
Canagliflozin: A review with specific focus on analytical methods in biological matrices and pharmaceuticals
-
Ajitha Azhakesan
 und
Sujatha Kuppusamy
und
Sujatha Kuppusamy


Abstract
Sodium-glucose transporter 2 inhibitor emerges as the latest group of oral hypoglycemic agents, which shows insulin-independent pathology and provides an upper hand to enhance renal glucose elimination. Canagliflozin (CGN) was the number one drug, approved by FDA on 29th March 2013 for the treatment of type 2 diabetes mellitus. By totting up to its glucose-lowering effects, it exhibits beneficial effects on the heart and potentially on the kidneys. The study aims to summarize various analytical techniques, such as chromatography, spectrophotometry, and hyphenated techniques, such as Liquid chromatography with tandem mass spectrometry (LC-MS/MS) and Ultra performance liquid chromatography with tandem mass spectrometer (UPLC-MS) for the analysis of CGN. In the proposed work, we have reviewed various analytical methods reported for the estimation of CGN in biological matrices and Pharmaceuticals from various databases like ScienceDirect, Springer, PubMed, Scopus, Taylor & Francis, and Web of Science for the estimation of CGN. Various analytical methods adapted were high-performance liquid chromatography, UPLC, LC-MS/MS, high-performance thin-layer liquid chromatography, Fourier-transform infrared spectroscopy, spectrofluorimetry, and UV spectrophotometry. This current review presented the determination of CGN using various analytical techniques and biological matrices either in single or in combination with other hypoglycemic agents, as per International Conference on Harmonization guidelines. Further, some future trends that can be integrated were also suggested.
Abbreviations
- CGN
-
canagliflozin
- DAD
-
diode array detector
- DGN
-
dapagliflozin
- EGN
-
empagliflozin
- HPLC
-
high performance liquid chromatography
- ICH
-
International Conference on Harmonization
- LC-MS/MS
-
liquid chromatography with tandem mass spectrometry
- LNG
-
linagliptin
- LLE
-
liquid–liquid extraction
- LOD
-
limit of detection
- LOQ
-
limit of quantitation
- MFN
-
metformin
- PDA
-
photo diode array
- PPE
-
protein precipitation extraction
- SGLT2
-
sodium glucose transporter 2
- SPE
-
solid-phase extraction
- T2DM
-
type 2 diabetes mellitus
- TLC
-
thin layer chromatography
- UPLC
-
ultra performance liquid chromatography
- UV-Vis
-
ultra violet-visible spectroscopy
- VWD
-
variable wavelength detector
1 Introduction
Type 2 diabetes mellitus (T2DM) resumes being a major non-communicable ailment with a universal burden of 366 million. India is considered the epicenter of diabetes with 77 million and is supposed to be 134 million by the year 2045. Sodium-glucose transporter 2 (SGLT2) inhibitor are the latest group of anti-diabetic medicines available in the market for the treatment of T2DM, preventing re-absorption of glucose from the blood, which is filtered through the kidneys and hence facilitating the excretion of glucose in urine. Canagliflozin (CGN) is the first approved member of the SGLT2 inhibitor class by the USFDA in March 2013 applicable to patients with T2DM as an adjunct to exercise and diet to enhance glycemic control [1]. SGLT2 inhibitors get inhibited in the proximal renal tubules by the action of CGN, lowering the urinary glucose threshold, which further enhances urinary glucose excretion. Cardiopathy is considered as a frequently developed etiology along with T2DM to cause morbidity and mortality among people with T2DM [2,3]. CGN was approved in the year 2018 to treat cardiovascular diseases, which includes blood pressure, body weight, and urinary function. CGN has superiority of SGLT2 inhibitors and cardiovascular treatment compared with other anti-diabetic drugs used to treat T2DM. CGN is chemically1-(beta-d-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate (Figure 1) with chemical formula C24H25FO5S·1/2H2O and molecular weight of 453.52 g·mol−1. It is freely soluble in methanol. It is yet to be monographed in any pharmacopoeias [4].

Structure of canagliflozin hemihydrate.
The threat of disease, access to medicine for the poor, and rising costs of medicine are the big challenges for the people of the world. For making affordable drugs available for people of the world that are extremely poor and prone to large epidemics, it is important to bring down the costs of research and development and thereby the production process and quality control. Simple, rapid (to reduce analytical down time in turn revenue), and cost-effective analytical method development is a thirst area of importance in the pharmaceutical industry. In view of the significance of CGN to the universal population, sensitive quality control methods are required for their analysis. Analytical methods have a major task in ensuring and assuring the quality of pharmaceuticals. Hence, currently, there is a need for the collection of the reported analytical methods of CGN. The main objective of the review is to summarize the multiple approaches available to estimate CGN in API, dosage form and biological matrix separately and drug combinations. The analytical methods were classified into four main methods: (1) UV-visible (UV-Vis) spectroscopy, (2) chromatography (Ultra performance liquid chromatography (UPLC), high-performance liquid chromatography (HPLC), high-performance thin-layer liquid chromatography (HPTLC)), and (3) bio-analytical. Figure 2 represents an overview of various analytical methods for the determination of CGN from various databases like ScienceDirect, Springer, PubMed, Scopus, Taylor & Francis, and Web of Science for the estimation of CGN.
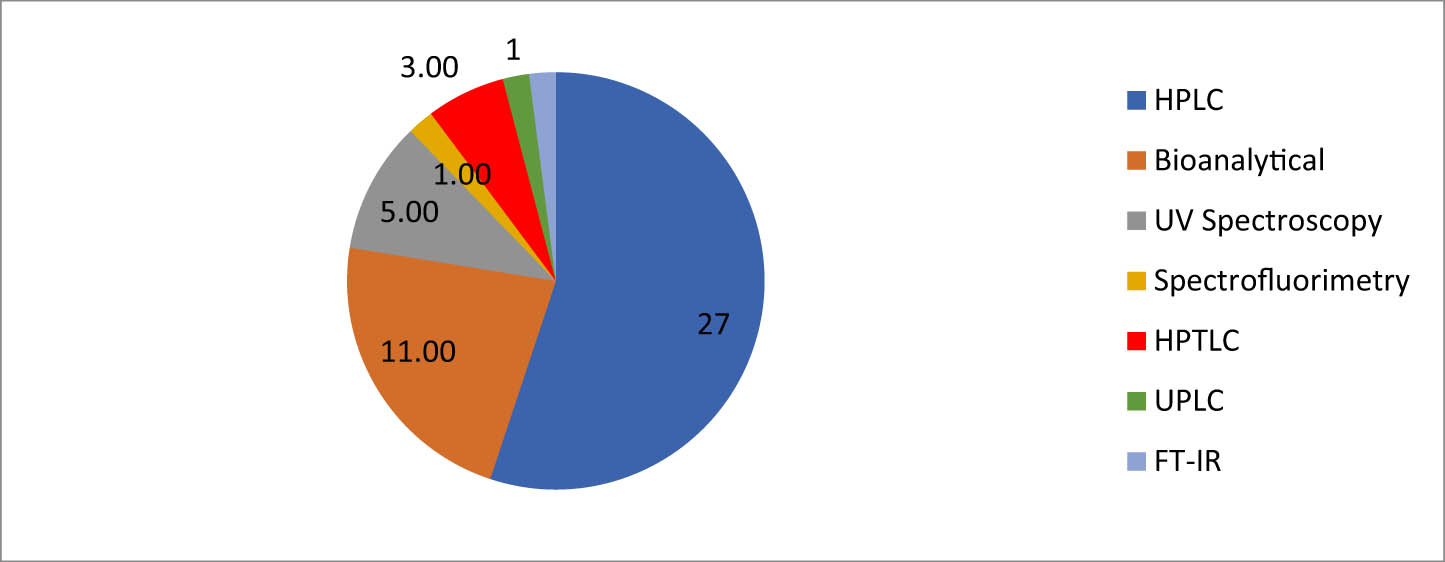
Analytical methods for the estimation of CGN.
Figure 3 provides the graphical representation of the number of articles published for the quantification of CGN from 2014 to 2022.

Number of analytical methods reported during 2014–2021. Database sources: ScienceDirect, Springer, PubMed, Scopus, Taylor & Francis, and Web of Science.
2 Spectroscopic methods
2.1 UV-Vis spectroscopic methods
UV-Vis spectroscopy methods are employed in pharmaceutical product estimation for their simplicity, versatility, accuracy, speed, and cost-effectiveness. Over a 40 years period, it has become the most important analytical instrument where expensive instruments like HPLC, Gas chromatography, Liquid chromatography with tandem mass spectrometry (LC-MS/MS) are not available. Singh et al. [5] developed a UV spectroscopic method using methanol as a diluent and measured the absorbance at 280 nm with a linearity of 5–50 µg·mL−1. Chinta et al. [6] reported a UV spectroscopic method using phosphate buffer as diluent at an absorbance wavelength of 289 nm and attained 99% purity with a linearity of 1–6 µg·mL−1. Ishpreet et al. [7] developed a UV spectroscopic method with a linearity of 5–10 µg·mL−1 at an absorbance of 290 nm and recovery within 80.00–120.00%. Vichare et al. [8] have reported two simultaneous methods, one based on absorbance correction UV spectroscopy (absorbance measurement at wavelengths 233 nm (λ max of metformin (MFN)) and 291 nm (λ max of CGN) and another based on first order derivative spectroscopy overlain spectra wavelengths 243 nm (zero absorbance of CGN) and 318 nm (zero absorbance of MFN) with a linearity of 0.75–4.5 µg·mL−1 for CGN and 2.5–15 µg·mL−1 for MFN, respectively. The percentage drug contents were found to be 98.48% ± 0.83% and 100.76% ± 1.29% for method A and 97.94 ± 0.96 and 97.22 ± 1.15 for CGN and MFN, respectively. The spectroscopic method existing for the determination of CGN as a single entity or in combination with other drugs [5,6,7,8] and the results (solvent, wavelength measured, linearity, accuracy, limit of detection (LOD), and limit of quantitation (LOQ)) obtained are represented in Table 1.
Overview of published UV-Vis spectroscopic methods
| Drug | Dosage form | Accuracy | LOD (µg·mL−1) | LOQ (µg·mL−1) | Reference |
|---|---|---|---|---|---|
| CGN | Bulk, tablets | 99.46–100.31% | 0.00945 | 2.8638 | Singh et al. [5] |
| CGN | Bulk, tablets | 80–120% | NA | NA | Chinta et al. [6] |
| CGN | Bulk, tablets | 80–120% | 0.084 | 0.255 | Ishpreet et al. [7] |
| CGN and MFN | Bulk, tablets | CGN: 98.48% and MFN: 100.76% | NA | NA | Vichare et al. [8] |
| CGN: 97.94% and MFN: 97.22% | NA | NA |
2.2 Spectrofluorimetry method
Spectrofluorimetry is a highly sensitive analytical method for the detection and determination of fluorescent compounds at ng or lower level. Nirav et al. [9] reported a specific, accurate, precise and robust spectrofluorometric method using methanol as the solvent, which emit excitation λ max at 293 nm and emission λ max at 349 nm. The method was linear over the range of 100–500 ng·mL−1 with a percentage recovery of 99.42–99.81%. The LOD and LOQ for the developed method were found to be 13.58 and 41.15 ng·mL−1, respectively. The method was novel, sensitive, and can be applied for the routine analysis of CGN by spectrofluorimetry [9].
3 Chromatographic methods
3.1 HPLC
HPLC is the most versatile and dominant separation technique in modern pharmaceutical and biomedical analysis due to its highly efficient separation and enhanced detection sensitivity. Most of the drugs are analyzed by HPLC method due to its accuracy, ease of automation, rapidity, specificity, and accuracy. Vymyslicky et al. [10] reported a new stability indicating HPLC method using electrochemical detection which significantly saves time and reduces the consumption of active ingredients and solvents, thereby minimizing the costs of forced degradation. Using ammonium phosphate buffer with methanol in a 50/50 volume ratio as a working electrolyte, the authors electrochemically oxidized samples and analyzed them by HPLC. Oxidation with hydrogen peroxide required 7 days, whereas electrochemical oxidation was completed in 3 h. Reddy et al. [11] reported a HPLC method which eluted CGN at a longer retention of 7.3 min using acetonitrile:water (50:50) as the mobile phase. The method was found to be sensitive over the other reported methods. Sadasivuni and Gundoju [12], using C18 column and acetonitrile:pH 2.5 with orthophosphoric acid (50:50) as the mobile phase, had developed an accurate method but did not mentioned the retention time (Rt). Mounika et al. [13] had developed a cost-effective, accurate, and sensitive method compared to other methods using a cost-effective mobile phase and eluted CGN at a Rt of 3.3 min. Another author had used methanol:water (90:10) and eluted CGN at 4.4 min (Sushil et al. [14]). Singh et al. [5] reported an accurate, precise, and linear method using acetonitrile:orthophosphoric acid (0.01 M) (50:50) and C18 column and eluted CGN at 4.7 min. Bhatt et al. [15] used acetonitrile:ammonium acetate buffer (pH 4.5) (70%:30% v/v) to elute CGN at 4.5 min. Parida et al. [16] reported a method using methanol:phosphate buffer with pH 4 (65:35) as mobile phase and C18 column using variable wavelength detector (VWD) at 293 nm and eluted CGN at 2.9 min. The Rt was comparatively less than the other methods. Sreenivasulu et al. [17] had reported a novel method using ion pair reagent, acetonitrile:1-octane sulphonic acid buffer (70:30) as the mobile phase and Rt of CGN was found at 3.4 min. The authors had achieved a good retention by using an ion pair reagent. Another author reported a method using orthophosphoric acid (0.1%):acetonitrile (60:40) which had a longer Rt at 8.7 min (Rahul et al. [18]) compared to the other reported methods. Vijaya et al. [19] developed a sensitive, selective method using 0.02% formic acid:acetonitrile (40:60) as the mobile phase and C18 column and achieved separation at 4.4 min. Goutam et al. [20] separated CGN from its process and degradation related substances using Ascentis Express RP-amide (150 mm × 4.6 mm, amide groups chemically bonded to porous silica particles of 2.7 µm) column, and ammonium acetate buffer:acetonitrile as the mobile phase at 290 nm. This method can be used to estimate the related substances of CGN. Triveni et al. [21], Ishpreet et al. [22], and Ladva et al. [23] have reported rapid, sensitive, accurate, and linear method using C18 as the stationary phase and various ratios of buffers (formic acid in water (0.1% v/v), ortho phosphoric acid, and 0.1% ammonium acetate:organic solvent (acetonitrile, methanol) as the mobile phase. The detectors used for the estimation were UV and PDA detectors. Suma et al. [24] had developed a cost effective, accurate method which eluted CGN at 2.8 min using HPLC grade water:acetonitrile (55:45 v/v) as the mobile phase. Al-Shdefat et al. [25] developed a simple, easy, and precise stability indicating HPLC method with diode array detector (DAD) for the simultaneous determination of CGN and MFN in a combined dosage formulation using a triple combination of mobile phase 0.05 M H3PO4, acetonitrile, and methanol in the 45:45:10 (v/v). The Rt for MFN and CGN were obtained at 8 and 9.45 min, respectively, which was a longer Rt compared to the other reported methods and found to be linear over the range of 5–100 g·mL−1. Authors Sunitha et al. [26], Bangaruthalli et al. [27], and Gandla et al. [28] performed simultaneous estimation of CGN and MFN using C18 as the stationary phase and wide choice of mobile phases. The methods were validated as per ICH guidelines. Moussa et al. [29] reported a simple and precise HPLC method for the quantitative estimation of CGN, empagliflozin (EGN), linagliptin (LNG), and MFN in their pharmaceutical formulation using a systematic and practical experimental design approach with minimum experimental trials. Authors selected three factors affecting the peaks significantly and determined using Plackett–Burman design. This method was found to be in good agreement with the observed value approving that design of experiment was simple methodology which was easy to instrument and chromatographically dependable in analyzing the single and multi-component drugs. Khalil et al., [30] developed the first reverse phase HPLC (RP-HPLC) method for simultaneous determination of CGN, dapagliflozin (DGN), EGN, and MFN on C18 column (250 mm × 4.6 mm, 5 µm p.s) Inertsil ODS through isocratic elution using acetonitrile and 0.05 M potassium dihydrogen phosphate buffer pH 4 in a ratio of 65:35, v/v as a mobile phase at flow rate of 1 mL·min−1. The method showed good linearity, accuracy, precision, and was successfully applied for determination of the four drugs in laboratory prepared mixtures and in the seven pharmaceutical dosage forms. Jyothi and Umadevi [31], Wafaa Zaghary et al. [32], Vinutha et al. [33], Kommineni et al. [34], Sonia et al. [35], and Panigrahy and Reddy [36] performed simultaneous estimation of CGN and MFN using C18 as the stationary phase and wide choice of mobile phases. The authors had developed RP-HPLC method for the estimation of CGN in single and in combined dosage forms by varying the chromatographic conditions and achieved better retention. The various HPLC methods (method, matrix, sample preparation, internal standard, column/mobile phase) available for quantification of CGN in pharmaceutical dosage forms as single and in combination [5,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36] and the results (Rt, detection, and linearity) obtained are presented in Table 2.
