Control of chirality inversion kinetics of triple-helical metallocryptands
-
Shigehisa Akine


Abstract
Dynamic helical molecules, which can undergo a reversible chirality inversion between the P and M forms, are useful as a platform for switchable chiral functional molecules. The chirality inversion of these molecules has been extensively studied. However, it has mostly been discussed from the viewpoint of the equilibrated P/M ratios before and after the inversion; control of the response speeds or kinetic profiles has rarely been explored. In order to construct helical structures with controllable kinetic profiles, triple-helical metallocryptands, LM3, have been designed and synthesized. These molecules can undergo a relatively slow dynamic P/M chirality inversion (helicity inversion) to produce an equilibrated mixture. The P/M equilibration was accelerated or decelerated based on the following two strategies. One is based on the guest binding in the cryptand cavity. The P/M racemization kinetics of LNi3 was significantly decelerated by recognition of guanidinium ion in the cavity. The other strategy is based on the ligand exchange reactions at the octahedral cobalt(III) centers in LCo3(amine)6. The P/M chirality inversion speeds were controlled by changing the initial and entering amine ligands. In addition, a unique transient chirality inversion behavior was observed when chiral amine ligands were removed from the metallocryptand by the ligand exchange reaction with piperidine.
Introduction
Helical structures have a useful chiral molecular scaffold with the right- and left-handed forms (P and M forms, respectively). In general, helical molecular scaffolds play an important role in providing a chiral environment for various kinds of chiral molecular functions, such as chiral discrimination [1], asymmetric catalysis in organic syntheses [2], chiroptical properties [3], etc. Typical helical molecules include biopolymers, such as DNA duplexes and α-helical peptides, as well as a variety of artificial helical molecules and polymers [4].
Some of the helical molecules can undergo a dynamic chirality inversion between the P and M forms; these are known as dynamic helical molecules [5]. The chirality inversion takes place if the structures have enough flexibility or are in equilibrium with a non-helical structure such as a random coil. These dynamic helical molecules have attracted increasing attention because they could be potentially useful as a molecular platform to enable stimuli-responsive switching of chiral functions [6]. If the stimulus converts a helical structure from P-rich to M-rich or vice versa, dynamic chirality inversion would take place (Fig. 1a). The stimuli-responsive P/M chirality inversion of a helical structure may lead to inversion of the chiral functions, such as chiroptical properties and enantioselectivities in asymmetric syntheses. In this regard, one can say that a helical molecule possesses the chirality information, i.e., the P/M isomeric ratio, which governs various kinds of chiral properties and functions.
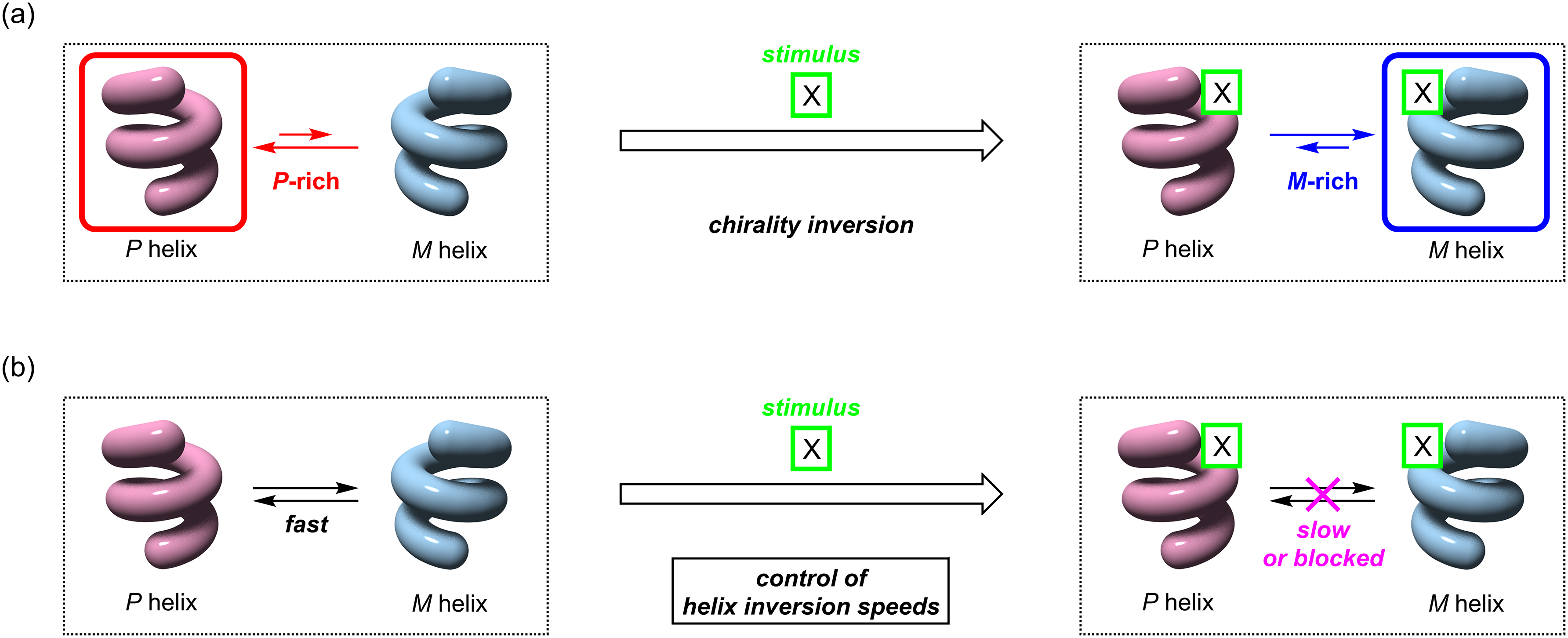
Schematic drawings of the chirality inversion of dynamic helical molecules between the P and M forms. (a) A stimulus inverts the chirality from the P-rich to M-rich if the stimulus changes the P/M preference of the P/M equilibrium. (b) Control of the P/M equilibration speeds of a dynamic helical molecule by an external stimulus.
From the viewpoint of the processing of the chiral information, there are two important aspects. One is the difference between the equilibrated P/M ratios before and after the stimulation, because the P/M ratio directly determines the chiral properties, such as the sign and amplitudes of the chiroptical signals and enantioselectivities, in various types of chemical events. If the P/M preference is reversed upon the stimulation, this reversal will lead to the opposite chiral properties or functions (Fig. 1a). The other one is the response speeds for the changes in the P/M ratios (Fig. 1b). A fast P/M interconversion is desired when chiral information is recorded or erased, but the P/M interconversion should be slow when the recorded information has to be kept [7]. In this context, it is important to develop a rational strategy to accelerate or decelerate the dynamic P/M chirality inversion depending on the circumstances. However, the P/M chirality inversion has been mostly discussed from the viewpoint of the equilibrated P/M ratios before and after the inversion; tuning of the response speeds or controlling the kinetic profiles has rarely been investigated. This is mainly because the response speeds are principally governed by the intrinsic P/M chirality inversion rate of the helical framework, which is difficult to be altered by external stimuli.
In order make a dynamic chiral structure with a controllable response speed, it is necessary to establish a strategy to efficiently stabilize or destabilize the transition state of the P/M interconversion. This can be achieved by the introduction of a binding site for a chemical species at a position where the transition state is directly influenced. From this structural viewpoint, the trinuclear metallocryptand, LM3, is advantageous (Fig. 2a) [8]. This molecule has a triple helical structure due to the rigid propeller-shaped triphenylbenzene scaffold, but it still has a dynamic feature that allows the interconversion between the P and M forms. The cryptand molecule has a three-dimensional cavity surrounded by three [M(saloph)] (H2saloph = N,N′-disalicylidene-o-phenylenediamine) arms connected with two pivotal benzene rings. Based on the structural features, there are two possible ways to alter the speed of the P/M chirality inversion (Fig. 2b). One is the guest binding in the cavity (strategy 1); the guest species accommodated in the cryptand cavity would significantly affect the transition state of the P/M chirality inversion. In fact, strong binding is expected because related oligo(metallosalen)-type structures are known to have an excellent binding affinity to cationic guest species [9]. The other one is the ligand exchange reactions at the constituent metal ions of the three [M(saloph)] arms (strategy 2); the reactivity of the ligand exchange can be altered by selecting a suitable combination of the initial and entering ligands [10]. In addition, if a chiral amine is introduced, removed, or exchanged with another one, the P/M ratio could be changed at the desired speed according to the reactivity.
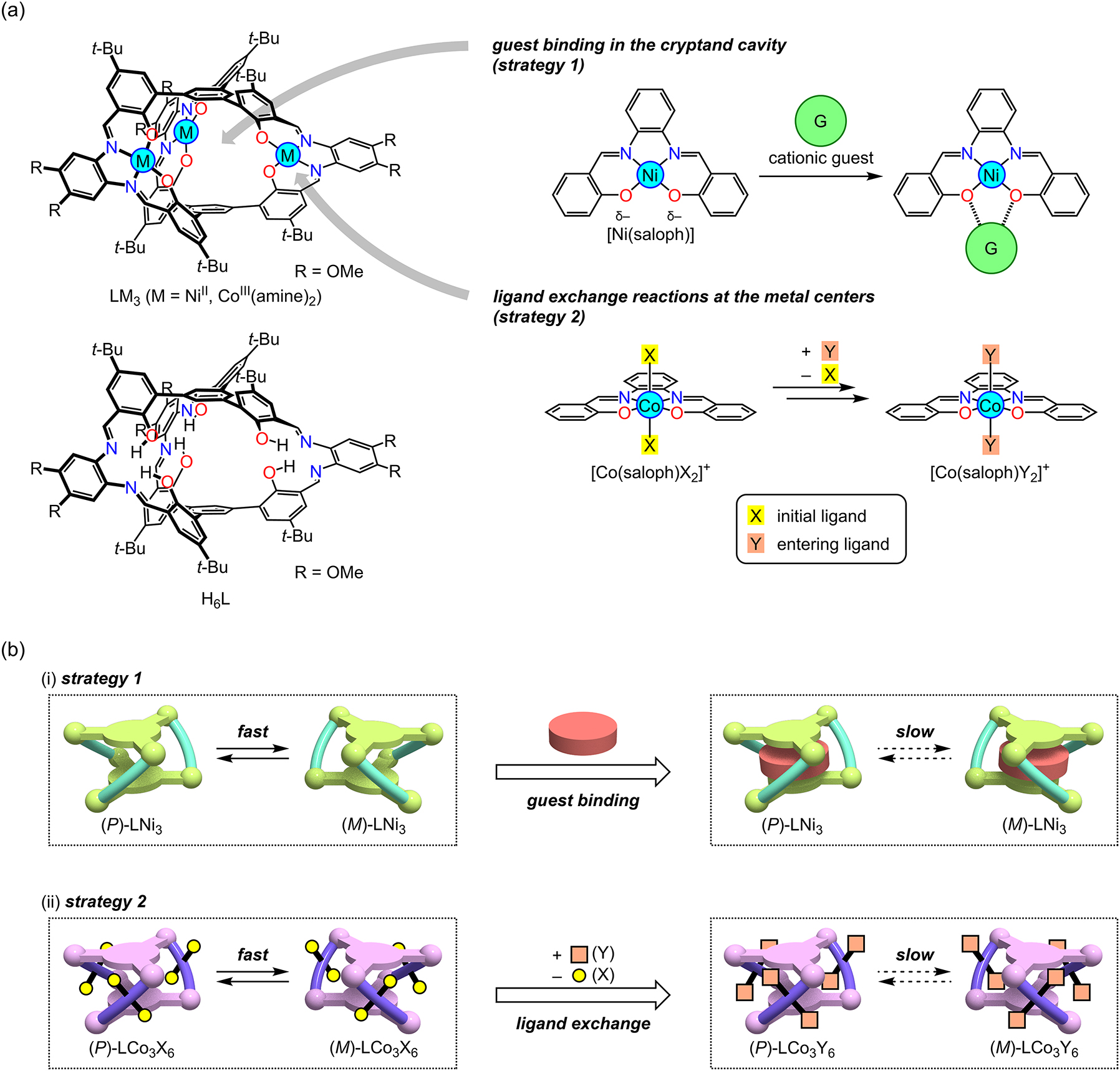
Design of triple-helical metallocryptands LM3 for controlling the P/M chirality inversion speeds. (a) Molecular structure of LM3 and the possible interaction/reaction with a chemical species. The structure of the precursor H6L is also shown. (b) Two strategies for controlling the helix inversion speeds: (i) guest binding in the cryptand cavity and (ii) ligand exchange reactions at the metal centers.
This article illustrates a series of the new P/M chirality inversion systems based on the helical LM3 metallocryptand structures. The chirality inversion speeds are effectively controlled by taking advantage of the guest binding in the LNi3 cryptand cavity or the ligand exchange reactions at the octahedral cobalt(III) centers in LCo3X6.
Control of P/M chirality inversion speeds by host–guest binding
In general, cryptand-like cage compounds show an excellent binding affinity to guest species in their three-dimensional cavity. The metallocryptand LM3 molecule would have a strong cation binding affinity because it has three [M(saloph)] motifs that can strongly interact with cationic species [9]. Since this binding site is located within the cage cavity of LM3, the guest binding would directly stabilize or destabilize the transition state of the P/M chirality inversion, which would effectively influence the P/M chirality inversion speed (Fig. 2b(i)). In this section, the design of a new helical cryptand molecule, LNi3, is described and the control of the P/M chirality inversion speed using the host–guest binding is discussed.
The triple-helical metallocryptand, LNi3 [8], was obtained by the reaction of the corresponding free H6L ligand with nickel(II) acetate. The X-ray crystal structural analysis unambiguously revealed the molecular structure of this trinickel(II) metallocryptand (Fig. 3a). The LNi3 molecule has a well-defined three-dimensional cavity surrounded by three [Ni(saloph)] arms arranged in a triple-helical fashion. The cavity is surrounded by six phenoxo oxygen atoms and two pivotal benzene rings, and this electron-rich nature was found to be suitable for the recognition of cationic guest species (see below). In addition, the flat-shaped cavity sandwiched by the two pivotal benzene rings was suitable for shape-complementary binding with a planar guest species. The distance between the two pivotal benzene rings is 5.523 Å, which is slightly narrower than the size appropriate for the recognition of a π-conjugate molecule. Therefore, when the guest is bound, the cavity should be expanded, which would influence the transition state of the P/M chirality inversion.
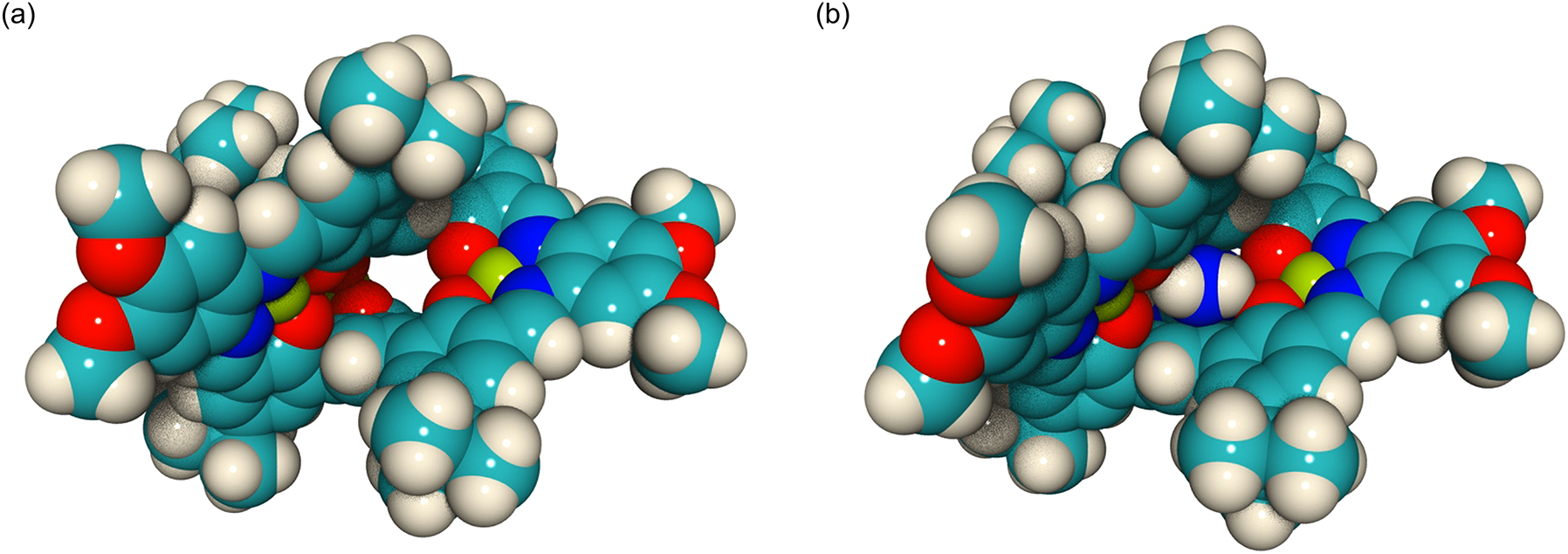
Crystal structures of (a) LNi3 and (b) LNi3 Gu.
The binding affinity of LNi3 to alkali metal ions was investigated by spectroscopic methods (Table 1) [11]. The binding constants logK a were in the range of 5.3 and 8.1, indicating the excellent binding affinity of LNi3 to these guest ions. In particular, the selectivity for larger ions such as Rb+ and Cs+ was clearly demonstrated. Importantly, the LNi3 metallocryptand binds alkali metal ions much stronger than the corresponding metal-free host, H6L, by 1–2 orders of magnitude. This can be explained by the more negatively charged phenoxo groups in the [Ni(saloph)] motifs, while the uncomplexed phenol groups in H6L are less negatively charged. This is the same trend observed for the already reported macrocyclic and acyclic oligo(metallosalen)-type compounds [9, 12, 13].
Binding constants logK a of cryptand hosts (H6L and LNi3) for alkali metal ionsa,b.
| Na+ | K+ | Rb+ | Cs+ | |
|---|---|---|---|---|
| Ionic radius (Å)c | 1.32 | 1.65 | 1.75 | 1.88 |
| H6L | 3.37 (3.29, 2.41)d | 4.94 | 5.66 | 6.67 |
| LNi3 | 5.32 | 6.85 | 8.04 | 7.83 |
This LNi3 complex also showed a particularly strong binding affinity to guanidinium ion (Gu); the binding constant was logK a > 8 [8]. This strong binding is probably due to the good shape complementarity of the guanidinium ion to the flat three-fold symmetrical cavity of LNi3. The 1H NMR signal of the bound guanidinium ion was observed at 5.06 ppm, which was significantly upfield shifted from that of the uncomplexed guest (6.98 ppm). This clearly indicated that the guanidinium ion was completely encapsulated inside the cavity sandwiched by the two pivotal benzene rings. The crystal structure of the guanidinium complex, LNi3 Gu, unambiguously demonstrated the complete encapsulation of the guanidinium ion in the LNi3 cavity (Fig. 3b). This guest binding undoubtedly expanded the cryptand cavity; the pivot–pivot distance was elongated from 5.523 Å to 6.059 Å. The distance between the guanidinium ion and the pivotal benzene rings was around 3 Å, which suggests the cation–π interactions between them. In addition, six-fold N–H⋯O hydrogen-bonding interactions were found between the phenoxo oxygen atoms and guanidinium hydrogen atoms, which would be the major interactions explaining the strong host–guest binding.
Since the LNi3 metallocryptand has a dynamic helical molecular framework with no chiral auxiliary, equimolar amounts of the P and M forms are present in the absence of any external chiral sources (Fig. 4a). However, if this helical metallocryptand LNi3 interacts with a chiral guest, the P and M forms become a diastereomer pair, which makes the thermodynamic stabilities of the two forms different from each other (Fig. 4b). This would shift the P/M equilibrium to one side. This is the principle of the dynamic helicity control [5], which has been observed in various kinds of dynamic chiral systems such as helical polymers and helical metal complexes [4, 15]. As the chiral guest, chiral ammonium ions, such as L-phenylalanine methyl ester (protonated form) (A), were found to be effective to shift the P/M equilibrium of LNi3, which was evidenced by the appearance of the CD signals. The binding constant was determined to be logK a = 3 for guest A, which is lower than that for the guanidinium ion.
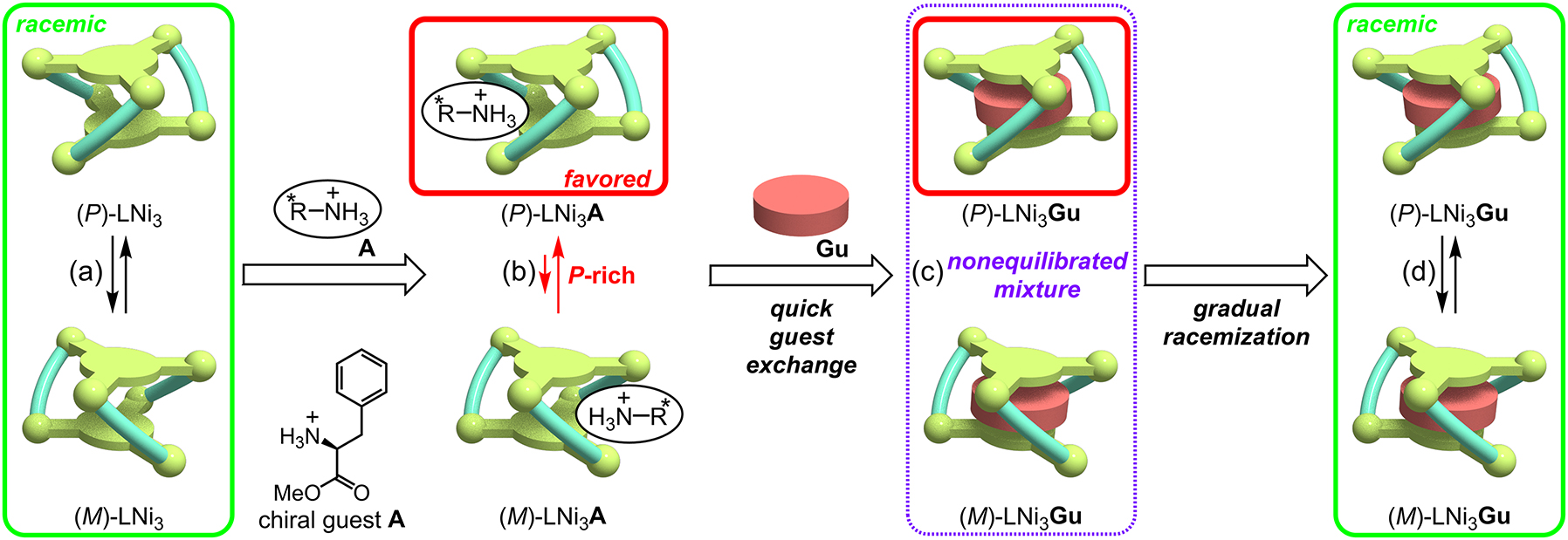
Deceleration of the P/M chirality inversion speed of LNi3 by binding with the guanidinium ion (Gu) in the cryptand cavity.
The P/M equilibrium shift caused by the binding with chiral guest A was found to be fast. The CD signal immediately appeared and became constant after the addition of the chiral guest A (t 1/2 < 12 s). Interestingly, this P/M chirality inversion rate of LNi3 was significantly decelerated by the very strong host–guest binding with the guanidinium ion. This was clearly demonstrated by the following guest exchange experiments. Since the chiral guest A efficiently shifted the P/M equilibrium (Fig. 4b) and guanidinium ion has a much higher binding affinity to LNi3, the biased P/M ratio might remain unchanged after the A → Gu guest exchange, even if the resultant host–guest complex LNi3 Gu has no chiral auxiliary (Fig. 4c).
The complete and rapid A → Gu guest exchange was confirmed by a 1H NMR experiment, which showed the characteristic upfield shifted signal of the bound guanidinium ion at 5.06 ppm 3 min after addition of the guanidinium ion. However, the induced CD signals remained almost unchanged even after the complete guest exchange with the achiral guanidinium ion. This indicated that the P/M equilibration of the guanidinium complex LNi3 Gu was significantly slow. The resultant nonequilibrated P/M mixture (Fig. 4c) underwent a slow racemization to give an equimolar P/M mixture (Fig. 4d), which was confirmed by the gradual decay of the CD signal to zero. The half-life was determined to be t 1/2 = 37 min, which was at least 180 times slower than that of the LNi3 metallocryptand in the presence of the chiral guest A. As a result, this LNi3 complex acted as a new chirality information storage system [7, 16], in which the P/M ratio remained unchanged after removal of the chiral source, based on the combination of the dynamic helical molecular scaffold with the strong host–guest binding.
Control of P/M chirality inversion rates by ligand exchange reactivity
As already described, the helical complex LNi3 can undergo a P/M chirality inversion upon suitable stimulation. If such an inversion behavior of LM3 is coupled with a slow chemical conversion, the P/M equilibrium ratio can be altered at the desired speed. For this purpose, the kinetic rate of the chemical conversion should be easily controllable as desired. If the conversion is slower than the intrinsic P/M chirality inversion, the P/M ratio would show a time-dependent change at the same speed as the chemical conversion that can be controllable [17]. The ligand exchange reaction at an inert low spin cobalt(III) center would be one of the suitable chemical conversions, because the reaction can be easily accelerated or decelerated depending on the initial and entering ligands [10, 18]. Thus, the triple helical complex LCo3X6, which has three octahedral cobalt(III) ions, was employed [19]. This complex undergoes a dynamic P/M chirality inversion as seen in the nickel(II) analogue, LNi3. In addition, each of the cobalt(III) centers has two additional monodentate ligands X to complete an octahedral geometry. This allows the LCo3X6 molecule to undergo a six-step ligand exchange reaction if each of the [Co(saloph)X2]+ sites undergoes the two-step X → Y ligand exchange reaction (Fig. 2b(ii)).
When the ligand X is achiral, the P and M forms of the helical metallocryptand LCo3X6 would be an enantiomer pair, giving rise to an equilibrated racemic mixture due to the dynamic P/M interconversion in solution (Fig. 5a). However, when this achiral amine ligand X is replaced with a chiral ligand Y, the P and M forms become a diastereomer pair. Therefore, this ligand exchange would cause a P/M equilibrium shift (Fig. 5b). In addition, the speed of the P/M equilibrium shift would be controllable because the reaction rate of the X → Y ligand exchange can be altered by changing the initially-coordinating amine X and added amine Y.
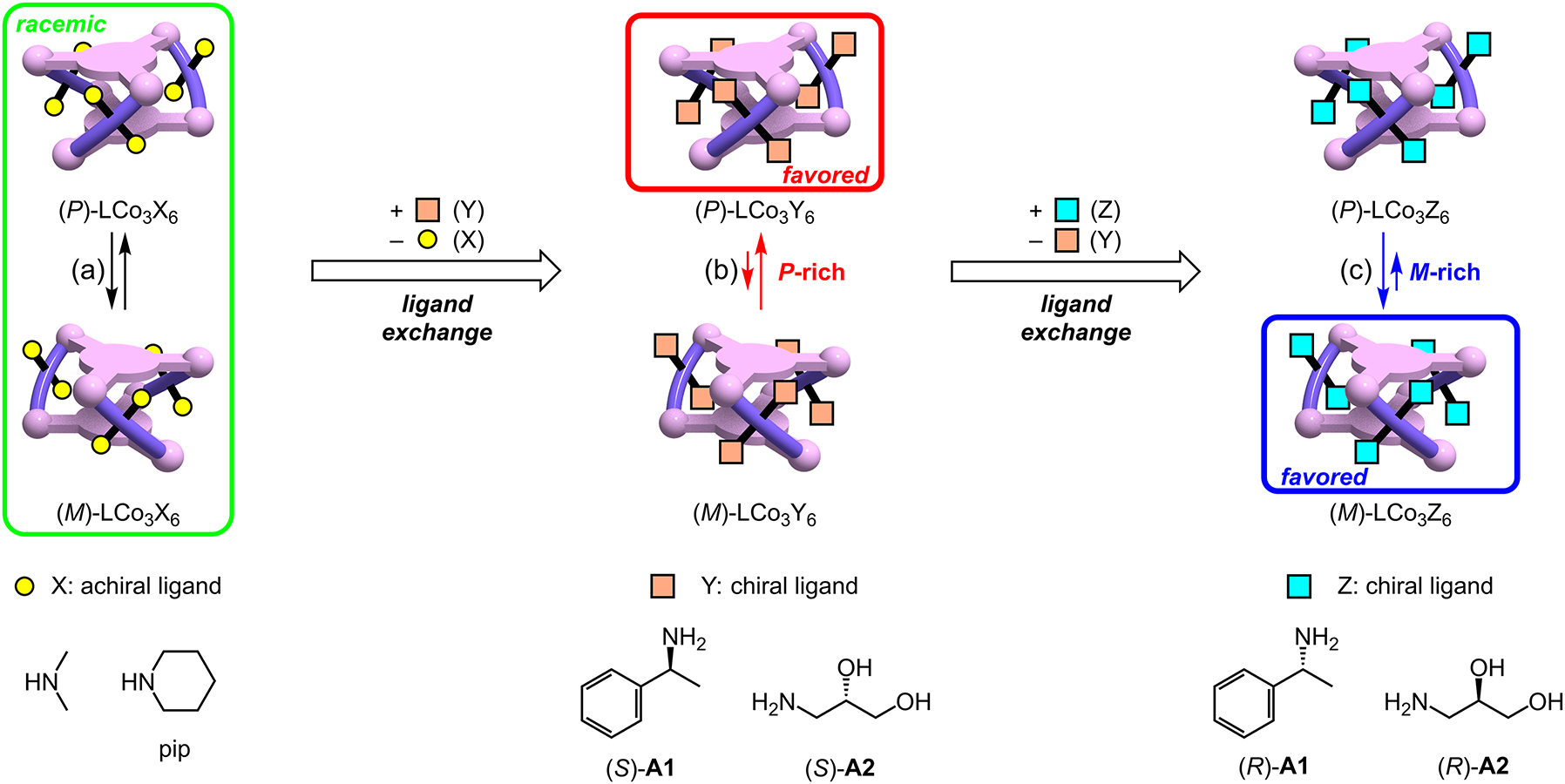
P/M equilibrium shifts of LCo3X6 driven by the X → Y → Z ligand exchange reactions at the cobalt(III) centers.
As the starting ligand X to be introduced into LCo3X6, monodentate achiral amines, such as dimethylamine and piperidine, were selected. If the achiral amine X is replaced by a chiral amine, such as A1 and A2, then the P/M equilibrium is shifted according to the progress of the ligand exchange (Fig. 5b).
The starting complexes LCo3X6, which have six achiral amine ligands X (X = Me2NH, pip), were obtained by the reaction of the free H6L ligand with cobalt(II) acetate in the presence of the achiral amines X under aerobic conditions [20]. These tricobalt(III) complexes also had a triple-helical structure as observed for the nickel(II) complex, which was confirmed by X-ray crystallography. Each of the cobalt(III) ions had an octahedral geometry, which had two amine ligands at the axial positions. The corresponding metallocryptands LCo3X6 having chiral amine ligands (X = A1, A2) were prepared in a similar manner. It is noteworthy that the metallocryptand LCo3((R)-A1)6, which has optically-active ligands (R)-A1, formed crystals only containing the left-handed form (M,R) due to the energy difference between the (M,R) and (P,R) diastereomers. This isolated left-handed helical complex showed a positive CD signal at around 550 nm in a methanol solution. Therefore, the P/M equilibrium shift can be monitored from the increase/decrease of the CD signal at 550 nm.
As expected, the achiral amine X in the helical metallocryptands LCo3X6 (X = Me2NH, pip) can be easily exchanged with the chiral amine, A1 or A2, just by mixing (Fig. 5a→b) [20]. The ligand exchange at the cobalt(III) centers gradually proceeded, which can be monitored by the time course change in the CD spectra. For example, the dimethylamine-coordinating helical complex, LCo3(Me2NH)6, was converted into LCo3((S)-A2)6 by the reaction with the chiral amine (S)-A2 (12 equiv). During the reaction, the intensity of the negative CD signals at 550 nm gradually increased and became almost constant after 3 h (Fig. 6a, b). This indicated that the P/M chirality inversion equilibrium was gradually shifted to give a P-rich mixture, which was driven by the ligand exchange reaction from dimethylamine to the chiral amine (S)-A2.
![Fig. 6:
Time-course CD spectral changes upon the ligand exchange of LCo3X6. (a) Full spectra for the exchange of Me2NH → (S)-A2. (b) Time-course changes in the CD intensity at 550 nm for the exchange of achiral amines with the chiral amines (12 equiv). (c) The P → M chirality inversion of LCo3X6 by the second amine, (R)-A1 or (R)-A2 (120 equiv). Reproduced with modification from ref. [20] with permission, Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA.](/document/doi/10.1515/pac-2023-0207/asset/graphic/j_pac-2023-0207_fig_006.jpg)
Time-course CD spectral changes upon the ligand exchange of LCo3X6. (a) Full spectra for the exchange of Me2NH → (S)-A2. (b) Time-course changes in the CD intensity at 550 nm for the exchange of achiral amines with the chiral amines (12 equiv). (c) The P → M chirality inversion of LCo3X6 by the second amine, (R)-A1 or (R)-A2 (120 equiv). Reproduced with modification from ref. [20] with permission, Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA.
The advantage of this system is that the speed of the P/M equilibrium shift can be altered by changing the combination of the initial achiral amine X and the added chiral amine Y (Fig. 6b). When a different chiral amine (S)-A1 was added instead of (S)-A2, the starting complex LCo3(Me2NH)6 underwent a slightly faster P/M equilibrium shift. On the other hand, when the initial amine was changed from Me2NH to piperidine, the equilibrium shift became significantly slower. These response speeds were quantitatively evaluated in terms of the initial rates (Table 2). The dimethylamine-coordinating complex, LCo3(Me2NH)6, underwent an up to 60 times faster P/M equilibrium shift than the piperidine-coordinating complex, LCo3(pip)6.
Initial rates of P/M equilibrium shift of LCo3X6 driven by the ligand exchange with the chiral amines.
| Starting complex | Added aminea | Initial rate (%de/min)b |
|---|---|---|
| LCo3(Me2NH)6 | (S)-A1 | 20 |
| LCo3(Me2NH)6 | (S)-A2 | 10 |
| LCo3(pip)6 | (S)-A1 | 0.33 |
| LCo3(pip)6 | (S)-A2 | 0.42 |
-
a12 equiv of amines were added. bReaction rates were determined in methanol by CD spectroscopy (ref. [20]).
If the initial and entering amines are both chiral but have opposite stereoconfigurations, the P/M equilibrium bias would be inverted from one side to the other side according to the reaction progress of the ligand exchange (Fig. 5b→c) [20]. Indeed, the P-rich mixture, which was produced by the addition of the first chiral amine (S)-A1 (12 equiv), was inverted to an M-rich mixture by the addition of an excess amount of (R)-A1 (120 equiv), i.e., the same chiral amine with the opposite stereoconfiguration. This inversion was clearly demonstrated by the time-dependent CD spectral changes (Fig. 6c); the negative CD signal at around 550 nm rapidly weakened and turned positive after 8 min, and became almost constant within 1 h. This P → M chirality inversion timescale can also be altered by changing the second chiral amine. When (R)-A2 was added instead of (R)-A1, the P → M chirality inversion became significantly slower. The CD signal inversion required 49 min, which is six times longer than in the case of (R)-A1. Therefore, this series of complexes worked as a controllable system in which the speeds of the P/M equilibrium shift and the P → M chirality inversion can be finely tuned by selecting the combination of the initial and entering amines.
The reverse reaction, i.e., the exchange of chiral amine Y in the helical complex LCo3Y6 with achiral amine X, showed unexpected time-dependent changes in the CD spectra. This reaction would finally give a P/M equimolar racemic mixture because the P and M forms of LCo3X6 constitute an enantiomer pair. During this reaction, the six chiral auxiliary ligands Y in LCo3Y6 are completely removed by the Y → X ligand exchange (Fig. 5b→a).
As already described, the LCo3((S)-A1)6 metallocryptand showed a negative CD signal at around 550 nm because the P/M chirality equilibrium is shifted to P-rich (Figs. 7a and 8a). If the (S)-A1 ligands are replaced by achiral amine ligands, this CD signal would gradually weaken and finally become silent according to the reaction progress. However, the reaction of LCo3((S)-A1)6 with piperidine (120 equiv) showed an unexpected non-trivial two-step feature during the time-dependent change [21]. The negative CD signal first turned positive within several minutes, then the resultant positive signal slowly decayed to zero for 2 d. This indicated that the chirality was inverted from P to M before the complete racemization. Usually, when a right-handed chiral compound undergoes racemization, the right-handed form is always dominant and the enantiomeric excess monotonically decreases to give a racemic mixture. The left-handed form will never be dominant. Consequently, the time-dependence observed in the racemization of LCo3((S)-A1)6 upon the addition of piperidine was really unexpected, because the M form was transiently generated during the racemization of the P form.
![Fig. 7:
Time course CD spectral changes of metallocryptand LCo3((S)-A1)6 upon the ligand exchange with piperidine (120 equiv). (a) Full CD spectra. (b) The CD intensity at 550 nm. Reproduced from ref. [21].](/document/doi/10.1515/pac-2023-0207/asset/graphic/j_pac-2023-0207_fig_007.jpg)
Time course CD spectral changes of metallocryptand LCo3((S)-A1)6 upon the ligand exchange with piperidine (120 equiv). (a) Full CD spectra. (b) The CD intensity at 550 nm. Reproduced from ref. [21].
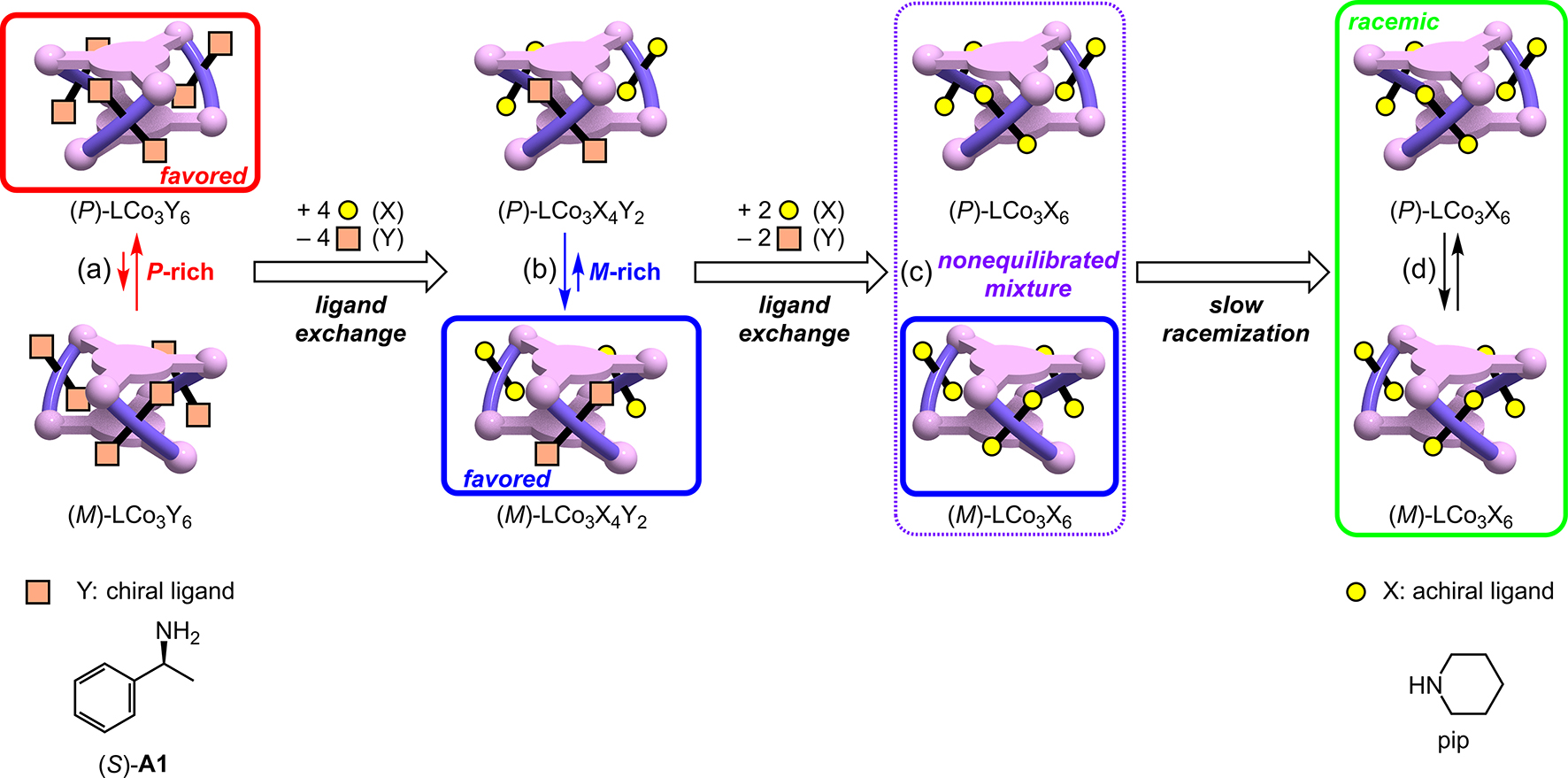
Transient P/M chirality inversion observed during the racemization of LCo3((S)-A1)6 by the ligand exchange with piperidine.
In order to clarify the reason for the unique time dependent changes, the intermediate state after 1 h, at which time the CD signal was transiently inverted to positive, was analyzed by spectroscopic methods [21]. The mass spectrometric measurements clearly demonstrated that the initial chiral amine ligands (S)-A1 were completely exchanged with the achiral piperidine ligands within 1 h, i.e., at an early stage of the overall CD spectral changes. Since the P and M forms of the LCo3(pip)6 are an enantiomer pair, i.e., the exact mirror images of each other, there is no thermodynamic preference to one of the two forms. Therefore, some of the intermediates containing one or more chiral ligands (S)-A1 should have a preference for the M form. A detailed analysis of the time-dependent mass spectra indicated that the tetra-exchanged intermediate LCo3((S)-A1)2(pip)4, which has only two chiral amine ligands (S)-A1, prefers the M form to give an M-rich mixture (Fig. 8b), which should be the origin of the unique transient chirality inversion. The P/M ratio of the M-rich mixture remained almost unchanged after 1 h, although the resultant species LCo3(pip)6 had no chiral auxiliary ligand, (S)-A1 (Fig. 8c). The complete racemization required 2 d; the very slow racemization can be explained by the six bulky piperidine ligands in the final product [LCo3(pip)6]. Due to this steric hindrance, the transition state of the P/M chirality inversion should be destabilized enough to practically maintain the P/M ratio of the nonequilibrated mixture (Fig. 8c→d).
It is noteworthy that this P→M transient chirality inversion was observed only during the racemization by the ligand exchange of (S)-A1 with piperidine (Fig. 5b→a) [21], whereas the P chirality was always dominant without showing the unique transient chirality inversion during the reverse reaction, i.e., the ligand exchange of piperidine with (S)-A1 (120 equiv) (Fig. 5a→b). Since the M chirality transiently appeared only in one of the forward and reverse steps, this can be regarded as a hysteretic cycle. This unique feature could originate from the reasonably slow P/M chirality inversion of the LCo3X6 metallocryptand scaffold, which occurs on a timescale comparable to the six-step ligand exchange reactions at the cobalt(III) center.
Conclusions
The metallocryptand structures, LM3, were found to act as a useful platform to cause a stimuli-responsive P/M chirality inversion because of the reasonably slow P/M chirality inversion kinetics. The response speed of the stimuli-responsive structural change can be controlled by two strategies; one is based on the guest binding in the LNi3 cryptand cavity that is surrounded by the three [Ni(saloph)] arms. The P/M chirality inversion equilibrium of LNi3 was decelerated by at least 180 times due to the very strong binding to the guanidinium ion with the binding constant of logK a > 8. The other strategy is based on the ligand exchange reactions at the cobalt(III) centers of LCo3X6, which occur on a timescale of minutes to hours. The unique time dependence of the P/M ratios can be explained by the relatively slow six-step ligand exchange reactions which simultaneously occur with the P/M chirality inversion.
The LM3 metallocryptand structures described in this article have metal complex moieties in their chiral framework, which can exhibit a variety of metal-derived properties and functions. In particular, the P/M ratio of this LM3 framework determines the sign and amplitudes of the chiroptical signals and enantioselectivities of various kinds of chemical events. Therefore, in combination with the time-dependent P/M chirality change of the LM3 metallocryptands, these chiral properties and functions can be changed over time as desired. Thus, advanced materials with unique time-dependent functions would be created by using a series of LM3 metallocryptand molecules as a basic backbone.
Funding source: Japan Society for the Promotion of Science
Award Identifier / Grant number: JP20K21206
Award Identifier / Grant number: JP16H06510
Award Identifier / Grant number: JP18H03913
Award Identifier / Grant number: JP21H05477
Award Identifier / Grant number: JP26288022
Funding source: University of Tsukuba
Award Identifier / Grant number: Unassigned
Acknowledgments
The author thanks Prof. Tatsuya Nabeshima (University of Tsukuba) and Dr. Yoko Sakata (Kanazawa University) for their valuable discussions during the research described in this article. Also, the author thanks Mr. Masato Miyashita, Ms. Shunjin Piao, and Mr. Shunsuke Chiba for performing the experiments described in this article.
-
Research funding: The research described in this article was supported by the Japan Society for the Promotion of Science (KAKENHI Grants JP16H06510 [Coordination Asymmetry], JP26288022, JP18H03913, JP20K21206, and JP21H05477 [Condensed Conjugation]) and the World Premier International Research Initiative, Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
[1(a)] Y. Okamoto, T. Nakano, S. Habaue, K. Shiohara, K. Maeda. J. Macromol. Sci., Pure Appl. Chem. A34, 1771 (1997), https://doi.org/10.1080/10601329708010307.Search in Google Scholar
(b) E. Yashima. Anal. Sci. 18, 3 (2002), https://doi.org/10.2116/analsci.18.3.Search in Google Scholar
(c) R. Amemiya, M. Yamaguchi. Org. Biomol. Chem. 6, 26 (2008), https://doi.org/10.1039/b713700a.Search in Google Scholar PubMed
[2(a)] Z. Peng, N. Takenaka. Chem. Rec. 13, 28 (2013), https://doi.org/10.1002/tcr.201200010.Search in Google Scholar PubMed
(b) S. Shirakawa, S. Liu, S. Kaneko. Chem.–Asian J. 11, 330 (2016), https://doi.org/10.1002/asia.201500951.Search in Google Scholar PubMed
[3(a)] J. M. Fox, T. J. Katz, S. van Elshocht, T. Verbiest, M. Kauranen, A. Persoons, T. Thongpanchang, T. Krauss, L. Brus. J. Am. Chem. Soc. 121, 3453 (1999), https://doi.org/10.1021/ja983633a.Search in Google Scholar
(b) S. K. Jha, K.-S. Cheon, M. M. Green, J. V. Selinger. J. Am. Chem. Soc. 121, 1665 (1999), https://doi.org/10.1021/ja983202s.Search in Google Scholar
(c) H. Fenniri, B.-L. Deng, A. E. Ribbe. J. Am. Chem. Soc. 124, 11064 (2002), https://doi.org/10.1021/ja026164s.Search in Google Scholar PubMed
(d) F. Sanda, K. Terada, T. Masuda. Macromolecules 38, 8149 (2005), https://doi.org/10.1021/ma051529p.Search in Google Scholar
(e) T. Biet, A. Fihey, T. Cauchy, N. Vanthuyne, C. Roussel, J. Crassous, N. Avarvari. Chem. Eur. J. 19, 13160 (2013), https://doi.org/10.1002/chem.201301095.Search in Google Scholar PubMed
[4(a)] E. Yashima, K. Maeda, H. Iida, Y. Furusho, K. Nagai. Chem. Rev. 109, 6102 (2009), https://doi.org/10.1021/cr900162q.Search in Google Scholar PubMed
(b) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai, K. Maeda. Chem. Rev. 116, 13752 (2016), https://doi.org/10.1021/acs.chemrev.6b00354.Search in Google Scholar PubMed
[5(a)] B. L. Feringa, A. M. Schoevaars, W. F. Jager, B. de Lange, N. P. M. Huck. Enantiomer 273, 325 (1996).Search in Google Scholar
(b) I. L. Karie. Biopolymers (Peptide Science) 60, 351 (2001), https://doi.org/10.1002/1097-0282(2001)60:5<351::AID-BIP10174>3.0.CO;2-U 10.1002/1097-0282(2001)60:5<351::AID-BIP10174>3.0.CO;2-USearch in Google Scholar
(c) K. Maeda, K. Morino, E. Yashima. Macromol. Symp. 201, 135 (2003), https://doi.org/10.1002/masy.200351116.Search in Google Scholar
(d) H. Miyake, H. Tsukube. Chem. Soc. Rev. 41, 6977 (2012), https://doi.org/10.1039/c2cs35192g.Search in Google Scholar
(e) H. K. Bisoyi, Q. Li. Angew. Chem., Int. Ed. 55, 2994 (2016), https://doi.org/10.1002/anie.201505520.Search in Google Scholar
(f) Z. Lv, Z. Chen, K. Shao, G. Qing, T. Sun. Polymers 8, 310 (2016), https://doi.org/10.3390/polym8080310.Search in Google Scholar
(g) S. Akine. Inorganics 6, 80 (2018), https://doi.org/10.3390/inorganics6030080.Search in Google Scholar
(h) S. Akine, H. Miyake. Coord. Chem. Rev. 468, 214582 (2022), https://doi.org/10.1016/j.ccr.2022.214582.Search in Google Scholar
(i) D. Sahoo, R. Benny, N. Kumar KS, S. De. ChemPlusChem 87, e202100322 (2022), https://doi.org/10.1002/cplu.202100322.Search in Google Scholar
[6(a)] M. Suginome, T. Yamamoto, Y. Nagata, T. Yamada, Y. Akai. Pure Appl. Chem. 84, 1759 (2012), https://doi.org/10.1351/pac-con-11-08-23.Search in Google Scholar
(b) K. Shimomura, T. Ikai, S. Kanoh, E. Yashima, K. Maeda. Nat. Chem. 6, 429 (2014), https://doi.org/10.1038/nchem.1916.Search in Google Scholar PubMed
[7(a)] K. Maeda, K. Morino, Y. Okamoto, T. Sato, E. Yashima. J. Am. Chem. Soc. 126, 4329 (2004), https://doi.org/10.1021/ja0318378.Search in Google Scholar PubMed
(b) L. Rosaria, A. d’Urso, A. Mammana, R. Purrello. Chirality 20, 411 (2008), https://doi.org/10.1002/chir.20464.Search in Google Scholar PubMed
[8] S. Akine, M. Miyashita, S. Piao, T. Nabeshima. Inorg. Chem. Front. 1, 53 (2014), https://doi.org/10.1039/c3qi00067b.Search in Google Scholar
[9] For reviews, see:.Search in Google Scholar
(a) S. Akine, T. Nabeshima. Dalton Trans., 10395 (2009), https://doi.org/10.1039/b910989g.Search in Google Scholar PubMed
(b) S. Akine. J. Inclusion Phenom. Macrocycl. Chem. 72, 25 (2012), https://doi.org/10.1007/s10847-011-0026-3.Search in Google Scholar
(c) S. Akine. The Chemistry of metal phenolates. in Patai’s Chemistry of Functional Groups, J. Zabicky (Ed.), Vol. 2, pp. 153–194, John Wiley and Sons, Ltd, Chichester, UK (2018), https://doi.org/10.1002/9780470682531.pat0909. Search in Google Scholar
(d) M. T. Chaudhry, S. Akine, M. J. MacLachlan. Chem. Soc. Rev. 50, 10713 (2021), https://doi.org/10.1039/d1cs00225b.Search in Google Scholar PubMed
[10] For a review, see: S. Akine. Dalton Trans. 50, 4429 (2021), https://doi.org/10.1039/d1dt00048a.Search in Google Scholar PubMed
[11] S. Akine, M. Miyashita, T. Nabeshima. Inorg. Chem. 60, 12961 (2021), https://doi.org/10.1021/acs.inorgchem.1c01376.Search in Google Scholar PubMed
[12] For ion recognition by macrocyclic oligo(metallosalen)-type compounds, see:Search in Google Scholar
(a) S. Akine, S. Sunaga, T. Taniguchi, H. Miyazaki, T. Nabeshima. Inorg. Chem. 46, 2959 (2007), https://doi.org/10.1021/ic062327s.Search in Google Scholar PubMed
(b) T. Nabeshima, H. Miyazaki, A. Iwasaki, S. Akine, T. Saiki, C. Ikeda, S. Sato. Chem. Lett. 35, 1070 (2006), https://doi.org/10.1246/cl.2006.1070.Search in Google Scholar
(c) S. Akine, F. Utsuno, T. Nabeshima. Chem. Commun. 46, 1029 (2010), https://doi.org/10.1039/b915722k.Search in Google Scholar PubMed
(d) S. Akine, S. Sunaga, T. Nabeshima. Chem. Eur. J. 17, 6853 (2011), https://doi.org/10.1002/chem.201100122.Search in Google Scholar PubMed
(e) S. Akine, Z. Varadi, T. Nabeshima. Eur. J. Inorg. Chem. 2013, 5987 (2013), https://doi.org/10.1002/ejic.201300917.Search in Google Scholar
(f) S. Akine, F. Utsuno, S. Piao, H. Orita, S. Tsuzuki, T. Nabeshima. Inorg. Chem. 55, 810 (2016), https://doi.org/10.1021/acs.inorgchem.5b02288.Search in Google Scholar PubMed
(g) Y. Sakata, S. Kobayashi, S. Akine. Chem. Commun. 53, 6363 (2017), https://doi.org/10.1039/c7cc02641b.Search in Google Scholar PubMed
(h) M. Cametti, Y. Sakata, J. Martí-Rujas, S. Akine. Inorg. Chem. 58, 14871 (2019), https://doi.org/10.1021/acs.inorgchem.9b02587.Search in Google Scholar PubMed
(i) M. T. Chaudhry, B. O. Patrick, S. Akine, M. J. MacLachlan. Org. Biomol. Chem. 20, 8259 (2022), https://doi.org/10.1039/d2ob01511k.Search in Google Scholar PubMed
[13] For ion recognition by acyclic oligo(metallosalen)-type compounds, see:Search in Google Scholar
(a) S. Akine, T. Taniguchi, T. Nabeshima. Angew. Chem., Int. Ed. 41, 4670 (2002), https://doi.org/10.1002/anie.200290011.Search in Google Scholar PubMed
(b) S. Akine, T. Taniguchi, T. Saiki, T. Nabeshima. J. Am. Chem. Soc. 127, 540 (2005), https://doi.org/10.1021/ja046790k.Search in Google Scholar PubMed
(c) S. Akine, T. Taniguchi, T. Nabeshima. J. Am. Chem. Soc. 128, 15765 (2006), https://doi.org/10.1021/ja0646702.Search in Google Scholar PubMed
(d) S. Akine, S. Kagiyama, T. Nabeshima. Inorg. Chem. 46, 9525 (2007), https://doi.org/10.1021/ic701585x.Search in Google Scholar PubMed
(e) S. Akine, T. Taniguchi, T. Nabeshima. Inorg. Chem. 47, 3255 (2008), https://doi.org/10.1021/ic702255s.Search in Google Scholar PubMed
(f) S. Akine, Y. Morita, F. Utsuno, T. Nabeshima. Inorg. Chem. 48, 10670 (2009), https://doi.org/10.1021/ic901372k.Search in Google Scholar PubMed
(g) S. Akine, S. Kagiyama, T. Nabeshima. Inorg. Chem. 49, 2141 (2010), https://doi.org/10.1021/ic9019926.Search in Google Scholar PubMed
(h) S. Akine, T. Matsumoto, S. Sairenji, T. Nabeshima. Supramol. Chem. 23, 106 (2011), https://doi.org/10.1080/10610278.2010.514906.Search in Google Scholar
(i) S. Akine, T. Tadokoro, T. Nabeshima. Inorg. Chem. 51, 11478 (2012), https://doi.org/10.1021/ic3012525.Search in Google Scholar PubMed
(j) S. Akine, T. Matsumoto, T. Nabeshima. Angew. Chem., Int. Ed. 55, 960 (2016), https://doi.org/10.1002/anie.201508065.Search in Google Scholar PubMed
[14] R. D. Shannon. Acta Crystallogr. A32, 751 (1976), https://doi.org/10.1107/s0567739476001551.Search in Google Scholar
[15] For helicity control of helical oligo(metallosalen)-type compounds, see:Search in Google Scholar
(a) S. Akine, T. Taniguchi, T. Nabeshima. Tetrahedron Lett. 47, 8419 (2006), https://doi.org/10.1016/j.tetlet.2006.09.125.Search in Google Scholar
(b) S. Akine, T. Matsumoto, T. Nabeshima. Chem. Commun., 4604 (2008), https://doi.org/10.1039/b810426c.Search in Google Scholar PubMed
(c) S. Akine, S. Hotate, T. Matsumoto, T. Nabeshima. Chem. Commun. 47, 2925 (2011), https://doi.org/10.1039/c0cc04998k.Search in Google Scholar PubMed
(d) S. Akine, S. Hotate, T. Nabeshima. J. Am. Chem. Soc. 133, 13868 (2011), https://doi.org/10.1021/ja205570z.Search in Google Scholar PubMed
(e) S. Akine, S. Sairenji, T. Taniguchi, T. Nabeshima. J. Am. Chem. Soc. 135, 12948 (2013), https://doi.org/10.1021/ja405979v.Search in Google Scholar PubMed
(f) S. Sairenji, S. Akine, T. Nabeshima. Tetrahedron Lett. 55, 1987 (2014), https://doi.org/10.1016/j.tetlet.2014.02.009.Search in Google Scholar
(g) S. Sairenji, S. Akine, T. Nabeshima. Chem. Lett. 43, 1107 (2014), https://doi.org/10.1246/cl.140263.Search in Google Scholar
(h) S. Sairenji, S. Akine, T. Nabeshima. Dalton Trans. 45, 14902 (2016), https://doi.org/10.1039/c6dt02635d.Search in Google Scholar PubMed
[16] Helicity inversion speeds of some helical metal complexes were controlled by changing its core metal ion, see:Search in Google Scholar
(a) S. Akine, T. Taniguchi, T. Matsumoto, T. Nabeshima. Chem. Commun., 4961 (2006), https://doi.org/10.1039/b610641b.Search in Google Scholar PubMed
(b) H. Miyake, H. Kamon, I. Miyahara, H. Sugimoto, H. Tsukube. J. Am. Chem. Soc. 130, 792 (2008), https://doi.org/10.1021/ja0768385.Search in Google Scholar PubMed
[17] Helicity changes driven by a chemical reaction, see: S. Sairenji, S. Akine, T. Nabeshima. Sci. Rep. 8, 137 (2018), https://doi.org/10.1038/s41598-017-16503-1.Search in Google Scholar PubMed PubMed Central
[18] Ligand exchange reactions at cobalt(III) centers in dinuclear macrocyclic complexes, see:Search in Google Scholar
(a) Y. Sakata, C. Murata, S. Akine. Nat. Commun. 8, 16005 (2017), https://doi.org/10.1038/ncomms16005.Search in Google Scholar PubMed PubMed Central
(b) Y. Sakata, M. Tamiya, M. Okada, S. Akine. J. Am. Chem. Soc. 141, 15597 (2019), https://doi.org/10.1021/jacs.9b06926.Search in Google Scholar PubMed
(c) Y. Sakata, M. Okada, M. Tamiya, S. Akine. Chem. Eur. J. 26, 7595 (2020), https://doi.org/10.1002/chem.202001072.Search in Google Scholar PubMed
(d) Y. Sakata, M. Okada, S. Akine. Chem. Eur. J. 27, 2284 (2021), https://doi.org/10.1002/chem.202004487.Search in Google Scholar PubMed
[19] The cobalt(III) metallocryptand framework was originally investigated from the viewpoint of its slow guest uptake/release behavior:Search in Google Scholar
(a) S. Akine, M. Miyashita, T. Nabeshima. J. Am. Chem. Soc. 139, 4631 (2017), https://doi.org/10.1021/jacs.7b00840.Search in Google Scholar PubMed
(b) S. Akine, M. Miyashita, T. Nabeshima. Chem. Eur. J. 25, 1432 (2019), https://doi.org/10.1002/chem.201805359.Search in Google Scholar PubMed
(c) S. Akine, Y. Sakata. Chem. Lett. 49, 428 (2020), https://doi.org/10.1246/cl.200017.Search in Google Scholar
[20] Y. Sakata, S. Chiba, M. Miyashita, T. Nabeshima, S. Akine. Chem. Eur. J. 25, 2962 (2019), https://doi.org/10.1002/chem.201805799.Search in Google Scholar PubMed
[21] Y. Sakata, S. Chiba, S. Akine. Proc. Natl. Acad. Sci. U. S. A. 119, e2113237119 (2022), https://doi.org/10.1073/pnas.2113237119.Search in Google Scholar PubMed PubMed Central
© 2023 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics