Synthesis of ferrocenyl aryl ethers via Cu(I)/phosphine catalyst systems
-
 und
und

Abstract
Ferrocenyl aryl ethers can be synthesized in good yields by Cu(I)/phosphine-catalyzed coupling reactions from iodoferrocene or 1,1′-dibromoferrocene and various phenols in toluene, using Cs2CO3 or K3PO4 as a base. For the first time a solid-state structure of a ferrocenyl-1,1′-diaryl ether [1,1′-di(4-tert-butylphenoxy)ferrocene] has been determined from single-crystal X-ray data. The mixed ferrocenyl aryl ether 1-(4-tert-butylphenoxy)-1′-(2,4-dimethylphenoxy)ferrocene was prepared in a two-step synthetic protocol.
1 Introduction
Ferrocenes are an important class of organometallic compounds for synthesis [1–7]. Current interest in ferrocenyl ethers lies in particular in the possibility for their use as organometallic ligands in homogeneous catalysts [8–13]. The synthetic availability of heteroatom-substituted ferrocenes is still limited because the routes for the introduction of heteroatoms at benzenes are not suitable for ferrocenes [12, 13]. Pertinent examples of successful syntheses are the amino- or hydroxyferrocenes functionalized via nucleophilic substitution with alkyl halides [14–17] or the synthesis of ferrocenyl alkyl ethers via a trialkylsilyl-protected hydroxyferrocene [18, 19].
Presently, only very few ferrocenyl aryl ethers are known. The first synthesis of such compounds date back to the 1960s, when Rausch [20] and Nefedova [21], employed Ullmann-type coupling resulting in product yields of ferrocenyl phenyl ether of 10–25%.
2 Results and discussion
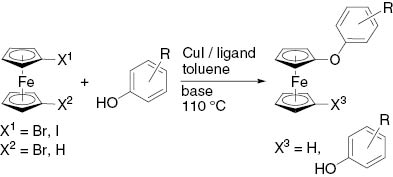
In a report by an der Heiden et al. [1], the high-yield synthesis of several ferrocenyl aryl ethers was described starting from iodo- and 1,1′-diiodoferrocene and phenols, mediated by Cu(I)/2,2,6,6-tetramethylheptane-3,5-dione (TMHD) or CuI/1,10-phenanthroline catalytic systems. The Cu(I)/TMHD system showed only half the activity when using brominated ferrocenes, and a much higher catalyst loading was required. The promising results obtained in this study with a CuI/phosphine system in toluene were not further evaluated. As a consequence, we used a CuI/phosphine system in toluene, a similar system to the Venkataraman catalyst (CuBr(PPh3)3) [22, 23], to further evaluate the potential of this coupling reaction of iodoferrocene and 1,1′-dibromoferrocene with phenols.
The results for this reaction are collected in Table 1. Entries 1 and 2 differ in a carbon–iodine vs. carbon–bromine bond, while the same catalyst was used. The more sophisticated reaction using 1,1′-dibromoferrocene resulted in a low yield (15%) compared with the results obtained with iodoferrocene [1]. A change to a less bulky alkylphosphine ligand did slightly improve the yield (up to 27%, Table 1; entry 3). In the present investigation, we have extended our previous studies and make use of different carbon–halogen bond strengths in the variation of product formation. According to the previous publication, we changed the ligand system back to arylphosphines, which provided a reasonable improvement in yield using Cs2CO3 as the base (up to 86%, Table 1; entries 4–7). A comparison of the two different bases Cs2CO3 and K3PO4 showed an additional improvement from 83 to 97% (Table 1; entries 7–8). Nearly identical results were also obtained for iodoferrocene in the previous work by an der Heiden et. al., where 85 and 99% were obtained, respectively [1]. The influence of the ligand system and the solvent does not play any specific role, contrary to previously published results using iodoferrocene and the NMP/TMHD/toluene catalytic systems for less sterically hindered phenols (substitution in meta or para position) (comparison of Tables 1–3 in reference [1] with the present Table 1 entries 9 and 10). A dramatic decrease in yield was only observed for ortho-substituted phenols, regardless of whether Cs2CO3 or K3PO4 was used as the base (Table 1, entries 11–13).
