Orthoamide und Iminiumsalze, LXXXVIII. Synthese N,N,N′,N′,N″,N″-persubstituierter Guanidiniumsalze aus N,N′-persubstituierten Harnstoff/Säurechlorid-Addukten**
-
Willi Kantlehner
 ,
Ralf Kreß
,
Ralf Kreß

Abstract
N,N,N′,N′,N″,N″-Hexamethylguanidinium chloride 9c was prepared by treating the reaction mixture formed from N,N,N′,N′-tetramethylurea (1a) and phthaloyl chloride (16) with dimethyltrimethylsilylamine 15. N,N,N′,N′-Tetramethyl-chloroformamidinium chloride (2a) is an intermediate in this synthesis. The chloroformamidinium chloride 2a can also be prepared by treating the urea 1a with thionyl chloride or phosphorus pentachloride, respectively. The guanidinium salt 9c can be obtained from the crude 2a thus prepared and the silylamine 15. From urea/phosphoryl chloride adducts and primary aromatic amines have been prepared guanidines 38, which are converted to N,N′-diaryl-N,N′,N″,N″-tetramethyl-guanidinium iodides 39 on treatment with methyl iodide. The N,N′,N″-trimethyl-N,N′,N″-triphenylguanidinium salt 44a was prepared from the chloroformamidinium salt 43 and N-methylaniline. The guanidinium salt 9c is the reaction product when the urea 1a/POCl3 adduct is treated with the silylamine 15.
1 Einleitung
1.1 Phosgenfreie Synthese für N,N,N′,N′,N″,N″-persubstituierte Guanidiniumsalze
N,N,N′,N′,N″,N″-persubstituierte Guanidiniumsalze haben in den letzten Jahren aus den verschiedensten Gründen das Interesse der Chemiker auf sich gezogen. Zum einen sind sie wertvolle Bausteine bei der Synthese von Orthoamid-Derivate der Kohlensäure [2, 3], der Ameisensäure [4, 5], aromatischer und heteroaromatischer Carbonsäuren [5, 6], Cyclopropan- sowie Alken- und Allencarbonsäuren [5] und Alkincarbonsäuren [5–10]. Zum anderen existiert eine Reihe von peralkylierten Guanidiniumsalzen, deren Schmelzpunkte unter 100 °C liegen, die daher definitionsgemäß auch als ionische Flüssigkeiten angesprochen werden können [11–16]. Peralkylierte Guanidiniumsalze lösen sich in vielen polaren und unpolaren aprotischen Lösungsmitteln sehr gut. So lassen sich z. B. Nitrile und Acylcyanide usw. aus Alkyl- bzw. Acylhalogeniden und N,N,N′,N′,N″,N″-Hexaethyl-guanidiniumcyanid in Benzol bzw. Dichlormethan herstellen [17]. Peralkylierte Guanidiniumsalze wurden auch als Phasentransferkatalysatoren in Betracht gezogen [13, 14, 17], und mittlerweile existiert eine reichhaltige Patenliteratur zu Umsetzungen, bei denen die katalytische Wirkung in zweiphasigen Systemen ausgenutzt wurde. Besondere katalytische Eigenschaften entfalten Guanidiniumchloride offenbar bei Carboxylierungsreaktionen mit Phosgen und Chlorameisensäurederivaten [18, 19]. Auch bei Halogenaustauschreaktionen an aromatischen und heteroaromatischen Systemen (Halex-Reaktionen) haben sich peralkylierte Guanidiniumsalze bewährt [20]. Peralkylierte Guanidiniumsalze wurden als nicht wässrige Elektrolyte in Batterien vorgeschlagen [21], sie sind auch mit gutem Erfolg als Elektrolyte in photoelektrochemischen Solarzellen verwendet worden [22].
Bei den effektivsten, nicht von Guanidin-Derivaten ausgehenden Synthesen, dienen N,N,N′,N′-Tetraalkylharnstoffe 1 bzw. Tetraalkylthiuramdisulfide 5 als Ausgangsmaterialien. Die Harnstoffe 1 werden aktiviert, indem sie z. B. mit Phosgen [11] bzw. Trifluormethansulfonsäureanhydrid [12] in Chlorformamidiniumchloride 2 bzw. Dikationenethersalze 4 übergeführt werden. Durch Anionenaustausch lassen sich aus den Chloriden 2 Chlorformamidiniumsalze 3 gewinnen [13]. Aus Tetraalkylthiuramdisulfiden 5 und elementarem Chlor sind die äußerst reaktiven Phosgeniminiumsalze 6 zugänglich [23–29] (Schema 1).
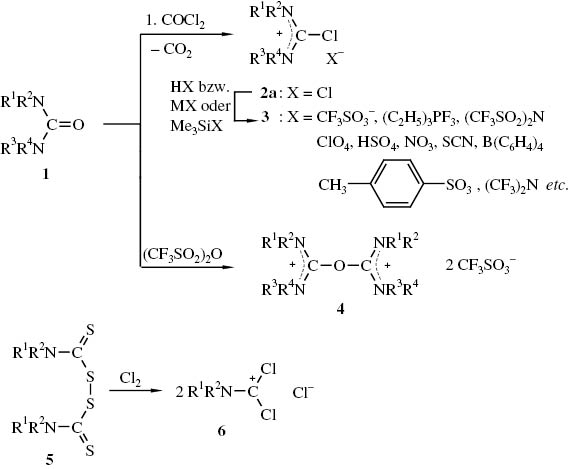
Darstellungsverfahren für Chlorformamidiniumsalze 2, 3, Dikationenethersalze 4 und Dichlormethylen-iminiumsalze 6.
Aus den Iminiumsalzen 2, 3, 4, 6 und sekundären Aminen bzw. Dialkyl-trimethylsilylaminen lassen sich N,N′,N″-peralkylierte Guanidiniumsalze 7 herstellen [11–13, 15, 16, 30–32] (Schema 2).
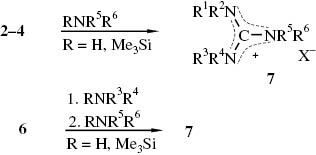
N,N,N′,N″-Peralkylierte Guanidiniumsalze 7 aus Iminiumsalzen 2–4, 6 und sekundären Aminen bzw. Dialkyl-trimethylsilylaminen.
Wir haben nun untersucht, ob sich N,N′-substituierte Harnstoffe durch andere Elektrophile in Iminiumsalze überführen lassen, die sich als Edukte zur Synthese von peralkylierten Guanidiniumsalzen eignen.
2 Ergebnisse und Diskussion
N,N,N′,N′-Tetramethylharnstoff (1a) kann sowohl mit Triethyloxonium-tetrafluoroborat [33] als auch mit Dimethylsulfat [34] am Sauerstoff alkyliert werden. Aus den dabei erhaltenen Alkoxymethyleniminiumsalzen 8a,b entstehen bei der Umsetzung mit Dimethylamin die Guanidiniumsalze 9a,b. Die entsprechende Reaktion von 8a mit Dipropylamin verläuft unter Entalkylierung von 8a, wogegen bei der Einwirkung von Pyrrolidin auf 8a neben der erwünschten Substitution der Ethoxygruppe noch eine Umaminierung stattfindet, was zur Bildung von Tris(pyrrolidino)carbenium-tetrafluoroborat (10) führt [3] (Schema 3).
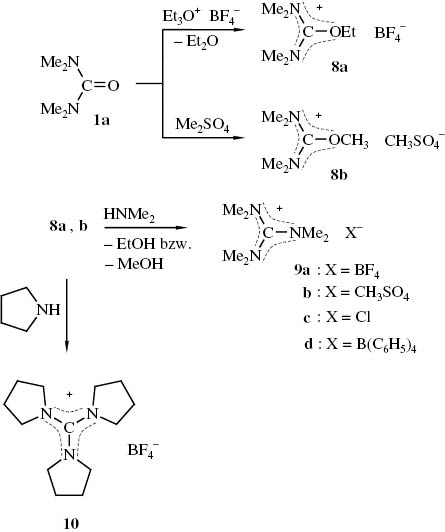
Darstellung von N,N-Dimethyl-alkoxy-dimethylaminomethylen-iminiumsalzen 8 und Guanidiniumsalzen 9a, b, 10.
Es erschien daher nicht besonders erfolgversprechend, dieses Verfahren zur Herstellung von Guanidiniumsalzen auszubauen.
Schon längere Zeit ist bekannt, dass sich aus N,N-Dimethylformamid bzw. -acetamid und N-Methyl-pyrrolidon mit Carbonsäurehalogeniden in Gleichgewichtsreaktionen Vilsmeier-Haack-analoge Addukte 11 bilden, die bei Verwendung von Carbonsäurebromiden isolierbar sind [35]. Werden N,N-disubstituierte Carbonsäureamide mit Carbonsäurechloriden in Gegenwart von Silbertrifluormethansulfonat umgesetzt, so entstehen nahezu quantitativ die entsprechenden Acyloxyiminiumsalze 12 [36] (Schema 4).
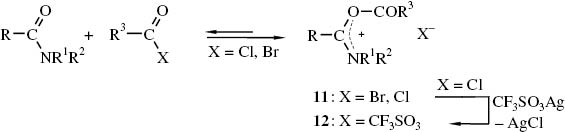
Acyloxyiminiumsalze 11, 12 aus N,N-Dialkyl-carbonsäureamiden und Carbonsäurehalogeniden.
Die sich aus N,N-Dimethylcarbonsäureamiden und Chlorameisensäurearylestern bildenden Addukte können sich durch CO2-Abspaltung stabilisieren [37]. Mit DMF-Aroylchlorid-Addukten wurden Indole formyliert [38–40] sowie Amine und Aminderivate in Amidine übergeführt [41, 42]. Addukte 11 reagieren mit α-Aryloxy-carbonsäuren und Aldiminen bzw. Bis(perfluorphenyl)zink zu β-Lactamen [43] bzw. Bis(pentafluor-phenyl)methyl-dimethylamin [44]. Gemische aus N,N-Dialkyl-carbonsäureamiden und Trifluoressigsäureanhydrid [45–47] bzw. Essigsäureanhydrid [48] besitzen formylierende bzw. acylierende Eigenschaften. Man kann daher vermuten, dass sich aus diesen Edukten im Gleichgewicht ebenfalls Addukte bilden, die analog wie die Iminiumsalze 11 gebaut sind.
Über entsprechende Addukte aus N,N,N′,N′-Tetraalkylharnstoffen und Carbonsäurehalogeniden scheinen bislang keine Erkenntnisse vorzuliegen. Wir haben deshalb 4-Nitrobenzoylchlorid (13) mit dem Harnstoff 1a 4 h auf ca. 80 °C erhitzt, wobei sich ein nicht näher untersuchter Feststoff bildet, bei dem es sich um das Addukt 14 handeln könnte. Bei der Umsetzung des Produkts mit Dimethyl-trimethylsilylamin (15) wurde jedoch anstelle des erwarteten N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorids (9c) N,N-Dimethyl-4-nitrobenzamid (16) erhalten (Schema 5).
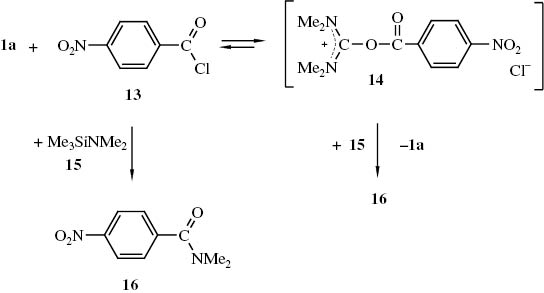
Addukt 14 aus N,N,N′,N′-Tetramethylharnstoff (1a) und 4-Nitrobenzoylchlorid (13) sowie dessen Umsetzung mit Dimethyl-trimethylsilylamin (15).
Das Amid 16 kann auf zwei Wegen entstehen. Zum einen kann das mit dem Addukt 14 im Gleichgewicht vorhandene Säurechlorid 13 mit dem Silylamin 15 zum Amid 16 reagieren; zum anderen kann das Amin 15 das Addukt 14 entacylieren, indem es an der Carbonylgruppe von 14 angreift, die ohnehin elektrophiler sein dürfte als der Kohlenstoff des Bis(dimethylamino)acyloxy-iminiumsystems von 14, an den sich, wohl auch aus sterischen Gründen das Amin 15 nur schwer addieren wird.
Addukte wie 11 und 14 können daher als ambidente Elektrophile aufgefasst werden. Der ambidente Charakter des Dimethylformamid-Acetylchlorid-Addukts 11a (R = H, R1 = R2 = R3 = Me) tritt auch bei der Umsetzung mit Anilin in Erscheinung, die zu einem Produktgemisch aus N-Phenylacetamid und N,N-Dimethyl-N-phenyl-formamidin führt [35].
Die Elektrophilie von Phthaloylchlorid (17) dürfte die des Säurechlorids 13 übertreffen, so dass bei der Umsetzung von 17 mit dem Harnstoff 1a, ein sich einstellendes Gleichgewicht stärker auf Seiten der Addukte 18a, 19a liegen müsste (Schema 6).
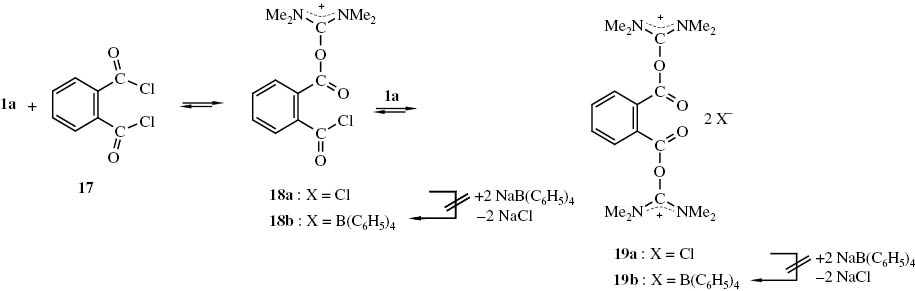
Möglicherweise aus N,N,N′,N′-Tetramethylharnstoff (1a) und Phthaloylchlorid (17) gebildete Addukte 18, 19.
In einem ersten Versuch wurde ein äquimolares Gemisch aus 1a und 17 5 h auf ca. 90 °C erhitzt. Das dabei gewonnene Produkt wurde in Acetonitril gelöst. Der Versuch, mit Hilfe von Natriumtetraphenylborat aus der Lösung Tetraphenylborate 18b bzw. 19b zu fällen, schlug fehl. Beim Einengen wurde zwar eine geringe Menge eines Feststoffs erhalten, bei dem es sich aber um durch Natriumphthalat verunreinigte Phthalsäure handeln dürfte.
Das Reaktionsprodukt, das beim sechsstündigen Erhitzen eines Gemisch aus 1a und 17 im Stoffmengenverhältnis 2:1 auf ca. 90–95 °C entsteht, reagiert in Acetonitril mit dem Silylamin 15 zu N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) (Ausb. 36 %) (Schema 7), das zur Kontrolle mit Natriumtetraphenylborat in N,N,N′,N′,N″,N″-Hexamethylguanidinium-tetraphenylborat (9d) übergeführt wurde.
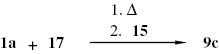
N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid 9c aus dem Harnstoff 1a, Phthaloylchlorid (17) und Dimethyl-trimethylsilylamin (15).
Wird der Harnstoff 1a mit dem Säurechlorid 17 und dem Amin 15 im Stoffmengenverhältnis 3:1:3 unter gleichen Bedingungen umgesetzt, so erhält man das Guanidiniumsalz 9c mit einer Ausbeute von 58 %. Lässt man 1a und 17 nur 4 h bei 90–95 °C reagieren und fügt dann das Amin 15 zu, so liefert die entsprechende Umsetzung im Stoffmengenverhältnis 6:1:6 das Guanidiniumsalz 9c mit etwas geringerer Ausbeute (48 %). In Tabelle 1 sind die Resultate der Versuche zusammengestellt.
N,N,N,N′,N″,N″-Hexamethyl-guanidiniumchlorid 9c aus N,N,N′,N′-Tetramethylharnstoff (1a), Phthaloylchlorid (17) und Dimethyl-trimethylsilylamin (15).
| Versuch Nr. | Edukte | Guanidiniumsalz 9c | |
|---|---|---|---|
| Stoffmengen [mmol] | Stoffmenge [mmol] | Ausb. [%]a | |
| 1a:17:15 | |||
| 1 | 100:50:100b | 18 | 36 |
| 2 | 150:50:150b | 28 | 57 |
| 3 | 300:50:150c | 24 | 48 |
aBezogen auf Phthaloylchlorid gemäß Gleichung 2, Schema 8; bvor der Zugabe von 15 wurde 1a mit 17 6 h bei 90–95 °C umgesetzt; c vor der Zugabe von 15 wurde 1a mit 17 4 h bei 90–95 °C umgesetzt.
Bei den Umsetzungen war das Säurechlorid 17 stets im Unterschuss vorhanden (50 mmol). Die dabei erzielte maximale Stoffmenge an dem Guanidiniumsalz 9c beträgt 28 mmol, selbst wenn der Harnstoff im 3- bzw. 6-fachen Überschuss eingesetzt wird. Daraus kann man schließen, dass bei der Bildung von 9c Addukte 19 nicht beteiligt sind. Andernfalls müssten die Stoffmengen des Salzes 9c ansteigen, wenn die Menge des zugesetzten Harnstoffs 1a gesteigert wird. Somit kommen als Intermediate nur die Addukte 18 in Frage. Nachdem aber aus dem Addukt 14 und dem Amin 15 kein Guanidiniumsalz 9c erhalten wurde, sind Zweifel angebracht, dass 9c unmittelbar aus dem Addukt 18 gebildet wird.
Bei einer schon längere Zeit bekannten, häufig angewandten Reaktion werden aus N,N,N′,N′-Tetraalkylharnstoffen und Oxalylchlorid Chlorformamidiniumsalze 2 erzeugt [49–61]. Phthaloylchlorid (17) kann als „phenyloges Oxalylchlorid“ aufgefasst werden. Er erscheint daher auch möglich, dass sich aus dem Harnstoff 1a und Phthaloylchlorid (17) das Chlorformamidiniumchlorid 2a bildet.
Dabei wandelt sich im ersten Schritt in einer Gleichgewichtsreaktion aus 1a und 17 entstehende Addukt 18 in das Chlorformamidiniumsalz 20 um, das unter Abspaltung von Phthalsäureanhydrid in das Chlorformamidiniumchlorid 2a übergeht. Aus 2a und dem Silylamin 15 entsteht dann in bekannter Weise das Guanidiniumsalz 9c (Schema 8). Mit dieser Annahme lassen sich die in Tabelle 1 zusammengestellten Versuchsergebnisse deuten. Die Verbindungen 1a und 15 sind im Überschuss vorhanden. Damit bestimmt allein die Stoffmenge des Phthaloylchlorids (17) die Stoffmenge des gebildeten Guanidiniumsalzes. Folgerichtig steigt die Ausbeute am Guanidiniumsalz 9c, wenn die Stoffmenge an 1a erhöht wird, weil sich aus 1a und 17 im Gleichgewicht mehr 18 bildet, das dann in 2a und weiter mit 15 in 9c übergehen kann (vgl. Versuche 1 und 2, Tabelle 1). Auf den ersten Blick passt dazu nicht das Ergebnis des Versuchs Nr. 3 in Tabelle 1; denn eine nochmalige Steigerung der Harnstoffmenge führt sogar zu einer Verringerung der Guanidiniumsalzausbeute. Wir nahmen jedoch an, dass die Gleichgewichtseinstellung 1a+ 17 ⇆ 18 nur langsam erfolgt, so dass bei dem Versuch infolge der Verkürzung der Reaktionszeit weniger 18 im Gleichgewicht und damit auch letztlich weniger 2a vorhanden war, das sich mit 15 zu 9c umsetzen konnte. Gestützt auf diese Annahme haben wir den Versuch 1 (Tabelle 1) wiederholt. Dabei aber 1a mit 17 zunächst 24 h auf 90–95 °C erhitzt, das gebildete 2a mit Ether digeriert und dann mit 15 in N,N-Dimethylformamid zu 9c umgesetzt. So lässt sich 9c mit einer Ausbeute von 55 % darstellen. In den etherischen Auszügen, die bei dem Versuch erhalten werden, befindet sich Phthalsäureanhydrid, was als weiterer Beleg für den im Schema 8 angegebenen Bildmechanismus für 2a angesehen werden kann. Das Chlorformamidiniumchlorid 2a kann auch isoliert werden, wenn der Harnstoff 1a mit Phthaloylchlord (17) im Stoffmengenverhältnis 1:1.1 48 h bei 90 °C umgesetzt wird (Ausb. 69 %). Aus diesen Ansätzen lässt sich mit Ether, THF oder Ethylacetat das gebildete Phthalsäureanhydrid extrahieren. Ermittelt man 1H NMR-spektroskopische den Gehalt an 2a in Gemischen aus 1a und 17 unter Variation der Reaktionstemperatur und -zeit (vgl. Tab. 3 im exp. Teil), so zeigt sich, dass 2a bei Temperaturen um 60 °C nur langsam gebildet wird (23 % nach 17 h). Das Ausbeutemaximum (70–90 %) wird bei Temperaturen zwischen 80 und 90 °C nach ca. 23 h erhalten.
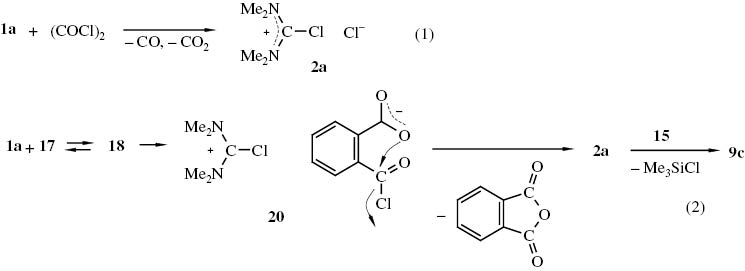
N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid 2a aus N,N,N′,N′-Tetramethylharnstoff (1a) und Oxalylchlorid (Gl. 1) bzw. Phthaloylchlorid 17 (Gl. 2).
In einer Publikation [62], von der wir erst geraume Zeit nach Abschluss unserer Untersuchungen [63] Kenntnis erhielten, werden u. Anderem die Umwandlungen des Harnstoffs 1a in das Chlorformamidiniumchlorid 2a sowie die von N,N-Dimethylformamid in N,N-Dimethyl-chlormethyleniminium-chlorid (Dimethylformamidchlorid) (21) mit Hilfe von Phthaloylchlorid (17) beschrieben (Schema 9).
![Schema 9 N,N-Dimethyl-chlormethyleniminium-chlorid (21) aus N,N-Dimethylformamid und Phthaloylchlorid (17) [62].](/document/doi/10.1515/znb-2014-0102/asset/graphic/znb-2014-0102_scheme9.jpg)
N,N-Dimethyl-chlormethyleniminium-chlorid (21) aus N,N-Dimethylformamid und Phthaloylchlorid (17) [62].
2.1 Aktivierung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Thionylchlorid
N,N-Dimethylformamid bildet mit Thionylchlorid ein Addukt 22, das beim Erwärmen unter reversibler Abspaltung von Schwefeldioxid in das Amidchlorid 21 übergeht [64] (Schema 10).
![Schema 10 N,N-Dimethyl-chlormethyleniminium-chlorid (21) aus N,N-Dimethylformamid und Thionylchlorid [64].](/document/doi/10.1515/znb-2014-0102/asset/graphic/znb-2014-0102_scheme10.jpg)
N,N-Dimethyl-chlormethyleniminium-chlorid (21) aus N,N-Dimethylformamid und Thionylchlorid [64].
Wir haben untersucht, ob sich, in Analogie zu dieser Reaktion, aus N,N,N′,N′-Tetramethylharnstoff (1a) und Thionylchlorid bzw. Phosphorpentachlorid das Chlorformamidiniumchlorid 2a und daraus das Guanidiniumsalz 9c herstellen lässt (Schema 11).
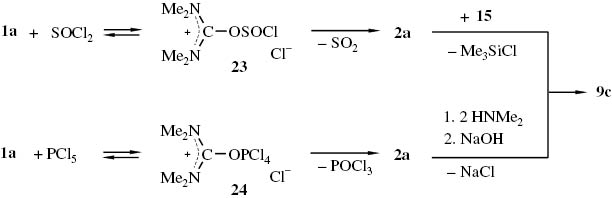
Bildung des Chlorformamidiniumchlorids 2a aus N,N,N′,N′-Tetramethylharnstoff (1a) und Thionylchlorid bzw. Phosphorpentachlorid sowie dessen Umwandlung in das Guanidiniumsalz 9c.
Wird ein Gemisch aus Thionylchlorid und N,N,N′,N′-Tetramethylharnstoff (1a) (Stoffmengenverhältnis ≥7:1) 24 h bei 80 °C gerührt, so erhält man das Iminiumsalz 2a mit 63 % Ausbeute. Die Ausbeute an 2a erhöht sich auf 88 %, wenn dem Ansatz ca. 8 ‰ Pyridin (bez. auf die Stoffmenge Thionylchlorid) zugesetzt werden. Zur Erzielung akzeptabler Ausbeuten an 2a scheint ein hoher Thionylchlorid-Überschuss erforderlich zu sein. Denn die Umsetzung von Thionylchlorid mit 1a im Stoffmengenverhältnis 1.3:1 in Gegenwart von 5 % Pyridin liefert 2a nur mit 25 % Ausbeute. Daneben entsteht ein nicht näher untersuchtes gelbes Öl, aus dem sich durch konsekutive Zugabe von trockenem Tetrahydrofuran noch weiteres 2a (ca. 15 %) ausfällen lässt. Wir nehmen an, dass bei der Bildung von 2a das Addukt 23 eine Zwischenstufe ist. Das daraus entstehende 2a lässt sich mit Dimethyl-trimethylsilylamin (15) in das Guanidiniumsalz 9c überführen. Das Guanidiniumsalz 9c lässt sich auch in einer Eintopfreaktion aus 1a, Thionylchlorid und Dimethylamin in Acetonitril herstellen. Dabei wird das bei der Umsetzung von 2a mit Dimethylamin neben 9c entstehende Dimethylaminhydrochlorid durch Zugabe äquivalenter Menge Natronlauge in das flüchtige Dimethylamin umgewandelt.
Bei der Umsetzung von Phosphorpentachlorid mit dem Harnstoff 1a in Acetonitril bei 0 bis 20 °C wird das Chlorformamidiniumchlorid 2a nicht kristallin, sondern – vermutlich durch Phosphorverbindungen verunreinigt – in Form eines hygroskopischen braunen Öls erhalten. Bei der entsprechenden, bei höheren Temperaturen in Tetrachlorkohlenstoff durchgeführten Umsetzung ließ sich rohes 2a mit einer Ausbeute von ca. 85 % gewinnen. Möglicherweise entsteht aus dem Harnstoff 1a und Phosphorpentachlorid primär das Addukt 24, das sich erst bei Temperaturerhöhung unter Abspaltung von POCl3 in 2a umwandelt. Die, bei Versuchen zur Herstellung des Guanidiniumsalzes 9c in einer Eintopfsynthese aus dem Harnstoff 1a, PCl5 und überschüssigem Silylamin 15 in Acetonitril, erzielten Ergebnisse, deuten ebenfalls in diese Richtung. Wird die Umsetzung von 1a mit PCl5 bei 0–20 °C durchgeführt und das Gemisch mit dem Silylamin 15 behandelt, so erhält man als Reaktionsprodukt ein schwarzes Öl, das ca. 38 % des Guanidiniumsalzes 9c enthält. Dagegen lieferte die Umsetzung eines Gemisches, das durch einstündiges Erhitzen von 1a mit PCl5 in Acetonitril erhalten wurde, mit dem Silylamin das Salz 9c mit einer Ausbeute um 70 %. Nach unseren Erfahrungen ist die Darstellung von 9c nach diesem Verfahren nicht empfehlenswert, weil die Ergebnisse nicht gut reproduzierbar sind. Möglicherweise sind dafür Wasserspuren verantwortlich, die eine gewisse Menge PCl5 unter Freisetzung von Chlorwasserstoff hydrolysieren was zu unerwünschten Nebenreaktionen mit dem Lösungsmittel führen kann (wie z. B. Amidchloridbildung, Selbstkondensation, Phosphorylierung etc.).
2.2 Guanidiniumsalze und Guanidine aus N,N,N′,N′-tetrasubstituierten Harnstoffen, Phosphorylchlorid und Aminen
Die Vilsmeier-Haack-Synthese spielt sowohl im Labor, als auch im technischen Maßstab eine wichtige Rolle zur Synthese aromatischer Aldehyde [65]. Bei dieser Reaktion fungieren als Elektrophile Addukte 25 aus N,N-disubstituierten Formamiden und Phosphorylchlorid, deren Natur lange Zeit umstritten war und die auch als Vilsmeier-Haack-Salze bezeichnet werden (Abb. 1). NMR-spektroskopische Untersuchungen des N,N-Dimethylformamid/POCl3-Addukts haben gezeigt, dass es sich dabei um ein Gleichgewichtsgemisch aus den Iminiumsalzen 27 und 28 handelt [66], wobei das Gleichgewicht nahezu vollständig auf der Seite von 28 liegt (Schema 12).
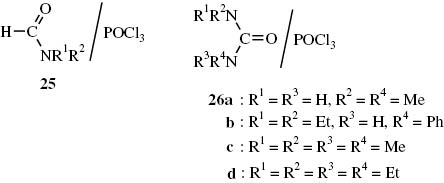
Allgemeine Formulierung von Vilsmeier-Haack-Addukten und von Addukten aus N,N,N′,N′-Tetraalkylharnstoffen und POCl3.
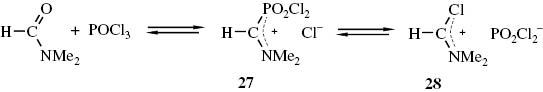
Verlauf der Bildung des Vilsmeier-Haack-Reagenzes.
Auch N,N,N′,N′-Tetraalkylharnstoffe bilden mit POCl3 Addukte 26, bei denen es sich nach NMR-spektroskopischen Untersuchungen ebenso um Gleichgewichtsgemische von Iminiumsalzen 29 und 30 handeln dürfte [66] (Schema 13). Dabei wurde festgestellt, dass die Gleichgewichtseinstellung zwischen dem Harnstoff 1a und POCl3 zu dem Addukt 29a langsam verläuft, wogegen sich das Gleichgewicht 29 ⇆ 30 schnell einstellt. In Methylenchlorid verlaufen die Reaktionen langsamer als im polaren Acetonitril. Die Reaktivität von Säureamiden bzw. Harnstoffen gegenüber POCl3 ist folgendermaßen abgestuft: N,N-Dimethylformamid > N,N-Dimethylacetamid > N,N,N′,N′-Tetramethylharnstoff > N,N,N′,N′-Tetrabutylharnstoff.
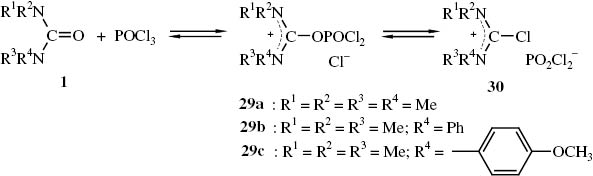
Iminiumsalzgemische 29/30 aus Harnstoffen 1 und Phosphorylchlorid.
Für die Existenz von Addukten 29, 30 spricht auch, dass aus dem Addukt 26c und HPF6 das Chlorformamidinium-hexafluorphosphat 31 entsteht, das sich mit Kaliumfluorid in das Fluorformamidinium-hexafluorphosphat 32 überführen lässt [67] (Schema 14).
![Schema 14 Halogenformamidiniumsalze 31, 32 aus dem N,N,N′,N′-Tetramethylharnstoff/POCl3-Addukt (26c) [67].](/document/doi/10.1515/znb-2014-0102/asset/graphic/znb-2014-0102_scheme14.jpg)
Halogenformamidiniumsalze 31, 32 aus dem N,N,N′,N′-Tetramethylharnstoff/POCl3-Addukt (26c) [67].
Die Addukte 26a–d wurden mit aromatischen Aminen umgesetzt. Aus den Kondensationsprodukten konnten mit Natronlauge die entsprechenden Guanidine 33 freigesetzt werden [68] (Schema 15). Später wurden aus dem Addukt 26c und aromatischen Aminen eine Vielzahl von N-Aryl-N′,N′,N″,N″-tetramethyl-guanidinen vom Typ 33 dargestellt [69, 70]. Diese Guanidinsynthese ist in der Folgezeit noch des öfteren genutzt worden [71–75].
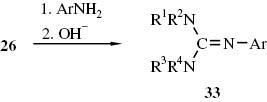
N-Arylguanidine 33 aus Harnstoff/POCl3-Addukten 26 und aromatischen Aminen.
In neuerer Zeit wurden vor allem Addukte aus heterocyclischen Harnstoffen wie Dihydroimidazolon [76–81] und Dihydro- und Tetrahydropyrimidon-Derivaten [82, 83] und POCl3 mit Aminen zu Guanidinen umgesetzt, auch C–C-Verknüpfungen sind mit solchen Addukten an Indolen gelungen [84]. Anstelle von POCl3 kann offenbar auch Phosphorsäuredimethylester-chlorid verwendet werden [85].
Wir haben nun untersucht, ob sich aus N,N,N′-Trialkyl-N′-aryl-harnstoffen (36a, b) N,N-Dialkyl-N′,N′-diaryl-harnstoffen 36c, d bzw. dem N,N,N′-triaryl-N′-methylharnstoff 36e und POCl3 Addukte bilden, die mit aromatischen Aminen zu den entsprechenden Guanidinen umgesetzt werden können. Die dazu benötigten Harnstoffe 36 wurden aus den Carbamoylchloriden 34 und den entsprechenden Aminen 35 hergestellt (Schema 16).
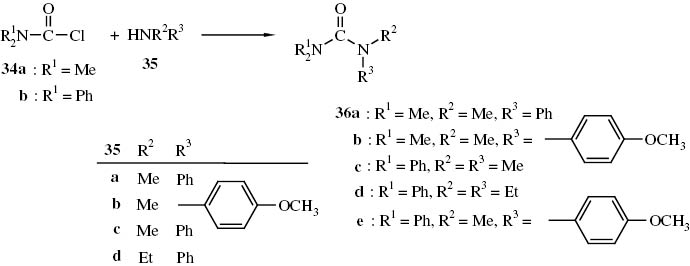
N,N′-Persubstituierte Harnstoffe 36 aus N,N-disubstituierten Carbaminsäurechloriden 34 und sekundären Aminen 35.
Die Umsetzung von POCl3 mit den Harnstoffen 36a, b in Benzol zu Addukten 29b, c verläuft nur unvollständig. Lässt man auf die Gleichgewichtsgemische die Aniline 37a–c einwirken, so erhält man nach der wässrig-alkalischen Aufarbeitung Gemische, die aus den Harnstoffen 36 den Guanidinen 38 und den Anilinen 37 bestehen. Aus diesen lassen sich die Aniline auswaschen, jedoch sind die Gemische aus dem Harnstoff 36a und den Guanidinen 38 nicht auf einfache Weise trennbar. Werden sie aber mit Methyliodid umgesetzt, so lassen sich die reinen Guanidiniumiodide 39a, b isolieren (Schema 17). Durch Auslesen wurden aus den Gemischen der Verbindungen 36a und 38b bzw. 36a und 38c Kristalle von 38b bzw. 38c erhalten, die Kristallstrukturanalysen ermöglichten, deren Ergebnisse bereits publiziert wurden (38b [86], 38c [87]).
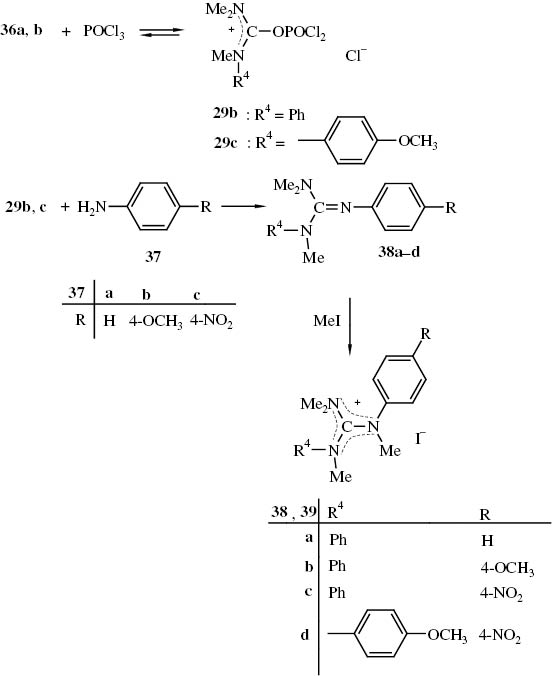
Darstellung N,N′,N″-persubstituierter Guanidine 38 und deren Umwandlung in Guanidiniumiodide 39.
Bei der Umsetzung von POCl3 mit den Harnstoffen 36c–e scheinen sich – wenn überhaupt, dann nur in geringem Umfang – Addukte vom Typ 29 zu bilden. Denn bei der Einwirkung von Anilin auf die Gemische wurde keine signifikante Guanidinbildung beobachtet. Die dabei zurückgewonnenen Harnstoffe könnten allenfalls eine sehr geringe Menge der erwarteten Guanidine enthalten.
Da Thioharnstoffe leichter und selektiver zu Chlorformamidiniumchloriden reagieren als die Sauerstoffanaloga [49, 88], haben wir die Thioharnstoffe 41a, b in bekannter Weise durch Schwefelung der kommerziell verfügbaren Sauerstoffanaloga 40 mit P4S10 hergestellt (Schema 18).
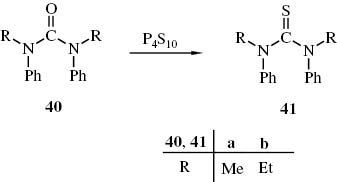
Darstellung der Thioharnstoffe 41a, b aus Harnstoffen 40a, b und P4S10.
Versuche, die Thioharnstoffe 41a, b mit POCl3 und Anilin zu den Guanidinen 42a, b umzusetzen, schlugen fehl (Schema 19).
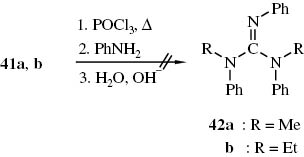
Fehlgeschlagene Versuche zur Synthese der Guanidine 42 aus Thioharnstoffen 41, POCl3 und Anilin.
Erfolgreicher verlief die Umsetzung von 41a mit Phosgen in Acetonitril. Das so erzeugte Chlorformamidiniumchlorid 43 wurde als Rohprodukt mit N-Methylanilin zum sehr hygroskopischen Guanidiniumchlorid 44a umgesetzt, das zur Charakterisierung in das Guanidiniumtetraphenylborat 44b überführt wurde (Schema 20).
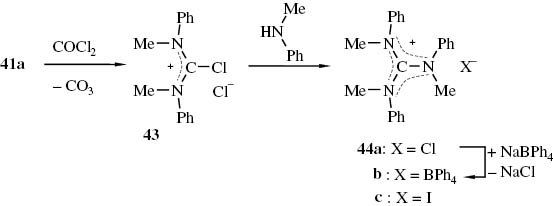
Darstellung der Guanidiniumsalze 44 aus dem Thioharnstoff 41a über das Chlorformamidiniumchlorid 43.
Das Iodid 44c ist seit geraumer Zeit bekannt. Das Salz wurde durch Umsetzung von Methyliodid mit N,N′,N′-Triphenyl-guanidin in Gegenwart von Natriumhydrid in N,N-Dimethylformamid dargestellt [89].
In einer Diplomarbeit [90] ist die Synthese von N,N,N′,N′,N″,N″-Hexamethylguanidiniumchlorid (9c) aus N,N,N′,N′-Tetramethylharnstoff (1a) Phosphorylchlorid und Dimethyl-trimethylsilylamin (15) beschrieben (Schema 21). Auch in der Patentliteratur [74, 75] finden sich Synthesen für N,N′,N″-peralkylierte Guanidiniumsalze, die von N,N,N′,N′-Tetraalkylharnstoffen und POCl3 ausgehen. Dabei werden die Harnstoff/POCl3-Addukte durch Erhitzen der Edukte in Toluol (entweder 2 h bei 60 °C oder 1 h bei 70 °C) hergestellt und mit sekundären Aminen umgesetzt. In den dabei entstehenden Salzgemischen werden die Ammoniumsalze durch Zugabe äquivalenter Mengen Natronlauge in freie Amine übergeführt. Aus den verbleibenden Salzgemischen werden die Guanidiniumchloride mit organischen Lösungsmitteln wie Methylenchlorid herausgelöst. Die sehr effektive Trennmethode für derartige Salzgemische wurde bereits früher bei der Synthese von Guanidiniumsalzen aus Chloroformamidinium-chloriden ausgearbeitet [11]. In einigen orientierenden Versuchen haben wir geprüft, in welchem Umfang die Ausbeuten, mit denen das Guanidiniumsalz 9c erhalten wird, von den Reaktionsbedingungen, insbesondere vom Stoffmengenverhältnis der Edukte, beeinflusst werden. Die Ergebnisse sind in Tabelle 2 zusammengestellt.
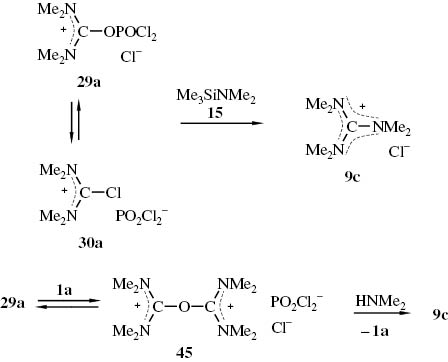
Bildung von N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) aus dem N,N,N′,N′-Tetramethylharnstoff/POCl3-Addukt (29a) und Dimethyl-trimethylsilylamin (15) – möglicherweise unter Beteiligung des Dikationenethersalzes 45.
N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) aus dem N,N,N′,N′-Tetramethylharnstoff/POCl3-Addukt (29a) und Dimethyl-trimethylsilylamin (15).
| Versuch Nr. | Stoffmenge [mol] N,N,N′,N′-Tetra-methylharnstoff (1a) | POCl3 | Dimethyl-trimethylsilylamin (15a) | N,N,N′,N′,N″,N″-Hexamethyl-guanidinium-chlorid (9c) Ausbeute [%]a |
|---|---|---|---|---|
| 1b | 1 | 1 | 2 | 45c |
| 2d | 1 | 1 | 1 | 55e,f |
| 3b | 2 | 1 | 2 | 38c |
| 4d | 2 | 1 | 2 | 76e,f |
| 5b | 1,5 | 1 | 2 | 35c |
aBezogen auf POCl3; bAddukt aus 1a und POCl3, bei 20 °C hergestellt, 1 h bei Raumtemperatur gerührt; cSchmp. 291–293 °C; dAddukt aus 1a und POCl3 bei 25–40 °C hergestellt und 0.5 h gerührt; ewässrige Aufarbeitung; fSchmp. 250–256 °C (9c enthält noch Kristallwasser).
Wie die Ergebnisse der Versuche 1, 3, 5 (Tabelle 2) – die alle unter gleichen Bedingungen durchgeführt wurden – zeigen, wirkt überschüssiger Harnstoff (1a) nicht ausbeutesteigernd. Bei den Versuchen 2 und 4 (Tabelle 2) bei denen hydrolytisch aufgearbeitet wurde, konnte das Guanidiniumsalz 9c mit besseren Ausbeuten gewonnen werden. Dies könnte bedeuten, dass bei einem Harnstoffüberschuss neben den Iminiumsalzen 29a, 30a im Gleichgewicht noch andere Iminiumsalze vorliegen – denkbar wären z. B. Dikationenethersalze 45, wie sie von Maas beschrieben wurden [12, 91], die sich dann mit dem bei der Hydrolyse freigesetzten Amin zu dem Guanidiniumsalz 9c umsetzen [12].
3 Schlussfolgerung und Ausblick
Es wurde gezeigt, dass es „Phosgen-freie“ Synthesen für N,N,N″-persubstituierte Guanidiniumsalze 9 gibt, bei denen als Zwischenstufen Chlorformamidinium-Salze auftreten, die aus Harnstoffen und Säurechloriden entstehen. Nachteilig an den Methoden ist, dass die Anionen dieser Intermediate häufig – insbesondere bei den PCl5 bzw. POCl3-Addukten – nicht einheitlich sind, was die Aufarbeitung der Ansätze erschwert. Die unter Verwendung von POCl3 vorgestellten Synthesemethoden könnten zur Herstellung N,N′,N″-peralkylierter Guanidiniumsalze, deren Alkylgruppen längerkettig sind, vorteilhaft sein. Zur Synthese von Guanidiniumsalzen deren Alkylgruppen nur wenige C-Atome umfassen, sind die Verfahren weniger geeignet, da sich diese Salze aus wässrigem Milieu mit org. Lösungsmitteln wie Methylenchlorid nur schwer extrahieren lassen.
4 Experimenteller Teil
4.1 Umsetzung von 4-Nitrobenzoylchlorid (13) mit N,N,N′,N′-Tetramethylharnstoff (1a) und Dimethyl-trimethylsilylamin
16.6 g (0.10 mol) 4-Nitrobenzoylchlorid (13) und 11.6 g (010 mol) Tetramethylharnstoff (1a) werden 4 h auf 80–85 °C erhitzt. Nach dem Abkühlen auf Raumtemperatur lässt man über Nacht stehen, wobei der Ansatz fest wird. Nach dem Abkühlen auf 0 °C gibt man 50 mL trockenes Acetonitril zu und tropft innerhalb von 1.5 h langsam 11.7 g (0.10 mol) Dimethyl-trimethylsilylamin hinzu, wobei eine klare Lösung entsteht. Es wird 12 h bei Raumtemperatur gerührt. Die flüchtigen Anteile werden im Rotationsverdampfer im Vakuum entfernt. Der feste Rückstand wird mit siedendem Tetrahydrofuran weitgehend gelöst, die Lösung wird filtriert, beim Erkalten scheidet sich das Produkt ab. Man erhält N,N-Dimethyl-4-nitrobenzamid als leicht gelblichen Feststoff mit Schmp. 93 °C (Lit. [92]: Schmp. 95–97 °C). – IR (ATR): ν = 1689, 1631, 1596 cm–1. – 1H NMR (250 MHz, CD3CN): δ = 2.88, 3.04 (je s, je 3 H, NMe2), 7.56–7.62, 8.15–8.28 (je m, je 2 H, Ar). – 13C NMR (63 MHz, CD3CN): δ = 35.2, 39.5 (NMe2), 124.6, 128.9, 144.3, 149.1 (Ar), 169.8 (CO). – C9H10N2O3 (194.19): ber. C 55.67, H 5.19, N 14.44; gef. C 55.67, H 5.24, N 14.31.
4.2 Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Phthaloylchlorid (17) – Versuch zur Fällung der Addukte 18 bzw. 19
5.81 g (50 mmol) N,N,N′,N′-Tetramethylharnstoff (1a) und 10.15 g (50 mmol) Phthaloylchlorid (17) werden 5 h auf 90–95 °C erhitzt. Nach dem Abkühlen auf Raumtemperatur werden dem Gemisch 1.98 g entnommen und mit 3.77 g (11 mmol) Kalignost in 10 mL Acetonitril versetzt. Es wird heiß abfiltriert und das Filtrat bis auf ungefähr die Hälfte eingeengt. Aus dem Filtrat scheiden sich 0.54 g rohe Phthalsäure in Form eines fast farblosen Feststoffs mit Schmp. 209 °C (ab 200 °C beginnende Zers.) (Lit. [93]: Schmp. 210–211 °C). – IR (ATR): ν = 1722, 1602, 1303, 1333 cm–1. – 1H NMR (250 MHz, CD3CN): δ = 7.93–8.07 (m, ArH). – 13C NMR (63 MHz, CD3CN): δ = 126.4, 132.4, 137.3 (CH-Ar), 164.3 (C=O).
4.3 Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Phthaloylchlorid (17) und Dimethyl-trimethylsilylamin (15) im Stoffmengenverhältnis 2:1:2
11.62 g (100 mmol) N,N,N′,N′-Tetramethylharnstoff (1a) und 10.15 g (50 mmol) Phthaloylchlorid (17) werden 6 h auf 90–95 °C erhitzt. Nach dem Abkühlen auf Raumtemperatur wird auf 0 °C abgekühlt, dann werden 50 mL Acetonitril zugegeben. Anschließend tropft man langsam (1.5 h) 11.72 g (100 mmol) N,N-Dimethyl-trimethylsilylamin (15) zu. Man lässt über Nacht bei Raumtemperatur rühren. Der Ansatz wird im Vakuum (Ölbad 40 °C) eingeengt und der Niederschlag abgetrennt. Das Rohprodukt wird aus DMF umkristallisiert. Man erhält 3.25 g (18 mmol), 36 %) N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) mit Schmp. 285–286 °C, (Lit. [11]: Schmp. 293 °C). – 1H NMR (250 MHz, CD3CN): δ = 2.92 (s, 18 H, NMe2). – 13C NMR (63 MHz, CD3CN): δ = 40.34 (NMe2), 164.13 (CN3).
4.4 N,N,N′,N′,N″,N″-Hexamethyl-guanidinium-tetraphenylborat (9d)
2.02 g (11 mmol) 9c werden in 5 mL Acetonitril gelöst und 3.42 g (10 mmol Natriumtetraphenylborat in 10 mL heißem Acetonitril hinzugegeben, wobei Natriumchlorid ausfällt. Die heiße Lösung wird filtriert. Aus dem Filtrat scheiden sich 2.92 g (63 %) N,N,N′,N′,N″,N″-Hexamethylguanidinium-tetraphenylborat mit Schmp. >300 °C (Zers.) ab. – 1H NMR (250 MHz, CD3CN): δ = 2.83 (s, 18 H, NMe2), 6.81–6.87 (m, 4 H, Ph), 6.96–7.02 (m, 8 H, Ph), 7.24–7.30 (m, 8 H, Ph). – C31H38BN3 (463.47): ber. C 80.34, H 8.26, N 9.07; gef. C 80.25, H 8.27, N 9.17.
4.5 N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) durch Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Phthaloylchlorid (17) und Dimethyl-trimethylsilylamin (15) im Stoffmengenverhältnis 3:1:3
17.40 g (0.150 mol) N,N,N′,N′-Tetramethylharnstoff (1a) und 10.15 g (0.050 mol) Phthaloylchlorid (17) werden 6 h auf 90–95 °C erhitzt. Nach dem Abkühlen auf Raumtemperatur wird auf 0 °C abgekühlt und es werden 30 mL Acetonitril zugegeben. Anschließend tropft man langsam 17.6 g (0.150 mol) N,N-Dimethyl-trimethylsilylamin (15) zu. Man lässt noch 2 h bei Raumtemperatur rühren. Der Ansatz wird im Vakuum (Ölbad 40 °C) eingeengt und der Niederschlag abgesaugt. Das Rohprodukt wird aus DMF umkristallisiert. Man erhält 5.08 g (28 mmol, 57 %) N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) mit Schmp. 285–286 °C, (Lit. [11]: Schmp. 293 °C). – 1H NMR (250 MHz, CD3CN): δ = 2.92 (s, 18 H, NMe2). – 13C NMR (63 MHz, CD3CN): δ = 40.3 (NMe2), 164.1 (CN3).
4.6 N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid 9c durch Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Phthaloylchlorid (17) und Dimethyl-trimethylsilylamin (15) im Stoffmengenverhältnis 6:1:6
34.8 g (0.30 mol) N,N,N′,N′-Tetramethylharnstoff (1a) und 10.15 g (0.050 mol) Phthaloylchlorid (17) werden 4 h auf 90–95 °C erhitzt. Nach dem Abkühlen auf Raumtemperatur wird auf 0 °C gekühlt und 50 mL Acetonitril zugegeben. Anschließend tropft man langsam 35.2 g (0.30 mol) N,N-Dimethyl-trimethylsilylamin (15) zu. Man lässt über Nacht bei Raumtemperatur rühren. Der Ansatz wird im Vakuum (Ölbad 40 °C) eingeengt und der Niederschlag abgesaugt. Das Rohprodukt wird aus DMF umkristallisiert. Man erhält 4.3 g (24 mmol, 48 %) N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) mit Schmp. 285 °C. – 1H NMR (250 MHz, CD3CN): δ = 2.92 (s, 18 H, NMe2). – 13C NMR (63 MHz, CD3CN): δ = 40.3 (NMe2), 164.1 (CN3).
4.7 In situ-Erzeugung des Chloroformamidiniumchlorids 2a aus 1a und 17 sowie dessen Umsetzung mit 15 zum Guanidiniumsalz 9c
9.63 g (83 mmol) N,N,N′,N′-Tetramethylharnstoff (1a) und 8.53 g (42 mmol) Phthaloylchlorid (17) werden 24 h auf 90–95 °C erhitzt. Nach dem Abkühlen auf Raumtemperatur wird dreimal mit Ether digeriert. Der Rückstand wird bei 0 °C mit 30 mL trockenem DMF versetzt. Zu dem Gemisch tropft man bei 60 °C unter Rühren 9.96 g (85 mmol) Dimethyl-trimethylsilylamin (15) zu. Das Gemisch wird noch 12 h bei Raumtemperatur gerührt. Das ausgefallene Guanidiniumsalz 9c wird abgetrennt und mit Aceton gewaschen. Ausb.: 4.15 g (55 %) mit Schmp. 284–286 °C.
4.8 N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid (2a) aus N,N,N′,N′-Tetramethylharnstoff (1a) und Phthaloylchlorid (17)
Zu 11.6 g (0.10 mol) N,N,N′,N′-Tetramethylharnstoff (1a) tropft man bei Ausschluss von Feuchtigkeit unter Rühren 22.3 g (0.11 mol) Phthaloylchlorid (17). Das Gemisch wird 48 h auf 90 °C erhitzt. Das stark hygroskopische Reaktionsprodukt wird dreimal mit je 40 mL trockenem THF intensiv digeriert, mit 50 mL trockenem Ether gewaschen und dann getrocknet. Ausb.: 11.7 g (68 %) 2a. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.28 (s, 6 H, NMe2). – 13C NMR (75 MHz, CDCl3, TMS): δ = 44.5 (NMe2), 158.8 (C+).
4.9 Verlauf der Bildung von N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid (2a) aus N,N,N′,N′-Tetramethylharnstoff (1a) und Phthaloylchlorid (17)
Die für die spektroskopischen Untersuchungen erforderlichen Gemische werden analog der nachstehenden typischen Arbeitsvorschrift hergestellt und gegebenenfalls präparativ aufgearbeitet, wobei die dabei erzielten Ausbeuten bis zu 15 % unter denen liegen können, die spektroskopisch ermittelt wurden. Tropft man 6.10 g (30 mmol) 17 bei 90 °C zu 3.50 g (30 mmol) 1a und rührt 17 h bei dieser Temperatur, so erhält man nach dreimaligem Waschen des Produkts mit je 50 mL trockenem THF und einmaligem Waschen mit 50 mL trockenem Ether nach Trocknen im Vakuum 3.4 g (66 %) 2a. Den äquimolaren Gemischen aus 1a und 17 werden zu den in Tabelle 3 genannten Zeiten Proben entnommen und darin 1H NMR-spektroskopisch der Gehalt an 2a bestimmt. Die Ergebnisse sind in Tabelle 3 zusammengestellt.
Chlorformamidiniumchlorid (2a) Ausbeuten (1H-NMR) bei der Umsetzung von 1a mit Phthaloylchlorid (17) bei unterschiedlichen Temperaturen und Reaktionszeiten.
| Temperatur [°C] | Reaktionszeit [h] | Chlorformamidiniumchlorid (2a) Ausbeute [%] |
|---|---|---|
| 60 | 17 | 23 |
| 80 | 26 | 80 |
| 85 | 17 | 60 |
| 85 | 23 | 90 |
| 90 | 17 | 66 |
| 90 | 23 | 72 |
4.10 N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid (2a) aus N,N,N′,N′-Tetramethylharnstoff (1a) und überschüssigem Thionylchlorid (Stoffmengenverhältnis 1:7) in Gegenwart katalytischer Mengen Pyridin
Bei Ausschluss von Feuchtigkeit werden zu 41.6 g (0.35 mol) Thionylchlorid und 0.25 mL (3 mmol) Pyridin unter Rühren 5.8 g (50 mmol) N,N,N′,N′-Tetramethylharnstoff (1a) getropft. Das Gemisch wird unter Rühren 24 h auf 80 °C erhitzt. Das überschüssige Thionylchlorid wird im Wasserstrahlpumpenvakuum (ca. 15 Torr) durch Erwärmen (60 °C) entfernt. Der farblose feste Rückstand wird dreimal mit je 50 mL trockenem Tetrahydrofuran gewaschen und im Ölpumpenvakuum getrocknet. Ausb.: 7.4 g (88 %) stark hygroskopisches N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid (2a).
Wird der Versuch ohne Pyridin durchgeführt, so erhält man 5.4 g (63 %) Chlorformamidiniumchlorid (2a).
4.11 Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Thionylchlorid (Stoffmengenverhältnis 1:1.3) in Gegenwart katalytischer Mengen Pyridin
7.7 g (65 mmol) Thionylchlorid und 0.25 mL (3 mmol) Pyridin werden mit 5.8 g (50 mmol) N,N,N′,N′-Tetramethylharnstoff (1a), wie vorstehend beschrieben, umgesetzt. Der nach dem Abdestillieren des überschüssigen Thionylchlorids (15 Torr, 60 °C) verbleibende ölige Rückstand wird mit 50 mL trockenem Tetrahydrofuran überschichtet. Nach ca. 2 h werden die ausgeschiedenen Kristalle abgetrennt und im Ölpumpenvakuum getrocknet. Ausb.: 1.5 g (25 %) 2a. Wird das Filtrat im Vakuum erneut eingeengt, so erhält man 4.9 g eines leicht gelb gefärbten öligen Rückstands, aus dem sich durch fortlaufende Zugabe von trockenem Tetrahydrofuran und anschließendem Absaugen noch ca. 1.1 g (15 %) weiteres 2a gewinnen lassen.
4.12 Umsetzung des aus Thionylchlorid und N,N,N′,N′-Tetramethylharnstoff (1a) gewonnenen N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorids (2a) mit Dimethyl-trimethylsilylamin (15) zu N,N,N′, N′,N″,N″-Hexamethyl-guanidiniumchlorid (9c) – Allgemeine Vorschrift
Zu einer Lösung von 5.1 g (30 mmol) N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid (2a) in 100 mL trockenem Acetonitril lässt man bei Eiskühlung unter Rühren und Feuchtigkeitsausschluss Dimethyl-trimethylsilylamin (15) tropfen. Das Gemisch wird 16 h gerührt, 0.5 h unter Rückfluss erhitzt und dann im Rotationsverdampfer im Vakuum eingedampft. Der feste Rückstand wird abgesaugt und mit 20 mL trockenem Tetrahydrofuran und 20 mL trockenem Aceton gewaschen. Ausb.: 4.5 g (84 %) N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid 9c, gelbstichige Kristalle mit Schmp. 285–286 °C.
4.13 N,N,N′,N′,N″,N″-Hexamethyl-guanidiniumchlorid 9c aus N,N,N′,N′-Tetramethylharnstoff (1a), Thionylchlorid und Dimethylamin
In einem Dreihalskolben bekannter Masse tropft man zu 833 g (7 mol) Thionylchlorid ein Gemisch aus 116 g (1.0 mol) N,N,N′,N′-Tetramethylharnstoff und 4 g (60 mmol) Pyridin und kocht das Gemisch bei Ausschluss von Feuchtigkeit unter Rühren 18 h unter Rückfluss. Danach destilliert man zunächst im Wasserstrahlvakuum bei 100 °C Badtemperatur und dann im Ölpumpenvakuum bei derselben Badtemperatur das überschüssige Thionylchlorid ab. Durch Wiegen des Kolbens wird überprüft, ob das überschüssige Thionylchlorid vollständig entfernt ist. Die zurückbleibenden gelb-braunen Kristalle werden in ca. 250 mL trockenem Acetonitril gelöst. Unter Rühren leitet man bei max. 10–20 °C ca. 100 g (ca. 2.2 mol) trockenes Dimethylamin ein. Das Gemisch wird 1 h unter Rückfluss erhitzt. Das Acetonitril wird abdestilliert und der Rückstand unter Rühren mit einer kalten Lösung von 10 g (1.0 mol) NaOH in 120 mL Wasser versetzt, wobei Dimethylamin entweicht. Danach wird im Vakuum im Rotationsverdampfer zur Trockene eingedampft. Der Rückstand wird mit 200 mL trockenem Acetonitril versetzt und das Gemisch auf 40 °C erwärmt. Das ausgeschiedene Kochsalz wird abgesaugt und das klare Filtrat im Vakuum eingedampft. Das so erhaltene rohe Guanidiniumsalz 9c wird aus trockenem Dimethylformamid umkristallisiert und nach Waschen mit trockenem Aceton und trockenem Diethylether im Vakuum getrocknet. Das wasserfreie Salz ist äußerst hygroskopisch und besitzt einen Schmp. von 293 °C. Für Umsetzungen genügt ein Produkt mit Schmp. 286–287 °C. Durch Einengen der Mutterlauge auf die Hälfte des Ausgangsvolumens erhält man weiteres 9c. Ausb.: 151 g (84 %) schwach gelbstichige Kristalle mit Schmp. 291–293 °C.
4.14 Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Phosphorpentachlorid in Acetonitril
Zu 10.41 g (50 mmol) PCl5 in 100 mL Acetonitril werden bei 0 °C 5.81 g (50 mmol) Tetramethylharnstoff 1a zugetropft und anschließend 3 h bei Raumtemperatur gerührt. Dann wird im Rotationsverdampfer bei 30 mbar und einer Temperatur von ∼90 °C eingeengt. Zu dem erhaltenen braunen Öl werden 100 mL Chloroform gegeben und nach 1 h Rühren der Feststoff abgetrennt. Das Filtrat wird im Rotationsverdampfer eingeengt und mit trockenem Tetrahydrofuran gewaschen. Man erhält 5.09 g eines hygroskopischen braunen Öls, das im Wesentlichen aus 2a besteht (1H-NMR), was einer Ausbeute von ca. 59 % entspricht.
4.15 Reaktion in Tetrachlorkohlenstoff
Zu einer Suspension aus 10.41 g (50 mmol) PCl5 in 100 mL Tetrachlorkohlenstoff werden bei 0 °C 5.81 g (50 mmol) des Harnstoffs 1a zugetropft, wobei sich sofort ein flockiger Niederschlag bildet. Zunächst wird 2 h bei Raumtemperatur gerührt und dann 1 h bei 80 °C. Der Niederschlag wird abgesaugt und mit 230 mL trockenem Chloroform aufgenommen. Die Lösung wird filtriert, das Filtrat eingedampft und der Rückstand mit 100 mL trockenem Tetrahydrofuran gewaschen, dann in Acetonitril aufgenommen und abermals filtriert. Nach dem Abdestillieren des Lösungsmittels erhält man 7.30 g (85 %) N,N,N′,N′-Tetramethyl-chlorformamidiniumchlorid (2a).
4.16 Umsetzung von N,N,N′,N′-Tetramethylharnstoff (1a) mit Phosphorpentachlorid und Dimethyl-trimethylsilylamin (15)
Umsetzung in Acetonitril bei Raumtemperatur Zu 20.82 g (100 mmol) PCl5 in 100 mL Acetonitril werden bei 0 °C 11.62 g (100 mmol) Tetramethylharnstoff (1a) zugetropft und anschließend 2 h bei Raumtemperatur gerührt. Dann werden bei 0 °C 46.91 g (40 mmol) N,N-Dimethyl-trimethylsilylamin (15) zugetropft und 1 h bei Raumtemperatur gerührt. Aus dem Reaktionsgemisch wird das gebildete Chlortrimethylsilan über eine Vigreux Kolonne abdestilliert und der Rückstand eingeengt. Man erhält ein schwarzes Öl, welches ca. 38 % 9c enthält (1H-NMR).
Umsetzung in Acetonitril bei erhöhter Temperatur Zu 20.82 g (100 mmol) Phosphorpentachlorid in 100 mL trockenem Acetonitril werden unter Rühren bei Eiskühlung 11.62 g (100 mmol) Tetramethylharnstoff (1a) zugetropft. Das Kühlbad wird entfernt und das Reaktionsgemisch 1 h unter Rückfluss erhitzt. Dann werden unter Eiskühlung 46.91 g (400 mmol) Dimethyl-trimethylsilylamin (15) zugetropft und anschließend 1 h bei Raumtemperatur gerührt. Aus dem Reaktionsgemisch wird das entstandene Chlortrimethylsilan über eine 30 cm Vigreux Kolonne abdestilliert und der Rückstand im Rotationsverdampfer zur Trockene eingeengt. Der gelb-braune Feststoff wird nochmals mit Aceton ausgekocht, nach dem Abkühlen abfiltriert und mit Diethylether nachgewaschen. Man erhält 12.6 g (70 %) 9c farbloser Feststoff mit Schmp. 290–291 °C, (Lit. [11]: Schmp. 293 °C). – IR (KBr): ν = 1595 (C=N)+ cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 2.92 (s, 18 H, NMe2). – 13C NMR (75 MHz, CDCl3, TMS): δ = 40.5 (NMe2), 164.3 (C+).
N,N,N′-Trimethyl-N′-phenylharnstoff (36a) Zu einer Lösung von 5.36 g (50 mmol) N-Methylanilin und 5.5 g (54 mmol) Triethylamin in 10 mL trockenem Acetonitril werden bei 0 °C langsam 5.4 g (50 mmol) Dimethylcarbamoylchlorid in 20 mL trockenem Acetonitril zugetropft. Nach 10-stündigem Rühren bei Raumtemperatur wird das Reaktionsgemisch auf 0 °C gekühlt, das ausgefallene Salz abfiltriert und das Filtrat im Rotationsverdampfer bei 50 °C/15 mbar von den flüchtigen Bestandteilen befreit. Der ölige Rückstand wird mit 100 mL Wasser versetzt und fünfmal mit 50 mL Diethylether ausgeschüttelt. Die vereinigten organischen Phasen werden über Magnesiumsulfat getrocknet und eingedampft. Der ölige Rückstand wird im Ölpumpenvakuum destilliert. Ausb.: 7.6 g (86 %), gelbliches Öl mit Sdp. 105 °C/0.005 mbar (Lit. [94]: Schmp.: 74 °C). Die spektroskopischen Daten sind im Einklang mit den Literaturangaben. – IR (ATR): ν = 1643 cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.68 (s, 6 H, NMe2), 3.19 (s, 3 H, NMe2), 6.94–7.08 (m, 3 H, ArH), 7.26–7.38 (m, 2 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 37.8 (2 NMe), 39.4 (NMe2), 123.4, 124.0, 129.3 (Ar-C), 146.8 (Ar-C), 161.6 (C=O).
N,N,N′-Trimethyl-N′-(4-methoxyphenyl)-harnstoff (36b) Wie bei 36a beschrieben, werden 6.9 g (50 mmol) N-Methyl-4-methoxyanilin und 5.5 g (54 mmol) Triethylamin 5.4 g (50 mmol) Dimethylcarbamoylchlorid in 20 mL trockenem Acetonitril umgesetzt. Der nach dem Entfernen der Lösungsmittel verbleibende feste Rückstand wird aus Methyl-tert.-butylether umkristallisiert. Ausb.: 8.2 g (80 %) 36b, farblose Kristalle mit Schmp. 59–60 °C. – IR (ATR): ν = 1638 (C=O) cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 2.63 (s, 6 H, NMe2), 3.14 (s, 3 H, NMe), 3.79 (s, 3 H, OMe), 6.86 (d, J = 4 Hz, 2 H, ArH), 7.01 (d, J = 4 Hz, 2 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 37.8 (NMe2), 40.1 (NMe), 55.2 (OMe), 114.5 (ArC), 125.4 (ArC), 127.1 (ArC), 139.9 (ArC), 156.4 (ArC), 162.1 (C=O). – MS ((+)-ESI): m/z = 231.1111 (ber. 231.1104 für C11H16N2O2Na, [M+Na]+).
N,N-Dimethyl-N′,N′-diphenyl-harnstoff (36c) Wie vorstehend beschrieben, erhält man bei der Umsetzung von 6.7 g (150 mmol) Dimethylamin und 5.5 g (54 mmol) Triethylamin in 30 mL trockenem Acetonitril mit 11.5 g (50 mmol) N,N-Diphenylcarbamoylchlorid in 20 mL trockenem Acetonitril nach dem Entfernen der Lösungsmittel einen öligen Rückstand, der im Vakuum destilliert wird (Sdp. 100 °C/30 mbar). Das ölige Destillat erstarrt beim Stehen zu einem farblosen Feststoff. Ausb.: 9.2 g (77 %) 36c mit Schmp. 71–74 °C (Lit: [95] Schmp.: 70–75 °C). – IR (ATR): ν = 1643 (C=O) cm–1. – 13C NMR (75 MHz, CDCl3, TMS): δ = 37.8 (NMe2), 40.1 (NMe), 125.4, 127.1, 139.9, 156.4 (ArC), 162.1 (C=O).
N,N-Diethyl-N′,N′-diphenyl-harnstoff (36d) Aus 3.65 g (50 mmol) Diethylamin und 5.5 g (54 mmol) Triethylamin in 30 mL trockenem Acetonitril und 11.5 g (50 mmol) N,N-Diphenylcarbamoylchlorid in 20 mL trockenem Acetonitril erhält man wie bei 36c beschrieben ein Öl, das im Ölpumpenvakuum destilliert wird. Das ölige Destillat erstarrt beim Stehen. Ausb.: 10.8 g (81 %) 36d mit Sdp. 119 °C (0.005 mbar), Schmp. 55 °C (Lit: [96] Schmp. 54 °C). – 1H NMR (300 MHz, CDCl3, TMS): δ = 1.04 (t, J = 6.6 Hz, 6 H, Me), 3.23 (q, J = 6.6 Hz, 4 H, NCH2), 6.87–6.96 (m, 2 H, ArH), 6.99–7.18 (m, 6 H, ArH), 7.22–7.30 (m, 2 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 13.0 (Me), 42.6 NCH2), 124.4, 126.1, 129.2, 145.0 (ArC), 160.3 (C=O).
N,N-Diphenyl-N′-methyl-N′-(4-methoxy)phenyl-harnstoff (36e) Wie bei der Synthese von 36b beschrieben werden 6.9 g (50 mmol) N-Methyl-4-methoxyanilin und 5.5 g (54 mmol) Triethylamin in 10 mL trockenem Acetonitril mit 11.5 g (50 mmol) Diphenylcarbamoylchlorid in 20 mL trockenem Acetonitril umgesetzt. Der nach dem Entfernen der flüchtigen Bestandteile verbleibende feste Rückstand wird aus Methyl-tert.-butylether umkristallisiert. Ausb.: 13.7 g (83 %) 36e. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.31 (s, 3 H, NMe), 3.62 (s, 3 H, OMe), 6.51–6.57 (m, 2 H, ArH), 6.71–6.79 (m, 2 H, ArH), 6.81–6.89 (m, 4 H, ArH), 6.91–7.01 (m, 2 H, ArH), 7.94–7.12 (m, 4 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 39.6 (NMe), 55.4 (OMe), 114.0 (ArC), 124.6 (ArC), 125.5 (ArC), 127.1 (ArC), 128.7 (ArC), 137.7 (ArC), 144.8 (ArC), 157.4 (ArC), 160.5 (C=O). – MS ((+)-ESI): m/z = 333.1605 (ber. 333.1598 für C21H21N2O2, [M+Na]+). – C21H20N2O2 (332.39): ber. C 75.88, H 6.06, N 8.43; gef. C 75.71, H 6.02, N 8.48.
N,N,N′,N″-Tetramethyl-N′,N″-diphenyl-guanidiniumiodid (39a) Zu einer Lösung von 8.90 g (50 mmol) N,N,N′-Trimethyl-N′-phenylharnstoff (36a) in 30 mL trockenem Benzol werden langsam 7.67 g (50 mmol) POCl3 in 10 mL Benzol getropft. Nach 8-stündigem Rühren bei Raumtemperatur werden langsam 4.65 g (50 mmol) Anilin in 20 mL trockenem Benzol zugetropft. Der Ansatz wird 18 Stunden bei 80 °C gerührt und danach die flüchtigen Bestandteile im Rotationsverdampfer im Vakuum bei 80 °C/15 mbar entfernt. Der ölige Rückstand wird mit 8.0 g (0.2 mol) NaOH in 50 mL Wasser versetzt und dreimal mit je 50 mL Benzol ausgeschüttelt. Die vereinigten organischen Phasen werden über MgSO4 getrocknet und im Rotationsverdampfer eingedampft. Der Rückstand wird mit Methyl-tert.-butylether gewaschen, um die Aminreste zu entfernen. Das Gemisch aus Guanidin und Harnstoff wird mit 14.2 g (0.1 mol) Methyliodid in 20 mL trockenem Acetonitril versetzt und 20 Stunden bei Raumtemperatur gerührt. Nach dem Abdampfen des Lösungsmittels wird der Rückstand dreimal mit je 20 mL Ethylacetat gewaschen und getrocknet. Ausb.: 12.1 g (61 %) 39a, farbloses Pulver mit Schmp. 112–113 °C. – IR (ATR): ν = 2998, 2929, 1601, 1574, 1521, 1452, 1402, 1253, 1115, 1023 cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.10 (br s, 6 H, NMe2), 3.56 (br s, 6 H, NMe2), 7.17–7.27 (m, 6 H, ArH), 7.33–7.48 (m, 2 H, ArH), 8.13–8.24 (m, 2 H, ArH. – 13C NMR (75 MHz, CDCl3, TMS): δ = 41.9, 42.1 (NMe), 122.6 (ArC), 127.0 (ArC), 130.0 (ArC), 141.6 (ArC), 161.4 (C=N+). – C17H22IN3 (395.29): ber. C 51.66, H 5.61, I 32.10, N 10.63; gef. C 51.78, H 5.62, I 31.95, N 10.63.
N,N,N′,N″-Tetramethyl-N′-(4-methoxyphenyl)-N″-phenyl-guanidiniumiodid (39b) Die gleiche Menge des Gemischs aus POCl3 und dem Harnstoff 36a werden, wie bei der Darstellung von 39a beschrieben, mit 6.15 g (50 mmol) 4-Methoxyanilin 13 h umgesetzt und aufgearbeitet. Der nach dem Abdampfen des Benzols erhaltene Rückstand wird über Kieselgel filtriert und mit Methyl-tert.-butylether ausgewaschen, um die Aminreste zu entfernen. Das Gemisch aus Guanidin und Harnstoff wird mit 14.2 g (0.1 mol) Methyliodid in 20 mL trockenem Acetonitril versetzt und 22 Stunden bei Raumtemperatur gerührt. Anschließend wird das Lösungsmittel im Rotationsverdampfer entfernt und der Rückstand dreimal mit je 20 mL Ethylacetat gewaschen und getrocknet. Ausb.: 8.7 g (41 %) 39b, farbloses Pulver mit Schmp. 134–135 °C. – IR (ATR): ν = 2996, 2910, 1598, 1574, 1507, 1447, 1297, 1248, 1112, 1028 cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.07 (br s, 6 H, NMe2), 3.53, 3.54 (je s, je 3 H, NMe2), 3.83 (s, 3 H, OMe), 6.87–6.91 (m, 2 H, ArH), 7.11–7.28 (m, 5 H, ArH), 7.38–7.48 (m, 2 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 41.8 (NMe), 42.6 (NMe), 55.7 (OMe), 115.2 (ArC), 122.5, 124.5 (ArC), 126.9 (ArC), 130.2 (ArC), 134.4 (ArC), 141.8 (ArC), 158.4, 161.3 (C=N+). – MS ((+)-ESI): m/z = 298.1913 (ber. 298.1914 für C18H24N3O, [M–I]+. – C18H24IN3O (425.32): ber. C 50.83, H 5.69, I 29.84, N 9.88; gef. C 50.73, H 5.69, I 29.83, N 9.65.
N,N,N′,N″-Tetramethyl-N′-(4-nitrophenyl)-N″-phenyl-guanidiniumiodid (39c) Wie bei 39b beschrieben wird das Reaktionsgemisch aus POCl3 und dem Harnstoff 36a mit 6.9 g (50 mmol) 4-Nitroanilin umgesetzt und aufgearbeitet. Das Gemisch aus Guanidin und Harnstoff wird mit 28.4 g (0.2 mol) Methyliodid in 20 mL Acetonitril versetzt und 48 Stunden bei Raumtemperatur gerührt. Anschließend wird das Lösungsmittel im Rotationsverdampfer entfernt und der Rückstand dreimal mit je 20 mL Ethylacetat gewaschen und getrocknet. Ausb.: 5.3 g (31 %) 39c, gelbes Pulver mit Schmp. 121–128 °C. – IR (ATR): ν = 2931, 2837, 1638, 1508, 1442, 1371, 1286, 1241, 1128, 1029 cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.19 (br s, 6 H, NMe2), 3.59, 3.69 (je s, je 3 H, NMe2), 7.14–7.29 (m, 5 H, ArH), 7.36–7.44 (m, 2 H, ArH), 8.18–8.24 (m, 2 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 41.5 (NMe), 42.8 (NMe), 122.3, 123.1, 125.6 (ArC), 127.8 (ArC), 130.3 (ArC), 141.2 (ArC), 144.8 (ArC), 147.1 (ArC), 161.2 (C=N+). – C17H21IN4O2 (440.29): ber. C 46.38, H 4.81, I 28.82, N 11.73; gef. C 46.49, H 4.93, I 29.32, N 11.33.
N,N,N′,N″-Tetramethyl-N′-(4-methoxy)phenyl)-N″-(4-nitro)phenyl-guanidiniumiodid (39d) Zu einer Lösung von 8.3 g (40 mmol) N,N,N′-Trimethyl-N′-(4-methoxyphenyl)-harnstoff 36b in 25 mL Benzol werden langsam 6.12 g (40 mmol) POCl3 in 10 mL Benzol zugetropft. Nach 6 Stunden bei Raumtemperatur werden langsam 5.5 g (40 mmol) 4-Nitroanilin in 20 mL Benzol zugetropft. Das Reaktionsgemisch wird bei 80 °C 20 Stunden gerührt; danach werden im Rotationsverdampfer im Vakuum (15 mbar) bei 80 °C die flüchtigen Bestandteile entfernt. Der ölige Rückstand wird mit 8.0 g (0.2 mol) NaOH in 50 mL Wasser versetzt, danach wird mit Benzol ausgeschüttelt. Die organische Phase wird über MgSO4 getrocknet und eingedampft. Der Rückstand wird über Kieselgel filtriert und mit Methyl-tert.-butylether ausgewaschen, um Aminreste zu entfernen. Das Gemisch aus Guanidin und Harnstoff wird mit 14.2 g (0.1 mol) Methyliodid in 20 mL trockenem Acetonitril versetzt. Nach 22-stündigem Rühren bei Raumtemperatur wird das Lösungsmittel im Rotationsverdampfer entfernt und der Rückstand dreimal mit je 20 mL Ethylacetat gewaschen und getrocknet. Ausb.: 6.7 g (36 %) 39d, farbloses Pulver, mit Schmp. 128–130 °C. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.18 (br s, 6 H, NMe2), 3.68 (br s, 6 H, NMe2), 3.81 (s, 3 H, OMe), 6.82–6.96 (m, 2 H, ArH), 7.12–7.32 (m, 2 H, ArH), 7.38–7.63 (m, 2 H, ArH), 8.18–8.26 (m, 2 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 42.6 (NMe), 42.8 (NMe), 43.3 (NMe), 55.6 (OMe), 115.3 (ArC), 122.0, 124.8 (ArC), 125.6, 133.8, 144.6, 147.3 (ArC), 158.9 (C=N+). – MS ((+)-ESI): m/z = 343.1749 (ber. 343.1765 für C18H23N4O3, [M–I]+). – C18H23IN4O3 (470.32): ber. C 45.97, H 4.93, I 26.98, N 11.91; gef. C 45.02, H 5.02, I 27.11, N 11.05.
N,N′-Dimethyl-N,N′-diphenylthioharnstoff (41a) Zu einer siedenden Suspension von 120 g (0.5 mol) N,N′-Dimethyl-N,N′-diphenylharnstoff (40a) in 1 L Toluol werden über 1 Stunde in fünf Portionen 111 g (0.25 mol) P4S10 zugegeben. Nach 24-stündigem Erhitzen unter Rückfluss wird das Reaktionsgemisch bei Raumtemperatur über Kieselgel filtriert. Das Filtrat wird im Rotationsverdampfer eingedampft und der Rückstand zweimal aus hochsiedendem Petrolether (Sdp. 110–140 °C) umkristallisiert. Ausb.: 88.5 g (69 %) 41a, gelbe Kristalle mit Schmp. 73.5 °C (Lit. [97]: Schmp.: 74 °C). – IR (ATR): ν = 3035, 2965, 2932, 1592, 1489, 1421, 1350, 1327, 1135, 1072 cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.51 (s, 6 H, NMe2), 6.62–6.69 (m, 4 H, ArH), 6.87–7.05 (m, 6 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 45.2 (NMe), 125.3, 125.4, 128.8, 147.5 (ArC), 190.8 (C=S). – C15H16N2S (256.43): ber. C 70.28, H 6.29, N 10.93, S 12.51; gef. C 70.16, H 6.31, N 11.06, S 12.52.
N,N′-Diethyl-N,N′-diphenylthioharnstoff (41b) Wie bei der Synthese von 41a beschrieben werden 134 g (0.5 mol) N,N′-Diethyl-N,N′-diphenylharnstoff (40b) in 1 L Toluol mit 111 g (0.25 mol) P4S10 umgesetzt und aufgearbeitet. Ausb.: 89 g (61 %) 41b, gelbe Kristalle mit Schmp. 76 °C (Lit: [97] Schmp.: 79 °C). – IR (ATR): ν = 3035, 2965, 2932, 1592, 1489, 1421, 1350, 1327, 1135, 1072 cm–1. – 1H NMR (300 MHz, CDCl3, TMS): δ = 1.34 (t, J = 6.8 Hz, 6 H, Me), 4.12 (q, J = 6.8 Hz, 4 H, NCH2), 6.58–6.64 (m, 4 H, ArH), 6.89–7.04 (m, 6 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 12.7 (Me), 51.2 (NCH2), 125.1, 126.0, 128.6, 145.9 (ArC), 189.3 (C=S). – C17H20N2S (284.43): ber. C 71.79, H 7.09, N 9.85, S 11.27; gef. C 71.46, H 7.06, N 9.73, S 11.49.
N,N′-Dimethyl-N,N′-diphenyl-chlorformamidiniumchlorid (43) In eine Lösung von 256 g (1.0 mol) N,N′-Dimethyl-N,N′-diphenylthioharnstoff (41a) in 1050 mL trockenem Acetonitril werden unter Ausschluss von Feuchtigkeit bei 0 °C 124 g (1.25 mol) Phosgen eingeleitet. Der Ansatz wird 72 h bei Raumtemperatur gerührt, wobei CO2 und Phosgen entweichen. Danach werden überschüssiges Phosgen und ca. 50 mL Acetonitril abdestilliert. Die so erhaltene ca. 1-molare Lösung von 43 wird bei den Umsetzungen verwendet.
N,N′,N″-Trimethyl-N,N′,N″-triphenyl-guanidiniumchlorid (44a) Zu einer Mischung aus Triethylamin (10 g, 0.10 mol) und N-Methylanilin (9.8 g, 0.10 mol) tropft man unter Rühren langsam bei 0 °C 29.5 g (0.1 mol) N,N′-Dimethyl-N,N′-diphenyl-chlorformamidiniumchlorid (43) in 100 mL Acetonitril. Nach 24 h Rühren bei Raumtemperatur wird das gebildete Hydrochlorid abfiltriert. Das Filtrat wird im Vak. eingedampft und der Rückstand in Tetrahydrofuran suspendiert. Der Feststoff wird abfiltriert und in Dichlormethan (ca. 100 mL) gelöst. Die Lösung wird mit Wasser ausgeschüttelt. Die organische Phase wird mit Natriumsulfat getrocknet und eingedampft. Ausb.: 20.8 g (57 %) stark hygroskopischer Feststoff. – 1H NMR (300 MHz, CDCl3, TMS): δ = 3.49 (s, 9 H, Me), 7.14–7.37 (m, 15 H, ArH). – 13C NMR (75 MHz, CDCl3, TMS): δ = 41.9 (Me), 123.3, 127.5, 130.0, 141.9 (ArC), 161.3 (C+).
N,N′,N″-Trimethyl-N,N′,N″-triphenyl-guanidiniumtetraphenylborat (44b) Zu einer Lösung von 1.0 g (2.7 mmol) N,N′,N″-Trimethyl-N,N′,N″-triphenyl-guanidiniumchlorid in 10 mL siedendem Acetonitril gibt man unter Rühren eine Lösung von 923 mg (2.7 mmol) Natriumtetraphenylborat in 5 mL Acetonitril. Die Lösung wird kurz erhitzt und filtriert. Das Filtrat wird eingedampft und der farblose Rückstand im Vak. getrocknet. Ausb.: 1.1 g (62 %) 47b farbloses Pulver, mit Schmp. 200–201 °C. – 1H NMR (250 MHz, CD3CN, TMS): δ = 3.26 (s. 9 H, Me), 6.76–6.88 (m, 4 H, ArH), 6.90–7.05 (m, 9 H, ArH), 7.06–7.14 (m, 5 H, ArH), 7.16–7.38 (m, 17 H, ArH). – 13C NMR (62.9 MHz, CD3CN, TMS): δ = 40.3 (Me), 121.4, 122.9, 125.2, 125.3, 127.1, 129.5, 135.4, 141.3 (ArC), 161.0 (C+), 163.8 (q, J = 49 Hz, C4B). – C46H44N3B (649.69): ber. C 85.04, H 6.83, N 6.47; gef. C 84.07, H 6.82, N 6.35.
N,N,N′,N′N″,N″-Hexamethyl-guanidiniumchlorid 9c aus dem N,N,N′,N′-Tetramethylharnstoff/POCl3-Addukt und Trimethylsilylmethylamin (15) – Allgemeine Vorschrift (Versuche 1, 3, 5, Tabelle 3) Zu 15.3 g (0.1 mol) Phosphorylchlorid in 20 mL trockenem Acetonitril tropft man unter Rühren bei Raumtemperatur 11.6 g (0.1 mol), 23.2 g (0.2 mol) bzw. 17.4 g (0.15 mol) N,N,N′,N′-Tetramethylharnstoff (1a) zu und rührt noch 1 h bei Raumtemperatur. Danach werden 23.5 g (0.2 mol) des Silylamins 15 unter Rühren zugetropft und das Gemisch noch 15 h bei 60 °C gerührt. Die flüchtigen Bestandteile mit Sdp. <82 °C werden über eine 30 cm lange Vigreux Kolonne abdestilliert und der Rückstand im Rotationsverdampfer im Vakuum zur Trockene eingedampft. Das Produkt wird mit ca. 50 mL Aceton versetzt, filtriert, mit Ether gewaschen und aus N,N-Dimethylformamid umkristallisiert.
Allgemeine Vorschrift (Versuche 2, 4, Tabelle 3) Wie vorstehend beschrieben wird Phosphorylchlorid mit 1a umgesetzt, wobei der Harnstoff so zügig zugetropft wird, dass die Temperatur bis maximal 40 °C ansteigt, danach wird noch 0.5 h gerührt. Anschließend werden 11.7 g (0.1 mol) bzw. 23.5 g (0.2 mol) des Silylamins 15 bei 40 °C unter Rühren zugetropft. Nach anschließendem 4-stündigem Rühren bei 60 °C wird der Ansatz mit 10 %-iger Sodalösung versetzt, bis die Mischung deutlich alkalisch ist. Das Gemisch wird zweimal mit je 25 mL Methylenchlorid ausgeschüttelt. Die Wasserphase wird im Vakuum eingedampft. Der Rückstand wird noch 1 h im Vakuum bei 70 °C belassen und danach in einer Soxhlet-Apparatur mit Methylenchlorid extrahiert. Durch Eindampfen des Extrakts erhält man das Guanidiniumsalz.
Dank: Herrn Dr. Bojan Iliev danken wir für die experimentelle Mitarbeit. Die Untersuchungen wurden vom Bundesministerium für Bildung und Forschung (BMBF) Bonn gefördert: BMBF-Projekt: Neuartige ionische Flüssigkeiten als innovative Reaktionsmedien für die Technische Organische Chemie, FKZ 01RI 05176. Der Firma BASF SE Ludwigshafen danken für Chemikalienspenden.
**Orthoamide und Iminiumsalze, LXXXVII: siehe Lit. [1]
References
[1] W. Kantlehner, J. Mezger, I. C. Ivanov, Z. Naturforsch., B: J. Chem. Sci. 2014, 69b, 519–524.Search in Google Scholar
[2] W. Kantlehner, W. W. Mergen, Synthesis 1979, 11, 343–344.10.1055/s-1979-28672Search in Google Scholar
[3] W. Kantlehner, L. Kienitz, H. Jaus, H. Bredereck, Liebigs Ann. Chem. 1979, 12, 2089–2095.Search in Google Scholar
[4] W. Kantlehner, P. Speh, H. J. Bräuner, Synthesis 1983, 1983, 905–906.10.1055/s-1983-30559Search in Google Scholar
[5] W. Kantlehner, R. Stieglitz, M. Hauber, E. Haug, C. Regele, J. Prakt. Chem. (Weinheim, Ger.), 2000, 342, 256–268.10.1002/(SICI)1521-3897(200003)342:3<256::AID-PRAC256>3.0.CO;2-GSearch in Google Scholar
[6] W. Kantlehner, E. Haug, R. Stieglitz, W. Frey, R. Kreß, J. Mezger, Z. Naturforsch., B: J. Chem. Sci. 2002, 57b, 399–419.Search in Google Scholar
[7] W. Kantlehner, E. Haug, W. W. Mergen, P. Speh, H. Lehmann, Chem.-Ztg. 1990, 114, 176–178.Search in Google Scholar
[8] W. Kantlehner, M. Hauber, M. Vettel, J. Prakt. Chem./Chem.-Ztg. 1996, 338, 403–413.Search in Google Scholar
[9] W. Kantlehner, M. Vettel, H. Lehmann, K. Edelmann, R. Stieglitz, I. C. Ivanov, J. Prakt. Chem./Chem.-Ztg. 1998, 340, 408–423.Search in Google Scholar
[10] W. Weingärtner, W. Kantlehner, G. Maas. Synthesis 2011, 43, 265–272.Search in Google Scholar
[11] W. Kantlehner, E. Haug, W. W. Mergen, P. Speh, T. Maier, J. J. Kapassakalidis, H.-J. Bräuner, H. Hagen, Liebigs Ann. Chem. 1984, 1984, 108–126.Search in Google Scholar
[12] H. Kunkel, G. Maas, Eur. J. Org. Chem. 2007, 22, 3746–3757.Search in Google Scholar
[13] N. Ignatyev, U. Welz-Biermann, G. Birsky, H. Willner, WO 2005/075413 (A1), 2004.Search in Google Scholar
[14] N. Ignatyev, U. Welz-Biermann, H. Willner, G. Birsky, DE 10325051 (A1), 2004.Search in Google Scholar
[15] N. M. Magalhaes Mateus, L. A. Almeida Fernandes Cobra Branco, C. A. Mateus Alfonso, J. M. Reis de Aguiar Navarro Y Rosa, P. M. Pimenta Gois, N. M. Torres Lourenso, EP 1466894 (A1), 2004.Search in Google Scholar
[16] W. Kantlehner, J. Mezger, R. Kreß, H. Hartmann, T. Moschny, I. Tiritiris, B. Iliev, O. Scherr, G. Ziegler, B. Souley, W. Frey, I. C. Ivanov, M. G. Bogdanov, U. Jäger, G. Dospil, T. Viefhaus, Z. Naturforsch., B: J. Chem. Sci. 2010, 65b, 873–906.Search in Google Scholar
[17] W. Kantlehner, J. J. Kapassakalidis, P. Speh, H.-J. Bräuner, Liebigs Ann. Chem. 1980, 1980, 389–393.Search in Google Scholar
[18] Übersicht: J.-P. Sennet, C. R. Acad. Sci., Ser. IIc: Chim. 2000, 3, 505–516.Search in Google Scholar
[19] J.-P. Senet, The Recent Advance in Phosgene Chemistry, Vol. 1, Groupe SNPE, Paris, 1997.Search in Google Scholar
[20] A. Pleschke, A. Marhold, M. Schneider, A. Kolomeitsev, G.-V. Röschenthaler, J. Fluorine Chem. 2004, 125, 1031–1038.Search in Google Scholar
[21] C. R. Schlaikjer, EP 1363345 (A2), 2003.Search in Google Scholar
[22] P. Wang, S. M. Zakeeruddin, M. Grätzel, W. Kantlehner, J. Mezger, E. V. Stoyanov, O. Scherr, Appl. Phys. A: Mater. Sci. Process. 2004, 79, 73–77.Search in Google Scholar
[23] H. G. Viehe, Z. Janousek, Angew. Chem. 1971, 83, 614–615; Angew. Chem., Int. Ed. Engl. 1971, 10, 573–574.Search in Google Scholar
[24] V. P. Kukhaŕ, V. I. Pasternak, A. V. Kirsanov, Zh. Org. Khim. 1971, 7, 2084–2086.Search in Google Scholar
[25] V. P. Kukhaŕ, V. I. Pasternak, G. V. Pesotskaya, Zh. Org. Khim. 1973, 9, 30–45.Search in Google Scholar
[26] D. F. Miranova, N. I. Vykhvestyuk, V. P. Kukhaŕ, Zh. Org. Khim. 1984, 20, 272–276.Search in Google Scholar
[27] C. Copeland, R. V. Stick, Aust. J. Chem. 1982, 35, 2541–2546.Search in Google Scholar
[28] G. Wieland, G. Simchen, Liebigs Ann. Chem. 1985, 1985, 2178–2193.Search in Google Scholar
[29] M. Vilkas, D. Qasmi, Synth. Commun. 1990, 20, 2769–2773.Search in Google Scholar
[30] T. Schlama, V. Gouverneur, A. Valleix, A. Greiner, L. Toupet, C. Mioskowski, J. Org. Chem. 1997, 62, 4200–4202.Search in Google Scholar
[31] N. M. M. Mateus, L. C. Branco, N. M. T. Lourenço, C. A. M. Afonso, Green Chem. 2003, 5, 347–352.Search in Google Scholar
[32] R. Weiss, R. Roth, Synthesis 1987, 49, 870–872.10.1055/s-1987-28106Search in Google Scholar
[33] H. Meerwein, W. Florian, N. Schön, G. Stopp, Justus Liebigs Ann. Chem. 1961, 641, 1–39.Search in Google Scholar
[34] H. Bredereck, F. Effenberger, H. P. Beyerlin, Chem. Ber. 1964, 97, 1834–1838.Search in Google Scholar
[35] H. K. Hall Jr., J. Am. Chem. Soc. 1956, 78, 2717–2719.Search in Google Scholar
[36] W. E. Bottomley, G. V. Boyd, J. Chem. Soc., Chem. Commun. 1980, 790–791.10.1039/c39800000790Search in Google Scholar
[37] M. Itoh, Chem. Pharm. Bull. 1970, 18, 784–788.Search in Google Scholar
[38] J. A. De Groot, R. Van der Steen, R. Fokkeus, J. Lugtenburg, Recl.: J. R. Neth. Chem. Soc. 1982, 101, 35–40.Search in Google Scholar
[39] A. G. H. Wee, E. Bunnenberg, C. Djerassi, Synth. Commun. 1984, 14, 383–390.Search in Google Scholar
[40] D. C. Miller, M. R. Johnson, J. J. Becker, J. A. Ibers, J. Heterocycl. Chem. 1993, 30, 1485–1490.Search in Google Scholar
[41] G. M. Iseitlin, B. V. Tokarev, O. V. Jlingin, Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol. 1979, 22, 1190–1193.Search in Google Scholar
[42] A. Ranise, S. Cesarini, A. Spallarossa, F. Sancassan, F. Bondavalli, O. Bruno, S. Schenone, G. Menozzi, P. Fossa, L. Mosti, Synthesis 2007, 39, 2495–2502.10.1055/s-2007-983811Search in Google Scholar
[43] M. Zarei, Bull. Chem. Soc. Jpn. 2012, 85, 360–368.Search in Google Scholar
[44] A. S. Vinogradov, V. I. Krasnov, V. E. Platonov, Mendeleev Commun. 2008, 18, 227–228.Search in Google Scholar
[45] C. Lemarre, L. Stella, Synlett 1999, 31, 725–726.10.1016/S0026-2692(00)00050-1Search in Google Scholar
[46] A. B. Charette, M. Grenon, Tetrahedron Lett. 2000, 41, 1677–1680.Search in Google Scholar
[47] A. Mekhalfia, R. Mutter, W. Heal, B. Chen, Tetrahedron 2006, 62, 5617–5625.10.1016/j.tet.2006.03.099Search in Google Scholar
[48] F. Würthner, Synthesis 1999, 1999, 2103–2113.10.1055/s-1999-3635Search in Google Scholar
[49] H. Eilingsfeld, M. Seefelder, H. Weidinger, Angew. Chem. 1960, 72, 836–845.Search in Google Scholar
[50] V. J. Bauer, S. R. Safir, J. Med. Chem. 1966, 9, 980–981.Search in Google Scholar
[51] T. Fujisawa, T. Mori, K. Fukumoto, T. Sato, Chem. Lett. 1982, 11, 1891–1894.Search in Google Scholar
[52] A. Hamed, E. Müller, J. C. Jochims, Tetrahedron 1986, 42, 6645–6656.10.1016/S0040-4020(01)82103-7Search in Google Scholar
[53] U. Schuchardt, R. M. Vargas, G. Gelbard, J. Mol. Catal. A: Chem. 1995, 99, 65–70.Search in Google Scholar
[54] T. Vojkovsky, B. Drake, Org. Prep. Proced. Int. 1997, 29, 497–498.Search in Google Scholar
[55] K. Edelmann, Dissertation, Universität Stuttgart, Stuttgart, 1998.Search in Google Scholar
[56] A. El-Faham, Chem. Lett. 1998, 27, 671–672.Search in Google Scholar
[57] T. Isobe, K. Fukuda, K. Yamaguchi, H. Seki, T. Tokunaga, T. Ishikawa, J. Org. Chem. 2000, 65, 7779–7785.Search in Google Scholar
[58] T. Ishikawa, T. Isobe, Chem. Eur. J. 2002, 8, 552–557.Search in Google Scholar
[59] M. A. Bailén, R. Chinchilla, D. J. Dodsworth, C. Nájera, Tetrahedron Lett. 2002, 43, 1661–1664.Search in Google Scholar
[60] T. Ishikawa, Arkivoc 2006, vii, 148–168.10.3998/ark.5550190.0007.713Search in Google Scholar
[61] J. Shah, J. Liebscher, Synthesis 2008, 40, 917–920.10.1055/s-2008-1032204Search in Google Scholar
[62] Y. Kimura, D. Matsuura, T. Hanawa, Y. Kobayashi, Tetrahedron Lett. 2012, 53, 1116–1118.Search in Google Scholar
[63] Die in dieser Arbeit beschriebenen Forschungsarbeiten wurden im Rahmen des BMBF-Projekts „Neuartige ionische Flüssigkeiten als innovative Reaktionsmedien für die Technische Organische Chemie‟ (Laufzeit 2005–2009) durchgeführt. Der bei der Technischen Informationsbibliothek Hannover dazu eingereichte Abschlussbericht (W. Kantlehner, G. Maas; Abschlussbericht zum BMBF-Verbundprojekt, FKZ 01RI05176, Juni 2010) ist seit Juni 2010 für jedermann einsehbar und erhältlich.Search in Google Scholar
[64] Übersicht: W. Kantlehner in Iminium Salts in Organic Chemistry (Adv. Org. Chem. 9/2), Vol. 2, (Eds.: H. G. Viehe, H. Böhme), John Wiley & Sons, New York, 1979, p. 5.Search in Google Scholar
[65] Übersicht: Ch. Jutz in Iminium Salts in Organic Chemistry (Adv. Org. Chem. 9/1), Vol. 1, (Eds.: H. G. Viehe, H. Böhme), John Wiley & Sons, New York, 1976, p. 225.Search in Google Scholar
[66] J.-R. Gavreau, G.-J. Martin, J. Chem. Soc., Perkin Trans. (1972–1999) 1983, 2, 1541–1543.10.1039/p29830001541Search in Google Scholar
[67] U. Boas, B. Pedersen, J. B. Christensen, Synth. Commun. 1998, 28, 1223–1231.Search in Google Scholar
[68] H. Bredereck, K. Bredereck, Chem. Ber. 1961, 94, 2278–2295.Search in Google Scholar
[69] H. Kessler, D. Leibfritz, Tetrahedron 1970, 26, 1805–1820.10.1016/S0040-4020(01)92757-7Search in Google Scholar
[70] H. Kessler, D. Leibfritz, Chem. Ber. 1971, 104, 2158–2169.Search in Google Scholar
[71] W. Galezowski, K. T. Leffek, P. Pruszynski, Can. J. Chem. 1992, 70, 2390–2393.Search in Google Scholar
[72] P. Pruszynski, Can. J. Chem. 1987, 65, 626–629.Search in Google Scholar
[73] D. J. Brunelle, US 5081298 (A), 1992.Search in Google Scholar
[74] D. J. Brunelle, US 5229482 (A), 1993.Search in Google Scholar
[75] D. J. Brunelle, D. H. Haitko, J. P. Barren, S. Singh, US 5132423 (A), 1992.Search in Google Scholar
[76] R. Iemura, M. Hori, T. Saito, H. Ohtaka, Chem. Pharm. Bull. 1989, 37, 2723–2726.Search in Google Scholar
[77] C. Anaya De Parrodi, L. Quintero-Cortés, J. Sandoval-Ramírez, Synth. Commun. 1996, 26, 3323–3329.Search in Google Scholar
[78] H. Göker, G. Ayhan-Kilcigil, M. Tunçbilek, C. Kus, R. Ertan, E. Kendi, S. Özbey, M. Fort, C. Garcia, A. J. Farré, Heterocycles 1999, 51, 2561–2573.10.3987/COM-99-8585Search in Google Scholar
[79] F. Chang Fong, K. Benamour, B. Szymonski, F. Thomasson, J.-M. Morand, M. Cussak. Chem. Pharm. Bull. 2000, 48, 729–733.Search in Google Scholar
[80] H. Xie, S. Zhang, H. Duan, Tetrahedron Lett. 2004, 45, 2013–2015.Search in Google Scholar
[81] S.-S. Hong, S. A. Bavadekar, S.-I. Lee, P. N. Patil, S. G. Lalchandani, D. R. Feller, D. D. Miller, Bioorg. Med. Chem. Lett. 2005, 15, 4691–4695.Search in Google Scholar
[82] P. Molina, M. Alajarín, A. Vidal, Tetrahedron 1995, 51, 5351–5360.10.1016/0040-4020(95)00196-FSearch in Google Scholar
[83] Y. Gao, S. W. Arritt, B. Twamley, J. M. Shreeve, Inorg. Chem. 2005, 44, 1704–1712.Search in Google Scholar
[84] U. Hary, U. Roettig, M. Paal, Tetrahedron Lett. 2001, 31, 5187–5189.Search in Google Scholar
[85] W. M. Kan, S.-H. Lin, C.-Y. Chern, Synth. Commun. 2005, 35, 2633–2639.Search in Google Scholar
[86] I. Tiritiris, W. Frey, W. Kantlehner, Acta Crystallogr., Sect. E: Struct. Rep. Online 2014, E70 (Pt 4), o460.10.1107/S1600536814005819Search in Google Scholar
[87] I. Tiritiris, W. Frey, W. Kantlehner, Acta Crystallogr., Sect. E: Struct. Rep. Online 2014, E70 (Pt 5), o516.10.1107/S160053681400693XSearch in Google Scholar
[88] H. Eilingsfeld, G. Neubauer, M. Seefelder, H. Weidincer, Chem. Ber. 1964, 97, 1232–1245.Search in Google Scholar
[89] A. Tanatani, K. Yamaguchi, I. Azumaya, R. Fukutomi, K. Shudo, H. Kagechika, J. Am. Chem. Soc. 1998, 120, 6433–6442.Search in Google Scholar
[90] G. Ziegler, Diplomarbeit, FH Aalen, Aalen, 1992.Search in Google Scholar
[91] P. J. Stang, G. Maas, D. L. Smith, J. A. McCloskey, J. Am. Chem. Soc. 1981, 103, 4837–4845.Search in Google Scholar
[92] G. P. Schiemenz, G. Stein, Tetrahedron 1970, 26, 2007–2026.10.1016/S0040-4020(01)92776-0Search in Google Scholar
[93] Handbook of Chemistry and Physics, 52. Aufl., (Ed.: R. C. Weast), The Chemical Rubber Co., Cleveland, Ohio, 1971.Search in Google Scholar
[94] J. Voß, Justus Liebigs Ann. Chem. 1971, 746, 92–101.Search in Google Scholar
[95] P. Hanson, D. A. R. Williams, J. Chem. Soc., Perkin Trans. 2 (1972–1999) 1973, 2162–2165.10.1039/P29730002162Search in Google Scholar
[96] W. Michler, Ber. Dtsch. Chem. Ges. 1876, 9, 716–718.Search in Google Scholar
[97] O. Billeter, Ber. Dtsch. Chem. Ges. 1887, 20, 1629–1632.Search in Google Scholar
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Editorial
- Zeitschrift für Naturforschung B now being published by De Gruyter
- Original Communications
- “Naked” S2O72– ions – the serendipitous formation of the disulfates [HPy]2[S2O7] and [bmim][HPy][S2O7] (HPy = pyridinium; bmim = 1-Butyl-3-methylimidazolium)
- Orthoamide und Iminiumsalze, LXXXVIII. Synthese N,N,N′,N′,N″,N″-persubstituierter Guanidiniumsalze aus N,N′-persubstituierten Harnstoff/Säurechlorid-Addukten**
- The acidic ionic liquid [BSO3HMIm]HSO4: a novel and efficient catalyst for one-pot, three-component syntheses of substituted pyrroles
- The influence of alkali-metal ions on the molecular and supramolecular structure of manganese(II) complexes with tetrachlorophthalate ligands
- New triazolothiadiazole and triazolothiadiazine derivatives as kinesin Eg5 and HIV inhibitors: synthesis, QSAR and modeling studies
- Two copper(I) complexes of bi- (or tri-)pyrazolyl ligands featuring Cu3pz3 or Cu4pz4 motifs
- Synthesis of ferrocenyl aryl ethers via Cu(I)/phosphine catalyst systems
- A prenylated acridone alkaloid and ferulate xanthone from barks of Citrus medica (Rutaceae)
- Synthesis and structural characterization of substituted phenols with a m-terphenyl backbone 2,4,6-R3C6H2OH (R=2,4,6-Me3C6H2, Me5C6)
- 2-Ethyl-1-phenylindazolium hexafluorophosphate. N-heterocyclic carbene formation, rearrangement, ring-cleavage reactions, and rhodium complex formation
Articles in the same Issue
- Frontmatter
- In this Issue
- Editorial
- Zeitschrift für Naturforschung B now being published by De Gruyter
- Original Communications
- “Naked” S2O72– ions – the serendipitous formation of the disulfates [HPy]2[S2O7] and [bmim][HPy][S2O7] (HPy = pyridinium; bmim = 1-Butyl-3-methylimidazolium)
- Orthoamide und Iminiumsalze, LXXXVIII. Synthese N,N,N′,N′,N″,N″-persubstituierter Guanidiniumsalze aus N,N′-persubstituierten Harnstoff/Säurechlorid-Addukten**
- The acidic ionic liquid [BSO3HMIm]HSO4: a novel and efficient catalyst for one-pot, three-component syntheses of substituted pyrroles
- The influence of alkali-metal ions on the molecular and supramolecular structure of manganese(II) complexes with tetrachlorophthalate ligands
- New triazolothiadiazole and triazolothiadiazine derivatives as kinesin Eg5 and HIV inhibitors: synthesis, QSAR and modeling studies
- Two copper(I) complexes of bi- (or tri-)pyrazolyl ligands featuring Cu3pz3 or Cu4pz4 motifs
- Synthesis of ferrocenyl aryl ethers via Cu(I)/phosphine catalyst systems
- A prenylated acridone alkaloid and ferulate xanthone from barks of Citrus medica (Rutaceae)
- Synthesis and structural characterization of substituted phenols with a m-terphenyl backbone 2,4,6-R3C6H2OH (R=2,4,6-Me3C6H2, Me5C6)
- 2-Ethyl-1-phenylindazolium hexafluorophosphate. N-heterocyclic carbene formation, rearrangement, ring-cleavage reactions, and rhodium complex formation