Summary of the reported HPLC methods for the determination of CGN in bulk and its dosage forms
| Drug | Column (dimension) | Mobile phase | Detector, detection wavelength (nm) | Flow rate (mL·min−1) | Rt (min) | Linearity (µg·mL−1) | Accuracy (%) | LOD (µg·mL−1) | LOQ (µg·mL−1) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| CGN | NA | Ammonium phosphate buffer:methanol (50:50) | Electrochemical detection | 0.1 mL·h−1 | NA | NA | NA | NA | NA | Vymyslicky et al. [10] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:water (50:50) | UV: 290 | 1 | 7.3 | From 98 ng·mL−1 to 50 µg·mL−1 | 98.9–99.8 | 0.048 | 0.098 | Reddy et al. [11] |
| CGN | C18 (100 mm × 4.6 mm, 5 µm) | Acetonitrile: orthophosphoric acid with pH 2.5 (50:50) | UV: 235 | 1 | NA | 10–200 | NA | NA | NA | Sadasivuni and Gundoju [12] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:0.1% sodium acetate (pH 4.6) (20:80) | 291 | 1 | 3.3 | 2–14 | 99.98 | NA | NA | Mounika et al. [13] |
| CGN | Grace C18 (4.6 mm × 250 mm, 5 µm) | Methanol:water (90:10) | UV: 290 | 0.9 | 4.4 | 1–5 | 99.95–106.18 | 0.7212 | 2.1854 | Sushil Patil et al. [14] |
| CGN | NA | Acetonitrile:orthophosphoric acid (0.01 M) (50:50) | 280 | 0.9 | 4.732 | 2–40 | 91.70–102.03 | NA | NA | Singh et al. [5] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:ammonium acetate buffer (pH 4.5) (70:30%, v/v) | UV: 252 | 1 | 4.5 | 5–30 | 98–101 | 0.01 | 0.04 | Bhatt et al. [15] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Methanol: phosphate buffer (pH 4) (65:35) | VWD: 293 | 1 | 2.9 | 10–125 | 99.33–99.92 | 0.9 | 2.7 | Parida et al. [16] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:1-octane sulphonic acid (70:30) | PDA: 245 | 1 | 3.4 | 10–100 | 98–102 | 0.017 | 0.1705 | Sreenivasulu et al. [17] |
| CGN | X-bridge C18 (4.6 mm × 150 mm, 5 µm) | Orthophosphoric acid (0.1%):acetonitrile (60:40) | 230 | 1 | 8.7 | 25–75 | 99.6–99.8 | 3.58 | 10.85 | Rahul et al. [18] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | 0.02% formic acid:acetonitrile (40:60) | UV: 230 | 1.2 | 4.4 | 10–50 | 98.44–100.31 | 0.00136 | 0.00414 | Vijayalakshmi et al. [19] |
| CGN, RS-1,2,3,4 | RP-amide (4.6 mm × 150 mm, 2.7 µm) | Ammonium acetate:acetonitrile (50:50) | UV: 290 | 0.7 | 23 | 0.055–1.177 | NA | 0.0005 | 0.015 | Goutam et al. [20] |
| CGN | C18 (4.6 mm × 150 mm, 5 µm) | Methanol:formic acid in water (0.1% v/v) (90:10) | UV: 220 | 1 | 2.4 | 100–300 | 99.9–100.8 | 0.0009 | 0.029 | Triveni et al. [21] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:ortho phosphoric acid (55:45) | PDA: 290 | 1 | 6.29 | 1–6 | 99.6–99.8 | 0.41 | 1.24 | Ishpreet et al. [22] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | Methanol:acetonitrile:0.1% ammonium acetate (40:40:20) | UV: 290 | 1.1 | 4.1 | 100–300 | 98.04–100.27 | 3.538 | 10.72 | Ladva et al. [23] |
| CGN | ODS (150 × 4.6 mm, 5 µm) | Water:acetonitrile (55:45, v/v) | PDA: 214 | 1 | 2.8 | 25–150 | NA | 0.037 | 0.112 | Suma et al. [24] |
| CGN | C18 (4.6 mm × 250 mm, 5 µm) | 0.05 M H3PO4:acetonitrile:methanol (45:45:10 (v/v)) | DAD: 238 | 1 | CGN: 8.00 | 5–100 | 98–102 | CGN: 0.653 | CGN: 2.167 MFN: 0.874 | Al-Shdefat et al. [25] |
| MFN | MFN: 9.45 | MFN: 0.261 | ||||||||
| CGN | Spolar C18 (4.6 mm × 250 mm, 5 µm) | pH 6 phosphate buffer:acetonitrile (55:45) | UV: 254 | 0.8 | CGN: 10.77 | CGN: 5–30 | NA | NA | NA | Sunitha et al. [26] |
| MFN | MFN: 3.24 | MFN: 50–300 | ||||||||
| CGN | Kromasil C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:phosphate buffer (pH 4.2):methanol (52:38:10) | 254 | 1 | CGN: 3.223 | CGN: 5–30 | NA | NA | NA | Bangaruthalli et al. [27] |
| MFN | MFN: 2.216 | MFN: 50–300 | ||||||||
| CGN | Primesil C18 (4.6 mm × 250 mm, 5 µm) | Methanol:phosphate buffer (70:30) | PDA: 220 | 1 | CGN: 3.47 | CGN: 50–250 | CGN: 99.38–99.53 | CGN: 4.13 | CGN: 0.112 | Gandla et al. [28] |
| MFN | MFN: 2.413 | MFN: 5–25 | MFN: 99.18–99.91 | MFN: 2.1 | MFN: 0.0372 | |||||
| CGN | C8 (250 mm × 4.6 mm, 5 μm) | pH 6 phosphate buffer (0.05 M):acetonitrile:methanol (50:25:25, v/v/v) | UV | 1.5 | CGN: 8.3 | 50–350 | 98–102 | 12.96 | 39.29 | Moussa et al. [29] |
| EGN | MFN: 2.2 | 500–3,500 | 57.51 | 174.29 | ||||||
| LNG | CGN/EMG: 3.6 | 10–70 | 1.1 | 3.33 | ||||||
| MFN | EMG: 6.0 | 20–140 | 1.73 | 5.24 | ||||||
| LNG/MFN: 3.6 | 1.25–8.75 | 0.11 | 0.32 | |||||||
| CGN | Inertsil, ODS C18 (250 mm × 4.6 mm, 5 µm) | Acetonitrile:0.05 M potassium dihydrogen phosphate (pH 4) (65:35) | UV: 212 | 1 | CGN: 4.414 | CGN: 7.5–225 | CGN: 2.1 | CGN: 6.5 | Khalil et al. [30] | |
| DGN | DGN: 3.560 | DGN: 5–150 | DGN: 0.7 | DGN: 2.2 | ||||||
| EGN | EGN: 3.004 | EGN: 6.5–187.5 | EGN: 1.4 | EGN: 2.3 | ||||||
| MFN | MFN: 1.898 | MFN: 10–1,000 | MFN: 3.2 | MFN: 9.6 | ||||||
| CGN | Inertsil ODS 3 VC18 (250 mm × 4.6 mm, 5 µm) | TFA in water (0.1% v/v):acetonitrile (20:80) | UV: 254 | 1 | CGN: 4.20 | CGN: 20–40 | NA | NA | NA | Jyothi and Umadevi [31] |
| MFN | MFN: 2.32 | MFN: 200–600 | ||||||||
| CGN | C18 (100 mm × 4.6 mm, 5 µm) | Methanol:0.03 M phosphate buffer (75:25) | 240 | 1.3 | CGN: 0.9 | CGN: 1–50 | CGN: 99.81 ± 0.73 | CGN: 0.7154 | CGN: 2.1075 | Wafaa et al. [32] |
| MFN | MFN: 0.5 | MFN: 0.5–90 | MFN: 99.37 ± 0.54 | MFN: 0.9327 | MFN: 2.8263 | |||||
| CGN | Kromasil C18 (4.6 mm × 250 mm, 5 µm) | 0.1% OPA (pH 2.8):acetonitrile (45:55) | PDA: 254 | 1 | CGN: 2.671 | CGN: 2.5–15 | 98.2–101.4 | CGN: 0.01 | CGN: 0.50 | Vinutha et al. [33] |
| MFN | MFN: 2.112 | MFN: 25–15 | MFN: 0.17 | MFN: 2.20 | ||||||
| CGN | Kromasil C18 (4.6 mm × 250 mm, 5 µm) | 0.1% orthophosphoric acid (pH 2.8):acetonitrile (45:55) | UV: 254 | 1 | CGN: 2.671 | CGN: 2.5–15 | 98.22–101.54 | CGN: 0.01 | CGN: 0.50 | Kommineni et al. [34] |
| MFN | MFN: 2.112 | MFN: 25–150 | MFN: 0.17 | MFN: 2.20 | ||||||
| CGN | Gracesmart C18 (4.6 mm × 250 mm, 5 µm) | Acetonitrile:ammonium acetate (pH 4.5) (45:55) | PDA: 252 | 1 | CGN: 5.76 | 1–80 | CGN: 99.19–100.57 | CGN: 0.124 | CGN: 0.376 | Sonia et al. [35] |
| MFN | MFN: 4.00 | MFN: 98.65–100.87 | MFN: 0.134 | MFN: 0.406 | ||||||
| CGN | Kromasil C18 (4.6 mm × 250 mm, 5 µm) | 0.01 M (pH 3.5) ammonium acetate:acetonitrile (65:55) | PDA: 254 | 1 | CGN: 3.713 | CGN: 5–30 | CGN: 99.45–100.65 MFN: 99.95–100.74 | NA | NA | Panigrahy and Reddy [36] |
| MFN | MFN: 2.440 | MFN: 50–300 |
3.2 UPLC technique
UPLC is a recent separation technique which reduces the cost and increases the rapidity and efficiency of analysis required for developing and validating the method. Wafaa et al. [32] validated an UPLC method for the determination of MFN and CGN in tablets using Hypersil Gold (50 mm × 3 mm, 1.9 µm) as the stationary phase and methanol:0.03 M phosphate buffer (80:20) considered as the mobile phase at 0.4 mL·min−1 flow rate. The Rt of CGN and MFN was 1 and 0.5 min using a detection wavelength of 240 nm. Accuracy of CGN and MFN was 99.47% ± 1.03% and 99.73% ± 0.89% with a linearity of 0.1–50 and 0.25–100 µg·mL−1 for CGN and MFN, respectively. LOD and LOQ were 0.463 and 1.406 µg·mL−1 for CGN and 0.614 and 1.862 µg·mL−1 for MFN, respectively [32].
