Chemically and Light-Driven Coordination-Induced Spin State Switching (CISSS) of a nonheme-iron complex
-
Kim Fischer


Abstract
The new Fe(II) complex [Fe(trident)(bmik)](ClO4)2 (1) (trident = bis(2-pyridylmethyl)benzylamine and bmik = bis(1-methylimidazole)ketone) exhibits a change of magnetic moment in dichloromethane (DCM) solution upon addition of pyridine which is attributed to the Coordination-Induced Spin State Switching effect (CISSS). By attaching a photoisomerizable azopyridine sidegroup to the tridentate ligand the complex [Fe(azpy-trident)(bmik)](ClO4)2 (2; azpy-trident = [N,N-bis(2-pyridylmethyl)]-3-(3-pyridylazo)benzylamine) is obtained. As detected by Evans NMR spectroscopy, 2 reversibly changes its magnetic moment in homogeneous solution upon photoirradiation which is attributed to intermolecular Light-Driven Coordination-Induced Spin State Switching (LD-CISSS). Further support for this interpretation is inferred from concentration-dependent Evans NMR measurements.
1 Introduction
Spin crossover denotes the transition between two spin states and is known for a range of transition metal complexes [1], [2], [3], [4], [5], [6]. It is accompanied by characteristic changes in structural, optical, and magnetic properties which renders such systems interesting for various applications in the fields of molecular electronics, data storage, and spintronics [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15]. Whereas most of the corresponding phenomena and switching mechanisms refer to the solid state [2, 5, 16], the investigation of spin-switchable systems in homogeneous solution has also become of significant interest in the recent years [6, 17], [18], [19], [20], [21], [22], [23], [24], [25]. An important means to switch between two stable spin states in solution is a change of the coordination number of the central ion (Coordination-Induced Spin State Switching [CISSS]) [17, 26]. While mostly nickel(II) systems have been investigated in this regard, the underlying effect is potentially applicable to a larger number of transition-metal ions, such as Fe(II), Fe(III), Mn(II), Mn(III), or Co(II). Ni(II) square-planar systems, e.g., are diamagnetic (low spin, S = 0) and may be converted through coordination of one or two axial ligands into paramagnetic, square-pyramidal or octahedral complexes exhibiting S = 1 ground states [17, 23], [24], [25], [26], [27], [28], [29].
The most straightforward way of inducing CISSS is to add ligands of sufficient binding affinity to a coordinatively unsaturated transition-metal complex in solution [17, 23, 24]. In order to make the ensuing spin switching process reversible, photoswitchable ligands may be employed whose isomers exhibit greatly different affinities to the metal center. Irradiation at different wavelengths generating one or the other isomer then leads to coordination/decoordination of the photoisomerizable ligand. In this context, the use of light is particularly attractive because it is associated with high selectivity, high temporal and spatial resolution and fast response. Application of photoswitchable ligands for coordination-induced spin switching in solution has been termed Light-Driven Coordination-Induced Spin State Switching (LD-CISSS) [18] by Herges et al. and implemented in two ways. On the one hand, these authors developed a Ni(II) porphyrin system that can be switched between low spin and high spin at room temperature in solution using photodissociable ligands (PDLs). Azopyridines and azoimidazoles were found to be suitable for that purpose, coordinating in their trans configuration to the Ni(II) porphyrin and decoordinating in their cis configuration due to steric hindrance [19, 30, 31]. On the other hand, the photoswitchable unit was also covalently linked to the porphyrin, leading to the so-called record player. In this case, the photoswitchable unit binds intramolecularly to the nickel center in its cis configuration and decoordinates in its trans configuration (Scheme 1, left). In the meantime, the record player could be significantly improved and optimized in many ways [18, 20, 32], [33], [34], [35], [36], [37], [38], but still represents the only known LD-CISSS system for spin switching in solution.
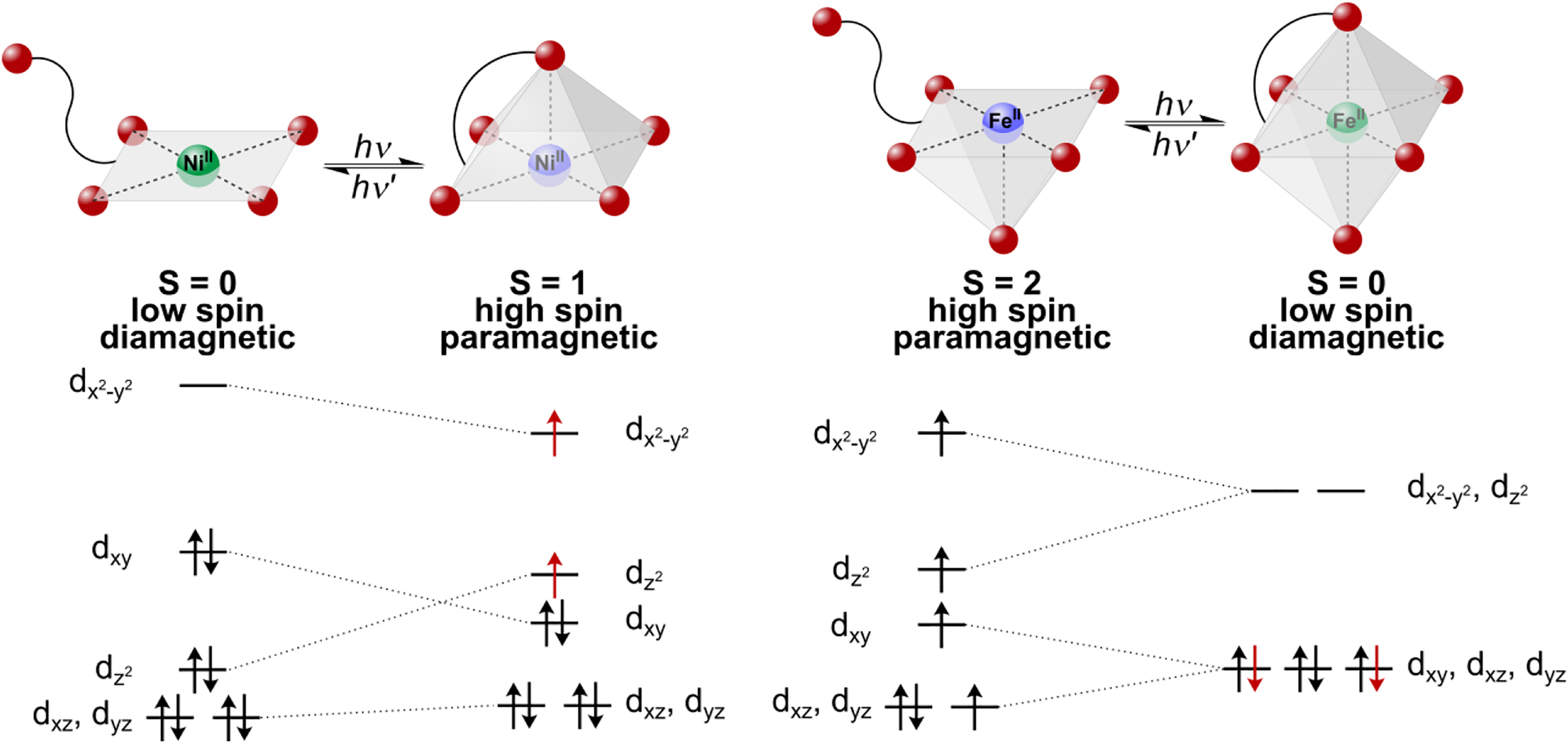
Principle of Light-Driven Coordination-Induced Spin State Switching (LD-CISSS) for nickel(II) (left) and iron(II) (right) and change of occupation of concerning orbitals.
Herein we present the first complex capable of LD-CISSS which is not based on a porphyrin platform and has an iron central atom. In the case of Fe(II), switching between a square-pyramidal, paramagnetic high spin complex (S = 2) and an octahedral, diamagnetic low spin complex (S = 0) is possible (Scheme 1, right), which corresponds to a larger change of spin as compared to Ni(II). For the square-pyramidal or octahedral environment of an Fe(II) LD-CISSS system, we decided to use a tridentate ligand being switchable to a tetradentate coordination as this is synthetically easier accessible than a pentadentate/hexadentate analog. In the former case, the octahedral coordination geometry is completed by a bidentate ligand. Notably, this strategy can also be used to finetune the ligand field strength. Following a rational approach to the synthesis of such a LD-CISSS system, it is meaningful to first prepare a complex suitable for (chemical) CISSS. If this effect is observed, the respective complex can then be furnished with a photoswitchable unit and examined with regard to LD-CISSS. Correspondingly, we have chosen to first synthesize complex 1 supported by the bis(2-pyridylmethyl)benzylamine (trident) ligand 3 and bis(1-methylimidazole)ketone (bmik, 4) as bidentate coligand as a CISSS model system.
Scheme 2 shows complex 1 in its three different possible isomers. There exist two possibilities for facial coordination (fac-1 and fac-2) and one possibility for meridional coordination (mer) of the tridentate ligand. Since the ligand-field stabilization energy is maximal if the strongest σ-donor is in axial position and the π-acceptor ligands are in equatorial positions, the fac-1 structure is assumed for complex 1. According to Scheme 3, the corresponding complex 1-py is formed when pyridine is added as an axial ligand. 1-py is assumed to be low spin (or close to low spin).
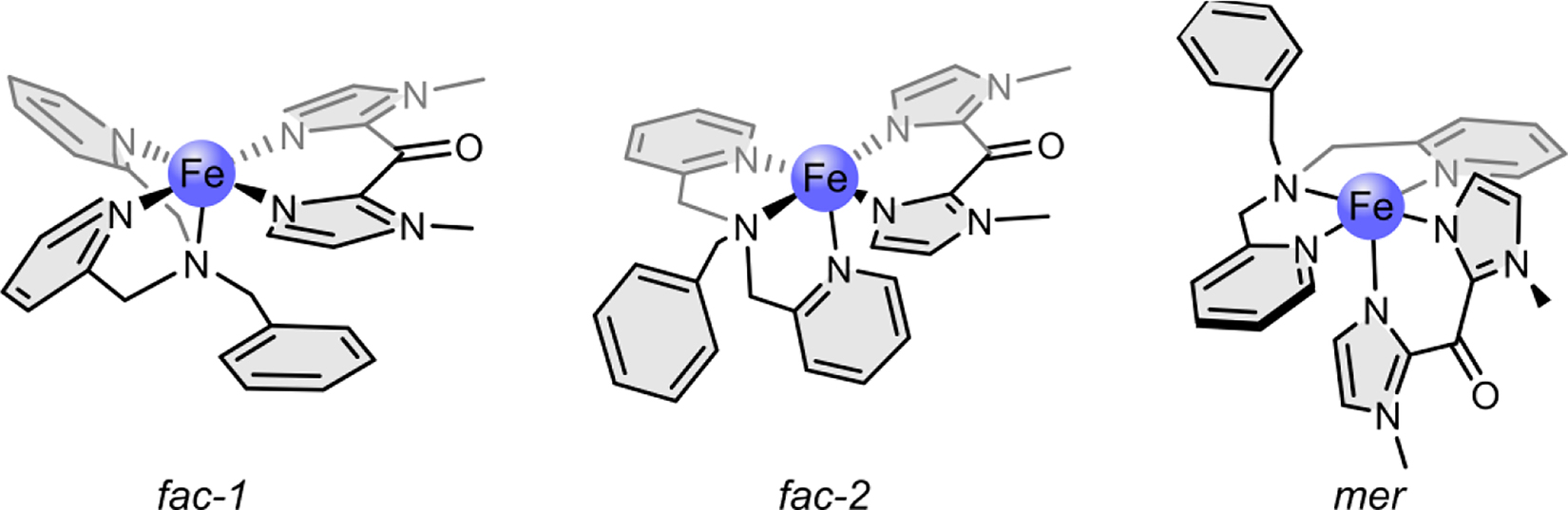
Complex 1 in its three possible isomers: fac-1, fac-2, and mer.