Screening for the coupling of ferrocene halides and phenols in toluene a.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Ferrocene | Ligand | Base | Yieldb (%) | Compound |
| 1 | 4-t-Bu | FcIc | (1-Ad)2PBn 2 eq. | Cs2CO3 | 84 [1] | 1 |
| 2 | 4-t-Bu | 1,1′-FcBr2d | (1-Ad)2PBn 2 eq. | Cs2CO3 | 15 | 2 |
| 3 | 4-t-Bu | 1,1′-FcBr2d | Cy2PBn 2 eq. | Cs2CO3 | 27 | 2 |
| 4 | 4-t-Bu | 1,1′-FcBr2d | dppm 1 eq. | Cs2CO3 | 86 | 2 |
| 5 | 4-t-Bu | 1,1′-FcBr2d | PPh2(o-i-propylphenyl) 2 eq. | Cs2CO3 | 86 | 2 |
| 6 | 4-t-Bu | 1,1′-FcBr2 | P(o-tolyl)3 2 eq. | Cs2CO3 | 64 | 2 |
| 7 | 4-t-Bu | 1,1′-FcBr2d | PPh3 2.5 eq. | Cs2CO3 | 83 | 2 |
| 8 | 4-t-Bu | 1,1′-FcBr2d | PPh3 2.5 eq. | K3PO4 | 97 | 2 |
| 9 | 4-Cl | FcIc | PPh3 3 eq. | Cs2CO3 | 96 | 3 |
| 10 | 3-t-Bu | FcIc | PPh3 3 eq. | Cs2CO3 | 94 | 4 |
| 11 | 2-t-Bu, 4-Me | FcIc,e | PPh3 3 eq. | K3PO4 | 27 | 5 |
| 12 | 2,4-t-Bu2 | FcIc,e | PPh3 3 eq. | K3PO4 | 23 | 6 |
| 13 | 2,4,6-Me3 | FcIc | PPh3 3 eq. | Cs2CO3 | 37 | 7 |
| 14 | 2,4,6-Me3 | 1,1′-FcBr2d | PPh3 3 eq. | Cs2CO3 | 23 | 8 |
| 15 | 3,5-Me2 | 1,1′-FcBr2d | PPh3 3 eq. | Cs2CO3 | 54 | 9 |
| 16 | 2-t-Bu, 4-OMe | FcIc | PPh3 3 eq. | Cs2CO3 | (24) | 10 |
| 17 | 2-NO2 | 1,1′-FcBr2d | PPh3 3 eq. | K3PO4 | <5 (none) | – |
| 18 | 2-NO2 | FcIc | PPh3 3 eq. | Cs2CO3 | <5 (none) | – |
| 19 | 2-NO2, 4-OMe | FcIc | (1-Ad)2PBn 2 eq. | Cs2CO3 | <5 (none) | – |
| 20 | 2-NO2, 4-OMe | FcIc | (1-Ad)2PBn 2 eq. | DBU | <5 (none) | – |
| 21 | 2-NO2, 4-OMe | FcIc | Cy2PBn 2 eq. | KOt-Bu | <5 (none) | – |
| 22 | 4-t-Bu | 1,1′-FcBr2f | PPh3 3 eq. | Cs2CO3 | (43)g | 11 |
| 23 | 2,4-Me2 | 11c | PPh3 3 eq. | Cs2CO3 | (56) | 12 |
aConditions: ferrocene (0.125 mmol), CuI (5 mol.%), ligand (10–15 mol.% according to CuI), base (0.25 mmol), T=110°C, toluene. Samples were taken after 26 h; byields determined by GC; in parentheses the isolated yield is given; cphenol (0.25 mmol); dphenol (0.35 mmol); ereaction time was 60 h instead of 26 h; fphenol (0.125 mmol); gresults in ∼62% yield from theory.
With this knowledge in hand, the less reactive 1,1′-dibromoferrocene was tested in entry 14, resulting in another decrease in yield (24% by GC) compared with the iodoferrocene reaction (37%). If the steric hindrance is reduced by a meta-substitution, with 1,1′-dibromoferrocene, a moderate yield could also be obtained (Table 1 entry 15). The reaction of iodoferrocene with a deactivated phenol such as 4-methoxyphenol resulted in a product yield of only 24% after column chromatography (see Table 1 entry 16). Reactions with phenols containing the strong electron-withdrawing substituent -NO2 failed completely (Table 1 entries 17–21), even if the phosphine or the ferrocene species were modified. Similar unsuccessful results were obtained in previous work [1].
Considering the obtained yields for these reactions, we found a suitable variation in starting material and side-product rates. For example, for entries 2 and 3, we obtained between 54 and 68% bromoferrocene and 1,1′-dibromoferrocene, ∼9% 1,1″-biferrocene, and 6–7% ferrocene after the given reaction time. For entry 4, we found 1% bromoferrocene, 3% 1,1′-dibromoferrocene, ∼3% 1,1″-biferrocene, and 7% ferrocene. For entry 17, where 1,1′-dibromoferrocene was used, we found a 3:1 ratio of bromoferrocene: 1,1′-dibromoferrocene after 26 h. In contrast to entries 18–21, where the more reactive iodoferrocene was used, we obtained after the 26-h reaction time mainly unreacted material (∼50–60%) and 1,1″-biferrocene and ferrocene in a ratio of ∼2:3.
If only one equivalent of phenol was used in the reaction with 1,1′-dibromoferrocene (Table 1, entry 22), a yield of 43% was obtained for product 11. In an additional attempt to prepare a mixed phenoxyferrocene, compound 11 was reacted with 2,4-dimethylphenol to obtain compound 12 in 56% yield after workup by column chromatography.
Crystals of 1,1′-di(4-tert-butylphenoxy)ferrocene (2) could be obtained by slow evaporation of the solvent (CDCl3), and their structure has been determined (Figs. 1 and 2). The result has provided data for the first structurally authenticated ferrocenyl diaryl ether complex. Selected bond lengths (in Å) and angles (in deg) are presented in the caption of Fig. 1.
![Fig. 1 diamond [24] plot of compound 2 in the solid state, showing 50% probability displacement ellipsoids and the atom numbering scheme. H atoms have been omitted for clarity. D1–Fe1 1.6549(4), D2–Fe1 1.6565(4), C1–O1 1.412(3), C6–O2 1.397(3), C11–O1 1.398(3), C21–O2 1.396(3), O1–C1–C6–O2 ∼74.12° (D1=centroid C1–C5, D2=centroid C6–C10).](/document/doi/10.1515/znb-2014-0178/asset/graphic/znb-2014-0178_fig1.jpg)
diamond [24] plot of compound 2 in the solid state, showing 50% probability displacement ellipsoids and the atom numbering scheme. H atoms have been omitted for clarity. D1–Fe1 1.6549(4), D2–Fe1 1.6565(4), C1–O1 1.412(3), C6–O2 1.397(3), C11–O1 1.398(3), C21–O2 1.396(3), O1–C1–C6–O2 ∼74.12° (D1=centroid C1–C5, D2=centroid C6–C10).
![Fig. 2 diamond [24] plot of compound 2, showing the conformation of the two Cp rings. H atoms have been omitted for clarity.](/document/doi/10.1515/znb-2014-0178/asset/graphic/znb-2014-0178_fig2.jpg)
diamond [24] plot of compound 2, showing the conformation of the two Cp rings. H atoms have been omitted for clarity.
The two Cp rings are rotated by 74.1(2)° against each other, with the two ether groups pointing in opposite directions out of the Cp planes to reduce steric hindrance. The molecules are connected by a single intermolecular hydrogen bond C9–H9···O2 with (H9···O2 2.51(3), H9–C9 0.97(4), C9···O2 3.398(4) Å). All other intermolecular hydrogen bonds establishing the three-dimensional framework are longer than 2.76(4) Å (H5···C12 in C5–H5···C12; H5–C5 0.95(4) Å, C5···C12 3.632(5) Å).
3 Conclusions
We present herein a simple high-yield synthesis of a variety of different ferrocenyl aryl ethers by an Ullmann-type coupling protocol. Using the more inexpensive bromo- and 1,1′-dibromo-ferrocenyl precursors instead of their iodo analogues, we have developed a more cost-efficient method for the synthesis of ferrocenyl aryl ethers. For the first time, a mixed 1,1′-diferrocenyl aryl ether could be prepared via a simple synthetic protocol.