3.3 HPTLC technique
HPTLC is a powerful technique for qualitative and quantitative analysis. It contains chromatographic layers with the highest separation efficiency, the possibility to use multiple detection methods on the same sample and plate. Accurate quantitative measurements and high resolution makes HPTLC to meet all quality requirements for today’s analytical labs.
Vichare et al. [37] reported a novel HPTLC method for the simultaneous estimation of CGN and MFN using toluene:methanol:triethyl amine:glacial acetic acid (7:2.6:0.2:0.2, v/v/v/v) as a mobile phase. The detection was done at 254 nm with an excellent resolution of retardation factor (R f) values 0.21 ± 0.02 and 0.50 ± 0.03 for MFN and CGN, respectively. Detection and quantitation limits were found to be 8.01 and 24.28 ng/band for MFN and 8.27 and 25.07 ng per band for CGN, respectively. The proposed method was applied to marketed formulation, the % drug contents were found to be 98.20% ± 0.24% and 99.33% ± 1.80% w/w for MFN and CGN, respectively. This HPTLC method was novel and found to be more linear, accurate, precise, and sensitive compared to the other reported methods [37].
Ishpreet et al. [38] validated a HPTLC method using toluene:ethyl acetate:methanol (2:2:1) as the mobile phase and silica gel 60 F254 as the stationary phase for the estimation of CGN in tablet dosage form. The densitometric detection was at 290 nm. The recovery was 99.04–99.82% with a linearity of 10–500 ng per spot. LOD and LOQ were 0.39 ng/spot and 119 ng/spot, respectively. The % assay of CGN tablets was found to be 99.8%. Forced degradation studies of CGN showed stability in acidic, alkaline, photolytic, and oxidation [38].
Bhole et al. [39] developed a combination of CGN and MFN using silica gel 60 F 254 (10 × 10 cm, 250 µm) as the stationary phase and toluene:ethyl acetate:methanol:ammonia (4:4:2:0.1) as mobile phase at a wavelength of 254 nm. The Rf of MFN and CGN was found to be 0.15 and 0.50 ng/band, respectively. The linearity of MFN and CGN was found to be 0.5–3 and 50–300 ng/band, respectively. The proposed HPTLC method selectively quantitated MFN and CGN in the presence of the degradation products [39].
4 Bioanalytical methods
Bio-analytical techniques are used for the quantitative determination of drugs and their metabolites in biological fluids, which plays a major role in the evaluation and interpretation of bioequivalence, pharmacokinetic, and toxicokinetic studies. Hence, the development of selective, sensitive, and reliable bio-analytical methods for the quantitative evaluation of drugs and their metabolites in biological matrices is important. An analytical method for the quantification of CGN in human plasma has been developed, validated, and enforced for the analysis of samples. The analytical method consists of extraction of the drug by protein precipitation method, the internal standard DGN was added to the plasma sample and extracted using protein denaturant acetonitrile followed by centrifugation. The supernatant was dried and reconstituted using the mobile phase and injected into the HPLC (Ajitha et al. [40]). Another method employed extraction of sample by liquid extraction using tertiary butyl methyl ether in human Plasma. In liquid–liquid extraction (LLE) the plasma was spiked with internal standard and tert-butyl methyl ether followed by centrifugation. The supernatant was dried and reconstituted with a solvent and then injected into the HPLC/LC-MS/MS. Determination was done using Zorbax XDB phenyl (7.5 × 4.6 mm, 3.5 mm) column and methanol:acetate (80:20) as the mobile phase by LC-MS/MS. The method used a deuterium labeled CGN as internal standard and eluted at 1.15 min with a linearity of 10–7,505 ng·mL−1 (Deepan et al. [41]). Udhayavani et al. [42] employed a LC-MS method to quantify CGN in human plasma using solid phase extraction. In solid-phase extraction (SPE), the plasma sample was spiked with ammonium acetate and internal standard into the SPE cartridge followed by conditioning and washing to extract CGN and injected into the HPLC/LC-MS/MS. Another author also followed a similar method but showed variation by using C18 (100 × 4.6 mm, 5 µm) as the stationary phase and 2 mM ammonium acetate:methanol (15:85) as the mobile phase (Somarouthu et al. [43]). Darshan et al. [44] estimated CGN by LC-MS/MS from rabbit plasma using EGN as internal standard. The author extracted CGN using LLE with tertiary butyl methyl ether using C18 (50 × 4.6 mm, 5 µm) column and 0.01 M ammonium acetate:methanol (30:70) as the mobile phase. CGN was extracted from rat plasma by LLE using LC-MS/MS (Kobuchi et al. [45]). Nalawade et al. [46] developed a simultaneous method and successfully applied for the determination of pharmacokinetics of MFN and CGN by using 100 μL of rat plasma. Mohamed et al. [47] performed the simultaneous estimation of CGN and MFN in biological samples (rat plasma) by protein precipitation extraction (PPE; acetonitrile) and LLE (ethyl acetate) using valsartan, CGN d4, MFN d6, tadalafil, and propranolol as the internal standard using LC-MS/MS. The authors estimated the plasma concentrations of the studied drugs in a pharmacokinetic study involving Egyptian health volunteers. Ramisetti et al. [48] reported a simple, rapid, and sensitive LC-MS/MS method for the simultaneous quantification of CGN and MFN. The authors used a one-step PPE for samples preparation and obtained highest recovery for the analytes. This method was successfully used for the in vivo plasma concentrations obtained from a pharmacokinetic study. Syeda et al. [49] used HPLC to estimate CGN and MFN in human plasma using PPE (2% v/v acetic acid in acetonitrile) with pioglitazone as the internal standard. The method was simple, accurate, precise, and can be applied in bioequivalence, pharmacokinetic, and toxicokinetic studies with desired precision and accuracy along with high-throughput. Another author also reported a sensitive, novel HPLC method using SPE as the extraction method (Deepan et al. [50]).The bio-analytical methods published for the quantitative analysis of CGN separately and in drug combination (40–50) and the results (method, matrix, sample preparation, internal standard, column/mobile phase, Rt, detection technique, and linearity) are represented in Table 3.
Summary of bio-analytical methods for the quantification of CGN alone and in combination with other drugs
| Drug | Matrix | Sample preparation | Internal standard | Column/mobile phase | Rt (min) | Detection | Linearity (ng·mL−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| CGN | Human plasma | PPE | DGN | Phenomenex Luna C18 (150 × 4.6 mm, 5 µm)/0.01 M phosphate buffer (pH 3.5):acetonirile (45:55) | 8.7 | UV: 222 nm | 60–2,400 | Ajitha et al. [40] |
| CGN | Human plasma | Tertiary butyl methyl ether | CGN d4 | Zorbax XDB Phenyl (7.5 mm × 4.6 mm, 3.5 mm) | 1.15 | m/z: 462.2–267.10 for CGN | 10–7,505 | Deepan et al. [41] |
| Methanol: acetate (80:20) | m/z: 466–267.2 for IS | |||||||
| CGN | Human plasma | SPE with ammonium acetate | CGN d4 | Zodiac C18 (100 mm × 4.6 mm, 5 µm) | 1.5 | MS/MS | 10.253–6019.311 | Udhayavani et al. [42] |
| Methanol:2 mM ammonium acetate (90:10) | ||||||||
| CGN | Human plasma | SPE with ammonium acetate | CGN d4 | C18 (100 mm × 4.6 mm, 5 µm) | 1.5 | m/z: 462.2–267.10 for CGN | 10.3–6,019 | Somarouthu et al. [43] |
| 2 mM ammonium acetate:methanol (15:85) | m/z: 466–267.1 for IS | |||||||
| CGN | Rabbit plasma | Tertiary butyl methyl ether | EGN | C18 (50 mm × 4.6 mm, 5 µm) | 2 | m/z: 462.1–267.10 for CGN | 5 × 105 to 600 | Darshan et al. [44] |
| 0.01 M ammonium acetate:methanol (30:70) | m/z: 451.2–71.10 for IS | |||||||
| CGN | Rat plasma | LLE with tertiary butyl methyl ether | EGN | Quicksorb ODS (150 mm × 2.1 mm, 5 µm) | NA | m/z: 462.1–191 for CGN | NA | Kobuchi et al. [45] |
| Acetonitrile:0.1% formic acid (90:10) | m/z: 451.2–71.0 for IS | |||||||
| CGN | Rat plasma | NA | Valsartan | Methanol:0.1% formic acid (65:35) | CGN: 8.2 | MS/MS | 200–8,000 for CGN | Vivek et al. [46] |
| MFN | MFN: 1.83 | 100–4,000 for MFN | ||||||
| IS: 6.2 | ||||||||
| CGN | NA | Protein precipitation | Internal standard 1-CGN d4 | Hypersil BDS C18 (100 mm × 4.6 mm, 5 µm) 5 mM ammonium acetate 0.01% formic acid:methanol (15:85) | CGN: 1.5 | MS/MS | 10–6,028 for CGN | Muralikrishna et al. [47] |
| MFN | Internal standard 2-MFN d6 | MFN: 1.8 | 10–3,027 for MFN | |||||
| CGN | Human plasma | PPE with acetonitrile | CGN-Tadalafil | C18 (50 mm × 4.6 mm, 5 µm) | CGN: 4.5 | m/z: 130.2–60.1 for MFN | 50–5,000 for MFN | Ramisetti et al. [48] |
| MFN | LLE with ethyl acetate | MFN-propranolol | 0.1% formic acid:acetonitrile | MFN: 0.9 | 462.3–191.0 for CGN | 10–1,000 for CGN | ||
| 260.2–183.0 for propranolol | ||||||||
| 390.2–268.8 for tadalafil | ||||||||
| CGN | NA | Protein precipitation | Pioglitazone | Spursil (Dikma) ODS C18 (4.6 mm × 250 mm, 5 µm) | CGN: 8.352 | NA | 250–1,250 for MFN | Syeda et al. [49] |
| MFN | 2% v/v acetic acid in acetonitrile | Acetonitrile:phosphate buffer (pH 3.0) (15:85% v/v) | MFN: 4.738 | 25–125 for CGN | ||||
| Pioglitazone: 10.995 | ||||||||
| CGN | Human plasma | SPE | Pioglitazone | Inertsil ODS C18 (4.6 mm × 250 mm, 5 µm) | CGN: 8.10 | NA | 5,000–25,000 for MFN | Deepan et al. [50] |
| MFN | (pH 3 with sodium hydroxide) phosphate buffer:acetonitrile (85:15) | MFN: 4.6 | 500,000–1,250,000 for CGN | |||||
| Pioglitazone: 10.64 |
5 Multiple spectroscopic methods
Elnadi et al. [51] reported three novel, simple, accurate, and sensitive methods for the determination of CGN using FTIR, spectrofluorimetry, a stability-indicating UV-Vis spectroscopic method for the simultaneous estimation of CGN and MET. Method A is a green FTIR method using KBr disc for CGN determination measuring alkyl halide C–F peak area centered on 1,230 cm−1. Method B is a spectrofluorimetry method using Δλ = 50 nm synchronous mode at a peak maximum of 291.8 nm for CGN determination using methanol as diluent. Method C is a stability-indicating MCRS method measuring the peak amplitude of CGN and MET at 306.2 and 246.6 nm, respectively, in their mixture with complete CGN oxidation degradation. All the three spectroscopic methods can be used efficiently for routine analysis in QC laboratories [51].
6 Discussion
CGN is considered as a primary member in the gliflozins family, which has a considerable contribution since 2013. Authors Singh et al. [5], Ishpreet et al. [7], and Vichare et al. [8] used methanol as the diluent to develop an accurate and linear method for the estimation of CGN separately and in combined dosage forms by UV-Vis spectroscopic technique. Vichare et al. [8] reported two analytical methods for the simultaneous estimation of CGN and MFN using methanol as the diluent. Both the methods showed good recovery with a linearity of 0.75–4.5 and 2.5–15 µg·mL−1 for CGN and MFN, respectively. Whereas Chinta et al. [6] developed a cost effective, sensitive method using phosphate buffer as the diluent which is a better method. HPLC method is utilized for the estimation of CGN separately and in combination with other drugs in bulk and pharmaceutical formulations. Rahul et al. [18] reported a cost-effective method for the estimation of CGN using X-Bridge C18 (4.6 × 150 mm, 5 µm) as stationary phase and orthophosphoric acid (0.1%):acetonitrile (60:40) as the mobile phase. Rt of CGN is about 8.7 min with a linearity of 25–75 µg·mL−1 and recovery of 99.6–99.8% at detection wavelength of 230 nm. This method had a higher Rt of 8.7 min compared to all the other reported methods. The authors would have tried by varying the mobile phase composition for a better method. Another author Reddy et al. [11] reported a HPLC method in which CGN eluted at 7.3 min, but achieved a linearity in the range from 98 ng·mL−1 to 50 µg·mL−1 indicating the sensitiveness of the method. Mounika et al. [13] developed a method which was cost effective method using ecofriendly solvents compared to the other reported methods. Panigrahy et al. [36] reported a cost-effective method for the simultaneous estimation of CGN and MFN using kromasil C18 (4.6 × 250 mm, 5 µm) as the stationary phase and 0.01 M (pH 3.5) ammonium acetate:acetonitrile (65:55) as the mobile phase at a detection wavelength of 254 nm. CGN eluted at 3.713 min with % recovery of 99.45–100.65%. MFN eluted at 2.44 min with % recovery of 99.85–100.74%. This method was validated as per ICH guidelines and found to be sensitive, accurate, and precise. Many bio-analytical methods using LC-MS/MS and a few HPLC methods were reported for CGN separately and in combination with other drugs. Deepan et al. [41] reported a LC-MS/MS method for the estimation of CGN using Zorbax XDB phenyl (7.5 × 4.6 mm, 3.5 mm) as the stationary phase and methanol:acetate (80:20) as the mobile phase with CGN d4 as the internal standard. LLE was performed by using tert butyl methyl ether as the extraction solvent. CGN eluted at 1.15 min with an m/z: 462.2–267.10 for CGN and m/z: 466–267.2 for CGN d4 and a linearity of 10–7505 ng·mL−1. The method was selective and sensitive compared to all the reported methods. Deepan et al. [50] reported a simultaneous method for the estimation of CGN and MFN by HPLC in human plasma using Inertsil ODS C18 (4.6 × 250 mm, 5 µm) as the stationary phase and (pH 3 with sodium hydroxide) phosphate buffer:acetonitrile (85:15) as the mobile phase. CGN eluted at 8.10 min and MFN eluted at 4.6 min with a linearity of 5–25 µg·mL−1 for MFN and 500–1,250 µg·mL−1 for CGN, respectively. Bio-analytical method using HPLC is always cost effective compared to the sophisticated LC-MS/MS techniques. But LC-MS/MS methods are sensitive and selective compared to the HPLC methods. All reviewed methods were validated as per ICH guidelines.
7 Conclusion
This detailed review explains the existing methods for the determination of CGN in pharmaceutical formulations and in biological fluids using spectroscopy, UPLC, HPLC, HPTLC, and LC-MS/MS. This review has presented the complete review of the methods reported right from the drug approval time to up-to-date. In future, researchers can take efforts to develop more methods using eco-friendly solvents for the determination of CGN in pharmaceutical dosage forms and employ sample extraction methods using green analytical techniques (single-drop micro-extraction, liquid-phase micro-extraction, liquid–liquid–liquid micro-extraction, single short column, solid-phase micro-extraction, stir-bar-sorptive extraction, matrix solid-phase dispersion, supercritical-fluid extraction, pressurized-liquid extraction, subcritical-water extraction, microwave-assisted extraction, sonication-assisted solvent extraction). More methods based on LC-MS/MS can pave the way for the toxicokinetic studies and therapeutic monitoring of CGN.
Acknowledgments
The authors are privileged to thank the Management of Sri Ramachandra Institute of Higher Education and Research (Deemed to be University) for helping us to carry out this work.
-
Funding information: Authors state no funding involved.
-
Author contributions: Ajitha Azhakesan: writing – original draft, writing – review and editing, methodology, and formal analysis; Sujatha Kuppusamy: writing – original draft and formal analysis.
-
Conflict of interest: Authors state no conflict of interest.
References
[1] Sha S, Devineni D, Ghosh A. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13:669–72.10.1111/j.1463-1326.2011.01406.xSuche in Google Scholar PubMed
[2] Polidori D, Sha S, Mudaliar S. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo-controlled study. Diabetes Care. 2013;36(8):2154–61.10.2337/dc12-2391Suche in Google Scholar PubMed PubMed Central
[3] Skelley JW, Carter BS, Roberts MZ. Clinical potential of canagliflozin in cardiovascular risk reduction in patients with type 2 diabetes. Vasc Health Risk Manag. 2018;14:419–28. 10.2147/vhrm.s168472.Suche in Google Scholar
[4] Budoff MJ, Wilding JPH. Effects of canagliflozin on cardiovascular risk factors in patients with type 2 diabetes mellitus. Int J Clin Pract. 2017;71(5):12948. 10.1111/ijcp.12948.Suche in Google Scholar PubMed PubMed Central
[5] Singh D, NeenaBedi, Tiwary Bedi AK. Comparison of UV Spectroscopic and HPLC method for estimating canagliflozin in bulk and tablet dosage form. Indian J Pharm Sci. 2019;81(1):39–44.10.4172/pharmaceutical-sciences.1000477Suche in Google Scholar
[6] Chinta P, Gurdeep S, Dharmesh S. Development and validation of UV spectrophotometric method for the estimation of canagliflozin in bulk and pharmaceutical dosage form. Int J Pharm. 2018;13(1):1–9.Suche in Google Scholar
[7] Ishpreet K, Sharad W, Harsharan PS. Development and validation of UV spectroscopic method for determination of canagliflozin in bulk and pharmaceutical dosage form. Pharm Method. 2015;6(2):82–6.10.5530/phm.2015.6.11Suche in Google Scholar
[8] Vichare VS, Choudhari VP, Venkat Reddy M. Development and validation of UV-visible spectroscopic methods for simultaneous estimation of canagliflozin and metformin in pharmaceutical formulation. Asian J Res Chem. 2019;12(1):16–20. 10.5958/0974-4150.2019.00004.Suche in Google Scholar
[9] Nirav P, Pratik T, Ashish M, Shailesh S. Development and validation of spectrofluorimetric method the estimation of canagliflozin in its pharmaceutical dosage form. Pharma Sci Monit. 2019;10(2):147–58.Suche in Google Scholar
[10] Vymyslicky F, Krizek T, Kozlik P, Kubickova A, Hert J, Bartosinska E. Alternative method for canagliflozin oxidation analysis using an electrochemical flow cell-comparative study. J Pharm Biomed Anal. 2022;207:114341.10.1016/j.jpba.2021.114341Suche in Google Scholar PubMed
[11] Reddy GS, Patel S, Nagappan KV, Bhavyasri K, Mounika C, Sumakanth M, et al. Development and validation of reverse phase high performance liquid chromatography method of the estimation of canagliflozin in bulk and its pharmaceutical formulation. J Young Pharm. 2020;12(4):321–6.10.5530/jyp.2020.12.85Suche in Google Scholar
[12] Sadasivuni H, Gundoju NR. Analytical Method Development and Validation of Canagliflozin hemihydrates in bulk and pharmaceutical dosage forms. Mukt Shabd J. 2020;IX(VI):1690–8.Suche in Google Scholar
[13] Mounika G, Bhavya Sri K, Swethasri R, Sumakanth M. Research article on development and validation of RP-HPLC method for estimation of canagliflozin. Samriddhi. 2019;11(2):85–92.10.18090/samriddhi.v11i02.2Suche in Google Scholar
[14] Sushil Patil D, Muqeet SA, Sanjay Shrisagar J. Development and validation of stability indicating RP-HPLC method for Canagliflozin. Asian JResearch Chem. 2019;12(1):11–5.10.5958/0974-4150.2019.00003.8Suche in Google Scholar
[15] Bhatt D, Thatavarthi P, Rajkamal B. Analytical method development and validation for the estimation of canagliflozin in bulk and formulation by RP-HPLC. Int J Pharm Sci Drug Res. 2018;10(3):139–43.10.25004/IJPSDR.2018.100306Suche in Google Scholar
[16] Parida AK, Rao KS, Patnaik AK. RP-HPLC method for the estimation of canagliflozin in bulk and pharmaceutical dosage forms. Int J Pharm. 2018;6(1):2308–13.Suche in Google Scholar
[17] Sreenivasulu S, Rameswara RM, Chandrasekhar KB. A validated stability indicating RP-HPLC method for the quantification of canagliflozin. Int J Res Pharm Sci. 2018;9(1):206–15.10.26452/ijrps.v9i1.1248Suche in Google Scholar
[18] Rahul KV, Amuthalakshmi S, Nalini CN. Validation of a stability indicating reverse phase HPLC method for determination of canagliflozin API. World J Pharm Res. 2018;7(3):459–68.Suche in Google Scholar
[19] Vijaya LM, Aisha S, Lakshmi P, Buchi N. A novel validated RP-HPLC method for the estimation of canagliflozin in bulk and pharmaceutical dosage forms. Int J Adv Pharm. 2017;7(3):24–7.Suche in Google Scholar
[20] Goutam S, Raghu BK, Annapurna N, Vekariy NA, Vundavilli JK, Pavan Kumar KSR, et al. Validation of stability-indicating reverse phase HPLC method for the determination of related substances in canagliflozin drug substance. IOSR J Pharm Biol Sci. 2017;12(6):86–94.Suche in Google Scholar
[21] Triveni V, Ram VD, Swathi M, Trivani N, Durga P, Rao T, et al. Method development and validation for the estimation of canagliflozin in drug substance by RP-HPLC method. Indo Am J Pharm Res. 2017;7(3):7973–8.Suche in Google Scholar
[22] Ishpreet K, Sharad W, Harsharan PS, Satish M. Development and validation of a stability indicating reverse phase HPLC-PDA method for determination of canagliflozin in bulk and pharmaceutical dosage form. Pharm Methods. 2016;7(1):54–62.10.5530/phm.2016.7.9Suche in Google Scholar
[23] Ladva BJ, Dobariya PV, Pancholi HD, Nayak BS, Jain S. RP-HPLC Development and validation of chromatographic method for estimation of canagliflozin in API and tablet dosage form. Int J Recent Sci Res. 2016;7:10976–79.Suche in Google Scholar
[24] Suma M, Manasa K, Rajakumari Ch, Lakshmaiah B. RP-HPLC method development and validation for the estimation of canagliflozin in tablet dosage form. Int J Pharm. 2014;5(4):1288–92.Suche in Google Scholar
[25] Al-Shdefat R, Al-Ani I, Tamimi L, Awad R, Rayyan WA, Dayyih WA, et al. Development and validation of a stability-indicating HPLC-DAD method for the determination of canagliflozin and metformin simultaneously in combination dosage form. Pharm Chem J. 2021;55(4):402–9.10.1007/s11094-021-02436-7Suche in Google Scholar
[26] Gurrala S, Shivaraj SC, Anumolu PD, Saraf G. Analytical quality by design assisted HPLC method for quantification of canagliflozin/metformin and stability studies. Indian J Pharm Educ. 2019;53(4):699–709.10.5530/ijper.53.4s.167Suche in Google Scholar
[27] Bangaruthalli J, Gowri Shankar D, Renuka MNL, Akhila P, Vanga D. Method Development and Validation of simultaneous estimation of metformin and canagliflozin by using RP-HPLC. Int J Sci Eng Res. 2018;9(11):1309–19.Suche in Google Scholar
[28] Gandla KSR, Lalitha Ch, Mounika B, Soumya D, Kumar S. A validated RP-HPLC method for simultanious determination of metformin and canagliflozin in pharmaceutical formulation. Asian J Pharm Ana. 2018;8(2):73–7. 10.5958/2231-5675.2018.00014.5.Suche in Google Scholar
[29] Moussa BA, Mahrouse MA, Fawzy MG. Application of experimental design in HPLC method optimization and robustness for the simultaneous determination of canagliflozin, empagliflozin, linagliptin, and metformin in tablet. Biomed Chromatogr. 2021;35(10):e5155.10.1002/bmc.5155Suche in Google Scholar PubMed
[30] Khalil GA, Salama I, Gomaa MS, Helal MA. Validated RP-HPLC method for simultaneous determination of canagliflozin, dapagliflozin, empagliflozin and metformin. Int J Pharm Chem Biol Sci. 2018;8(1):1–13.Suche in Google Scholar
[31] Jyothi U, Umadevi P. Analytical method development and validation for the simultaneous estimation of canagliflozin and metformin in drug product by RP-HPLC. Eur J Biomed Pharm. 2017;4:298–303.Suche in Google Scholar
[32] Wafaa Zaghary A, Mowaka S, Moataz Hendy S. Comparative liquid chromatographic study for concurrent determination of canagliflozin and metformin in combined tablets. J Anal Methods Chem. 2017;2017:1–9.10.1155/2017/9197230Suche in Google Scholar PubMed PubMed Central
[33] Vinutha K, Chowdary KP, Prasad SV. Development and validation of a new RP-HPLC method for simultaneous estimation of metformin hydrochloride and canagliflozin and its comparison with the reported methods. World J Pharm Pharm Sci. 2017;6(3):696–713.Suche in Google Scholar
[34] Kommineni V, Chowdary KPR, Prasad MUVS. Development of a new stability indicating RP-HPLC method for simultaneous estimation of metformin hydrochloride and canagliflozin and its validation as per ICH guidelines. Int J Pharm Sci Res. 2017;8(8):3427–5.Suche in Google Scholar
[35] D'souza S, Krishna M, Sushmitha GS, Vasantharaju SG. Stability indicating assay method development and validation to simultaneously estimate metformin hydrochloride and canagliflozin by RP-HPLC. Curr Trends Biotechnol Pharm. 2016;10(4):334–42.Suche in Google Scholar
[36] Panigrahy UP, Reddy AS. A novel validated RP-HPLC-DAD method for the simultaneous estimation of metformin hydrochloride and canagliflozin in bulk and pharmaceutical tablet dosage form with forced degradation studies. Orient J Chem. 2015;31(3):1489–507.10.13005/ojc/310328Suche in Google Scholar
[37] Vichare VS, Choudhari VP, Reddy MV. Development of new validated HPTLC Method for simultaneous estimation of canagliflozin and metformin in tTablet formulation. Res J Pharm Technol. 2022;15(6):2599–604.10.52711/0974-360X.2022.00434Suche in Google Scholar
[38] Kaur I, Wakode S, Singh HP. Development and validation of a stability-indicating high performance thin layer chromatography (HPTLC) method for estimation of canagliflozin in bulk and pharmaceutical dosage form. J Appl Pharm Sci. 2016;6(5):51–7.10.7324/JAPS.2016.60508Suche in Google Scholar
[39] Bhole RP, Gandhi JS, Wankhode SB. Development and validation of HPTLC method for simultaneous estimation of canagliflozin and metformin hydrochloride in bulk and tablet dosage form. Basic Pharm Science/Pharmacognosy. 2017;84(6):65–71.Suche in Google Scholar
[40] Ajitha A, Sujatha K, Abbulu K. A novel and rapid RP-HPLC quantitative method for the estimation of canagliflozin in human plasma. Ijrps. 2021;12:93–101.10.26452/ijrps.v12i1.3940Suche in Google Scholar
[41] Deepan T, Rao MB, Dhanaraju MD. Bioanalytical method development and validation of Canagliflozin in human plasma by Liquid Chromatography-tandem mass spectrometry. Asian J Pharm Clin Res. 2019;12(8):46–51.10.22159/ajpcr.2019.v12i18.33228Suche in Google Scholar
[42] Udhayavani S, Girija Sastry V, Govinda Rajan R, Tejaswi DKJ, Rajani R. Method development and validation of canagliflozin in human plasma by liquid chromatography tandem mass spectrometry (LC-MS/MS). Int J Pharma Bio Sci. 2018;9(2):140–7.10.22376/ijpbs.2018.9.2.p140-147Suche in Google Scholar
[43] Somarouthu VS, Nageswara RP, Bhavani PK, Bimireddy PSP. A novel and rapid LC-MS/MS assay method for the determination of canagliflozin in human plasma by solid phase extraction technique and its application to a pharmacokinetic study. Future J Pharm Sci. 2018;4(2):131–8.10.1016/j.fjps.2017.12.003Suche in Google Scholar
[44] Bhatt DA, Rajkamal B. A validated LC-MS/MS method for pharmacokinetic study of canagliflozin in healthy rabbits. Int J Pharm Pharm. 2018;10(2):80–6.10.22159/ijpps.2018v10i2.23245Suche in Google Scholar
[45] Kobuchi S, Yano K, Ito Y, Sakaeda T. A validated LC-MS/MS method for the determination of canagliflozin, a glucose co-transporter 2 (SGLT-2) inhibitor, in a lower volume of rat plasma; application to pharmacokinetic studies in rats. Biomed Chromatogr. 2016;30(10):1549–55.10.1002/bmc.3720Suche in Google Scholar PubMed
[46] Nalawade V, Dixit VA, Vora A, Zade H. Development and validation of an LC-MS/MS method for simultaneous determination of canagliflozin and metformin HCl in rat plasma and its application. Curr Pharm Anal. 2020;16(6):752–62.10.2174/1573412915666190312161823Suche in Google Scholar
[47] Mohamed D, Elshahed MS, Nasr T, Aboutaleb N, Zakaria O. Novel LC-MS/MS method for analysis of metformin and canagliflozin in human plasma application to a pharmacokinetic study. BMC Chem. 2019;13(82):1–11.10.1186/s13065-019-0597-4Suche in Google Scholar PubMed PubMed Central
[48] Ramisetti M, Atmakuri LR, Venkata BR, Adireddy V. Simulataneous determination of canagliflozin and metformin in human plasma by LC-MS/MS assay and its application to a human pharmacokinetic study. Indian J Pharm Educ Res. 2019;53(3 suppl 2):s304–72.10.5530/ijper.53.3s.107Suche in Google Scholar
[49] Syeda K, VidyaSagar G, Senthil Kumar R, Kazim SM. Bioanalytical method development and validation for metformin and canagliflozin by RP-HPLC. World J Pharm and Pharm Sci. 2017;6(12):371–81.Suche in Google Scholar
[50] Deepan T, Rao B, Dhanaraju MD. Bioanalytical method development and validation for metformin and canagliflozin drugs in human plasma by RP-HPLC method. Middle-East J Sci Res. 2017;25(7):1451–7.Suche in Google Scholar
[51] Elnadi S, Abdalsabour S, Farouk M, Trabik YA. Fourier transform infrared spectroscopic, spectrofluorimetric assays of canagliflozin, and stability-indicating UV-spectrophotometric method for the simultaneous determination of canagliflozin and metformin. J AOAC Int. 2022;105(4):964–71.10.1093/jaoacint/qsac030Suche in Google Scholar PubMed
© 2022 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Research Articles
- Abnormal retention of s-triazine herbicides on porous graphitic carbon
- One-factor-at-a-time method combined with ICP-MS for determining 11 elements in soy sauce and their migration from the containing glass bottles
- Analysis of initiator content of prepreg by near-infrared spectroscopy
- Simultaneous MEKC-DAD and smart spectrophotometric assays of thiocolchicoside and etoricoxib in challenging concentration ratio mixtures
- Alternative analytical methods for ibrutinib quantification in pharmaceutical formulation: A statistical comparison
- Chemometric determination of common cold infection drugs in human urine
- An effective, novel, and cheap carbon paste electrode for naproxen estimation
- Fabrication of ultra-sensitive carbon paste electrode with nanocomposite CdS modification for electroanalysis of rafoxanide in dosage form and biological fluids
- Purification and characterisation of phytochemicals extracted from Rhizophora mucronata: Their efficacy against Pseudomonas aeruginosa infection in Catla catla
- Review Articles
- Recent applications of quantitative analytical FTIR spectroscopy in pharmaceutical, biomedical, and clinical fields: A brief review
- Review of characteristics and analytical methods for determination of indomethacin
- A review of the application of comprehensive two-dimensional gas chromatography MS-based techniques for the analysis of persistent organic pollutants and ultra-trace level of organic pollutants in environmental samples
- Enrichment and analysis of glycated proteins
- Round robin tests of secondary raw materials: A systematic review of performance parameters
- Paper-based microfluidic devices: Fabrication, detection, and significant applications in various fields
- Applications of headspace solid-phase microextraction in human biological matrix analysis
- Chemometrics and infrared spectroscopy – A winning team for the analysis of illicit drug products
- Canagliflozin: A review with specific focus on analytical methods in biological matrices and pharmaceuticals
- RNA-based isothermal amplification technology and its clinical application in pathogen infection
- Detection of diarrheal shellfish toxins
- Special Issue: Nanomaterials with mimetic enzymatic properties for label-free sensors and biosensors (Guest Editors: Gang Wei and Zhiqiang Su)
- Preparation of cuprous oxide-supported silver-modified reduced graphene oxide nanocomposites for non-enzymatic electrochemical sensor
- Recent advances in matrix metalloproteinases-responsive nanoprobes for cancer diagnosis and therapy
- A simple salicylaldehyde-bearing pyrazine as a turn-on fluorescent chemosensor for Al3+ and Zn2+ recognition and its applications
- Preparation of silver nanosheet-assembled film as a surface-enhanced Raman scattering substrate
- Synthesis of group I–III–VI semiconductor quantum dots and its application in food safety testing
Artikel in diesem Heft
- Research Articles
- Abnormal retention of s-triazine herbicides on porous graphitic carbon
- One-factor-at-a-time method combined with ICP-MS for determining 11 elements in soy sauce and their migration from the containing glass bottles
- Analysis of initiator content of prepreg by near-infrared spectroscopy
- Simultaneous MEKC-DAD and smart spectrophotometric assays of thiocolchicoside and etoricoxib in challenging concentration ratio mixtures
- Alternative analytical methods for ibrutinib quantification in pharmaceutical formulation: A statistical comparison
- Chemometric determination of common cold infection drugs in human urine
- An effective, novel, and cheap carbon paste electrode for naproxen estimation
- Fabrication of ultra-sensitive carbon paste electrode with nanocomposite CdS modification for electroanalysis of rafoxanide in dosage form and biological fluids
- Purification and characterisation of phytochemicals extracted from Rhizophora mucronata: Their efficacy against Pseudomonas aeruginosa infection in Catla catla
- Review Articles
- Recent applications of quantitative analytical FTIR spectroscopy in pharmaceutical, biomedical, and clinical fields: A brief review
- Review of characteristics and analytical methods for determination of indomethacin
- A review of the application of comprehensive two-dimensional gas chromatography MS-based techniques for the analysis of persistent organic pollutants and ultra-trace level of organic pollutants in environmental samples
- Enrichment and analysis of glycated proteins
- Round robin tests of secondary raw materials: A systematic review of performance parameters
- Paper-based microfluidic devices: Fabrication, detection, and significant applications in various fields
- Applications of headspace solid-phase microextraction in human biological matrix analysis
- Chemometrics and infrared spectroscopy – A winning team for the analysis of illicit drug products
- Canagliflozin: A review with specific focus on analytical methods in biological matrices and pharmaceuticals
- RNA-based isothermal amplification technology and its clinical application in pathogen infection
- Detection of diarrheal shellfish toxins
- Special Issue: Nanomaterials with mimetic enzymatic properties for label-free sensors and biosensors (Guest Editors: Gang Wei and Zhiqiang Su)
- Preparation of cuprous oxide-supported silver-modified reduced graphene oxide nanocomposites for non-enzymatic electrochemical sensor
- Recent advances in matrix metalloproteinases-responsive nanoprobes for cancer diagnosis and therapy
- A simple salicylaldehyde-bearing pyrazine as a turn-on fluorescent chemosensor for Al3+ and Zn2+ recognition and its applications
- Preparation of silver nanosheet-assembled film as a surface-enhanced Raman scattering substrate
- Synthesis of group I–III–VI semiconductor quantum dots and its application in food safety testing