Clifford Gaussians as Atomic Orbitals for periodic systems: one and two electrons in a Clifford Torus
-

 and
and

Abstract
We recently proposed a new approach that permits to apply the methods of Quantum Chemistry to infinite periodic systems. This is based on the transformation of the topology of a finite fragment of the periodic system into a Clifford Torus, and a redefinition of the distances in this new manifold. The standard Gaussian orbitals, that are so common in Quantum-Chemistry calculations, are also redefined, in order to obtain smooth orbitals whose behavior is compatible with the periodicity of the torus. We call these new functions “Clifford Gaussians”. Finally, after a set of calculations on systems having increasing size, an extrapolation to a system having infinite size is performed, in order to obtain the crystal properties at the thermodynamic limit. In the present paper, we apply this formalism to the study of two model systems: the free particle in a ring (a 1D system), and a pair of electrons in a square (2D) or a cube (3D). In the first case, for which the spectrum of the Hamiltonian is analytically known, we show that a set of s-type Clifford Gaussians is able to reproduce the exact eigenvalues of the system with an extremely high accuracy, suggesting that the Clifford Gaussians, under some circumstances, could form a complete basis set for the torus. In the case of the second system, that had already be studied by our group by using a large number of ordinary Gaussians, we show that two Clifford Gaussians only are able to reproduce the behavior of the system with a good accuracy. In particular, they are able to describe the Wigner localization of the electrons that occurs at very low density. We believe that this formalism could open a new approach for the study of infinite periodic systems within the powerful and well established framework of Quantum Chemistry.
Introduction
The use of first-principle formalisms, Quantum Mechanics in particular, in order to investigate the behavior of atoms, molecules and clusters is nowadays a well established practice in science. The resulting field is Quantum Chemistry, a very successful branch of Science. Therefore, it would be tempting to apply the same philosophy to crystals and other “infinite” periodic systems. In principle, one could consider a crystal as a large molecular system, and apply the powerful Quantum-Chemistry machinery to the study of its properties. Two main obstacles make this strategy not straightforward at all:
The long-range nature of the Coulomb interaction: The decrease of the inter-charge potential as the inverse of their distance implies that an enormous number of atoms should be considered before the effect of the interaction between a distant pair of them could be neglected.
The presence of surface effects: The surface of a cluster has properties that are totally different from its interior. The use of dangling atoms to terminate a crystal fragment does not completely remove the surface effects.
For this reason, the application of Quantum Chemistry to the study of extended systems is not trivial, and presents problems that are far from being totally solved. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 Things are relatively simple in the case of mean-field methods, like Hartree–Fock (HF) or Density-Functional Theory (DFT). In this case, the Bloch Theorem can be used in order to take advantage of the periodicity of the system, and reduce the calculation to the elementary unit cell only. On the other hand, the situation becomes much more problematic if the electron-electron repulsion is explicitly taken into account, in order to include the effects of the electron correlation in the description of the system.
In recent years, in order to eliminate the surface effects and the problems with the Coulomb interaction, we investigated the possibility of changing the topology of the cluster, and give the system the structure of a Clifford Torus, because of the reasons that will become clear in the following. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 For this reason, we named our approach as based on Clifford Boundary Conditions. Notice that the use of a somehow similar topology was propose by J. Noga and coworkers, working on semi-empirical Hamiltonians. 26 They called their approach as the “Cyclic Cluster” formalism. In their work, however, the connection with the Clifford-Torus topology was not explicitly stated. Moreover, the distance involved in the Coulomb interaction was not modified.
A Clifford Torus (CT) is a border-less and flat surface having a finite-extension. It was first conceived and discussed by William Kingdon Clifford in 1873. Although originally proposed as a 2D manifold, a CT can be straightforwardly generalized to any number of dimension n. We report here part of the introduction of Klaus Volkert’s historical article on the Bulletin of the Manifold Atlas: 27 , 28
Clifford-Klein space forms entered the history of mathematics in 1873 during a talk which was delivered by W. K. Clifford at the meeting of the British Association for the Advancement of Sciences (Bradford, in September 1873) and via an article he published in June 1873. 29 The title of Clifford’s talk was ‘On a surface of zero curvature and finite extension’, the proceedings of the meeting only provide this title. But we know a bit more about it from F. Klein who attended Clifford talk and who described it on several occasions (for example, see 30 ). The mathematical properties of Clifford Torus make it very appealing in order to describe a finite fragment of an infinite periodic system. In particular, its finite extension is suited to describe a finite portion of the infinite system. Moreover, the absence of borders eliminates undesired surface effects. Finally, the flatness of the manifold implies that a crystal fragment (or supercell) can be accommodated into the torus without any distortion, contrary to what happens if we would fold, for instance, a rectangle into an ordinary torus. The price one has to pay is that this type of tori do not exist in our ordinary 3D space, except for the trivial case of a 1D torus, when the torus is simply a circle. However, a Clifford Torus having dimension n can be naturally embedded into a complex space having the same number of dimensions. In this article, the cases n = 1, 2 or 3 will be considered.
The embedding of an n-dimension torus

Three different distances between two points, A and B, on a 1D Clifford Torus (equivalent to a circle): two Geodesic distances (the absolute minimum one, in green color) and a longer one (blue); the unique Euclidean distance (red). Notice that, strictly speaking, there are infinite many other Geodesic distances, obtained by adding kL (with k being a positive integer) to the two ones shown in the figure.
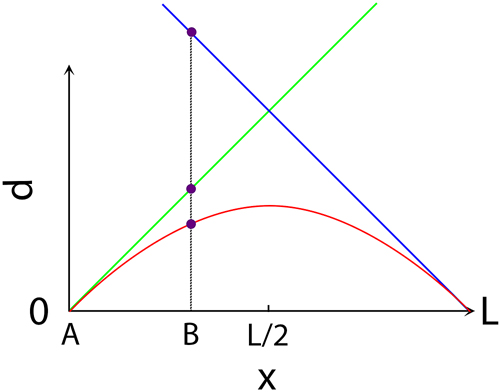
The plot of the three distances of the previous figure, as a function of the position x of the point B on the circle, the point A being fixed in the origin. The maximum value of the shortest one among the two Geodesic distances is L/2, while the maximum value of the Euclidean distance is L/π.
Once we have set the topology of the supercell and the Coulomb interaction between the particles, we need a wavefunction for the system. In the Quantum-Chemistry language, this means that we need the orbitals that describe the electrons. In the “Linear Combination of Atomic Orbitals” (LCAO) scheme, the Molecular Orbitals (MO) of the system are obtained as linear combinations of the Atomic Orbitals (AO) of the atoms. In the overwhelming majority of this type of calculations, the AO of the system are linear combinations of Gaussian functions, originally proposed by Boys as suitable functions to describe AOs.
