance of CGPCs and thus answering the third research question.
2 Poly(acrylic acid) in conventional glass polyalkenoate cements: historical background and basic behavior
2.1 Background and general characteristics of the PAA component of CGPCs
Poly(acrylic acid) was patented in 1966 by Gene Harper of Dow Chemical and Carlyle Harmon of Johnson & Johnson and has been used in a range of healthcare applications including diapers, cosmetics and paint, ion-exchange resins and adhesives, and as a thickening, dispersing, suspending and emulsifying agent in pharmaceuticals [27]. The most commonly used polymeric materials for GPC formulations are PAA and copolymers of acrylic and itaconic acid [poly(AA-co-IA], or acrylic and maleic acid [poly(AA-co-MA)] with other monomers such as 3-butene 1,2,3-tricarboxylic acid (Figure 2) also being employed from time to time [3]. Each repeat unit of PAA has an ionizable group, carboxylic acid (COOH). Partially neutralized PAA has a sufficient charge density making it a superabsorbent (water soluble) polymer. In the presence of water at neutral pH, the PAA chains will lose their protons and acquire a negative charge, giving them the ability to absorb and retain water molecules [2, 28, 29], as shown in Eq. (1):
![Figure 2: The structure of poly(alkenoic acid)s containing (A) acrylic, (B) maleic, (C) itaconic, (D) methacrylic and (E) 3-butene-1,2,3-tricarboxylic acid units (reprinted with permission from [2]).](/document/doi/10.1515/polyeng-2015-0079/asset/graphic/j_polyeng-2015-0079_fig_002.jpg)
The structure of poly(alkenoic acid)s containing (A) acrylic, (B) maleic, (C) itaconic, (D) methacrylic and (E) 3-butene-1,2,3-tricarboxylic acid units (reprinted with permission from [2]).
Commercial PAA solutions are usually prepared by free radical polymerization of the polymer/monomer (such as acrylic acid) in aqueous solution and concentrated to 40–50% for clinical use [2]. Depending on the production process of PAA, the final product can be either in liquid or anhydrous (freeze-dried) form. Although the PAA used in the formation of early GPC compositions was in aqueous form and almost all CGPCs available from suppliers utilize liquid polyacid, Hill et al. [30] reported the advantage of using powdered, anhydrous PAA over aqueous PAA. Anhydrous PAA is better suited for prolonging the setting time of CGPCs. The finely ground PAA will have to dissolve in the water first and then attack the glass particles; this slows the setting reaction to some extent, allowing for the use of high-molecular-weight PAA and consequently, improved strength of the GPC matrix [30]. The majority of available studies [21, 30–33] detail anhydrous CGPC systems which contained anhydrous PAAs, confirming the theory presented by Hill et al. [30].
2.2 The role of PAA in the setting reaction of CGPCs
CGPCs are set by an acid-base reaction between a polyelectrolyte such as PAA and an acid-degradable glass. The cement-forming reaction consists of a number of overlapping stages, including the following [2, 34, 35]:
The attack by the PAA protons on the glass cations.
The release and migration of the liberated ions from the glass into the aqueous phase.
Neutralization and ionization of the polyacid resulting in unwinding, or relaxation, of the “twisted” polymer chains contributing to increasing the cement viscosity.
Ion binding between the charged polyacid chains and glass cations.
Gelation due to an increase in the pH of the pre-formed cement.
Continuous ion binding leading to the hardening phase.
The PAA component of CGPCs plays a significant role in the cement-forming reaction. Additionally, changes in the cement properties, ranging from molecular transport to mechanical properties, result from several physio-chemical processes including, but not restricted to [2, 36], the following:
Conformational changes in the polymer chain (changes in their ordered structure): PAA is a conformation changing polymer that undergoes unwinding/relaxation processes during the cement-forming reaction. This may modulate ion release and control the network properties [37].
Binding of the polymer chains to the glass cations: as cations become bound along the polymer chain, the polymer becomes in effect a polyelectrolyte. Cations already bound to the polymer chains may introduce charge repulsions reducing the cation binding. These repulsions may be decreased by counterions in the vicinity of the polymer shielding the charges. Further, dipole-dipole interactions in the polymer domain between cation pairs on the chain and cation pairs in solution may also influence cation binding to the polymer [2, 38]. In effect, changes in GPC properties would be expected.
Hydration surrounding polyanion and cation regions: as mentioned earlier in this article, PAA is a superabsorbent polymer that has the ability to absorb and retain water molecules at neutral pH. The extent and rate of interaction between hydrated cations and polyanions disrupts the hydration regions surrounding both, therefore changing the setting reaction of the CGPC and probably its long-term strength.
The extent of ion binding (between PAA anions and glass cations) depends on a number of characteristics of the polyion including, but not restricted to [2, 22, 39]: the following:
Polymer structure: longer chain PAA results in long-range entanglements and hence stronger cation-anion bonds. Further discussions will be provided later in this article.
Acid strength: strength of the PAA refers to its ability to ionize and lose protons in a solution, i.e. stronger PAAs will provide more sites for cations to bind resulting in stronger bonds.
Degree of dissociation: PAA neutralizes in water due to its ability to dissociate. Degree of dissociation refers to the fraction of the original polymer structure that has dissociated. Higher degree of PAA dissociation will result in higher rates of cation attack for binding. Further, the quicker the PAA dissociation in the GPC matrix, quicker is the setting reaction and stronger is the structure.
Degree of conformation: the polymer conformational changes can determine the proximity of binding sites to one another and hence affecting the binding process [38].
The ability of PAA to bind to basic cations released from the glass is determined largely by the ionic radius and complexation constant (the strength of the cation-releasing base, in interacting with the acid polyion, for the formation of a complex in aqueous solution) of the cations involved. Put another way, the smaller the ionic radius of the glass cation, the greater the binding strength. Similarly, the greater the complexation constant, the stronger the acid-base complex. For example, zinc ions (Zn2+) are likely to be more stable than calcium ions (Ca2+) when bonded to PAA, because zinc has a smaller ionic radius and greater complexation constant. Apart from its ability to bond to bone, the main advantages of PAA are low toxicity coupled with high solubility in water which allows solutions of 50% by mass to be produced [2, 3, 40, 41]. Some of the PAA factors proportionally affect the rate of the setting reaction of GPCs [Eq. (2)].
where M is the molar mass or molecular weight of the polyacid, C is the concentration of the polymeric solution, R is the powder:liquid ratio and H is the presence of additives or chelating agents.
2.3 The role of PAA during CGPC maturation
The setting reaction of CGPCs is regarded as continuous and the extent to which the ionic network develops has a direct impact on mechanical properties with strengths usually developing rapidly in the first 24 h [2, 42–44]. The strength of CGPCs increases quickly in the early stages of the reaction, within the first few days of maturation, and then increases at a slower rate over the following year [45, 46]. It was first postulated by Crisp and Wilson [44] that this hardening process is based on the gelation of the polymeric acid by cross-linking, or forming inter- and intramolecular salt bridges, of the polymeric carboxyl groups with Al3+ and Ca2+ from the glass phase. Later in 1976, Crisp et al. [46] attributed the increase in strength with time up to 1 year, to an increase in cross-link density. Crisp’s conclusions were later confirmed by Matsuya et al. [45], who used infrared (IR) spectroscopy and nuclear magnetic resonance (NMR) to show that an increase in cross-link density was the reason for increased strength. Wasson and Nicholson have otherwise found that the strength of the cement increased with time even when the glass powder was mixed with acetic acid which did not form insoluble salts with Al3+ and Ca2+ ions. Meanwhile, they attributed the hardening reaction to the silica component which leached from the glass powder and formed a hydrated silicate in the matrix [34, 35]. These results were supported by Cattani-Lorente et al. [47] and Hatton and Brook [48], who concluded that the strengthening of CGPCs during maturation resulted from additional cross-linking and the development of a silica gel phase. It is presumed that restricted chain pullout and increased cross-linking are the most likely causes of increased strength while the inorganic phase acts as a reinforcing phase in the strength of CGPCs [2, 31]. Maturation of a CGPC does not always result in increased strength in all modalities [47]. The literature has considered the effect of maturation on both compressive and biaxial flexural strength. Pearson and Atkinson [49] showed that the flexural strength increased with maturation up to a period of about 3 months but decreased thereafter. The flexural strength of Opus-fil (Davis Schottlander & Davis Ltd., Letchworth, Herts, UK), a conventional restorative GPC, increased to 73 MPa at 56 days, and then decreased to 53 MPa after 100 days of maturation. This phenomenon of decreasing the strength of some GPC formulations with maturation has been attributed to hydration of the ionic bonds within the cement matrix resulting in the loss of matrix-forming ions into solution, and consequently decreasing the integrity of the GPC [50, 51]. This phenomenon has also been attributed to the erosion and the plasticizing effect of water on these materials and to the slower rate of the reaction, as the cement ages in aqueous solution, resulting from the lesser number of COOH groups available to form ionic bonds [35, 52].
3 The influence of PAA properties on the physical characteristics of CGPCs
3.1 Effect of the PAA molar mass
Hill et al. [53] showed, using dynamic mechanical thermal analysis and dielectric thermal analysis, that GPCs exhibit sharp loss peaks similar to thermoplastics. Berry [54] demonstrated that the fracture surface energy of a thermoplastic polymer was much greater than the energy required to break all the polymer chains crossing the crack plane. Hence, GPCs may be classed as thermoplastic polymer composites with ionic cross-links liable to continual breaking and reforming. The strength of thermoplastic polymers is related to long-range entanglements that serve to restrict chain motion. Originally, these entanglements were viewed as physical knots. However, most polymer chains are too inflexible to form a physical knot and a model has been developed that views a chain as being trapped in a tube of entanglements formed by neighboring chains, as shown in Figure 3A. This model, known as the reptation model, assumes that a polymer chain only crosses the fracture plane once. This theory was first proposed in 1986 by Doi and Edwards [55] and was based on the theories postulated previously by Edwards [56] that polymer chains were contained within a hypothetical tube constructed by the forces imposed by the chain’s nearest neighbors. By calculating simple Brownian motion of a polymer chain within this hypothetical tube, Edwards proposed a mathematical expression of the rate of motion of the chain along its tube and postulated that the long-chain polymers become solid once a certain cross-link density is exceeded [55–59]. This proved to fit for many thermoplastics and explained the variation of chain interaction with temperature. Several studies have elaborated on the Edwards model, and variations are still being produced for specific applications [60, 61].
![Figure 3: Schematic illustration of the polymer reptation model (A) Reptating entangled chain, (B) chain scission (reprinted with permission from [31]).](/document/doi/10.1515/polyeng-2015-0079/asset/graphic/j_polyeng-2015-0079_fig_003.jpg)
Schematic illustration of the polymer reptation model (A) Reptating entangled chain, (B) chain scission (reprinted with permission from [31]).
In 1998, Griffin and Hill [31] derived an equation [Eq. (3)], based on a previous study by Prentice [59], relating the molar mass of the PAA (M) to the fracture surface energy per unit area of fracture plane (τ). τ can also be identified as the work that needs to be done to remove chains from a unit area of crack plane, whereas Mc is the critical molar mass required for entanglements to occur. Detailed derivation of Eq. (3) can be found in the literature [31, 59].
Further, based on the reptation theory, as the molar mass increases, a critical molar mass (typically about 105; however, its value is generally lower) will be reached where the stress to extract a chain from its tube is greater than that required for homolytic chain scission, as depicted in Figure 3B. In other words, for molar masses greater than a critical value, the force required to remove the chain will be greater than the force required to break the carbon-carbon bonds of the polymer backbone, making the surface fracture energy independent of molar mass, and hence the matrix toughness is no longer related to the molar mass of the PAA. Similarly, at low molar masses, below approximately 2.7×104, chain entanglements will not form, making the fracture surface energy independent of molar mass because the chain length is too short to form entanglements and the tube concept no longer applies. Griffin and Hill [31] conducted a study which evaluated the influence of the PAA molar mass on the mechanical properties of GPCs. Their results are summarized in Table 1, which shows that the molar mass of PAA affects a range of mechanical properties, including compressive strength, flexural strength, fracture toughness, toughness and plastic zone size, but will have no significant effect on Young’s modulus of the cement matrix. These results (Table 1) are in good agreement with those later reported by Fennell and Hill [32, 33, 62].
Influence of PAA molar mass on the fracture properties of 4.5SiO2-1.5P2O5-3Al2O3-4CaO-CaF2 GPCs (*E5, E7, E9 and E11 PAAs have number average molar masses of 3.25×103, 6.66×103, 2.29×104 and 1.08×105, respectively) [31].
| Property | Formula | Cement preparation | *E5 | E7 | E9 | E11 |
|---|---|---|---|---|---|---|
| Compressive strength (σc) (MPa) (SD) | Cements were prepared by mixing the glass powder with different molar mass PAAs concentrated at 40% in a weight ratio of 5:1 and then adding this mixture to water containing 10% m/v (+) tartaric acid, in a weight ratio of 4:1. Samples were aged for 1 day. | 42.63 (2.14) | 53.75 (3.16) | 54.84 (1.61) | 70.33 (3.17) | |
| Young’s modulus (GPa) | Independent of molar mass | |||||
| Flexural strength (σf) (MPa) (SD) | 10.90 (0.84) | 14.00 (1.07) | 14.36 (1.32) | 22.50 (0.36) | ||
| Fracture toughness (KI) (MPa m1/2) (SD) | 0.41 (0.02) | 0.52 (0.02) | 0.54 (0.01) | 0.98 (0.04) | ||
| Toughness (GI) (J m-2) | 23 | 37 | 49 | 167 | ||
| Plastic zone size (Rp) (μm) | 9.2 | 9.9 | 13.0 | 42.3 | ||
The compressive strength of CGPCs increased with increasing PAA molar mass (Table 1). This was found to be in good agreement with the literature [21, 46]. Wilson noted that higher molar mass cements failed with marked plastic deformation, while lower molar mass cements failed in a brittle fashion [21], confirming the Edwards model. Additionally, it was postulated [35], and later supported in the literature [45, 63, 64], that the change in the mechanical properties of GPCs, by changing the molar mass, results from the formation of the silicate network during the maturation period. Other studies however have illustrated that the role of the silicate phase is small and confirmed on the significant role of the increased cross-linking of the polyacrylate chains in dominating the fracture behavior of GPCs [31]. Hence, the cross-linking reaction of PAA chains is not only important in the early stages of the setting process of GPCs but also in dominating the long-term fracture behavior [31].
Griffin and Hill [31] evaluated the influence of PAA molar mass on the fracture properties of GPCs being investigated (Table 1). They have indicated that Young’s modulus was independent of the molar mass of the polymer phase of GPCs, agreeing with the literature [30, 65]. Young’s modulus is not predicted to rise with increased molar mass, according to the reptation theory [62, 66], but is, instead, dominated by the strength and number of interactions in the polymeric phase, in particular the number and strength of ionic cross-links. Altering the molar mass of the PAA will not affect the concentration or number of functional carboxylate groups present for cross-linking [62, 66], which provides evidence of why Young’s modulus is independent of the molar mass of the polymer phase.
In several studies, flexural strength has been observed to increase with molar mass [62, 66, 67], as evident from the data obtained by Griffin and Hill (Table 1). However, this increase is more pronounced for GPCs produced using higher molar mass acids [31, 66]. This may indicate that, although chain pullout is the predominant mechanism involved in fracture, it may not be the only one. CGPCs produced from PAA with molar masses lower than the critical molar mass result in flexural strengths far lower than that predicted by reptation theory, as the carbon-carbon bonding strength determines the overall strength. This may reflect the fact that cements utilizing low molar mass PAA are more brittle and the sensitivity to inherent flaws on the tensile edge of the sample will be increased. However, cements based on PAA with molar masses higher than the critical molar mass have a greater degree of plasticity, which reduces the sensitivity of the samples to surface flaws and consequently increases the flexural strength [62].
Fracture toughness, toughness and plastic zone size have also been observed to increase with molar mass (Table 1). This was attributed to the continuing cross-linking reaction of the polymer phase of GPCs, restricting chain motion and/or molecular flow taking place at the crack tip [31, 33]. The application of reptation theory to GPCs has been criticized, but it is capable of making quantitative predictions for analysis of GPC’s fracture behavior [31].
3.2 Effect of the PAA molecular weight
The weight average (Mw) and number average (Mn) molecular weight of the polyelectrolyte also affect the rheological and mechanical properties of GPCs, as shown in Eq. (4a). The molecular weight averages are defined mathematically using Eqs. (4b) and (4c):
where Mi is the mass of a specific isotope and Ni is the number of molecules whose weight is Mi.
The simplicity of Eq. (4a) has resulted in a number of studies that have found similar trends with regard to the influence of the PAA molecular weight (Mn or Mw) on the mechanical and rheological properties of CGPCs [21, 23, 30, 68]. Increasing the PAA molecular weight, in order to improve cement strength, increases the cement viscosity for clinical handling and decreases working and setting times [21, 22] (see Table 2). In particular, it can be seen that the use of low-Mw PAA results in non-measurable rheological properties. Very low Mw (≤3.5 K) results in very low viscosity resulting in a weakened cement and, at the same time, very long rheological properties when compared with the minimum requirements noted by ISO 9917-1:2007 for dental-based cements. In general, higher Mw PAA would provide more unbonded carboxylic (−COO) groups to bind with glass cations resulting in quicker interaction and hence shorter setting time; i.e. this occurs due to the faster ability of cross-linking between the polymer chains and the released cations while the low viscosity allowed for longer working time [22]. In such a case, the resulting cement is expected to be putty-like. Results presented by Wilson et al. have been later confirmed by a number of studies in this field [69, 70]. In the clinic, a long working time (sufficient for placement of the cement before its workability diminishes) and a sharp setting time (to prevent unfavorable acid-tissue interactions) are desirable properties.
Effect of PAA Mw on the rheological properties of CGPCs.
| PAA (Mw) (K) | Viscosity (cP) | Working time (min) | Setting time (min) |
|---|---|---|---|
| 3.5 | 16 | – | – |
| 27 | 50 | 9.25 | 6.50 |
| 76 | 200 | 4.75 | 5.00 |
| 230 | 3.5 K | 2.75 | 3.25 |
The effect of the PAA molecular weight on the fracture properties of CGPCs is controlled, to a large extent, by the PAA molar mass. The conceptual framework of reptation theory and the ideas of entanglements help in illustrating the effect of polymer molecular weight on the properties of CGPCs. The application of the reptation model to CGPCs was successful in describing the dependence of the fracture properties of CGPCs on the PAA molecular weight. Wilson et al. [21] conducted a study investigating the influence of the PAA molecular weight on the mechanical properties of GPCs. Table 3 brings together their results. As shown in Table 3, the compressive strength of GPCs increases with the molecular weight. In the literature [20, 21, 31, 32], there were little differences in compressive strengths reported with Mw of PAA in excess of 100 K which confirmed the suggestion by Hill et al. [30] that a critical Mw for PAA was approximately 100 K when anhydrous PAA was investigated. A recent study by Gomes et al. [71] considered Mw for PAA between 50 K and 1250 K and observed that the highest compressive strength and compressive elastic modulus (E) were associated with an Mw of PAA of 50 K. Young’s modulus was also evaluated by Wilson et al.; values in Table 3 show little variation, as expected, with PAA molecular weight, confirming that molecular weight does not influence the chemistry of the setting reaction. In a similar fashion to compressive strength, the flexural strength was markedly increased with increased molecular weight. However, it was realized that the flexural strength ceases to rise at very high Mw (>5×105). This behavior is similar to that of thermoplastics; at Mw higher than 5×105, the flexural strength becomes independent of the molecular weight as a result of reaching a critical stress sufficient to cause chain scission. This explanation agrees with the reptation chain pullout model presented by Prentice [59]. Further, fracture toughness, toughness and flaw size measurements also increased with PAA molecular weight. It was illustrated that the increase in toughness resulted from both PAA cross-linking reactions and the interface between the glassy phase and the polymer matrix. Additional testing included wear and erosion as a function of molecular weight, both decreased with increased molecular weight (values are not tabulated in the original article) [21]. Hence, it can be seen that a large number of mechanical properties are dependent on the PAA molecular weight since the longer the polyacid chain, the larger the number of cross-links required to be broken to free the chain. It is relatively easier to pull out a low-molecular-weight chain than to pull out a high-molecular-weight chain. In addition, increased strength can be attributed to limited motion of the bonded polyacid molecules due to their long-chain entanglements [21, 72]. Values in Table 3 were compared to other experimental data in the literature [22, 30, 73] and were found to have similar patterns.
Dependence of the fracture properties and acid erosion of 12.39Si-16.44Al-7.14Ca-10.40F-7.26Na-4.54P-41.83O GPCs on PAA molecular weight [*E5, E7, E9, E11, E13 and E15 PAAs have weight average molecular weights (Mw) of 1.15×104, 2.27×104, 1.14×105, 3.83×105, 1.08×106 and 1.49×106, respectively] [21].
| Property | Cement preparation | *E5 | E7 | E9 | E11 | E13 | E15 |
|---|---|---|---|---|---|---|---|
| Compressive strength (σc) (MPa) (SD) | Cements were prepared by mixing the glass powder (50 g) with 7 g of different molecular weight anhydrous PAAs and then adding 11 g of this mixture to water containing 10% by mass (+) tartaric acid. Samples were aged for 1 day. | 30.40 (2.53) | 41.95 (3.30) | 45.80 (8.66) | 50.14 (1.48) | 65.88 (5.39) | 57.48 (2.05) |
| Young’s modulus (GPa) (SD) | 1750 (314) | 1754 (374) | 1220 (150) | 1313 (132) | 1429 (132) | 1580 (145) | |
| Flexural strength (σf) (MPa) (SD) | 7.06 (1.05) | 8.05 (0.61) | 9.63 (0.41) | 10.98 (0.78) | 13.74 (0.57) | 13.25 (1.90) | |
| Fracture toughness (KI) (MPa m1/2) (SD) | 0.13 (0.01) | 0.10 (0.01) | 0.23 (0.02) | 0.26 (0.04) | 0.33 (0.04) | – | |
| Toughness (GI) (J m-2) | 10 | 15 | 30 | 38 | 61 | – | |
| Flaw size (Rp) (μm) | 91 | 106 | 142 | 171 | 155 | – | |
| Mechanical wear | Both mechanical wear and acid erosion decreased as the molecular weight of the PAA increased. | ||||||
| Acid erosion | |||||||
In 2011, Dowling and Fleming [23, 68] investigated the ways of improving the mechanical properties of GPCs, in particular compressive strength and elastic modulus, using PAA molecular weight mixtures, different blend ratios and different PAA concentrations without impacting viscosity of the PAA solution. The PAAs in their studies were conventional aqueous solutions with Mn ranging between 5 K and 200 K. They have suggested, in line with the observations of Martin et al. [74], that the critical Mw would be approximately 80 K, lower than that (100 K) suggested by Hill et al. [30] and based on work by Prentice [59]. Additionally, Dowling and Fleming [23] suggested the entanglement Mw for PAA to be below 5 K, for aqueous PAAs, compared with the 7 K suggested by Hill et al. [30] for anhydrous PAAs. Their results have confirmed the aforementioned discussions by Wilson and Hill groups. By replacing 10–30% of a lower Mw PAA (~15 K) with a higher Mw PAA (~80 K), significant increases in compressive strength and elastic modulus were observed with minimal increases in the viscosity of the PAA solutions. It is important to note that although the approach of mixing different molecular weights at different ratios to improve the mechanical properties is encouraging, no marked increases in compressive strength and elastic modulus were observed compared with those for the control cements with highest Mw PAA (~80 K) investigated. The use of 50 K Mw PAA with a particle size <50 μm would result in the optimum polymerization and end-use characteristics of GPCs for clinical use. The use of Mw PAA higher than 50 K is favorable as it results in marked plastic deformation due to the wider distribution of the polymer chain lengths. It is important, though, to note that the use of Mw>500 K would not improve the mechanical properties since the cement matrix reaches a point of critical stress sufficient to cause chain scission and the mechanical properties become independent of Mw. Attention must also be paid to the use of different molecular weight PAAs as they result in different molecular weight distributions. The short chains in the high-molecular-weight PAA would contribute to the quick setting reaction with the released cations through faster disentanglement and dissolution and hence resulting in improved mechanical properties, while the long chains in the low-molecular-weight PAA would contribute to slightly delaying the matrix from quick setting [2, 21, 22].
3.3 Effect of the PAA concentration
Continuing our earlier discussions on the reptation model, the theory also considers individual polymer chains and states that when the concentration of a polymer solution is high enough to produce dense chain entanglements, each chain is forced to wriggle in an anisotropic curvilinear motion, called “reptation”. Increasing the PAA concentration involves reducing water content, i.e. increasing the relative amount of chemically bonded atoms with stable electronic configuration, alternatively called covalent bonds, or the number of COOH groups resulting in better distribution of stresses through the structure. In other words, higher concentration of PAA results in cements with higher numbers of polyacid chains, thus affecting GPC characteristics. Figure 4 illustrates this concept and shows that the strength of the cement depends on the stress transfer between volume elements, which would improve if the PAA molecular weight or concentration were increased [75, 76].
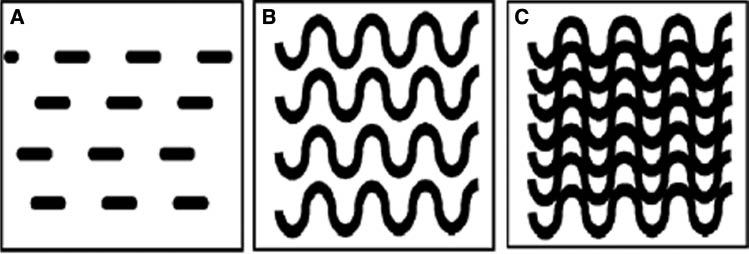
Schematic illustration of the (A) Cement volume, (B) effect of increasing molecular weight and (C) effect of increasing concentration.
CGPCs have utilized the PAA at a concentration of about 45% [40]. Since then, various studies have considered the effect of changing the PAA concentration on the physical (rheological and mechanical) properties of GPCs. The effect of the PAA concentration on rheological properties was studied by Crisp et al. [20]. Table 4 provides the active region (a region in which identifying the properties of GPCs is possible) of PAA concentration for different rheological properties and comments on each. It is clear that increasing the PAA concentration increases the cement viscosity resulting in a quicker setting reaction. In general, the more concentrated PAA results in improved matrix formation through the assimilation of metallic ions and formation of salt bridges; however, this improvement happens at the expense of rheological properties [2, 20]. It was also postulated that increasing polymer concentration results in higher polymer conductivity and lower pH. This is expected to increase intra- and intermolecular interactions resulting in compressed macromolecular chains and hence increased surface reactivity [76–78]. Thus, it can be noted that the concentration of the polymer phase is an important factor affecting the setting reaction, whereas a suitable balance is important to allow the cement components to react and to attain optimum properties [20]. These results observed by Crisp et al. were confirmed by similar studies in the literature [2, 23, 35, 68].
Effect of the PAA concentration (% w/w) on the rheological properties and handling characteristics of CGPCs [20].
| Parameter | Active region | Comment |
|---|---|---|
| Viscosity | 28–52 | Linear relationship; however, the viscosity increases rapidly at PAA concentrations >48% w/w. |
| Powder:liquid ratio | 38–50 | Increasing the PAA concentration corresponds to a reduction in the P:L ratio resulting in increased viscosity maintaining the cement consistency. PAA concentration <38% w/w resulted in no effects on the P:L ratio. |
| Working time | 38–50 | Within this active region, increasing the PAA concentration decreased the working time. This corresponds to the increased viscosity of the cement. Below 38% w/w, there is no effect on the working time, while the use of ratios >50 is not recommended for clinical purposes. |
| Setting time | 28–48 | The setting time decreased for PAA concentrations between 28% and 38% w/w. However, an increase in the setting time was recorded corresponding to concentrations between 29% and 43% w/w. Subsequently, the setting time drops until a plateau is reached. |
The literature [23, 31, 68, 79, 80] also researched and commented on the effect of PAA concentration on the mechanical properties of GPCs. Table 5 brings together the results observed by a series of studies by Fennell and Hill [32, 33, 62], which focused on the influence of PAA concentration and molar mass on the fracture properties of GPCs. It can be seen that, generally, cement strength increases with PAA concentration (30–50%). Further increases in PAA concentration (up to approximately 45–50%) increases the number of chains crossing the fracture plane leading to more energy being expanded and hence higher toughness and strength [62]. However, a slight fall and variation in some mechanical properties can be seen for PAA concentrations higher than 50% offering justification for why a PAA concentration of 45% is preferred for the preparation of GPCs in the dental clinic. This slight fall/variation for cements prepared with PAA concentrations higher than 50% can be attributed to the deficiency of metal cations in the polysalt matrix present for cross-linking and/or due to incomplete dissolution of the polyacid particles [32]. Moreover, Young’s modulus was observed to be independent of PAA molar mass (Table 1); however, it was found (Table 5) to increase with the PAA concentration up to approximately 50%. The increase in Young’s modulus was due to the reduced water content as the PAA concentration increases. Water molecules are likely to cause a plasticizing action, spacing the polymer chains apart, and hence lower chain entanglement density or decreased number of COOH groups for cross-linking would be expected resulting in lower or unpredictable Young’s modulus [62]. Generally, the viscosity and mechanical properties of GPCs increase with the PAA concentration regardless of the PAA molecular weight. However, an optimum PAA concentration (ranging between 35% and 50%) exists for each PAA molecular weight, above which the mechanical properties decrease [23].
Influence of the PAA concentration on the mechanical properties of GPCs [32, 33, 62].
| Property | Cement preparation | PAA concentration | ||||||
|---|---|---|---|---|---|---|---|---|
| 30% | 35% | 40% | 45% | 50% | 55% | 60% | ||
| Compressive strength (σc) (MPa) (SD) | Cements were prepared by mixing the glass powder (4.5 SiO2-1.5P2O5-3Al2O3-4CaO-CaF2) with E7 PAAs concentrated between 30% and 60% m/m and then adding this mixture to water containing 10% m/v (+) tartaric acid. Cements were prepared with 0.4 glass volume fraction and were aged for 1 day | 34 (2) | 44 (3) | 50 (3) | 73 (7) | 76 (6) | 88 (5) | 73 (4) |
| Young’s modulus (GPa) | 2.93 (0.23) | 3.07 (0.28) | 4.05 (0.47) | 5.04 (0.75) | 5.67 (0.59) | 4.90 (0.37) | 3.55 (0.27) | |
| Flexural strength (σf) (MPa) (SD) | 5.14 (1.28) | 5.53 (1.14) | 8.65 (1.31) | 10.08 (1.83) | 11.01 (1.38) | 19.18 (0.65) | 18.14 (0.72) | |
| Fracture toughness (KI) (MPa m1/2) (SD) | 0.25 (0.03) | 0.27 (0.10) | 0.40 (0.05) | 0.42 (0.08) | 0.52 (0.16) | 0.50 (0.04) | 0.51 (0.04) | |
| Toughness (GI) (J m-2) | 21 | 24 | 39 | 35 | 48 | 51 | 73 | |
3.4 Effect of the PAA polydispersity index
The PDI is a measure of molecular weight distribution (MWD) of the PAA [Eq (5)]. MWD results from the unequal growth of the polymer chains during polymerization [23, 81].
Most studies in the field have considered the effect of changing molecular weight, while less attention is paid to PDI. Studies by Hill et al. [30], Wilson et al. [21] and Dowling and Fleming [23] have shown that increasing PDI results in increased strength. Dowling and Fleming, for example, investigated the influence of PDI of a series of PAA solutions, with number average molecular weights ranging from 5 K to 200 K and concentrations ranging from 10% to 60%. PDI values between 1.5 and 2.2 were used in the study of Dowling and Fleming and were considered narrow. Otherwise, high PDI (>3) is indicative of a wide distribution of PAA chain lengths and can be called “polydisperse PAA”, which is generally advantageous for processing purposes since low-molecular-weight fractions or low Mn result in higher viscosities and behave like lubricants [21, 23]. This behavior was attributed to the fact that longer PAA chains, associated with higher Mw PAA, have a disproportionate effect on the viscosity, while a mono-disperse PAA, with a narrower distribution of PAA chain lengths, would provide a proportionate effect on the viscosity when concentrated in solution [31].
The chain length of the polyacid is known to be an important parameter affecting a wide range of clinically relevant parameters such as rheology, acid erosion, solubility, mechanical properties and abrasive wear. Increasing the chain length of the polymer results in lower viscosity and hence higher strength because the narrow distribution of PAA chain length results in larger chain entanglements and hence lower stress cracking sensitivity [21, 30].
3.5 Effect of the mixing ratio of PAA solution and glass powder
Variations in the powder:liquid (P:L) ratio can, understandably, influence both mechanical and rheological properties of GPCs. Fleming et al. [26] manipulated the ratio for ChemFil (Dentsply, Germany), a commercially available restorative cement, and examined the effects on rheological and mechanical properties. Using the manufacturer’s recommended ratio as a baseline (100% powder), other formulations were mixed at 90%, 80% and 50% of the recommended powder content, with a constant volume (1 ml) of aqueous polyacid. Table 6 summarizes key results from the literature. Decreasing powder content, while keeping liquid content constant, results in reduced strength but longer rheological properties. Xie et al. [82] studied the effect of increasing glass content on the GPC strength and rheological properties. They showed that increasing the P:L ratio in the cement resulted in increased compressive strength but decreased setting time. Further, they showed that increasing the P:L ratio from 1 to 2 increased the compressive strength from ~40 to ~140 MPa, while a non-significant increase resulted when the P:L ratio increased further from 2 to 2.5. Beyond 2.5, the compressive strength dropped significantly. Results of Xie et al. [82] are in good agreement with those presented by Fleming et al. [26].
Properties of ChemFil mixed with 100%, 90%, 80% and 50% of the recommended powder content [26].
| Property | 100% | 90% | 80% | 50% |
|---|---|---|---|---|
| Compressive strength (MPa) | 102 | 94 | 83 | 56 |
| Working time (s) | 90 | 90 | 108 | 120 |
| Net setting time (s) | 150 | 168 | 186 | 210 |
The higher the amount of glass powder, the higher the cement strength [83]. This correlation has been explained through the particles of the glass powder that remain unchanged due to the lower level of acid. The “unreacted” glass cations, within the cement structure, act as reinforcing filler particles and prevent crack propagation within the cement matrix, resulting in improved strength [24, 83]. Reducing the volume of reinforcing glass particles (or increasing the amount of liquid used for a specific formulation) reduces the ability of GPCs to resist compressive forces during loading and consequently failure occurs at lower compressive loads. Variations in the rheology of the cements with P:L ratio can be explained in similar terms. First, the increased PAA ratio will inevitably provide a more fluid cement upon mixing, and decreasing the glass volume fraction provides fewer available matrix-forming ions relative to active bonding sites on the polyacid chain [2, 24, 84]. There must be an optimum P:L ratio, providing that the cement has sufficient working and setting times for the application of the cement at this optimum ratio.
3.6 Effect of additives/chelating agents
It has been postulated that the polymer phase is responsible for the strength of the cement while the incorporation of chelating agents was assumed to affect various properties of GPCs [85, 86]. Tartaric acid (Figure 5), as a co-additive to the PAA backbone, is often added to GPCs as a rate-controlling additive [2]. Wilson et al. [85] showed that tartaric acid was effective in the extraction of ions from the alumino-silicate glass. The expected effect would be of shortening the setting time and accelerating the rate of cement hardening. However, the paper reported that d-tartaric acid was also an effective complexing agent for improving working and setting times of cement pastes [86] and postulated that the addition of tartaric acid first delays the setting reaction and then enhances ion bridging as a result of withholding cations from the polyanion chains. (+)-Tartaric acid (5–10 wt.%) (Figure 5A) improves the rheological properties of GPCs by extending the working time and sharpening the onset of the setting time [40]. Results of Wilson et al. were confirmed by Prosser et al. [87], who showed by means of 13C Fourier transform NMR spectroscopy that tartaric acid reacts preferentially with the glass and prevents the early binding of cations to the polyanion chains, resulting in increased working time. Tartaric acid is fully complexed at pH≈3, and complexing by PAA then occurs with the pH of the set cement rising to pH≈5 [87]. This change in complexing species occurs due to the stability of calcium and aluminum tartrate compounds at low pH (pH≈3–4). In the presence of (+)-tartaric acid, calcium salts form more slowly and aluminum salts form more rapidly. It has also been noticed that the conformation of the acid is also of paramount importance as neither (−) (Figure 5B) nor (±)-tartaric acids influence the setting process in the same way as the (+)-isomer (meso form shown in Figure 5C) does, whilst their use is contraindicated. A modest increase in strength has also been noticed with the addition of (+)-tartaric acid. These strengthening effects have been attributed to reduction in bulk homogeneities and indirectly improving the surface of the specimen by increasing flow properties [40, 86, 87].
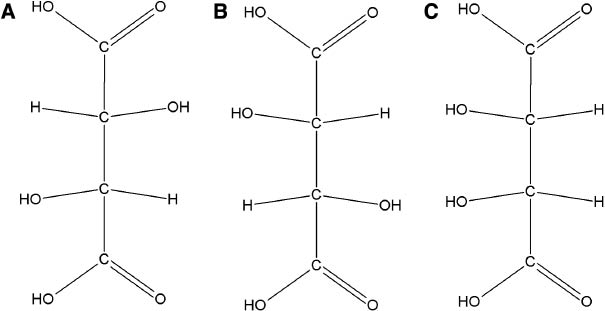
Structure of different tartaric acids (A) (+)-Tartaric acid, (B) (−)-tartaric acid and (C) meso-tartaric acid.
Other additives including, but not restricted to, phosphoric [88], amino [89], maleic [90], itaconic [91] and oxalic acids [92] have been studied and discussed in other review papers [3, 7, 40]. Yet, they are not the focus of this review article.
4 The influence of the PAA component on the clinical performance of CGPCs
This review considers the role of PAA in the setting chemistry and maturation of CGPCs and then discusses the effect of changing various aspects of the PAA phase, including molar mass, molecular weight, concentration, PDI, content and inclusion of chelating agents on the mechanical and rheological properties of CGPCs. This section focuses on the clinical aspects of these biomaterials.
4.1 Biocompatibility
Biocompatibility refers to “the ability of a biomaterial to perform with an appropriate host response in a specific application” [93]. This definition was later modified by Williams, who redefined biocompatibility of a biomaterial as “the ability to perform as a substrate that will support the appropriate cellular activity in order to optimize tissue regeneration, without eliciting any undesirable local or systemic responses in the eventual host” [94]. Many studies have been performed in vitro and in vivo to evaluate the biocompatibility of PAA-based systems. A study by Brodbeck et al. [95] demonstrated that PAA could prevent the failure of implanted biomedical devices by limiting macrophage fusion and monocyte adhesion in vivo using the rat cage implant system. Other studies [96, 97] have also commented on the anti-corrosion performance of the PAA, the ability to functionalize such water-soluble polymers with bioactive molecules and their compatibility towards human osteoblast-like cells.
The biocompatibility of CGPCs has been investigated and reported over the past several decades. There are no studies reporting the systemic toxicity of CGPCs, attributed to their positive preclinical biocompatibility results [98]. A single study [99] reported a case of allergy reaction to CGPCs, in which a generalized urticaria occurred after the application of CGPCs. In general, CGPCs do not pose an acute systemic risk or chronic toxic behavior; however, it is important to assess their systemic toxicity prior to their market launch. A number of studies [100–105] intensively investigated the cytotoxicity of CGPCs. It was consistently illustrated that the cytotoxic behavior of CGPCs depends on their setting reaction. A freshly mixed cement may exhibit an antibacterial activity or cytotoxicity, both diminishing as the cement matrix hardens [106–109]. The literature postulated that this behavior could result from the release of metal ions such as aluminum and fluoride and/or free PAAs when freshly mixed [110, 111]. The cytotoxic effect of freshly mixed CGPC was also attributed to the high acidity of the freshly mixed cement (pH 1.6–3.7) when compared to that (pH 5.4–7.3) of the completely set cement [112]. The antimicrobial properties of CGPCs have been investigated in vitro and in vivo [113–120]. It was found that freshly mixed CGPCs inhibited bacterial growth, whereas completely set cement samples revealed no antimicrobial effect. In addition to the antimicrobial effect of CGPCs, these adhesive materials have resulted in decreased microbial adhesion when compared with other dental materials, for instance, resin-based composites [98].
A number of clinical studies on CGPCs have questioned their biocompatibility. An implantation study by Steinbrunner et al. [121] revealed that very thin mixes of CGPC, when used as a pit and fissure sealant, generated much more pronounced tissue reactions than a thick mix of the same product when used as a filling material. Other studies [104, 105, 122] showed that the exposure of pulp to CGPCs has resulted in severe pulp reactions, including abscess formation. CGPCs used as luting agents caused severe pain in certain cases [123]. The possible causes of these clinical reactions can be the incorrect handling (e.g. pronounced drying of the prepared tooth prior to cementation), insufficient remaining dentin thickness, excessive pressure during cementation or increased solubility resulting from inhibited setting reaction [120, 124, 125]. Further details on the biocompatibility of CGPCs are presented elsewhere [9, 126, 127].
4.2 Rheological and mechanical properties
The mechanical and rheological properties of GPCs are often interrelated. Generally, increasing the rheological properties has been shown to decrease the mechanical properties, while strength has increased with a shortened working and setting times. The strength of GPCs increases with time as a result of cross-linking in the polysalt matrix. The previous sections of this article have commented on the chemistry of these novel materials and discussed the effect of various properties of PAA on the mechanical and rheological properties of CGPCs. GPCs are not only required to set to give a strong material, but they must also remain viscous for a sufficient time to allow manipulation by the clinician after which they must present a degree of hardness following placement to avoid failure.
4.3 Adhesion to substrates
CGPCs adhere to both dentin and enamel without prior treatment [2, 128]. Many studies have considered the mechanism of adhesion using information from spectroscopic techniques such as IR spectroscopy. Smith [129] suggested that PAA carboxylate groups in the GPC chelate Ca2+ ions in hydroxyapatite. Beech [130] postulated, from IR absorption spectra, that adhesion is a result of ionic attraction between carboxylate groups and the surface of the tooth. Wilson [131] considered that metal ions could form a salt bridge between pendant carboxylate groups in the cement and the negatively charged apatite surface of enamel. Further, Wilson indicated that effective adhesion results from excellent wetting which is attributed to the ability of the free COOH groups present in fluid cement to form hydrogen bonds as shown in Figure 6. These hydrogen bonds are replaced by ionic bridges as the cement sets. Belton and Stupp [132] showed that ionized rather than unionized polyacrylate is responsible for adhesion. Another possibility remains is that the polyacrylate chains affect adhesion by crossing the interface and interacting with the surface layer of the enamel apatite. Wilson et al. [133] showed that the surface layer of the adhering cement becomes enriched in phosphate and calcium ions as these diffuse from the enamel surface. A more recent laboratory study by van Meerbeek et al. [134] described a micromechanical interlocking mechanism between the self-etching effect of the polyacid component of the glass ionomer and the hydroxyl apatite coated collagen fibril network of dentine. Therefore, it was concluded that ionic bonds are formed between the poly carboxyl groups of the glass ionomers poly acid and the Ca2+ ions of the tooth, confirming the results presented by Smith [129]. Furthermore, a study by Yoshida et al. [135] showed the capability of using novel biomaterial characterization techniques such as x-ray photoelectron spectroscopy (XPS) in identifying the chemical bonding at biomaterial-hard tissue interfaces. They demonstrated, using XPS, that the PAA component of the glass ionomer system significantly influences the chemical bonding potential. Further, they have shown that a PAA based upon 10:1 acrylic/maleic acid units has about two thirds of its carboxyl groups bonded to hydroxyapatite vs. half of the carboxyl groups of pure PAA. It is therefore important to note that adhesion is not solely a result of ion bridging, but of interactions between the polyacrylate chains and the enamel surface of the tooth, displacing phosphate for example. In other words, adhesion is dynamic in nature with bond interchange since ion exchange is continually taking place between the oral fluids and the cement interface [8, 133].
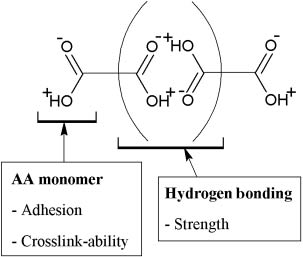
A schematic diagram illustrating the properties of the acrylic acid monomer in the copolymer and its ability to form hydrogen bonds resulting in effective adhesion.
Adhesion of CGPCs to surfaces improved through chemical treatment [8]. Although CGPCs can adhere to substrates chemically, surface treatment is of great importance since effective adhesion can only occur when an adhesive and a substrate are brought into molecular contact. Some work on this topic has been reported. Hotz [136] recommended pre-treatment with citric acid. Levine et al. [137] and Causton and Johnson [138] studied the use of mineralizing solutions. The use of citric acid was found unfavorable as, although it resulted in improved adhesion [136], it also opens up dentinal tubules and causes loss of the smear layer. An experimental study [8] assessed the effectiveness of adhesion of GPCs to substrates pre-treated with chemical reagents that are less aggressive than citric acid, which include PAA, ferric acid and tannic acid. Carboxylic acid hydroxyl containing PAA was found as one of the most effective conditioning solutions, for both enamel and dentin. The functional group of the PAA has the ability to form a multiplicity of hydrogen bonds to the substrate surface. These bonds promote wetting, cleansing and, probably, sorption of the conditioning agent by chelation. Enamel treated with 25% PAA resulted in a bond strength of 7.1 MPa, which was found significantly higher than that of the untreated enamel (3.2 MPa) [8]. Similar results were achieved for dentine. There was no significant modification in the morphology of the enamel surfaces. Although surface modification improves the cement adhesion to the substrate, the substrate surface must be treated with caution. Surface modifications could lead to the creation of air voids, which could act as foci for high stress, hence lowering the strength of the adhesive joint [8].
GPCs have been used in dentistry for over 40 years and have been considered as promising biomaterials in orthopedics. GPCs have been reported to have great potential for use as bone cements as an alternative to the conventional acrylic cements [139]. The importance of the adhesion property of CGPCs cannot be neglected; it plays an important role in both dental and orthopedic therapies. Hence, the following important factors must be considered for improving the GPCs’ adhesion properties [2, 140]:
Good initial wetting of the surfaces, achieved through a low contact angle, high surface energy and/or low viscosity.
Excellent intermolecular and interatomic forces between the adhesive and the substrate to avoid cohesive or adhesive failure in the substrate or within the adhesive; both may compromise the applicability of the material for clinical use.
The polymer chain molecules must possess sufficient mobility and be mutually soluble.
The material must have comparable strength to the tissue it replaces to prevent material failure.
The solubility of both the adhesive and the substrate must match to provide stronger interaction.
Air voids in cements must be avoided as they act as stress raisers and result in unsatisfactory adhesive strength attributed to the reduced ability of the adhesive to penetrate into the substrate surface.
4.4 Ion release
The physical properties of CGPCs are dominated by the polymer matrix, with the residual glass particles simply acting as a filler [21]. Ion release takes part in the formation of the cement matrix and contributes to the therapeutic activity, giving these materials the potential to be used for various clinical applications. The fluoro-alumino-silicate glass-based CGPCs are known for their sustained release of clinically beneficial amounts of fluoride [51, 141, 142], as shown by Wilson et al. [143], who found that the release of fluoride continued for at least 18 months. Fluoride plays an important biological role, particularly in dentistry, and has the effect of improving the resistance of the tooth material to acid attack, decreasing demineralization and increasing remineralization, inhibiting dental decays, and making the cement translucent [144–146]. Although the therapeutic activity of CGPCs depends mainly on the glass phase [144, 147–150], the polymer phase plays an important role in attacking the glass cations and releasing them or complexing them within its network. PAA or copolymers of acrylic and itaconic or maleic acid have been the most commonly used polymers in the preparation of GPCs. Towler et al. [151] showed that increasing the concentration of PAA minimizes ion release from GPCs. They also showed that agitation (sample rotation at 1000 rounds per minute) or aging samples at higher temperature (70°C) significantly increases the ion release of Zn2+ ions from the cement matrix into the medium, when compared to those aged at 37°C in static conditions. Wren et al. [152] investigated titanium (Ti)-containing glasses and found that the immersion of GPCs based on such glasses into distilled water resulted in an increased surface area after 1 day which then decreased over the next 29 days of incubation. The increase in the surface area was attributed to the dissolution of PAA within the cement matrix (hydration processes) resulting in open porosity and thus liberating ions bound within the cement structure, which then sought anionic sites. The formation of the siliceous hydrogel, during the hydration processes of GPCs, might be another reason for the increased surface area observed in their study [2]. The release of ions was also influenced by the cross-linked PAA matrix. Studies [152, 153] on Ti-containing glasses reported that there was no Ti4+ release. Shen et al. [153] showed that sodium was dissolved at higher rates than calcium and strontium. These results were attributed to the anion-cation reactions, suggesting that Ca2+ and Sr2+ are complexed more strongly by the PAA matrix than Na2+ ions. Ti, on the other hand, is expected to be complexed by the PAA at higher rates than those of the Na, Sr and Ca. Further, these results illustrate that the PAA matrix continues its degradation, post-setting and while immersed into water, resulting in selective retention of cations. This is in agreement with the literature [154], where it was indicated that PAA readily complexes alkali metal cations. A study [141] on fluoride release showed that acidity of the PAA component of a CGPC and/or the powder:liquid mixing ratio may affect the rate of fluoride release from CGPCs. Xu and Burgess [155] proposed a mathematical equation [Eq. (6)] to model fluoride release from both conventional and resin-modified glass ionomers,
where [F]c is the cumulative fluoride concentration, [F]I is the initial fluoride concentration, t is the time, and b and β are the mathematically derived constants.
4.5 Acid erosion and clinical durability
Clinical durability of a CGPC can be defined as the ability of the cement matrix to withstand long-term clinical use and resist failure [2]. Clinical durability depends on the resistance to acid erosion. CGPCs have generally shown failure rates between 20% and 30% after 2 years [156–159]. However, a clinical study by Mount [160] showed a failure rate of only 2% over 7 years. The significant difference in the failure rate can be attributed to the fact that the extent of acid erosion varies inversely with the time allowed for the cement to set prior to exposure [161]. It was reported [2] that GPCs based on copolymers of acrylic and maleic acids are less durable than those based on PAA, suggesting that the extent of erosion depends on the type of the polyelectrolyte used. Crisp et al. [52] performed a chemical study of the erosion of a GPC under acid attack. They found that the chief species eluted were sodium and fluoride ions and silicic acid, suggesting that the polyacid attack occurred mainly on the glass particles rather than on the matrix. They [52] also reported that GPCs begin to erode at pH=4.0; however, a study by Wilson et al. [162] showed that one brand of GPCs did not erode at all at this pH. In general, the susceptibility of GPCs to acid erosion is low even when pH is 2.7 [2].
5 Summary
PAA has been the most commonly used acid for the preparation of GPCs. This review has critically summarized and evaluated the role of PAA in the performance of CGPCs. The authors suggest that this critical review is crucial for dental material scientists for building proper understanding of the chemistry and properties of the PAA component in GPCs, and hence, facilitating the development of new cements that may overcome various disadvantages of the commercial GPCs currently used in the clinic.
The current review has shown that PAA, due to its ability to neutralize and ionize in the presence of water, initiates the GPC forming and setting/hardening reactions. This review has also shown that the molar mass, molecular weight, concentration and PDI of the PAA and the powder:liquid ratio of the GPC system were found to have a similar effect on the mechanical and rheological properties of the cement being investigated. Increasing any of these factors would increase the strength of the cement; however, the rheological properties of the material are shortened, representing a challenge in this field. The use of additives such as tartaric acid improves the GPC rheological properties by increasing the workability of the cement. PAA contributes to the biocompatibility of the CGPC system and controls its adhesion to the substrate. It was also found that ion release is restricted as the molecular weight, molar mass or concentration of the PAA increases.
Acknowledgments
The authors would like to thank the Atlantic Innovation Fund, Canada (Grant No. 57-02-03-1035).
References
[1] Kent B, Wilson A. Br. Dent. J. 1973, 135, 322–326.10.1038/sj.bdj.4803083Suche in Google Scholar
[2] Wilson A, Nicholson J. Acid-Base Cements: Their Biomedical and Industrial Applications, Cambridge University Press: Cambridge, 1993.10.1017/CBO9780511524813Suche in Google Scholar
[3] Culbertson B. Prog. Polym. Sci. 2001, 26, 577–604.10.1016/S0079-6700(01)00006-5Suche in Google Scholar
[4] Wilson A, Kent B. J. Dent. Res. 1970, 49, 7–13.10.1177/00220345700490013301Suche in Google Scholar
[5] Kenny S, Buggy M. J. Mater. Sci. Mater. Med. 2003, 14, 923–938.10.1023/A:1026394530192Suche in Google Scholar
[6] Alhalawani A, Curran D, Pingguan-Murphy B, Boyd D, Towler M. J. Funct. Biomater. 2013, 4, 329–357.10.3390/jfb4040329Suche in Google Scholar
[7] Moshaverinia A, Roohpour N, Chee W, Schricker S. J. Mater. Chem. 2012, 22, 2824–2833.10.1039/c2jm14880cSuche in Google Scholar
[8] Powis D, Follerås T, Merson S, Wilson A. J. Dent. Res. 1982, 61, 1416–1422.10.1177/00220345820610120801Suche in Google Scholar
[9] Nicholson J, Braybrook J, Wasson E. J. Biomater. Sci. Polym. Ed. 1991, 2, 277–285.10.1163/156856291X00179Suche in Google Scholar
[10] Peltola M, Suopää J, Aitasalo K, Varpula M, Yli-Urpo A, Happonen R. Head Neck 1998, 20, 315–319.10.1002/(SICI)1097-0347(199807)20:4<315::AID-HED6>3.0.CO;2-1Suche in Google Scholar
[11] Cancian D, Hochuli-Vieira E, Marcantonio R, Garcia RJ. Int. J. Oral Maxillofac. Implants 2004, 1, 73–79.Suche in Google Scholar
[12] Snik A, Cremers C. Clin. Otolaryngol. 2004, 29, 5–9.10.1111/j.1365-2273.2004.00749.xSuche in Google Scholar
[13] Babu S, Seidman M. Otol. Neurotol. 2004, 25, 98–101.10.1097/00129492-200403000-00003Suche in Google Scholar
[14] Ellis J, Anstice M, Wilson A. Clin. Mater. 1991, 7, 341–346.10.1016/0267-6605(91)90079-USuche in Google Scholar
[15] Moshaverinia A, Roohpour N, Chee W, Schricker S. J. Mater. Chem. 2011, 21, 1319–1328.10.1039/C0JM02309DSuche in Google Scholar
[16] Bertolini M, Palma-Dibb R, Zaghete M, Gimenes R. J. Non. Cryst. Solids 2005, 351, 466–471.10.1016/j.jnoncrysol.2005.01.040Suche in Google Scholar
[17] Crisp S, Wilson A. J. Dent. Res. 1974, 53, 1408–1413.10.1177/00220345740530061901Suche in Google Scholar
[18] Fennell B, Hill R, Akinmade A. Dent. Mater. 1998, 14, 358–364.10.1016/S0109-5641(99)00006-8Suche in Google Scholar
[19] Darling M, Hill R. Biomaterials 1994, 15, 299–306.10.1016/0142-9612(94)90055-8Suche in Google Scholar
[20] Crisp S, Lewis BG, Wilson AD. J. Dent. 1977, 5, 51–56.10.1016/S0300-5712(77)80025-0Suche in Google Scholar
[21] Wilson A, Hill R, Warren C, Lewis B. J. Dent. Res. 1989, 68, 89–94.10.1177/00220345890680021401Suche in Google Scholar PubMed
[22] Wilson AD, Crisp S, Abel G. J. Dent. 1977, 5, 117–120.10.1016/0300-5712(77)90070-7Suche in Google Scholar
[23] Dowling A, Fleming G. Dent. Mater. 2011, 27, 535–543.10.1016/j.dental.2011.02.003Suche in Google Scholar
[24] Crisp S, Lewis BG, Wilson AD. J. Dent. 1976, 4, 287–290.10.1016/S0300-5712(76)80008-5Suche in Google Scholar
[25] Mitsuhashi A, Hanaoka K, Teranaka T. Dent. Mater. 2003, 19, 747–757.10.1016/S0109-5641(03)00022-8Suche in Google Scholar
[26] Fleming G, Farooq A, Barralet J. Biomaterials 2003, 24, 4173–4179.10.1016/S0142-9612(03)00301-6Suche in Google Scholar
[27] Orwoll R, Chong Y. In Polymer Data Handbook, Mark J, Ed., Oxford University Press: New York, 1999, pp. 252–253.Suche in Google Scholar
[28] Wilson A, Kent B. Appl. Chem. Biotechnol. 1971, 21, 313.10.1002/jctb.5020211101Suche in Google Scholar
[29] Kent B, Wilson A. J. Dent. Res. 1969, 48, 412–418.10.1177/00220345690480031401Suche in Google Scholar PubMed
[30] Hill R, Wilson A, Warrens C. J. Mater. Sci. 1989, 24, 363–371.10.1007/BF00660982Suche in Google Scholar
[31] Griffin S, Hill R. J. Mater. Sci. 1998, 33, 5383–5396.10.1023/A:1004498217028Suche in Google Scholar
[32] Fennell B, Hill R. J. Mater. Sci. 2001, 36, 5193–5202.10.1023/A:1012445928805Suche in Google Scholar
[33] Fennell B, Hill R. J. Mater. Sci. 2001, 36, 5185–5192.10.1023/A:1012493811967Suche in Google Scholar
[34] Wasson E, Nicholson J. Clin. Mater. 1991, 7, 289–293.10.1016/0267-6605(91)90072-NSuche in Google Scholar
[35] Wasson E, Nicholson J. J. Dent. Res. 1993, 72, 481–483.10.1177/00220345930720020201Suche in Google Scholar
[36] Flory P. Principles of Polymer Chemistry, Cornell University Press: New York, 1953.Suche in Google Scholar
[37] King W, Murphy W. Polym. Chem. 2011, 2, 476–491.10.1039/C0PY00244ESuche in Google Scholar
[38] Morawetz H. Macromolecules in Solution, Interscience: New York, 1965.Suche in Google Scholar
[39] Wilson A, Crisp S, Ferner A. J. Dent. Res. 1976, 55, 489–495.10.1177/00220345760550033101Suche in Google Scholar
[40] Nicholson J. Biomaterials 1998, 19, 485–494.10.1016/S0142-9612(97)00128-2Suche in Google Scholar
[41] Gregor H, Luttinger L, Lobel E. J. Phys. Chem. 1955, 59, 34–39.10.1021/j150523a011Suche in Google Scholar
[42] Tanner D, Rushe N, Towler M. J. Mater. Sci. Mater. Med. 2006, 17, 313–318.10.1007/s10856-006-8229-7Suche in Google Scholar PubMed
[43] Hanting C, Hanxing L, Guoqing Z. J. Wuhan. Univ. Technol. Mater. Sci. Ed. 2005, 20, 110–112.10.1007/BF02841298Suche in Google Scholar
[44] Crisp S, Wilson A. J. Dent. Res. 1974, 53, 1420–1424.10.1177/00220345740530062101Suche in Google Scholar PubMed
[45] Matsuya S, Maeda T, Ohta M. J. Dent. Res. 1996, 75, 1920–1927.10.1177/00220345960750120201Suche in Google Scholar
[46] Crisp S, Lewis B, Wilson A. J. Dent. 1976, 4, 162–166.10.1016/0300-5712(76)90025-7Suche in Google Scholar
[47] Cattani-Lorente M, Godin C, Meyer J. Dent. Mater. 1994, 10, 37–44.10.1016/0109-5641(94)90020-5Suche in Google Scholar
[48] Hatton P, Brook I. Br. Dent. J. 1992, 173, 275–277.10.1038/sj.bdj.4808026Suche in Google Scholar
[49] Pearson G, Atkinson A. Biomaterials 1991, 12, 658–660.10.1016/0142-9612(91)90113-OSuche in Google Scholar
[50] Williams J, Billington R. J. Oral Rehabil. 1991, 18, 163–168.10.1111/j.1365-2842.1991.tb00044.xSuche in Google Scholar
[51] Causton B. Biomaterials 1981, 2, 112–115.10.1016/0142-9612(81)90008-9Suche in Google Scholar
[52] Crisp S, Lewis B, Wilson A. J. Dent. 1980, 8, 68–74.10.1016/S0300-5712(80)80046-7Suche in Google Scholar
[53] Hill R. J. Mater. Sci. Lett. 1989, 8, 1043–1047.10.1007/BF01730481Suche in Google Scholar
[54] Berry J. J. Polym. Sci. 1961, 50, 313–321.10.1002/pol.1961.1205015405Suche in Google Scholar
[55] Doi M, Edwards S. The Theory of Polymer Dynamics, Oxford University Press: New York, 1986.Suche in Google Scholar
[56] Edwards S. Proc. Phys. Soc. London 1967, 92, 9–16.10.1088/0370-1328/92/1/303Suche in Google Scholar
[57] de Gennes P. Scaling Concepts in Polymer Physics, Cornell University Press: New York, 1979.Suche in Google Scholar
[58] Prentice P. Polymer 1983, 24, 344–350.10.1016/0032-3861(83)90275-6Suche in Google Scholar
[59] Prentice P. J. Mater. Sci. 1985, 20, 1445–1454.10.1007/BF01026342Suche in Google Scholar
[60] Leygue A, Beris A, Keunings R. J. Non-Newtonian Fluid 2001, 101, 95–111.10.1016/S0377-0257(01)00143-4Suche in Google Scholar
[61] Yan L, Guo B, Xu J, Xie X. Polymers 2006, 47, 3696–3704.10.1016/j.polymer.2006.03.032Suche in Google Scholar
[62] Fennell B, Hill R. J. Mater. Sci. 2001, 36, 5177–5183.10.1023/A:1012441727897Suche in Google Scholar
[63] Milne K, Calos N, O’Donnell J, Kennard C, Vega S, Marks D. J. Mater. Sci. Mater. Med. 1997, 8, 349–356.10.1023/A:1018576715479Suche in Google Scholar
[64] Wilson A. J. Mater. Sci. Lett. 1996, 15, 275–276.10.1007/BF00274473Suche in Google Scholar
[65] Hill R. J. Mater. Sci. 1993, 28, 3851–3858.10.1007/BF00353190Suche in Google Scholar
[66] Sullivan A, Hill R. J. Mater. Sci. 2000, 35, 1125–1134.10.1023/A:1004763815097Suche in Google Scholar
[67] Boyd D, Towler M. J. Mater. Sci. Mater. Med. 2005, 16, 843–850.10.1007/s10856-005-3578-1Suche in Google Scholar PubMed
[68] Dowling A, Fleming G. Dent. Mater. 2011, 27, 1170–1179.10.1016/j.dental.2011.08.398Suche in Google Scholar PubMed
[69] Majekodunmi A, Deb S, Nicholson J. J. Mater. Sci. Mater. Med. 2003, 14, 747–752.10.1023/A:1025028119787Suche in Google Scholar
[70] Majekodunmi A, Deb S. J. Mater. Sci. Mater. Med. 2007, 18, 1883–1888.10.1007/s10856-007-3026-5Suche in Google Scholar PubMed
[71] Gomes F, Pires R, Reis R. Mater. Sci. Eng. C 2013, 33, 1361–1370.10.1016/j.msec.2012.12.037Suche in Google Scholar PubMed
[72] Kaufman H, Falcetta J. Introduction to Polymer Science and Technology, John Wiley & Sons: New York, 1977.Suche in Google Scholar
[73] Prosser H, Powis D, Wilson A. J. Dent. Res. 1986, 65, 146–148.10.1177/00220345860650021101Suche in Google Scholar PubMed
[74] Martin J, Johnson J, Cooper A. Polym. Rev. 1972, 8, 57–199.10.1080/15321797208068169Suche in Google Scholar
[75] de Gennes P. Phys. Today 1983, 36, 33–39.10.1063/1.2915700Suche in Google Scholar
[76] Darvel B. Materials Science for Dentistry, Woodhead Publishing: Cambridge, UK, 2009.10.1533/9781845696672Suche in Google Scholar
[77] Abdiyev K, Shaikhutdinov E, Zhursumbaeva M, Khussain S. Colloid. J. 2003, 65, 399–402.10.1023/A:1025105631381Suche in Google Scholar
[78] van Noort R. Introduction to Dental Materials, Mosby: London, UK, 2014.Suche in Google Scholar
[79] de Barra E, Hill R. J. Mater. Sci. 1998, 33, 5487–5497.10.1023/A:1004479108367Suche in Google Scholar
[80] Guggenberger R, May R, Stefan K. Biomaterials 1998, 19, 479–483.10.1016/S0142-9612(97)00127-0Suche in Google Scholar
[81] Akay M. Introduction to Polymer Science and Technology, Bookboon Ventus Publishing: Denmark, 2012.Suche in Google Scholar
[82] Xie D, Feng D, Chung I-, Eberhardt AW. Biomaterials 2003, 24, 2749–2757.10.1016/S0142-9612(03)00090-5Suche in Google Scholar
[83] Yap A, Mudambi S, Chew C, Neo J. Oper. Dent. 2001, 26, 295–301.Suche in Google Scholar
[84] Neve A, Piddock V, Combe E. Clin. Mater. 1992, 9, 7–12.10.1016/0267-6605(92)90004-DSuche in Google Scholar
[85] Crisp S, Wilson A. J. Dent. Res. 1976, 55, 1023–1031.10.1177/00220345760550060401Suche in Google Scholar
[86] Crisp S, Lewis BG, Wilson AD. J. Dent. 1979, 7, 304–312.10.1016/0300-5712(79)90143-XSuche in Google Scholar
[87] Prosser H, Richards C, Wilson A. J. Biomed. Mater. Res. 1982, 16, 431–445.10.1002/jbm.820160411Suche in Google Scholar
[88] Prentice L, Tyas M, Burrow M. Dent. Mater. 2006, 22, 94–97.10.1016/j.dental.2005.04.004Suche in Google Scholar
[89] Kao E, Culbertson B, Xie D. Dent. Mater. 1996, 12, 44–51.10.1016/S0109-5641(96)80063-7Suche in Google Scholar
[90] Rivas B, Seguel G. Polyhedron 1999, 18, 2511–2518.10.1016/S0277-5387(99)00149-7Suche in Google Scholar
[91] Rodríguez E, Katime I. Macromol. Mater. Eng. 2003, 288, 607–612.10.1002/mame.200350006Suche in Google Scholar
[92] Prentice L, Tyas M. Acta Biomater. 2006, 2, 109–112.10.1016/j.actbio.2005.08.007Suche in Google Scholar
[93] Williams D. Proceedings of a Consensus Conference of the European Society for Biomaterials, Chester, England, 3–5 March 1986. Elsevier: New York, 1987.Suche in Google Scholar
[94] Williams D. Biomaterials 2008, 29, 2941–2953.10.1016/j.biomaterials.2008.04.023Suche in Google Scholar
[95] Brodbeck W, Patel J, Voskerician G, Christenson E, Shive M, Nakayama Y, Matsuda T, Ziats N, Anderson J. Proc. Natl Acad. Sci. USA 2002, 99, 10287–10292.10.1073/pnas.162124199Suche in Google Scholar
[96] de Giglio E, Cafagna D, Ricci M, Sabbatini L. J. Bioact. Compat. Pol. 2010, 25, 374–391.10.1177/0883911510372290Suche in Google Scholar
[97] de Giglio E, Cometa S, Cioffi N, Torsi L, Sabbatini L. Anal. Bioanal. Chem. 2007, 389, 2055–2063.10.1007/s00216-007-1299-7Suche in Google Scholar
[98] Schmalz G, Arenholt Bindslev D. Biocompatibility of Dental Materials, Springer-Verlag: Berlin, 2009.Suche in Google Scholar
[99] Mjör I. Adv. Dent. Res. 1992, 6, 7–16.10.1177/08959374920060012001Suche in Google Scholar
[100] Caughman W, Caughman G, Dominy W, Schuster G. J. Prosthet. Dent. 1990, 63, 513–521.10.1016/0022-3913(90)90067-MSuche in Google Scholar
[101] Geurtsen W, Spahl W, Leyhausen G. J. Dent. Res. 1998, 77, 2012–2019.10.1177/00220345980770121001Suche in Google Scholar
[102] Müller J, Bruckner G, Kraft E, Hörz W. Dent. Mater. 1990, 6, 172–177.10.1016/0109-5641(90)90024-9Suche in Google Scholar
[103] Peltola M, Salo T, Oikarinen K. Endod. Dent. Traumatol. 1992, 8, 120–124.10.1111/j.1600-9657.1992.tb00448.xSuche in Google Scholar PubMed
[104] Schmalz G, Schmalz C, Rotgans J. Dtsch. Zahnärztl. Z 1986, 41, 806–812.10.1037/0003-066X.41.7.806Suche in Google Scholar
[105] Schmalz G, Thonemann B, Riedel M, Elderton R. Dent. Mater. 1994, 10, 4–13.10.1016/0109-5641(94)90038-8Suche in Google Scholar
[106] Tobias R, Browne R, Wilson C. Int. Dent. Res. 1985, 18, 161–167.10.1111/j.1365-2591.1985.tb00435.xSuche in Google Scholar
[107] Tyas M. J. Dent. Res. 1977, 56, 1285.10.1177/00220345770560103401Suche in Google Scholar
[108] Meryon S, Browne R. Cell Biochem. Funct. 1984, 2, 43–48.10.1002/cbf.290020112Suche in Google Scholar
[109] Brook I, Craig G, Lamb D. Biomaterials 1991, 12, 179–186.10.1016/0142-9612(91)90197-ISuche in Google Scholar
[110] Kawahara H, Imanishi Y, Oshima H. J. Dent. Res. 1979, 58, 1080–1086.10.1177/00220345790580030901Suche in Google Scholar
[111] Nakamura M, Kawahara H, Imia K, Tomoda S, Kawata Y, Hikari S. Dent. Mater. 1983, 1, 100–101.10.4012/dmj.2.100Suche in Google Scholar
[112] Tam L, Pulver E, McComb D, Smith D. Dent. Mater. 1989, 5, 145–149.10.1016/0109-5641(89)90001-8Suche in Google Scholar
[113] De Schepper E, White R, von der Lehr W. Am. Dent. J. 1989, 2, 51–56.Suche in Google Scholar
[114] Palenik C, Behnen M, Setcos J, Miller C. Dent. Mater. 1992, 8, 16–20.10.1016/0109-5641(92)90047-GSuche in Google Scholar
[115] Forsten L. Scand. J. Dent. Res. 1990, 98, 179–185.10.1111/j.1600-0722.1990.tb00958.xSuche in Google Scholar
[116] Hatibovic-Kofman S, Koch G. Swed. Dent. J. 1991, 15, 253–258.Suche in Google Scholar
[117] Kawase T, Suzuki A. Arch. Oral Biol. 1989, 34, 103–107.10.1016/0003-9969(89)90133-7Suche in Google Scholar
[118] Li Y, Noblitt T, Dunipace A, Oshima H. J. Dent. Res. 1990, 69, 1188–1192.10.1177/00220345900690051301Suche in Google Scholar PubMed
[119] Min K, Kim H, Park H, Pi S, Hong C, Kim E. J. Endod. 2007, 33, 163–166.10.1016/j.joen.2006.07.022Suche in Google Scholar
[120] Six N, Lasfargues J, Goldberg M. J. Dent. 2000, 28, 413–422.10.1016/S0300-5712(00)00015-4Suche in Google Scholar
[121] Steinbrunner R, Setcos J, Kafrawy A. Am. J. Dent. 1991, 4, 281–284.Suche in Google Scholar
[122] Paterson R, Watts A. Br. Dent. J. 1981, 151, 228–230.10.1038/sj.bdj.4804668Suche in Google Scholar
[123] Council on Dental Materials, Instruments and Equipment. J. Am. Dent. Assoc. 1984, 109, 476.Suche in Google Scholar
[124] Andersson O, Dahl J. Biomaterials 1994, 15, 882–888.10.1016/0142-9612(94)90111-2Suche in Google Scholar
[125] Smith D, Ruse N. J. Am. Dent. Assoc. 1986, 112, 654–657.10.14219/jada.archive.1986.0069Suche in Google Scholar
[126] Sasanaluckit P, Albustany K, Doherty P, Williams D. Biomaterials 1993, 14, 906–916.10.1016/0142-9612(93)90132-LSuche in Google Scholar
[127] Hatton P, Hurrell-Gillingham K, Brook I. J. Dent. 2006, 34, 598–601.10.1016/j.jdent.2004.10.027Suche in Google Scholar
[128] Lacefield W, Reindl M, Retied D. J. Prosthet. Dent. 1985, 53, 194–198.10.1016/0022-3913(85)90108-8Suche in Google Scholar
[129] Smith D. Br. Dent. J. 1968, 125, 381–384.Suche in Google Scholar
[130] Beech D. Arch. Oral Biol. 1972, 17, 907–911.10.1016/0003-9969(72)90035-0Suche in Google Scholar
[131] Wilson A. Br. Polym. J. 1974, 6, 165–179.10.1002/pi.4980060303Suche in Google Scholar
[132] Belton D, Stupp S. Biomedical and Dental Applications of Polymers, Springer: New York, 1980, pp. 427–439.10.1007/978-1-4757-9510-3_32Suche in Google Scholar
[133] Wilson A, Prosser H, Powis D. J. Dent. Res. 1983, 62, 590–592.10.1177/00220345830620051801Suche in Google Scholar
[134] van Meerbeek B, Yoshida Y, Inoue S, de Munck J, van Landuyt K, Lambrechts P. J. Dent. 2006, 34, 614–622.10.1016/j.jdent.2006.01.012Suche in Google Scholar
[135] Yoshida Y, Van Meerbeek B, Nakayama Y, Snauwaert J, Hellemans L, Lambrechts P, Vanherle G, Wakasa K. J. Dent. Res. 2000, 79, 709–714.10.1177/00220345000790020301Suche in Google Scholar
[136] Hotz P, McLean J, Sced I, Wilson A. Br. Dent. J. 1977, 142, 41–47.10.1038/sj.bdj.4803864Suche in Google Scholar
[137] Levine R, Beech D, Garton B. Br. Dent. J. 1977, 143, 275–277.10.1038/sj.bdj.4803990Suche in Google Scholar
[138] Causton B, Johnson N. J. Dent. Res. 1979, 58, 1383–1393.10.1177/00220345790580041401Suche in Google Scholar
[139] Wood D, Hill R. Biomaterials 1991, 12, 164–170.10.1016/0142-9612(91)90195-GSuche in Google Scholar
[140] Akinmade A, Nicholson J. J. Mater. Sci. Mater. Med. 1993, 4, 95–101.10.1007/BF00120376Suche in Google Scholar
[141] Cranfield M, Kuhn A, Winter G. J. Dent. 1982, 10, 333–341.10.1016/0300-5712(82)90028-8Suche in Google Scholar
[142] Forsten L. Scand. J. Dent. Res. 1977, 85, 503–504.10.1111/j.1600-0722.1977.tb00586.xSuche in Google Scholar
[143] Wilson A, Groffman D, Kuhn A. Biomaterials 1985, 6, 431–433.10.1016/0142-9612(85)90107-3Suche in Google Scholar
[144] Horowitz H. Community Dent. Oral Epidemiol. 1973, 1, 104–114.10.1111/j.1600-0528.1973.tb01326.xSuche in Google Scholar
[145] Maldonado A, Swartz M, Phillips R. J. Am. Dent. Assoc. 1978, 96, 785–791.10.14219/jada.archive.1978.0195Suche in Google Scholar
[146] Retief D, Bradley E, Denton J, Switzer P. Caries Res. 1984, 18, 250–257.10.1159/000260773Suche in Google Scholar
[147] Yamaguchi M, Oishi H, Suketa Y. Biochem. Pharmacol. 1987, 36, 4007–4012.10.1016/0006-2952(87)90471-0Suche in Google Scholar
[148] Ito A, Kawamurab H, Otsukac M, Ikeuchia M, Ohgushia H, Ishikawad K, Onumaa K, Kanzakia N, Sogoa Y, Ichinosee N. Mater. Sci. Eng. C 2002, 22, 21–25.10.1016/S0928-4931(02)00108-XSuche in Google Scholar
[149] Guida A, Towler M, Wall J. J. Mater. Sci. Lett. 2003, 22, 1401–1403.10.1023/A:1025794927195Suche in Google Scholar
[150] Coughlan A, Scanlon K, Mahon B, Towler M. Biomed. Mater. Eng. 2010, 20, 99–106.10.3233/BME-2010-0620Suche in Google Scholar
[151] Towler M, Kenny S, Boyd D, Pembroke T, Buggy M, Hill R. Biomed. Mater. Eng. 2004, 14, 565–572.Suche in Google Scholar
[152] Wren A, Cummins N, Laffir F, Hudson S, Towler M. J. Mater. Sci. Mater. Med. 2011, 22, 19–28.10.1007/s10856-010-4184-4Suche in Google Scholar PubMed
[153] Shen L, Coughlan A, Towler M, Hall M. J. Mater. Sci. Mater. Med. 2014, 25, 965–973.10.1007/s10856-014-5143-2Suche in Google Scholar PubMed
[154] Gregor H, Frederick M. J. Polym. Sci. 1957, 23, 451–465.10.1002/pol.1957.1202310338Suche in Google Scholar
[155] Xu X, Burgess J. Biomaterials 2003, 24, 2451–2461.10.1016/S0142-9612(02)00638-5Suche in Google Scholar
[156] Forsten L, Karjalainen S. Scand. J. Dent. Res. 1990, 98, 70–73.10.1111/j.1600-0722.1990.tb00942.xSuche in Google Scholar
[157] Welbury R, Walls A, Murray J, McCabe J. Br. Dent. J. 1991, 170, 177–181.10.1038/sj.bdj.4807465Suche in Google Scholar
[158] Holst A. Swed. Dent. J. 1996, 20, 209–214.10.1007/BF00043310Suche in Google Scholar
[159] Anderson-Wenckert I, Dijken van J, Stenberg R. J. Dent. Child 1995, 62, 197–200.Suche in Google Scholar
[160] Mount G. In: Clinical Dentistry, Clarke J, Ed., Harper & Row: Philadelphia, 1984.Suche in Google Scholar
[161] Walls A, McCabe J, Murray J. Br. Dent. J. 1988, 164, 141–144.10.1038/sj.bdj.4806382Suche in Google Scholar
[162] Wilson A, Groffman D, Powis D, Scott R. Biomaterials 1986, 7, 217–220.10.1016/0142-9612(86)90106-7Suche in Google Scholar
©2016 by De Gruyter
Artikel in diesem Heft
- Frontmatter
- Review
- The role of poly(acrylic acid) in conventional glass polyalkenoate cements
- Original articles
- Development of a polystyrene latex-based reagent for rheumatoid factor detection
- Biodegradation study of bio-corn flour filled low density polyethylene composites assessed by natural soil
- Efficacy of PU foam materials for scientific investigation in footwear research
- Degradation of PVDF-based composite membrane and its impacts on membrane intrinsic and separation properties
- Fabrication of cellulose fine fiber based membranes embedded with silver nanoparticles via Forcespinning
- Influence of wax content on the electrical, thermal and tribological behaviour of a polyamide 6/graphite composite
- In situ formation of copper nanoparticles in a p(NIPAM-VAA-AAm) terpolymer microgel that retains the swelling behavior of microgels
- Possible application of lead sulfide quantum dot in memory device
- Solution properties of polyaniline/carbon particle composites
- Preparation and characterization of SiO2/fluoroalkyl-trialkoxysilane/2-hydroxyethyl methacrylate/trimethylolpropane triacrylate and SiO2/3-(trimethoxysilyl)propyl methacrylate/ 2-hydroxyethyl methacrylate/trimethylolpropane triacrylate coatings on glass substrates using the sol-gel method
- The performance and morphology of PMMA/SAN/ABS blends
Artikel in diesem Heft
- Frontmatter
- Review
- The role of poly(acrylic acid) in conventional glass polyalkenoate cements
- Original articles
- Development of a polystyrene latex-based reagent for rheumatoid factor detection
- Biodegradation study of bio-corn flour filled low density polyethylene composites assessed by natural soil
- Efficacy of PU foam materials for scientific investigation in footwear research
- Degradation of PVDF-based composite membrane and its impacts on membrane intrinsic and separation properties
- Fabrication of cellulose fine fiber based membranes embedded with silver nanoparticles via Forcespinning
- Influence of wax content on the electrical, thermal and tribological behaviour of a polyamide 6/graphite composite
- In situ formation of copper nanoparticles in a p(NIPAM-VAA-AAm) terpolymer microgel that retains the swelling behavior of microgels
- Possible application of lead sulfide quantum dot in memory device
- Solution properties of polyaniline/carbon particle composites
- Preparation and characterization of SiO2/fluoroalkyl-trialkoxysilane/2-hydroxyethyl methacrylate/trimethylolpropane triacrylate and SiO2/3-(trimethoxysilyl)propyl methacrylate/ 2-hydroxyethyl methacrylate/trimethylolpropane triacrylate coatings on glass substrates using the sol-gel method
- The performance and morphology of PMMA/SAN/ABS blends