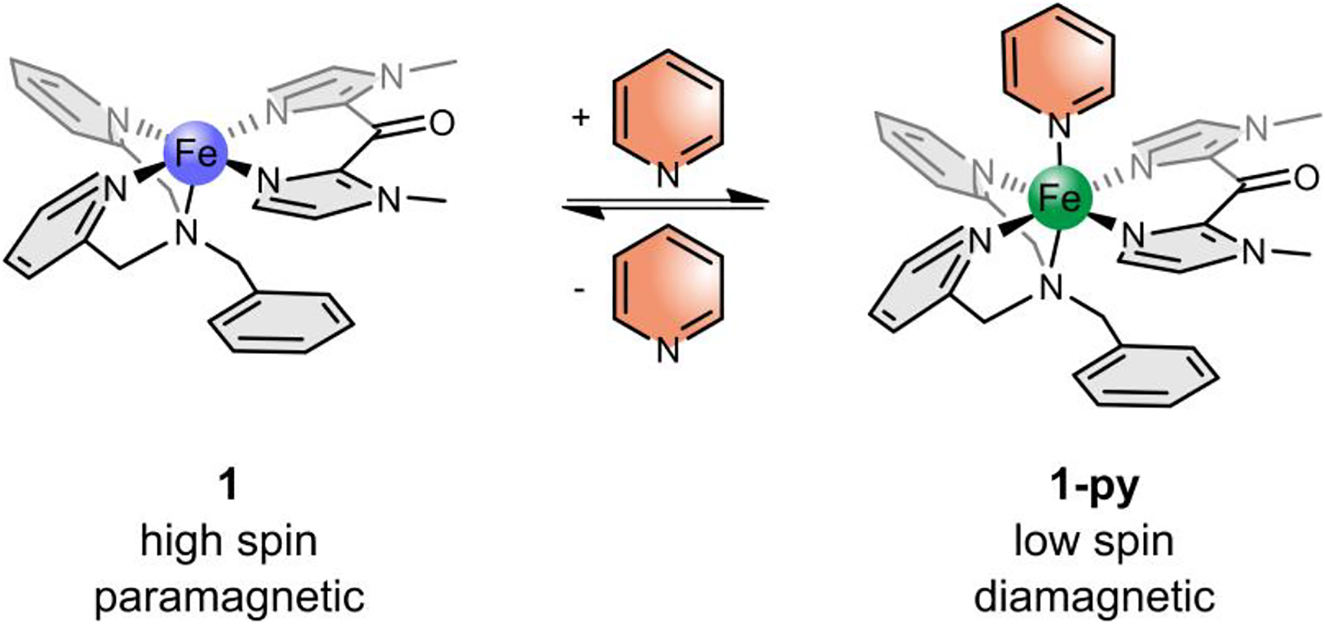
Coordination-Induced Spin State Switching (CISSS) complex 1 in its paramagnetic square-pyramidal isomer (left, 1) and its diamagnetic octahedral isomer (right, 1-py). By coordination or decoordination of pyridine the spin state changes.
Having investigated complex 1 with regard to CISSS in solution, the benzyl unit of the tridentate ligand is replaced by an azopyridine as photoswitchable unit in order to realize the analogous LD-CISSS system 2.
For this purpose [N,N-bis(2-pyridylmethyl)]-3-(3-pyri-dylazo)benzylamine (azpy-trident, 5) is prepared as photoswitchable tridentate/tetradentate ligand. As discussed below, a different coordination geometry of 5 as compared to that of 3 in complex 1 is attained in complex 2 (Scheme 4). If the attached pyridine is axially bound, 2 is assumed to be low spin or close to low spin. The synthesis, characterization and CISSS/LD-CISSS properties of complexes 1 and 2 are reported in the following sections.
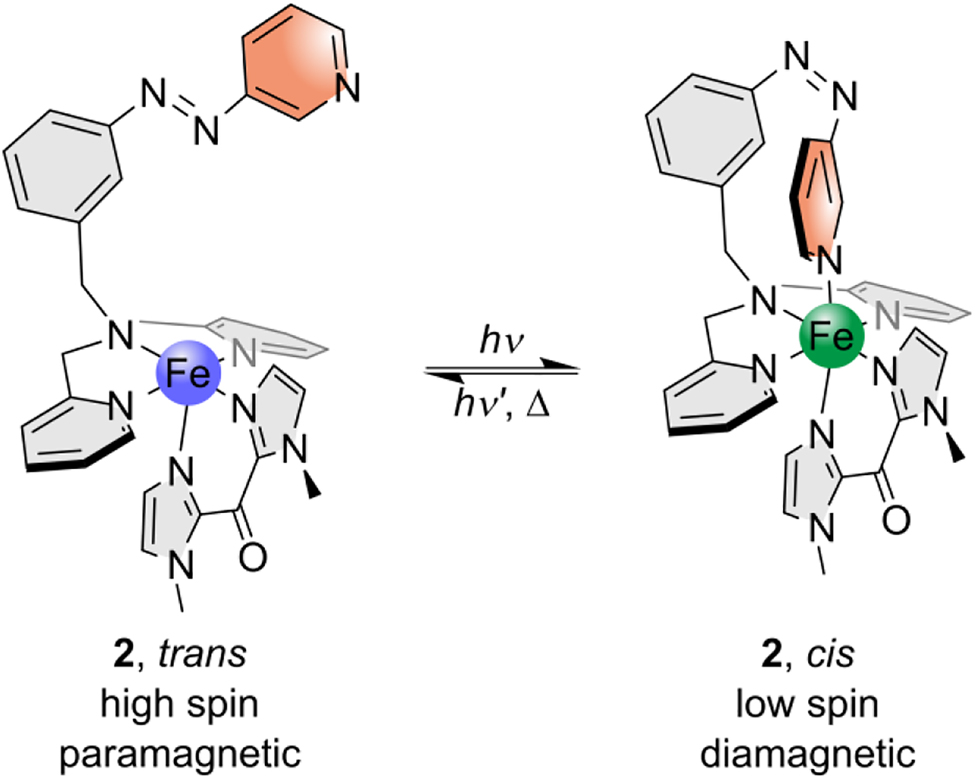
Target complex 2 in its paramagnetic square-pyramidal trans isomer (left) and its diamagnetic octahedral cis isomer (right). By irradiation with different wavelengths switching between these two isomers should be possible.
2 Results and discussion
2.1 Synthesis of complexes 1 and 2
To obtain the target CISSS complex 1 the general method of Scheme 5 was used. Iron(II) perchlorate hydrate and the literature-known tridentate ligand bis(2-pyridylmethyl)benzylamine (3) [39] were stirred for four days until addition of the bidentate ligand bis(1-methylimidazole)ketone (bmik, 4) [40].
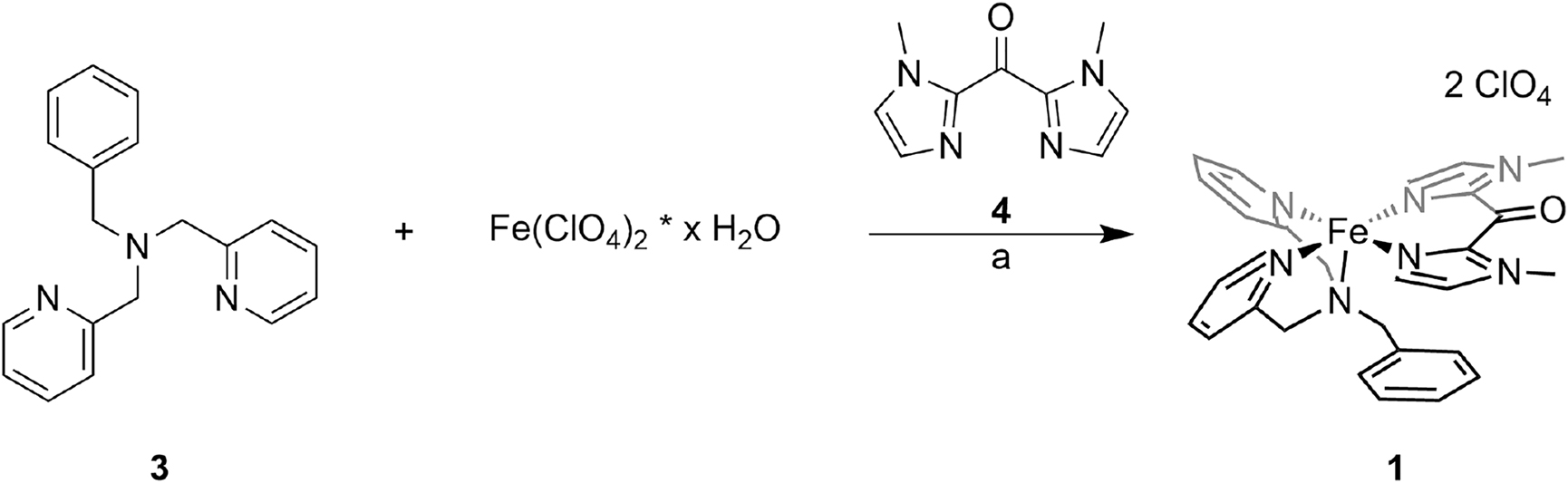
Synthesis of complex 1: (a) 1. MeOH, rt, four days, 2. 4, rt, 20 h.
Adding 4 to a solution of Fe(ClO4)2 * x H2O in MeOH immediately led to precipitation of the homoleptic bmik complex, which was separated from the reaction solution. The desired complex 1 could then be obtained with a yield of 51%.
A six-step synthetic route was developed to generate the azpy-trident ligand 5 (Scheme 6). We started with the literature-known synthesis of 3-nitrosobenzoic acid (6) [41]. Azopyridine carbonic acid 7 was synthesized via the Mills reaction under basic conditions. Esterification of 7 led to the azo ester 8, and subsequent reduction with lithium aluminum hydride gave the corresponding azo alcohol 9. Azo chloride 10 could be obtained by refluxing in thionyl chloride. It turned out that the azo chloride is very unstable and has to be converted further directly. For this reason, we only removed excess thionyl chloride in vacuo and then carried out an in situ Finkelstein reaction with the literature-known bis(2-pyridylmethyl)amine (11) [39]. The photoswitchable azpy-trident ligand 5 could be generated this way with moderate yields.
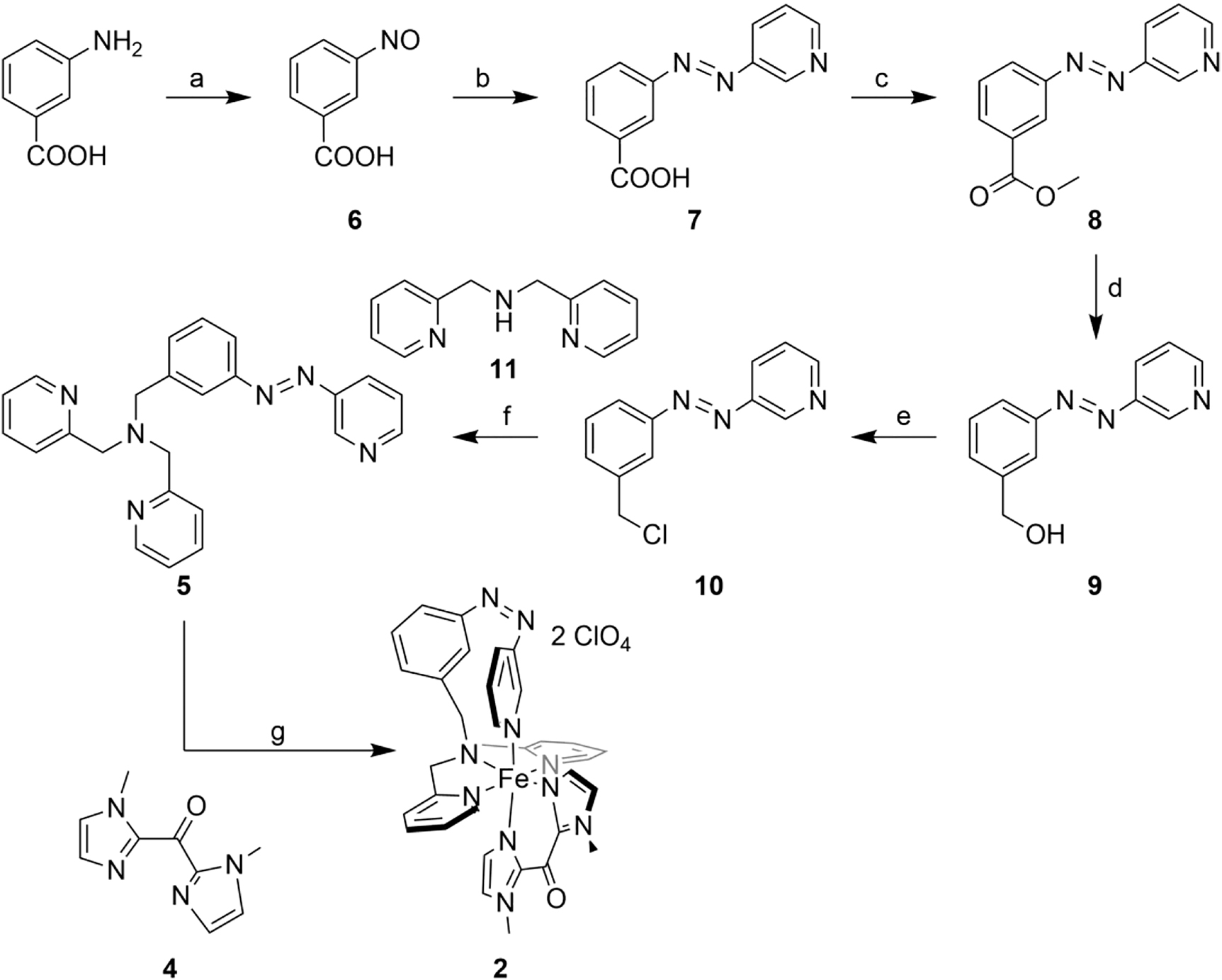
Synthesis of complex 2: (a) Oxone, DCM/H2O, rt, 4.5 h; (b) 3-aminopyridine, TMAH, pyridine, 1. 120 °C, 4.5 h, 2. rt, 18 h; (c) H2SO4, MeOH, reflux, 18 h; (d) 1 M LAH solution in THF, 1. 0 °C, 2. rt, two days; (e) thionyl chloride, 90 °C, 20 h; (f) Cs2CO3, TBAI, MeCN, 1. 90 °C, 24 h, 2. rt, three days; (g) 1. iron(II) perchlorate hydrate, MeOH, rt, 20 h, 365 nm, 2. 4, rt, 20 h.
The switching properties of azo compounds 5, 8, and 9 were investigated by UV/Vis and NMR spectroscopy (Supporting Information Figures S1–S3, S8–S10).
For the synthesis of complex 2, iron(II) perchlorate hydrate was first stirred with the azpy-trident ligand 5 for 20 h. In order to promote κ4-coordination of 5 (i.e., by the cis isomer), the reaction flask was irradiated with a wavelength of 365 nm. The bidentate ligand bmik 4 [40] was then added in a second step. Again, formation of the homoleptic bmik complex could not be avoided, such that always a mixture of both complexes was obtained. However, while the homoleptic complex precipitated at the bottom of the flask, the desired complex 2 crystallized at the top of the flask, allowing the two complexes to be separated manually. Moreover, due to irradiation with light with 365 nm during the reaction (see above) and a longer reaction time, the yield of the desired complex could be increased up to 31%.
We could not obtain single crystals for either complex 1 or complex 2 and therefore no scXRD data are available. It should therefore be mentioned that the structures given are based on those of related compounds and/or spectroscopic/physicochemical evidence.
2.2 Investigation of complex 1
Since the coordination of an axial ligand to 1 should lead to a diamagnetic octahedral complex (cf. Scheme 3), structural and electronic changes are to be expected. As a result, corresponding electronic absorption spectra should change as well. In order to obtain information on this issue, UV/Vis spectra of 1 were measured in both dichloromethane (DCM) and pyridine, as shown in Figure 1.
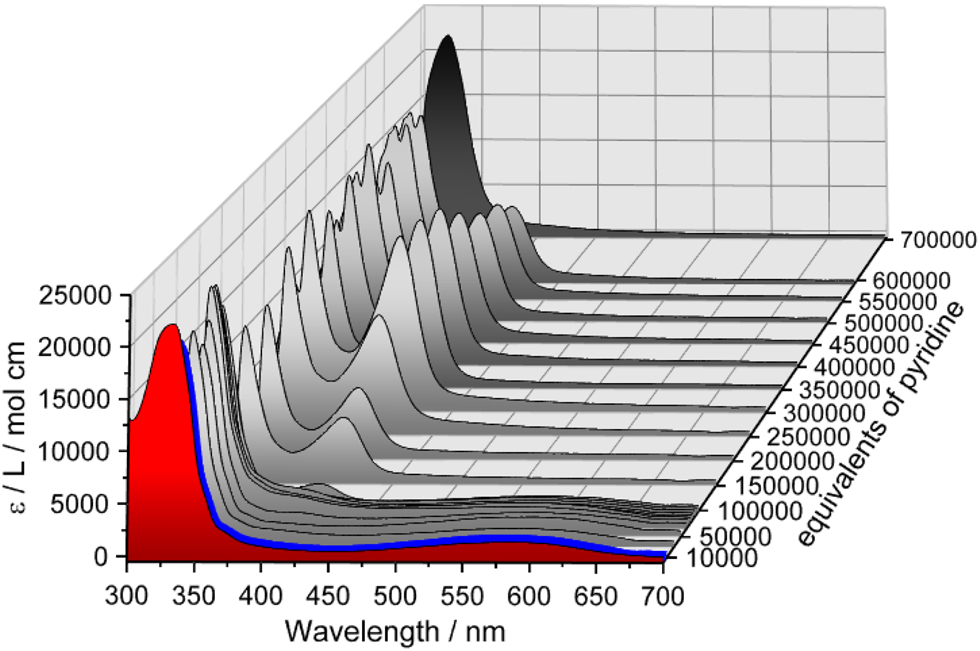
Electronic absorption spectra of 1 in dichloromethane (DCM) (red, c = 0.034 mM) and in pyridine (black, c = 0.034 mM); pyridine titration: stepwise addition of pyridine leads to a decrease of the absorption band at 569 nm and evolution of an absorption band at 410 nm.
The spectrum of 1 in DCM (Figure 1, red) shows an absorption band at 330 nm (22,153 L mol−1 cm−1) and two weak shoulders at 359 and 375 nm (cf. Figure S5a in Supporting Information). For iron(II) complexes containing pyridine-based ligands MLCT absorption bands in this range are common [42]. In addition, an absorption band at 569 nm (1392 L mol−1 cm−1) is observed, which is attributed to MLCT transitions typical for iron(II) bmik complexes [43]. The spectrum of 1 in pure pyridine (Figure 1, black and Figure S5a), on the other hand, shows no absorption band at 569 nm, and the absorption band at 330 nm just shifts to 322 nm (21,936 L mol−1 cm−1).
In order to obtain information regarding spectral changes resulting from coordination of pyridine to complex 1, pyridine titration experiments were performed. Gradual addition of pyridine up to 10,000 equivalents of 1 in DCM results in a slight intensity increase of the MLCT band at 569 nm (Figure 1, blue, and Figure S6). Further addition of pyridine up to 100,000 equivalents (Figure 1, gray), however, leads to an intensity decrease of the same band, and upon adding larger amounts of pyridine this band disappears completely. On the other hand, above 50,000 equivalents of pyridine, an additional band appears at 410 nm, reaching its maximum intensity at about 300,000 equivalents (Figure 1, gray, and Figure S7). In the case of pyridine-containing N-donor ligands, this band has been observed before. Specifically, it was found that its position and intensity depend on the number of pyridine groups [42]. Together with the disappearance of the MLCT band at 569 nm, it thus can be assumed that the bmik ligand is replaced by pyridine. Above 300,000 equivalents of pyridine, the 410 nm band decreases in intensity and completely disappears in pure pyridine, indicating formation of a polypyridine complex (Figure 1, black). Taken together, it appears that the desired six-coordinate complex 1-py (Scheme 3) forms initially upon addition of pyridine to its five-coordinate precursor 1, but at higher amounts of pyridine, ligand exchange occurs.
To get insight into the change of the spin state of 1 upon addition of pyridine, Evans NMR susceptibility measurements were performed. This method takes advantage of the fact that the shift of a signal depends on the susceptibility of the surrounding solution [44, 45]. Two tubes, one inside the other, are used, both containing solvent and a probe, here tetramethylsilane. One tube also contains the paramagnetic sample, which induces a downfield shift of the TMS probe signal. Two TMS signals are then obtained due to the use of two tubes inside one another, and the magnetic gram susceptibility χ g,para can be calculated according to Equation (1).
Here, Δf is the frequency shift of the reference (TMS, Hz), f is the frequency of the NMR spectrometer (Hz), m is the concentration of the sample (g/cm3), 3/4π is the demagnetization factor for a cylindric sample parallel to the high field of a cryomagnet [46], and χ g,dia is the diamagnetic correction for the sample calculated from Pascal’s constants [47]. From (1), the molar magnetic susceptibility χ m (cm3/mol) and the effective magnetic moment μ eff (B.M.) are in turn obtained according to Equations (2) and (3):
where M is the molar mass and T the temperature.
It is important that both tubes are completely sealed airtight. It turned out that even the smallest amounts of oxygen lead to a paramagnetic shift and that different amounts of oxygen in the two tubes falsify the obtained shift. We therefore used a conventional J. Young NMR tube as the outer tube and employed a capillary as the inner tube, which was fused airtight. In order to apply this method to complex 1, we transferred a solution of this complex in DCM or pyridine into the outer sample tube and added 3% of TMS. The inner reference capillary only contained solvent and 3% of TMS.
Table 1 and Figure 2 summarize the 1H NMR shifts resulting from a pyridine titration of 1 in DCM. For 1 dissolved in pure DCM a signal shift of 26.96 Hz is obtained. According to Equations (2) and (3), this corresponds to an effective magnetic moment of 5.22 B.M. which fits very well to a paramagnetic Fe(II) complex. Gradual addition of pyridine leads to a decrease in μ eff; i.e., the samples become more diamagnetic. With the addition of 9000–10,000 equivalents of pyridine, a minimum value 3.30 B.M. is attained, and upon adding higher amounts of pyridine, the sample becomes more paramagnetic again. These results agree with the UV/Vis data and confirm that the desired six-coordinate complex 1-py is formed first, inducing a change of spin state. This way a CISSS effect of complex 1 could be demonstrated by Evans NMR measurements. Further addition of pyridine, however, leads to a ligand exchange probably going along with loss of the bmik coligand.
1H NMR shifts of complex 1 and different equivalents of pyridine obtained by Evans susceptibility measurements: Equations (1)–(3) were used to determine magnetic gram susceptibility and effective magnetic moment.
| Eq. pyridine | Δf [Hz] | Χ g,para [·10−6 ± 0.36·10−6 cm3/g] | μ eff [± 0.02 B.M.] |
|---|---|---|---|
| 0 | 26.96 | 15.4552 | 5.22 |
| 1000 | 24.31 | 13.9830 | 4.96 |
| 2000 | 23.30 | 13.4220 | 4.86 |
| 4000 | 20.03 | 11.6054 | 4.52 |
| 6000 | 15.08 | 8.8556 | 3.94 |
| 8500 | 11.26 | 6.7334 | 3.44 |
| 9000 | 9.73 | 6.1790 | 3.30 |
| 10,000 | 8.75 | 6.1744 | 3.30 |
| 11,000 | 9.67 | 7.4029 | 3.61 |
| 12,000 | 12.30 | 10.0869 | 4.21 |
| 20,000 | 12.50 | 16.7531 | 5.68 |
| 50,000 | 4.20 | 14.1491 | 5.37 |
-
For the determination of standard deviations see Supporting Information.
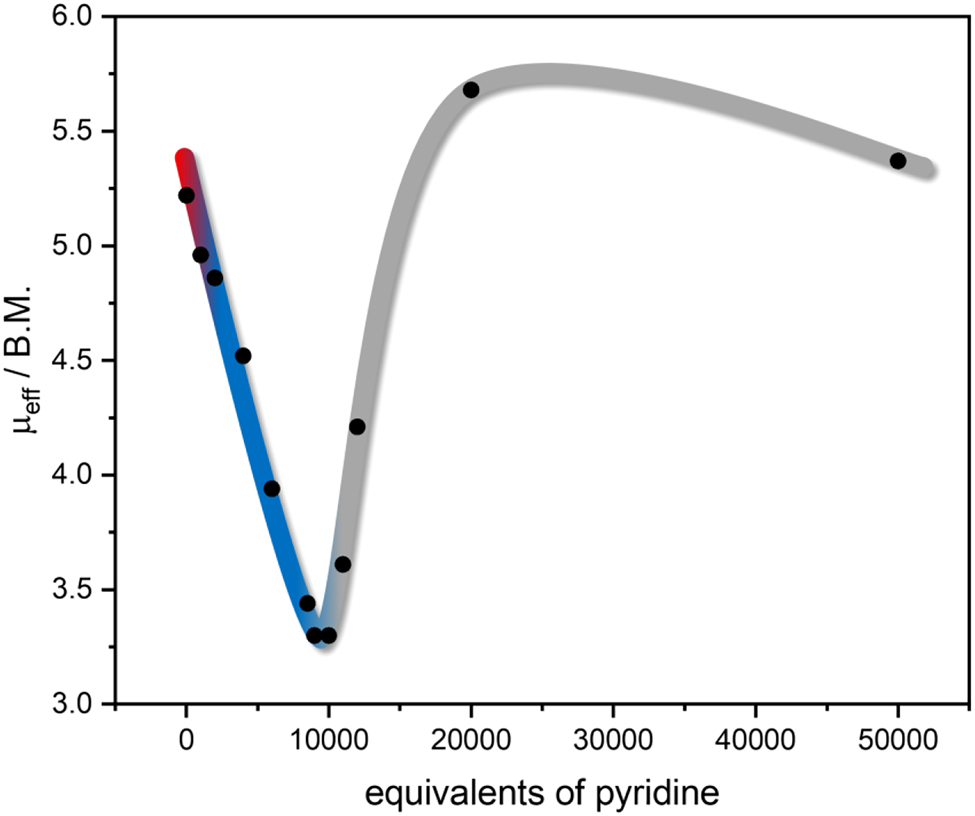
Change of μ eff during the Evans NMR pyridine titration of complex 1: plot of μ eff as a function of added pyridine equivalents. The colors were chosen according to Figure 1: red corresponds to the initial spectrum, blue represents the coordination of pyridine to 1, and gray represents the ligand exchange with pyridine.
2.3 Investigation of complex 2
As the π–π* and n-π* bands of azo compounds change characteristically upon transition from the trans to the cis isomer, UV/Vis spectroscopy was employed to examine the switching behavior of complex 2 and compare it with that of the free ligand 5. In addition, structural changes due to coordination/decoordination should influence the electronic structure, so that a possible LD-CISSS effect should also be detectable by optical absorption spectroscopy.
Figure 3 shows the UV/Vis absorption spectra of photoswitchable ligand 5 (a) and the derived complex 2 (b) in DCM. In case of the free ligand 5, the initial spectrum (Figure 3a, black) shows the π–π* band at 319 nm (12,493 L mol−1 cm−1) and the n-π* band at 436 nm (319 L mol−1 cm−1), typical for the thermodynamically more stable trans isomer of azo compounds. As expected, after irradiation with light with 324 nm (Figure 3a, red; trans → cis) the intensity of the π–π* band decreases while that of the n-π* band increases (641 L mol−1 cm−1). The initial spectrum is obtained again by irradiation with a wavelength of 440 nm (Figure 3a, blue; cis → trans). A total of five switching cycles (trans → cis → trans) were run and no fatigue was detected. Moreover, by measuring the thermal relaxation after photoexcitation at 324 nm, a half-life of the cis isomer of 180 h was determined (Supporting Information, Figure S3).
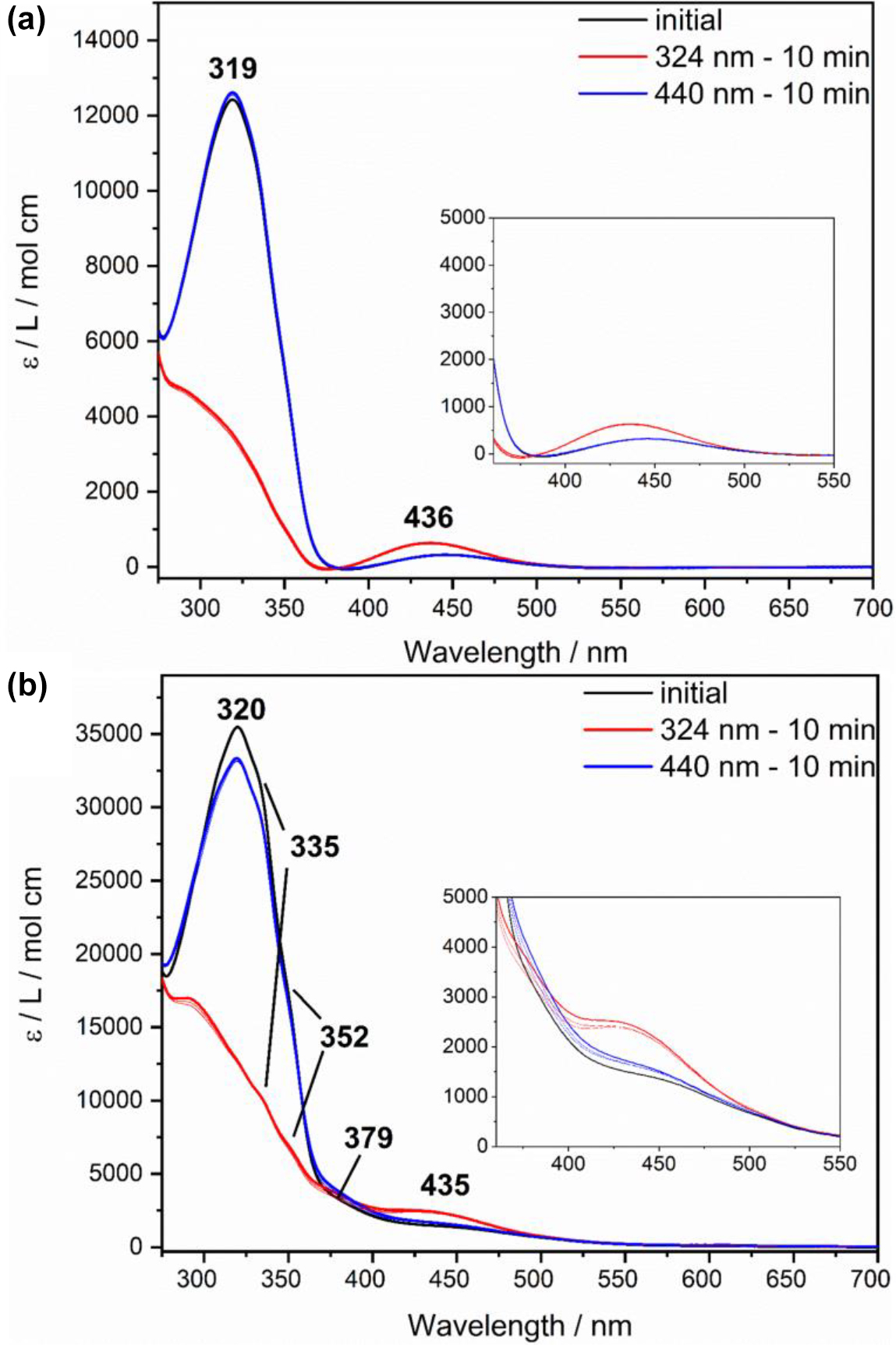
Electronic absorption spectra of azpy-trident ligand 5 (a) (c = 0.051 mM) and of complex 2 (b) (c = 0.024 mM) in DCM: initial spectra are shown in black. After irradiation, spectra of the respective cis isomers (324 nm, 10 min, red) and trans isomers (440 nm, 10 min, blue) are obtained.
For comparison, electronic absorption spectra of complex 2 were recorded under identical conditions. Here, the initial spectrum of complex 2 (Figure 3b, black) also shows the typical bands (π–π*: 320 nm, 35,747 L mol−1 cm−1; n-π*: 435 nm, 2545 L mol−1 cm−1) of the azo group. In addition, shoulders at 335, 352, and 379 nm indicate CT bands of the metal complex. Compared to complex 1, the typical MLCT band for the bmik ligand appears to be missing in the range of 550–600 nm. However, the intensity in the region of the n-π* band is quite high (ε = 1500–3000 L mol−1 cm−1; see Insets). We suspect that the MLCT transition associated with the bmik ligand (see above) is, at least partially, shifted to higher energy with respect to 1 and gets superimposed with the n-π* transition. This could be due to the fact that complex 2, unlike complex 1, does not exhibit a fac-1 geometry of the tridentate ligand (cf. Scheme 2). The change in coordination geometry of the tridentate ligand may be induced by the replacement of the phenyl group by a more bulky azopyridine moiety. Positioning this group cis to the equatorial bmik ligand (cf. Scheme 2) possibly becomes sterically unfavorable in complex 2 whence the tridentate ligand in the latter complex adopts a fac-2 or mer coordination. In, e.g., the mer-configuration of the tridentate ligand the bmik ligand exhibits an axial-equatorial coordination (cf. Scheme 2) and the energy of one of the d-orbitals which are involved in a MLCT transition into the π*-orbitals of the bmik ligand is lowered by backbonding interactions with two pyridine moieties. In complex 1 no such interactions exist for these d-orbitals. The corresponding MLCT transition thus is shifted to higher energy. Similar considerations apply to the fac-2 geometry such that we cannot distinguish between fac-2 and mer. For simplicity, however, it is assumed in the following that the latter geometry applies to complex 2.
By irradiation of 2 with light with 324 nm, the corresponding cis isomer can be obtained (Figure 3b, red) whereby no impairment of switching capability due to coordination of the azpy-trident ligand 5 is noticeable. This could be due to the fact that in complex 2 the photoswitchable unit is linked via a methylene bridge, which precludes electronic coupling. In previous work, we have demonstrated that coordination of an azopyridine-functionalized ligand reduces the switchability of the latter which was ascribed to electronic effects [48]. Employing light of 440 nm wavelength it was also possible to switch the complex back to trans (Figure 3b, blue), with 94% trans isomer being obtained in comparison to the initial spectrum. No further decrease in trans fraction was observed over 5 switching cycles. Finally, a half-life of 84 h of the cis isomer of complex 2 was determined by following thermal relaxation after photoexcitation at 324 nm (Supporting Information, Figure S4), about 50% of the value obtained for the free ligand. The comparatively small reduction of the half-life also suggests a weak influence of the metal coordination on the photophysical properties of the azo group [49].
To conclude, the photoswitchable unit of complex 2 can be reversibly switched between its trans and its cis isomer. However, MLCT bands, which could indicate a LD-CISSS effect, are superimposed by bands of the azo unit and, therefore, UV/Vis spectroscopy does not provide information about whether the ligand is coordinating/decoordinating and/or whether the system is changing its spin state. In order to obtain insight into these issues, we investigated system 2 using Evans NMR susceptibility measurements. For this, we dissolved complex 2 (4.85 mM) in DCM in the outer sample tube and added 3% of TMS. The inner reference capillary only contained DCM and 3% of TMS. Since the trans configuration corresponds to the thermodynamically more stable isomer, a paramagnetic complex and a downfield shift of the TMS signal, respectively, are expected for the initial measurement according to Schemes 1 and 4.
Figure 4 shows the initial spectrum in black at the bottom. It exhibits a shift of 48.48 Hz, which according to Equations (2) and (3) results in μ eff value of 3.92 B.M., clearly below the value of complex 1 in solution (see above). Therefore, it can be assumed that not only the paramagnetic species of 2 is present, but also partially a diamagnetic octahedral complex. Subsequently, we exposed the sample to 365 nm for 30 min to obtain the cis isomer. Due to an intramolecular LD-CISSS a highfield shift and a decrease of μ eff were expected. However, with 61.80 Hz and resulting µ eff of 4.38 B.M. a slight downfield shift and an increase of paramagnetism were observed. Switching back to the trans isomer using light of 440 nm for 30 min should yield a more paramagnetic complex. Again, the opposite was observed, if only to a small extent: with a total shift of 60.55 Hz and a resulting μ eff value of 4.34 B.M. the sample apparently became more diamagnetic.