31
Unfortunately, ordinary Gaussians have a major problem if used on a Torus: they are defined on the whole space
In this article, the Clifford formalism (i.e., Clifford boundary conditions and Clifford Gaussians) is used in order to investigate the behavior of two simple, model systems:
One electron on a 1-Dimensional (1D) torus (actually, a ring).
two electrons on a 2-Dimensional (2D) or 3-Dimensional (3D) torus.
The first system is analytically solvable. In atomic units, the eigenvalues of the Hamiltonian are given by
This article is organized as follows. In Section “The Clifford formalism” we describe the Clifford formalism for periodic extended systems. In Section “Gaussian orbitals on a torus: Clifford Gaussians” we define the Clifford Gaussians on a torus, and some of their properties are discussed. In Section “One electron on a ring: an analytically solvable model” we study 1D system composed of one electron confined on 1D Torus, which is nothing but a ring. In Section “Two electrons in square and cubic Clifford Tori” we study the case of two electrons on a square or cubic torus. In Section “Conclusions and future perspectives”, we draw some conclusions, and discuss the future perspectives of our work. Finally, in the Appendix we prove the product theorem between two Clifford Gaussians, which is similar to the corresponding property holding for ordinary Gaussian functions.
The Clifford formalism
In the last few years, we have been developing an original formalism for the treatment of extended systems. The aim of the proposed formalism is to study a periodic infinite system by considering it as a large molecule, to which the standard ab initio methods of Quantum Chemistry can be applied. To this purpose, the following procedure is set up:
Extract a fragment (we call it a supercell, of the type discussed later) out of the infinite system, and give to the supercell the topology of a Clifford Torus. Then replace the ordinary geodesic distance that appears in the Coulomb interaction by a “renormalized” distance, given by the Euclidean distance in the complex space in which the Torus can be embedded.
Use periodic Gaussians, or “Clifford Gaussians”, possibly contracted ones, as the Atomic Orbitals of the system, compute the one- and two-electron integrals for these orbitals, and perform the desired ab initio calculation.
Repeat the two previous steps two by using toroidal supercells of similar shape but increasing sizes, in order to extrapolate the desired properties from finite oligomers to the infinite system at the thermodynamic limit.
As already anticipated, some caution must be used in the choice of the supercell. This must be a connected fragment of the whole system, composed of an integer number N of elementary unit cells, N = n 1 ⋅ n 2 ⋅ n 3 in the 3D case, with similar expressions for the 1D and 2D crystals. Here n i is an integer positive number that denotes the number of unit cells in the ith principal axis of the supercell. The extrapolation mentioned at the previous point 3. must be performed on supercells containing λn 1 ⋅ λn 2 ⋅ λn 3 = λ 3 N unit cells, with λ a positive integer. Notice that the supercell λn 1 ⋅ λn 2 ⋅ λn 3 becomes the whole system in the limit λ → ∞. At the moment, our formalism is able to treat right supercells only, whose shape is a rectangle in 2D and a right parallelepiped in 3D. Work is in progress to overcome this limitation.
We notice that, with respect to the ordinary molecular Quantum-Chemistry calculation, our approach for solids only requires rather limited modifications. The crucial advantage of our procedure is that these modifications only concern the calculation of the one- and two-electron integrals and the inter-nuclear repulsion, but are completely independent from the Quantum-Chemistry method used in the calculation. Therefore, once these modifications have been applied, any Quantum-Chemistry method (like Hartree–Fock (HF), Configuration Interaction (CI), Coupled Cluster (CC), Perturbation Theory (PT), etc.) can be used to perform calculations on periodic solids. Notice, however, that the use of Size-Consistent methods, like HF, CC or PT, is strongly recommended in order to extrapolate the results to the thermodynamic limit, step 3.
In summary, the proposed approach reduces the calculation of a solid to several calculations on Clifford supercells of increasing size. This is followed by the extrapolation to the thermodynamic limit. The smoothness and efficiency of the extrapolation are guaranteed by the topology of the Clifford Torus: the absence of boundaries, which eliminates border effects; and the zero Gaussian curvature, which insures that a crystal supercell can be fitted into the torus without any distortion. In a recent work, 25 by using the example of ground-state energy of a chain of hydrogen atoms, we have shown that in the thermodynamic limit the results obtained within our approach are strictly equivalent to those obtained for a ring of the same atoms.
Gaussian orbitals on a torus: Clifford Gaussians
Generally speaking, a Gaussian Function
where x
0 and x are two points that belong to
Equation (1) defines an unnormalized Gaussian. The corresponding normalized function,
where
In order to distinguish the Gaussian functions that are defined on different types of manifolds, we will use the notation
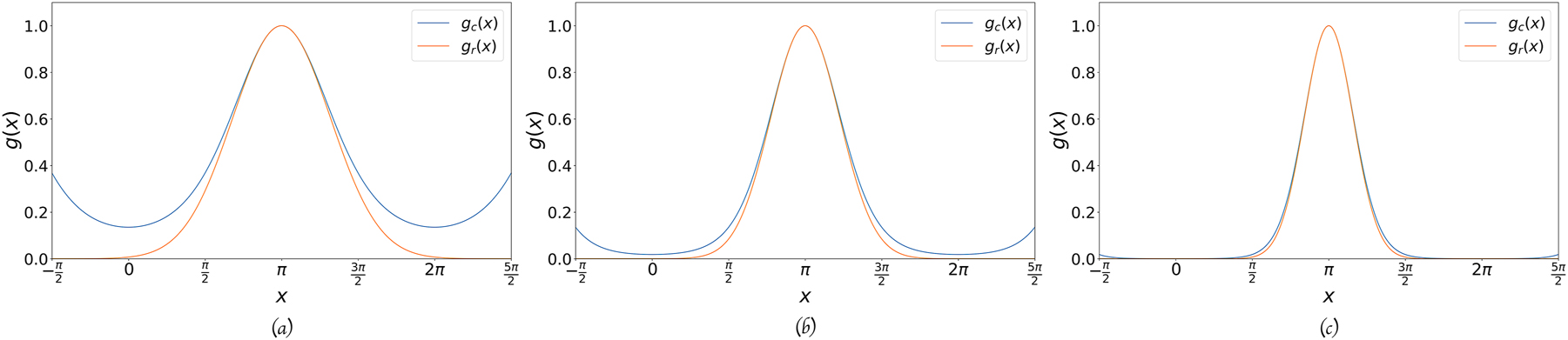
Comparison between the usual
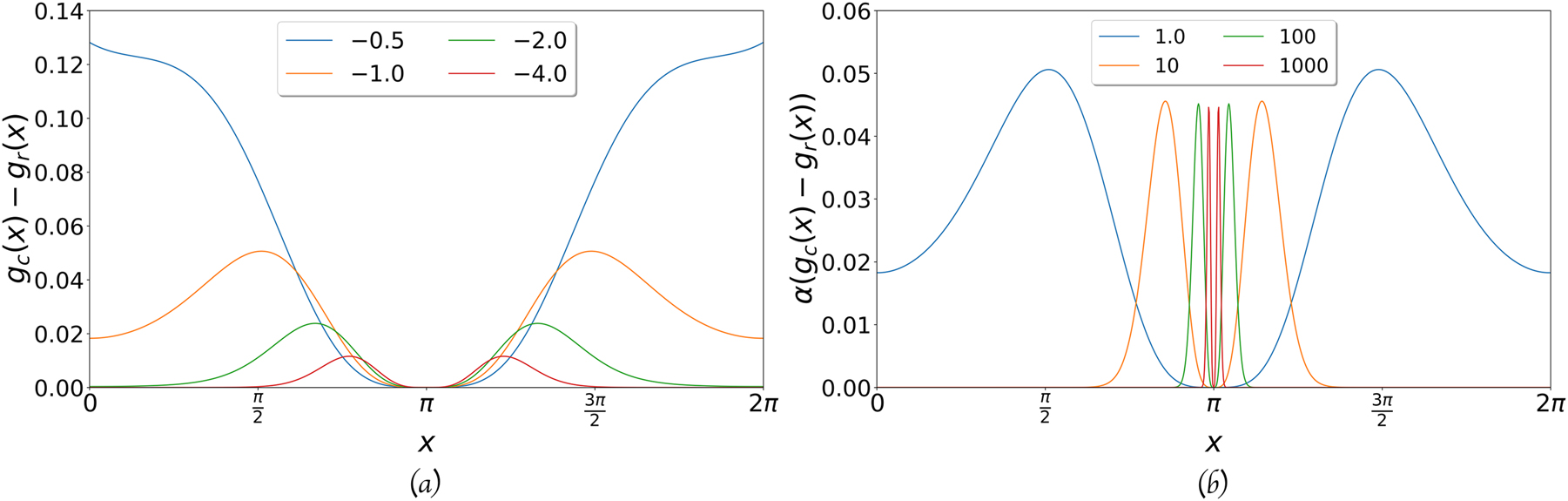
The difference between the two types of Gaussians for different values of the exponent. The center is in x = π. (a) The difference between the two functions, for α equal to 0.5, 1.0, 2.0, and 4.0. (b) The difference between the two functions times α, for α equal 1.0, 10.0, 100.0, and 1000.0.
The most common example of Atomic Orbitals is given by the Gaussian Atomic Orbitals, introduced by Boys in order to perform Molecular-Orbital calculations of atoms, molecules and periodic systems. 31 Today, the large majority of Quantum-Chemistry calculations is indeed performed within this type of orbitals. An s-type Gaussian Orbital is given, in 1D, by the function
where N
α
is a normalization constant. The exponent α determines the Gaussian width. In fact, the two inflection points of the curve are located at positions satisfying
It is possible to define Gaussian functions on other types of manifolds. For instance, Spherical-Gaussian Orbitals have been recently introduced by Loos, Gill and coworkers in order to describe the behavior of electrons on a 2D or 3D sphere.
32
,
33
,
34
,
35
On a Clifford Torus, continuous and derivable Gaussian Orbitals can be defined by using the Euclidean distance. For a Torus
where x 0 is the center of the Gaussian, x 0 ∈ [0, L x ]. In a similar way, in two dimensions one can define Clifford Gaussians as the product of two 1D Gaussians,
and, in three dimensions, one has
In principle, it is possible to have different exponential parameters for the different space directions, α
x
, α
y
, and α
z
. However, we will not take the opportunity of this freedom. Notice that Gaussian functions on the torus defined by using the Riemannian distance are continuous but not differentiable functions of the point positions, x and x
0. On the other hand, the Clifford Gaussians that will be considered in the article are defined by using the Euclidean distance, and they are
In actual calculations on atomic systems, Gaussians functions of higher angular momentum, p, d, f, and so on, are also considered, in order to describe the shell structure of the atoms. This definition can be extended to Clifford Gaussians, by replacing the function x and its powers in the polynomial part of orbital by the corresponding powers of
where
and
while
![Fig. 5:
The center and exponent of the Gaussian product of two Clifford
T
$\mathbb{T}$
-Gaussians. The first Gaussian has α = 1 and center in x = 0, and the second one, with variable α, is centered in x ∈ [0, 2π]. The four curves correspond to the value of the second exponent of 1.0, 0.99; 0.9, 0.1. (a) The center x
c
. (b) The exponent α
c
.](/document/doi/10.1515/pac-2025-0617/asset/graphic/j_pac-2025-0617_fig_005.jpg)
The center and exponent of the Gaussian product of two Clifford
The factorization property of the
One electron on a ring: an analytically solvable model
The model of an electron freely moving in a 1D Torus (or equivalently, a ring) is exactly solvable with an analytic approach. It is essentially coincident with the well known rigid-rotator system. For the sake of simplicity, and since the physics of the system do not depend on the size of the ring, we consider an electron confined on a ring of length 2π. In atomic units, the energy levels of the system are given by the expression
Therefore, all the energy levels are positive and doubly degenerate, except for the ground-state one, E 0 = 0. We describe the system through a set of N equally spaced 1D Clifford Gaussians, having a common exponent α. The spacing d between two neighboring Gaussians is therefore 2π/N. In our previous investigations by using ordinary Gaussians, we found that a crucial parameter that must be considered is the quantity ξ = αd 2, as we already mentioned in a previous Section. Indeed, if ξ is significantly lower than one, the system of Gaussians becomes almost linearly dependent. On the other hand, if ξ is much greater than one, the Gaussians are very sharp, and therefore are not able to describe a smooth function.
We computed the overlap and kinetic matrix elements on this basis set, and diagonalized the resulting Hamiltonian. The resulting approximated eigenvalues are denoted as
which is the mean quadratic deviation of the first N/2 approximate eigenvalues with respect to the exact ones. Notice that in the last equation the notations for the exact eigenvalues in eqs. (12) and (13) are slightly different, and no negative labels are considered for the approximate eigenvalues.
