Individual variability in clinical effect and tolerability of opioid analgesics – Importance of drug interactions and pharmacogenetics
-
Vigdis Solhaug


Abstract
Background
As pain is often a comorbid condition, many patients use opioid analgesics in combination with several other drugs. This implies a generally increased risk of drug interactions, which along with inherent pharmacogenetic variability and other factors may cause differences in therapeutic response of opioids.
Aim
To provide an overview of interactions and pharmacogenetic variability of relevance for individual differences in effect and tolerability of opioid analgesics, which physicians and other healthcare professionals should be aware of in clinical practice.
Methods
The article was based on unsystematic searches in PubMed to identify literature highlighting the clinical impact of drug interactions and pharmacogenetics as sources of variable response of opioid analgesics.
Results
Cytochrome P450 (CYP)-mediated metabolism is an important process for both clinically relevant interactions and pharmacogenetic variability of several opioids. Concomitant use of CYP inhibitors (e.g. paroxetine, fluoxetine and bupropion) or inducers (e.g. carbamazepine, phenobarbital and phenytoin) could counteract the clinical effect or trigger side effects of analgesics in the same manner as genetically determined differences in CYP2D6-mediated metabolism of many opioids. Moreover, combination treatment with drugs that inhibit or induce P-glycoprotein (ABCB1), a blood-brain barrier efflux transporter, may alter the amount (‘dose’) of opioids distributed to the brain. At the pharmacodynamic level, it is crucial to be aware of the potential risk of interaction causing serotonergic syndrome when combining opioids and serotonergic drugs, in particular antidepressants inhibiting serotonin reuptake (SSRIs and SNRIs). Regarding pharmacogenetics at the receptor level of pain treatment, the knowledge is currently scarce, but an allelic variant of the μ1 opioid receptor (OPRM1) gene has been associated with higher dosage requirement to achieve analgesia.
Conclusions and implications
Drug interactions and pharmacogenetic differences may lead to therapeutic failure or serious side effects of opioid analgesics. Many interactions involve combinations with antidepressants and antiepileptics, which are highly relevant drugs in patients suffering from pain. To prevent unfavourable drug interactions it is important that clinicians pay close attention and use electronic drug interaction checkers when treatments are initiated or discontinued. For the management of issues related to pharmacogenetic differences, blood-based CYP genotyping is available as routine test at many laboratories, and provide a valuable tool for proper choice of drugs and doses for treatment of pain and other diseases.
1 Introduction
Pain is a common condition accompanying many different diseases. In the pharmacotherapeutic management of pain, drugs either act peripherally by reducing pain stimuli or centrally by suppressing the perception of pain. Opioids are key drugs used for suppressing pain perception. Although opioids generally are effective drugs, their use is associated with extensive individual differences in clinical effect and tolerability.
Individual differences in effect and tolerability of opioids may be the result of many different factors, including age, gender, organ functions, dietary habits, smoking habits, drug interactions and inherent pharmacogenetic differences. These factors may affect concentrations (pharmacokinetics) or receptor/target responsiveness (pharmacodynamics) of opioids. In later years, there has been a growing knowledge on the importance of drug interactions and pharmacogenetics for individual variability in therapeutic response of opioids. The aim of the present topical review was therefore to provide an overview of key drug interactions and pharmacogenetic differences affecting opioids, including possible actions to prevent therapeutic failure or adverse effects, which physicians and other healthcare professionals should be aware of in clinical practice. The review was based on literature identified by unsystematic searches in PubMed.
2 Drug interactions involving opioids
Drug interactions may alter pharmacokinetics (exposure) or pharmacodynamics (sensitivity) when medications are combined. For a drug-drug interaction pair, there will generally be one perpetrator drug creating the interaction and one victim drug being affected by the interaction. Occasionally, however, the drugs may interact mutually and therefore be perpetrators and victims at the same time.
Opioids are usually victims in pharmacokinetic interactions, but may act as perpetrators in pharmacodynamic interactions. The following subsections highlight the most relevant interactions involving opioids, i.e. cytochrome P450, P-glycoprotein and serotonergic interactions. The two former represent pharmacokinetic interactions, while the latter is pharmacodynamic in nature.
2.1 Cytochrome P450 interactions
Enzymes within the cytochrome P450 (CYP) super family are the most important ones in drug metabolism, and many opioids are subject to metabolism via CYP enzymes. The CYP enzymes mainly involved in metabolism of opioids are CYP2D6 and CYP3A4/5 [1]. Methadone, which has been tested in treatment of cancer pain [2], is mainly metabolized by CYP2B6, while CYP3A4/5 play a secondary role [3]. In Table 1, opioids being substrates of the various CYP enzymes are listed along with interacting inhibitors and inducers of the respective enzymes.
Overview of opioids that are substrates of cytochrome P450 (CYP) enzymes and/or P-glycoprotein, including inhibitors and inducers that may change exposure of the various opioids.
| Agents | CYP2B6 | CYP2D6 | CYP3A4/5 | Pgp |
|---|---|---|---|---|
| Opioid | Methadone | Codeine[a] | Alfentanil | Fentanyl |
| substrates | Hydrocodone[a] | Buphrenorphine | Loperamide | |
| Oxydocone | Codeine | Methadone | ||
| Tramadol[a] | Fentanyl | Morphine | ||
| Methadone | (Norbuprenorphine) | |||
| Tramadol | ||||
| Inhibitors | Memantine | Bupropion | Amiodarone | Amiloride |
| Sertraline | Citalopram | Amprenavir | Amiodarone | |
| Ticlopidine | Esciatlopram | Atazanavir | Atorvastatin | |
| Fluoxetine | Clarithromycin | Azithromycin | ||
| Levome- | Delavirdine | Clarithromycin | ||
| promazine | Diltiazem | Cyclosporine | ||
| Methadone | Efavirenz | Diltiazem | ||
| Quinidine | Erythromycin | Dronedarone | ||
| Paroxetine | Indinavir | Erytromycin | ||
| Ritonavir | Itraconazole | Esomeprazole | ||
| Terbinafine | Ketoconazole | Indinavir[b] | ||
| Lopinavir | Itrakonazol | |||
| Nelfinavir | Ketokonazol | |||
| Nevirapine | Klaritromycin | |||
| Posaconazole | Lanzoprazole | |||
| Ritonavir | Meflokin | |||
| Saquinavir | Nelfinavir[b] | |||
| Tipranavir | Omeprazole | |||
| Verapamil | Pantoprazole | |||
| Voriconazole | Ritonavir[b] | |||
| Grapefruit juice | Indinavir[b] | |||
| Simvastatin | ||||
| Sirolimus | ||||
| Probenecid | ||||
| Tacrolimus | ||||
| Telapravir | ||||
| Verapamil | ||||
| Inducers | Carbamazpine | None identified | Carbamazpine | Dexamethasone |
| Phenytoin | Phenytoin | Indinavir[b] | ||
| Phenobarbital | Phenobarbital | Nelfinavir[b] | ||
| Rifampicin | Rifampicin | Rifampicin | ||
| Hypericum p. | Ritonavir[9] | |||
| (St John’s wort) | Saquinavir[10] | |||
| Hypericum p. | ||||
| (StJohn’s wort) |
Interactions via CYP enzymes occur when a substrate of an enzyme is combined with an inhibitor or inducer of the same enzyme. Generally, combined use of inhibitor-substrate pairs of the same CYP enzyme, results in increased concentration (exposure) and reinforced clinical effect and side effect risk of the substrate. However, in cases where the substrate is a prodrug the opposite scenario will occur.
2.1.1 CYP inhibitors
Two of the most commonly used analgesic opioids, codeine and tramadol, are both prodrugs activated by the enzyme CYP2D6 [1] (Fig. 1). Codeine’s analgesics properties are due to its conversion via CYP2D6 to morphine, which has a 200-fold higher affinity for μ-opioid receptors than codeine, and subsequently via glucuronidation to morphine-6-glucuronide, a phase II metabolite with potent analgesic activity. Tramadol is also converted by CYP2D6-mediated oxidation to an active metabolite, O-desmethyltramadol. This metabolite is a high-affinity ligand and produce potent analgesia via opioid receptors [4]. Unmetabolized tramadol might have some impact on pain perception by inhibiting reuptake of the neurotransmitters serotonin and noradrenaline, but this mechanism is mainly responsible for adverse effects and unfavourable interactions (see Section 2.3).

Structural bio-transformation of the prodrugs codeine and tramadol to their active opioid metabolites via the enzyme CYP2D6, which differs in phenotype due to concomitant treatment with interacting enzyme inhibitors or inherent (pharmaogenetic) variability.
In the case of codeine, about 10-15% of the dose is normally converted by CYP2D6 to morphine, which is responsible for the analgesic effect. Thus, if patients are comedicated with CYP2D6 inhibitors, bioactivation of codeine to morphine will be reduced or totally blocked. The degree of interaction mainly depends on the CYP2D6 inhibitor potency, and inhibitors classified as the most potent ones comprise fluoxetine, paroxetine, bupropion and orally administered terbinafine, and antimycotic agent [5,6,7,8]. Coadministration of these potent inhibitors will completely block CYP2D6-mediated O-desmethylation of codeine to morphine (Fig. 1), while more moderate inhibitors, such as citalopram, escitalopram, methadone and levomepromazine will reduce bioactivation of codeine in a dose-dependent manner [5,9,10].
The interaction scenario described above for codeine is similar for tramadol, where combined use of CYP2D6 inhibitors results in reduced bioactivation to its active metabolite, O-desmethyltramadol (Fig. 1). However, due to the serotonergic properties of the parent drug, concurrent use of CYP2D6 inhibitors with tramadol both reduce the analgesic effect and increase the risk of serotonergic side effects. The latter might be severe if patients are simultaneously comedicated with other serotonergic drugs (see Section 2.3).
Oxycodone is metabolized by CYP2D6, but it is unclear to which extent the metabolite play a role for the overall analgesic effects [1,11]. Oxycodone is also a CYP3A4 substrate, and in cases where both inhibitors of CYP2D6 and CYP3A4 are used simultaneously with oxycodone, dangerous overdosing may occur. Typical CYP3A4 inhibitors include macrolide antibiotics (clarithromycin and erythromycin), azole antifungal agents (itraconzaole, fluconazole, ketoconazole, posaconazole and voriconazole) and grapefruit juice [12,13] (Table 1).
Fentanyl, alfentanil and methadone are other opioids subjected to metabolism via CYP3A4. For fentanyl, a potent opioid, systemic exposure during intravenous administration has been reported to increase 1.5-fold by potent CYP3A4 inhibitors [14], and several cases of respiratory depression with fatal or non-fatal outcomes of fentanyl in combination with CYP3A4 inhibitors have been reported [1516,17,18]. Methadone, on the other hand, is much less sensitive towards CYP3A4 inhibition, possibly due to the secondary role of this enzyme in its metabolism and involvement of multiple CYP. The risk of side effect of methadone in combination with CYP3A4 inhibitors is therefore limited, but some patients might be more vulnerable if other metabolic pathways are reduced, e.g. via CYP2B6. With regard to alfentanil, less is known about the effect of CYP3A4 inhibitors on change in exposure, but a case report has described a prolonged alfentanil effect during concurrent administration of erythromycin [19].
2.1.2 CYP inducers
While several compounds have been described as potent inducers of CYP3A4 metabolism, no inducers of CYP2D6 have so far been identified. This probably reflects that gene expression of CYP2D6 is non-inducible, which might be related to the strong and conserved impact of genetics on variability in CYP2D6 phenotype (see Section 3.1). In a clinical context, this means that drug interactions involving enzyme induction do not occur via CYP2D6, but is an issue of great importance for CYP3A4 metabolism.
Although CYP3A4 is not involved in the bioactivation of codeine and tramadol, concurrent use of CYP3A4 inducers will affect the levels of the active metabolites in terms of increased metabolism via inactivation pathways and thereby reduced availability of parent drugs for formation of active metabolites. The most clinically relevant CYP3A4 inducers include the antiepileptics carbamazepine, phenytoin and phenobarbital [20]. In addition, it is important to be aware of the CYP3A4-inducing effect of hypericum perforatum (St John’s wort) [21], a herbal drug used against depression.
The CYP3A4 inducers mentioned above will also substantially reduce exposure and clinical effect of fentanyl and oxycodone, which are inactivated by CYP3A4. In interaction studies, the dose requirements of fentanyl have been shown to be 1.5-2-fold higher combined with antiepileptic enzyme inducers [22], while the concentration of oxycodone is reported to be reduced by 80-90% in combination with the potent CYP3A4 inducer rifampicin [23]. For methadone respiratory depression has been reported after withdrawal of the enzyme inducer carbamazepine due to dose-escalation during combination therapy without corresponding dose reduction after discontinuation of the enzyme inducer [24]. Thus, despite that dose increases of the victim drug might be an option to manage the interaction, this could be a dangerous strategy because future discontinuation of the inducing drug will lead to overdosing if concomitant dose reductions (e.g. of the opioids) are not performed.
2.2 Permeability-glycoprotein interactions
Permeability-glycoprotein (P-glycoprotein) is an ATP-binding cassette-binding (ABC) efflux transporter that was originally discovered by Juliano and Ling in the 1970s as a key protein involved in multidrug resistance of anticancer agents [25]. Today, it is known that P-glycoprotein (ABCB1) is expressed in several biological interfaces/membrane barriers (Fig. 2), where it acts as pump restricting entry of many xenobiotics into the body and tissues that might be particularly vulnerable to drugs and other chemicals.
One of the key functions of P-glycoprotein is to limit the entry of xenobiotics into the brain, which may have an important impact of the exposure of centrally-acting drugs at their target site of action. For opioids that are P-glycoprotein substrates, i.e. fentanyl, loperamide, methadone and morphine [26], concurrent use of agents that inhibit or induce P-glycoprotein activity may alter the amount of the bioavailable dose reaching the brain. An overview of agents described as inhibitors or inducers of P-glycoprotein is provided in Table 1.
2.2.1 P-glycoprotein inhibitors
Several opioids are substrates of P-glycoprotein (Table 1). Among opioids that are P-glycoprotein substrates, loperamide is probably most effectively effluxed at the BBB. Due to the effective P-glycoprotein-mediated efflux, loperamide normally does not exhibit opioid effects in the brain, and is only used in treatment of diarrhoea to reduce GI motility, i.e. a kind of therapeutic application of a typical side effect. However, a study in healthy volunteers showed that combination of loperamide with the P-glycoprotein inhibitor quinidine resulted in moderate respiratory depression as an indication of loperamide-mediated stimulation of μ-opioid receptors in the brain [27]. This shows that loperamide, if combined with potent P-glycoprotein inhibitors, possess some risk of respiratory side effects, particularly in cases where another opioid is used concomitantly. Moreover, it is important to be aware that loperamide in combination with P-glycoprotein inhibitors may have some abuse potential [28].
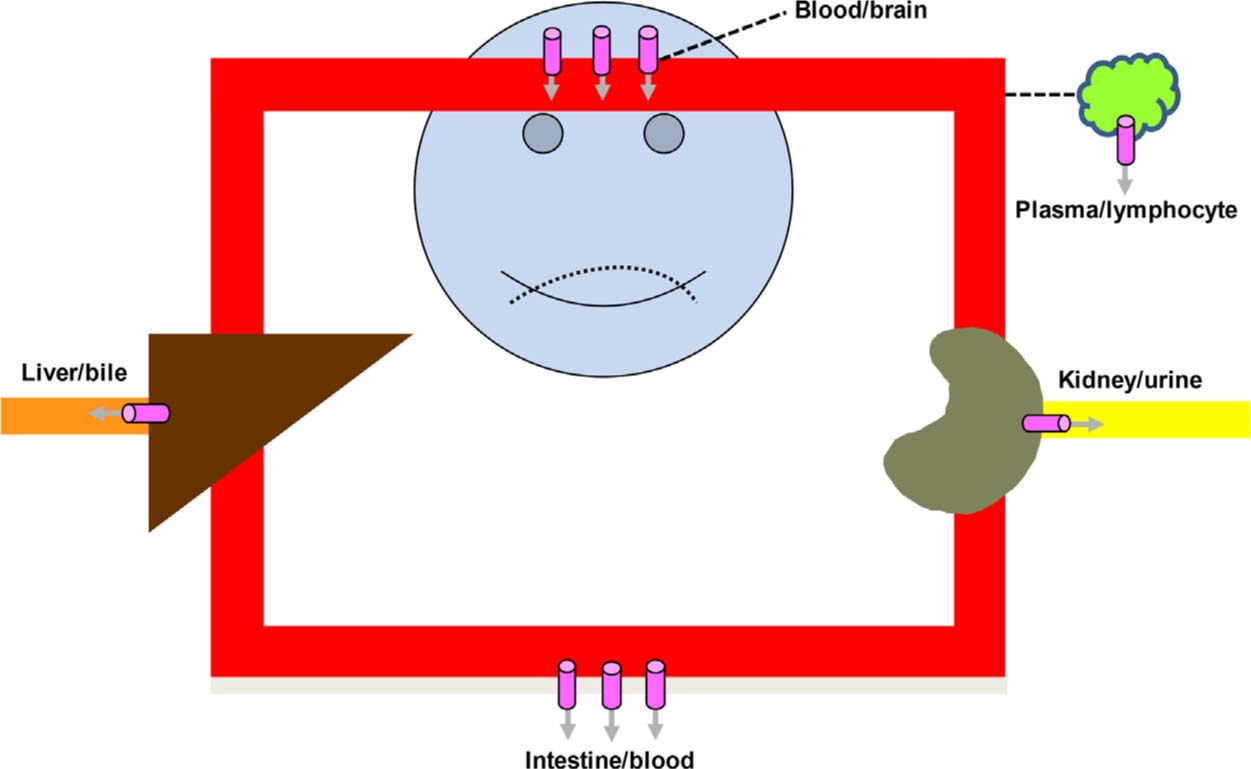
Localization of the efflux transporter P-glycoprotein ( ) in body interfaces (barriers).
) in body interfaces (barriers).
Not all opioid analgesics are P-glycoprotein substrates, and codeine, tramadol and oxycodone are examples not being effluxed by this transporter [26]. In the case of buprenorphine, the parent compound is not a P-glycoprotein substrate, but its CYP3A4-mediated metabolite, norbuprenorphine, is subjected to efflux by P-glycoprotein. Interestingly, norbuprenorphine is a potent respiratory depressor in rodents [29], which imply that concurrent use of P-glycoprotein inhibitors may increase the toxicity of buprenorphine therapy. This is supported by a study of Megarbane et al., where pupil diameter was shown to be significantly decreased without relevant alterations in plasma concentrations, when buprenorphine was coadministered with the P-glycoprotein inhibitor telaprevir [30].
2.2.2 P-glycoprotein inducers
While inhibition of P-glycoprotein will increase brain exposure of many opioids, the opposite is the case for P-glycoprotein induction. Increased expression of P-glycoprotein, possibly due to auto-induction over time, has been associated to development of central tolerance to opioids [28], although the evidence for this is limited. For well-characterized P-glycoprotein inducers, however, it is clear evidence that combination therapy over time will increase efflux of opioid being P-glycoprotein substrates. Carbamazepine is a particularly relevant inducer, as it used in treatment in neuropathic pain. Chronic administration of carbamazepine would be expected to double the expression of P-glycoprotein [31], and thereby substantially reduce entry of several opioids into the brain. Due to its ability to induce CYP3A4 metabolism as well, carbamazepine may decrease analgesic effects of several opioids via a dual interacting mechanism, and should generally be avoided in opioid-treated patients.
In a study using rats, it has been shown that administration of diclofenac, a non-steroidal anti-inflammatory drug (NSAID) upregulates P-glycoprotein expression and reduces both brain exposure and analgesic effect of morphine [32]. A similar effect on P-glycoprotein in the BBB in a rat model has also been reported after administration of paracetamol [33]. However, the molecular mechanism behind the upregulation of P-glycoprotein by diclofenac and paracetamol in the rat studies is not fully understood, and it is unclear to which extent NSAIDs and paracetamol might induce P-glycoprotein in humans at therapeutic doses. Thus, human studies on the possible P-glycoprotein-mediated interaction between opioids and NSAIDs/paracetamol are required before it could be considered of potential relevance for clinical practice.
2.3 Serotonergic interactions
Several opioids display serotonergic activity (Table 2). This might be of relevance for their clinical effect, but is mainly regarded as a safety issue due to the risk of serotonin syndrome. The risk is particularly of relevance in combination with other agents that increase serotonergic transmission in the brain (Table 2). Most antidepressants increase serotonergic transmission, and there are many reports of serotonergic syndrome when for example selective serotonin reuptake inhibitors (SSRIs) are combined with tramadol [34,35], fentanyl [36] or buprenorphine [37]. Among these tramadol featured as the most frequent drug involved in deaths due to serotonergic toxicity in an article by Pilgrim et al. summarizing cases reported on serotonergic syndrome [38]. Thus, tramadol should only be used with great caution in combination with SSRIs and serotonin-noradrenaline reuptake inhibitors (SNRIs).
Opioids and other agents with serotonergic activities that in combination may trigger serotonin syndrome.
| Drug class | Substances |
|---|---|
| Opioids | Buprenorphine, cyclobenzaprine, fentanyl, hydrocodone, meperidine, morphine, oxycodone, pethidine, pentazocine, tramadol |
| Selective serotonin reuptake inhibitors (SSRI) | Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline |
| Serotonin and noradrenaline reuptake inhibitors (SNRI) | Duloxetine, venlafaxine |
| Tricyclic antidepressants (TCA) | Amitriptyline, clomipramine, desipramine, doxepine, imipramine, trimipramine |
| Selective MAO-A hemmere | Moclobemide |
| Selective MAO-B inhibitors | Rasagiline, selegiline |
| CNS stimulants | Amphetamine, methylphenidate, 3,4-methylenedioxymethamphetamine (MDMA; ecstasy) |
| Antibiotics | Linezolid |
| Triptans | Almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan, zolmitriptan |
| Others | St John’s wort (hypericum perforatum), metoclopramide, psychedelics (LSD) |
The risk of serotonergic interactions depends on treatment intensity of the involved drugs. Thus, dosing regimens of the interacting drugs are of relevance for the interaction risk. The risk of trigging a serotonergic syndrome in combination with SSRIs or SNRI is likely to be higher at increasing daily dosing of tramadol, but in case reports tramadol doses have ranged between 50 and 800 mg/day [35]. Interestingly, serotonergic syndrome has also been reported for tramadol in a case of monotherapy where 200 mg was taken orally four hours before admission with severe symptoms [39]. This illustrates the pronounced risk of serotonergic side effects associated with use tramadol, and it is crucial that physicians are aware of this issue when prescribing tramadol in combination with other drugs with serotonergic activity.
3 Pharmacogenetics affecting opioid response
Several genes encoding different receptors, transporters and enzymes involved in the mechanisms of action and metabolism of opioids have been studied in the effort to detect genetic variation responsible for the heterogeneity in the response to opioids (gene-drug interactions). Gene-drug interactions are in many ways similar to drug-drug interactions in terms pharmacological mechanisms and clinical consequences. Gene-drug interactions also affect pharmacokinetic and pharmacodynamic variability of opioids and may have strong impact on clinical effect and tolerability. The below sections provide brief overviews of selected pharmacological proteins where inherent mutations in the encoding genes have been associated with individual variability in pharmacokinetics or pharmacodynamics of opioids.
3.1 CYP enzymes
Among several enzymes involved in the metabolism of opioids, CYP2D6 is the most important related to pharmacogenetic variation. Genotype of CYP2D6 strongly predicts metabolic phenotype and divides the population into inherent metabolizer subgroups, i.e. poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs) and ultra-rapid metabolizers (UMs). The distribution of various inherent metabolizer subpopulations differs substantially between ethnic groups. In Caucasians, 5-10% and 1-2% of the population are PMs and UMs, respectively, while approximately opposite distributions are present among Africans [4].
