White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
-
Hung-Cheng Chen
 ,
Chen-Yen Hung
,
Chen-Yen Hung

Abstract
In this work, we report an energy upconversion system based on triplet-triplet annihilation comprised of rose bengal dye as the sensitizer and a highly fluorescent 9-(triphenyl)ethynyl-10-phenylanthracene (TPE-PAn) as the triplet annihilator. The energy upconversion can be observed with a low-power laser excitation at 532 nm or noncoherent Xe arc lamp excitaion at 540 nm. A delayed fluorescence from (TPE-PAn) at 418 nm was observed with an energy upconversion up to 0.64 eV and quantum yield of 0.8 %. A white-light emission was achieved as a result of combination of delayed fluorescence from TPE-PAn and residual fluorescence from rose bengal dye.
Introduction
Rose bengal (RB) [1] has been frequently used as a singlet oxygen generator in both synthetic chemistry [2] and in photodynamic therapy [3]. Other application of RB including metal-enhanced phosphorescence [4], dye-sensitized solar cells [5], photosentisized hydrogen evolution from water [6], and organic memory devices [7]. Recently, energy upconversion through triplet-triplet annihilation (TTA) has drawn much attention [8]. The system for energy upconversion through TTA is composed of two molecular components, a triplet sensitizer and a triplet annihilator. The working principle of energy upconversion through TTA is briefly summarized. First, a long-wavelength excitation is applied to pump the triplet sensitizer to singlet manifold and quickly relax to lowest triplet excited state (T 1) through intersystem crossing. An annihilator with lower T 1 state is sensitized by the triplet sensitizer through triplet-triplet energy transfer (TT-EnT). Subsequently, two triplet annihilators collide through TTA and lead to one ground-state and one singlet excited-state annihilator where an upconverted emission is generated. The advantage of TTA-based upconversion is its intrinsic independence on the coherence and its low power-driven nature, with the required power typically in the order of mW cm−2. Previously, we have reported the white light emission by low power photon upconversion involving a purely organic pair of triplet sensitizer of 2,4,5,7-tetraiodo-6-hydroxy-3-fluorone (TIHF) and triplet acceptor/annihilator of 9,10-diphenylanthracene (DPA) [9].
Dinnocenzo et al. had observed a weak upconverted blue emission from poly(methyl methacrylate) (PMMA) film blended with a derivative of RB and DPA [10]. Since the lowest triplet state of RB (T 1 = 1.83–1.78 eV) [11] and DPA (T 1 = 1.77 eV) [12] are almost degenerate and the triplet–triplet energy transfer (TT-EnT) rate highly depends on the driving-force [13], it is possible to increase the upconversion efficiency by choosing an annihilator with a lower T 1 state than that of DPA. In this work, we have employed a new highly fluorescent molecule, 9-(triphenyl)ethynyl-10-phenylanthracene (TPE-PAn), as the triplet energy acceptor as well as the annihilator. The structures of the two molecules employed in the energy upconversion system are shown in Chart 1. The TPE-PAn extends the π-conjugation in the 9-position of anthracene linked with (triphenyl)ethynyl group. The bulky triphenyl group is incorporated to avoid molecular aggregation that quenches fluorescence and shift the energy of TPE-PAn.

Chemical structures of the sensitizer rose bengal (RB) and the triplet annihilator 9-(triphenyl)ethynyl-10-phenylanthracene (TPE-PAn).
Experimental
TPE-PAn was prepared according to the synthetic procedures in supporting information. Sample solutions were degassed by several freeze-pump-thaw cycles before each measurement. The molecular geometry of TPE-PAn was fully optimized at the DFT-B3LYP/6-31G(d) level without imposing any symmetry constraints. Time dependent-density functional theory using the B3LYP functional (referred to throughout as TD-B3LYP) was employed for excitation energies using B3LYP optimized ground state geometries. All calculations were done using the Q-Chem 03 program. Sample solutions were degassed by several freeze-pump-thaw cycles before each measurement. The sample solutions were excited by a diode pumped solid-state laser at a wavelength of 532 nm. The output power of the laser was about 50 mW, which was attenuated by neutral density filters. The emission spectra were measured at the right angle through a fiber coupled spectrometer. The acquisition time of each emission spectrum was 500 ms. Power dependence experiments were carried out with an argon ion laser operated at 514.5 nm. The excitation power was measured by a thermopile energy and power meter. The laser line filter was used to attenuate the background plasma and to improve the monochromaticity of the light. The luminescence spectra were taken using an Andor Shamrock spectrograph with grating 600 l/mm and an EMCCD.