4 Experimental section
4.1 General methods
Phenols, bases, CuI, and ligands were purchased and used as received. All reactions and experiments were performed under an atmosphere of dry argon using standard Schlenk techniques. Column chromatography: Silica MN60 (63–200 μm), TLC on Merck plates coated with silica gel 60, F254. Gas chromatography: Perkin-Elmer Autosystem with a Varian CP-SIL-8 column; ferrocene samples were applied to the column using the sandwich technique to obtain reproducible results. NMR spectroscopy: Spectra were recorded at 293 K with Bruker Avance 500 (1H NMR 500 MHz, 13C NMR 125 MHz) and Bruker AC 300 (1H NMR 300 MHz, 13C NMR 75 MHz) spectrometers. 1H NMR spectra were referenced to residual protonated impurities in the solvent (CDCl3 7.24 ppm), 13C NMR spectra to the solvent signal (CDCl3 77.0 ppm). Starting materials were commercially available or prepared according to literature procedures: iodoferrocene [17, 25], 1,1′-dibromoferrocene [26, 27]. For compounds 3, 4, 5, 6, 7, and 9, 1H and 13C NMR data as well as Rf values were found to be identical with those reported in the literature [1].
4.2 General procedure for the coupling reactions
In a Schlenk tube CuI (1.2 mg, 6.3 μmol, 5 mol.%), the respective ligand (10–15 mol.%), the respective ferrocenyl halide (0.125 mmol), the respective phenol (0.25–0.35 mmol), and a base (0.25 mmol) were dissolved in toluene (7.5 mL), and the reaction mixture was stirred at 110°C for a given time (26–60 h). After evaporation of the volatiles the crude products were purified by column chromatography in cyclohexane-ethyl acetate.
4-tert-Butylphenoxyferrocene (1) 1H NMR (300 MHz, CDCl3): δ=1.22 (s, 9 H, t-BuH), 3.86 (m, J=1.9 Hz, 2 H, C5H4), 4.13 (t, J=1.9 Hz, 2 H, C5H4), 4.19 (s, 5 H, C5H5), 6.85 (d, J=8.8 Hz, 2 H, C6H4), 7.21 ppm (d, J=8.8 Hz, 2 H, C6H4). – 13C NMR (75 MHz, CDCl3): δ=31.6 (CH3), 34.3 (CCH3), 59.8 (C5H4), 62.9 (C5H4), 69.4 (C5H5), 116.7 (o-ArC), 126.2 (m-ArC), 141.6 (C(C5H4)O), 145.3 (t-Bu-ArC), 156.5 ppm (O-ArC). – MS (FAB) m/z (%)=334.1 (100) [M+H]+. – IR (KBr): v=2965, 2867, 1604, 1510, 1455, 1410, 1374, 1363, 1242, 1213, 1172, 1116, 1105, 1022, 1011, 998, 930, 855, 835, 815, 718, 686, 591, 550, 528, 503 cm–1. – C20H22FeO (334.24): calcd. C 71.87, H 6.63; found C 71.24, H 6.90.
1,1 ′-Di(4-tert-butylphenoxy)ferrocene (2) 1H NMR (500 MHz, CDCl3): δ=1.22 (s, 18 H, t-BuH), 3.95 (m, J=2.2 Hz, 4 H, C5H4), 4.21 (t, J=2.2 Hz, 4 H, C5H4), 6.91 (d, J=8.6 Hz, 4 H, C6H4), 7.23 ppm (d, J=8.6 Hz, 4 H, C6H4). – 13C NMR (125 MHz, CDCl3): δ=31.6 (CH3), 34.3 (C(CH3)3), 60.8 (C5H4), 64.2 (C5H4), 117.0 (ArC), 126.2 (ArC), 141.7 (C(C5H4)O), 145.4 (t-Bu-ArC), 156.4 ppm (ArC). – IR (KBr): v=2961, 2897, 2862, 1604, 1509, 1456, 1361, 1289, 1247, 1215, 1172, 1115, 1026, 1011, 931, 829, 752, 724, 686, 591, 549, 510, 503 cm–1. – C30H34FeO2·0.5 C4H8O2 (526.50): calcd. C 73.00, H 7.27; found C 72.88, H 7.53.
1,1′-Di(2,4,6-trimethylphenoxy)ferrocene (8) Rf=0.57 [cyclohexane-ethyl acetate (20:1)]. – 1H NMR (500 MHz, CDCl3): δ=2.18 (s, 12H, o-CH3), 2.22 (s, 6H, p-CH3), 3.83 (“t”, J=1.9 Hz, 2 H, C5H4), 4.09 (t, J=1.9 Hz, 4 H, C5H4), 6.78 ppm (s, 4 H, C6H2). – 13C NMR (125 MHz, CDCl3): δ=17.0 (ArCH3), 20.8 (ArCH3), 59.1 (C5H4), 62.7 (C5H4), 122.6 (C(C5H4)O), 129.4, 134.4 (H3C-ArC), 142.0 (H3C-ArC), 154.4 ppm (O-ArC).
2-tert-Butyl-4-methoxyphenoxyferrocene (10) In a Schlenk tube CuI (10 mg, 53 μmol), PPh3 (160 μmol), iodoferrocene (0.125 mmol), 2-tert-butyl-4-methoxyphenol (45 mg, 2 eq.), and Cs2CO3 (82 mg, 2 eq.) were dissolved in toluene (8 mL), and the reaction mixture was stirred at 110°C for 26 h. After evaporation of the volatiles, the crude product was purified by silica column chromatography (cyclohexane-ethyl acetate). Of a light-yellow fraction, the solvent was removed, providing 11 mg (24%) of a yellow solid. – Rf=0.60 [cyclohexane-ethyl acetate (20:1)]. – 1H NMR (CDCl3): δ=1.36 (s, 9H; CH3), 3.55 (m, 2H; Cp), 3.66 (s, 5H; Cp), 3.83 (m, 2H, Cp), 4.20 (s, 3H, OCH3), 6.51 (s, 1H, phenyl), 6.78 (s, 1H, phenyl), 7.18 ppm (s, 1H, phenyl H3).
1-(4-tert-Butylphenoxy)-1′-bromoferrocene (11) In a Schlenk tube CuI (100 mg, 0.53 mmol), PPh3 (1.60 mmol), 1,1′-dibromoferrocene (1.8 g, 5.2 mmol), 4-t-butylphenol (1.02 g, 1.3 eq.), and Cs2CO3 (1.86 g, 1.1 eq.) were dissolved in toluene (35 mL), and the reaction mixture was stirred at 110°C for 26 h. After evaporation of the volatiles, the crude product was purified by silica column chromatography (cyclohexane-ethyl acetate). Of a light-yellow fraction, the solvent was removed, which resulted in 930 mg (43%) of a yellow-brownish solid. – Rf=0.65 [cyclohexane-ethyl acetate (20:1)]. – 1H NMR (CDCl3): δ=1.22 (s, 9 H; CH3), 3.95 (t, J=1.9 Hz, 4 H, Cp), 4.21 (t, J=1.9 Hz, 4 H, Cp), 6.87 (d, J=8.6 Hz, 2 H, phenyl H2, H6), 7.19 ppm (d, J=8.6 Hz, 2 H, phenyl H3, H5). – 13C NMR (75 MHz, CDCl3): δ=31.7 (CH3), 34.4 (CCH3), 60.8 (C5H4), 64.2 (C5H4), 68.2, 68.8, 77.6, 117.0, 123.2, 126.2, 145.4, 156.4 ppm.
1-(4-tert-Butylphenoxy)-1′-(2,4-dimethylphenoxy)ferrocene (12) In a Schlenk tube CuI (10 mg, 53 μmol), PPh3 (160 μmol), compound 11 (206 mg, 0.5 mmol), 2,4-dimethylphenol (120 μL, 1 mmol), and Cs2CO3 (350 mg) were dissolved in toluene (8 mL), and the reaction mixture was stirred at 110°C for 26 h. After evaporation of the volatiles, the crude product was purified by silica column chromatography (cyclohexane – ethyl acetate). Of a light-yellow fraction, the solvent was removed, providing 126 mg (56%) of a yellow solid. – Rf=0.55 [cyclohexane-ethyl acetate (20:1)]. – 1H NMR (300 MHz, CDCl3): δ=1.22 (s, 9 H, t-butyl), 2.12 (s, 3 H, CH3), 2.18 (s, 3 H, CH3), 3.84 (t, J=1.8 Hz, 2 H, Cp(1)), 3.94 (t, J=2.2 Hz, 2 H, Cp(2)), 4.14 (t, J=2.2 Hz, 2 H, Cp(2)), 4.19 (t, J=1.8 Hz, 2 H, Cp(1)), 6.81–6.91 (m, 5 H, phenyl), 7.20 ppm (d, J=7.7 Hz, 2 H). – 13C NMR (75 MHz, CDCl3): δ=16.5 (CH3-phenyl C2), 20.7 (CH3-phenyl C4), 31.6 (C(CH3)3), 34.3 (C(CH3)3), 59.8 (Cp(2)), 60.8 (Cp(1)), 62.9 (Cp(2)), 64.2 (Cp(1)), 116.3, 116.7, 117.0, 126.2, 127.2, 128.7, 131.8, 132.0, 132.6, 145.4, 150.4 (phenyl(2) C1), 156.4 ppm (phenyl(1) C1).
4.3 Single-crystal X-ray structure determination of compound 2
Crystal data and details of the structure determination are presented in Table 2. Single crystals suitable for the X-ray diffraction study were grown from chloroform. A clear brown prism (0.30×0.20×0.10 mm3) was stored under perfluorinated ether and transferred into a Lindemann capillary for data collection. Data were corrected for Lorentz, polarization, and, arising from the scaling procedure, for latent decay and absorption effects [28]. The structure was solved by a combination of Direct Methods and difference Fourier syntheses [29]. All nonhydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were found in the final difference Fourier map and allowed to refine freely with isotropic displacement parameters. Full-matrix least-squares refinements with 434 parameters were carried out by minimizing ∑w(Fo2–Fc2)2 with the shelxl-97 [30] weighting scheme and stopped at shift/err<0.001.
Summary of the crystallographic data of compound 2.
| Empirical formula | C30H34FeO2 |
| Molecular weight | 482.42 |
| Crystal color/shape | brown/fragment |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a, Å | 6.2588(5) |
| b, Å | 38.0747(16) |
| c, Å | 11.6187(6) |
| β, deg | 116.005(7) |
| V, Å3 | 2488.4(3) |
| Z | 4 |
| ρcalcd., g cm–3 | 1.29 |
| μ, mm–1 | 0.6 |
| Diffractometer | Oxford, Xcalibur 3 CCD |
| Wavelength; λ, Å | MoKα; 0.71073 |
| T, K | 150(2) |
| θ range, deg | 3.32–25.32 |
| Index ranges hkl | –7≤h≤4 |
| –45≤k≤45 | |
| –13≤l≤13 | |
| Reflections integrated | 14 870 |
| Independent reflections/Rint | 4533/0.038 |
| Observed reflections [I>2σ(I)] | 4533 |
| Parameters refined | 434 |
| R1 (observed/all data)a | 0.0496/0.1026 |
| wR2 (observed/all data)b | 0.1026/0.1058 |
| GOFc | 1.143 |
| Largest diff. peak/hole, e Å–3 | 0.59/–0.56 |
aR1=∑||Fo|–|Fc||/∑|Fo|; bwR2=[∑w(Fo2–Fc2)2/∑w(Fo2)2]1/2, w=[σ2(Fo2)+ (AP)2+BP]–1, where P=(Max(Fo2, 0)+2Fc2)/3; cGoF=[∑w(Fo2–Fc2)2/(nobs–nparam)]1/2.
CCDC 1017347 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
G.D.F. thanks Prof. Dr. Herbert Plenio and the Eduard-Zintl Institute of Inorganic and Physical Chemistry of the Technische Universität Darmstadt for laboratory support and cooperation, Prof. Dr. Wolfgang W. Schoeller and Dr. Rian D. Dewhurst for helpful discussions, and Dr. Eberhardt Herdtweck for assistance with the single-crystal X-ray structure determination.
References
[1] M. R. an der Heiden, G. D. Frey, H. Plenio, Organometallics 2004, 23, 3548–3551.10.1021/om0497893Suche in Google Scholar
[2] P. D. Beer, P. A. Gale, G. Z. Chen, Coord. Chem. Rev. 1999, 186, 3–36.Suche in Google Scholar
[3] I. Manners, Macromol. Symp. 2003, 196, 57–62.Suche in Google Scholar
[4] Y. Y. Luk, N. L. Abbott, Science 2003, 301, 623–626.10.1126/science.1084527Suche in Google Scholar
[5] H. Plenio, C. Aberle, Chem. Eur. J. 2001, 7, 4438–4446.Suche in Google Scholar
[6] P. Štěpnička (Ed.), Ferrocenes: Ligands, Materials and Biomolecules, John Wiley & Sons, Chichester, 2008.10.1002/9780470985663Suche in Google Scholar
[7] E. S. Phillips (Ed.), Ferrocenes: Compounds, Properties and Applications, Nova Science Publishers, New York, 2011.Suche in Google Scholar
[8] T. J. Colacot, Chem. Rev. 2003, 103, 3101–3118.Suche in Google Scholar
[9] H.-U. Blaser, Adv. Synth. Catal. 2002, 344, 17–31.Suche in Google Scholar
[10] A. Togni, T. Hayashi (Eds.), Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, VCH, Weinheim, 1995.10.1002/9783527615599Suche in Google Scholar
[11] L. X. Dai, T. Tu, S. L. You, W. P. Deng, X. L. Hou, Acc. Chem. Res. 2003, 36, 659–667.Suche in Google Scholar
[12] D. Schaarschmidt, H. Lang, Eur. J. Inorg. Chem. 2010, 4811–4821.10.1002/ejic.201000722Suche in Google Scholar
[13] H. Lang, D. Schaarschmidt. DE 10 2010 001364 B4.Suche in Google Scholar
[14] M. Herberhold in Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, (Eds.: A. Togni, T. Hayashi), VCH, Weinheim, 1995, pp. 219–279.Suche in Google Scholar
[15] A. Shafir, M. P. Power, G. D. Whitener, J. Arnold, Organometallics 2000, 19, 3978–3982.10.1021/om0004085Suche in Google Scholar
[16] M. Herberhold, H.-D. Brendel, A. Hofmann, B. Hofmann, W. Milius, J. Organomet. Chem. 1998, 556, 173–187.Suche in Google Scholar
[17] B. Bildstein, M. Malaun, H. Kopacka, K. Wurst, M. Mitterböck, K.-H. Ongania, G. Opromolla, G. Zanello, Organometallics 1999, 18, 4325–4336.10.1021/om990377hSuche in Google Scholar
[18] H. Plenio, C. Aberle, Organometallics 1997, 16, 5950–5957.10.1021/om970681fSuche in Google Scholar
[19] H. Plenio, C. Aberle, Chem. Commun. 1996, 2123–2124.10.1039/cc9960002123Suche in Google Scholar
[20] M. D. Rausch, J. Org. Chem. 1961, 26, 1802–1805.Suche in Google Scholar
[21] V. A. Nefedov, M. N. Nefedova, Zh. Obs. Khim. 1966, 36, 122–127.Suche in Google Scholar
[22] R. K. Gujadhur, C. M. Bates, D. Venkataraman, Org. Lett. 2001, 3, 4315–4317.Suche in Google Scholar
[23] R. K. Gujadhur, D. Venkataraman, Synth. Commun. 2001, 31, 2865–2879.Suche in Google Scholar
[24] K. Brandenburg, diamond (version 3.2i), Crystal and Molecular Structure Visualization, Crystal Impact-H. Putz & K. Brandenburg GbR, Bonn (Germany) 2012. See also: http://www.crystalimpact.com/diamond/.Suche in Google Scholar
[25] F. S. Kamounah, J. B. Christensen, J. Chem. Res. Synop. 1997, 150.10.1039/a607056fSuche in Google Scholar
[26] T.-Y. Dong, L.-L. Lai, J. Organomet. Chem. 1996, 509, 131–134.Suche in Google Scholar
[27] M. S. Inkpen, S. Du, M. Driver, T. Albrecht, N. J. Long, Dalton Trans. 2013, 42, 2813–2816.Suche in Google Scholar
[28] Z. Otwinowski, W. Minor in Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, (Eds.: C. W. Carter Jr, R. M. Sweet), Academic Press, New York, 1997, pp. 307–326.10.1016/S0076-6879(97)76066-XSuche in Google Scholar
[29] A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori, M. Camalli, J. Appl. Crystallogr. 1994, 27, 435–436.Suche in Google Scholar
[30] G. M. Sheldrick, Acta Crystallogr. 2008, A64, 112–122.Suche in Google Scholar
©2015 by De Gruyter
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Editorial
- Zeitschrift für Naturforschung B now being published by De Gruyter
- Original Communications
- “Naked” S2O72– ions – the serendipitous formation of the disulfates [HPy]2[S2O7] and [bmim][HPy][S2O7] (HPy = pyridinium; bmim = 1-Butyl-3-methylimidazolium)
- Orthoamide und Iminiumsalze, LXXXVIII. Synthese N,N,N′,N′,N″,N″-persubstituierter Guanidiniumsalze aus N,N′-persubstituierten Harnstoff/Säurechlorid-Addukten**
- The acidic ionic liquid [BSO3HMIm]HSO4: a novel and efficient catalyst for one-pot, three-component syntheses of substituted pyrroles
- The influence of alkali-metal ions on the molecular and supramolecular structure of manganese(II) complexes with tetrachlorophthalate ligands
- New triazolothiadiazole and triazolothiadiazine derivatives as kinesin Eg5 and HIV inhibitors: synthesis, QSAR and modeling studies
- Two copper(I) complexes of bi- (or tri-)pyrazolyl ligands featuring Cu3pz3 or Cu4pz4 motifs
- Synthesis of ferrocenyl aryl ethers via Cu(I)/phosphine catalyst systems
- A prenylated acridone alkaloid and ferulate xanthone from barks of Citrus medica (Rutaceae)
- Synthesis and structural characterization of substituted phenols with a m-terphenyl backbone 2,4,6-R3C6H2OH (R=2,4,6-Me3C6H2, Me5C6)
- 2-Ethyl-1-phenylindazolium hexafluorophosphate. N-heterocyclic carbene formation, rearrangement, ring-cleavage reactions, and rhodium complex formation
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Editorial
- Zeitschrift für Naturforschung B now being published by De Gruyter
- Original Communications
- “Naked” S2O72– ions – the serendipitous formation of the disulfates [HPy]2[S2O7] and [bmim][HPy][S2O7] (HPy = pyridinium; bmim = 1-Butyl-3-methylimidazolium)
- Orthoamide und Iminiumsalze, LXXXVIII. Synthese N,N,N′,N′,N″,N″-persubstituierter Guanidiniumsalze aus N,N′-persubstituierten Harnstoff/Säurechlorid-Addukten**
- The acidic ionic liquid [BSO3HMIm]HSO4: a novel and efficient catalyst for one-pot, three-component syntheses of substituted pyrroles
- The influence of alkali-metal ions on the molecular and supramolecular structure of manganese(II) complexes with tetrachlorophthalate ligands
- New triazolothiadiazole and triazolothiadiazine derivatives as kinesin Eg5 and HIV inhibitors: synthesis, QSAR and modeling studies
- Two copper(I) complexes of bi- (or tri-)pyrazolyl ligands featuring Cu3pz3 or Cu4pz4 motifs
- Synthesis of ferrocenyl aryl ethers via Cu(I)/phosphine catalyst systems
- A prenylated acridone alkaloid and ferulate xanthone from barks of Citrus medica (Rutaceae)
- Synthesis and structural characterization of substituted phenols with a m-terphenyl backbone 2,4,6-R3C6H2OH (R=2,4,6-Me3C6H2, Me5C6)
- 2-Ethyl-1-phenylindazolium hexafluorophosphate. N-heterocyclic carbene formation, rearrangement, ring-cleavage reactions, and rhodium complex formation