In Table 1, we report the results obtained for different values of N, and, for each value of N, for a set of different values of α. In order to simplify the comparison between the different cases, both N and α have been chosen as powers of two. However, a finer sampling of α could have been explored in order to better minimize Δ. For each value of N and α, we report in the table the value of ξ = α d
2, the lowest eigenvalue of the overlap, S
0, and the mean quadratic error
The resolution parameter, ξ, the lowest eigenvalue of the metric, S
0, end the mean quadratic error,
| N | α | ξ | S 0 |
|
|---|---|---|---|---|
| 16 | ||||
| 0.5 | 7.71 × 10−2 | 1.14 × 10−13 | 1.14 × 10−12 | |
| 1.0 | 1.54 × 10−1 | 2.17 × 10−9 | 3.68 × 10−16 | |
| 2.0 | 3.08 × 10−1 | 7.20 × 10−6 | 1.24 × 10−16 | |
| 4.0 | 6.17 × 10−1 | 2.55 × 10−3 | 3.40 × 10−10 | |
| 8.0 | 1.23 × 100 | 8.03 × 10−2 | 1.62 × 10−4 | |
| 16.0 | 2.47 × 100 | 4.25 × 10−1 | 4.18 × 10−1 | |
| 32.0 | 4.93 × 100 | 8.27 × 10−1 | 4.26 × 101 | |
| 64.0 | 9.87 × 100 | 9.85 × 10−1 | 7.66 × 102 | |
| 32 | ||||
| 16.0 | 6.17 × 10−1 | 2.24 × 10−3 | 3.04 × 10−10 | |
| 32.0 | 1.23 × 100 | 8.21 × 10−2 | 1.21 × 10−3 | |
| 64.0 | 2.47 × 100 | 0.43 × 100 | 4.97 × 100 | |
| 64 | ||||
| 32.0 | 3.08 × 10−1 | 1.24 × 10−6 | 1.52 × 10−14 | |
| 64.0 | 6.17 × 10−1 | 2.17 × 10−3 | 1.31 × 10−9 | |
| 128.0 | 1.23 × 100 | 8.25 × 10−2 | 1.28 × 10−2 | |
| 128 | ||||
| 64.0 | 1.54 × 10−1 | 2.72 × 10−13 | 3.31 × 10−11 | |
| 128.0 | 3.08 × 10−1 | 1.07 × 10−6 | 2.26 × 10−13 | |
| 256.0 | 6.17 × 10−1 | 2.15 × 10−3 | 1.18 × 10−8 | |
| 256 | ||||
| 128.0 | 7.71 × 10−2 | l.d. | / | |
| 256.0 | 1.54 × 10−1 | 2.73 × 10−13 | 3.12 × 10−10 | |
| 512.0 | 3.08 × 10−1 | 1.03 × 10−6 | 3.49 × 10−12 | |
| 512 | ||||
| 256.0 | 3.51 × 10−2 | l.d. | / | |
| 512.0 | 7.71 × 10−2 | l.d. | / | |
| 1024.0 | 1.54 × 10−1 | 3.93 × 10−13 | 1.28 × 10−9 | |
| 2048.0 | 3.08 × 10−1 | 1.02 × 10−6 | 5.85 × 10−11 | |
| 4096.0 | 6.17 × 10−1 | 2.14 × 10−3 | 2.06 × 10−6 | |
| 8192.0 | 1.23 × 100 | 8.27 × 10−2 | 3.61 × 101 |
Two electrons in square and cubic Clifford Tori
In this section, we use the Clifford formalism to describe the behavior of two electrons confined into a torus. The strictly 1D system is ill defined, because of the singularity of the 1D Coulomb potential at short range. Therefore, we limit our investigation to the 2D and 3D cases. In both the 2D and 3D cases, a single Clifford Gaussian is enough to describe one unpaired electron, so a total of two orbitals for the entire system is qualitatively enough.
The model
We consider square and cubic Clifford Tori of dimensions L
x
= L
y
= L and L
x
= L
y
= L
z
= L, respectively. In the torus, two Clifford-Gaussian orbitals having the same exponent α are considered. They are located in such a way to minimize the mutual Coulomb repulsion. This is obtained by maximizing the Euclidean distance d
L
between the two centers: x
1 ≡ (x
1, y
1) for g
1 and x
2 ≡ (x
2, y
2) for g
2, respectively, for
and
that, in a more compact way, can be written as
We obtain similar relations for the case of a 3D cubic Clifford Torus. Notice that the systems is translationally invariant, which means that only the difference between the centers x 1 and x 2 is relevant in the calculation.
The one- and two-electron integrals that are relevant for this calculation and involve Clifford Gaussians g i (x) are the following ones (notice that the orbitals are real):
Results
We considered several square and cubic Tori of different size, going from relatively high electron density (L = 1 bohr) to very low density (L = 106 bohr). Notice that with two electrons only and periodic boundary conditions, the high-density limit of the kinetic energy is zero, and one doubly occupied constant orbital minimizes the total energy. Therefore, the high-density limit is not particularly interesting, and we did not explore values of α lower than one. In each case, by spanning a large range of values of the exponent α, we performed the Self-Consistent Field (SCF) and Full Configuration Interaction (FCI) calculations. In order to do this, we numerically computed the 1-e and 2-e integrals, as discussed in ref. 25]. Then, for each value of the Torus size L, we computed the values of the exponents α that minimize the SCF and FCI energies for the ground 1Σ g state, in order to find the best two-orbital wavefunction that describes the system at these two levels.
The SCF wave-function
The exponent corresponding to the SCF energy minimum has been numerically computed for each value of L. The minimum is found for very small values of α, an this confirms the fact that the exact minimum position is located at α = 0. Since in this case the Gaussian orbital becomes a constant, the two orbitals are identical, so only one is kept for the calculation. This fact means that the two electrons are uniformly distributed in the Torus, and completely delocalized for any value of the system size L. This is not a surprising result, that is to be compared to the plane waves that describe the SF solution for the homogeneous infinite free-electron gas. In our case, since the two orbitals g 1 and g 2 are linearly dependent, the whole SCF + FCI procedure breaks down, since a single orbital must be used to describe the system. In this case, the SCF energy can be computed by simply considering that the kinetic energy vanishes, so the total SCF energy is given just by the electron-electron repulsion (11|11), evaluated for a normalized constant function. For this value of α, the FCI result coincide with the SCF one.
The FCI wave-function
Things are much more interesting at FCI level. In this case, due to the Coulomb repulsion, the two electrons tend to localize at opposite positions in the Torus. Therefore, we restricted our investigation to this position of the orbitals. As discussed in a previous article,
15
the shape of the wavefunction is the result of two different, competing, phenomena. The kinetic energy tends to distribute the electrons as much as possible, and reaches its minimum (T
min = 0) for a constant orbital. The electron-electron repulsion, on the other hand, reaches its minimum for two delta-like orbitals located as far apart as possible one from the other one in the Torus. In this case we have
The results for several values of L are reported in Table 2. For each L, the value of α which minimizes the ground-state 1Σ
g
energy is numerically obtained. At large values of L, the electrons are completely localized in one orbital, and the wave-function is totally neutral in the Valence-Bond language. In this case, the singlet and triplet lowest states,
The FCI energies (Hartree) of the different states as a function of the system size L, in Bohr, for the square
| L | E FCI (N*Gauss) ⋅ L | α | E(1Σ g ) ⋅ L | E(3Σ u ) ⋅ L | E(1Σ u ) ⋅ L |
|---|---|---|---|---|---|
| 1 | 3.830402736 | 0.6482352941 | 4.013014260 | 22.748626240 | 24.8102329 |
| 10 | 3.341151624 | 0.0551427593 | 3.806440756 | 4.956076291 | 7.21759770 |
| 100 | 2.696524187 | 0.0024538105 | 2.940311452 | 2.937831178 | 8.46944608 |
| 1000 | 2.367285277 | 0.0000614009 | 2.436287842 | 2.436287795 | 13.6487884 |
| 10 000 | 2.264144855 | 0.0000017332 | 2.288165632 | 2.288165632 | 23.1742091 |
| 100 000 | 2.231970680 | 0.0000007700 | 2.244004858 | 2.244004858 | 49.3331443 |
| 1 000 000 | 2.221862760 | 0.0000000190 | 2.222820000 | 2.222820000 | 78.8513000 |
|
|
|||||
| 1 | 2.872009610 | 0.1962500180 | 2.857732292 | 22.239451744 | 22.961051 |
| 10 | 2.652234404 | 0.0186927727 | 2.829355521 | 4.470156885 | 5.206218 |
| 100 | 2.248181503 | 0.0015801231 | 2.543355955 | 2.553775202 | 4.430400 |
| 1000 | 1.921838799 | 0.0000107829 | 2.034108452 | 2.034108453 | 11.905348 |
| 10 000 | 1.810452655 | 0.0000012884 | 1.887462369 | 1.887462369 | 12.727726 |
| 100 000 | / | 0.0000000662 | 1.829425683 | 1.829425683 | 39.288272 |
| 1 000 000 | / | 0.0000000041 | 1.817885691 | 1.817885691 | 52.637321 |
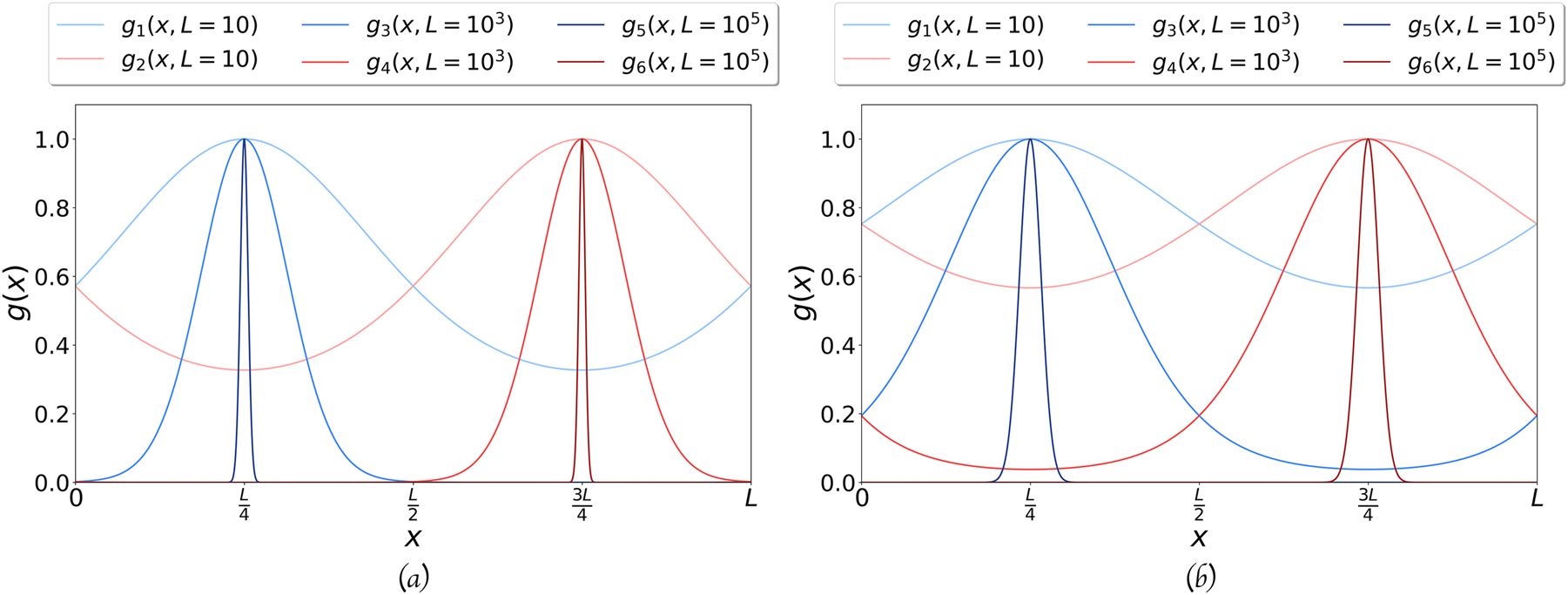
Comparison of the shape of the optimal Gaussians for different values of the side L. (a) The two Gaussian orbitals on the square: optimum value of α, for the square side L equal to 10 (g
1, g
2), 1000 (g
3, g
4), and 100 000 (g
5, g
6). A factor
It is interesting to compare these results, obtained by using a pair of Clifford Gaussians only, with the corresponding values from reference, for which the FCI energies by using a relatively large number of ordinary Gaussians were computed. 15 The present energy values are only a few percent higher than the FCI “exact” results for intermediate values of the density, probably because of dynamical-correlation effects, and only a fraction of percent at very low densities. This confirms the well known fact that the orbitals of the electrons in a Wigner Crystal have a Gaussian shape. 37 More importantly, since a small number of orbitals per electron is required to describe the system (just one, or a few ones if the effects of correlation are to be taken into account), this could open the way to a full quantum treatment of Wigner Crystals by using the methods of Quantum Chemistry.
Conclusions and future perspectives
We presented a general paradigm for the treatment of infinite periodic systems within the framework of Quantum Chemistry. Our approach is based on three main steps.
Associate the topology of a Clifford Torus to a supercell of the system, and choose the Euclidean distance in the torus as the distance that appears in the Coulomb interaction, and, more generally, in any distance-dependent property of the system.
Define the AOs of the system as a combination of Clifford Gaussians in the torus, and build the MOs from this set of AOs; use your favorite Quantum Chemistry method (if possible, a size-consistent one) to investigate the system.
Repeat this procedure for a set of supercells of increasing size and similar shape, in order to extrapolate the desired properties of the system to the infinite thermodynamical limit.
In the present work, in particular, we numerically investigated two important model systems: one particle in a box (a 1D system), and two electrons confined in a Clifford Torus (2D and 3D).
In the first case, we showed that a set of N Clifford Gaussians having a suitable exponent is able to reproduce the lowest N/2 eigenvalues of the Hamiltonian to an extremely high accuracy, comparable with the machine precision of the computer (all the calculations have been done in double precision). This fact strongly suggests that these functions could form, under certain circumstances, a complete basis set for the one-particle Hilbert Space that is used in Quantum Mechanics.
The second case is particularly pertinent for the description of free electrons at very low density, a situation that gives rise to the so called Wigner Localization. Our investigation shows that one single Clifford Gaussian per electron is able to qualitatively describe the localization of the electrons. By comparison, a large number of
We are presently working on a mathematical proof of the completeness property of Clifford Gaussians for one-particle systems, that at the moment we have only guessed. The treatment of systems composed of a large number of free electrons by using this type of Gaussians, in order to investigate the behavior of Wigner Crystals, is also being considered.
Our investigation is certainly preliminary, and we are currently working to the application of our scheme to realistic systems. We think, in particular, to real 2D and 3D systems, like graphene, diamond, ionic crystals, and so on. Also rigid quasi-1D systems, that cannot be treated by arranging the atoms on a circle, like Carbon or Boron-Nitride nanotubes, would be particularly suitable for our approach. The treatment of such systems would require the introduction of high angular-momentum orbitals in the atomic basis set. At the moment, we are testing the calculation of the two-electron integrals for p functions, and implementing the integrals for d orbitals. Preliminary tests indicate that the computer time required to obtain the integrals over Clifford Gaussians is comparable to the same time needed in the case of ordinary Gaussians. This is a very encouraging result, also considering the rather preliminary state of our investigation. If these results will be confirmed by further investigations, we believe that our approach could open the road to an ab initio treatment of periodic systems, in the same way as molecules and clusters are presently treated by Quantum Chemistry.
Article note:
A collection of invited papers to celebrate the UN’s proclamation of 2025 as the International Year of Quantum Science and Technology.
Funding source: The French “Agence Nationale de la Recherche” (ANR)
Award Identifier / Grant number: ANR-22-CE29-0001
Acknowledgments
We thank the French “Agence Nationale de la Recherche” (ANR) for financial support (Grant Agreement ANR-22-CE29-0001).
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: The authors state no conflict of interest.
-
Research funding: The French “Agence Nationale de la Recherche” (ANR) (Grant Agreement ANR-22-CE29-0001).
-
Data availability: Not applicable.
Appendix: Product theorem
A non-normalized toroidal Gaussian centered in the point x i and having exponent α i (with i = 1, 2) can be written as
We consider now the product of two toroidal Gaussians, centered in two points x 1 and x 2 and having exponents equal to α 1 and α 2, respectively. We will show that a Product Theorem holds.
Theorem:
The product of two non-normalized Toroidal-Gaussian functions,
where α c , x c and C are for the moment unknown constants.
Proof:
We can rewrite the previous equality as
By taking the logarithm of both sides of this equation, we have that the exponents must fulfill the following relation:
where α c and x c are the unknown exponent and center of the new Gaussian, respectively, and C is a multiplicative constant, also to be determined. By expanding the previous expression, we get:
Since the three functions 1 = x 0, sin x and cos x are linearly independent in the [0, L x ] interval, their coefficients must be separately identically zero. Therefore we obtain the three independent equations:
and
that must hold simultaneously.
From these equations, we can compute the parameters (x c , α c , C) of the product Gaussian. In particular, by dividing the two members of eq. (23) by the corresponding terms of eq. (24) we obtain the relation
Therefore we can obtain the expression of the new center, which is given by
if
if
where θ(x) is the Heaviside step function: θ(x) = 0 if x < 0 and θ(x) = 1 if x ≥ 0.
On the other hand, by making the square of the two members in each equations and then summing the two equations tem by term, we get the new exponential parameter, which is given by
Finally, once α c has been computed, we can express ln C as
or
This completes the proof.
References
1. Sun, J.-Q.; Bartlett, R. J. Second-Order Many-Body Perturbation-Theory Calculations in Extended Systems. J. Chem. Phys. 1996, 104, 8553–8565; https://doi.org/10.1063/1.471545.Search in Google Scholar
2. Ayala, P. Y.; Kudin, K. N.; Scuseria, G. E. Atomic Orbital Laplace-Transformed Second-Order Møller–Plesset Theory for Periodic Systems. J. Chem. Phys. 2001, 115, 9698–9707; https://doi.org/10.1063/1.1414369.Search in Google Scholar
3. Hirata, S.; Podeszwa, R.; Tobita, M.; Bartlett, R. J. Coupled-Cluster Singles and Doubles for Extended Systems. J. Chem. Phys. 2004, 120, 2581–2592; https://doi.org/10.1063/1.1637577.Search in Google Scholar PubMed
4. Pisani, C.; Busso, M.; Capecchi, G.; Casassa, S.; Dovesi, R.; Maschio, L.; Zicovich-Wilson, C.; Schütz, M. Local-MP2 Electron Correlation Method for Nonconducting Crystals. J. Chem. Phys. 2005, 122, 094113; https://doi.org/10.1063/1.1857479.Search in Google Scholar PubMed
5. Katagiri, H. Equation-Of-Motion Coupled-Cluster Study on Exciton States of Polyethylene with Periodic Boundary Condition. J. Chem. Phys. 2005, 122, 224901; https://doi.org/10.1063/1.1929731.Search in Google Scholar PubMed
6. Marsman, M.; Grüneis, A.; Paier, J.; Kresse, G. Second-Order Møller–Plesset Perturbation Theory Applied to Extended Systems. I. Within the Projector-Augmented-Wave Formalism Using a Plane Wave Basis Set. J. Chem. Phys. 2009, 130, 184103; https://doi.org/10.1063/1.3126249.Search in Google Scholar PubMed
7. Grüneis, A.; Booth, G. H.; Marsman, M.; Spencer, J.; Alavi, A.; Kresse, G. Natural Orbitals for Wave Function Based Correlated Calculations Using a Plane Wave Basis Set. J. Chem. Theory Comput. 2011, 7, 2780–2785; https://doi.org/10.1021/ct200263g.Search in Google Scholar PubMed
8. Pisani, C.; Schütz, M.; Casassa, S.; Usvyat, D.; Maschio, L.; Lorenz, M.; Erba, A. Cryscor: A Program for the Post-Hartree–Fock Treatment of Periodic Systems. Phys. Chem. Chem. Phys. 2012, 14, 7615; https://doi.org/10.1039/c2cp23927b.Search in Google Scholar PubMed
9. Shepherd, J. J.; Grüneis, A.; Booth, G. H.; Kresse, G.; Alavi, A. Convergence of Many-Body Wave-Function Expansions Using a Plane-Wave Basis: From Homogeneous Electron Gas to Solid State Systems. Phys. Rev. B 2012, 86, 035111; https://doi.org/10.1103/physrevb.86.035111.Search in Google Scholar
10. Booth, G. H.; Grüneis, A.; Kresse, G.; Alavi, A. Towards an Exact Description of Electronic Wavefunctions in Real Solids. Nature 2013, 493, 365–370; https://doi.org/10.1038/nature11770.Search in Google Scholar PubMed
11. McClain, J.; Sun, Q.; Chan, G. K.-L.; Berkelbach, T. C. Gaussian-Based Coupled-Cluster Theory for the Ground-State and Band Structure of Solids. J. Chem. Theory Comput. 2017, 13, 1209–1218; https://doi.org/10.1021/acs.jctc.7b00049.Search in Google Scholar PubMed
12. Wang, X.; Berkelbach, T. C. Excitons in Solids from Periodic Equation-of-Motion Coupled-Cluster Theory. J. Chem. Theory Comput. 2020, 16, 3095–3103; https://doi.org/10.1021/acs.jctc.0c00101.Search in Google Scholar PubMed
13. Erba, A.; Desmarais, J. K.; Casassa, S.; Civalleri, B.; Donà, L.; Bush, I. J.; Searle, B.; Maschio, L.; Edith-Daga, L.; Cossard, A.; Ribaldone, C.; Ascrizzi, E.; Marana, N. L.; Flament, J.-P.; Kirtman, B. CRYSTAL23: A Program for Computational Solid State Physics and Chemistry. J. Chem. Theory Comput. 2023, 19, 6891–6932; https://doi.org/10.1021/acs.jctc.2c00958.Search in Google Scholar PubMed PubMed Central
14. Valença Ferreira de Aragão, E.; Moreno, D.; Battaglia, S.; Bendazzoli, G. L.; Evangelisti, S.; Leininger, T.; Suaud, N.; Berger, J. A. A Simple Position Operator for Periodic Systems. Phys. Rev. B 2019, 99, 205144; https://doi.org/10.1103/physrevb.99.205144.Search in Google Scholar
15. Escobar Azor, M.; Brooke, L.; Evangelisti, S.; Leininger, T.; Loos, P.-F.; Suaud, N.; Berger, A. A Wigner Molecule at Extremely Low Densities: A Numerically Exact Study. SciPost Phys. Core 2019, 1, 001; https://doi.org/10.21468/scipostphyscore.1.1.001.Search in Google Scholar
16. Tavernier, N.; Bendazzoli, G. L.; Brumas, V.; Evangelisti, S.; Berger, J. A. Clifford Boundary Conditions: A Simple Direct-Sum Evaluation of Madelung Constants. J. Phys. Chem. Lett. 2020, 11, 7090–7095; https://doi.org/10.1021/acs.jpclett.0c01684.Search in Google Scholar PubMed
17. Tavernier, N.; Bendazzoli, G. L.; Brumas, V.; Evangelisti, S.; Berger, J. A. Clifford Boundary Conditions for Periodic Systems: The Madelung Constant of Cubic Crystals in 1, 2 and 3 Dimensions. Theor. Chem. Acc. 2021, 140, 106; https://doi.org/10.1007/s00214-021-02805-1.Search in Google Scholar
18. Alves, E.; Bendazzoli, G. L.; Evangelisti, S.; Berger, J. A. Accurate Ground-State Energies of Wigner Crystals from a Simple Real-Space Approach. Phys. Rev. B 2021, 103, 245125; https://doi.org/10.1103/physrevb.103.245125.Search in Google Scholar
19. Angeli, C.; Bendazzoli, G. L.; Evangelisti, S.; Berger, J. A. The Localization Spread and Polarizability of Rings and Periodic Chains. J. Chem. Phys. 2021, 155, 124107; https://doi.org/10.1063/5.0056226.Search in Google Scholar PubMed
20. Escobar Azor, M.; Alves, E.; Evangelisti, S.; Berger, J. A. Wigner Localization in Two and Three Dimensions: An Ab Initio Approach. J. Chem. Phys. 2021, 155, 124114; https://doi.org/10.1063/5.0063100.Search in Google Scholar PubMed
21. Evangelisti, S.; Abu-Shoga, F.; Angeli, C.; Bendazzoli, G. L.; Berger, J. A. Unique One-Body Position Operator for Periodic Systems. Phys. Rev. B 2022, 105, 235201; https://doi.org/10.1103/physrevb.105.235201.Search in Google Scholar
22. François, G.; Angeli, C.; Bendazzoli, G. L.; Brumas, V.; Evangelisti, S.; Berger, J. A. Mapping of Hückel Zigzag Carbon Nanotubes onto Independent Polyene Chains: Application to Periodic Nanotubes. J. Chem. Phys. 2023, 159, 094106; https://doi.org/10.1063/5.0153075.Search in Google Scholar
23. Alrakik, A.; Escobar Azor, M.; Brumas, V.; Bendazzoli, G. L.; Evangelisti, S.; Berger, J. A. Solution to the Thomson Problem for Clifford Tori with an Application to Wigner Crystals. J. Chem. Theory Comput. 2023, 19, 7423–7431; https://doi.org/10.1021/acs.jctc.3c00550.Search in Google Scholar
24. Escobar Azor, M.; Alrakik, A.; De Bentzmann, L.; Telleria-Allika, X.; Sánchez de Merás, A.; Evangelisti, S.; Berger, J. A. The Emergence of the Hexagonal Lattice in Two-Dimensional Wigner Fragments. J. Phys. Chem. Lett. 2024, 15, 3571–3575; https://doi.org/10.1021/acs.jpclett.4c00453.Search in Google Scholar
25. Alrakik, A.; Bendazzoli, G. L.; Evangelisti, S.; Berger, J. A. Quantum Chemistry for Solids Made Simple with the Clifford Formalism, 2025. Available from: https://arxiv.org/abs/2508.03917.Search in Google Scholar
26. Noga, J.; Baňack, P.; Biskupič, S.; Boča, R.; Pelikán, P.; Svrčeký, M.; Zajac, A. Approaching Bulk Limit for Three-Dimensional Solids Via the Cyclic Cluster Approximation: Semiempirical INDO Study. J. Comput. Chem. 1999, 20, 253–261; https://doi.org/10.1002/(sici)1096-987x(19990130)20:2<253::aid-jcc7>3.0.co;2-9.10.1002/(SICI)1096-987X(19990130)20:2<253::AID-JCC7>3.0.CO;2-9Search in Google Scholar
27. Volkert, K. Space Forms: A History. Bull. Manifold Atlas 2013, 1–5.Search in Google Scholar
28. McIntosh, A.; Mitrea, M. Clifford Algebras and Maxwell’s Equations in Lipschitz Domains. Math. Methods Appl. Sci. 1999, 22, 1599–1620; https://doi.org/10.1002/(sici)1099-1476(199912)22:18<1599::aid-mma95>3.3.co;2-d.10.1002/(SICI)1099-1476(199912)22:18<1599::AID-MMA95>3.3.CO;2-DSearch in Google Scholar
29. Clifford. Preliminary Sketch of Biquaternions. Proc. Lond. Math. Soc. 1871 (s1–4), 381–395; https://doi.org/10.1112/plms/s1-4.1.381.Search in Google Scholar
30. Klein, F. Zur Nicht-Euklidischen Geometrie. Math. Ann. 1890, 37, 544–572; https://doi.org/10.1007/bf01724772.Search in Google Scholar
31. Boys, S. F. Electronic Wave Functions - I. A General Method of Calculation for the Stationary States of Any Molecular System. Proc. Roy. Soc. Lond. Ser. A. Math. Phys. Sci. 1950, 200, 542–554.10.1098/rspa.1950.0036Search in Google Scholar
32. Loos, P.-F.; Gill, P. M. W. Uniform Electron Gases. I. Electrons on a Ring. J. Chem. Phys. 2013, 138, 164124; https://doi.org/10.1063/1.4802589.Search in Google Scholar
33. Loos, P.-F.; Ball, C. J.; Gill, P. M. W. Uniform Electron Gases. II. the Generalized Local Density Approximation in One Dimension. J. Chem. Phys. 2014, 140, 18A524; https://doi.org/10.1063/1.4867910.Search in Google Scholar PubMed
34. Gill, P. M. W.; Loos, P.-F.; Agboola, D. Basis Functions for Electronic Structure Calculations on Spheres. J. Chem. Phys. 2014, 141, 244102; https://doi.org/10.1063/1.4903984.Search in Google Scholar PubMed
35. Agboola, D.; Knol, A. L.; Gill, P. M. W.; Loos, P.-F. Uniform Electron Gases. III. Low-density Gases on Three-Dimensional Spheres. J. Chem. Phys. 2015, 143, 084114; https://doi.org/10.1063/1.4929353.Search in Google Scholar PubMed
36. Szabo, A.; Ostlund, N. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Dover Books on Chemistry; Dover Publications: New York, USA, 1996.Search in Google Scholar
37. Giuliani, G.; Vignale, G. Quantum Theory of the Electron Liquid, 1st ed.; Cambridge University Press: Cambridge, UK, 2005.10.1017/CBO9780511619915Search in Google Scholar
© 2025 IUPAC & De Gruyter
Articles in the same Issue
- Frontmatter
- IUPAC Recommendations
- Experimental methods and data evaluation procedures for the determination of radical copolymerization reactivity ratios from composition data (IUPAC Recommendations 2025)
- IUPAC Technical Reports
- Kinetic parameters for thermal decomposition of commercially available dialkyldiazenes (IUPAC Technical Report)
- FAIRSpec-ready spectroscopic data collections – advice for researchers, authors, and data managers (IUPAC Technical Report)
- Review Articles
- Are the Lennard-Jones potential parameters endowed with transferability? Lessons learnt from noble gases
- Quantum mechanics and human dynamics
- Quantum chemistry and large systems – a personal perspective
- The organic chemist and the quantum through the prism of R. B. Woodward
- Relativistic quantum theory for atomic and molecular response properties
- A chemical perspective of the 100 years of quantum mechanics
- Methylene: a turning point in the history of quantum chemistry and an enduring paradigm
- Quantum chemistry – from the first steps to linear-scaling electronic structure methods
- Nonadiabatic molecular dynamics on quantum computers: challenges and opportunities
- Research Articles
- Alzheimer’s disease – because β-amyloid cannot distinguish neurons from bacteria: an in silico simulation study
- Molecular electrostatic potential as a guide to intermolecular interactions: challenge of nucleophilic interaction sites
- Photophysical properties of functionalized terphenyls and implications to photoredox catalysis
- Combining molecular fragmentation and machine learning for accurate prediction of adiabatic ionization potentials
- Thermodynamic and kinetic insights into B10H14 and B10H14 2−
- Quantum origin of atoms and molecules – role of electron dynamics and energy degeneracy in atomic reactivity and chemical bonding
- Clifford Gaussians as Atomic Orbitals for periodic systems: one and two electrons in a Clifford Torus
- First-principles modeling of structural and RedOx processes in high-voltage Mn-based cathodes for sodium-ion batteries
- Erratum
- Erratum to: Furanyl-Chalcones as antimalarial agent: synthesis, in vitro study, DFT, and docking analysis of PfDHFR inhibition
Articles in the same Issue
- Frontmatter
- IUPAC Recommendations
- Experimental methods and data evaluation procedures for the determination of radical copolymerization reactivity ratios from composition data (IUPAC Recommendations 2025)
- IUPAC Technical Reports
- Kinetic parameters for thermal decomposition of commercially available dialkyldiazenes (IUPAC Technical Report)
- FAIRSpec-ready spectroscopic data collections – advice for researchers, authors, and data managers (IUPAC Technical Report)
- Review Articles
- Are the Lennard-Jones potential parameters endowed with transferability? Lessons learnt from noble gases
- Quantum mechanics and human dynamics
- Quantum chemistry and large systems – a personal perspective
- The organic chemist and the quantum through the prism of R. B. Woodward
- Relativistic quantum theory for atomic and molecular response properties
- A chemical perspective of the 100 years of quantum mechanics
- Methylene: a turning point in the history of quantum chemistry and an enduring paradigm
- Quantum chemistry – from the first steps to linear-scaling electronic structure methods
- Nonadiabatic molecular dynamics on quantum computers: challenges and opportunities
- Research Articles
- Alzheimer’s disease – because β-amyloid cannot distinguish neurons from bacteria: an in silico simulation study
- Molecular electrostatic potential as a guide to intermolecular interactions: challenge of nucleophilic interaction sites
- Photophysical properties of functionalized terphenyls and implications to photoredox catalysis
- Combining molecular fragmentation and machine learning for accurate prediction of adiabatic ionization potentials
- Thermodynamic and kinetic insights into B10H14 and B10H14 2−
- Quantum origin of atoms and molecules – role of electron dynamics and energy degeneracy in atomic reactivity and chemical bonding
- Clifford Gaussians as Atomic Orbitals for periodic systems: one and two electrons in a Clifford Torus
- First-principles modeling of structural and RedOx processes in high-voltage Mn-based cathodes for sodium-ion batteries
- Erratum
- Erratum to: Furanyl-Chalcones as antimalarial agent: synthesis, in vitro study, DFT, and docking analysis of PfDHFR inhibition