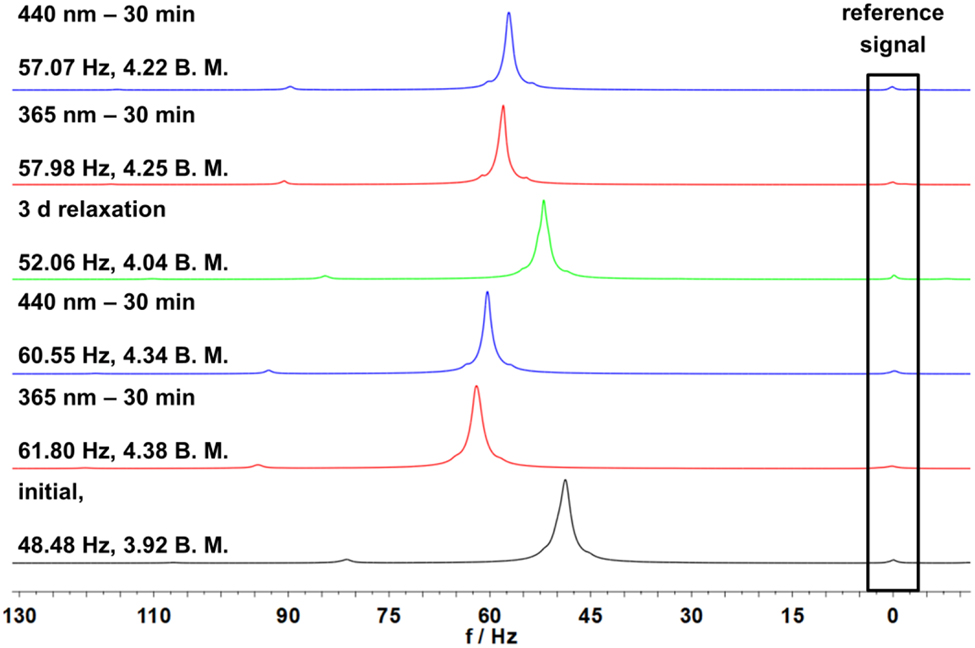
Shift of the TMS signals in 1H NMR spectra measured by the Evans method of complex 2 with a concentration of 4.85 mM in DCM. From bottom to top: black shows the initial spectrum, red spectra are obtained after irradiation with light with 365 nm for 30 min, blue spectra are obtained after irradiation with light with 440 nm for 30 min and green spectrum after three days of relaxation time.
More trans isomer could be obtained after thermal relaxation for three days (Figure 4, green), leading to a more diamagnetic sample (52.06 Hz, 4.04 B.M.). Subsequent irradiation with wavelengths of 365 nm and 440 nm gave spectra following the previous observations (cf. Figure 4).
The obtained data suggest that the complex does not exhibit an intramolecular, but an intermolecular LD-CISSS effect. Here, the azopyridine of the switching unit coordinates in its trans configuration to another complex and decoordinates in its cis configuration (Scheme 7). The complex is then present as an octahedral diamagnetic trans isomer and as a square-pyramidal, paramagnetic cis isomer. Since an intermolecular LD-CISSS should be concentration-dependent, in contrast to the intramolecular LD-CISSS, we carried out Evans susceptibility measurements at different concentrations.
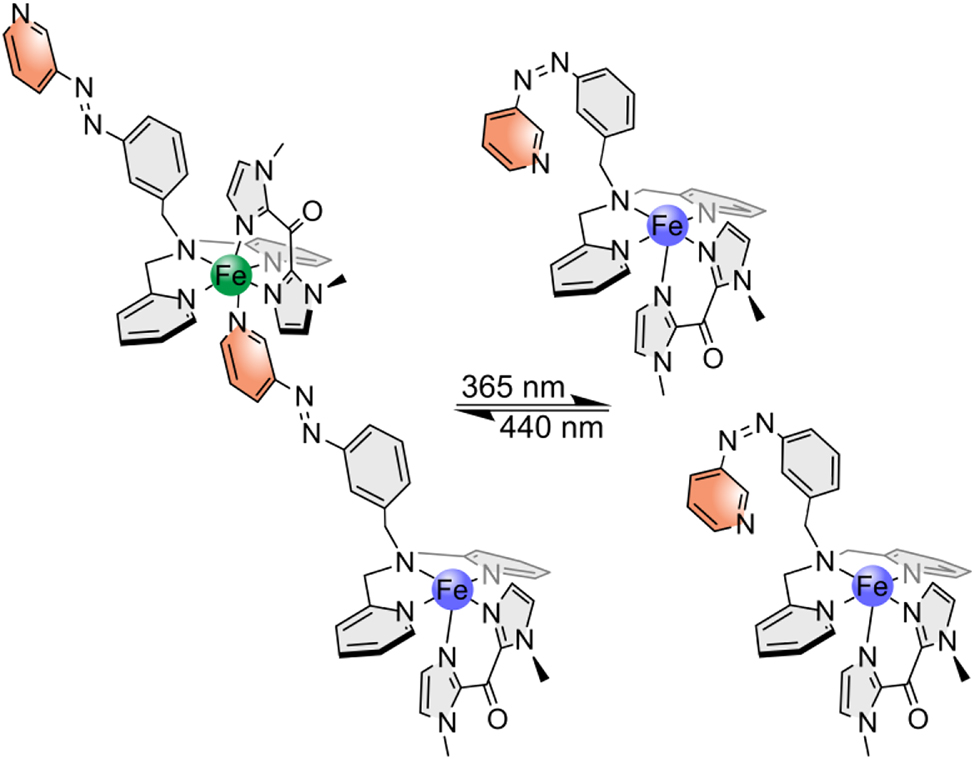
Assumed intermolecular LD-CISSS of complex 2: in trans configuration the switching unit coordinates to another complex and octahedral diamagnetic complexes are created (left); in cis configuration, the azo unit decoordinates and square pyramidal paramagnetic complexes are generated (right).
In case of a concentration-dependent intermolecular LD-CISSS, it is to be expected that the proportion of diamagnetic octahedral complex increases with increasing concentration. Table 2 collects the shifts obtained for the four different concentrations used in this experiment. As expected, the proportion of diamagnetic complex increases with increasing concentration. While at the lowest concentration of 1.25 mM with 4.87 B.M. mainly the uncoordinated paramagnetic complex appears to be present, less paramagnetic species is present at the highest concentration of 4.85 mM, leading to a µ eff of 3.92 B.M. At each concentration we carried out irradiation and relaxation in analogy to Figure 4; i.e., after the initial measurement, the sample was irradiated with light with 365 nm for each concentration to obtain the cis isomer (Figure 5 and Supplementary Information). In agreement with Scheme 7, the concentration of uncoordinated, paramagnetic complex increased in all cases.
1H NMR shifts of different concentrations of complex 2, obtained by Evans susceptibility measurements: Equations (1)–(3) were used to determine magnetic gram susceptibility and effective magnetic moment for each concentration.
| c [mM] | Δf [Hz] | Χ g,para [·10−6 ± 0.36·10−6 cm3/g] | μ eff [± 0.02 B.M.] |
|---|---|---|---|
| 1.25 | 18.91 | 11.7713 | 4.87 |
| 1.86 | 27.16 | 10.8765 | 4.68 |
| 2.43 | 31.20 | 9.63185 | 4.40 |
| 4.85 | 48.48 | 7.64502 | 3.92 |
-
For the determination of standard deviations see Supporting Information.
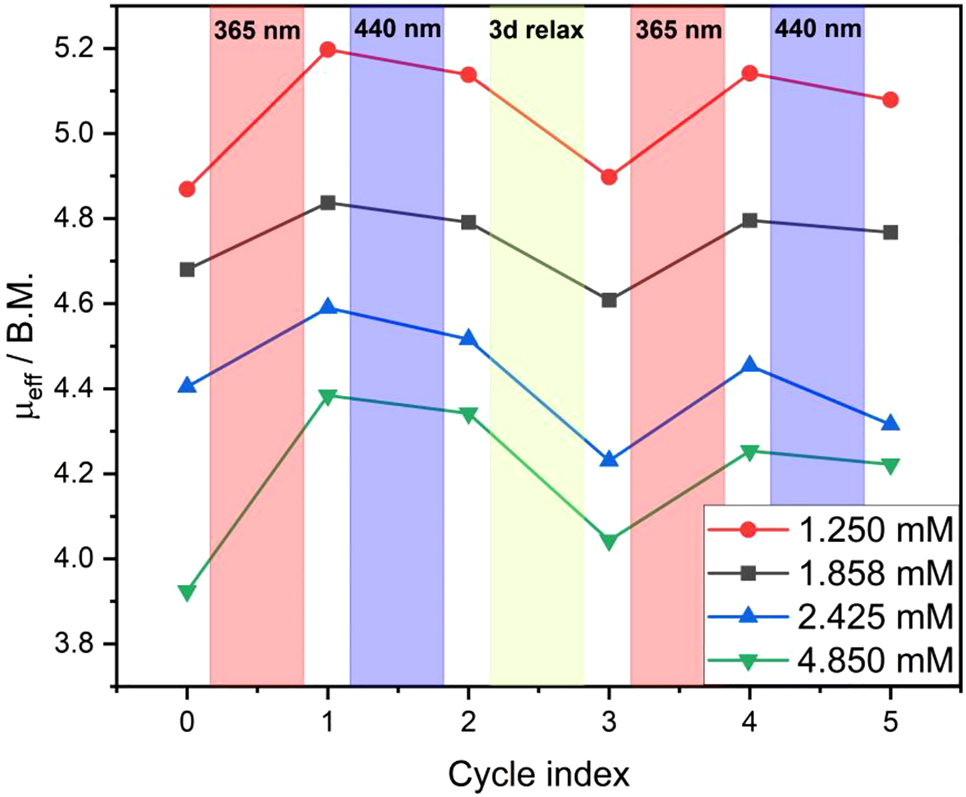
Plot of μ eff changes upon irradiation and relaxation for each concentration of complex 2: values for irradiation with light with 365 nm are shown in red, for irradiation at 440 nm in blue and for three days relaxation time in green respectively.
Subsequent exposure to 440 nm resulted again in more diamagnetic species. However, the μ eff value of the respective initial measurement was not reached at any concentration (see above). Instead, after three days of thermal relaxation more diamagnetic species and correspondingly smaller values of μ eff were obtained. At two concentrations (1.858 mM and 2.425 mM) even lower values compared to the initial ones were determined. Further irradiation with light with 365 nm and back switching at 440 nm corroborated the previous results. The Evans NMR data thus confirm the presence of a concentration-dependent, intermolecular LD-CISSS effect.
3 Conclusions
In order to apply the concept of LD-CISSS to non-porphyrinoid ligands and iron centers, we first synthesized the complex [Fe(trident)(bmik)](ClO4)2 (1). UV/Vis spectroscopic investigations of 1 in solution demonstrated that the addition of pyridine as an axial ligand leads to a change in complex geometry and, as a result, to changes in the electronic absorption spectrum. Higher amounts of pyridine caused ligand exchange and loss of the bidentate ligand bmik. Upon adding pyridine, a spin state change from 5.22 B.M. to 3.30 B.M. could be detected by Evans NMR spectroscopy. This confirmed the coordination of an axial ligand and the presence of a CISSS effect in this system. Higher equivalents of pyridine resulted in more paramagnetic values, corroborating the results of the UV/Vis measurements and supporting the assumption that the complex undergoes ligand exchange at higher concentrations of pyridine.
To obtain information regarding a possible LD-CISSS effect in our system, we generated the new photoswitchable tridentate/tetradentate ligand azpy-trident 5 and examined its switching behavior using NMR and UV/Vis spectroscopy. With a seven-step synthetic route we were able to prepare the complex [Fe(azpy-trident)(bmik)](ClO4)2 (2). By means of UV/Vis switching experiments, it could be shown that the azo unit of 2 can still be switched between its cis and trans isomer, in contrast to an earlier complex where partial loss of the switching capability was observed upon coordination of the azo-functionalized ligand to an iron(II) center [48]. Evans NMR switching experiments were carried out to investigate the LD-CISSS effect in 2. In fact, a partial, but fully reversible change of the spin state was detected. However, it turned out that, contrary to our expectations, the system does not form a low spin complex in its cis but rather in its trans isomer. This led to the conclusion that 2 does not mediate intramolecular, but intermolecular LD-CISSS. We could successfully prove such an intermolecular LD-CISSS by performing concentration-dependent Evans NMR measurements. All in all, by changing its concentration and applying light, the µ eff value of complex 2 could be switched between a minimum value of 3.9 and a maximum value of 5.2 B.M., lying within the range of µ eff values (3.3–5.2 B.M.) attainable to complex 1 by chemically driven CISSS. While this switching efficiency is well below that of Ni(II) porphyrin systems [18], [19], [20], it renders 2 the first nonheme-iron complex to exhibit both chemically and light-driven CISSS in homogeneous solution.
4 Experimental section
Water and oxygen sensitive reagents were handled in an M. Braun Labmaster 130 Glovebox under N2 or Ar. Moisture and air sensitive reactions were carried out in dried solvents under N2-atmosphere using Schlenk techniques. Et2O was dried over LiAlH4 under Ar, MeOH, and THF were dried over CaH2 under N2 and were distilled prior to use, pyridine was dried over potassium hydroxide under N2 and distilled prior to use. All other commercially available starting materials and solvents were used as received. NMR spectra were recorded in deuterated solvents at 300 K on a Bruker Avance 400 Pulse Fourier Transform spectrometer equipped with a cryo-probehead Prodigy BBO400S1 BB-H&F-D-05-Z operating at a 1H frequency of 400.1 MHz and a 13C frequency of 100.6 MHz. Referencing was performed using the solvent residue signal. Signals were assigned with the help of DEPT-135 and two-dimensional correlation spectra (1H,1H-COSY, 1H,13C-HSQC, 1H,13C-HMBC). Signal multiplicities are abbreviated as s (singlet), d (doublet), m (multiplet). The UV/Vis spectroscopic experiments were performed with a Cary5000 spectrometer in transmission geometry. Infrared spectra were recorded with an ALPHA-P ATR spectrometer from Bruker. Signal intensities are marked as s (strong), m (medium), w (weak) and br (broad). Elemental analyses were measured using a vario MICRO cube element analyzer (Elementar). EI mass spectra were recorded on a JEOL AccuTOF GCv 4G spectrometer. High-resolution ESI mass spectra (HR-ESI) were measured with a Q Exactive Plus (Thermo Scientific). Silica gel (Merck, particle size 0.040–0.063 mm) was used for column chromatography purifications. Flash column chromatography were carried out on Isolera™ One from Biotage using Biotage SNAP Ultra cartridges. For switching experiments light sources of Sahlmann Photochemistry Solutions (365 nm [5400 mW] for NMR; 440 nm [4800 mW] for NMR and UV/Vis) and of Thorlabs (324 nm [50 mW] for UV/Vis) were used.
Syntheses of bis(2-pyridylmethyl)benzylamine [39] (3), bis(1-methylimidazole)ketone (bmik) [40] (4), 3-nitrosobenzoic acid [41] (6) and bis(2-pyridylmethyl)amine [39] (9) were prepared according to literature procedures.
4.1 Synthesis of 3-(3-pyridylazo)benzoic acid (7)
To a solution of 3-aminopyridine (6.39 g, 67.9 mmol) in 60 mL pyridine 90 mL of a tetramethylammonium hydroxide solution (25 wt% in water) was added and the mixture was heated to 80 °C. 3-nitrosobenzoic acid (10.26 g, 67.9 mmol) was dissolved in 280 mL pyridine at 40 °C and then added dropwise to the 3-aminopyrdine solution. The reaction mixture was stirred for 4.5 h at 120 °C and additional 18 h at room temperature. After adjusting a pH of 3–4 by adding conc. hydrochloric acid the precipitated product was washed with water until the filtrate remained colorless and dried in vacuo. 3-(3-pyridylazo)benzoic acid was obtained as an orange solid. Yield: 14.32 g (63.0 mmol, 93%). Anal. calculated for C12H9N2O3: C, 63.43; H, 3.99; N, 18.49; found: C, 62.61; H, 4.19; N, 17.85. 1H NMR (400.1 MHz, DMSO-d 6): δ = 13.30 (s, 1H, OH), 9.16 (dd, 4 J = 2.4 Hz 5 J = 0.7 Hz, 1H, py H-2), 8.77 (dd, 3 J = 4.7 Hz, 4 J = 1.6 Hz, 1H, py H-6), 8.41–8.39 (m, 1H, benzyl H-2), 8.22 (ddd, 3 J = 8.2 Hz, 4 J = 2.4 Hz, 4 J = 1.6 Hz, 1H, py H-4), 8.19–8.13 (m, 2H, benzyl H-4,6), 7.76 (td, 3 J = 7.9 Hz, 3 J = 7.9 Hz, 5 J = 0.5 Hz, 1H, benzyl H-5), 7.64 (ddd, 3 J = 8.2 Hz, 3 J = 4.7 Hz, 5 J = 0.8 Hz, 1H, py H-5) ppm. 13C NMR (100.6 MHz, DMSO-d 6): δ = 166.6 (Cq, 1C, C=O), 152.5 (CH, 1C, py C-6), 151.8 (Cq, 1C, benzyl C-1), 147.2 (Cq, 1C, py C-3), 146.5 (CH, 1C, py C-2), 132.4 (CH, 1C, benzyl C-6), 132.2 (Cq, 1C, benzyl C-3), 130.1 (CH, 1C, benzyl C-5), 127.6 (CH, 1C, C-4), 127.1 (CH, 1C, py C-4), 124.7 (CH, 1C, py C-5), 122.4 (CH, 1C, benzyl C-2) ppm. HR-ESI-MS: m/z calculated for C12H10N3O3 [M+H]+: 228.07675, 229.08011, found: 228.07644, 229.07983. IR (ATR): ṽ/cm−1 = 2432 (br), 1695 (s), 1601 (w), 1425 (m), 1294 (s), 1272 (s), 1147 (m), 1039 (m), 819 (s), 759 (s), 692 (s), 678 (s), 639 (s).
4.2 Synthesis of 3-(3-pyridylazo)methyl benzoate (8)
To a suspension of 3-(3-pyridylazo)benzoic acid (14.32 g, 63.0 mmol) in dry MeOH (210 mL) 12 mL conc. sulfuric acid was added. After heating for 18 h under reflux the solvent was removed in vacuo. The residue was dissolved in 20 mL water and a pH of nine was adjusted by using a saturated sodium hydrogen carbonate solution. After extracting seven times with 50 mL DCM the combined organic phases were dried over MgSO4 and the solvent removed in vacuo. The product was obtained as a red solid. Yield: 10.61 g (44.0 mmol, 70%). Anal. calculated for C13H11N3O3: C, 64.72; H, 4.60; N, 17.42; found: C, 64.40; H, 4.61; N, 16.99. 1H NMR (400.1 MHz, DMSO-d 6): δ = 9.14 (dd, 4 J = 2.4 Hz, 5 J = 0.6 Hz, 1H, py H-2), 8.76 (dd, 3 J = 4.7 Hz, 4 J = 1.6 Hz, 1H, py H-6), 8.37–8.35 (m, 1H, benzyl H-2), 8.20 (ddd, 3 J = 8.1 Hz, 4 J = 2.4 Hz, 4 J = 1.5 Hz, 1H, py H-4), 8.17 (ddd, 3 J = 7.9 Hz, 4 J = 2.2 Hz, 4 J = 1.3 Hz, 1H, benzyl H-4), 8.14 (ddd, 3 J = 7.7 Hz, 4 J = 1.7 Hz, 4 J = 1.2 Hz, 1H, benzyl H-6), 7.76 (td, 3 J = 7.9 Hz, 3 J = 7.9 Hz, 5 J = 0.5 Hz, 1H, benzyl H-5), 7.63 (ddd, 3 J = 8.2 Hz, 3 J = 4.7 Hz, 5 J = 0.5 Hz, 1H, py H-5) 3.91 (s, 3H, CH 3) ppm. 13C NMR (100.6 MHz, DMSO-d 6): δ = 165.5 (Cq, 1C, C=O), 152.5 (CH, 1C, py C-6), 151.7 (Cq, 1C, benzyl C-1), 147.1 (Cq, 1C, py C-3), 146.5 (CH, 1C, py C-2), 132.1 (CH, 1C, benzyl C-6), 130.9 (Cq, 1C, benzyl C-3), 130.2 (CH, 1C, benzyl C-5), 127.8 (CH, 1C, benzyl C-4), 127.0 (CH, 1C, py C-4), 124.6 (CH, 1C, py C-5), 122.2 (CH, 1C, benzyl C-2), 52.5 (CH3, 1C, CH3) ppm. HR-ESI-MS: m/z calculated for C13H12N3O3 [M+H]+: 242.09240, 243.09576, 244.09911, found: 242.09286, 243.09607, 244.09941. IR (ATR): ṽ/cm−1 = 3075 (w), 3024 (w), 2964 (w), 2852 (w), 1725 (s), 1580 (m), 1425 (s), 1296 (s), 1274 (s), 1192 (s), 1149 (s), 1072 (s), 968 (m), 913 (m), 811 (m), 751 (s), 694 (s), 676 (s), 543 (m). UV/Vis: 0.060 mM in MeCN, 316 nm (π–π*, 12,833 L mol−1 cm−1), 430 nm (n-π*, 783 L mol−1 cm−1).
4.3 Synthesis of 3-(3-pyridylazo)benzyl alcohol (9)
Under N2-atmosphere, 3-(3-pyridylazo)methyl benzoate (10.61 g, 44.0 mmol) was dissolved in 150 mL dry tetrahydrofuran. A 1 M LiAlH4-THF-solution (30.8 mL, 30.8 mmol, 0.7 eq.) was added dropwise while cooling with ice. The reaction mixture was stirred for two days at room temperature until quenching dropwise with water. The precipitate was filtered and washed with THF and the filtrate was extracted five times with 60 mL brine and five times with 60 mL THF. The combined organic phases were dried over MgSO4 and the solvent removed in vacuo. The crude product was purified on silicagel by column chromatography (DCM/MeOH 10:1) and 3-(3-pyridylazo)benzyl alcohol was obtained as a red-orange solid. Yield: 4.29 g (20.1 mmol, 46%). Anal. calculated for C12H11N2O: C, 67.59; H, 5.20; N, 19.71; found: C, 67.44; H, 5.29; N, 19.45. 1H NMR (400.1 MHz, DMSO-d 6): δ = 9.13 (d, 4 J = 2.1 Hz, 1H, py H-2), 8.75 (dd, 3 J = 4.7 Hz, 4 J = 1.5 Hz, 1H, py H-6), 8.19 (ddd, 3 J = 8.2 Hz, 4 J = 2.4 Hz, 4 J = 1.6 Hz, 1H, py H-4), 7.89 (ddd, 4 J = 2.1 Hz, 4 J = 1.6 Hz, 5 J = 0.7 Hz 1H, benzyl H-2), 7.82 (dt, 3 J = 7.4 Hz, 4 J = 1.7 Hz, 4 J = 1.7 Hz 1H, benzyl H-4), 7.63 (ddd, 3 J = 8.2 Hz, 3 J = 4.7 Hz, 5 J = 0.8 Hz, 1H, py H-5), 7.60–7.55 (m, 1H, benzyl H-5), 7.56–7.52 (m, 1H, benzyl H-6), 5.40 (t, 3 J = 5.8 Hz, 1H, OH), 4.63 (d, 3 J = 5.8 Hz, 2H, CH 2) ppm. 13C NMR (100.6 MHz, DMSO-d 6): δ = 152.1 (CH, 1C, py C-6), 151.9 (Cq, 1C, benzyl C-3), 147.3 (Cq, 1C, py C-3), 146.3 (CH, 1C, py C-2), 144.3 (Cq, 1C, benzyl C-1), 130.0 (CH, 1C, benzyl C-5), 129.2 (CH, 1C, benzyl C-6), 126.9 (CH, 1C, py C-4), 124.6 (CH, 1C, py C-5), 122.0 (CH, 1C, benzyl C-4), 119.7 (CH, 1C, benzyl C-2), 62.4 (CH2, 1C, CH2) ppm. HR-ESI-MS: m/z calculated for C12H12N3O [M+H]+: 214.09749, 215.10084, 216.10420, found: 214.09762, 215.10076, 216.11301. IR (ATR): ṽ/cm−1 = 3197 (br), 2922 (w), 2871 (w), 2832 (w), 1595 (w), 1574 (w), 1462 (w), 1435 (m), 1419 (m), 1329 (w), 1245 (w), 1123 (w), 1082 (w), 1047 (m), 1025 (m), 792 (m), 686 (s), 627 (m), 527 (m). UV/Vis: 0.085 mM in MeCN, 319 nm (π–π*, 16,868 L mol−1 cm−1), 430 nm (n-π*, 1112 L mol−1 cm−1).
4.4 Synthesis of 3-(3-pyridylazo)benzyl chloride (10)
Under N2-atmosphere, a solution of 3-(3-pyridylazo)benzyl alcohol (3.83 g, 18.0 mmol) in thionyl chloride (200 mL) was heated for 20 h at 90 °C. Excess thionyl chloride was removed in vacuo to give 3-(3-pyridylazo)benzyl chloride as a dark red solid. Yield: a quantitative yield was assumed. 1H NMR (400.1 MHz, DMSO-d 6): δ = 9.32 (d, 4 J = 2.3 Hz, 5 J = 0.6 Hz, 1H, py H-2), 8.95 (dd, 3 J = 5.2 Hz, 4 J = 1.4 Hz, 1H, py H-6), 8.61 (ddd, 3 J = 8.3 Hz, 4 J = 2.3 Hz, 4 J = 1.4 Hz, 1H, py H-4), 8.02 (d, 4 J = 2.1 Hz, 1H, benzyl H-2), 8.00 (ddd, 3 J = 8.3 Hz, 3 J = 5.2 Hz, 5 J = 0.5 Hz, 1H, py H-5), 7.95 (ddd, 3 J = 7.8 Hz, 4 J = 2.0 Hz, 4 J = 1.4 Hz, 1H, benzyl H-4), 7.75–7.71 (m, 1H, benzyl H-6) 7.69–7.64 (m, 1H, benzyl H-5), 4.92 (s, 2H, CH 2) ppm. 13C NMR (100.6 MHz, DMSO-d 6): δ = 151.8 (Cq, 1C, benzyl C-3), 148.2 (Cq, 1C, py C-3), 147.3 (CH, 1C, py C-6), 141.7 (CH, 1C, py C-2), 139.5 (Cq, 1C, benzyl C-1), 133.1 (CH, 1C, benzyl C-6), 132.6 (CH, 1C, py C-4), 130.1 (CH, 1C, benzyl C-5), 126.7 (CH, 1C, py C-5), 123.6 (CH, 1C, benzyl C-4), 122.7 (CH, 1C, benzyl C-2), 45.3 (CH2, 1C, CH2) ppm. HR-ESI-MS: m/z calculated for C12H11ClN3 [M+H]+: 232.06360, 233.06696, 234.06065, 235.06401, 236.06736, found: 232.06373, 233.06708, 234.06079, 235.06413, 235.95401. IR (ATR): ṽ/cm−1 = 3040 (br), 2358 (br), 2084 (br), 1964 (br), 1725 (m), 1603 (m), 1529 (m), 1456 (m), 1264 (m), 1135 (m), 1000 (m), 915 (m), 813 (m), 684 (s), 619 (s), 529 (s).
4.5 Synthesis of [N,N-bis(2-pyridylmethyl)]-3-(3-pyridylazo)benzylamine (azpy-trident ligand) (5)
A solution of 3-(3-pyridylazo)benzyl chloride (0.32 g, 1.36 mmol) in 10 mL acetonitrile was added to a solution of bis(2-pyridylmethyl)amine (0.27 g, 1.36 mmol) and cesium carbonate (0.80 g, 2.45 mmol, 1.8 eq.) in 5 mL acetonitrile. After adding TBAI (0.17 g, 0.45 mmol, 0.33 eq.) the reaction mixture was stirred for 24 h at 90 °C and then three days at room temperature. The precipitate was filtered and washed with acetonitrile and the collected filtrates extracted three times with 20 mL H2O and the aqueous phase three times with 20 mL DCM. The collected organic phases were dried over MgSO4 and the solvent removed in vacuo. The crude product was purified using flash column chromatography (DCM/MeOH, MeOH 0 → 20%) to give the azpy-trident ligand as a red-brown oil. Yield: 0.10 g (0.25 mmol, 18%). Anal. calculated for C24H22N6: C, 73.07; H, 5.62; N, 21.30; found: C, 72.60; H, 6.06; N, 21.08. 1H NMR (400.1 MHz, DMSO-d 6): δ = 9.12 (dd, 4 J = 2.4 Hz, 5 J = 0.7 Hz, 1H, azo-py H-2), 8.75 (dd, 3 J = 4.6 Hz, 4 J = 1.6 Hz, 1H, azo-py H-6), 8.50 (ddd, 3 J = 4.9 Hz, 4 J = 1.8 Hz, 5 J = 0.9 Hz, 2H, py H-6), 8.19 (ddd, 3 J = 8.2 Hz, 4 J = 2.4 Hz, 4 J = 1.6 Hz, 1H, azo-py H-4), 7.95 (t, 4 J = 1.6 Hz, 4 J = 1.6 Hz 1H, benzyl H-2), 7.79 (td, 3 J = 7.6 Hz, 3 J = 7.6 Hz, 4 J = 1.8 Hz, 2H, py H-4), 7.82–7.77 (m, 1H, benzyl H-4), 7.68–7.64 (m, 1H, benzyl H-6), 7.63 (ddd, 3 J = 8.2 Hz, 3 J = 4.7 Hz, 5 J = 0.8 Hz, 1H, azo-py H-5), 7.61–7.57 (m, 1H, benzyl H-5), 7.59 (d, 3 J = 7.8 Hz, 2H, py H-3), 7.26 (ddd, 3 J = 7.5 Hz, 3 J = 4.9 Hz, 4 J = 1.2 Hz, 2H, py H-5), 3.79 (s, 2H, benzyl CH 2), 3.78 (s, 4H, py CH 2) ppm. 13C NMR (100.6 MHz, DMSO-d 6): δ = 158.9 (Cq, 2C, py C-2), 152.1 (CH, 1C, azo-py C-6), 152.0 (Cq, 1C, benzyl C-3), 148.8 (CH, 2C, py C-6), 147.3 (Cq, 1C, azo-py C-3), 146.3 (CH, 1C, azo-py C-2), 140.6 (Cq, 1C, benzyl C-1), 136.6 (CH, 2C, py C-4), 132.3 (CH, 1C, benzyl C-6), 129.4 (CH, 1C, benzyl C-5), 126.9 (CH, 1C, azo-py C-4), 124.6 (CH, 1C, azo-py C-5), 122.8 (CH, 1C, benzyl C-2), 122.6 (CH, 2C, py C-3), 122.2 (CH, 2C, py C-5), 121.5 (CH, 1C, benzyl C-4), 59.1 (CH2, 2C, py CH2), 57.0 (CH2, 1C, benzyl CH2) ppm. EI-MS: m/z calculated for C24H23N6 [M+H]+: 395.1, 396.1, 397.2, found: 395.1, 396.1, 397.1. HR-ESI-MS: m/z calculated for C24H23N6 [M+H]+: 395.19787, 396.20123, 397.20458, found: 395.19777, 396.20111, 397.20440. IR (ATR): ṽ/cm−1 = 3054 (w), 3038 (w), 3001 (w), 2984 (w), 1605 (m), 1579 (w), 1480 (m), 1445 (m), 1428 (m), 1313 (w), 1246 (w), 1161 (w), 1130 (w), 1063 (w), 1018 (w), 845 (m), 766 (s), 733 (s), 705 (s), 612 (s). UV/Vis: 0.051 mM in MeCN, 319 nm (π–π*, 12,493 L mol−1 cm−1), 436 nm (n-π*, 641 L mol−1 cm−1).
4.6 Synthesis of [Fe(trident)(bmik)](ClO4)2 (1)
Under N2-atmosphere a solution of bis(2-pyridylmethyl)benzylamine (0.58 g, 2 mmol) in 2.5 mL dry methanol was added to a solution of iron(II) perchlorate hydrate (0.51 g, 2 mmol) in 1 mL dry methanol. After stirring for four days at room temperature a solution of bis(1-methylimidazole)ketone (0.19 mg, 1 mmol) in 2.5 mL dry methanol was added. The reaction mixture was stirred for additional 20 h at room temperature. The resulting light blue solid was filtered and washed with dry methanol. The filtrate was reduced to 2 mL and stored at 8 °C until the product precipitated as a dark blue solid. The product was filtered, washed with dry MeOH and dry diethyl ether and dried in vacuo. Yield: 0.38 g (0.52 mmol, 52%). Anal. calculated for C28H29Cl2FeN7O9: C, 45.80; H, 3.98; N, 13.35; found: C, 46.21; H, 4.03; N, 13.05. HR-ESI-MS: m/z calculated for C28H29FeN7O [M]2+: 267.58860, 268.09028, 268.59196, 269.09363; found: 267.58846, 268.09013, 268.59181, 269.08756. IR (ATR): ṽ/cm−1 = 3135 (w), 2966 (w), 2928 (w), 2865 (w), 1768 (w), 1640 (w), 1608 (w), 1487 (w), 1427 (w), 1292 (w), 1168 (w), 1082 (m), 1025 (m), 900 (m), 766 (m), 707 (w), 620 (m), 603 (w), 419 (w), 289 (w). UV/Vis: 0.034 mM in DCM, 330 nm (22,153 L mol−1 cm−1), 569 nm (1392 L mol−1 cm−1).
4.7 Synthesis of [Fe(azpy-trident)(bmik)](ClO4)2 (2)
Under N2-atmosphere a solution of the azpy-trident ligand (0.10 g, 0.25 mmol) in 3 mL dry methanol was added to a solution of iron(II) perchlorate hydrate (0.064 g, 0.25 mmol) in 1 mL dry methanol. After stirring for 20 h at room temperature, a solution of bis(1-methylimidazole)ketone (0.024 mg, 0.13 mmol) in 1 mL dry methanol was added. The reaction mixture was stirred for additional 20 h at room temperature. The both resulting blue and red-brown solids were separated manually. The red-brown product was washed with dry methanol and dry diethyl ether and dried in vacuo. Yield: 33.4 mg (0.040 mmol, 31%). Anal. calculated for C33H32Cl2FeN10O9: C, 47.22; H, 3.84; N, 16.69; found: C, 47.69; H, 4.22; N, 16.24. HR-ESI-MS: m/z calculated for C33H32FeN10O [M]2+: 319.10729, 320.10495, 320.60662, 321.10829; found: 319.10691, 320.10452, 320.60617, 321.10472. IR (ATR): ṽ/cm−1 = 3136 (w), 3073 (w), 2946 (w), 1642 (w), 1607 (w), 1573 (w), 1485 (w), 1424 (w), 1294 (w), 1193 (w), 1164 (w), 1079 (m), 1022 (m), 901 (w), 815 (w), 766 (m), 705 (m), 621 (m), 605 (w), 418 (w), 286 (w), 214 (w). FT-Raman (solid): ṽ/cm−1 3068 (w), 1585 (w), 1493 (m), 1458 (s), 1200 (s), 1140 (w), 1039 (w), 1024 (w), 1001 (w), 933 (w). UV/Vis: 0.024 mM in DCM, 320 nm (π–π*, 35,747 L mol−1 cm−1), 435 nm (n-π*, 2545 L mol−1 cm−1).
Dedicated to Professor Christian Näther on the occasion of his 60th birthday.
Funding source: Deutsche Forschungsgemeinschaft
Award Identifier / Grant number: CRC 677
Acknowledgment
The authors thank Spectroscopic Department of the Institute of Inorganic Chemistry for Spectroscopic Measurements. We thank Nicolai Burzlaff for the provision of bmik.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: This study was funded by Deutsche Forschungsgemeinschaft (DFG) through Sonderforschungsbereich CRC 677 (Function by Switching).
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
1. Cambi, L., Szegö, L. Ber. Dtsch. Chem. Ges. 1931, 64, 2591–2598; https://doi.org/10.1002/cber.19310641002.Suche in Google Scholar
2. Gütlich, P., Hauser, A., Spiering, H. Angew. Chem., Int. Ed. Engl. 1994, 33, 2024–2054.10.1002/anie.199420241Suche in Google Scholar
3. Gütlich, P., Goodwin, H. A., Eds. Spin Crossover in Transition Metal Compounds I, Topics in Current Chemistry, Vol. 233; Springer: Berlin, Heidelberg, 2004.10.1007/b96439Suche in Google Scholar
4. Gütlich, P. Eur. J. Inorg. Chem. 2013, 2013, 581–591.10.1002/ejic.201300092Suche in Google Scholar
5. Gütlich, P., Garcia, Y., Goodwin, H. A. Chem. Soc. Rev. 2000, 29, 419–427.10.1039/b003504lSuche in Google Scholar
6. Halcrow, M. A. Spin-Crossover Materials. Properties and Applications; Wiley: Chichester, 2013.10.1002/9781118519301Suche in Google Scholar
7. Kahn, O., Kröber, J., Jay, C. Adv. Mater. 1992, 4, 718–728; https://doi.org/10.1002/adma.19920041103.Suche in Google Scholar
8. Kröber, J., Codjovi, E., Kahn, O., Groliere, F., Jay, C. J. Am. Chem. Soc. 1993, 115, 9810–9811.10.1021/ja00074a062Suche in Google Scholar
9. Kahn, O. Science 1998, 279, 44–48; https://doi.org/10.1126/science.279.5347.44.Suche in Google Scholar
10. Sanvito, S. Chem. Soc. Rev. 2011, 40, 3336–3355; https://doi.org/10.1039/c1cs15047b.Suche in Google Scholar PubMed
11. Létard, J.-F., Guionneau, P., Goux-Capes, L. Towards spin crossover applications. In Spin Crossover in Transition Metal Compounds III; Gütlich, P., Goodwin, H. A., Eds. Springer: Berlin, Heidelberg, 2004, pp. 221–249.10.1007/b95429Suche in Google Scholar
12. Bousseksou, A., Molnár, G., Salmon, L., Nicolazzi, W. Chem. Soc. Rev. 2011, 40, 3313–3335; https://doi.org/10.1039/c1cs15042a.Suche in Google Scholar PubMed
13. Jureschi, C.-M., Linares, J., Boulmaali, A., Dahoo, P., Rotaru, A., Garcia, Y. Sensors 2016, 16, 187–195; https://doi.org/10.3390/s16020187.Suche in Google Scholar PubMed PubMed Central
14. Ruben, M., Breuning, E., Lehn, J.-M., Ksenofontov, V., Renz, F., Gütlich, P., Vaughan, G. B. M. Chem. Eur J. 2003, 9, 4422–4429; https://doi.org/10.1002/chem.200304933.Suche in Google Scholar PubMed
15. Kumar, K. S., Ruben, M. Angew. Chem. Int. Ed. 2021, 60, 7502–7521; https://doi.org/10.1002/anie.201911256.Suche in Google Scholar PubMed PubMed Central
16. Weber, B., Bauer, W., Obel, J. Angew. Chem. Int. Ed. 2008, 47, 10098–10101; https://doi.org/10.1002/anie.200802806.Suche in Google Scholar PubMed
17. Thies, S., Bornholdt, C., Köhler, F., Sönnichsen, F. D., Näther, C., Tuczek, F., Herges, R. Chem. Eur J. 2010, 16, 10074–10083; https://doi.org/10.1002/chem.201000603.Suche in Google Scholar PubMed
18. Venkataramani, S., Jana, U., Dommaschk, M., Sönnichsen, F. D., Tuczek, F., Herges, R. Science 2011, 331, 445–448; https://doi.org/10.1126/science.1201180.Suche in Google Scholar PubMed
19. Thies, S., Sell, H., Schütt, C., Bornholdt, C., Näther, C., Tuczek, F., Herges, R. J. Am. Chem. Soc. 2011, 133, 16243–16250; https://doi.org/10.1021/ja206812f.Suche in Google Scholar PubMed
20. Dommaschk, M., Schütt, C., Venkataramani, S., Jana, U., Näther, C., Sönnichsen, F. D., Herges, R. Dalton Trans. 2014, 43, 17395–17405; https://doi.org/10.1039/c4dt03048f.Suche in Google Scholar PubMed
21. Ni, Z., Fiedler, S. R., Shores, M. P. Dalton Trans. 2011, 40, 944–950; https://doi.org/10.1039/c0dt01079k.Suche in Google Scholar PubMed
22. Salaam, J., Rivat, M., Fogeron, T., Hasserodt, J. Anal. Sens. 2021, 1, 11–29; https://doi.org/10.1002/anse.202000015.Suche in Google Scholar
23. Brandenburg, H., Krahmer, J., Fischer, K., Schwager, B., Flöser, B., Näther, C., Tuczek, F. Eur. J. Inorg. Chem. 2018, 2018, 576–585. https://doi.org/10.1002/ejic.201701281.Suche in Google Scholar
24. Klaß, M., Krahmer, J., Näther, C., Tuczek, F. Dalton Trans. 2018, 47, 1261–1275.10.1039/C7DT03952BSuche in Google Scholar PubMed
25. Kurz, H., Schötz, K., Papadopoulos, I., Heinemann, F. W., Maid, H., Guldi, D. M., Köhler, A., Hörner, G., Weber, B. J. Am. Chem. Soc. 2021, 143, 3466–3480; https://doi.org/10.1021/jacs.0c12568.Suche in Google Scholar PubMed
26. Dommaschk, M., Gutzeit, F., Boretius, S., Haag, R., Herges, R. Chem. Commun. 2014, 50, 12476–12478; https://doi.org/10.1039/c4cc05525j.Suche in Google Scholar PubMed
27. Dommaschk, M., Thoms, V., Schütt, C., Näther, C., Puttreddy, R., Rissanen, K., Herges, R. Inorg. Chem. 2015, 54, 9390–9392; https://doi.org/10.1021/acs.inorgchem.5b01756.Suche in Google Scholar PubMed
28. Kurz, H., Lochenie, C., Wagner, K. G., Schneider, S., Karg, M., Weber, B. Chem. Eur J. 2018, 24, 5100–5111; https://doi.org/10.1002/chem.201704632.Suche in Google Scholar PubMed
29. Lochenie, C., Wagner, K. G., Karg, M., Weber, B. J. Mater. Chem. C 2015, 3, 7925–7935; https://doi.org/10.1039/c5tc00837a.Suche in Google Scholar
30. Thies, S., Sell, H., Bornholdt, C., Schütt, C., Köhler, F., Tuczek, F., Herges, R. Chem. Eur J. 2012, 18, 16358–16368; https://doi.org/10.1002/chem.201201698.Suche in Google Scholar PubMed
31. Schütt, C., Heitmann, G., Wendler, T., Krahwinkel, B., Herges, R. J. Org. Chem. 2016, 81, 1206–1215.10.1021/acs.joc.5b02817Suche in Google Scholar PubMed
32. Dommaschk, M., Näther, C., Herges, R. J. Org. Chem. 2015, 80, 8496–8500; https://doi.org/10.1021/acs.joc.5b01524.Suche in Google Scholar PubMed
33. Dommaschk, M., Peters, M., Gutzeit, F., Schütt, C., Näther, C., Sönnichsen, F. D., Tiwari, S., Riedel, C., Boretius, S., Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555; https://doi.org/10.1021/jacs.5b00929.Suche in Google Scholar PubMed
34. Ludwig, J., Helberg, J., Zipse, H., Herges, R. Beilstein J. Org. Chem. 2020, 16, 2119–2126; https://doi.org/10.3762/bjoc.16.179.Suche in Google Scholar PubMed PubMed Central
35. Ludwig, J., Moje, T., Röhricht, F., Herges, R. Beilstein J. Org. Chem. 2020, 16, 2589–2597; https://doi.org/10.3762/bjoc.16.210.Suche in Google Scholar PubMed PubMed Central
36. Dommaschk, M., Gröbner, J., Wellm, V., Hövener, J.-B., Riedel, C., Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299; https://doi.org/10.1039/c9cp04156g.Suche in Google Scholar PubMed
37. Wellm, V., Groebner, J., Heitmann, G., Sönnichsen, F. D., Herges, R. Angew. Chem. Int. Ed. 2021, 60, 8220–8226; https://doi.org/10.1002/anie.202015851.Suche in Google Scholar PubMed PubMed Central
38. Wellm, V., Näther, C., Herges, R. J. Org. Chem. 2021, 86, 9503–9514; https://doi.org/10.1021/acs.joc.1c00806.Suche in Google Scholar PubMed
39. Hamann, J. N., Rolff, M., Tuczek, F. Dalton Trans. 2015, 44, 3251–3258; https://doi.org/10.1039/c4dt03010a.Suche in Google Scholar PubMed
40. Elgafi, S., Field, L. D., Messerle, B. A., Hambley, T. W., Turner, P. J. Chem. Soc., Dalton Trans. 1997, 2341–2346; https://doi.org/10.1039/a700474e.Suche in Google Scholar
41. Priewisch, B., Rück-Braun, K. J. Org. Chem. 2005, 70, 2350–2352; https://doi.org/10.1021/jo048544x.Suche in Google Scholar PubMed
42. Britovsek, G. J. P., England, J., White, A. J. P. Inorg. Chem. 2005, 44, 8125–8134; https://doi.org/10.1021/ic0509229.Suche in Google Scholar PubMed
43. De, S., Tewary, S., Garnier, D., Li, Y., Gontard, G., Flambard, A., Breher, F., Boillot, M., Rajaraman, G., Lescouezec, R. Eur. J. Inorg. Chem. 2018, 2018, 414–428; https://doi.org/10.1002/ejic.201701013.Suche in Google Scholar
44. Evans, D. F. J. Chem. Soc. 1959, 2003–2005; https://doi.org/10.1039/jr9590002003.Suche in Google Scholar
45. Grant, D. H. J. Chem. Educ. 1995, 72, 39–40; https://doi.org/10.1021/ed072p39.Suche in Google Scholar
46. Live, D. H., Chan, S. I. Anal. Chem. 1970, 42, 791–792; https://doi.org/10.1021/ac60289a028.Suche in Google Scholar
47. Bain, G. A., Berry, J. F. J. Chem. Educ. 2008, 85, 532–536; https://doi.org/10.1021/ed085p532.Suche in Google Scholar
48. Megow, S., Fitschen, H.-L., Tuczek, F., Temps, F. J. Phys. Chem. Lett. 2019, 10, 6048–6054; https://doi.org/10.1021/acs.jpclett.9b02083.Suche in Google Scholar PubMed
49. Schlimm, A., Löw, R., Rusch, T., Röhricht, F., Strunskus, T., Tellkamp, T., Sönnichsen, F., Manthe, U., Magnussen, O., Tuczek, F., Herges, R. Angew. Chem. Int. Ed. 2019, 58, 6574–6578; https://doi.org/10.1002/ange.201814342.Suche in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/znb-2022-0011).
© 2022 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this issue
- Laudatio/Preface
- Christian Näther zum 60. Geburtstag gewidmet
- Research Articles
- Bismuth-rich bimetallic clusters (CuBi8)3+ and [MBi10]4+ (M = Pd, Pt) from ionothermal synthesis
- Crystal structure of phenanthrenide salts stabilized by 15-crown-5 and 18-crown-6
- Structure and properties of two new heteroleptic bismuth(III) dithiocabamates of the general composition Bi(S2CNH2)2X (X = Cl, SCN)
- Synthesis and structural characterization of three new mixed ligand alkaline-earth metal picrates
- Dimorphism of MnHAsO4(H2O): natural monoclinic krautite and its synthetic triclinic modification
- Synthesis, crystal structure, and topology of a polycatenated bismuth coordination polymer
- The unexpected crystal structure of thallium(I) tricyanomethanide Tl[C(CN)3]
- Synthesis, structure characterization and properties of a new oxidovanadium(IV) coordination polymer incorporating bridging (MoO4)2– and (Mo8O26)4– ligands
- Crystal structure of Dy11Ge4.33In5.67 and Tm11Ge4In6 from X-ray single-crystal and powder data
- Crystallisation of phosphates revisited: a multi-step formation process for SrHPO4
- Oxygen evolving reactions catalyzed by different manganese oxides: the role of oxidation state and specific surface area
- Synthesis and structural characterization of a new heterometallicmolybdate coordination polymer based on a µ3-bridging amino alcohol
- Chemically and Light-Driven Coordination-Induced Spin State Switching (CISSS) of a nonheme-iron complex
- Extracting information from X-ray diffraction patterns containing Laue oscillations
- Gadolinium trisilicide − a paramagnetic representative of the YbSi3 type series
Artikel in diesem Heft
- Frontmatter
- In this issue
- Laudatio/Preface
- Christian Näther zum 60. Geburtstag gewidmet
- Research Articles
- Bismuth-rich bimetallic clusters (CuBi8)3+ and [MBi10]4+ (M = Pd, Pt) from ionothermal synthesis
- Crystal structure of phenanthrenide salts stabilized by 15-crown-5 and 18-crown-6
- Structure and properties of two new heteroleptic bismuth(III) dithiocabamates of the general composition Bi(S2CNH2)2X (X = Cl, SCN)
- Synthesis and structural characterization of three new mixed ligand alkaline-earth metal picrates
- Dimorphism of MnHAsO4(H2O): natural monoclinic krautite and its synthetic triclinic modification
- Synthesis, crystal structure, and topology of a polycatenated bismuth coordination polymer
- The unexpected crystal structure of thallium(I) tricyanomethanide Tl[C(CN)3]
- Synthesis, structure characterization and properties of a new oxidovanadium(IV) coordination polymer incorporating bridging (MoO4)2– and (Mo8O26)4– ligands
- Crystal structure of Dy11Ge4.33In5.67 and Tm11Ge4In6 from X-ray single-crystal and powder data
- Crystallisation of phosphates revisited: a multi-step formation process for SrHPO4
- Oxygen evolving reactions catalyzed by different manganese oxides: the role of oxidation state and specific surface area
- Synthesis and structural characterization of a new heterometallicmolybdate coordination polymer based on a µ3-bridging amino alcohol
- Chemically and Light-Driven Coordination-Induced Spin State Switching (CISSS) of a nonheme-iron complex
- Extracting information from X-ray diffraction patterns containing Laue oscillations
- Gadolinium trisilicide − a paramagnetic representative of the YbSi3 type series