A clean and simple method for deprotection of phosphines from borane complexes
-
Oleg M. Demchuk

 ,
Radomir Jasinski
,
Radomir Jasinski

Abstract
Simple, efficient, clean, and stereospecific protocols of protection of phosphorus atom with borane and deprotection from the borane complexes of the tertiary phosphines in mild conditions are reported. The proposed protection/deprotection reactions tolerate a range of functional groups and lead to pure products with excellent yield with no need for application of chromatographic or crystallisation purification procedures. For the first time mechanisms of the reactions of phosphine protection and deprotection have been studied based on experimental kinetic data as well as quantumchemical calculations, which allows designing reaction conditions suitable for a given substrate.
Introduction
Catalytic reactions, especially enantioselective ones, are widely recognised as an environmentally beneficial alternative to well defined classical industrial synthetic approaches based on utilisation of stoichiometric amounts of highly reactive organic substrates. They are also frequently considered as greener methods [1], [2], [3]. The transition metal-catalysed reactions constitute the core of modern synthetic tools used in the synthesis of fine organic compounds, pharmaceuticals, functional materials, and plant protective chemicals [4], [5], [6], [7], [8]. These reactions are mediated by certain transition metal complexes with organic ligands bearing coordinating heteroatoms such as N, O, P, and S at lower oxidation stages. The selection of the most appropriate ligand depends on the reaction type, and in many cases, transition metal complexes of phosphines are superior. The phosphine ligand based complexes are widely applied in cross-coupling reactions [5], [6], [9], [10], [11], in asymmetric hydrogenation [12], [13], [14], [15], [16], hydrocarboxylation [17], [18], and many other crucially important processes [19], [20]. In many cases, the syntheses of appropriate phosphines are complicated by their sensitivity to air exposure. Thus, protection of the phosphorus atom during the ligand synthesis is a common practice. Phosphine-borane complexes are regarded as a convenient alternative to free phosphines [21], [22], [23], [24]. Borane protection is easily introduced with several complementary methods [25], [26], [27], [28], [29], [30], [31], [32]. It is also considered to be easy to remove from the phosphorus(III) atom. Only in very rare cases can the phosphine borane complexes be directly used in the syntheses of catalysts [33]. Nevertheless, in general, deprotection of phosphines is required and it is based on only two reactions: (a) less electron-rich phosphines can be liberated in reaction with the excess of strongly basic amines, such as TMEDA, morpholine, diethylamine, or DABCO, run at elevated temperature; (b) more inert P–B bonds of electron-rich phosphines can be efficiently cleaved by treatment with strong acids such as MeSO3H, CF3SO3H, or HBF4 [24], [34], [35]. Both methods require a post reaction workup and impose significant limitation on the nature of substituents which the phosphines possess. Thus, a new efficient method of deprotection of phosphine from borane complexes is still pursued.
Experimental
General all reagents were purchased from Sigma-Aldrich, Strem, TCI, and Alfa Aesar chemical companies and used without further purification. Analytical thin-layer chromatography (TLC) was performed using silica gel 60 F254 precoated plates (0.25 mm thickness) with a fluorescent indicator. Visualisation of TLC plates was performed by means of UV light and either KMnO4 or I2 stains. NMR spectra were recorded on Bruker Avance 500 MHz spectrometers, and chemical shifts are reported in ppm, and calibrated to residual solvent peaks at 7.27 ppm and 77.00 ppm for 1H and 13C in CDCl3 or internal reference compounds (H3PO4 capillary). The following abbreviations are used in reporting the NMR data: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad). Coupling constants (J) are in Hz. Spectra are reported as follows: chemical shift (d, ppm), multiplicity, integration, coupling constants (Hz). Products were purified by flash chromatography on silica gel 60 (230–400 mesh) using a BUCHI MPLC chromatograph. MS spectra were recorded on a Shimadzu LCMS ITTOF spectrometer. Commercially unavailable compounds were obtained by known literature procedures. The additional experimental procedures, spectral and physical data of presented compounds as well as mol2 files of the calculated critical structures are available in ESI.
General procedure for synthesis of phosphine borane complexes
1.0 M solution of borane in THF (2 mL or 4 mL for diphosphines) was added to an evacuation-degassed and argon-refilled solution of phosphine (1 mmol) in dry THF (2 mL). The mixture was heated to 40°C and stirred for 0.5 or 1 h. After that time, the reaction was cooled down to r.t. and the solvent was removed in vacuum to afford a crude product. The solid products were simply washed with a minimal amount of dry degassed cyclohexane. The liquid and hexane-soluble boranes were purified by flash column chromatography.
General procedure for deprotection of phosphine borane complexes with trimethylphosphine
The phosphine borane complex (0.5 mmol) and 2 mL of CPME (cyclopenthyl methyl ether) were placed in a Schlenk tube. Then an amount of chromatographic silica gel 60 equal to borane mass was added, and the reactor was evacuated and refilled with argon. The 1.0 M solution of trimethylphosphine in toluene (1.5 mL and 3 mL for diphosphine borane complexes) was added under an argon atmosphere, and the mixture was heated at 100°C for 12 h. The mixture was cooled down to r. t. After that, the 31P NMR analysis of the reaction mixture indicated the complete conversion of the starting complexes. The solvents were removed by a rotary evaporator. The trace amounts of volatile impurities were removed during an additional 16 h evacuation of the obtained crude products under 5 torr pressure. The resulting products did not need further purification.
Kinetic measurements
The starting reaction mixtures were prepared in a Schlenk-type reactor charged with a dicyclohexylphenylphosphine borane complex (576 mg, 2 mmol) and 5.5 mL of toluene; the reactor was evacuated and refilled with argon and 2 mL of a 1 M solution of trimethylphosphine in toluene was added. One milliliter of the obtained solution was introduced into an argon-filled NMR tube, which contained an internal standard capillary. The tube was next sealed with a PTFE screw and placed into oil bath preheated up to the required temperature (80, 95, 110°C). Progress of the reactions was monitored by means of 31P NMR spectroscopy. The conversion of Cy2PhPBH3 was determined at 1 h periods. Since the initial concentrations of the reactants were known, the areas of NMR signals were correlated to the instantaneous concentration (c) of the reactants. Next, on the basis of plots 1/c vs. reaction time, second order rate constants were calculated using standard equations [36]. Finally, on the basis of the rate constants measured at different temperatures, the activation enthalpy (ΔH≠) and activation entropy (ΔS≠) were calculated using the Eyring equation in the form:
where ΔG≠ is the Gibbs energy of activation, kB is Boltzmann’s constant, h is Planck’s constant, ΔS≠ is the entropy of activation, and ΔH≠ is the enthalpy of activation.
Quatumchemical calculations
The calculations reported in this paper were performed both on the “Prometheus” cluster in the CYFRONET regional computational centre in Cracow and on a personal desktop PC (Intel® Core™ I7 – 3930 K CPU @ 3.20 GHz, 16 GB RAM). Hybrid functional B3LYP with the 6−31+G(d) basis set included in the GAUSSIAN 09 [37] package and Spartan 10 [38] were used. In particular, optimizations of the stable structures were performed with the Berny algorithm, whereas the transition states were calculated using the QST2 procedure followed by the TS method. Stationary points were characterised by frequency calculations. All reactants and products had positive Hessian matrices. All transition states showed only one negative eigenvalue in their diagonalized Hessian matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration. For all reactions, intrinsic reaction coordinate (IRC) calculations were performed to connect previously computed transition structures (TS) with suitable minima. For the calculations of the solvent effect (GAUSSIAN 09 package) on the reaction paths, the polarizable continuum model (PCM) [39] in which the cavity is created via a series of overlapping spheres was used.
Results and discussion
In continuation of our studies in the field of synthesis and application of tertiary phosphine ligands [40], [41], [42], [43], [44], [45], [46], [47], [48], herein we present a new method of deprotection of phosphines from corresponding borane complexes. The main aim of our studies is to present a simple and inexpensive synthetic approach leading to sensitive target phosphines with high purities and high yields with no need to apply post reaction purification. To achieve this challenge, we decided to use the simplest exchange reaction of low molecular weight volatile nucleophile with phosphine boranes. Low molecular weight amines are not nucleophilic enough to shift the equilibrium of the reactions with phosphine boranes in the direction of products. Phosphines are more reactive and the reaction between small trialkyl phosphines and phosphine borane complexes has been assessed. In analogy to the reaction of phosphine with amine boranes, it is reasonable that an exchange of the BH3 group is possible in a mixture of free phosphine and a phosphine borane complex (Scheme 1).
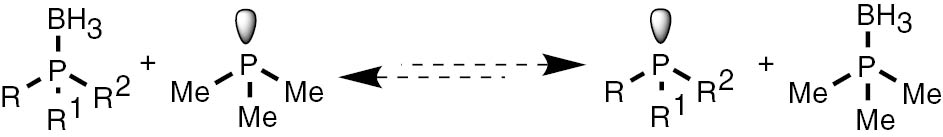
The exchange reaction between phosphine and phosphine boranes.
Nevertheless, for the synthesis of phosphines, this possibility has not been explored yet. We decided to use this equilibrium-driven reaction to have direct access to unprotected phosphines.
The trimethylphosphine was chosen as a reagent of choice to be used for the deportation of phosphines. It has low molecular weight (76.08 g/mol), low boiling point (38°C), small size, and marginal steric hindrance at an electron-rich phosphorus atom. In addition, it is commercially available in individual form and in a solution. According to the MSDS data, the environmental and toxicological threats of trimethylphosphine are not critically high [49]. Moreover, its strong stench is a good indicator of possible contamination. Upon rapid oxidation when exposed on air, a trace contamination with trimethylphosphine is self-converted to a much safer trimethylphosphine oxide. On the other hand, borane complex of dicyclohexylphenylphosphine (obtained in high yield according to the classical approach) [50], [51] was selected as a model partner for the reaction with trimethylphosphine because of its high basicity and significant steric hindrance, which are known to complicate the deprotection of the phosphorus site.
In the preliminary experiments run in a toluene solution at 80 and 110°C, an exchange reaction between the model borane 1 and trimethylphosphine 2, used in an equimolar ratio, was observed (Scheme 2). The progress of the reaction was monitored by means of 31P NMR spectroscopy; the spectra were recorded in the presence of an internal standard – a capillary with a solution of phosphoric acid in deuterium oxide.
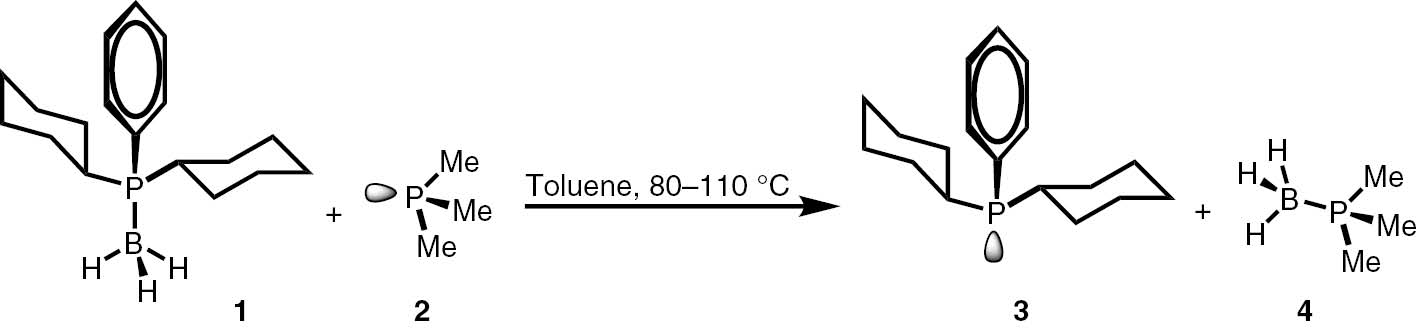
Model reaction between 1 and 2.
The reaction approaches the equilibrium at conversion of 1=32% in less than 5 h at 110°C. It allows calculating the Gibbs free energy of the reaction (ΔG=−0.52 kcal/mol) based on the equation:
were K is the equilibrium constant, and it is equal to 1.97 at 110°C
At the same time, the mechanism of this reaction remains unknown. It has been postulated that the reaction may proceed via a dissociation stage [24], but no mechanistic studies of this issue have ever been published.
According to a few previous reports, the exchange reaction of phosphine boranes with amines [52], as well as the reaction of amine boranes with phosphines [53], [54] has the SN2-like mechanism. Some other nucleophilic substitution reactions at the tetrahedral boron atom are also known [55], [56]. To elucidate the mechanism of the reaction studied herein, series kinetic experiments were performed at the temperatures of 80, 95, and 110°C, molar ratio of 1:2=1:1, and initial concentration of 1=0.25 M (Table 1, Fig. 1). To obtain reliable results, all kinetic experiments were conducted under an argon atmosphere in sealed NMR tubes. Additionally, to exclude possible oxidization and evaporation effects, which may influence the quantitative results, the initial reaction mixtures were prepared under an argon atmosphere in Schlenk-type reactors. The progress of the reactions was monitored by means of 31P NMR spectroscopy with a H3PO4 capillary used as an internal standard. The corresponding signals in the 31P NMR spectra appear as follows (δ, ppm): 1 at 27.0÷27.7 (m), 2 at −62.6 (s), 3 at 1.9 (s), internal standard at 0 (s), and 4 at −1.3÷−2.5 (m, JP-B=59.7 Hz). Since the signal of 2 appears very far from the other signals, its accurate integration was problematic; similarly, the integration of borane 4 was not especially accurate because in the 31P NMR spectra 4 appears as a wide multiple of four separate signals. For this reason, the kinetic calculation was based on the signal of starting borane 1. In the same time, in the control experiment, left at 20°C for 2 days, we did not observe progress of the reaction.
The linear dependence in the coordinate 1/c vs time with the acute angle slope clearly indicates the second order of the reaction. The reaction rate constants calculated on the basis of the relationships mentioned above were 1.24·10−6 dm3/(mol·s), 4.85·10−6 dm3/(mol·s), and 9.67·10−6 dm3/(mol·s) at 80, 95, and 110°C, respectively.
The activation parameters of the reaction activation were calculated next, based on the Eyring equation:
where ΔG≠ is the Gibbs energy of activation, kB is Boltzmann’s constant, h is Planck’s constant, ΔS≠ is the entropy of activation, and ΔH≠ is the enthalpy of activation.
ΔH≠=17.72 kcal/mol and ΔS≠=−35.5 cal/(mol·K) were calculated from the Eyring plot [ln (k/T) vs. 1/T], where slope=ΔH≠/R; intercept=ln (kB/h)+ΔS≠/R.
On the basis of activation parameters, it is possible to make a conclusion about the reaction mechanism. In particular, a high negative ΔS≠ value suggests a high degree of order within the transition state (TS). In addition, a relatively low value of ΔH≠ shows that the changes in the system energy resulting from breakage of the existing σ-bond are compensated to a large extent by the energy changes resulting from the formation of a new σ-bond. Thus, interpretation of Eyring parameters fully complies with the expected SN2-like mechanism.
To shed a deeper light on the molecular mechanism of this reaction, a theoretical study was carried out based on the DFT calculation data. In particular, the reaction pathway was explored using the hybrid, B3LYP functional [37], [38], [57] and 6-31+G(d) basis set. A similar level of theory was successfully verified with regard to a number of organophosphorus compounds, including phosphines and their oxides [45], [58], [59]. For the simulations of the solvent effect, the polarizable continuum model (PCM) was used [39].
The results of the DFT (PCM) computational study prove that the reaction between 1 and 2 should be considered as a one-step, synchronous process. In particular, on the reaction profile, a single transition state (TS) is localised between the minimum linked with the existence of individual substrates 1, 2 and the minimum linked with the existence of products 3, 4. Conversion of the reaction system into TS involves an increase in the enthalpy by 21.61 kcal/mol. At the same time, the entropy of a more ordered system is reduced by −24.6 cal/(mol·K) (Fig. 2). The transition state was almost symmetrical with the distance between Me3P–BH3 equal to 2.684 Å and the distance between Cy2PhP–BH3 equal to 2.699 Å. Both distances were significantly bigger than those appearing in substrate 1 (1.951 Å) and product 4 (1.935 Å) optimized under identical conditions. The slightly higher degree of development of the Me3P–BH3 bond suggests higher stability of the less hindered reaction product 4 over substrate 1. In the TS, the BH3 moiety adopts the flat geometry of the sp2 hybridised boron atom. The reliability of the localised transition state was confirmed by IRC calculations and supported by vibrational analysis (one imaginary frequency corresponding to movement along P–B–P bonds was detected at −300.78 L/cm).
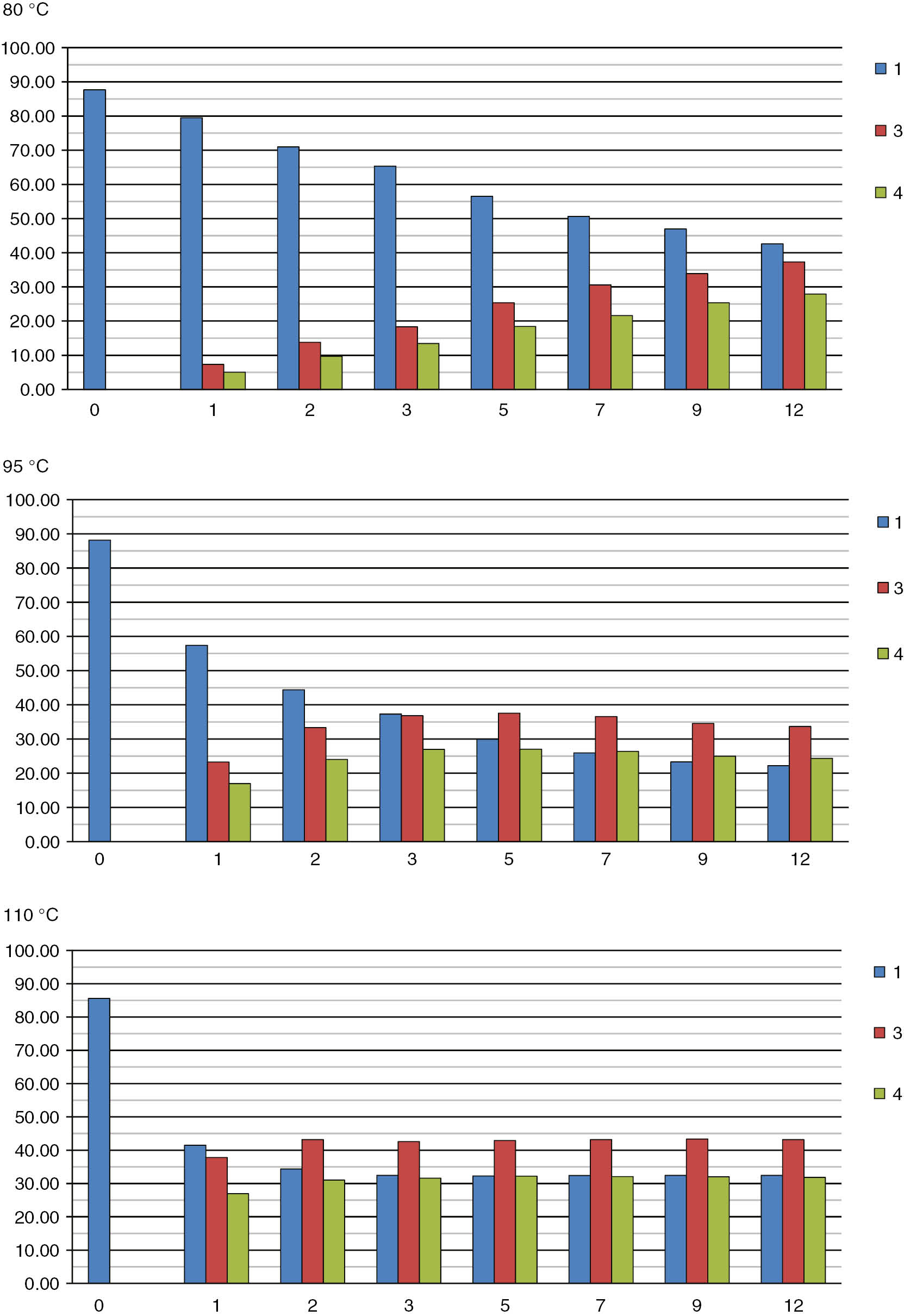
Visualisation of the reaction progress in time at 80, 95, and 110°C.
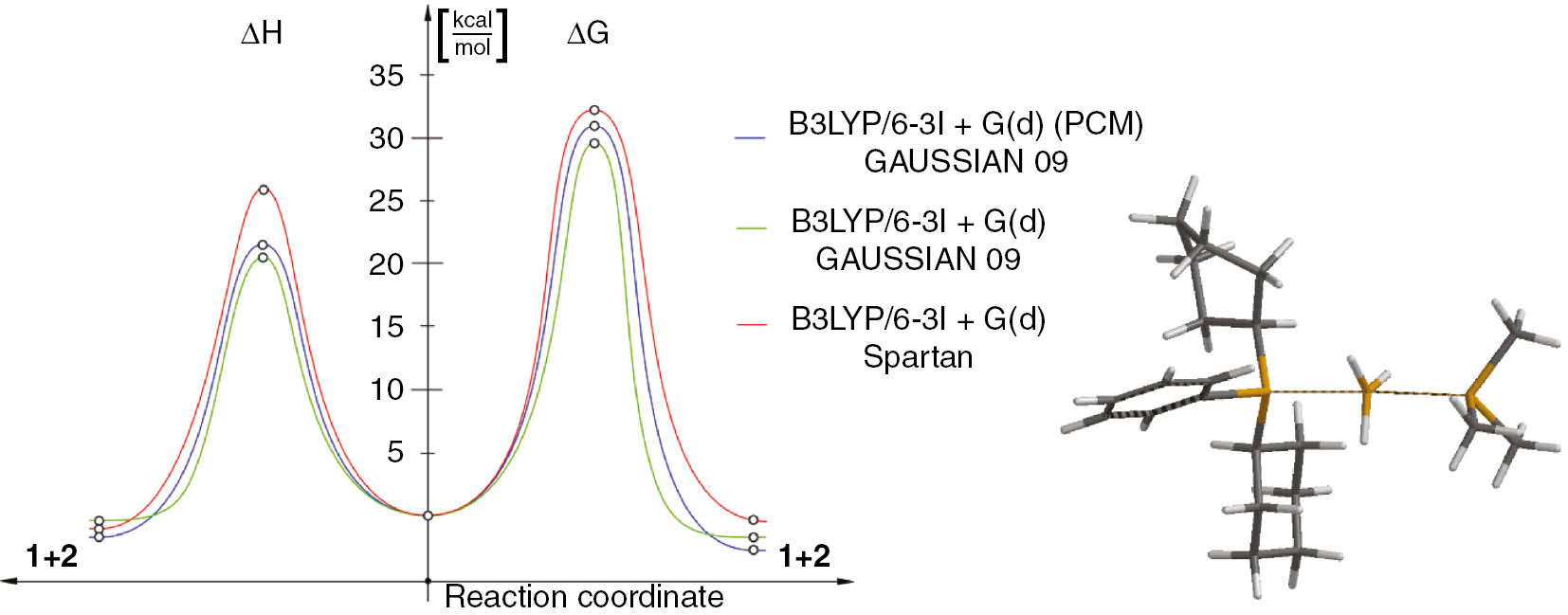
Enthalpy profile of the reaction between 1 and 2 and view of its TS structure.
Relative intensity of the signals in the 31P NMR spectra of the reaction mixture.
| T (h) | Integration of 31P signalsa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
3 |
4 |
|||||||
| 80°C | 95°C | 110°C | 80°C | 95°C | 110°C | 80°C | 95°C | 110°C | |
| 0 | 87.66 | 88.19 | 85.60 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 79.50 | 57.38 | 41.47 | 7.31 | 23.26 | 37.81 | 5.05 | 16.97 | 26.95 |
| 2 | 70.97 | 44.39 | 34.37 | 13.81 | 33.34 | 43.19 | 9.69 | 24.03 | 31.04 |
| 3 | 65.33 | 37.30 | 32.47 | 18.32 | 36.82 | 42.60 | 13.48 | 26.98 | 31.60 |
| 5 | 56.51 | 29.95 | 32.28 | 25.38 | 37.55 | 42.92 | 18.44 | 27.00 | 32.19 |
| 7 | 50.64 | 25.95 | 32.44 | 30.60 | 36.55 | 43.18 | 21.63 | 26.37 | 32.10 |
| 9 | 46.97 | 23.31 | 32.46 | 33.90 | 34.55 | 43.36 | 25.37 | 24.96 | 32.02 |
| 12 | 42.60 | 22.20 | 32.48 | 37.30 | 33.68 | 43.18 | 27.91 | 24.33 | 31.83 |
| 0 | 87.66 | 88.19 | 85.60 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 79.50 | 57.38 | 41.47 | 7.31 | 23.26 | 37.81 | 5.05 | 16.97 | 26.95 |
| 2 | 70.97 | 44.39 | 34.37 | 13.81 | 33.34 | 43.19 | 9.69 | 24.03 | 31.04 |
| 3 | 65.33 | 37.30 | 32.47 | 18.32 | 36.82 | 42.60 | 13.48 | 26.98 | 31.60 |
| 5 | 56.51 | 29.95 | 32.28 | 25.38 | 37.55 | 42.92 | 18.44 | 27.00 | 32.19 |
| 7 | 50.64 | 25.95 | 32.44 | 30.60 | 36.55 | 43.18 | 21.63 | 26.37 | 32.10 |
| 9 | 46.97 | 23.31 | 32.46 | 33.90 | 34.55 | 43.36 | 25.37 | 24.96 | 32.02 |
| 12 | 42.60 | 22.20 | 32.48 | 37.30 | 33.68 | 43.18 | 27.91 | 24.33 | 31.83 |
-
Conditions: initial molar ratio of 1:2=1:1 and initial concentration of 1=0.25 M. aIntegration of the internal standard capillary was assigned as 100.
The calculations were performed for simulated presence of toluene (PCM solvation model was used) and, subsequently, for reaction in the gas phase, using the GAUSSIAN 09 package [37] allocated on supercomputer cluster and Spartan PC [38] package on a regular desktop PC. The obtained energetic parameters provide similar energy profiles and the geometrical parameters of critical structures (Table 2). This indicates that the reaction run through non-polar TS is not sensitive to the solvent effects and, therefore, could be accomplished not only in toluene, but also in some other solvents, compatible with the BH3 and functional groups, which the phosphine borane complex bear. The non-polar nature of TSs is additionally confirmed by analysis of their dipole moments, which are less than 0.4 D in the gas phase and about 0.5 D in the simulated presence of toluene). For comparison, typical polar TSs are characterised by dipole moments which are higher than 10 D [60], [61].
Kinetic and thermodynamic parameters of reaction 1+2 calculated at the B3LYP/6-31+G(d) theory level and measured experimentally.
| Transition | GAUSSIAN 09 |
Spartan |
Experimental |
|||||
|---|---|---|---|---|---|---|---|---|
| Toluene (PCM) |
Vacuum |
Vacuum |
Toluene |
|||||
| ΔH (kcal/mol) | ΔS [cal/(mol·K)] | ΔH (kcal/mol) | ΔS [cal/(mol·K)] | ΔH (kcal/mol) | ΔS [cal/(mol·K)] | ΔH (kcal/mol) | ΔS (cal/(mol·K)] | |
| 1+2→TS | 21.61 | −25.3 | 19.99 | −24.6 | 25.98 | −46.25 | 17.72 | −35.5 |
| 1+2→3+4 | −1.80 | 3.5 | −0.96 | 3.4 | −1.19 | +1.7 | – | – |
| ΔG=−3.12 | ΔG=−2.25 | ΔG=−0.54 | ΔG=−0.52 | |||||
-
ΔG was calculated for the reaction run at 110°C (kcal/mol).
A similar B3LYP/6-31+G(d) (PCM) simulation-based approach was applied to elucidate the mechanism of the reaction between the THF–BH3 complex and trimethylphosphine (Scheme 3, Table 3). In this case, we have also found that the reaction has an SN2-like mechanism with ordered transition state TS1, which is significantly symmetrical in relation to bonds P–B and O–B, forming and backing during the reaction with distances 2.837 and 2.122 Å, respectively. The shorter distance of the O–B bond in the TS1 corresponds to the geometry of the earlier transition state. The BH3 group adopts almost flat geometry of the sp2 hybridised boron atom, with only 2.5° deviation outside the plane of three hydrogens. Despite the reaction of trimethylphosphine with phosphine boranes, the reaction trimethylphosphine with tetrahydrofuran borane complex is rapid and exothermic.
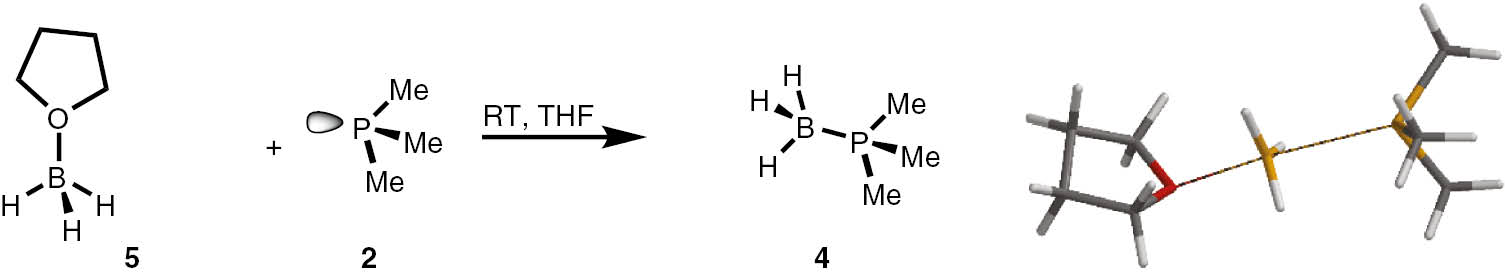
Model reaction between THF–BH35 and trimethylphosphine 2 and view its transition state (TS).
Thermodynamic parameters of reaction 5+2 calculated at the B3LYP/6-31+G(d) theory level.
| Transition | GAUSSIAN 09 |
Spartan |
||||
|---|---|---|---|---|---|---|
| THF (PCM) |
Vacuum |
Vacuum |
||||
| ΔH (kcal/mol) | ΔS [cal/(mol·K)] | ΔH (kcal/mol) | ΔS [cal/(mol·K)] | ΔH (kcal/mol) | ΔS [cal/(mol·K)] | |
| 5+2→TS1 | 10.43 | −26.3 | 7.04 | −26.5 | 5.88 | −41.4 |
| 5+2→6+THF | −15.93 | 0.7 | −15.29 | 1.4 | −11.04 | 0.0 |
| ΔG=−16.14 | ΔG=−15.74 | ΔG=−11.04 | ||||
-
ΔG was calculated for the reaction run at 40°C (kcal/mol).
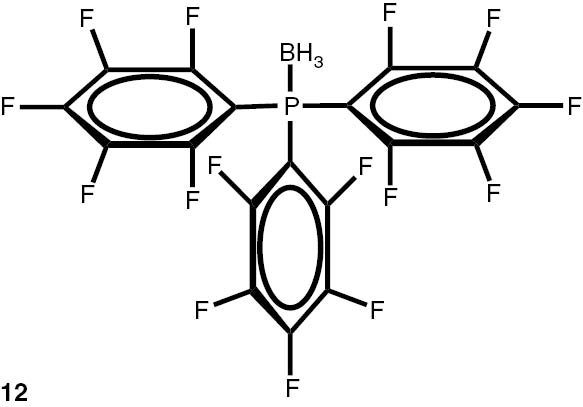
In comparison with non-hindered and electron-rich trimethylphosphine, the reactions of triphenylphosphine and electron-deficient perfluorotriphenylphosphine with the THF–BH3 complex were less favourable. Thus, in the case of the triphenylphosphine complex, the thermodynamic parameters of formation ΔH, ΔS, and ΔG were −10.42 kcal/mol, 0.5 cal/(mol·K), and −10.56 kcal/mol (at 25°C), correspondingly. At the same time, the perfluorotriphenylphosphine borane was not stable at room temperature, with analogous thermodynamic parameters of formation equal to 3.86 kcal/mol, −8.6 cal/(mol·K), and 6.43 kcal/mol (at 25°C), respectively.
The analysis of the mechanism and kinetic parameters of the protection/deprotection reactions indicates that, in the case of non-electron deficient phosphines, the protection reactions with the THF–BH3 complex are fast and only a minor excess of borane is required for complete conversion of phosphine at room temperature. At the same time, the reaction of phosphine boranes with trimethylphosphine is much slower and a few-fold excess of trimethylphosphine as well as elevated temperature are required to achieve high yields of the products.
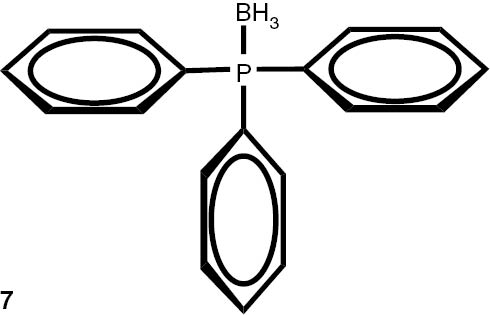
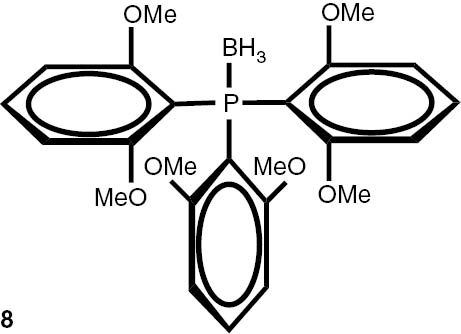
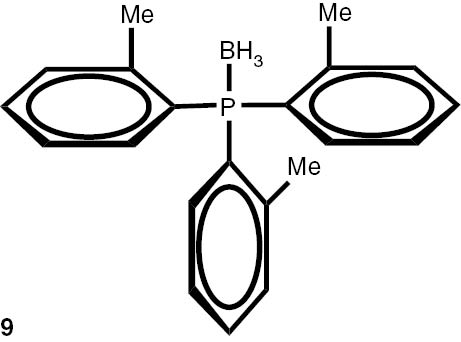
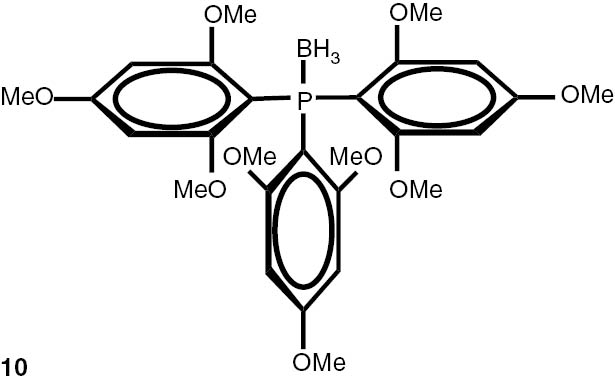
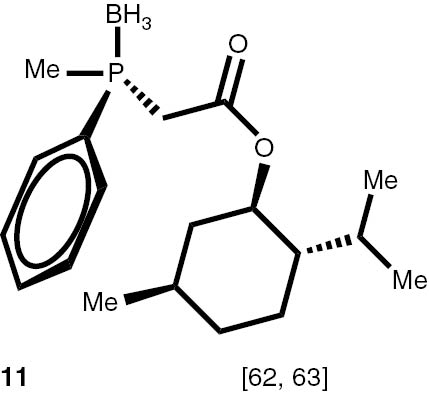
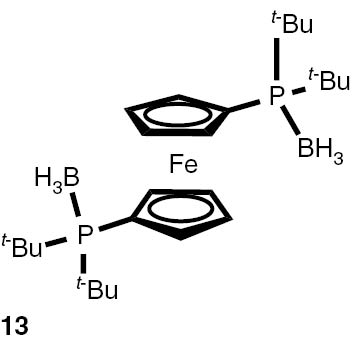
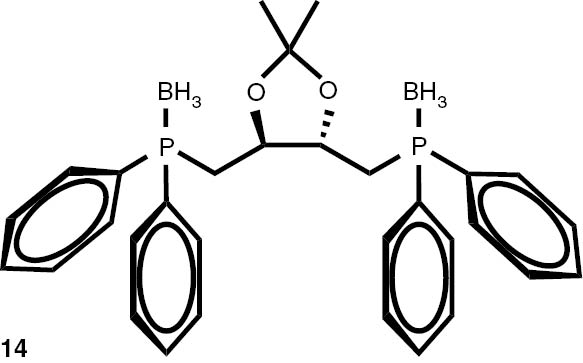
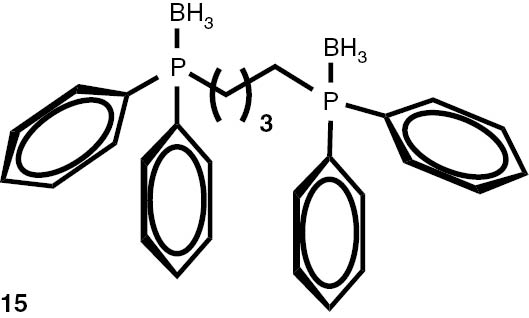
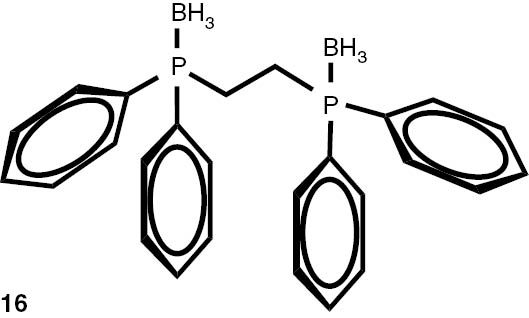
With these considerations, series phosphine boranes 1, 6–11 and diphosphine bisboranes 13–16 were obtained in the reaction of corresponding phosphines with the THF–BH3 complex in tetrahydrofurane at 40°C. Utilisation of a two-fold excess of borane resulted in a very clean fast reaction. The spectrally pure products were obtained after evaporation of the solvent under reduced pressure and washing the crude products with a small amount of cyclohexane. Only in the cases of a few boranes, soluble in cyclohexane, flash column chromatography was applied (Table 4).
Syntheses of phosphine boranes 1, 6–16.
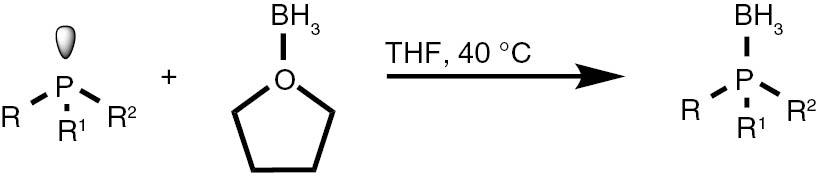
| Phosphine borane | Yield (%) | Phosphine borane | Yield (%) |
|---|---|---|---|
 |
75, purified by flash chromatography |
 |
82, washed with cyclohexane |
 |
92, washed with cyclohexane |
 |
80, washed with cyclohexane |
 |
85, purified by flash chromatography |
 |
85, washed with cyclohexane |
 |
90, purified by flash chromatography |
 |
0 |
 |
59, purified by flash chromatography |
 |
39, washed with cyclohexane |
 |
81, washed with cyclohexane |
 |
97, washed with cyclohexane |
-
Conditions: solution of phosphine (1 mmol) in dry THF (2 mL), 1.0 M solution of borane in THF (2 mL per mmol of phosphine), argon atmosphere, 40°C, 1 h.
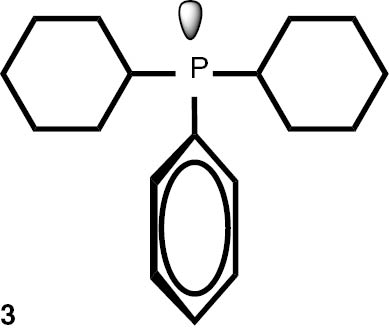
The obtained boranes were next subjected to the deprotection procedure. The theoretic consideration, based on the measured reaction rate constant and equilibrium constants, suggested that a three-fold excess of trimetylphosphine should be enough to shift the equilibrium into the direction of the product at the 100°C reaction temperature. In preliminary experiments, we observed the formation of trace amounts of weakly soluble boric acid derivatives, which contaminated the products. To eliminate the need for further purification of sensitive phosphines, the silica gel sorbent was added to the reaction mixture. A Schlenk-type reactor was charged with phosphine borane and an equal weight amount of silica gel 60. Cyclopenthylmethyl ether (usually considered as a green alternative to the ethereal and aromatic solvents) was added and the reactor was degassed and filled back with argon. Next, the solution of trimethylphosphine in toluene was added and the reactor was closed with a glass stopper and heated at 100°C for 16 h. Upon the cooling down to room temperature, the solution of the products was taken with a needle; then, the solvents and volatile products were evaporated off under pressure reduced down to 5 torr. Application of a dry ice cooled trap allowed collecting the reagents to be reused further. The products were subjected to additional 12 h vacuum degassing, which allowed obtaining spectrally pure (contaminated with less than 5% of inert Me3PO and Me3PBH3) phosphines in high yields (Table 5).
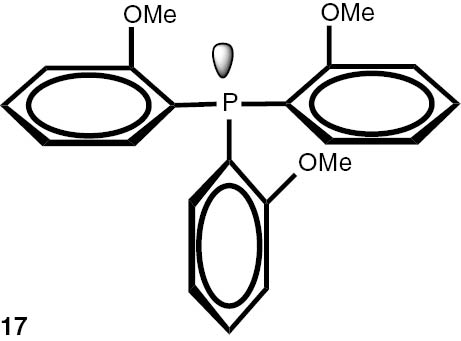
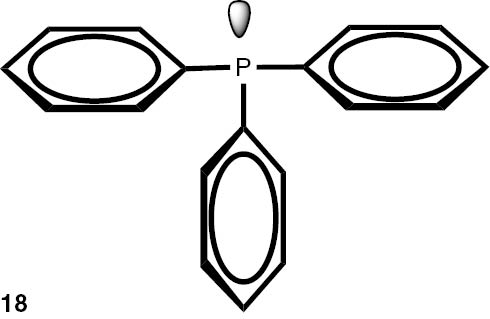
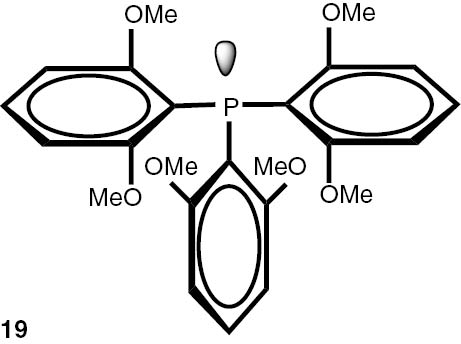
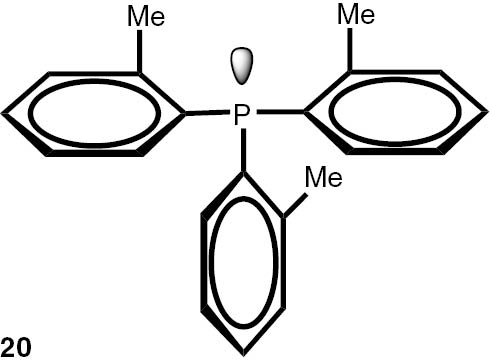
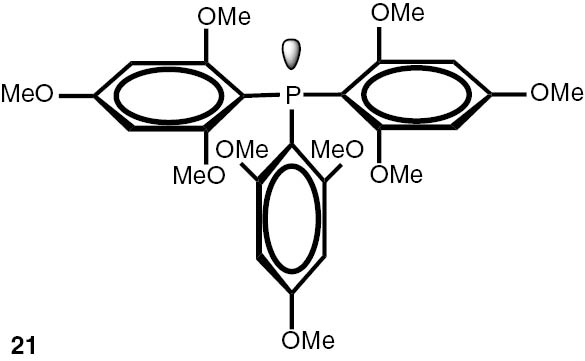
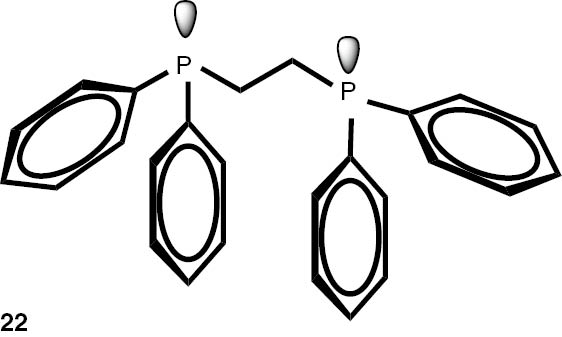
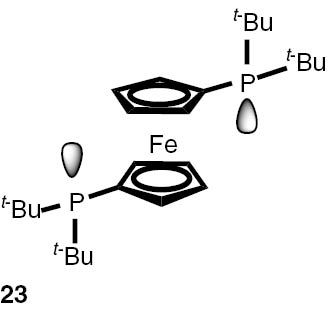
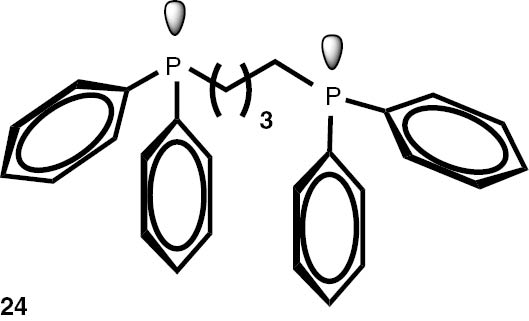
Synthesis of phosphines from the phosphine borane complexes 3, 17–29.
| Phosphine | Yield (conversion), (%) | Phosphine | Yield (conversion), (%) |
|---|---|---|---|
 |
86 (96) |
 |
99 (100) |
 |
96 (99)a |
 |
96 (100) |
 |
96 (100) |
 |
91 (91) |
 |
93 (93)a |
 |
86 (87) |
 |
98 (99)a |
 |
97 (99) |
 |
95 (99) |
 |
97 (99) |
 |
97 (99) |
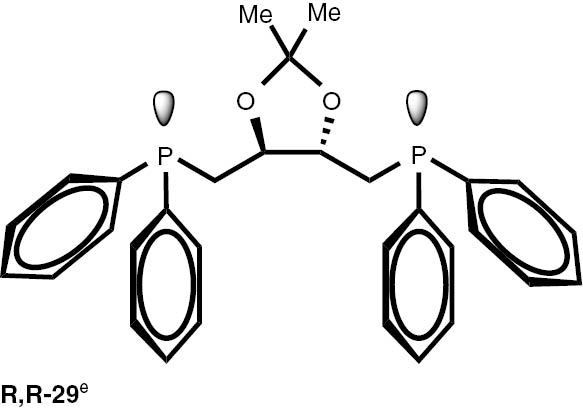 |
93 (99) |
-
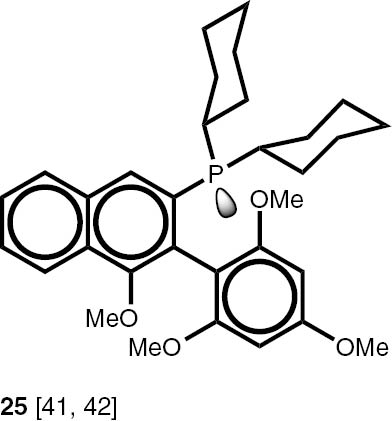
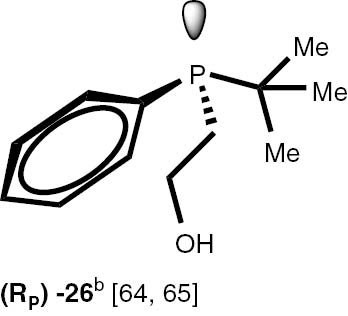
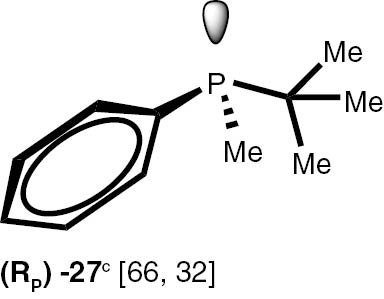
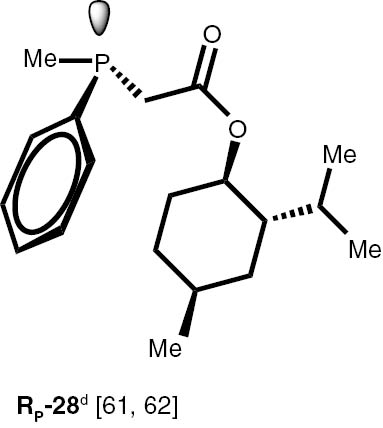
Conditions: phosphine borane complex (0.5 mmol), 2 mL of CPME (cyclopenthyl methyl ether), 200 mg SiO2, 1.0 M solution of trimethylphosphine in toluene (1.5 mL per 0.5 mmol of borane complex), argon atmosphere, 100°C, 12 h. Conversion was calculated from 31P NMR spectra of the reaction mixtures. aTHF was used instead of CPME. bborane complex was prepared according to known procedure [64], [65] >75% ee substrate was used, >71% ee product was obtained. cborane complex was prepared according to known procedure [32], [66] >92% ee substrate was used, >90% ee product was obtained (borane complex: HPLC, Chiralcel ad-h column; phosphine converted to oxide: 1H, 31P NMR in the presence of Naproxen) [62]. dborane complex was prepared according to known procedure [40], [63] Conditions: 60°C, 18 h, 2 equiv. Me3P, 48% de substrate was used, 46% de product was obtained (1H, 31P NMR). e>98% de substrate was used, >99% de product was obtained (ORP).
The stereoselectivity of the transformations of chiral non-racemic phosphines are crucially important because the chiral purification of the sensitive products is not usually possible, and, due to the well known non-linear effects in asymmetric catalysis, even small contamination of the chiral catalysts with the minor stereoisomer may significantly influence the stereochemical outcome of the reaction. To check the assumption that the configuration of the stereogenic centre located at phosphorus and carbon atoms is not changed during the reaction, several chiral P-stereogenic 26–28 as well as C-stereogenic phosphine boranes 28, 29 were obtained. The stereo composition of the used boranes and obtained phosphines were measured by means of HPLC-MS chromatography and NMR spectroscopy. To simplify the procedure of determination of samples stereocomposition, the enriched in one of the stereoisomers mixture of the chiral substrates were used. The experiments with chiral substrates were also repeated with corresponding racemic mixtures. In all the cases, no inversion of the configuration at the stereogenic centres or racemisation above the measurement tolerance was observed. Nevertheless we have found that chiral phosphines 26–28 undergo a partial thermal racemisation at the temperature above 100°C, thus low reaction temperature of 60°C and prolonged time were applied to prevent the racemisation. Notable, the sensitive to acid/base conditions, phosphines possessing unprotected hydroxy, di-O-isopropylidene, and ester functional groups were obtained in excellent yields.
Conclusions
The straightforward stereospecific protocols of deprotection of phosphorus atom of phosphine boranes in reaction with trimethylphosphine run in mild conditions have been reported for the first time. The proposed reactions tolerate a range of function groups and lead to pure products in excellent yields. The unselective transformations of starting phosphine boranes were not observed, and the isolation of target products do not require the application of post reaction workup and chromatographic or crystallisation purification procedures. The approach to protection of phosphorus atom of phosphines in reaction with THF–BH3 adduct was also studied. The SN2-like mechanisms of both reactions were elucidated. The general conclusions that can be made upon the analysis of the mechanisms of the reactions of protection of phosphines with borane and deprotection of the obtained complexes are as follows:
both reactions are reversible and undergo a second-order nucleophilic substitution mechanism (on the boron atom) with retention of the configuration at phosphorus;
the reaction of non-electron deficient phosphines with the THF–BH3 complex is fast and the equilibrium is shifted into the direction of products. The small excess of borane may be favourable for complete conversion of phosphine;
the reaction of phosphine boranes with trimethylphosphine is much slower and leads to less hindered trimetilphosphine borane; thus, elevated temperature and a few-fold excess of trimethylphosphine is required to achieve sufficient conversions of the substrates.
The reaction could be performed in different aprotic solvents such as THF, toluene or CPME, considered as a green alternative to the ethereal solvents and aromatic hydrocarbons.
The computer simulation of the reaction is quite simple and can be done on a regular desktop PC in reasonable time by reoptimalisation of critical structures of the reactions with consideration of given substituents they bear (see ESI). This allows designing the optimal conditions of the reactions and minimising the cost of syntheses and waste emission.
Article note
A collection of invited papers based on presentations at the 6th international IUPAC Conference on Green Chemistry (ICGC-6), Venice (Italy), 4–8 September 2016.
Acknowledgment
The generous allocation of computing time by the regional computer centre “Cyfronet” in Cracow (Grant No.: MNiSW/Zeus_lokalnie/PK/009/2013) and support from the Polish National Science Centre (National Science Center, Poland grant number 2012/05/B/ST5/00362) are gratefully acknowledged. The part of experimental works was financially supported by the National Science Center, Poland fund awarded based on the decision UMO-2012/06/A/ST5/00227 (MAESTRO). The authors are also deeply thankful to mgr. Magdalena Okła (UMCS, Lublin) for valuable editorial assistance.
References
[1] P. Tundo. Chemistry Beyond Chlorine. Springer Berlin Heidelberg, New York (2016).10.1007/978-3-319-30073-3Search in Google Scholar
[2] P. Tundo, P. T. Anastas. Green Chemistry: Challenging Perspectives. Oxford University Press, Oxford, New York (2000).Search in Google Scholar
[3] P. Tundo, A. Perosa, F. Zecchini. Methods and Reagents for Green Chemistry: An Introduction. Wiley-Interscience, Hoboken, New Jersey (2007).10.1002/9780470124086Search in Google Scholar
[4] X. Ribas. C-H and C-X Bond Functionalization: Transition Metal Mediation. RSC Publishing, Cambridge, UK (2013).10.1039/9781849737166Search in Google Scholar
[5] J. Magano, J. R. Dunetz. Transition Metal-Catalyzed Couplings in Process Chemistry: Case Studies from the Pharmaceutical Industry. Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, Germany (2013).Search in Google Scholar
[6] M. L. Crawley, B. M. Trost. Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective. John Wiley & Sons, Hoboken, New Jersey (2012).10.1002/9781118309872Search in Google Scholar
[7] A. Behr, P. Neubert. Applied Homogeneous Catalysis. Wiley-VCH, Weinheim, Germany (2012).Search in Google Scholar
[8] L. S. Hegedus, B. R. C. G. S öderberg. Transition Metals in the Synthesis of Complex Organic Molecules. University Science Books, Sausalito, California (2010).Search in Google Scholar
[9] A. D. Meijere, F. O. Diederich. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH, Weinheim, Germany (2004).10.1002/9783527619535Search in Google Scholar
[10] Y. Nishihara. Applied Cross-Coupling Reactions. Springer, Berlin, New York (2013).10.1007/978-3-642-32368-3Search in Google Scholar
[11] T. J. Colacot. New Trends in Cross-Coupling: Theory and Applications. The Royal Society of Chemistry, Cambridge, UK (2015).10.1039/9781782620259Search in Google Scholar
[12] J. M. Brown, P. A. Chaloner. “Asymmetric Hydrogenation Reactions Using Chiral Diphosphine Complexes of Rhodium”, in Homogeneous Catalysis with Metal Phosphine Complexes, M. L. Pignolet (Ed.), pp. 137–165. Plenum Press, New York (1983).10.1007/978-1-4613-3623-5_4Search in Google Scholar
[13] B. Štefane, F. Požgan. “Asymmetric Hydrogenation and Transfer Hydrogenation of Ketones”, in Hydrogenation, I. Karamé (Ed.), Open Acess, EN-FIST Centre of Excellence, Slovenia, Doi: 10.5772/3208 (2012).10.5772/3208Search in Google Scholar
[14] E. I. Klabunovskiĭ, G. V. Smith, A. G. Zsigmond. Heterogeneous Enantioselective Hydrogenation: Theory and Practice. Springer, Dordrecht, Netherlands (2006).10.1007/978-1-4020-4296-6Search in Google Scholar
[15] H.-U. Blaser. “Industrial Asymmetric Hydrogenation”, in Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, M. L. Crawley, B. M. Trost (Eds.), pp. 315–342, Wiley, New York (2012) Doi: 10.1002/9781118309872.10.1002/9781118309872Search in Google Scholar
[16] T. Ohkuma, M. Kitamura, R. Noyori. “Asymmetric Hydrogenation”, in Catalytic Asymmetric Synthesis, I. Ojima (Ed.), pp. 1–100, Wiley, New York (2005) DOI: 10.1002/0471721506.ch1.10.1002/0471721506.ch1Search in Google Scholar
[17] R. C. J. Gautreau-Service. Carbon Dioxide Fixation in Asymmetric Hydrocarboxylation Reactions and the Formatino of Acylureas. Queen’s University, Canada (2007).Search in Google Scholar
[18] K. Nozaki, I. Ojima. “Asymmetric Carbonylations”, in Catalytic Asymmetric Synthesis, I. Ojima (Ed.), pp. 429–463, Wiley, New York (2005) Doi: 10.1002/0471721506.10.1002/0471721506Search in Google Scholar
[19] Q.-L. Zhou. Privileged Chiral Ligands and Catalysts. Wiley-VCH, Weinheim, Germany (2011).10.1002/9783527635207Search in Google Scholar
[20] A. Grabulosa. P-Stereogenic Ligands in Enantioselective Catalysis. Royal Society of Chemistry, Cambridge, UK (2011).10.1039/9781849732703Search in Google Scholar
[21] A. Staubitz, A. P. M. Robertson, M. E. Sloan, I. Manners. Chem. Rev.110, 4023 (2010).10.1021/cr100105aSearch in Google Scholar
[22] J. M. Brunel, B. Faure, M. Maffei. Coord. Chem. Rev.178–180, 665 (1998).10.1016/S0010-8545(98)00072-1Search in Google Scholar
[23] E. Fernández, A. Whiting, eds. “Synthesis and Application of Organoboron Compounds”, in Topics in Organometallic Chemistry, M. Beller (Ed.), Springer Cham Heidelberg New York Dordrecht London (2015).10.1007/978-3-319-13054-5Search in Google Scholar
[24] M. Ohff, J. Holz, M. Quirmbach, A. Börner. Synthesis. 1998, 1391 (1998).10.1055/s-1998-2166Search in Google Scholar
[25] A. B. Burg, R. I. Wagner. J. Am. Chem. Soc.75, 3872 (1953).10.1021/ja01112a002Search in Google Scholar
[26] W. A. G. Graham, F. G. A. Stone. J. In. Nuc. Chem.3, 164 (1956).10.1016/0022-1902(56)80014-6Search in Google Scholar
[27] H. Schmidbaur, E. Weiss. Angew. Chem. Int. Ed. Engl.18, 781 (1979).10.1002/anie.197907811Search in Google Scholar
[28] J. McNulty, Y. Zhou. Tetrahedron Lett.45, 407 (2004).10.1016/j.tetlet.2003.10.145Search in Google Scholar
[29] A. B. Burg, P. J. Slota. J. Am. Chem. Soc.82, 2145 (1960).10.1021/ja01494a014Search in Google Scholar
[30] R. Köster, Y. Morita. Angew. Chem. Int. Ed. Engl.4, 593 (1965).10.1002/anie.196505931Search in Google Scholar
[31] T. Imamoto, T. Kusumoto, N. Suzuki, K. Sato. J. Am. Chem. Soc.107, 5301 (1985).10.1021/ja00304a061Search in Google Scholar
[32] K. V. Rajendran, D. G. Gilheany. Chem. Commun.48, 817 (2012).10.1039/C1CC14856GSearch in Google Scholar
[33] D. B. G. Williams, P. D. R. Kotze, A. C. Ferreira, C. W. Holzapfel. J. Ir. Chem. Soc.8, 240 (2012).10.1007/BF03246221Search in Google Scholar
[34] D. B. G. Williams, H. Lombard, M. van Niekerk, P. P. Coetzee, C. W. Holzapfel. Phosphorus Sulfur Silicon Relat. El.177, 2799 (2002).10.1080/10426500214885Search in Google Scholar
[35] M. Pericàs, S. Sayalero. Synlett.2006, 2585 (2006).10.1055/s-2006-950439Search in Google Scholar
[36] K. Schwietlick. Kinetische Metoden zur Untersuchung von Reaktionsmechanismen. VEB Deutscher Verlag der Wissenschaften, Berlin, Germany (1971).Search in Google Scholar
[37] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. J. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, Y. Nakajima, O. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, M. C. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, F. D. J., K. T., A.-L. M. A., P. C. Y., N. A., C. M., P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople. Gaussian 09. Gaussian, Inc., Wallingford, UK (2009).Search in Google Scholar
[38] Spartan’10, Wavefunction, Inc., Irvine, CA.Search in Google Scholar
[39] M. Cossi, N. Rega, G. Scalmani, V. Barone. J. Comput. Chem.24, 669 (2003).10.1002/jcc.10189Search in Google Scholar
[40] O. M. Demchuk, W. Świerczyńska, K. Dziuba, S. Frynas, A. Flis, K. M. Pietrusiewicz. Phosphorus Sulfur Silicon Relat. El. 192, 64 (2016).10.1080/10426507.2016.1225052Search in Google Scholar
[41] O. M. Demchuk, K. Kapłon, L. Mazur, D. Strzelecka, K. M. Pietrusiewicz. Tetrahedron72, 6668 (2016).10.1016/j.tet.2016.08.087Search in Google Scholar
[42] K. Szwaczko, O. M. Demchuk, B. Mirosław, D. Strzelecka, K. M. Pietrusiewicz. Tetrahedron Lett.57, 3491 (2016).10.1016/j.tetlet.2016.06.104Search in Google Scholar
[43] O. M. Demchuk, R. Jasiński. Phosphorus Sulfur Silicon Relat. El.191, 245 (2015).10.1080/10426507.2015.1064921Search in Google Scholar
[44] O. M. Demchuk, K. Kapłon, A. Kącka, K. M. Pietrusiewicz. Phosphorus Sulfur Silicon Relat. El.191, 180 (2016).10.1080/10426507.2015.1079197Search in Google Scholar
[45] O. M. Demchuk, R. Jasiński, K. M. Pietrusiewicz. Heteroatom Chem.26, 441 (2015).10.1002/hc.21279Search in Google Scholar
[46] O. M. Demchuk, K. Kielar, K. M. Pietrusiewicz. Pure and Appl. Chem.83, 633 (2011).10.1351/PAC-CON-10-08-06Search in Google Scholar
[47] V. Snieckus, O. Demchuk, B. Yoruk, T. Blackburn. Synlett.2006, 2908 (2006).10.1055/s-2006-951538Search in Google Scholar
[48] Z. Pakulski, O. M. Demchuk, J. Frelek, R. Luboradzki, K. M. Pietrusiewicz. Eur. J. Org. Chem.2004, 3913 (2004).10.1002/ejoc.200400186Search in Google Scholar
[49] M. L. Luetkens, A. P. Sattelberger, H. H. Murray, J. D. Basil, J. P. Fackler, R. A. Jones, D. E. Heaton. “Trimethylphosphine”, in Inorganic Syntheses: Reagents for Transition Metal Complex and Organometallic Syntheses, R. J. Angelici (Ed.), John Wiley & Sons, Inc., Hoboken, NJ (1990).10.1002/9780470132593.ch76Search in Google Scholar
[50] E. A. Standley, T. F. Jamison. J. Am. Chem. Soc.135, 1585 (2013).10.1021/ja3116718Search in Google Scholar
[51] C. A. Busacca, R. Raju, N. Grinberg, N. Haddad, P. James-Jones, H. Lee, J. C. Lorenz, A. Saha, C. H. Senanayake. J. Org. Chem.73, 1524 (2008).10.1021/jo7024064Search in Google Scholar
[52] G. C. Lloyd-Jones, N. P. Taylor. Chem. Eur. J.21, 5423 (2015).10.1002/chem.201406585Search in Google Scholar
[53] M. F. Hawthorne, W. L. Budde. J. Am. Chem. Soc.93, 3147 (1971).10.1021/ja00742a008Search in Google Scholar
[54] M. F. Hawthorne, D. E. Walmsley, W. L. Budde. J. Am. Chem. Soc.93, 3150 (1971).10.1021/ja00742a009Search in Google Scholar
[55] C. Carra, J. C. Scaiano. Eur. J. Org. Chem.2008, 4454 (2008).10.1002/ejoc.200800187Search in Google Scholar
[56] S. A. Genchur, G. L. Smith, H. C. Kelly. Can. J. Chem.49, 3165 (1971).10.1139/v71-527Search in Google Scholar
[57] Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O’Neill, R. A. DiStasio, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C. P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill, M. Head-Gordon. Phys. Chem. Chem. Phys.8, 3172 (2006).10.1039/B517914ASearch in Google Scholar
[58] P. Camps, G. Colet, S. Segura, S. Vazquez. Arkivoc.2007, 8 (2007).10.3998/ark.5550190.0008.402Search in Google Scholar
[59] O. V. Prezhdo, B. Gawdzik, V. V. Zubkova, V. V. Prezhdo. J. Mol. Struct.919, 146 (2009).10.1016/j.molstruc.2008.08.026Search in Google Scholar
[60] R. Jasiński, M. Kubik, A. Łapczuk-Krygier, A. Kącka, E. Dresler, A. Boguszewska-Czubara. React. Kinet. Mech. Catal.113, 333 (2014).10.1007/s11144-014-0753-8Search in Google Scholar
[61] R. Jasiński. Comp. Theor. Chem.1046, 93 (2014).10.1016/j.comptc.2014.08.002Search in Google Scholar
[62] O. M. Demchuk, W. Świerczynska, K. M. Pietrusiewicz, M. Woźnica, D. Wójcik, J. Frelek. Tetrahedron Asymm.19, 2339 (2008).10.1016/j.tetasy.2008.10.001Search in Google Scholar
[63] T. Imamoto, T. Oshiki, T. Onozawa, T. Kusumoto, K. Sato. J. Am. Chem. Soc.112, 5244 (1990).10.1021/ja00169a036Search in Google Scholar
[64] S. Lemouzy, D. H. Nguyen D. Gatineau, L. Giordano, D. Hérault, G. Buono. Pure Appl. Chem.88, 333 (2016).10.1515/pac-2016-0103Search in Google Scholar
[65] S. Lemouzy, D. H. Nguyen, V. Camy, M. Jean, D. Gatineau, L. Giordano, J.-V. Naubron, N. Vanthuyne, D. Hérault, G. Buono. Chem. Eur. J.21, 15607 (2015).10.1002/chem.201502647Search in Google Scholar
[66] B. Wolfe, T. Livinghouse. J. Am. Chem. Soc.120, 5116 (1998).10.1021/ja973685kSearch in Google Scholar
Supplemental Material
The online version of this article (DOI: https://doi.org/10.1515/pac-2017-0313) offers supplementary material, available to authorized users.
©2018 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers
- Papers from the 6th International IUPAC Conference on Green Chemistry (ICGC-6)
- Microwave assisted synthesis of glycerol carbonate from glycerol and urea
- Silica gel mediated oxidative C–O coupling of β-dicarbonyl compounds with malonyl peroxides in solvent-free conditions
- Definition of green synthetic tools based on safer reaction media, heterogeneous catalysis, and flow technology
- Heavy metal removal from waste waters by phosphonate metal organic frameworks
- A clean and simple method for deprotection of phosphines from borane complexes
- Development and treatment procedure of arsenic-contaminated water using a new and green chitosan sorbent: kinetic, isotherm, thermodynamic and dynamic studies
- Bio-adsorbent derived from papaya peel waste and magnetic nanoparticles fabricated for lead determination
- 5-Membered cyclic ethers via phenonium ion mediated cyclization through carbonate chemistry
- Synergy in food, energy and advanced materials production from biomass
- Step economy strategy for the synthesis of amphoteric aminoaldehydes, key intermediates for reduced hydantoins
- Separation technology meets green chemistry: development of magnetically recoverable catalyst supports containing silica, ceria, and titania
- Green chemistry and sustainable development: approaches to chemical footprint analysis
- Greener solvents for solid-phase organic synthesis
- Photocatalytic hydrogenolysis of allylic alcohols for rapid access to platform chemicals and fine chemicals
- IUPAC Recommendations
- Definition of the mole (IUPAC Recommendation 2017)
- Terminology of separation methods (IUPAC Recommendations 2017)
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers
- Papers from the 6th International IUPAC Conference on Green Chemistry (ICGC-6)
- Microwave assisted synthesis of glycerol carbonate from glycerol and urea
- Silica gel mediated oxidative C–O coupling of β-dicarbonyl compounds with malonyl peroxides in solvent-free conditions
- Definition of green synthetic tools based on safer reaction media, heterogeneous catalysis, and flow technology
- Heavy metal removal from waste waters by phosphonate metal organic frameworks
- A clean and simple method for deprotection of phosphines from borane complexes
- Development and treatment procedure of arsenic-contaminated water using a new and green chitosan sorbent: kinetic, isotherm, thermodynamic and dynamic studies
- Bio-adsorbent derived from papaya peel waste and magnetic nanoparticles fabricated for lead determination
- 5-Membered cyclic ethers via phenonium ion mediated cyclization through carbonate chemistry
- Synergy in food, energy and advanced materials production from biomass
- Step economy strategy for the synthesis of amphoteric aminoaldehydes, key intermediates for reduced hydantoins
- Separation technology meets green chemistry: development of magnetically recoverable catalyst supports containing silica, ceria, and titania
- Green chemistry and sustainable development: approaches to chemical footprint analysis
- Greener solvents for solid-phase organic synthesis
- Photocatalytic hydrogenolysis of allylic alcohols for rapid access to platform chemicals and fine chemicals
- IUPAC Recommendations
- Definition of the mole (IUPAC Recommendation 2017)
- Terminology of separation methods (IUPAC Recommendations 2017)