Results and discussion
The normalized absorption and emission spectra of TPE-PAn and RB in a 2:1 (v/v) THF/MeOH solution are illustrated in Fig. 1. RB exhibits an orange color emission with quantum yield of 0.08 in MeOH. The quantum yield of triplet state formation was reported as 0.90 and it has a long-lived triplet excited-state lifetime of 130 μs in MeOH [14]. The protic MeOH was used as a co-solvent to ensure the fully dissociation of the acidic proton in RB because the deprotonated RB is the desired species with the appropriate spectral properties for the purposes of selective photo-excitation and subsequent energy transfer. The same absorption spectrum as the one in MeOH was obtained by adding tetrabutylammonium hydroxide into a THF solution of RB. It is mandatory to employ THF as a second co-solvent in order to achieve a high solubility of TPE-PAn in solution. Thus, the subsequent measurements were all carried out in a mixture of 2:1 (v/v) THF/MeOH solution. The lower limit of RB triplet energy level in a 2:1 (v/v) THF/MeOH solution is estimated to be 1.78 eV, whereas the triplet energy level of TPE-PAn is estimated at 1.57 eV by theoretical calculations, which is located in the appropriate position for an efficient triplet energy transfer.

Normalized absorption and luminescence spectra of TPE-PAn (5.0 × 10−6 M) and RB (1.0 × 10−5 M) mixture in 2:1 (v/v) THF/MeOH at room temperature.
The energy level diagram of the upconversion process in RB and TPE-PAn is depicted in Fig. 2. RB is first excited to the Franck-Condon state by a green light at 532 nm followed by rapid vibrational relaxation to its lowest singlet excited state S 1 and emits with a yellow fluorescence at 571 nm or by intersystem crossing to its lowest triplet excited state T 1. The triplet state level of RB (1.78 eV) is high enough to facilitate a triplet-triplet energy transfer to populate the triplet state of TPE-PAn (1.57 eV). Two of such triplet 3TPE-PAn* molecules may undergo a TTA and generate one singlet 1TPE-PAn* molecule which emits blue light with a high quantum yield. It is possible to simultaneously observe the orange emission from RB and the blue emission from TPE-PAn in the entire photophysical processes and this gives us a chance to yield a white-light emitting system.

Qualitative energy states involve in the upconversion process of RB and TPE-PAn. Solid colored lines represent radiative processes. Acronyms: VR, vibrational relaxation; ISC, intersystem crossing; T-T EnT, triplet-triplet energy transfer; and TTA, triplet-triplet annihilation.
A solution composed of TPE-PAn and RB at concentrations of 1.4 × 10−2 M and 1.4 × 10−5 M, respectively, dissolved in deaerated 2:1 (v/v) THF and MeOH mixture was typically employed for energy up-conversion studies. A 540 nm noncoherent Xe arc lamp or a 532 nm coherent laser has been employed as the excitation source. As shown in Fig. 3, an upconverted fluorescence signal at 450 nm was observed in a 2:1 (v/v) THF/MeOH mixed solvent. The emission profile is very similar to the TPE-PAn fluorescence but now with a long-wavelength excitation. The nature of the delayed fluorescence at 450 nm was confirmed by its long lifetime of 732.9 µs, which is in contrast to the prompt fluorescence lifetime of 4.87 ns? To confirm the nonlinear property of the upconversion process, we have measured the power dependence of the upconversion signal. A degassed THF/MeOH solution containing RB (1.0 × 10−5 M) and TPE-PAn (1.8 × 10−2 M) was excited at 532 nm with a number of different incident powers ranging from 2.7 to 45.3 mW. The acquired delayed fluorescence spectra are shown in Fig. 4(a). The power dependent upconversion fluorescence intensity is illustrated in Fig. 4(b). The presence of a quadratic power dependence to the upconversion fluorescence intensity supports the triplet-triplet energy transfer and triplet–triplet annihilation model as shown in Fig. 2 since the process requires two sensitized triplet excited-state RB molecule [15]. The calculated upconversion quantum based on the delayed fluorescence is estimated to be 0.8 %. The low upconversion efficiency is mainly attributed to the inefficient intersystem crossing of RB from singlet to triplet excited state as judged by the strong fluorescence from RB at 571 nm [16].
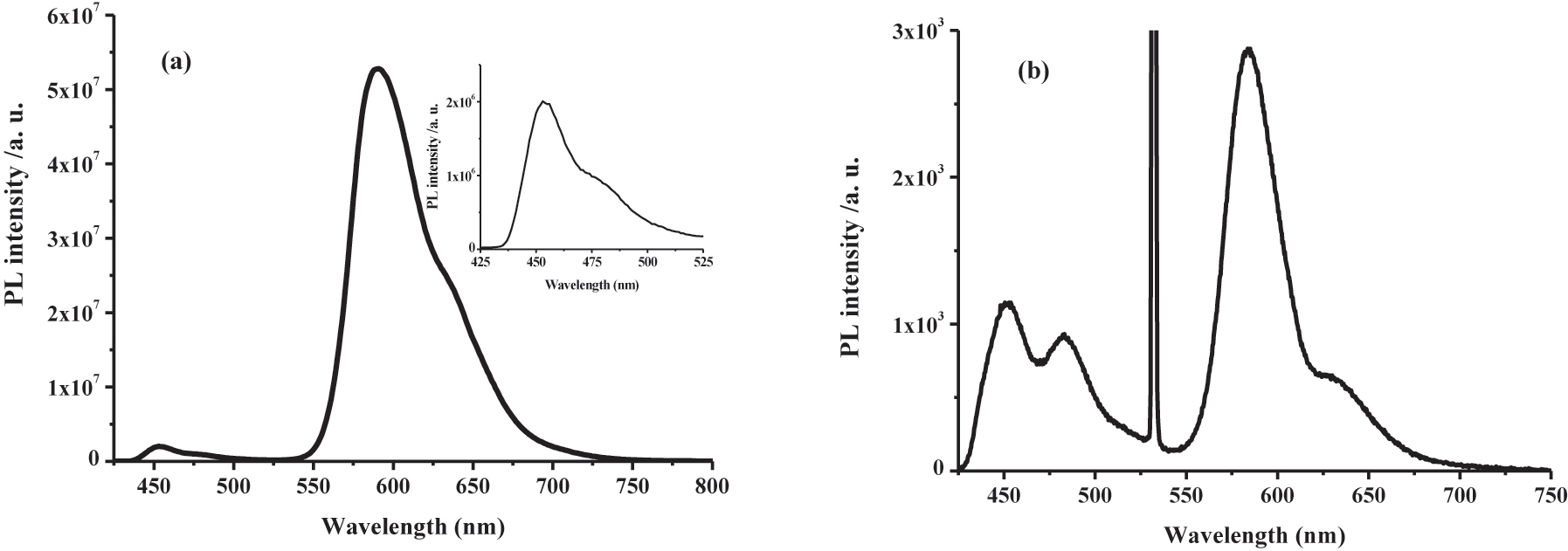
Up-conversion fluorescence spectrum of TPE-PAn (1.4 × 10−2 M) and RB (1.4 × 10−5 M) in 2:1 THF/MeOH solutions. The emission spectra were measured by (a) 540 nm noncoherent Xe lamp excitation and (b) a 532 nm laser excitation. The peak at 532 nm is from the scattered laser light.

(a) Photoluminescence intensity profile of TPE-PAn (1.8 × 10−2 M) and RB (1.0 × 10−5 M) in 2:1 THF/MeOH solutions excited at 532 nm with different laser power densities. (b) Normalized integrated emission intensity plotted as a function of the normalized incident power density of the laser. The red curve is fitted by a quadratic function, x 2.
The green-to-blue photon upconversion can be easily visualized in the RB/TPE-PAn system as illustrated in Fig. 5(a) where a commercial green laser pointer (λ ex = 532 nm with peak power 40 mW) was used to selectively excite the RB in the mixture. A white light emission is clearly visualized along the direction of incident laser beam. On the other hand, only an orange fluorescence from RB was observed in the absence of TPE-PAn in solution (Fig. 5b).

Digital photographs of (a) upconverted white light emission produced in a deaerated 2:1 THF-MeOH solution of TPE-PAn (1.8 × 10−2 M) and RB (1 × 10−5 M) and (b) orange emission of RB (1.0 × 10−5 M) in a 2:1 THF-MeOH solution. Solutions were excited by a commercial green laser pointer (λ ex = 532 nm with 40 mW peak power).
Energy upconversions with white light emission were reported from Tm3+/Yb3+/Er3+-doped Lu3Ga5O12 nanocrystals, following a near-infrared excitation (λ ex = 980 nm) with a 500 mW laser power [17]. In another work, a broad-band, white visible light was generated with a ruthenium polypyridyl complex as the triplet sensitizer [18]. The observed white-light emission in the current RB/TPE-PAn upconversion pair is a result of dual emission from both the upconverted blue delayed fluorescence from TPE-PAn and the orange fluorescence from RB. To quantify the color of the emission that human eye perceives, the Commission internationale de l’eclairage (CIE) coordinates were calculated and shown in Fig. 6. We measured the upconverted spectrum excited by three different continuous laser power of 3.6, 18.5 and 39.7 mW with a 532 nm diode laser. The CIE coordinate for the emission spectrum with 3.6 mW excitation power was in the orange region (0.40, 0.34), whereas for the 39.7 mW laser power it is in the region of (0.36, 0.34), which is close to an optimum white color. Therefore, we have demonstrated a low-power white light upconversion using only organic compounds. It is indeed possible to develop organic white-light-emitting devices with low drive voltages based on such simple triplet energy donor-acceptor pairs.
![Fig. 6:
CIE diagram indicating the color coordinates calculated from the upconverted emission spectra of 2:1 THF/MeOH solutions of TPE-PAn and RB, [RB] = 1.36 × 10−5 M, [TPE-PAn] = 1.4 × 10−2 M, 532 nm excitation with ▲: 3.6 mW, ×: 18.5 mW, and ●: 39.7 mW laser power.](/document/doi/10.1515/pac-2023-0210/asset/graphic/j_pac-2023-0210_fig_007.jpg)
CIE diagram indicating the color coordinates calculated from the upconverted emission spectra of 2:1 THF/MeOH solutions of TPE-PAn and RB, [RB] = 1.36 × 10−5 M, [TPE-PAn] = 1.4 × 10−2 M, 532 nm excitation with ▲: 3.6 mW, ×: 18.5 mW, and ●: 39.7 mW laser power.
Conclusions
In summary, we have devised a simple triplet energy donor-acceptor pair capable of producing energy upconversion emission with low power excitation sources. The combination of the upconverted blue emission and the residual orange emission from the sensitizer produces a white light emission spectrum. To our knowledge, this is the first report on RB to be designed in a low power photon upconversion system through triplet-triplet annihilation with a white light emission.
Supporting information
Detailed synthetic procedures and characterization data for TPE-PAn, additional figures of spectroscopic measurements.
Funding source: National Science and Technology Council of Taiwan
Award Identifier / Grant number: MOST 111-2113-M-001-041
Funding source: Academia Sinica
Acknowledgments
We are grateful to the National Science and Technology Council of Taiwan and Research Program on Nanoscience and Nanotechnology of Academia Sinica for support of this research.
References
[1(a)] D. C. Neckers. J. Chem. Educ. 64, 649 (1987), https://doi.org/10.1021/ed064p649.Search in Google Scholar
(b) D. C. Neckers. Photochem. Photobiol. 47, 1 (1989).Search in Google Scholar
[2(a)] M. Oelgemöller, C. Jung, J. Ortner, J. Mattayc, E. Zimmermann. Green Chem. 7, 35 (2005), https://doi.org/10.1039/b414230f.Search in Google Scholar
(b) K. Jähnisch, U. Dingerdissen. Chem. Eng. Technol. 28, 426 (2005), https://doi.org/10.1002/ceat.200407139.Search in Google Scholar
(c) R. C. R. Wootton, R. Fortt, A. J. de Mello. Org. Process Res. Dev. 60, 187 (2002), https://doi.org/10.1021/op0155155.Search in Google Scholar
[3(a)] W. Mark. Photosensitisers in Biomedicine, p. 97, John Wiley & Sons Ltd, Chichester, UK (2009).Search in Google Scholar
(b) C. Soldani, A. C. Croce, M. G. Bottone, A. Fraschini, M. Biggiogera, G. Bottiroli, C. Pellicciari. Histochem. Cell Biol. 128, 485 (2007), https://doi.org/10.1007/s00418-007-0333-3.Search in Google Scholar PubMed
(c) E. Wachter, C. Dees, J. Harkins, T. Scott, M. Petersen, R. E. Rush, A. Cada. Laser Surg. Med. 32, 101 (2003), https://doi.org/10.1002/lsm.10138.Search in Google Scholar PubMed
[4(a)] Y. Zhang, K. Aslan, S. N. Malyn, C. D. Geddes. Chem. Phys. Lett. 427, 432 (2006), https://doi.org/10.1016/j.cplett.2006.06.078.Search in Google Scholar
[5(a)] B. Pradhan, S. K. Batabyal, A. J. Pal. Sol. Energy mater. Sol. Cells 91, 769 (2007)10.1016/j.solmat.2007.01.006Search in Google Scholar
(b) M. S. Roy, P. Balraju, M. Kumar, G. D. Sharma. Sol. Energy Mater. Sol. Cells 92, 909 (2008);10.1016/j.solmat.2008.02.022Search in Google Scholar
(c) C. Simpson, T. M. Clarke, R. W. MacQueen, Y. Y. Cheng, A. J. Trevitt, A. J. Mozer, P. Wagner, T. W. Schmidt, A. Nattestad. Phys. Chem. Chem. Phys. 17, 24826 (2015), https://doi.org/10.1039/c5cp04825g.Search in Google Scholar PubMed
[6] X. Zhang, Z. Jin, Y. Li, S. Li, G. Lu. J. Phys. Chem. C 113, 2630 (2009), https://doi.org/10.1021/jp8085717.Search in Google Scholar
[7(a)] F. L. E. Jakobsson, X. Crispin, M. Cölle, M. Buöchel, D. M. de Leeuw, M. Berggren. Org. Electron. 8, 559 (2007), https://doi.org/10.1016/j.orgel.2007.04.002.Search in Google Scholar
(b) A. Bandhopadhyay, A. J. Pal. J. Phys. Chem. B 107, 2531 (2003), https://doi.org/10.1021/jp027369q.Search in Google Scholar
[8(a)] R. R. Islangulov, D. V. Kozlov, F. N. Castellano. Chem. Commun. 3776 (2005).10.1039/b506575eSearch in Google Scholar PubMed
(b) S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda, G. Wegner. Phys. Rev. Lett. 97, 143903 (2006), https://doi.org/10.1103/physrevlett.97.143903.Search in Google Scholar
(c) S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen, G. Wegner. Angew. Chem., Int. Ed. 46, 7693 (2007), https://doi.org/10.1002/anie.200700414.Search in Google Scholar PubMed
(d) T. N. Singh-Rachford, F. N. Castellano (Coord.) Chem. Rev. 254, 2560 (2010), https://doi.org/10.1016/j.ccr.2010.01.003.Search in Google Scholar
(e) T. Dilbeck, K. Hanson. J. Phys. Chem. Lett. 9, 5810 (2018), https://doi.org/10.1021/acs.jpclett.8b02635.Search in Google Scholar PubMed
(f) B. Albinsson. A. Olesund. Nat. Photonics 14, 528 (2020), https://doi.org/10.1038/s41566-020-0681-2.Search in Google Scholar
[9] H.-C. Chen, C.-Y. Hung, K.-H. Wang, H.-L. Chen, W.-S. Fann, F.-C. Chien, P. Chen, T. J. Chow, C.-P. Hsu, S.-S. Sun. Chem. Commun. 4064 (2009).10.1039/b905572jSearch in Google Scholar PubMed
[10] P. B. Merkel, J. P. Dinnocenzo. J. Lumin. 129, 303 (2009), https://doi.org/10.1016/j.jlumin.2008.10.013 .Search in Google Scholar
[11(a)] J. Tran. J. Olmsted III. Photochem. Photobiol., A 71, 45 (1993), https://doi.org/10.1016/1010-6030(93)87007-a.Search in Google Scholar
(b) T. Shen, Z. G. Zhao, Q. Yu, H. J. Xu. Photochem. Photobiol. 47, 203 (1989), https://doi.org/10.1016/1010-6030(89)87066-2.Search in Google Scholar
(c) C. R. Lambert, I. E. Kochevar, R. W. Redmond. J. Phys. Chem. B 103, 3737 (1999), https://doi.org/10.1021/jp983812e.Search in Google Scholar
[12(a)] S. K. Chattopadhyay, C. V. Kumar, P. K. Das. Chem. Phys. Lett. 98, 250 (1983), https://doi.org/10.1016/0009-2614(83)87160-7.Search in Google Scholar
(b) S. L. Murov, I. Carmichael, G. L. Hug. Handbook of Photochemistry, Marcel Dekker, New York, 2nd ed. (1993).Search in Google Scholar
[13(a)] G. L. Closs, M. D. Johnson, J. R. Miller, P. Piotrowiak. J. Am. Chem. Soc. 111, 3751 (1989), https://doi.org/10.1021/ja00192a044.Search in Google Scholar
(b) M. E. Sigman, G. L. Closs. J. Phys. Chem. 95, 5012 (1991), https://doi.org/10.1021/j100166a022.Search in Google Scholar
(c) Z. Murtaza, A. P. Zipp, L. A. Worl, D. Graff, W. E. JonesJr., W. D. Bates, T. J. Meyer. J. Am. Chem. Soc. 113, 5113 (1991), https://doi.org/10.1021/ja00013a085.Search in Google Scholar
[14] P. M. Suardi, E. Gassmann, A. M. Braun, E. Oliveros. Helv. Chim. Acta 70, 1760 (1987).10.1002/hlca.19870700712Search in Google Scholar
[15] C. Bohne, E. B. Abuin, J. C. Scaiano. J. Am. Chem. Soc. 112, 4226 (1990), https://doi.org/10.1021/ja00167a018.Search in Google Scholar
[16(a)] N. Yanai, K. Suzuki, T. Ogawa, Y. Sasaki, N. Harada, N. Kimizuka. J. Phys. Chem. A 123, 10197 (2019), https://doi.org/10.1021/acs.jpca.9b08636.Search in Google Scholar PubMed
(b) Y. Zhou, F. N. Castellano, T. W. Schmidt, K. Hanson. ACS Energy Lett. 5, 2322 (2020), https://doi.org/10.1021/acsenergylett.0c01150.Search in Google Scholar
[17] V. Mahalingam, F. Mangiarini, F. Vetrone, V. Venkatramu, M. Bettinelli, A. Speghini. J. A. Capobianco. J. Phys. Chem. C. 112, 17745 (2008), https://doi.org/10.1021/jp8076479.Search in Google Scholar
[18] T. N. Singh-Rachford, R. R. Islangulov, F. N. Castellano. J. Phys. Chem. A 112, 3906 (2008), https://doi.org/10.1021/jp712165h.Search in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/pac-2023-0210).
© 2023 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics