Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
-
Takeharu Haino


Abstract
Octaphosphonate biscavitand and self-folding deep biscavitand show strong positive and negative cooperativity, respectively. The mechanism of the cooperativity is discussed in terms of thermodynamic parameters and the detailed structure of the host-guest complexes. The two cavitand units of both biscavitands are tightly connected via four butylene linkers; thus, they are conformationally coupled, with the first guest binding information transferred to the resting-state cavities. This preorganization modulates the successive guest binding process in strong positive and negative cooperative manners, even though they display structural similarity. The first guest complexation always preorganizes the resting-state cavities where an existing water cluster and a toluene molecule are enthalpically stabilized. Successive guest complexation competes with the water cluster or a toluene molecule, reducing enthalpy gains. However, the desolvation upon successive guest binding processes liberate the solvents within the resting-state cavities. The water cluster is composed of 12 water molecules that are released upon successive guest complexation, resulting in a large entropy benefit. In contrast, toluene desolvation results in a limited entropy benefit. The difference in entropy benefits directs the strong positive or negative cooperativity of the structurally similar biscavitands.
Introduction
In biological regulation, enzymes and receptors with multiple binding sites often show allostery (cooperativity) [1]. Ligand binding to one of the multiple binding sites enhances or hinders successive ligand binding, known as positive or negative cooperativity, respectively [2]. A typical example of positive cooperativity is oxygen binding to hemoglobin [3]. There are four heme molecules in hemoglobin. All the heme molecules show fairly low oxygen affinities. When an oxygen molecule ligates at one of the four heme molecules, the remaining heme molecules are activated; then, their oxygen affinity is increased, thus facilitating the second, third, and fourth oxygen binding. However, the oxygen-binding affinity of hemoglobin is reduced by the complexation of 2,3-biphosphoglycerate (2,3-BPG) in cells [4]. 2,3-BPG is a negative allosteric effector that binds in the positively charged binding pocket of hemoglobin, reducing the oxygen binding affinity of the heme molecules. Therefore, oxygen molecules are released from hemoglobin in cells. Overall, oxygen molecules are captured by hemoglobin through positive cooperativity in the lung and then transferred and released in tissue cells through negative cooperativity by the binding of 2,3-BPG. Therefore, oxygen molecules are supplied to tissue cells through positive and negative allosteric regulation.
During the history of supramolecular chemistry, developing allosteric receptors by mimicking biological allosteric systems has been an extremely active research area [5]. The essential requirement for achieving cooperativity is that guest-binding information can be delivered among multiple binding sites [6]. Therefore, artificial allosteric receptors possess multiple guest binding sites that are conformationally coupled, such that binding of an effector to a remote site induces a conformational change of an active site that facilitates or deactivates successive substrate binding, known as the preorganization concept [7]. When an effector is not identical to a substrate molecule, a receptor shows heterotropic cooperativity. When the substrate acts as an effector that modulates substrate binding, it is known as homotropic cooperativity.
Recently, we developed a new feet-to-feet bisresorcinarene 1 in which two resorcinarene units are tightly connected via four butylene linkers (Fig. 1a) [8]. Four equivalents of dialdehyde 2a and two equivalents of resorcinol were mixed in acidic ethanol under reflux conditions, which gave rise to 1 in poor yield. Given that 12 chemical components must be assembled to form 1, a long time and fairly high temperature are required under acidic conditions. Dialdehyde 2a can be easily cyclized through an aldol reaction under acidic conditions to afford a five-membered cyclic aldehyde, which obviously interferes with the reaction with resorcinol. Bis(ethylene acetal) 2b most likely suppresses this undesired aldol reaction even under acidic conditions. Thus, the condensation reaction of 2b and resorcinol proceeded remarkably better to afford desired bisresorcinarene 1 in 81 % yield [9].
![Fig. 1:
(a) Synthesis of bisresorcinarene 1 and (b) the crystal structure of 1 with two DMF molecules. Adapted from Ref. [9] with permission. Copyright 2017 American Chemical Society.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_001.jpg)
(a) Synthesis of bisresorcinarene 1 and (b) the crystal structure of 1 with two DMF molecules. Adapted from Ref. [9] with permission. Copyright 2017 American Chemical Society.
The crystal structure of 1 includes two dimethylformamide molecules within the two resorcinarene cavities (Fig. 1b). The steric requirement of the four butylene linkers determines the orientation of the two resorcinarene cavities. A helically twisted conformation is observed, with a dihedral angle of 32° [10]. The two resorcinarene cavities are tightly connected; therefore, the guest binding information can be transferred to the resting-state cavity upon modulation of its successive guest binding. Accordingly, 1 fulfills the essential requirement to be an ideal platform for the development of homoditopic allosteric host molecules.
Cavitands are facilely constructed by connecting the phenolic hydroxyl groups of resorcinarenes with interannular bridges, facilitating the encapsulation of various large guests within the deepened cavity [11]. We envisioned introducing cavitand structures onto bisresorcinarene 1. The biscavitand structures possess two firmly connected deep cavities; therefore, cooperative guest binding can be realized through the steric communication between them. Herein, we introduce two biscavitands, 3a and 4: the former possesses two tetraphosphonate cavitands and displays strong positive cooperativity, and the latter contains two Rebek’s deep cavitands and illustrates strong negative cooperativity (Fig. 2). Specific solvent molecules located at the cavities effectively participate in directing the positive and negative cooperativity.
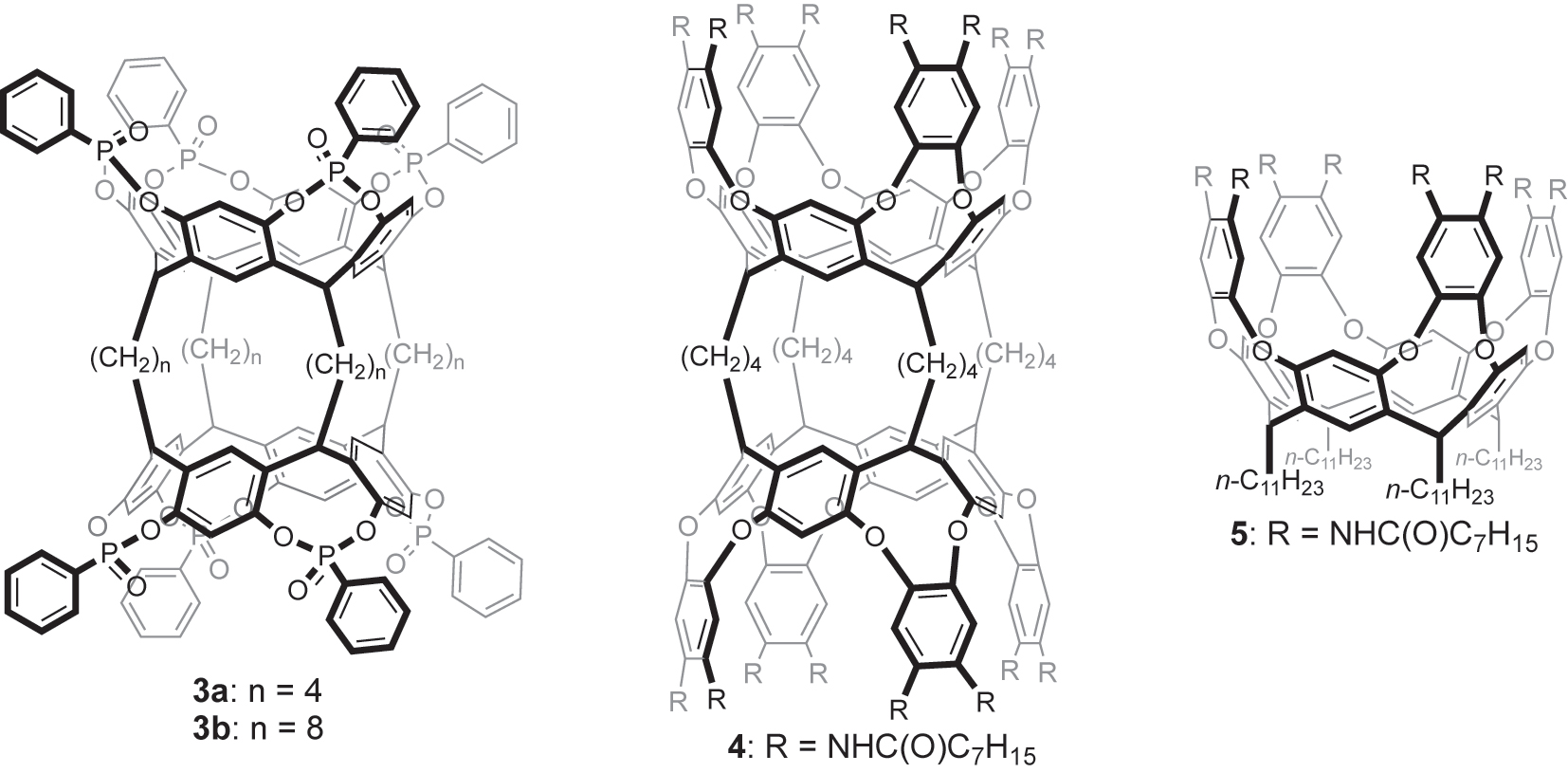
Structures of biscavitands 3a,b and 4, and self-folding cavitand 5.
Results and discussion
Entropy-driven positive cooperative effect in the molecular recognition of an octaphosphonate biscavitand
Bisresorcinarene 1 was functionalized with interannular phosphonate bridges to afford octaphosphonate biscavitands 3a (Fig. 2) [12]. A tetraphosphonate cavitand is known to encapsulate various ammonium guests through CH/π interactions [13]. Guest molecules G1 and G2 are potential candidates, being encapsulated within the two cavities of 3a in a cooperative manner (Fig. 3). We carried out titration experiments with guest molecule G1 in methanol and chloroform using isothermal titration calorimetry (ITC). Figure 4 shows the heat changes upon successive additions of the guest solution. The enthalpy contribution corresponds to the peak area in guest binding. The titration in methanol resulted in downward peaks, illustrating that 3a encapsulated G1 in a host-guest ratio of 1:2, which is an exothermic enthalpy-favored process. In contrast, the host-guest complexation in chloroform gave rise to upward peaks; thus, the host-guest complexation became an endothermic enthalpy-opposed process. The host-guest complexation process is surprisingly influenced by solvent properties.
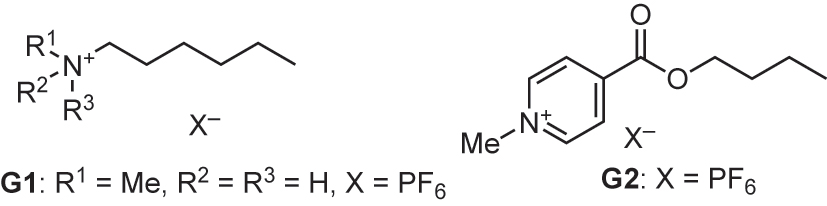
Guest molecules G1 and G2.
![Fig. 4:
ITC titration experiments of 3a with G1 in (a) methanol and (b) chloroform. The top graph shows the instrumental power function vs. time. The bottom graph shows the plot obtained from the integration of the calorimetric titration. Reproduced from Ref. [12b] with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_004.jpg)
ITC titration experiments of 3a with G1 in (a) methanol and (b) chloroform. The top graph shows the instrumental power function vs. time. The bottom graph shows the plot obtained from the integration of the calorimetric titration. Reproduced from Ref. [12b] with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
The cooperative effects for multiple guest binding can be discussed in terms of an interaction parameter (α) calculated based as α = 4K 2/K 1 where strong positive cooperativity results in a value above 3.1, whereas a value less than 0.3 indicates strong negative cooperativity [14]. 3a encapsulates the guest in methanol in a noncooperative or slightly negatively cooperative manner (Table 1). However, the guest binding behavior of 3a is strong positive cooperativity in chloroform, whereas that of 3b is in noncooperative or slightly negative cooperativity. Given that 3b possesses the two cavities that are linked with more flexible octylene chains, the rigidity of 3a has a meaningful impact on the cooperativity in the host-guest complexation in chloroform. The thermodynamics of the host-guest complexation of 3a provide detailed insights into the cooperative regulation. The first and second guest binding behaviors of 3a receive certain enthalpy gains in methanol, which is a common enthalpy-favored association process. Guests G1 and G2 possess methyl groups connected to the cationic nitrogen, which are encapsulated within the cavities of 3a through CH/π interactions with the cavity walls, resulting in enthalpy benefits. The enthalpy gains in the first guest binding behaviors are smaller than those in the second binding behaviors. Thus, the charge repulsion between the guest molecules in the 1:2 host-guest complexes is unlikely due to the strong, hydrogen bonding accepting ability of the P=O groups. The guest binding processes are also slightly entropy-favored. The host-guest complexation process is commonly entropy-unfavorable due to the loss of translational entropy of the host and guest upon complexation. Dalcanale reported that a methanol molecule is tightly complexed within the phosphonate cavitand [15]. Therefore, a methanol molecule solvates each cavity of 3a, which must be liberated upon guest binding. The translational entropy penalty is compensated by releasing the methanol molecule, which is responsible for the entropy gain upon host-guest complexation. The enthalpy contributions indicate that the first guest binding facilitates successive guest binding processes; thus, the two cavity units are conformationally coupled through the four butylene chains. In particular, the complexation of G2 remarkably preorganizes the resting-state cavity, which provides an enthalpy benefit to successive guest binding. However, the enthalpy gain in the second guest binding process is counteracted by the entropy penalty, which results in negative cooperativity. In contrast, the chloroform solvent system results in excellent positive cooperativity, with an interaction parameter far above 3.1 and thermodynamics quite different from those for the methanol system. The first guest binding process is associated with large enthalpy gains that result from the CH/π interaction of the methyl groups connected to the cationic nitrogen. However, the successive guest binding processes are enthalpy-opposed and entropy-driven.
Binding constants and thermodynamic parameters for the molecular associations of biscavitands 3a,b and guests G1 and G2 in methanol and chloroform [12b].
| Solvent | Host | Guest |
K
1/104
L mol−1 |
ΔH
1
/kcal mol−1 |
ΔS
1
/cal mol−1 K−1 |
K
2/104
L mol−1 |
ΔH
2
/kcal mol−1 |
ΔS
2
/cal mol−1 K−1 |
α (4K 2/K 1) |
|---|---|---|---|---|---|---|---|---|---|
| MeOH | 3a | G1 | 11 ± 1 | −4.55 ± 0.04 | 7 ± 2 | 3.4 ± 0.1 | −4.74 ± 0.04 | 4.6 ± 0.6 | 1.2 ± 0.1 |
| G2 | 2.5 ± 0.3 | −3.38 ± 0.08 | 9 ± 2 | 0.12 ± 0.01 | −6.1 ± 0.4 | −6 ± 2 | 0.19 ± 0.03 | ||
| CHCl3 | 3a | G1 | 43 ± 9 | −9 ± 3 | −6 ± 11 | 70 ± 10 | 26 ± 2 | 113 ± 7 | 7 ± 1 |
| G2 | 82 ± 24 | −7.1 ± 0.2 | 3 ± 8 | 115 ± 28 | 27 ± 2 | 118 ± 9 | 6 ± 2 | ||
| 3b | G1 | 157 ± 37 | −27.1 ± 0.6 | −64 ± 6 | 11 ± 2 | −12.2 ± 0.8 | −18 ± 5 | 0.28 ± 0.08 |
To discuss the strong positive cooperativity in chloroform, the structures of 3a in the absence and presence of G1 in chloroform-d 1 were studied using 1H NMR and diffusion-ordered NMR spectroscopy (DOSY). The spectrum in Fig. 5a displays a broad resonance at 4.6 ppm, corresponding to water molecules, as established by a deuterated water shake experiment [16]. On the basis of the integration, the broad resonance corresponded to the 12 water molecules that generated the NOE correlation with the aromatic protons at the upper rims of 3a. The diffusion coefficient of the 12 water molecules was perfectly consistent with that of 3a; thus, the water molecules are located at the upper ends of the two cavitand units, participating hydrogen bonding interactions with the phosphonate oxygens. When G1 was added to the solution of 3a, the broad water resonance vanished, and a new resonance appeared at −0.5 ppm, which was assigned to the N-methyl resonance of the bound G1 (Fig. 5b). The new methyl resonance corresponded to a diffusion coefficient identical to those of 3a. Accordingly, the 1:2 host-guest complex G1 2@3a was established by simultaneously releasing water molecules.
![Fig. 5:
1H NMR and 2D DOSY spectra of (a) 3a (2.7 mmol L−1) and (b) 3a (2.7 mmol L−1) with G1 (5.4 mmol L−1) at 273 K in chloroform-d
1. Reproduced from Ref. [12b] with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_005.jpg)
1H NMR and 2D DOSY spectra of (a) 3a (2.7 mmol L−1) and (b) 3a (2.7 mmol L−1) with G1 (5.4 mmol L−1) at 273 K in chloroform-d 1. Reproduced from Ref. [12b] with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Based on our structural study of biscavitand 3a in chloroform-d 1, a hydrogen-bonded water cluster consisting of six water molecules is located at each of the upper rims. Figure 6 illustrates the plausible cooperative binding mechanism of 3a in chloroform. The first guest binding process is associated with a large enthalpy gain arising from the effective CH/π interaction between the guest and the cavity, as well as simultaneous liberation of the water molecules at the upper rim. As a result, the first guest binding process shows limited entropy changes, most likely due to a trade-off between the translational entropy lost upon host-guest complexation and the entropy gained upon the liberation of water molecules. The first guest binding process preorganizes the resting-state cavity, which most likely enhances the hydrogen bonding interaction of the six water molecules located at the upper rim of the resting-state cavity. The successive guest binding to the resting-state cavity is thought to destroy the hydrogen-bonded water cluster, resulting in a higher enthalpy penalty than in the first guest binding. Thus, the second guest binding process must pay the enthalpy penalty. The six released water molecules receive translational freedom, resulting in a substantial entropy benefit. Subsequently, the local desolvation of the water clusters located at the upper rims directs the entropy-driven cooperative effect.
![Fig. 6:
Plausible picture of the positive cooperative host–guest complexation of 3a. Reproduced from Ref. [12b] with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_006.jpg)
Plausible picture of the positive cooperative host–guest complexation of 3a. Reproduced from Ref. [12b] with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Negative cooperative effect in molecular recognition of self-folding biscavitands
Bisresorcinarene 1 was functionalized with Rebek’s self-folding cavitands [17] to afford biscavitand 4 possessing deep cavities (Fig. 2). Each of the cavitands possessed eight amide groups that form interannular head-to-tail hydrogen bonding along the periphery, maintaining a deep open-ended cavity. The cavities were surrounded by aromatic walls, where cationic guest G3 was accommodated through CH/π interactions (Fig. 7). We studied the guest binding behavior of biscavitand 4 in toluene-d 8, chloroform-d 1, and THF-d 8 at 263 K using 1H NMR spectroscopy. 4 encapsulated G3 in toluene-d 8. The signals of free and bound G3 separately appeared. The presence of a substantial energetic barrier slowed the in-and-out guest exchange process on the NMR time scale [18].
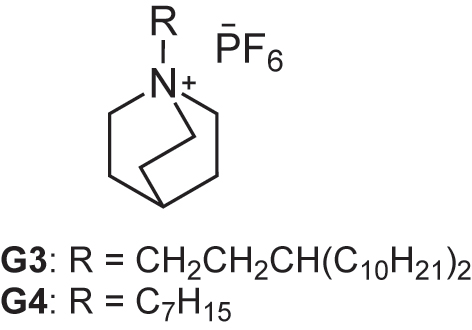
Guest molecules G3 and G4.
Large upfield shifts were observed for the guest protons H1, H2, and H3, appearing at −2.0, −1.2, and 1.3 ppm (Fig. 8) [19]. Given that each of the cavities was walled with the four aromatic rings, the encapsulated guest G3 experienced a strong shielding effect and exhibited guest inclusion characteristics. The integration ratio of 4 to G3 was determined to be 1:1 even in the presence of 20 equivalents of G3. Although a 1:2 host-guest complex G3 2⊂3 was formed even when 80 equivalents of G3 were added, the 1:1 host-guest complex G3⊂3 remained dominant. In contrast, the 1:1 and 1:2 host-guest complexes G3⊂3 and G3 2⊂3 coexisted in the presence of 20 equivalents of G3 when chloroform-d 1 and THF-d 8 were employed as solvent systems. Negative cooperativity was observed in toluene-d 8.
![Fig. 8:
1H NMR spectra of (a) G3 and (b) 4 with 20 equiv. of G3, (c) 4 with 80 equiv. of G3 at 263 K in toluene-d
8, (d) G3, (e) 4 with 20 equiv. of G3 at 263 K in chloroform-d
1, (f) G3 and (g) 4 with 20 equiv. of G3 at 263 K in THF-d
8: [4] = 2.00 mmol L−1. Adapted from Ref. [20] with permission. Copyright 2021 American Chemical Society.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_008.jpg)
1H NMR spectra of (a) G3 and (b) 4 with 20 equiv. of G3, (c) 4 with 80 equiv. of G3 at 263 K in toluene-d 8, (d) G3, (e) 4 with 20 equiv. of G3 at 263 K in chloroform-d 1, (f) G3 and (g) 4 with 20 equiv. of G3 at 263 K in THF-d 8: [4] = 2.00 mmol L−1. Adapted from Ref. [20] with permission. Copyright 2021 American Chemical Society.
The crystal structures of 4 and G4 2⊂4 were analyzed to determine their structural characteristics (Fig. 9). The structure of 4 shows that the interannular head-to-tail cyclic hydrogen bonds among the 16 amide groups maintain two deep cavities. The two deep cavities are firmly linked by the butylene linkers with a lack of flexibility in conformation. A toluene molecule is located within each cavity. The host-guest complex G4 2⊂4 is symmetric, with the nitrogen atoms pointed to the upper ends of the cavities. The guest methylene groups are close to the aromatic walls, making attractive contacts with CH/π interactions. Accordingly, the deep cavities are always occupied by solvent molecules or guest molecules, which indicates that guest complexation competes with solvents.
![Fig. 9:
Crystal structures of (a and b) 4 and (c and d) G4
2⊂4. (a and c) Side views and (b and d) top views. The aliphatic side chain, hydrogen atoms and crystal solvents are omitted for the sake of clarity. Adapted from Ref. [20] with permission. Copyright 2021 American Chemical Society.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_009.jpg)
Crystal structures of (a and b) 4 and (c and d) G4 2⊂4. (a and c) Side views and (b and d) top views. The aliphatic side chain, hydrogen atoms and crystal solvents are omitted for the sake of clarity. Adapted from Ref. [20] with permission. Copyright 2021 American Chemical Society.
ITC titration experiments provided detailed insights into the host-guest complexation of 4 with G3 in toluene, chloroform, and THF (Table 2). The guest-binding behaviors were found to depend on the solvent properties. Biscavitand 4 showed remarkably high binding constants for the first and second guest binding processes in toluene. Chloroform and THF reduced the stabilities of the host-guest complexes of 4 and G3. The two deep cavities were maintained by the cyclic hydrogen bonding of the amide groups; thus, more hydrogen bonding competition in chloroform and THF destabilized the deep cavity structures, which reduced the extent of the preorganization. Therefore, the host-guest complexations of G3⊂4 and G3 2⊂4 preferred the nonpolar solvent toluene over the slightly polar solvents chloroform and THF.
Binding constants and thermodynamic parameters for the molecular associations of biscavitand 4 with G3 at 295 K [20].
| Solvent | K 1/L mol−1 | ΔH
1
/kcal mol−1 |
ΔS
1
/cal mol−1 K−1 |
K
2
/L mol−1 |
ΔH
2
/kcal mol−1 |
ΔS
2
/cal mol−1 K−1 |
α (4K 2/K 1) |
|---|---|---|---|---|---|---|---|
| Toluene | 440 000 ± 20 000 | −5.16 ± 0.02 | 8 ± 1 | 10 000 ± 500 | −0.34 ± 0.2 | 17.23 ± 0.9 | 0.089 ± 0.006 |
| CHCl3 | 970 ± 90 | −1.46 ± 0.02 | 9 ± 1 | 27 ± 2 | −0.6 ± 0.1 | 4.3 ± 0.7 | 0.110 ± 0.001 |
| THF | 341 ± 7 | −3.72 ± 0.02 | −1.0 ± 0.2 | 171 ± 3 | 0.50 ± 0.02 | 12.0 ± 0.2 | 2.00 ± 0.05 |
Cooperativity in the multiple guest complexation of 4 can be discussed in terms of the interaction parameters. Strong negative cooperativity in the guest complexations was observed in toluene and chloroform, with very small interaction parameters of 0.089 and 0.11, respectively. The host-guest complexation was noncooperative in THF. Accordingly, the solvent polarity considerably influenced the cooperative effects in the host-guest complexations. The host-guest complexation processes can be discussed in terms of the thermodynamic parameters. The first guest binding processes are more enthalpy-favored than the second binding processes in all the solvents. This implies that the electrostatic repulsion between the guests encapsulated at both ends of 4 might be involved in the host-guest complexations, which does not play a role in determining the positive or negative cooperativity. The first and second host-guest complexation processes in toluene and chloroform are enthalpy- and entropy-favored. Given that a toluene molecule fills each of the cavities in the crystal structure, the solvent molecules in the cavities and structured around guest G3 are desolvated upon guest complexation, resulting in positive entropy contributions. As observed in the solid state, the effective CH/π interactions between the quinuclidine and the cavitand aromatic rings drive the host-guest complexation. However, THF is a hydrogen-bonding-competitive solvent, resulting in a slightly different thermodynamic picture. Although the first guest binding process is enthalpy-favored, successive guest binding to the resting-state cavity becomes enthalpy-opposed and entropy-favored due to the desolvation of THF molecules hydrogen-bonded to the amide groups.
The conformational behavior of the tightly linked two cavities of 4 was studied by dynamic NMR spectroscopy. The interannular head-to-tail hydrogen bonding of the eight amide groups at each of the biscavitand rims maintains the deep, open-ended cavities. The clockwise and anticlockwise cycloenantiomers of the cyclic hydrogen bonding were in equilibrium at room temperature (Fig. 10). Table 3 shows that a sizable energetic barrier exists in the clockwise/anticlockwise interconversion process, which was discussed with the activation enthalpies (ΔH ⧧) and entropies (ΔS ⧧) in toluene-d 8, chloroform-d 1, and THF-d 8. The interconversion process must pay enthalpy costs as a result of the breakage of the hydrogen bonds at the transition state. The enormous entropy penalties most likely come from the highly symmetric nature of the transition state, where the breaking of the eight hydrogen bonds occurs simultaneously. A meaningful difference in the enthalpy costs of activation between 4 and 5 illustrates the cooperative cooperation in the two cyclic hydrogen bonds of 4. Given that 4 requires the enthalpy cost of activation, 1.7 times as high as that of 5 in toluene-d 8, the clockwise/anticlockwise interconversions are conformationally coupled with each other owing to the tight connection of the two cavitand units with four butylene linkages. Accordingly, the cyclic hydrogen bonding at each of the biscavitand rims is cooperatively strengthened. In contrast, hydrogen bond donor solvents chloroform and THF resulted in smaller differences in the enthalpy costs of activation between 4 and 5. Competitive solvation on the cyclic hydrogen bonds weakens the connection between the two cavities, presumably diminishing cooperativity in the interconversion. Therefore, the solvation determines the conformational communication of the two cavities tightly connected with the four butylene linkages, directing the cavity preorganization. The negative cooperativity is probably driven by the conformational communication of the two cavities, where the complexation of the guest molecule within one of the cavities induces a tiny conformational change that is transmitted to the resting-state cavity, being less prone to capture a consecutive guest.
![Fig. 10:
Clockwise/anticlockwise interconversion of the cyclic hydrogen bonding at the cavitand units of 4 and 5. Reproduced from Ref. [20] with permission. Copyright 2021 American Chemical Society.](/document/doi/10.1515/pac-2023-0206/asset/graphic/j_pac-2023-0206_fig_010.jpg)
Clockwise/anticlockwise interconversion of the cyclic hydrogen bonding at the cavitand units of 4 and 5. Reproduced from Ref. [20] with permission. Copyright 2021 American Chemical Society.
Activation parameters of clockwise/anticlockwise interconversions of 4 and 5.
| Solvent | Host | ΔH ‡/kJ mol−1 | ΔS ‡/J mol−1 K−1 |
|---|---|---|---|
| Toluene | 4 | 43.6 ± 0.9 | −95(3) |
| 5 | 25.3 ± 0.8 | −158(3) | |
| Chloroform | 4 | 37.6 ± 0.4 | −102.9(1) |
| 5 | 26.5 ± 0.2 | −134.1(8) | |
| THF | 4 | 21.9 ± 0.2 | −132.4(8) |
| 5 | 17.5 ± 0.1 | −140.2(2) |
Figure 11 illustrates a plausible explanation of the strong negative cooperativity proposed from the thermodynamic behaviors of the host-guest complexation. The enthalpy gains of the first guest binding processes in all solvents exceeded those of the successive guest binding processes. Given that the two cavitand units are tightly connected via butylene linkers, the conformationally coupled resting-state cavity is preorganized by the first guest binding process, where a toluene molecule preserved within the resting-state cavity is stabilized with a certain enthalpy gain. The desolvation of the toluene molecule within the cavity must compete with the successive guest binding, compensating for the additional enthalpy penalty. Even though the entropy contribution is positive, the extent of desolvation is too small to compensate for the enthalpy penalty associated with the desolvation of the toluene molecule. Accordingly, the strong negative cooperativity is directed by the desolvation of the stabilized toluene molecule. However, the amide groups are solvated with THF molecules, which reduces the extent of preorganization. The first guest binding process slightly facilitates the solvation of the resting-state cavity. The successive guest binding must liberate multiple THF molecules, which most likely results in the enthalpy penalty that is compensated for by the entropy gain. Accordingly, the solvation determines the degree of preorganization that defines the cooperativity in the host-guest complexation of 4.
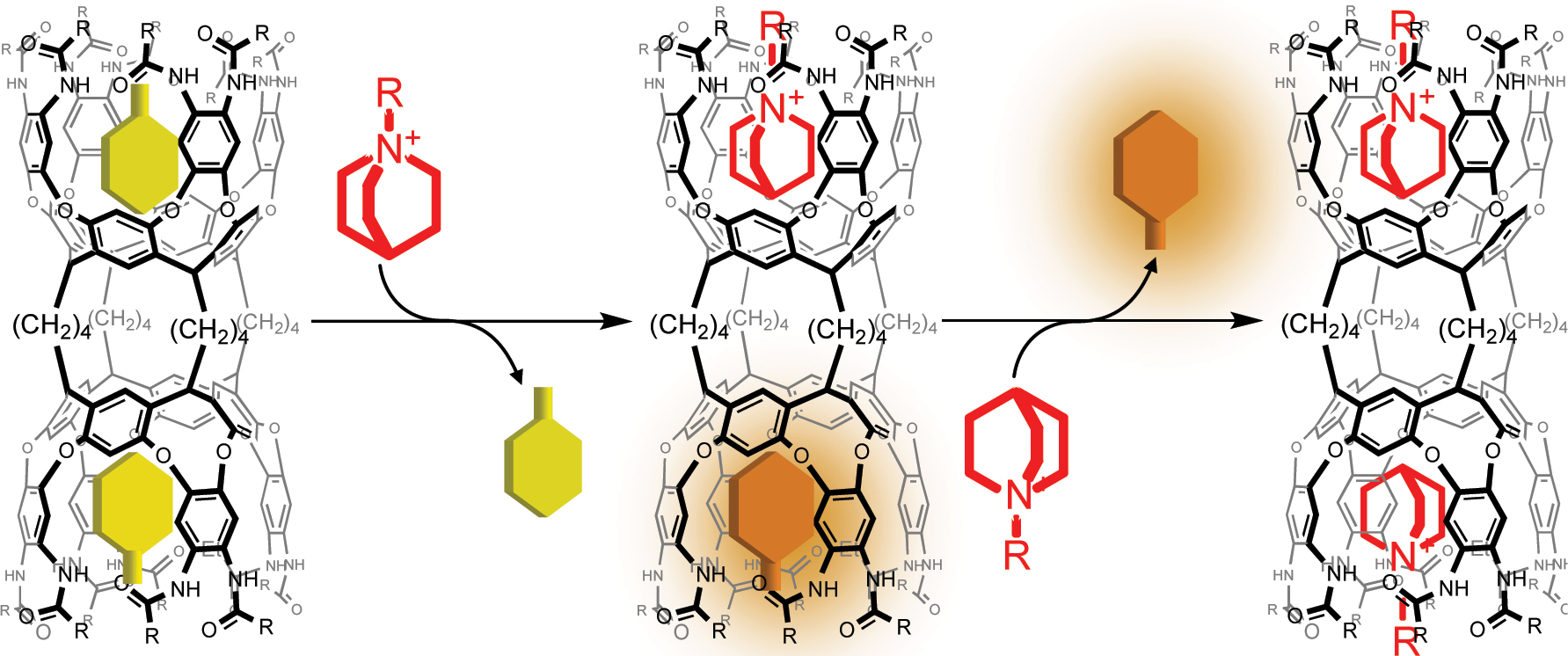
Plausible explanation of the negative cooperative host–guest complexation of 4.
Conclusion
In conclusion, the cavitand units of a phosphonate biscavitand and a self-folding biscavitand are locally solvated with water clusters and toluene molecules, respectively. Both cavitand units encapsulate cationic guests through CH/π interactions between the guests and the aromatic walls. The two cavitand units are tightly bridged with four butylene linkers in both biscavitands. Guest binding occurs at one of the cavitand units, which results in the preorganization of the resting-state cavity, where a water cluster or a toluene molecule left is greatly stabilized with an enthalpy gain. However, the enthalpy gains are counteracted by the cost of successive guest binding due to desolvation. From the entropic point of view, the water cluster within the cavity is composed of six water molecules, which are liberated with a large entropy benefit, whereas the toluene molecule within the resting-state cavity is replaced by the guest molecule without an entropy benefit. These entropy gains compensate for the enthalpy penalty of the successive guest binding. Accordingly, the number of solvent molecules released determines the positive or negative cooperativities in the biscavitands.
In general, the preorganization concept in allosteric receptors might be discussed in view of conformation behaviors of flexibly connected multiple-guest binding sites, which is applicable to many cases. In the present systems, the two cavities are firmly connected; however, the preorganization concept is operative for multiple guest binding. The preorganization effect stabilizes specific solvents located within the resting-state cavity. Accordingly, entropy-directed positive or negative cooperativity becomes obvious in such deep cavities. Many chemists understand the contribution of desolvation; however, these contrastive cooperativities are directed by the desolvation of explicit solvated molecules, reminding us of the contribution of desolvation to both enthalpy and entropy from a quantitative viewpoint.
Article note:
A collection of invited papers based on presentations at the 25th International Conference on Physical Organic Chemistry which was held in Hiroshima, Japan on 10–15th July 2022.
Funding source: Japan Society for the Promotion of Science
Award Identifier / Grant number: 21H04685
Award Identifier / Grant number: 21H05491
Acknowledgment
This work was supported by Grant-in-Aid for Scientific Research (A) JSPS KAKENHI Grant Number JP21H04685 and Grant-in-Aid for Transformative Research Areas “Condensed Conjugation” JSPS KAKENHI Grant Number JP21H05491 from Japan Society for the Promotion of Science (JSPS).
References
[1] A. Whitty. Nat. Chem. Biol. 4, 435 (2008), https://doi.org/10.1038/nchembio0808-435.Search in Google Scholar PubMed
[2] D. H. Williams, E. Stephens, D. P. O’Brien, M. Zhou. Angew. Chem. Int. Ed. 43, 6596 (2004), https://doi.org/10.1002/anie.200300644.Search in Google Scholar PubMed
[3] M. F. Perutz, G. Fermi, B. Luisi, B. Shaanan, R. C. Liddington. Acc. Chem. Res. 20, 309 (1987), https://doi.org/10.1021/ar00141a001.Search in Google Scholar
[4] R. Macdonald. Anaesthesia 32, 544 (1977), https://doi.org/10.1111/j.1365-2044.1977.tb10002.x.Search in Google Scholar PubMed
[5a)] L. Kovbasyuk, R. Krämer. Chem. Rev. 104, 3161 (2004), https://doi.org/10.1021/cr030673a.Search in Google Scholar PubMed
b) C. Kremer, A. Lützen. Chem. Eur. J. 19, 6162 (2013), https://doi.org/10.1002/chem.201203814.Search in Google Scholar PubMed
c) C. A. Hunter, H. L. Anderson. Angew. Chem. Int. Ed. 48, 7488 (2009), https://doi.org/10.1002/anie.200902490.Search in Google Scholar PubMed
d) T. Nabeshima, S. Akine. Chem. Rec. 8, 240 (2008), https://doi.org/10.1002/tcr.20153.Search in Google Scholar PubMed
e) S. Shinkai, M. Ikeda, A. Sugasaki, M. Takeuchi. Acc. Chem. Res. 34, 494 (2001), https://doi.org/10.1021/ar000177y.Search in Google Scholar PubMed
f) J. Rebek. Acc. Chem. Res. 17, 258 (1984), https://doi.org/10.1021/ar00103a006.Search in Google Scholar
[6] J. Rebek, T. Costello, L. Marshall, R. Wattley, R. C. Gadwood, K. Onan. J. Am. Chem. Soc. 107, 7481 (1985), https://doi.org/10.1021/ja00311a043.Search in Google Scholar
[7a)] D. J. Cram. Angew. Chem. Int. Ed. 27, 1009 (1988), https://doi.org/10.1002/anie.198810093.Search in Google Scholar
b) D. J. Cram. Angew. Chem. Int. Ed. 25, 1039 (1986), https://doi.org/10.1002/anie.198610393.Search in Google Scholar
[8] H. Yamada, T. Ikeda, T. Mizuta, T. Haino. Org. Lett. 14, 4510 (2012), https://doi.org/10.1021/ol301996q.Search in Google Scholar PubMed
[9] D. Shimoyama, T. Ikeda, R. Sekiya, T. Haino. J. Org. Chem. 82, 13220 (2017), https://doi.org/10.1021/acs.joc.7b02301.Search in Google Scholar PubMed
[10] D. Shimoyama, T. Haino. J. Org. Chem. 84, 13483 (2019), https://doi.org/10.1021/acs.joc.9b01730.Search in Google Scholar PubMed
[11a)] R. Pinalli, F. Boccini, E. Dalcanale. Isr. J. Chem. 51, 781 (2011), https://doi.org/10.1002/ijch.201100057.Search in Google Scholar
b) Y. Yu, J. M. Yang, J. Rebek. Chem. 6, 1265 (2020), https://doi.org/10.1016/j.chempr.2020.04.014.Search in Google Scholar
c) D. J. Cram. Nature 356, 29 (1992), https://doi.org/10.1038/356029a0.Search in Google Scholar
d) R. Pinalli, E. Dalcanale, F. Ugozzoli, C. Massera. CrystEngComm 18, 5788 (2016), https://doi.org/10.1039/c6ce01010e.Search in Google Scholar
e) J. Murray, K. Kim, T. Ogoshi, W. Yao, B. C. Gibb. Chem. Soc. Rev. 46, 2479 (2017), https://doi.org/10.1039/c7cs00095b.Search in Google Scholar PubMed PubMed Central
f) J. H. Jordan, B. C. Gibb. Chem. Soc. Rev. 44, 547 (2015), https://doi.org/10.1039/c4cs00191e.Search in Google Scholar PubMed
g) S. Liu, B. C. Gibb. Chem. Commun. 44, 3709 (2008), https://doi.org/10.1039/b805446k.Search in Google Scholar PubMed PubMed Central
h) A. Galan, P. Ballester. Chem. Soc. Rev. 45, 1720 (2016), https://doi.org/10.1039/c5cs00861a.Search in Google Scholar PubMed
i) K. Kobayashi, M. Yamanaka. Chem. Soc. Rev. 44, 449 (2015), https://doi.org/10.1039/c4cs00153b.Search in Google Scholar PubMed
[12a)] D. Shimoyama, H. Yamada, T. Ikeda, R. Sekiya, T. Haino. Eur. J. Org. Chem. 2016, 3300 (2016), https://doi.org/10.1002/ejoc.201600410.Search in Google Scholar
b) D. Shimoyama, T. Haino. Chem. Eur. J. 26, 3074 (2020), https://doi.org/10.1002/chem.201905036.Search in Google Scholar PubMed
[13a)] R. M. Yebeutchou, F. Tancini, N. Demitri, S. Geremia, R. Mendichi, E. Dalcanale. Angew. Chem. Int. Ed. 47, 4504 (2008), https://doi.org/10.1002/anie.200801002.Search in Google Scholar PubMed
b) P. Delangle, J. C. Mulatier, B. Tinant, J. P. Declercq, J. P. Dutasta. Eur. J. Org. Chem. 2001, 3695 (2001), https://doi.org/10.1002/1099-0690(200110)2001:19<3695::aid-ejoc3695>3.0.co;2-9.10.1002/1099-0690(200110)2001:19<3695::AID-EJOC3695>3.0.CO;2-9Search in Google Scholar
[14] Z. Huang, K. Qin, G. Deng, G. Wu, Y. Bai, J.-F. Xu, Z. Wang, Z. Yu, O. A. Scherman, X. Zhang. Langmuir 32, 12352 (2016), https://doi.org/10.1021/acs.langmuir.6b01709.Search in Google Scholar
[15] R. Pinalli, F. F. Nachtigall, F. Ugozzoli, E. Dalcanale. Angew. Chem. Int. Ed. 38, 2377 (1999), https://doi.org/10.1002/(sici)1521-3773(19990816)38:16<2377::aid-anie2377>3.0.co;2-o.10.1002/(SICI)1521-3773(19990816)38:16<2377::AID-ANIE2377>3.0.CO;2-OSearch in Google Scholar
[16a)] C. Massera, M. Melegari, E. Kalenius, F. Ugozzoli, E. Dalcanale. Chem. Eur. J. 17, 3064 (2011), https://doi.org/10.1002/chem.201003407.Search in Google Scholar
b) C. Massera, M. Melegari, F. Ugozzoli, E. Dalcanale. Chem. Commun. (Camb.) 46, 88 (2010), https://doi.org/10.1039/b917931c.Search in Google Scholar
[17a)] A. Shivanyuk, K. Rissanen, S. K. Korner, D. M. Rudkevich, J. Rebek. Helv. Chim. Acta 83, 1778 (2000), https://doi.org/10.1002/1522-2675(20000809)83:8<1778::aid-hlca1778>3.0.co;2-j.10.1002/1522-2675(20000809)83:8<1778::AID-HLCA1778>3.0.CO;2-JSearch in Google Scholar
b) D. M. Rudkevich, G. Hilmersson, J. Rebek. J. Am. Chem. Soc. 120, 12216 (1998), https://doi.org/10.1021/ja982970g.Search in Google Scholar
[18] L. C. Palmer, J. Rebek. Org. Biomol. Chem. 2, 3051 (2004), https://doi.org/10.1039/b412510j.Search in Google Scholar
[19] T. Haino, D. M. Rudkevich, J. Rebek. J. Am. Chem. Soc. 121, 11253 (1999), https://doi.org/10.1021/ja993365w.Search in Google Scholar
[20] H. Fujimoto, D. Shimoyama, K. Katayanagi, N. Kawata, T. Hirao, T. Haino. Org. Lett. 23, 6217 (2021), https://doi.org/10.1021/acs.orglett.1c01837.Search in Google Scholar
© 2023 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics