
Step economy strategy for the synthesis of amphoteric aminoaldehydes, key intermediates for reduced hydantoins
-
Júlia L. Monteiro


Abstract
Despite of the orthogonal reactivity of the N–H aziridines aldehyde, these compounds exist as an equilibrium of three different forms – whereas the dimeric one is mostly observed in a variety of solvents. In this work, we have developed an alternative protocol for the aminoaldehyde dimers synthesis in two steps starting with an organocatalyzed aziridination between α,β-unsaturated aldehydes and a protected amine to afford known isolable and stable N-protected aziridine aldehydes. After Boc-deprotection, dimeric species were immediately formed from monomeric N–H aziridine aldehydes. From this building-block new reduced hydantoins were prepared via [3+2]-annulation with isocyanates.
Introduction
N-heterocycles are of major interest in medicinal chemistry and related areas due to their pharmacological properties. In particular, the hydantoin scaffold is an important structural motif with a broad range of biological activities [1], [2]. Natural hydantoins have been isolated from marine organisms, for example (Z)-5-(4-hydroxybenzylidene)-hydantoin, from the Red Sea sponge Hemimycale arabica, showed potent in vitro anti-growth and anti-invasive properties against PC-3M prostate cancer cells in MTT and spheroid disaggregation assays [3]. Cachet et al. in 2009 isolated five new hydantoin alkaloids, named parazoanthines A−E, from the Mediterranean Sea anemone Parazoanthus axinellae. All compounds were evaluated for their natural toxicity (microtox assay), and parazoanthine C exhibited the highest values [4].
Additionally, several others biological and pharmacological activities have also been described for hydantoins, including androgen receptor modulators [5], anticonvulsant [6], antidiabetic [7], and anticancer agents [8]. Furthermore, the activity of hydantoins against neurodegenerative diseases such as Parkinson and Alzheimer’s has also been described [9], [10], [11].
Therefore, a great variety of synthetic strategies is available for the construction of the hydantoin synthon from several precursor molecules [12]. Among those, the Bucherer–Bergs reaction is regarded as one of the most straightforward approaches to achieve hydantoins from aldehydes or ketones. In 2016, we reported the continuous flow synthesis of hydantoins employing this reaction [13]. Later on, a two-step semi-continuous flow synthesis of hydantoins from amines was described by Gilmore et al. [14]. The microwave irradiation has also been applied as an alternative energy source in the Bucherer-Bergs [15]. Moreover, Colacino et al. described a mechanochemical preparation of 5- and 5,5-disubstituted hydantoins from various amino ester hydrochlorides and potassium cyanate in a planetary ball-mill [16].
Conformationally constrained fused bicyclic amino acids have attracted much attention, as their incorporation into a native peptide or peptidomimetics induce conformational restriction and provide significant structural effects. That can lead to improvements in the efficiency and selectivity of compounds toward a specific receptor, resistance to chemical, and enzymatic degradation, and thus the bioavailability [17]. With this in mind, Šmit et al. reported the synthesis of angularly fused bicyclic [18] and tricyclic [19] hydantoins as suitable precursors to cis-fused bicyclic α-amino acids by combining a diastereoselective Bucherer–Bergs reaction of 2-alkenylcycloalkanones and a regiospecific selenium-induced closure of pyrrolidine ring (Scheme 1a).
![Scheme 1:
Synthesis of angularly fused bicyclic hydantoins. (a) Bucherer–Bergs reaction and selenium-induced closure of pyrrolidine ring; (b) Cascade reaction of amino esters with in situ-generated isocyanates obtained from N-benzyl carbazates; (c) [3+2] Cycloaddition between aziridine aldehydes and isocyanates.](/document/doi/10.1515/pac-2017-0705/asset/graphic/j_pac-2017-0705_fig_001.jpg)
Synthesis of angularly fused bicyclic hydantoins. (a) Bucherer–Bergs reaction and selenium-induced closure of pyrrolidine ring; (b) Cascade reaction of amino esters with in situ-generated isocyanates obtained from N-benzyl carbazates; (c) [3+2] Cycloaddition between aziridine aldehydes and isocyanates.
Beauchemin et al. described a cascade synthesis of aminohydantoins using in situ-generated N-substituted isocyanates that can be obtained from N-benzyl carbazates or hydrazones. These reactive amphoteric intermediates can be accessed via an equilibrium that allows controlled reactivity in the presence of bifunctional partners such as α-amino esters. Thus, by employing this method the authors were able to rapid assembly complex aminohydantoins, including a fused bicyclic derivative [20] (Scheme 1b).
Yudin et al. [21] have shown that 1,3-amphoteric aziridine aldehydes participate in a highly efficient annulation with readily accessible isocyanates. The [3+2] chemistry delivered an aziridine-fused reduced hydantoin equipped with three contiguous stereocenters. Reduced hydantoins were first described by Ponsold et al. [22] as steroid derivatives. Mild oxidation with TPAP/NMO lead to the hydantoin derivative (Scheme 1c).
The reduced hydantoins have been prepared from 2-formyl aziridines, which dimerize in pH>5 in a variety of solvents [23]. Crystallographic studies have shown that this dimerization is stereoselective, since two S enantiomers react faster than a racemic mixture due to the stabilization by intermolecular hydrogen bonding. Furthermore, only the five-membered ring is formed and none of the six-membered one is observed (Scheme 2).
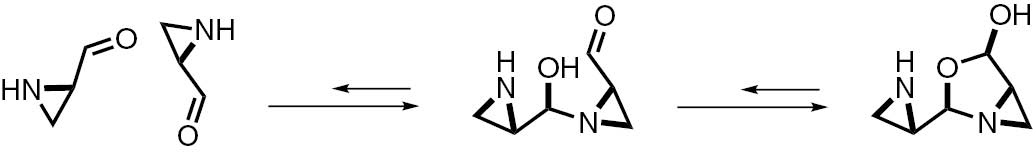
Equilibrium between the monomeric and dimeric forms of aziridine aldehydes.
The synthesis of aziridine aldehydes was first described by Hili and Yudin [24] via a three-step racemic route starting from α-epoxyesters (Scheme 3a). The same authors reported that enantiomeric enriched aziridine aldehyde dimers can be obtained from natural α-amino acids in five steps and 49% overall yield. The main drawbacks associated with that methodology is the use of hazardous starting materials such as alkyl chloroformate, n-BuLi and DIBAL, and the requirement of purifying all intermediates (Scheme 3b). In this work, we describe an alternative synthetic route to α-aziridine aldehydes starting from α,β-unsaturated aldehydes through an asymmetric organocatalytic aziridination followed by Boc deprotection, under mild conditions in two steps (Scheme 3c).

Synthetic routes to aziridine aldehyde dimers. (a) Racemic synthesis; (b) asymmetric synthesis from α-aminoacids; (c) this work.
Materials and methods
All commercially available reagents were purchased from Sigma-Aldrich. The synthesized products were purified through chromatographic column using silica gel 60, 230–400 mesh. TLC were performed in silica gel 60 F254 supported in aluminum sheets. 1H and 13C NMR spectra were recorded on a Bruker DRX 400 MHz spectrometer. Chemical shifts (δ) were presented in ppm units and the coupling constants (J) in Hertz (Hz). Signals multiplicity were expressed by the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). UltraPerformance Convergence Chromatography™ was carried using a ACQUITY UPC2™ system (Waters Corp., Milford, MA, USA), equipped with stationary phases Trefoil™ CEL1 (2.5 μm, 3 mm×150 mm), Trefoil™ CEL2 (2.5 μm, 3 mm×150 mm) and Trefoil™ AMY1 (2.5 μm, 3 mm×150 mm). HPLC analysis were performed using a Shimadzu equipment, equipped with UV-vis detector, and Daicel Chiralpak OD-H column. The exact mass measurement was carried out using a micrOTOF Q IITOF Mass Spectrometer (Bruker Daltonics, Billerica, MA, USA) equipped with an ESI ion source (positive ionization mode).
Synthesis of 2-formyl aziridines 4
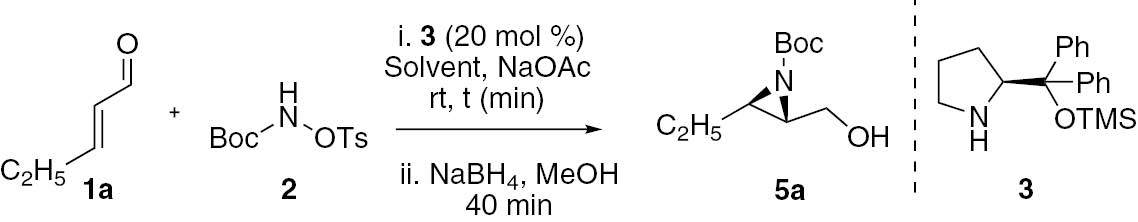
To a vial equipped with a magnetic stir bar were added the organocatalyst 3 (20 mol%, 26.0 mg), the corresponding α,β-unsaturated aldehydes (0.48 mmol, 1.2 equiv.) dissolved in AcOEt (2.0 mL), NaOAc (1.2 mmol, 3.0 equiv., 98 mg) and TsONHBoc (0.40 mmol, 1.0 equiv., 114 mg). The reaction was stirred for 40 min at room temperature or at −20°C, depending on the substrate. After this period, the reaction was extracted with EtOAc (3×15 mL) and the combined organic layers were dried with anhydrous Na2SO4 and concentrated under reduced pressure. The crude residue was purified by column chromatography using silica flash and a mixture of hexane: ethyl acetate as eluent.
The enantiomeric excess of compounds 4a–b were obtained after reduction and benzoylation, while the enantiomeric excess of compounds 4c–e were determined by reduction to the corresponding alcohols. Thus, the crude 2-formyl aziridine 4 was dissolved in methanol (2.0 mL) and NaBH4 (0.8 mmol, 2.0 equiv., 30 mg) was added to the solution. The reaction was allowed to react over 40 min, at 0°C. After this period, the reaction mixture was extracted with EtOAc (2×15 mL) and the product 5 was isolated by flash chromatography on silica gel, using a mixture of hexane: ethyl acetate (20–30%) as solvent. Aliphatic alcohols 5a and 5b (0.25 mmol) were further dissolved in dichloromethane (18 mL) and submitted to react with benzoyl chloride (1.3 equiv.), DMAP (0.5 equiv.) and Et3N (4.75 equiv.) over 2 h, at room temperature. The reaction mixture was extracted with dichloromethane (3×15 mL) and washed with saturated solution of NH4Cl (2×15 mL). The combined organic layers were dried using Na2SO4, filtered and concentrated. The crude residue was purified by flash chromatography using a solution of hexane: ethyl acetate (5%) affording benzoate 6.
(2S,3R)-tert-butyl 2-formyl-3-ethylaziridine-1-carboxylate (4a)
Purification by flash chromatography using a solution of hexane: ethyl acetate (97:3). Yield: 66% (52.6 mg, 0.26 mmol). 1H NMR (400 MHz, CDCl3) δ: 9.09 (d, J=5.2 Hz, 1H), 2.92 (dd, J=5.2, 2.7 Hz, 1H), 2.79 (td, J=6.0, 2.6 Hz, 1H), 1.73–1.64 (m, 1H), 1.59–1.50 (m, 1H), 1.46 (s, 9H), 1.05 (t, J=7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 195.86, 158.91, 82.56, 46.89, 44.94, 28.00, 24.32, 10.89.
(2S,3R)-tert-butyl 2-((benzoyloxy)methyl)-3-ethylaziridine-1-carboxylate (6a)
1H NMR (400 MHz, CDCl3) δ: 8.06–8.04 (m, 1H), 8.04–8.03 (m, 1H), 7.56 (tt, J=7.4, 1.3 Hz, 1H), 7.44 (tt, J=7.8, 1.3 Hz, 2H), 4.43 (d, J=4.9 Hz, 2H), 2.62 (td, J=4.9, 3.3 Hz, 1H), 2.51–2.47 (m, 1H), 1.78–1.68 (m, 1H), 1.42 (s, 9H), 1.37 (dd, J=14.1, 7.2 Hz, 1H), 1.07 (t, J=7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 166.29, 160.44, 133.32, 129.90, 128.55, 81.43, 63.76, 42.88, 40.66, 28.12, 27.99, 24.21, 11.28. The enantiomeric excess was determined by HPLC analysis (OD-H- column, n-hexane/iPrOH=99:1, λ=230 nm, 1.0 mL/min) tR (major enantiomer)=7.6 min tR (minor enantiomer)=8.6 min. ee=98%.
(2S,3R)-tert-butyl 2-formyl-3-penthylaziridine-1-carboxylate (4b)
Purification by flash chromatography using a solution of hexane: ethyl acetate (97:3). Yield: 67% (64.6 mg, 0.27 mmol). 1H NMR (400 MHz, CDCl3) δ: 9.06 (d, J=5.2 Hz, 1H), 2.88 (dd, J=5.2, 2.7 Hz, 1H), 2.80–2.76 (m, 1H), 1.63–1.45 (m, 4H), 1.44 (s, 9H), 1.36–1.24 (m, 4H), 0.87 (t, J=7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 195.71, 158.76, 82.40, 47.08, 43.64, 31.22, 30.87, 27.86, 26.41, 22.46, 13.93.
(2S,3R)-tert-butyl 2-((benzoyloxy)methyl)-3-penthylaziridine-1-carboxylate (6b)
1H NMR (400 MHz, CDCl3) δ: 8.05 (dd, J=8.4, 1.3 Hz, 2H), 7.56 (tt, J=7.4, 1.4 Hz, 1H), 7.48–7.40 (tt, J 8.0, 1.5 Hz, 2H), 4.45 (dd, J=11.7, 4.9 Hz, 1H), 4.40 (dd, J=11.6, 4.3 Hz, 1H), 2.61 (td, J=5.0, 3.3 Hz, 1H), 2.50 (ddd, J=7.0, 5.7, 3.2 Hz, 1H), 1.77–1.70 (m, 1H), 1.57–1.43 (m, 2H), 1.42 (s, 9H), 1.37–1.23 (m, 5H), 0.87 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 166.31, 160.45, 133.33, 129.90, 128.55, 81.45, 63.87, 41.77, 40.93, 31.56, 30.87, 28.13, 26.82, 22.70, 14.10. The enantiomeric excess was determined by UPC2 (Trefoil column, CEL2 (90% MeOH, gradient, 1.2 mL/min, 2000 psi, 35°C), tR (major enantiomer)=5.3 min. ee=99%.
(2S,3S)-tert-butyl 2-((benzyloxy)methyl)-3-formylaziridine-1-carboxylate (4c)
Purification by flash chromatography using a solution of hexane: ethyl acetate (8:2). Yield: 74% (87.4 mg, 0.30 mmol). 1H NMR (400 MHz, CDCl3) δ: 9.16 (d, J=5.0 Hz, 1H), 7.36–7.26 (m, 5H), 4.55 (d, J=3.7 Hz, 2H), 3.74 (dd, J=11.2, 4.1 Hz, 1H), 3.70 (dd, J=11.3, 3.9 Hz, 1H) 3.21 (dd, J=4.9, 2.6 Hz, 1H), 3.02 (dd, J=6.5, 3.7 Hz, 1H), 1.44 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 195.96, 158.29, 137.46, 128.57, 128.00, 127.78, 82.62, 73.14, 66.71, 43.85, 41.82, 27.94.
(2S,3S)-tert-butyl 2-((benzyloxy)methyl)-3-(hydroxymethyl)aziridine-1-carboxylate (5c)
Purification by flash chromatography using a solution of hexane: ethyl acetate (8:2). 1H NMR (400 MHz, CDCl3) δ: 7.30–7.17 (m, 5H), 4.49 (s, 2H), 3.91 (dd, J=12.5, 2.9 Hz, 1H), 3.57 (ddd, J=22.7, 11.0, 4.4 Hz, 2H), 3.45 (dd, J=12.5, 6.3 Hz, 1H), 2.63 (dt, J=6.2, 3.1 Hz, 1H), 2.56 (dd, J=7.7, 4.3 Hz, 1H), 1.38 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 160.97, 137.88, 128.49, 127.83, 127.72, 81.86, 72.95, 68.24, 61.87, 41.46, 39.24, 28.03. The enantiomeric excess was determined by UPC2 (Trefoil column, AMY1, 40% MeCN, gradient, 2.0 mL/min, 2000 psi, 35°C)=3.3 min, tr (minor isomer)=3.5 min. ee=93%.
(2S,3R)-tert-butyl 2-formyl-3-phenethylaziridine-1-carboxylate (4d)
Purification by flash chromatography using a solution of hexane: ethyl acetate (97:3). Yield: 55% (151 mg, 0.22 mmol). 1H NMR (400 MHz, acetone-d6) δ: 9.12 (d, J=4.8 Hz, 1H), 7.29–7.25 (m, 4H), 7.22–7.18 (m, 1H), 2.96–2.93 (m, 1H), 2.87–2.79 (m, 2H), 2.79 (s, 1H), 2.05 (dt, J=4.4, 2.2 Hz, 1H), 1.96–1.81 (m, 2H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 195.50, 158.82, 140.51, 128.73, 128.58, 126.46, 82.69, 47.12, 43.28, 33.11, 32.95, 28.02.
(2S,3R)-tert-butyl 2-(hydroxymethyl)-3-phenethylaziridine-1-carboxylate (5d)
Purification by flash chromatography using a solution of hexane: ethyl acetate (9:1). 1H NMR (400 MHz, CDCl3) δ: 7.31–7.27 (m, 2H), 7.22–7.19 (m, 3H), 3.90 (dd, J=12.5, 2.6 Hz, 1H), 3.38 (dd, J=12.5, 6.8 Hz, 1H), 2.87 (ddd, J=14.0, 8.6, 5.6, 1H), 2.77–2.70 (m, 1H), 2.37 (dtd, J=3.2, 6.1, 9.3, 2H), 1.90 (ddt, J=11.0, 8.5, 6.1, 1H), 1.76–1.67 (m, 1H), 1.49 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 161.61, 141.18, 128.59, 128.56, 126.24, 81.89, 62.65, 44.65, 40.46, 33.31, 33.08, 28.11. The enantiomeric excess was determined by UPC2 (Trefoil column, CEL1, 40% MeCN, gradient, 1.0 mL/min, 2000 psi, 35°C)=4.2 min. ee=99%.
(2S,3R)-tert-butyl 2-formyl-3-(2-nitrophenyl)aziridine-1-carboxylate (4e)
Purification by flash chromatography using a solution of hexane: ethyl acetate (9:1). Yield: 72% (84.2 mg, 0.29 mmol). 1H NMR (400 MHz, CDCl3) δ: 9.54 (d, J=3.4 Hz, 1H), 8.18 (dd, J=8.2, 1.0 Hz, 1H), 7.73 (dd, J=7.8, 1.5 Hz, 1H), 7.68 (td, J=7.6, 1.0 Hz, 1H), 7.53 (ddd, J=8.6, 7.1, 1.6, 1H), 4.37 (d, J=2.5 Hz, 1H), 3.18 (dd, J=3.4, 2.6 Hz, 1H), 1.51 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 193.33, 158.35, 134.47, 131.48, 129.61, 129.37, 125.18, 83.43, 48.78, 44.44, 28.03.
(2S,3S)-tert-butyl 2-(hydroxymethyl)-3-(2-nitrophenyl)aziridine-1-carboxylate (5e)
Purification by flash chromatography using a solution of hexane: ethyl acetate (8:2). 1H NMR (400 MHz, CDCl3) δ: 8.10 (dd, J=8.2, 1.2 Hz, 1H), 7.71 (dd, J=7.8, 1.5 Hz, 1H), 7.63 (td, J=7.5, 1.2 Hz, 1H), 7.45 (ddd, J= 8.6, 7.2, 1.6 Hz, 1H), 4.23 (ddd, J=12.6, 8.8, 2.6, 1H), 3.94 (d, J=3.2 Hz, 1H), 3.82 (ddd, J=12.7, 6.2, 3.4, 1H), 2.84 (dd, J=8.8, 4.2 Hz, 1H), 2.62 (dt, J=6.2, 3.0 Hz, 1H), 1.49 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 161.14, 148.00, 134.37, 133.30, 129.39, 128.76, 124.89, 82.64, 62.16, 47.58, 40.69, 28.11. The enantiomeric excess was determined by UPC2 (Trefoil column, AMY1, 40% iPrOH, gradient, 2.0 mL/min, 2000 psi, 35°C) tR (major isomer)=4.9 min. ee=99%.
Synthesis of the amino aldehyde dimers 8
A solution of TBAF (0.25 mmol, 1.0 M in THF, 0.25 mL) was added to a solution of the 2-formyl aziridines 4a or 4b (0.25 mmol, 1.0 equiv.) in 2-MeTHF (1.25 mL). The reaction was heated to reflux and stirred over 2 h. After cooling, the crude mixture was extracted with ethyl acetate (3×15 mL) and washed with saturated aqueous NaHCO3. The products were isolated by flash chromatography, using a solution of hexane: ethyl acetate 40–50%, affording the desired products as colorless oils.
(2R,4R,5S,6R)-6-Pentyl-2-((2S,3R)-3-pentylaziridin-2-yl)-3-oxa-1-azabicyclo[3.1.0]hexan-4-ol (8a)
Yield: 67% (70.6 mg, 0.25 mmol). 1H NMR (400 MHz, CDCl3) δ: 5.24 (s, 1H), 4.92 (s, 1H), 2.41 (d, J=1.6 Hz, 1H), 2.13 (dd, J=3.4, 0.9 Hz, 1H), 1.98–1.94 (m, 1H), 1.49–1.22 (m, 24H), 0.88 (t, J=7,0 Hz, 6H), 13C NMR (100 MHz, CDCl3) δ: 96.47, 94.51, 50.49, 39.32, 38.70, 34.27, 33.68, 31.69, 31.58, 31.21, 27.27, 26.96, 22.72, 22.69, 14.15, 14.09.
(2R,4R,5S,6R)-6-Ethyl-2-((2S,3R)-3-ethylaziridin-2-yl)-3-oxa-1-azabicyclo[3.1.0]hexan-4-ol (8b)
Yield: 65% (48.6 mg, 0.24 mmol). 1H NMR (400 MHz, CDCl3) δ: 5.24 (s, 1H), 4.92 (s, 1H), 2.42 (d, J=2.8 Hz, 1H), 2.15 (dd, J=3.5, 1.1 Hz, 1H), 1.96 (td, J=6.1, 3.6 Hz, 1H), 1.56–1.40 (m, 5H), 0.98 (dt, J=8.6, 7.4 Hz, 6H), 13C NMR (100 MHz, CDCl3) δ: 96.46, 94.58, 50.31, 40.66, 38.35, 35.57, 26.54, 24.42, 11.50, 11.32.
Sequential synthesis of the reduced hydantoins 9
The first step of the sequential synthesis of reduced hydantoins was performed as mentioned above (synthesis of the amino aldehyde dimers). The reduced hydantoin synthesis was carried out following the procedure previously reported by Yudin et al. [21], with slight modifications. In a vial equipped with a magnetic stir bar, isocyanate (1.5 equiv.) was added to the crude solution of amino aldehyde dimers in HFIP:H2O (8:2). The reaction was stirred over 1 hour, at room temperature. After total consumption of the starting material, the reaction was extracted with ethyl acetate (3×15 mL) and the organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by flash chromatography using a solution of hexane: ethyl acetate 40–60%, affording the desired compounds in good to moderate yields, after two steps.
(4S,5S,6R)-4-Hydroxy-6-pentyl-3-phenyl-1,3-diazabicyclo[3.1.0]hexan-2-one (9a)
The product was isolated in 59% yield (49 mg, 0.19 mmol) as colorless solid. Mp: 118°C. 1H NMR (400 MHz, CDCl3) δ: 7.54 (t, J=1.6 Hz, 1H), 7.52 (t, J=1.7 Hz, 1H ), 7.33 (tt, J=7.4, 1.9 Hz, 2H), 7.19 (tt, J=1.2, 7.4 Hz, 1H), 5.66 (s, 1H), 4.60 (s, 1H), 2.80 (d, J=3.6 Hz, 1H), 2.25–2.21 (m, 1H), 1.71–1.62 (m, 1H), 1.58–1.43 (m, 3H), 1.35–1.30 (m, 4H), 0.90 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 165.31, 136.66, 129.19, 126.16, 122.75, 83.12, 49.88, 46.18, 31.48, 31.21, 26.28, 22.62, 14.09. HRMS (ESI): m/z: calcd for C15H20N2O2 [(M+H)]+: 261.1598, found: 261.1594. The enantiomeric excess was determined by UPC2 (Trefoil column, CEL2, 70% EtOH, gradient, 0.4 mL/min, 2000 psi, 40°C) tR (major isomer)=6.6 min. ee=99%.
(4S,5S,6R)-6-Ethyl-4-hydroxy-3-phenyl-1,3-diazabicyclo[3.1.0]hexan-2-one (9b)
The product was isolated in 43% yield (35 mg, 0.16 mmol) as colorless solid. Mp: 108°C. 1H NMR (400 MHz, CDCl3) δ 7.55–7.53 (m, 1H), 7.52 (dd, J=2.0, 1.0 Hz, 1H), 7.36–7.31 (m, 2H), 7.19 (tt, J=7.4, 1.1 Hz, 1H), 5.66 (s, 1H), 4.71 (s, 1H), 2.82 (d, J=3.6 Hz, 1H), 2.23 (td, J=6.0, 3.6 Hz, 1H), 1.73–1.59 (m, 2H), 1.06 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 165.43, 136.67, 129.26, 126.23, 122.72, 83.15, 50.81, 45.73, 24.46, 10.61. HRMS (ESI): m/z: calcd for C12H14N2O2 [(M+H)]+: 219.1128, found: 219.1117.
(4S,5S,6R)-6-((Benzyloxy)methyl)-4-hydroxy-3-phenyl-1,3-diazabicyclo[3.1.0]hexan-2-one (9c)
The product was isolated in 40% yield (32 mg, 0.10 mmol) as yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.50 (dd, J=8.6, 1.1 Hz, 2H), 7.35–7.28 (m, 7H), 7.19 (tt, J=7.4, 1.3, Hz, 1H), 5.62 (s, 1H), 4.84 (s, 1H), 4.55 (2×d, J=26.5, 2.8 Hz, 2H), 3.68 (dd, J=11.2, 4.7 Hz, 1H), 3.56 (dd, J=11.2, 4.6 Hz, 1H), 2.95 (d, J=3.5 Hz, 1H), 2.45 (dd, J=8.2, 4.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 165.20, 137.62, 136.45, 129.29, 128.66, 128.12, 128.08, 126.38, 122.84, 83.06, 73.59, 68.42, 47.18, 43.55. HRMS (ESI): m/z: calcd for C18H18N2O3 [(M+H)]+: 311.1390, found: 311.1389.
(4S,5S,6R)-4-Hydroxy-6-phenethyl-3-phenyl-1,3-diazabicyclo[3.1.0]hexan-2-one (9d)
The product was isolated in 44% yield (37 mg, 0.12 mmol) as yellow solid. Mp: 153°C. 1H NMR (400 MHz, CDCl3) δ: 7.51 (d, J=8.8 Hz, 2H), 7.33 (dd, J=11.2, 4.6 Hz, 2H), 7.29 (d, J=7.3 Hz, 2H), 7.24–7.17 (m, 4H), 5.61 (d, J=10.4 Hz, 1H), 4.01 (d, J=10.6 Hz, 1H), 2.91–2.76 (m, 2H), 2.76 (d, J=3.5 Hz, 1H), 2.22 (td, J=6.3, 3.5, 1H), 2.00–1.86 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 165.38, 140.54, 136.64, 129.19, 128.73, 128.62, 126.44, 126.15, 122.61, 83.06, 48.94, 46.27, 32.95, 32.82. HRMS (ESI): m/z: calcd for C18H18N2O2 [(M+H)]+: 295.1441, found: 295.1440. The enantiomeric excess was determined by UPC2 (Trefoil column, CEL2, 40% MeOH, gradient, 0.8 mL/min, 2000 psi, 40°C) tR (major isomer)=9.1 min. ee=99%.
(4S,5S,6R)-3-Allyl-4-hydroxy-6-pentyl-1,3-diazabicyclo[3.1.0]hexan-2-one (9e)
The product was isolated in 51% yield (47 mg, 0.21 mmol) as yellow oil. 1H NMR (400 MHz, CDCl3) δ: 5.76–5.66 (m, 1H), 5.24 (d, J=1.4 Hz, 1H), 5.20–5.17 (m, 2H), 4.58 (s, 1H), 4.00 (ddt, J=15.2, 4.9, 1.3 Hz, 1H), 3.74 (ddt, J=15.3, 7.6, 1.2, 1H), 2.69 (d, J=3.6 Hz, 1H), 2.02 (ddd, J=6.4, 5.4, 3.6 Hz, 1H), 1.61–1.39 (m, 5H), 1.33–1.36 (m, 3H), 0.89 (t, J=7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 166.89, 132.19, 118.94, 80.89, 49.96, 46.52, 43.05, 31.49, 31.33, 26.23, 22.61, 14.09. HRMS (ESI): m/z: calcd for C12H20N2O2 [(M+H)]+: 295.1598, found: 295.1594. The enantiomeric excess was determined by UPC2 (Trefoil column, CEL2, 40% iPrOH, gradient, 0.8 mL/min, 2000 psi, 40°C) tR (major isomer)=5.3 min, tR (minor isomer)=5.8 min. ee=98%.
(4S,5S,6R)-3- Benzyl-6-ethyl-4-hydroxy-1,3-diazabicyclo[3.1.0]hexan-2-one (9f)
The product was isolated in 43% yield (34 mg, 0.15 mmol) as yellow oil (mixture of diastereomers (1:0.5). 1H NMR (400 MHz, CDCl3) δ: 7.28–7.17 (m, 5H), 4.93 (s, 1H), 4.59 e 4.61 (2 d, J=14.9 e J=14.8 Hz, 1H), 4.12 (d, J=14.8 Hz, 1H), 2.60 (d, J=3.6 Hz, 1H), 1.85 (td, J=6.0, 3.6 Hz, 1H), 1.55–1.45 (m, 3H), 0.92 (t, J=7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 166.93 e 166.51, 136.35 e 135.88, 128.98, 128.89, 128.74, 128.61, 128.51, 128.01, 127.95, 80.36, 79.35, 50.60, 48.43, 46.28, 44.39, 44.04, 43.38, 24.70, 24.44, 10.67, 10.54. The enantiomeric excess was determined by UPC2 (Trefoil column, CEL2, 60% ACN, gradient, 1.0 mL/min, 2000 psi, 40°C) tR (major isomer)=5.7 min, tR (minor isomer)=7.1 min. ee=90%. HRMS (ESI): m/z: calcd for C13H16N2O2 [(M+Na)]+: 255.1104, found: 255.1106.
(4S,5S,6R)-4-Hydroxy-3-(4-methoxyphenyl)-6-pentyl-1,3-diazabicyclo[3.1.0]hexan-2-one (9g)
The product was isolated in 49% yield (32 mg, 0.11 mmol) as yellow solid. Mp: 101°C. 1H NMR (400 MHz, CDCl3) δ: 7.38 (dt, J=9.1, 3.4 Hz, 2H), 6.87 (dt, J=9.1, 3.4 Hz, 2H), 5.55 (s, 1H), 4.39 (sl, 1H), 3.78 (s, 3H), 2.78 (d, J=3.6 Hz, 1H), 2.23 (td, J=5.6, 3.8 Hz, 1H), 1.72–1.28 (m, 8H), 0.90 (t, J=6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 165.71, 158.14, 129.22, 125.27, 114.50, 83.56, 55.60, 49.81, 46.13, 31.51, 31.28, 26.30, 22.64, 14.10. The enantiomeric excess was determined by UPC2 Trefoil column, CEL2, 50% iPrOH, gradient, 0.6 mL/min, 2000 psi, 40°C). tR (major isomer)=10.1 min. ee=99%. HRMS (ESI): m/z: calcd for C16H22N2O3 [(M+Na)]+: 291.1703, found: 291.1703.
Results and discussion
Organocatalyzed synthesis of 2-formyl aziridines
Several methods have been described for the asymmetric synthesis of aziridines [25], including the use of organometallic catalysts [26]. Vesely et al. [27] were pioneers to develop an organocatalyzed synthesis of 2-formyl aziridines from α,β-unsaturated aldehydes and protected amines, using dichloromethane as solvent. Thus, based on this work, we started our studies evaluating alternative solvents [28], [29] for this organocatalyzed protocol. Initially, we have examined MeCN, which furnished the desired product in 70% yield (Table 1, entry 1). Polar protic solvents as EtOH delivered the desired product in lower yield (Table 2, entry 2). The bio-based 2-MeTHF [30] also provided a lower yield, in spite of the excellent enantio- and diastereoselectivity (Table 1, entry 3). At 40°C, we observed a higher yield, but a decrease in the diastereoisomeric ratio (dr) (Table 1, entry 4).
Solvent screening in the aziridination reaction.
| Entry | Solvent | t (min) | Yield (%)a | ee (%)b | drc |
|---|---|---|---|---|---|
| 1 | MeCN | 20 | 70 | 97 | 98:2 |
| 2 | EtOH | 20 | 52 | 96 | 90:10 |
| 3 | 2-MeTHF | 120 | 36 | 99 | 96:4 |
| 4 d | 2-MeTHF | 20 | 56 | 98 | 88:12 |
| 5 | Toluene | 20 | 52 | 99 | 88:12 |
| 6 | EtOAc | 40 | 85 | 98 | 91:9 |
| 7 | TFE | 120 | – | – | – |
| 8 | GVL | 20 | nd | – | – |
-
Reactions were carried out using 1a (1.2 equiv), 2 (0.4 mmol), NaOAc (3.0 equiv), catalyst 3 (20 mol%) and solvent (2.0 mL). aIsolated yield. bDetermined by chiral HPLC after reduction and esterification of the aziridine aldehyde. cDetermined by 1H NMR analysis of the crude. dReaction was carried out at 40°C.
Synthesis of 2-formyl aziridines from aliphatic α,β-unsaturated aldehydes.

| Entry | Aldehyde | Structure | Yield (%) | ee (%) | drd |
|---|---|---|---|---|---|
| 1 | 4a |
 |
85a | 97b | 92:8 |
| 2 | 4b |
 |
66a | 99b | 92:8 |
| 3 | 4c |
 |
74 | 93c | 88:12 |
| 4 | 4d |
 |
55 | 99c | 92:8 |
-
Reactions were carried out using 1a (1.2 equiv), 2 (0.4 mmol), NaOAc (3.0 equiv), catalyst 3 (20 mol%) and EtOAc (2.0 mL). aIsolated yield after reduction. bDetermined by chiral HPLC after reduction and esterification of the aziridine aldehyde. cDetermined by chiral HPLC after reduction of the aziridine aldehyde. dDetermined by 1H NMR of the crude product.
Other solvents as toluene, ethyl acetate, trifluoroethanol (TFE) and γ-valerolactone (GVL) were also evaluated (Table 1, entries 5–8). In the case of toluene, low yield and dr were observed. For our delight, EtOAc furnished an excellent yield, ee and dr. Using TFE, however, no product was observed, and the starting materials were recovered after 2 h. When GVL was used, the desired product was observed, but was not isolable due to the high boiling point of the solvent. Therefore, EtOAc has been selected to continue our study.
In order to evaluate the enantiomeric excess (ee) using chiral HPLC, the aziridine aldehyde 4 was reduced with NaBH4 to the corresponding alcohol 5 followed by esterification with benzoyl chloride, giving the benzoate derivative 6.
With the optimal conditions in hand (Table 1, entry 6), the scope and limitations of this reaction were explored with different α,β-unsaturated aldehydes. Firstly, trans-2-octenal (1b) was employed, furnishing 4b in 66% yield over two steps with excellent ee and dr (Table 2, entry 2). Aldehydes 1c and 1d were prepared from cis-2-butenediol and 3-phenyl-1-propanol, respectively [31], [32], and then submitted to the aziridination reaction affording the corresponding aziridine aldehydes 4c and 4d in good yields and high ee and dr (Table 2, entries 3 and 4).
In order to further explore this reaction, we then evaluated aromatic α,β-unsaturated aldehydes. trans-Cinnamaldehyde (1e) was the first to be tested, but only the α-aminoaldehyde 7e, which is formed via deprotonation and opening of the aziridine ring, was observed [33]. Aiming to obtain the aforementioned desired aziridine aldehyde, we have carried out the reaction at −20°C and also decreasing the base amount (1.5 equiv.), but once more, only the 7e was observed as product (Table 3, entries 1 and 2). The same result was observed when trans-p-methoxycinnamaldehyde (1f) and trans-p-fluorocinnamaldehyde (1g) were employed, affording the corresponding aminoaldehydes 7f and 7g (Table 3, entries 6–8).
Synthesis of 2-formyl aziridines from aromatic α,β-unsaturated aldehydes.

| Entry | R | Base (equiv.) | Temp. (°C) | Product (yield %) | ee (%) | dr |
|---|---|---|---|---|---|---|
| 1 | H | 3.0 | −20 | 7a | nd | nd |
| 2a | H | 1.5 | −20 | 7a | nd | nd |
| 3 | o-MeO | 3.0 | 25 | 7b | nd | nd |
| 4 | o-MeO | 3.0 | −20 | 7b | nd | nd |
| 5a | o-MeO | 1.5 | −20 | 7b | nd | nd |
| 6 | p-F | 3.0 | 25 | 7c | nd | nd |
| 7 | p-F | 3.0 | −20 | 7c | nd | nd |
| 8a | p-F | 1.5 | −20 | 7c | nd | nd |
| 9b | o-NO2 | 1.5 | −20 | 4e (57%) | 99% | 72:22 |
| 10b | o-NO2 | 3.0 | −20 | 4e (72%) | 99% | 82:18 |
-
Reactions were carried out using 1a (1.2 equiv), 2 (0.4 mmol), NaOAc (3.0 equiv), catalyst 3 (20 mol%) and EtOAc (2.0 mL). aReaction time=2 h. bIsolated yield, ee determined by chiral HPLC after reduction of the aziridine aldehyde.
Nevertheless, the reaction with trans-o-nitrocinnamaldehyde (1h) at −20°C with 1.5 equiv. of base furnished the desired product 4h in 57% yield, 99% ee and 78:22 dr (Table 3, entry 9), while using 3.0 equiv. of the base, a better yield and selectivity were attained (Table 3, entry 10). The observed moderate diastereoseletivity can be a consequence of the corresponding dr of the starting material 1h, which was 88:12. These results suggested that only when strong electron-withdrawing groups are present in the aromatic ring of the α,β-unsaturated aldehydes are able to deliver the aziridine aldehyde products in good yields and stereoselectivities.
Having the aziridine aldehydes in hands, we turned our attention to the one-pot Boc deprotection followed by dimerization to generate the amphoteric aminoaldehydes. We initiated this study by evaluating conventional conditions, as shown in Table 4 (entries 1–3). However, in all cases we observed low yields and the formation of several byproducts – which probably can be related to the excess of the aldehyde, which may react via an aza-Michael aldol reaction [34]. Fusco et al. [35] reported the asymmetric synthesis of aziridines via Boc deprotection using TBAF under reflux of THF. Thus, using this condition, we were able to deprotect the Boc group of 4a and isolated the corresponding dimer 8a in 68% yield (Table 4, entry 4). As an alternative, this reaction also occurred using 2-MeTHF as solvent with full conversion, however in 42% isolated yield.
Optimization of the reaction conditions of the tandem Boc deprotection – dimerization.

| Entry | Conditions | Yield (%)c |
|---|---|---|
| 1a | DCM, (C2H5)3SiH, DIPEA, 0°C, 40 min | – |
| 2a | 4M HCl, Dioxane, NaHCO3, rt, 2 h | – |
| 3a | Anisole, TFA, H2SO4, rt, 40 min | – |
| 4b | TBAF, THF, 60°C, 2 h | 68 |
| 5b | TBAF, 2-MeTHF, 60°C, 2 h | 42 |
-
aReaction carried out one-pot. bReaction carried out with the isolated aziridine aldehyde 4a (0.25mmol), TBAF (1.0 equiv.), solvent (1.25 mL). cIsolated yield after purification by column chromatography.
The use of fluoride in Boc deprotection has been reported for other N-heterocycles [36]. According to the literature, this anion can act either by a nucleophilic attack leading to Boc-F or as a base via hydrogen abstraction, furnishing the desired product, isobutene, CO2 and HF [34]. Due to the low acidity of the hydrogen of compound 4a, we believe that the fluoride could act as a nucleophile.
Synthesis of reduced hydantoins
Since the aziridine aldehyde do not present a high stability and its alkyl-dimer do not show high tolerance to column chromatography purification, which could explain the moderate yields obtained in this deprotection, we decided to submit this crude product to the [3+2] cycloaddition reaction with isocyanates without any further purification [21]. Fortunately, the formyl aziridines readily reacted with isocyanates in hexafluoroisopropanol (HFIP) as solvent [34]. Thus, using the conditions described by Yudin and co-workers, we carried out the reaction of the crude aziridine aldehyde dimers 8 with different isocyanates using a 8:2 mixture of HFIP:H2O [21]. The reaction of phenylisocyanate with the crude aliphatic dimers 8a–d afforded the corresponding hydantoins 9a–d in good yields over two steps (Scheme 4). Unfortunately, in our hands the trans-o-nitrocinnamaldehyde dimer derivative did not produce the desired reduced hydantoins as previously reported [21].

Scope of the synthesis of reduced hydantoins.a
aReactions were carried out using 4 (0.25–0.35 mmol, 0.2M), TBAF (1.0 equiv), 2-MeTHF then, after extraction with EtOAc and concentration, HFIP:H2O (8:2) and isocyanate (1.5 equiv.) were added. Isolated yield over two steps after column chromatography purification. bThe separation of compounds 9a and 9c could not be accomplished using the tested chiral columns.
To further explore the scope of this protocol, aromatic and aliphatic isocyanates were employed. Functionalized aliphatic isocyanates containing the benzyl or allyl group afforded the corresponding hydantoins 9e and 9f in good overall yields. Moreover, electron-donating groups in the aromatic ring of the isocyanate gave the corresponding hydantoin 9g in 49% yield (Scheme 4).
In conclusion, in this work we have developed a two-step organocatalyzed-synthetic route to amphoteric aziridine aldehydes. Then, these compounds were employed in the synthesis of new reduced hydantoins in good overall yields, scaffolds with numerous potential applications as buildings blocks in organic synthesis. In both processes, bio-based and/or greener solvents were used. Although there is still room for improvement in this protocol, especially in what regards the use of safer solvents in the purification steps (which already are fewer when compared to the previously reported approaches), we believe that this work is an important step towards the development of more sustainable routes for the synthesis of important heterocyclic compounds.
Article note
A collection of invited papers based on presentations at the 6th international IUPAC Conference on Green Chemistry (ICGC-6), Venice (Italy), 4–8 September 2016.
Acknowledgments
The authors are grateful to FAPESP (grants 2012/07284-1, 2013/07600-3, 2014/50249-8 and 2015/17141-1), CNPq and CAPES (Brazil) and GSK for financial support and fellowships.
References
[1] M. Meusel, M. Gütschow. Org. Prep. Proc. Int.36, 391 (2004).10.1080/00304940409356627Search in Google Scholar
[2] E. Ware. Chem. Rev.46, 403 (1950).10.1021/cr60145a001Search in Google Scholar PubMed
[3] M. Mudit, M. Khanfar, A. Muralidharan, S. Thomas, G. V. Shah, R. W. M. Soest, K. A. Sayed. Bioorg. Med. Chem.17, 1731 (2009).10.1016/j.bmc.2008.12.053Search in Google Scholar PubMed
[4] N. Cachet, G. Genta-Jouve, E. L. Regalado, R. Mokrini, P. Amade, G. Culioli, O. P. Thomas. J. Nat. Prod.72, 1612 (2009).10.1021/np900437ySearch in Google Scholar PubMed
[5] F. Nique, S. Hebbe, C. Peixoto, D. Annoot, J.-M. LeFrancois, E. Duval, L. Michoux, N. Triballeau, J.-M. LeMoullec, P. Mollat, M. Thauvin, T. Prange, D. Minet, P. Clement-Lacroix, C. Robin-Jagerschimidt, D. Fleury, D. Guedin, P. Deprez. J. Med. Chem.55, 8225 (2012).10.1021/jm300249mSearch in Google Scholar PubMed
[6] M. Dhanawat, A. G. Banerjee. Med. Chem. Res.21, 2807 (2012).Search in Google Scholar
[7] Z. Iqbal, S. Ali, J. Iqbal, Q. Abbas, I. Z. Qureshi, S. Hameed. Bioorg. Med. Chem. Lett.23, 488 (2013).10.1016/j.bmcl.2012.11.039Search in Google Scholar PubMed
[8] A. A. Sallam, M. M. Mohyeldin, A. I. Foudah, M. R. Akl, S. Nazzal, S. A. Meyer, Y.-Y. Liu, K. A. El Sayed. Org. Biomol. Chem.12, 5295, 2014.10.1039/C4OB00553HSearch in Google Scholar
[9] M. A. Khanfar, B. A. Asal, M. Mudit, K. Kaddoumi, K. A. Sayed. Bioorg. Med. Chem. 17, 6032 (2009).10.1016/j.bmc.2009.06.054Search in Google Scholar PubMed PubMed Central
[10] K. Y. Cheoul, L. G. Eun, P. J. Hui. KR2012086957-A (2012).Search in Google Scholar
[11] G. S. Hamilton, M. D. Catonsville. US 20020058685 (2002).Search in Google Scholar
[12] D. Zhang, X. Xing, G. Cuny. J. Org. Chem.71, 1750 (2006).10.1021/jo052474sSearch in Google Scholar PubMed
[13] J. L. Monteiro, B. Pieber, A. G. Correa, C. O. Kappe. Synlett27, 83 (2016).10.1055/s-0035-1560317Search in Google Scholar
[14] S. Vukelic, B. Koksch, P. H. Seeberger, K. Gilmore. Chem. Eur. J. 22, 13451 (2016).10.1002/chem.201602609Search in Google Scholar PubMed
[15] H. Prevet, M. Flipo, P. Roussel, B. Deprez, N. Willand. Tetrahedron Lett.57, 2888 (2016).10.1016/j.tetlet.2016.05.065Search in Google Scholar
[16] L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez, E. Colacino. J. Org. Chem. 79, 10132 (2014).10.1021/jo5017629Search in Google Scholar PubMed
[17] L. Belvisi, L. Colombo, L. Manzoni, D. Potenza, C. Scolastico. Synlett15, 1449 (2004).Search in Google Scholar
[18] B. M. Šmit, R. Z. Pavlović. Tetrahedron71, 1101 (2015).10.1016/j.tet.2014.12.088Search in Google Scholar
[19] B. Šmit, M. Rodić, R. Z. Pavlović. Synthesis48, 387 (2016).10.1055/s-0035-1561285Search in Google Scholar
[20] J.-F. Vincent-Rocan, C. Clavette, K. Leckett, A. M. Beauchemin. Chem. Eur. J.21, 3886 (2015).10.1002/chem.201405648Search in Google Scholar PubMed
[21] L. l. W. Cheung, Z. He, S. M. Decker, A. K. Yudin. Angew. Chem. Int. Ed.50, 1178 (2011).10.1002/anie.201106024Search in Google Scholar PubMed
[22] K. Ponsold, B. Schoenecker, P. Grosse. Chem. Berich. 99, 3485 (1966).10.1002/cber.19660991115Search in Google Scholar PubMed
[23] R. Hili, A. K. Yudin. J. Am. Chem. Soc.131, 6404 (2009).10.1021/ja9072194Search in Google Scholar PubMed
[24] R. Hili, A. K. Yudin. J. Am. Chem. Soc.128, 14772 (2006).10.1021/ja065898sSearch in Google Scholar PubMed
[25] G. Righi, S. Pietrantonio, C. Bonini. Tetrahedron57, 10039 (2001).10.1016/S0040-4020(01)01038-9Search in Google Scholar
[26] Y. Cui, C. He. J. Am. Chem. Soc. 125, 16202 (2003).10.1021/ja038668bSearch in Google Scholar PubMed
[27] J. Vesely, I. Ibrahem, G. Zhao, R. Rios, A. Córdova. Angew. Chem. Int. Ed. 46, 778 (2007).10.1002/anie.200603810Search in Google Scholar PubMed
[28] H. Arai, N. Sugaya, N. Ssaki, K. Makino, S. Lectard, Y. Hamada. Tetrahedron Lett. 50, 3329 (2009).10.1016/j.tetlet.2009.02.095Search in Google Scholar
[29] C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster, H. F. Sneddon. Green Chem. 18, 3879 (2016).10.1039/C6GC00611FSearch in Google Scholar
[30] A. G. Corrêa, M. W. Paixão, R. S. Schwab. Curr. Org. Synth. 12, 675 (2015).10.2174/157017941206150828102108Search in Google Scholar
[31] V. Jeena, R. S. Robinson. Org. Biomol. Chem. 13, 8958 (2015).10.1039/C5OB01308ASearch in Google Scholar
[32] A. Pollex, A. Millet, J. Müller, M. Hiersemann, L. Abraham. J. Org. Chem.70, 5579 (2005).10.1021/jo0505270Search in Google Scholar PubMed
[33] L. Deiana, P. Dziedzic, G. Zhao, J. Vesely, I. Ibrahem, R. Rios, J. Sun, A. Córdova. Chem. Eur. J. 17, 7904 (2011).10.1002/chem.201100042Search in Google Scholar PubMed
[34] N. Assem, R. Hili, Z. He, T. Kasahara, B. L. Inman, S. Decker, A. K. Yudin. J. Org. Chem. 77, 5613 (2012).10.1021/jo3007418Search in Google Scholar PubMed
[35] C. De Fusco, T. Fuoco, G. Croce, A. Lattanzi. Org. Lett. 14, 4078 (2012).10.1021/ol3017066Search in Google Scholar PubMed
[36] S. Routier, L. Saugé, N. Ayerbe, G. Coudert, J. Mérour. Tetrahedron Lett. 43, 589 (2002).10.1016/S0040-4039(01)02225-0Search in Google Scholar
Supplemental Material
The online version of this article (https://doi.org/10.1515/pac-2017-0705) offers supplementary material, available to authorized users.
©2018 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers
- Papers from the 6th International IUPAC Conference on Green Chemistry (ICGC-6)
- Microwave assisted synthesis of glycerol carbonate from glycerol and urea
- Silica gel mediated oxidative C–O coupling of β-dicarbonyl compounds with malonyl peroxides in solvent-free conditions
- Definition of green synthetic tools based on safer reaction media, heterogeneous catalysis, and flow technology
- Heavy metal removal from waste waters by phosphonate metal organic frameworks
- A clean and simple method for deprotection of phosphines from borane complexes
- Development and treatment procedure of arsenic-contaminated water using a new and green chitosan sorbent: kinetic, isotherm, thermodynamic and dynamic studies
- Bio-adsorbent derived from papaya peel waste and magnetic nanoparticles fabricated for lead determination
- 5-Membered cyclic ethers via phenonium ion mediated cyclization through carbonate chemistry
- Synergy in food, energy and advanced materials production from biomass
- Step economy strategy for the synthesis of amphoteric aminoaldehydes, key intermediates for reduced hydantoins
- Separation technology meets green chemistry: development of magnetically recoverable catalyst supports containing silica, ceria, and titania
- Green chemistry and sustainable development: approaches to chemical footprint analysis
- Greener solvents for solid-phase organic synthesis
- Photocatalytic hydrogenolysis of allylic alcohols for rapid access to platform chemicals and fine chemicals
- IUPAC Recommendations
- Definition of the mole (IUPAC Recommendation 2017)
- Terminology of separation methods (IUPAC Recommendations 2017)
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers
- Papers from the 6th International IUPAC Conference on Green Chemistry (ICGC-6)
- Microwave assisted synthesis of glycerol carbonate from glycerol and urea
- Silica gel mediated oxidative C–O coupling of β-dicarbonyl compounds with malonyl peroxides in solvent-free conditions
- Definition of green synthetic tools based on safer reaction media, heterogeneous catalysis, and flow technology
- Heavy metal removal from waste waters by phosphonate metal organic frameworks
- A clean and simple method for deprotection of phosphines from borane complexes
- Development and treatment procedure of arsenic-contaminated water using a new and green chitosan sorbent: kinetic, isotherm, thermodynamic and dynamic studies
- Bio-adsorbent derived from papaya peel waste and magnetic nanoparticles fabricated for lead determination
- 5-Membered cyclic ethers via phenonium ion mediated cyclization through carbonate chemistry
- Synergy in food, energy and advanced materials production from biomass
- Step economy strategy for the synthesis of amphoteric aminoaldehydes, key intermediates for reduced hydantoins
- Separation technology meets green chemistry: development of magnetically recoverable catalyst supports containing silica, ceria, and titania
- Green chemistry and sustainable development: approaches to chemical footprint analysis
- Greener solvents for solid-phase organic synthesis
- Photocatalytic hydrogenolysis of allylic alcohols for rapid access to platform chemicals and fine chemicals
- IUPAC Recommendations
- Definition of the mole (IUPAC Recommendation 2017)
- Terminology of separation methods (IUPAC Recommendations 2017)