The weak opioids, codeine and tramadol, are both metabolized by CYP2D6 to the active compounds mediating their main analgesic effect (Fig. 1). While CYP2D6 PMs will not obtain sufficient analgesic effect of codeine and tramadol, the UM subpopulation are at risk of excessive formation of active metabolites [4]. The latter has been highlighted in several case reports, where increased conversion to primary, active metabolites has been accompanied by immature or reduced renal function resulting in accumulation of subsequent active phase II metabolites (glucuronides) and respiratory toxicity. The most tragic cases were a newborn baby who died from breastfeeding of a codeine-treated mother being CYP2D6 UM [40], and a 2-year-old boy who died of standard codeine doses, after tonsillectomy [41]. Another child, also ultra-rapid metabolizer, developed respiratory depression after taking tramadol for pain relief related to tonsillectomy [42]. Cases of respiratory toxicity of codeine and tramadol have also been described in elderly CYP2D6 UMs with reduced renal function [43,44]. In one of the latter cases, concurrent use of CYP3A4 inhibitors worsened the risk, as more of the prodrug was available for activation via the ultra-rapid CYP2D6 pathway [43].
While genetic polymorphisms in the CYP3A4 gene have minor impact on metabolic activity, the sister enzyme, CYP3A5, is strongly determined by genetics. CYP3A5 metabolizes many of the same agents as CYP3A4, but CYP3A5 is only expressed in the minority of most ethnic populations. In Caucasians, 5-10% express CYP3A5, while higher proportions of expressers are present in Asians and Africans. The functional consequence of CYP3A5 genetics is that those expressing the enzyme display a rapid CYP3A phenotype. Several opioids are substrates of CYP3A4/5, and patients expressing CYP3A5 therefore obtain lower levels of drugs such as fentanyl, alfentanil and methadone than those lacking enzyme function [45,46,47]. Although this may have an influence on effect and tolerability of respective drugs, the literature is too scarce for firm statements of the clinical relevance of CYP3A5 genetics for therapeutic response of affected opioids.
3.2 P-glycoprotein transporter
Individual variability in P-glycoprotein expression is extensive [48,49], which may affect entry of many opioids and other drugs into the brain [28]. Many studies have investigated the phenotypic impact of mutations in the ABCB1 gene encoding P-glycoprotein, but most of them have reported conflicting findings. About 50 SNPs (single nucleotide polymorphisms) in the ABCB1 gene have been identified, and a complicating issue is that many of these exist on different haplotypes [50]. Thus, investigating the phenotypic impact of one or a few SNPs limits the validity of the findings.
The three SNPs that have attracted most attention in pharmacogenetic studies on P-glycoprotein phenotype are 1236C> T (rs1128503), 3435C> T (rs1045642), and 2677G> T/A (rs2032582). These SNPs has been associated with lower dosage requirement, as well as higher frequencies of opioid-related adverse events [48,49]. However, findings from studies performed on these three SNPs are inconsistent, and the current knowledge is therefore not sufficient for them to be implemented as clinical routine analyses.
Recently, a less investigated and less frequent SNP in the ABCB1 gene (4036A>G, rs9282564) was significantly associated with increased risk of respiratory depression in morphine-treated children undergoing elective outpatient tonsillectomy [51]. This could have implication for management of postoperative pain in paediatric patients, but additional validation of this finding is needed. Hopefully, future studies of this and other SNPs in the gene encoding the P-glycoprotein transporter will provide knowledge that can be implemented in clinical practice for improvement of opioid treatment and tolerability.
3.3 μ-Opioid receptor
The gene coding for the μ-opioid receptor, OPRM1, has gained great interest as a potential source of variable opioid response. A genetic variant in OPRM1 with impact on the receptor function is the SNP 118A>G (rs1799971). The nucleotide switch from adenine (A) to guanine (G) in position 118 in exon 1 of the OPRM1 gene results in an amino acid change in the extracellular part of the protein, which seems to reduce receptor binding of opioids. In some clinical trials, mainly postoperative pain studies with morphine or fentanyl, carriers of the G allele have been reported to require higher doses to achieve sufficient analgesic effect compared to patients homozygous for the A allele (AA, wildtype) [49].
The 118A> G SNP has a frequency of around 15% in Caucasians, 1% in Africans an up to 40% in Asians [52]. Thus, the frequencies of homozygous carriers of the mutated G allele (GG) are about 0.1% in Africans, 2-3% in Caucasians and 15% in Asians. The higher frequency of this variant in Asians is probably the reason why the clinical impact of this SNP apparently is strongest in this population. This was shown in a meta-analysis by Hwang et al. where they included 18 studies that explored the impact of the OPRM1 118A> G polymorphism on postoperative opioid requirement. The G allele was associated with higher opioid requirement, especially in Asian patients [53]. In another, more recent meta-analysis, including 29 studies, it was also found that patients homozygous of the mutated G allele required significantly higher doses of post-operative opioids than homozygous carriers of wild type A allele [54].
Most of the studies on OPRM1 genotype are related to opioid requirement for surgical or postsurgical pain management, and the two meta-analyses referred above focused on opioid dose requirements for acute pain. There are also studies relating OPRM1 genotypes to opioid requirement in patient with chronic pain or cancer pain, but these results are more inconsistent. This probably reflects the complexity of pain, that the nature of pain varies between for instance postoperative pain and cancer pain, and that other factors, such as tolerance mechanism, are more important for dose requirement than genetic variability during longer treatment periods.
4 Actions to prevent problems due to interactions and pharmacogenetic variability
Concomitant use of drugs interacting with opioids and individual pharmacogenetic differences affecting their response may have major impact on clinical effect and safety of the treatment. In order to prevent unfavourable therapeutic outcome, it is important that physicians and other health care professionals are aware of potential problems and practical solutions on how to prevent unwanted reactions.
4.1 Management of drug interactions involving opioid analgesics
Many countries, including Sweden, Denmark and Norway, have developed drug interactions databases, which are freely available via Internet [55,56,57]. Identification of drug interactions could easily be achieved by uploading/writing the patient’s list of prescribed drugs into the search function of the databases. If drug interactions are present within the medication regimen, hits on interacting drugs will appear on the screen with a short description of mechanism and clinical relevance, including recommendations on how the interaction should be managed in clinical practice (e.g. dose adjustments or use of alternative drugs).
Several electronic medical record systems have interaction databases to provide automatic alerts during drug prescription. This simplifies the management of drug interactions, but may also imply a risk that important interactions are overlooked if there are too many alerts. Moreover, as interactions are relevant both when drugs are initiated and discontinued, automatic alerts will miss the latter. Thus, it will often be valuable to check for potential interactions manually in electronic databases when drugs are initiated or discontinued.
4.2 Management of pharmacogenetic variability affecting opioid analgesics
In cases where interindividual variability in drug response is determined by pharmacogenetic factors, it will be necessary to have access to the patient’s genotype to make treatment adjustments. Many Scandinavian hospitals offer blood sample-based genotyping of cytochrome P450 enzymes where inherent mutations in encoding genes determine drug metabolic capacity. The coverage of the various analytical methods is fairly the same at different laboratories, but the extent to which the analytical results are interpreted to be clinically useful vary between laboratories. Analysis rates and funding arrangements for genotyping may vary between different countries, but prices will usually be regulated by public authorities. In Norway, genotyping of CYP2D6, which is generally most relevant for opioids, currently has a total cost of about 600 NOK, i.e. approximately equal to the current monthly treatment cost of 40 mg oxycodone administered as depot tablets once daily.
For patients where long-term treatment with opioid analgesics is expected, it is our opinion that CYP genotyping should be considered as a routine test during treatment initiation to optimize selection of drug type and dose intensity for each patient. The indication for CYP genotyping is particularly strong for vulnerable patient groups with multiple risk factors for adverse side effects, i.e. children, elderly, patients with renal failure decline and pregnant or breast-feeding women. Importantly, the CYP genotyping test result can be used in a lifelong perspective to adjust drug selection and dose across different therapeutic areas, since these enzymes metabolize a wide range of agents. For example, if a patient has been genotyped as a CYP2D6 poor metabolizer (PM), and hence will not respond to the prodrug codeine for pain relief, he or she will also be at high risk of adverse effects of drugs primarily metabolized to inactive metabolites by CYP2D6 (e.g. some beta blockers, antidepressants and antipsychotics).
While CYP genotyping is widespread as a routine test available in clinical practice, genotyping of OPRM1 has so far mainly been performed in experimental studies. However, recently OPRM1 genotyping was established as a routine test at the authors’ institution (Diakonhjemmet Hospital in Oslo).The experience with OPRM1 genotyping is so far limited, but is expected to be mainly used in association with postoperative treatment with opioids. In this context, OPRM1 genotyping can predict dose requirement of opioids for proper pain control.
5 Summary and conclusion
Drug interactions and pharmacogenetic differences represent major sources to individual variability in clinical effect and safety of opioids. It is crucial that physicians and health care professionals are aware of these issues and make preventive actions to avoid therapeutic failure and adverse reactions of opioids. An important action is to check for drug interactions and make possible adjustments when treatment changes are performed (drug initiation or discontinuation). Moreover, blood-based genotyping of CYP enzymes, and in particular CYP2D6, may provide valuable information in a lifelong perspective for individualization of treatment with opioids and many other drugs from various therapeutic areas. Costs for CYP genotyping are limited compared to resources required in the follow-up of chronic pain patients and should therefore be considered as a routine test when long-term treatment with opioid analgesics is expected.
DOI of refers to article: http://dx.doi.org/10.1016/j.sjpain.2017.09.023.
-
Ethical issues: In line with its status as a review article, this project did not require any approval from an ethical committee.
-
Conflicts of interest: The authors work in a hospital department performing pharmacogenetic analyses as a public-funded service for clinicians, but have no other conflicts of interest to declare in relation to this paper.
References
[1] Smith HS. The metabolism of opioid agents and the clinical impact of their active metabolites. ClinJ Pain 2011;27:824–38.Suche in Google Scholar
[2] Nicholson AB, Watson GR, Derry S, Wiffen PJ. Methadone for cancer pain. Cochrane Database Syst Rev 2017;2.CD003971.Suche in Google Scholar
[3] Kringen MK, Chalabianloo F, Bernard JP, Bramness JG, Molden E, Hoiseth G. The combined effect of CYP2B6 genotype and other candidate genes on a steady-state serum concentration of methadone in opioid maintenance treatment. Ther Drug Monit 2017;39:550–5.Suche in Google Scholar
[4] Crews KR, Gaedigk A, Dunnenberger HM, Leeder JS, Klein TE, Caudle KE, Haidar CE, Shen DD, Callaghan JT, Sadhasivam S, Prows CA, Kharasch ED, Skaar TC, Clinical Pharmacogenetics Implementation C. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther 2014;95:376–82.Suche in Google Scholar
[5] Molden E, Spigset O. [Interactions between metoprolol and antidepressants]. Tidsskr Nors Laegeforen 2011;131:1777–9.Suche in Google Scholar
[6] Mannheimer B, von Bahr C, Pettersson H, Eliasson E. Impact of multiple inhibitors or substrates of cytochrome P450 2D6 on plasma risperidone levels in patients on polypharmacy. Ther Drug Monit 2008;30:565–9.Suche in Google Scholar
[7] Abdel-Rahman SM, Marcucci K, Boge T, Gotschall RR, Kearns GL, Leeder JS. Potent inhibition of cytochrome P-450 2D6-mediated dextromethorphan O-demethylation by terbinafine. Drug Metab Dispos 1999;27:770–5.Suche in Google Scholar
[8] Saarikoski T, Saari TI, Hagelberg NM, Backman JT, Neuvonen PJ, Scheinin M, Olkkola KT, Laine K. Effects of terbinafine and itraconazole on the pharmacokinetics of orally administered tramadol. Eur J Clin Pharmacol 2015;71:321–7.Suche in Google Scholar
[9] Coller JK, Michalakas JR, James HM, Farquharson AL, Colvill J, White JM, Somogyi AA. Inhibition of CYP2D6-mediated tramadol O-demethylation in methadone but not buprenorphine maintenance patients. Br J Clin Pharmacol 2012;74:835–41.Suche in Google Scholar
[10] Vevelstad M, Pettersen S, Tallaksen C, Brors O. O-demethylation of codeine to morphine inhibited by low-dose levomepromazine. Eur J Clin Pharmacol 2009;65:795–801.Suche in Google Scholar
[11] Soderberg Lofdal KC, Andersson ML, Gustafsson LL. Cytochrome P450-mediated changes in oxycodone pharmacokinetics/pharmacodynamics and their clinical implications. Drugs 2013;73:533–43.Suche in Google Scholar
[12] Hiemke C, Baumann P, Bergemann N, Conca A, Dietmaier O, Egberts K, Fric M, Gerlach M, Greiner C, Grunder G, Haen E, Havemann-Reinecke U, Jaquenoud Sirot E, Kirchherr H, Laux G, Lutz UC, Messer T, Muller MJ, Pfuhlmann B, Rambeck B, Riederer P, Schoppek B, Stingl J, Uhr M, Ulrich S, Waschgler R, Zernig G. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry 2011;44:195–235.Suche in Google Scholar
[13] Nieminen TH, Hagelberg NM, Saari TI, Neuvonen M, Neuvonen PJ, Laine K, Olkkola KT. Grapefruit juice enhances the exposure to oral oxycodone. Basic Clin Pharmacol Toxicol 2010;107:782–8.Suche in Google Scholar
[14] Saari TI, Laine K, Neuvonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole and fluconazole on the pharmacokinetics of intravenous fentanyl. Eur J Clin Pharmacol 2008;64:25–30.Suche in Google Scholar
[15] Cronnolly B, Pegrum H. Fentanyl-clarithromycin interaction. BMJ Case Rep 2012;2012.Suche in Google Scholar
[16] Horton R, Barber C. Opioid-induced respiratory depression resulting from transdermal fentanyl-clarithromycin drug interaction in a patient with advanced COPD. J Pain Symptom Manag 2009;37:e2–5.Suche in Google Scholar
[17] Mercadante S, Villari P, Ferrera P. Itraconazole-fentanyl interaction in a cancer patient. J Pain Symptom Manag 2002;24:284–6.Suche in Google Scholar
[18] Hallberg P, Marten L, Wadelius M. Possible fluconazole-fentanyl interaction - a case report. Eur J Clin Pharmacol 2006;62:491–2.Suche in Google Scholar
[19] Bartkowski RR, McDonnell TE. Prolonged alfentanil effect following erythromycin administration. Anesthesiology 1990;73:566–8.Suche in Google Scholar
[20] Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol 2006;61:246–55.Suche in Google Scholar
[21] Roby CA, Anderson GD, Kantor E, Dryer DA, Burstein AH. St John’s Wort: effect on CYP3A4 activity. Clin Pharmacol Ther 2000;67:451–7.Suche in Google Scholar
[22] Tempelhoff R, Modica PA, SpitznagelJr EL. Anticonvulsant therapy increases fentanyl requirements during anaesthesia for craniotomy. Can J Anaesth 1990;37:327–32.Suche in Google Scholar
[23] The Norwegian Medicines Agency. Summary of product characteristics (SPC), Oxycodone Actavis [last update: 03.01.17].Suche in Google Scholar
[24] Benitez-Rosario MA, Salinas Martin A, Gomez-Ontanon E, Feria M. Methadone-induced respiratory depression after discontinuing carbamazepine administration. J Pain Symptom Manag 2006;32:99–100.Suche in Google Scholar
[25] Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamsterovary cell mutants. Biochim Biophys Acta 1976;455:152–62.Suche in Google Scholar
[26] Drewes AM, Jensen RD, Nielsen LM, Droney J, Christrup LL, Arendt-Nielsen L, Riley J, Dahan A. Differences between opioids: pharmacological, experimental, clinical and economical perspectives. Br J Clin Pharmacol 2013;75:60–78.Suche in Google Scholar
[27] Sadeque AJ, Wandel C, He H, Shah S, Wood AJ. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther 2000;68:231–7.Suche in Google Scholar
[28] Mercer SL, Coop A. Opioid analgesics and P-glycoprotein efflux transporters: a potential systems-level contribution to analgesic tolerance. Curr Top Med Chem 2011;11:1157–64.Suche in Google Scholar
[29] Alhaddad H, Cisternino S, Decleves X, Tournier N, Schlatter J, Chiadmi F, Risede P, Smirnova M, Besengez C, Scherrmann JM, Baud FJ, Megarbane B. Respiratory toxicity of buprenorphine results from the blockage of P-glycoprotein-mediated efflux of norbuprenorphine at the blood-brain barrier in mice. Crit Care Med 2012;40:3215–23.Suche in Google Scholar
[30] Megarbane B, Alhaddad H, Decleves X. Reduced pupil diameter in volunteers on stable buprenorphine maintenance therapy with telaprevir: a drug-drug interaction involving p-glycoprotein at the blood-brain barrier? Antimicrob Agents Chemother 2012;56:6070.Suche in Google Scholar
[31] McCance-Katz EF, Sullivan LE, Nallani S. Drug interactions of clinical importance among the opioids, methadone and buprenorphine, and other frequently prescribed medications: a review. AmJ Addict 2010;19:4–16.Suche in Google Scholar
[32] Sanchez-Covarrubias L, Slosky LM, Thompson BJ, Zhang Y, Laracuente ML, DeMarco KM, Ronaldson PT, Davis TP. P-glycoprotein modulates morphine uptake into the CNS: a role for the non-steroidal anti-inflammatory drug diclofenac. PLOS ONE 2014;9:e88516.Suche in Google Scholar
[33] Slosky LM, Thompson BJ, Sanchez-Covarrubias L, Zhang Y, Laracuente ML, Vanderah TW, Ronaldson PT, Davis TP. Acetaminophen modulates P-glycoprotein functional expression at the blood-brain barrier by a constitutive androstane receptor-dependent mechanism. Mol Pharmacol 2013;84:774–86.Suche in Google Scholar
[34] Sansone RA, Sansone LA. Tramadol: seizures, serotonin syndrome, and coadministered antidepressants. Psychiatry (Edgmont (Pa: Township)) 2009;6:17–21.Suche in Google Scholar
[35] Nelson EM, Philbrick AM. Avoiding serotonin syndrome: the nature of the interaction between tramadol and selective serotonin reuptake inhibitors. Ann Pharmacother 2012;46:1712–6.Suche in Google Scholar
[36] Greenier E, Lukyanova V, Reede L. Serotonin syndrome: fentanyl and selective serotonin reuptake inhibitor interactions. AANA J 2014;82:340–5.Suche in Google Scholar
[37] Isenberg D, Wong SC, Curtis JA. Serotonin syndrome triggered by a single dose of suboxone. Am J Emerg Med 2008;26, 840.e3-5.Suche in Google Scholar
[38] Pilgrim JL, Gerostamoulos D, Drummer OH. Deaths involving serotonergic drugs. Forensic Sci Int 2010;198:110–7.Suche in Google Scholar
[39] Aminiahidashti H, Shafiee S, Mousavi SJ, Hajiaghaei G. Tramadol pill alone may cause serotoninsyndrome. Chin MedJ 2016;129:877–8.Suche in Google Scholar
[40] Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006;368:704.Suche in Google Scholar
[41] Ciszkowski C, Madadi P, Phillips MS, Lauwers AE, Koren G. Codeine, ultrarapid-metabolism genotype, and postoperative death. N Engl J Med 2009;361:827–8.Suche in Google Scholar
[42] Orliaguet G, Hamza J, Couloigner V, Denoyelle F, Loriot MA, Broly F, Garabedian EN. A case of respiratory depression in a child with ultrarapid CYP2D6 metabolism after tramadol. Pediatrics 2015;135:e753–5.Suche in Google Scholar
[43] Stamer UM, Stuber F, Muders T, Musshoff F. Respiratory depression with tramadol in a patient with renal impairment and CYP2D6 gene duplication. Anesth Analg 2008;107:926–9.Suche in Google Scholar
[44] Gasche Y, Daali Y, Fathi M, Chiappe A, Cottini S, Dayer P, Desmeules J. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med 2004;351:2827–31.Suche in Google Scholar
[45] Takashina Y, Naito T, Mino Y, Yagi T, Ohnishi K, Kawakami J. Impact of CYP3A5 and ABCB1 gene polymorphisms on fentanyl pharmacokinetics and clinical responses in cancer patients undergoing conversion to a transdermal system. Drug Metab Pharmacokinet 2012;27:414–21.Suche in Google Scholar
[46] Klees TM, Sheffels P, Thummel KE, Kharasch ED. Pharmacogenetic determinants of human liver microsomal alfentanil metabolism and the role of cytochrome P450 3A5. Anesthesiology 2005;102:550–6.Suche in Google Scholar
[47] De Fazio S, Gallelli L, De Siena A, De Sarro G, Scordo MG. Role of CYP3A5 in abnormal clearance of methadone. Ann Pharmacother 2008;42:893–7.Suche in Google Scholar
[48] Matic M, de Wildt SN, Tibboel D, van Schaik RHN. Analgesia and opioids: a pharmacogenetics shortlist for implementation in clinical practice. Clin Chem 2017;63:1204–13.Suche in Google Scholar
[49] Nielsen LM, Olesen AE, Branford R, Christrup LL, Sato H, Drewes AM. Association between human pain-related genotypes and variability in opioid analgesia: an updated review. Pain Pract 2015;15:580–94.Suche in Google Scholar
[50] Kroetz DL, Pauli-Magnus C, Hodges LM, Huang CC, Kawamoto M, Johns SJ, Stryke D, Ferrin TE, DeYoung J, Taylor T, Carlson EJ, Herskowitz I, Giacomini KM, Clark AG. Sequence diversity and haplotype structure in the human ABCB1 (MDR1, multidrug resistance transporter) gene. Pharmacogenetics 2003;13: 481-94.Suche in Google Scholar
[51] Sadhasivam S, Chidambaran V, Zhang X, Meller J, Esslinger H, Zhang K, Martin LJ, McAuliffe J. Opioid-induced respiratory depression: ABCB1 transporter pharmacogenetics. Pharmacogenomics J 2015;15:119–26.Suche in Google Scholar
[52] National Center for Biotechnology Information, National Library of Medicine. Database of single nucleotide polymorphisms (dbSNP). Available from:https://www.ncbi.nlm.nih.gov/projects/SNP/ [accessed August 2017].Suche in Google Scholar
[53] Hwang IC, Park JY, Myung SK, Ahn HY, Fukuda K, Liao Q. OPRM1 A118G gene variant and postoperative opioid requirement: a systematic review and metaanalysis. Anesthesiology 2014;121:825–34.Suche in Google Scholar
[54] Choi SW, Lam DM, Wong SS, Shiu HH, Wang AX, Cheung CW. Effects of single nucleotide polymorphisms on surgical and post-surgical opioid requirements - a systematic review and meta-analysis. Clin J Pain 2017,http://dx.doi.org/10.1097/AJP.0000000000000498.Suche in Google Scholar
[55] The Norwegian Medicines Agency.www.legemiddelinteraksjoner.no.Suche in Google Scholar
[56] Danish Medicines Agency.http://www.interaktionsdatabasen.dk/.Suche in Google Scholar
[57] Janusinfo, Drug Therapeutic Committee and the Health and Medical Care Administration of the Stockholm County Council, Sweden.http://www.janusinfo.se.Suche in Google Scholar
© 2017 Scandinavian Association for the Study of Pain
Artikel in diesem Heft
- Observational study
- Perceived sleep deficit is a strong predictor of RLS in multisite pain – A population based study in middle aged females
- Clinical pain research
- Prospective, double blind, randomized, controlled trial comparing vapocoolant spray versus placebo spray in adults undergoing intravenous cannulation
- Clinical pain research
- The Functional Barometer — An analysis of a self-assessment questionnaire with ICF-coding regarding functional/activity limitations and quality of life due to pain — Differences in age gender and origin of pain
- Clinical pain research
- Clinical outcome following anterior arthrodesis in patients with presumed sacroiliac joint pain
- Observational study
- Chronic disruptive pain in emerging adults with and without chronic health conditions and the moderating role of psychiatric disorders: Evidence from a population-based cross-sectional survey in Canada
- Educational case report
- Management of patients with pain and severe side effects while on intrathecal morphine therapy: A case study
- Clinical pain research
- Behavioral inhibition, maladaptive pain cognitions, and function in patients with chronic pain
- Observational study
- Comparison of patients diagnosed with “complex pain” and “somatoform pain”
- Original experimental
- Patient perspectives on wait times and the impact on their life: A waiting room survey in a chronic pain clinic
- Topical review
- New evidence for a pain personality? A critical review of the last 120 years of pain and personality
- Clinical pain research
- A multi-facet pain survey of psychosocial complaints among patients with long-standing non-malignant pain
- Clinical pain research
- Pain patients’ experiences of validation and invalidation from physicians before and after multimodal pain rehabilitation: Associations with pain, negative affectivity, and treatment outcome
- Observational study
- Long-term treatment in chronic noncancer pain: Results of an observational study comparing opioid and nonopioid therapy
- Clinical pain research
- COMBAT study – Computer based assessment and treatment – A clinical trial evaluating impact of a computerized clinical decision support tool on pain in cancer patients
- Original experimental
- Quantitative sensory tests fairly reflect immediate effects of oxycodone in chronic low-back pain
- Editorial comment
- Spatial summation of pain and its meaning to patients
- Original experimental
- Effects of validating communication on recall during a pain-task in healthy participants
- Original experimental
- Comparison of spatial summation properties at different body sites
- Editorial comment
- Behavioural inhibition in the context of pain: Measurement and conceptual issues
- Clinical pain research
- A randomized study to evaluate the analgesic efficacy of a single dose of the TRPV1 antagonist mavatrep in patients with osteoarthritis
- Editorial comment
- Quantitative sensory tests (QST) are promising tests for clinical relevance of anti–nociceptive effects of new analgesic treatments
- Educational case report
- Pregabalin as adjunct in a multimodal pain therapy after traumatic foot amputation — A case report of a 4-year-old girl
- Editorial comment
- Severe side effects from intrathecal morphine for chronic pain after repeated failed spinal operations
- Editorial comment
- Opioids in chronic pain – Primum non nocere
- Editorial comment
- Finally a promising analgesic signal in a long-awaited new class of drugs: TRPV1 antagonist mavatrep in patients with osteoarthritis (OA)
- Observational study
- The relationship between chronic musculoskeletal pain, anxiety and mindfulness: Adjustments to the Fear-Avoidance Model of Chronic Pain
- Clinical pain research
- Opioid tapering in patients with prescription opioid use disorder: A retrospective study
- Editorial comment
- Sleep, widespread pain and restless legs — What is the connection?
- Editorial comment
- Broadening the fear-avoidance model of chronic pain?
- Observational study
- Identifying characteristics of the most severely impaired chronic pain patients treated at a specialized inpatient pain clinic
- Editorial comment
- The burden of central anticholinergic drugs increases pain and cognitive dysfunction. More knowledge about drug-interactions needed
- Editorial comment
- A case-history illustrates importance of knowledge of drug-interactions when pain-patients are prescribed non-pain drugs for co-morbidities
- Editorial comment
- Why can multimodal, multidisciplinary pain clinics not help all chronic pain patients?
- Topical review
- Individual variability in clinical effect and tolerability of opioid analgesics – Importance of drug interactions and pharmacogenetics
- Editorial comment
- A new treatable chronic pain diagnosis? Flank pain caused by entrapment of posterior cutaneous branch of intercostal nerves, lateral ACNES coined LACNES
- Clinical pain research
- PhKv a toxin isolated from the spider venom induces antinociception by inhibition of cholinesterase activating cholinergic system
- Clinical pain research
- Lateral Cutaneous Nerve Entrapment Syndrome (LACNES): A previously unrecognized cause of intractable flank pain
- Editorial comment
- Towards a structured examination of contextual flexibility in persistent pain
- Clinical pain research
- Context sensitive regulation of pain and emotion: Development and initial validation of a scale for context insensitive avoidance
- Editorial comment
- Is the search for a “pain personality” of added value to the Fear-Avoidance-Model (FAM) of chronic pain?
- Editorial comment
- Importance for patients of feeling accepted and understood by physicians before and after multimodal pain rehabilitation
- Editorial comment
- A glimpse into a neglected population – Emerging adults
- Observational study
- Assessment and treatment at a pain clinic: A one-year follow-up of patients with chronic pain
- Clinical pain research
- Randomized, double-blind, placebo-controlled, dose-escalation study: Investigation of the safety, pharmacokinetics, and antihyperalgesic activity of L-4-chlorokynurenine in healthy volunteers
- Clinical pain research
- Prevalence and characteristics of chronic pain: Experience of Niger
- Observational study
- The use of rapid onset fentanyl in children and young people for breakthrough cancer pain
- Original experimental
- Acid-induced experimental muscle pain and hyperalgesia with single and repeated infusion in human forearm
- Original experimental
- Swearing as a response to pain: A cross-cultural comparison of British and Japanese participants
- Clinical pain research
- The cognitive impact of chronic low back pain: Positive effect of multidisciplinary pain therapy
- Clinical pain research
- Central sensitization associated with low fetal hemoglobin levels in adults with sickle cell anemia
- Topical review
- Targeting cytokines for treatment of neuropathic pain
- Original experimental
- What constitutes back pain flare? A cross sectional survey of individuals with low back pain
- Original experimental
- Coping with pain in intimate situations: Applying the avoidance-endurance model to women with vulvovaginal pain
- Clinical pain research
- Chronic low back pain and the transdiagnostic process: How do cognitive and emotional dysregulations contribute to the intensity of risk factors and pain?
- Original experimental
- The impact of the Standard American Diet in rats: Effects on behavior, physiology and recovery from inflammatory injury
- Educational case report
- Erector spinae plane (ESP) block in the management of post thoracotomy pain syndrome: A case series
- Original experimental
- Hyperbaric oxygenation alleviates chronic constriction injury (CCI)-induced neuropathic pain and inhibits GABAergic neuron apoptosis in the spinal cord
- Observational study
- Predictors of chronic neuropathic pain after scoliosis surgery in children
- Clinical pain research
- Hospitalization due to acute exacerbation of chronic pain: An intervention study in a university hospital
- Clinical pain research
- A novel miniature, wireless neurostimulator in the management of chronic craniofacial pain: Preliminary results from a prospective pilot study
- Clinical pain research
- Implicit evaluations and physiological threat responses in people with persistent low back pain and fear of bending
- Original experimental
- Unpredictable pain timings lead to greater pain when people are highly intolerant of uncertainty
- Original experimental
- Initial validation of the exercise chronic pain acceptance questionnaire
- Clinical pain research
- Exploring patient experiences of a pain management centre: A qualitative study
- Clinical pain research
- Narratives of life with long-term low back pain: A follow up interview study
- Observational study
- Pain catastrophizing, perceived injustice, and pain intensity impair life satisfaction through differential patterns of physical and psychological disruption
- Clinical pain research
- Chronic pain disrupts ability to work by interfering with social function: A cross-sectional study
- Original experimental
- Evaluation of external vibratory stimulation as a treatment for chronic scrotal pain in adult men: A single center open label pilot study
- Observational study
- Impact of analgesics on executive function and memory in the Alzheimer’s Disease Neuroimaging Initiative Database
- Clinical pain research
- Visualization of painful inflammation in patients with pain after traumatic ankle sprain using [11C]-D-deprenyl PET/CT
- Original experimental
- Developing a model for measuring fear of pain in Norwegian samples: The Fear of Pain Questionnaire Norway
- Topical review
- Psychoneuroimmunological approach to gastrointestinal related pain
- Letter to the Editor
- Do we need an updated definition of pain?
- Narrative review
- Is acetaminophen safe in pregnancy?
- Book Review
- Physical Diagnosis of Pain
- Book Review
- Advances in Anesthesia
- Book Review
- Atlas of Pain Management Injection Techniques
- Book Review
- Sedation: A Guide to Patient Management
- Book Review
- Basics of Anesthesia
Artikel in diesem Heft
- Observational study
- Perceived sleep deficit is a strong predictor of RLS in multisite pain – A population based study in middle aged females
- Clinical pain research
- Prospective, double blind, randomized, controlled trial comparing vapocoolant spray versus placebo spray in adults undergoing intravenous cannulation
- Clinical pain research
- The Functional Barometer — An analysis of a self-assessment questionnaire with ICF-coding regarding functional/activity limitations and quality of life due to pain — Differences in age gender and origin of pain
- Clinical pain research
- Clinical outcome following anterior arthrodesis in patients with presumed sacroiliac joint pain
- Observational study
- Chronic disruptive pain in emerging adults with and without chronic health conditions and the moderating role of psychiatric disorders: Evidence from a population-based cross-sectional survey in Canada
- Educational case report
- Management of patients with pain and severe side effects while on intrathecal morphine therapy: A case study
- Clinical pain research
- Behavioral inhibition, maladaptive pain cognitions, and function in patients with chronic pain
- Observational study
- Comparison of patients diagnosed with “complex pain” and “somatoform pain”
- Original experimental
- Patient perspectives on wait times and the impact on their life: A waiting room survey in a chronic pain clinic
- Topical review
- New evidence for a pain personality? A critical review of the last 120 years of pain and personality
- Clinical pain research
- A multi-facet pain survey of psychosocial complaints among patients with long-standing non-malignant pain
- Clinical pain research
- Pain patients’ experiences of validation and invalidation from physicians before and after multimodal pain rehabilitation: Associations with pain, negative affectivity, and treatment outcome
- Observational study
- Long-term treatment in chronic noncancer pain: Results of an observational study comparing opioid and nonopioid therapy
- Clinical pain research
- COMBAT study – Computer based assessment and treatment – A clinical trial evaluating impact of a computerized clinical decision support tool on pain in cancer patients
- Original experimental
- Quantitative sensory tests fairly reflect immediate effects of oxycodone in chronic low-back pain
- Editorial comment
- Spatial summation of pain and its meaning to patients
- Original experimental
- Effects of validating communication on recall during a pain-task in healthy participants
- Original experimental
- Comparison of spatial summation properties at different body sites
- Editorial comment
- Behavioural inhibition in the context of pain: Measurement and conceptual issues
- Clinical pain research
- A randomized study to evaluate the analgesic efficacy of a single dose of the TRPV1 antagonist mavatrep in patients with osteoarthritis
- Editorial comment
- Quantitative sensory tests (QST) are promising tests for clinical relevance of anti–nociceptive effects of new analgesic treatments
- Educational case report
- Pregabalin as adjunct in a multimodal pain therapy after traumatic foot amputation — A case report of a 4-year-old girl
- Editorial comment
- Severe side effects from intrathecal morphine for chronic pain after repeated failed spinal operations
- Editorial comment
- Opioids in chronic pain – Primum non nocere
- Editorial comment
- Finally a promising analgesic signal in a long-awaited new class of drugs: TRPV1 antagonist mavatrep in patients with osteoarthritis (OA)
- Observational study
- The relationship between chronic musculoskeletal pain, anxiety and mindfulness: Adjustments to the Fear-Avoidance Model of Chronic Pain
- Clinical pain research
- Opioid tapering in patients with prescription opioid use disorder: A retrospective study
- Editorial comment
- Sleep, widespread pain and restless legs — What is the connection?
- Editorial comment
- Broadening the fear-avoidance model of chronic pain?
- Observational study
- Identifying characteristics of the most severely impaired chronic pain patients treated at a specialized inpatient pain clinic
- Editorial comment
- The burden of central anticholinergic drugs increases pain and cognitive dysfunction. More knowledge about drug-interactions needed
- Editorial comment
- A case-history illustrates importance of knowledge of drug-interactions when pain-patients are prescribed non-pain drugs for co-morbidities
- Editorial comment
- Why can multimodal, multidisciplinary pain clinics not help all chronic pain patients?
- Topical review
- Individual variability in clinical effect and tolerability of opioid analgesics – Importance of drug interactions and pharmacogenetics
- Editorial comment
- A new treatable chronic pain diagnosis? Flank pain caused by entrapment of posterior cutaneous branch of intercostal nerves, lateral ACNES coined LACNES
- Clinical pain research
- PhKv a toxin isolated from the spider venom induces antinociception by inhibition of cholinesterase activating cholinergic system
- Clinical pain research
- Lateral Cutaneous Nerve Entrapment Syndrome (LACNES): A previously unrecognized cause of intractable flank pain
- Editorial comment
- Towards a structured examination of contextual flexibility in persistent pain
- Clinical pain research
- Context sensitive regulation of pain and emotion: Development and initial validation of a scale for context insensitive avoidance
- Editorial comment
- Is the search for a “pain personality” of added value to the Fear-Avoidance-Model (FAM) of chronic pain?
- Editorial comment
- Importance for patients of feeling accepted and understood by physicians before and after multimodal pain rehabilitation
- Editorial comment
- A glimpse into a neglected population – Emerging adults
- Observational study
- Assessment and treatment at a pain clinic: A one-year follow-up of patients with chronic pain
- Clinical pain research
- Randomized, double-blind, placebo-controlled, dose-escalation study: Investigation of the safety, pharmacokinetics, and antihyperalgesic activity of L-4-chlorokynurenine in healthy volunteers
- Clinical pain research
- Prevalence and characteristics of chronic pain: Experience of Niger
- Observational study
- The use of rapid onset fentanyl in children and young people for breakthrough cancer pain
- Original experimental
- Acid-induced experimental muscle pain and hyperalgesia with single and repeated infusion in human forearm
- Original experimental
- Swearing as a response to pain: A cross-cultural comparison of British and Japanese participants
- Clinical pain research
- The cognitive impact of chronic low back pain: Positive effect of multidisciplinary pain therapy
- Clinical pain research
- Central sensitization associated with low fetal hemoglobin levels in adults with sickle cell anemia
- Topical review
- Targeting cytokines for treatment of neuropathic pain
- Original experimental
- What constitutes back pain flare? A cross sectional survey of individuals with low back pain
- Original experimental
- Coping with pain in intimate situations: Applying the avoidance-endurance model to women with vulvovaginal pain
- Clinical pain research
- Chronic low back pain and the transdiagnostic process: How do cognitive and emotional dysregulations contribute to the intensity of risk factors and pain?
- Original experimental
- The impact of the Standard American Diet in rats: Effects on behavior, physiology and recovery from inflammatory injury
- Educational case report
- Erector spinae plane (ESP) block in the management of post thoracotomy pain syndrome: A case series
- Original experimental
- Hyperbaric oxygenation alleviates chronic constriction injury (CCI)-induced neuropathic pain and inhibits GABAergic neuron apoptosis in the spinal cord
- Observational study
- Predictors of chronic neuropathic pain after scoliosis surgery in children
- Clinical pain research
- Hospitalization due to acute exacerbation of chronic pain: An intervention study in a university hospital
- Clinical pain research
- A novel miniature, wireless neurostimulator in the management of chronic craniofacial pain: Preliminary results from a prospective pilot study
- Clinical pain research
- Implicit evaluations and physiological threat responses in people with persistent low back pain and fear of bending
- Original experimental
- Unpredictable pain timings lead to greater pain when people are highly intolerant of uncertainty
- Original experimental
- Initial validation of the exercise chronic pain acceptance questionnaire
- Clinical pain research
- Exploring patient experiences of a pain management centre: A qualitative study
- Clinical pain research
- Narratives of life with long-term low back pain: A follow up interview study
- Observational study
- Pain catastrophizing, perceived injustice, and pain intensity impair life satisfaction through differential patterns of physical and psychological disruption
- Clinical pain research
- Chronic pain disrupts ability to work by interfering with social function: A cross-sectional study
- Original experimental
- Evaluation of external vibratory stimulation as a treatment for chronic scrotal pain in adult men: A single center open label pilot study
- Observational study
- Impact of analgesics on executive function and memory in the Alzheimer’s Disease Neuroimaging Initiative Database
- Clinical pain research
- Visualization of painful inflammation in patients with pain after traumatic ankle sprain using [11C]-D-deprenyl PET/CT
- Original experimental
- Developing a model for measuring fear of pain in Norwegian samples: The Fear of Pain Questionnaire Norway
- Topical review
- Psychoneuroimmunological approach to gastrointestinal related pain
- Letter to the Editor
- Do we need an updated definition of pain?
- Narrative review
- Is acetaminophen safe in pregnancy?
- Book Review
- Physical Diagnosis of Pain
- Book Review
- Advances in Anesthesia
- Book Review
- Atlas of Pain Management Injection Techniques
- Book Review
- Sedation: A Guide to Patient Management
- Book Review
- Basics of Anesthesia