Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
-
Victor I. Saloutin
 ,
Marina V. Goryaeva
,
Marina V. Goryaeva

Abstract
The competitive routes were found for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones with 3-amino alcohols. It was shown that the reactions with 3-aminopropanol in 1,4-dioxane predominantly lead to hexahydropyrido[2,1-b][1,3]oxazin-6-ones, and in ethanol to 3-hydroxypropylaminocyclohexenones. In contrast, cyclizations with 2-aminoethanol and its analogues, regardless of the reaction conditions, yield hexahydrooxazolo[3,2-a]pyridin-5-ones as the main products. The trans- and cis-diastereomeric structure of heterocycles was established using X-ray and 1H, 19F, 13C NMR spectroscopy, 2D 1H-13C HSQC and HMBC experiments. The mechanism is proposed for competitive transformations of polyfluoroalkyl-3-oxo esters, methyl ketones with 3-amino alcohols.
Introduction
Multicomponent reactions (MCR) play an important role in generating a variety of compounds whose structure is formed from three or more reagents in one step. MCR are extremely efficient, atom-economical, and technically feasible processes [1], [2], [3], [4]. In the literature there are many examples illustrating the use of trifluoroacetoacetic ester in MCR to obtain different heterocyclic products [5], [6], [7], [8], [9], [10], [11], [12].
Generally speaking, trifluoroacetoacetic ester belongs to the universally acknowledged building blocks of organic synthesis for obtaining a variety of open-chain, carbo- and hetero-cyclic molecules [13], [14], [15], including practically important compounds. Synthesis of trifluoromethyl-substituted heterocycles is especially promising for pharmaceutical chemistry, because the heterocycles have unique physical and biological properties due to the presence of the trifluoromethyl group [16], [17], [18]. It is an established fact that the replacement of methyl group by trifluoromethyl moiety leads to an increase in metabolic stability and lipophilicity of organic molecules, which can contribute to improving the pharmacokinetic characteristics of bioactive compounds [19], [20], [21]. A great achievement in this field is the trifluoroacetoacetic ester-based production of the medicinal drug celecoxib as a selective COX-2 inhibitor [22], [23].
Our research team [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34] and other researchers [35], [36], [37], [38] have found numerous examples illustrating the synthesis of various heterocyclic systems from polyfluoroalkyl-containing 3-oxo esters and their derivatives in a way which is uncharacteristic of the non-fluorinated analogues.
Recently, we discovered a new three-component reaction of polyfluoroalkyl-3-oxo esters, methyl ketones with 1,2-di- and monoamines. It was found that the use of 1,2-ethanediamines in such reactions leads to chemo-, regio- and stereo-selective formation of hexahydroimidazo[1,2-a]pyridine-5-ones [39], which under dehydration conditions are transformed into 1-(2-aminoethyl)-6-alkyl-4-polyfluoroalkylpyridinones, including tuberculostatic active ones [40]. The introduction of di- and monoalkyl amines into this three-component reaction changed the direction of cyclization, which allowed cyclohex-2-en-1-ones to be obtained [41]. Thus, varying the amine component in the new three-component reaction can result in both heterocyclic and carbocyclic compounds (Scheme 1).
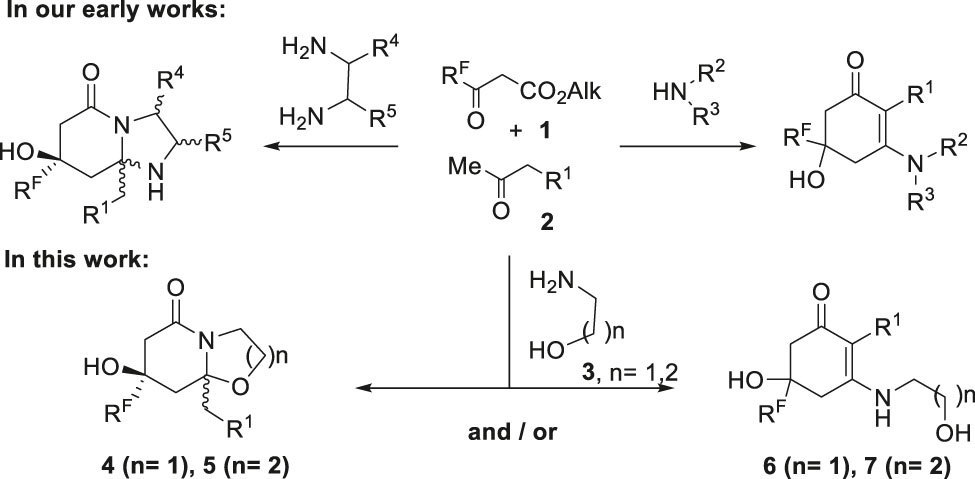
The routes of the reactions polyfluoroalkyl 3-oxo esters 1 with methyl ketones 2 and amines 3.
In this work, the three-component reactions of polyfluoroalkyl-3-oxo esters 1 and methyl ketones 2 with 1,2- and 1,3-amino alcohols 3 were investigated. Amino alcohols are commercially available reagents that contain two nonequivalent reaction centres, whereby they can react as N,O-dinucleophiles generating a bicyclic backbone of hexahydrooxazolo(azino)pyridines 4, 5, and/or as N-mononucleophiles giving a cyclohexenone core 6, 7 (Scheme 1).
Oxazolo(azino)piperidine moiety is present in some natural products and drug-like molecules [42], [43], [44], [45], [46]. In addition, these compounds are used as intermediate products for the synthesis of bioactive alkaloids, for example, (R)-(–)-coniine, (–)-anabasine, (–)-solenopsin A, (±)-adalinine, etc. [47], [48], [49], as well as of a wide range of other optically pure carbo- and hetero-cycles [50], [51]; therefore, it shows promise to develop new approaches to the synthesis of these heterocycles derivatives.
The main method used for preparing hexahydrooxazolo[3,2-a]pyridinones and hexahydropyrido[2,1-b]oxazinones involves two-component cyclizations of 1,2- and 1,3-aminо alcohols with acids having a ketone [52], [53], [54], [55], [56], [57] or aldehyde [58], [59], [60] group at the C5 position [61]. Glutaric anhydride can be used instead of acids in condensation with amino alcohols, but in this case it is necessary to carry out subsequent reduction [62], [63]. The formation of the hexahydrooxazolo(azino)pyridine backbone is described as a result of four-component cyclization of butenoic acid, a 1,2- or 1,3-amino alcohol and gaseous CO and H2 [64], [65]. Oxazolopiperidones were also prepared by condensation of Ser-OMe and 3-(5-oxo-1,3-oxazolidin-4-yl)propanal [66] or by cyclization of 2,4-dimethyl-4-(3-oxo-1-phenylbutyl)-1,3-oxazol-5(4H)-one with alanine [67]. The only example found in literature, which illustrates the synthesis of 3-phenyl-8a-(trifluoromethyl)hexahydro-5H-[1,3]oxazolo[3,2-a]pyridin-5-one, is by the reaction of 6,6,6-trifluoro-5-oxohexanoic acid with (S)-(+)-phenylglycinol [68].
Previously, we have reported on the value of synthesis of new cyclohexenone structures [41].
Results and discussion
In the present work, possible alternative ways were studied for the recently discovered three-component cyclization of polyfluoroalkyl-3-oxo esters 1 and methyl ketones 2 with 1,2- and 1,3-amino alcohols 3 as amine component into bicyclic heterocycles (hexahydrooxazolo[3,2-a]pyridin-5-ones 4 and hexahydro-2H,6H-pyrido[2,1-b][1,3]oxazin-6-ones 5) or cyclohex-2-en-1-ones 6, 7. The probable formation of diastereomeric bicycles cannot be overlooked, because the cyclization of polyfluoroalkyl-3-oxo esters 1 and methyl ketones 2 with 1,2-ethanediamines resulted in 7-hydroxy-7-(polyfluoroalkyl)hexahydroimidazo[1,2-a]pyridin-5(1H)-ones in trans- and/or cis-forms depending on their structure and the reaction conditions [39].
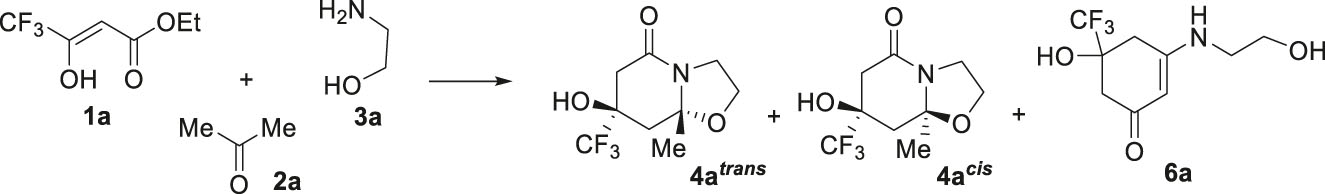
Indeed, when we examined the 1H, 19F NMR spectra of the reaction mixture of trifluoroacetoacetic ester 1a, acetone 2a and 2-aminoethanol 3a in acetonitrile after three-day ageing, we detected the formation of two hexahydrooxazolo[3,2-a]pyridin-5-ones 4a cis and 4a trans as well as cyclohex-2-en-1-ones 6a as a minor product. Although the reaction in acetonitrile runs with the high total yield, the diastereoselectivity was low in these transformations (Table 1, entry 1), so we decided to optimize the reaction conditions. To control their course, we used the 19F NMR spectroscopy, since all three products bearing CF3-group had singlet signals with determined chemical shifts (4a trans at δ 79.73 ppm, 4а cis at δ 80.79 ppm and 6a at δ 80.38 ppm). It turned out that in aprotic low-polar solvents (THF, 1,4-dioxane) a high conversion of the initial reagents was achieved (Table 1, entries 2, 3), but in 1,4-dioxane the reaction took place with high diastereoselectivity to give predominantly one bicycle 4a trans (Table 1, entry 3). Under all these conditions, the formation of cyclohexenone 6a was observed only in trace amounts. Previously, for the effective formation of cyclohexenones in similar three-component synthesis, it was proposed to carry out the reaction in absolute ethanol with zeolites [41]. However, even under these conditions, we could not obtain product 6a with an acceptable yield (table 1, entries 4, 5). According to 19F NMR spectrum, its formation was recorded only with the yield at 8% (entry 5). Attempts to isolate cyclohexenone 6a as an individual compound were unsuccessful. Instead, it turned out that the proportion of cis-isomer 4a cis in ethanol increases significantly. It should be noted that three-component cyclization of ester 1a, acetone 2a with aminoethanol 3a were sensitive to heating, because under these conditions there was a strong tarring of the reaction mass in contrast to similar reactions with 1,2-ethanediamines [39].
Optimization of the reaction conditions for the 2-aminoethanol 3a.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditionsa | Time, days | Products, yield [%]b | Total yield [%]b | ||
| 4a trans | 4a cis | 6a | ||||
| 1 | CH3CN, HPLC | 3 | 55 | 35 | 3 | 93 |
| 2 | THF, 99+% | 3 | 61 | 25 | 2 | 88 |
| 3 | 1,4-dioxane, 99+% | 3 | 80 | 9 | Trace | 89 |
| 4 | EtOH (abs) | 3 | 42 | 33 | 7 | 82 |
| 5 | EtOH (abs), zeolites | 3 | 33 | 25 | 8 | 66 |
aConditions: Reactions performed with 1a (1 mmol), 2a (1 mmol) and 3a (1 mmol) in 5 mL of the solvent at room temperature.
bDetermined by 19F NMR analysis of the mixture.
Thus, in order to obtain the trans-isomers of hexahydrooxazolo[3,2-a]pyridin-5-ones 4 trans regioselectively, we carried out the reactions of esters 1 with methyl ketones 2 and aminoethanol 3a in 1,4-dioxane at room temperature, whereas cis-isomers 4 cis were isolated from reactions in absolute ethanol.
As expected, in the three-component reaction of trifluoroacetoacetic ester 1a with 2-aminoethanol 3a in 1,4-dioxane, varying the methyl ketone component 2 by using 2-butanone 2b and 2-hexanone 2c instead of acetone 2a led to the formation of bicycles trans-diastereomers 4b trans , 4c trans (Scheme 2). Cis-isomers 4b cis , 4c cis were isolated from a mixture of isomers that formed in approximately equal ratios in ethanol. The isomers were separated by fractional crystallization or column chromatography. However, we failed to find conditions for the isolation of 4b cis as an individual compound.
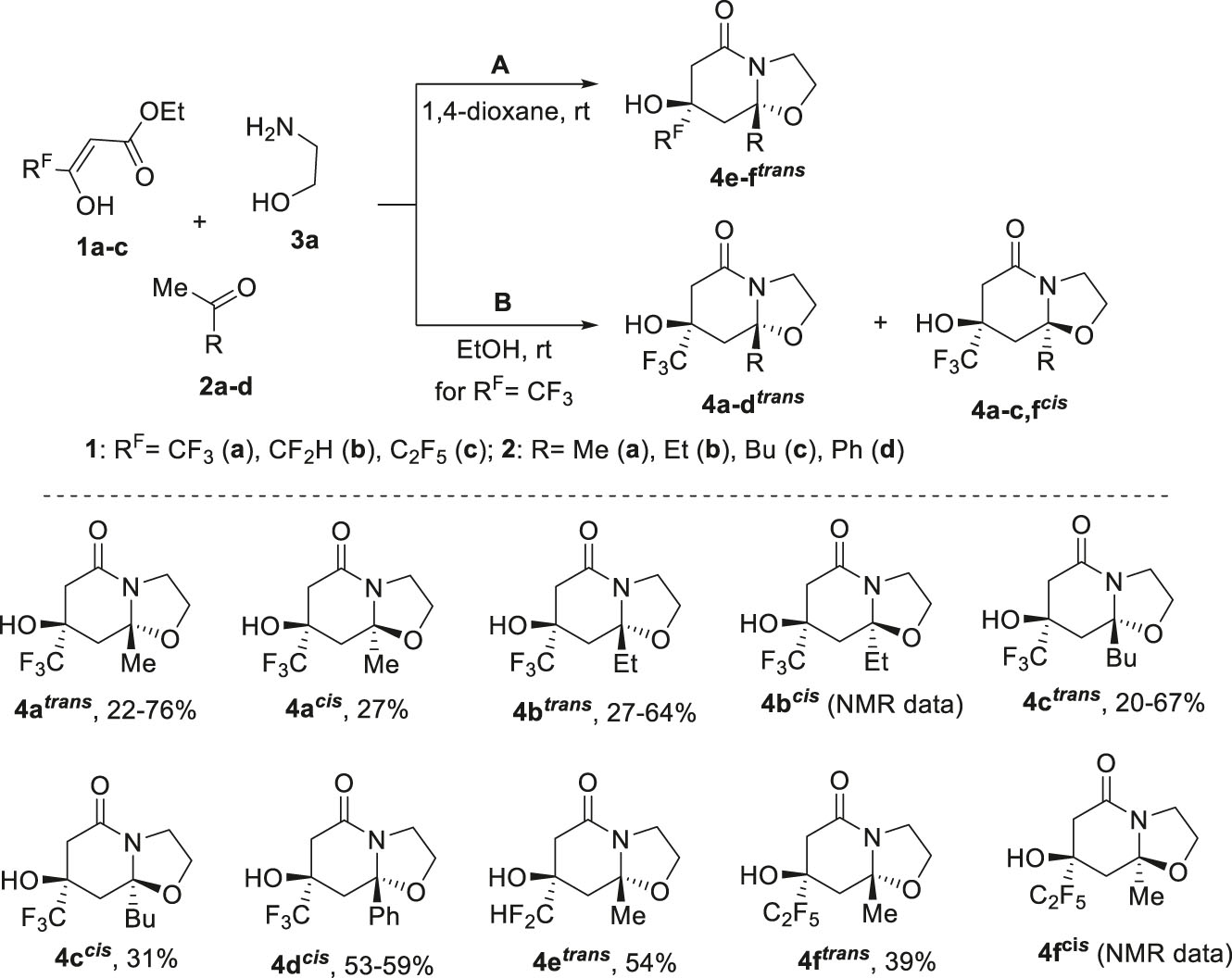
Three-component reaction of polyfluoroalkyl-3-oxo esters 1, methyl ketones 2 and 2-aminoethanol 3a.
Unlike alkyl methyl ketones 2a–c, the three-component reaction of acetophenone 2d with ester 1a and 2-aminoethanol 3a even in ethanol resulted in one cis-diastereomer of hexahydrooxazolo[3,2-a]pyridin-5-ones 4d cis . The change and increase in stereoselectivity of this reaction may be due to the presence of a bulk phenyl substituent, which plays the role of a conformational anchor.
Further, it was found that the replacement of trifluoroacetoacetic ester 1a by difluoromethyl- and pentafluoroethyl-containing 3-oxo esters 1b and 1c in reaction with acetone 2a and aminoethanol 3a in 1,4-dioxane gives trans-diastereomers of bicycles 4e trans and 4f trans . At the same time, their yields are reduced compared to the trifluoromethyl analogue. Carrying out these reactions in ethanol led to a significant tarring of the reaction mass. We were able to isolate only the cis-isomer 4f cis in the mixture of 3 : 5 with the trans-isomer 4f trans from the reaction of pentafluoroethyl ester 1c.
We failed to isolate cyclohexenone 6a as an individual substance from the three-component reaction of trifluoroacetoacetic ester 1a with methyl ketone 2a and aminoethanol 3a, so we tried to realize its synthesis in ethanol via a two-component reaction of aminoethanol 3a and aldol 8a obtained from ester 1a with methyl ketone 2a according to the method [41]. However, under these conditions, cyclohexenone 6a was formed as a byproduct along with heterocycle 4a trans (Scheme 3). Nevertheless, a similar reaction of ethyl-substituted aldol 8b with aminoethanol 3a allowed cyclohexenone 6b to be isolated from a mixture with bicycle 4b trans . Note that the heterocycles 4a trans , 4b trans were obtained from aldol 8a,b in 1,4-dioxane, albeit with lower yields compared to the three-component synthesis.
![Scheme 3:
Two-component synthesis of cyclohexenones 6 and hexahydrooxazolo[3,2-a]pyridin-5-ones 4.](/document/doi/10.1515/pac-2019-1216/asset/graphic/j_pac-2019-1216_scheme_003.jpg)
Two-component synthesis of cyclohexenones 6 and hexahydrooxazolo[3,2-a]pyridin-5-ones 4.
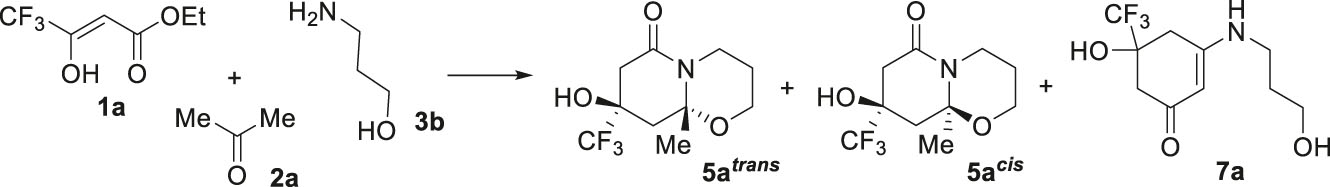
In contrast to the reactions with aminoethanol 3a, three-component cyclization of ester 1a and acetone 2a with 3-amino-1-propanol 3b in aprotic solvents proceeded stereoselectively to form one diastereomer of hexahydropyrido[2,1-b][1,3]oxazin-6-one 5a trans , whereas the isomer 5a cis was either formed in trace amounts or not at all (Table 2, entries 1–4). The highest yields of bicycle 5a trans were achieved in THF and 1,4-dioxane (Table 2, entries 2–4), but in 1,4-dioxane the selectivity of the reaction was higher. Another feature of the transformations with aminopropanol 3b was the obtaining of cyclohexenone 7a when ethanol was used as a solvent (Table 2, entries 5, 6). It should also be noted that cyclizations with amino alcohol 3b took more time (7–10 days), and they occurred with moderate yields with incomplete conversion of the starting ester 1a. The attempts to increase the reaction rate and yields by adding triethylamine as a catalyst did not lead to the desired result (Table 2, entries 4, 6).
Optimization of the reaction conditions for the 3-amino-1-propanol 3b.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditionsa | Time, days | Products, yield [%]b | Yield total | ||
| 5a trans | 5a cis | 7a | ||||
| 1 | CH3CN, HPLC | 10 | 27 | 8 | 6 | 41 |
| 2 | THF, 99+% | 10 | 60 | trace | 7 | 67 |
| 3 | 1.4-dioxane, 99+% | 10 | 58 | trace | trace | 58 |
| 4 | 1.4-dioxane, 99+%, NEt3 | 7 | 50 | - | 3 | 53 |
| 5 | EtOH (abs), zeolites | 10 | 8 | 4 | 53 | 61 |
| 6 | EtOH (abs), NEt3, zeolites | 7 | 9 | - | 51 | 60 |
aConditions: Reactions performed with 1a (1 mmol), 2a (1 mmol) and 3b (1 mmol) in 5 mL of the solvent at room temperature.
bDetermined by 19F NMR analysis of the mixture.
The reactions were monitored using 19F NMR spectra, in which all three products can be identified by singlet signals of their CF3 groups (5a trans at δ 79.76 ppm, 5а cis at δ 80.21 ppm and 7a at δ 80.38 ppm).
Realization of three-component reactions of trifluoroacetoacetic ester 1a with methyl ketones 2a–d and 3-amino-1-propanol 3b in 1,4-dioxane allowed hexahydro-2H,6H-pyrido[2,1-b][1,3]oxazin-6-ones 5a–d to be obtained as a single trans-diastereomer (Scheme 4). Introduction of 3-oxoesters 1b (RF = CF2H) or 1c (RF = C2F5) instead of ester 1a in the reaction with acetone 2a and 3-amino-1-propanol 3b also led to the formation of hexahydro-2H,6H-pyrido[2,1-b][1,3]oxazin-6-ones 5e trans and 5f trans , albeit with a small yield.
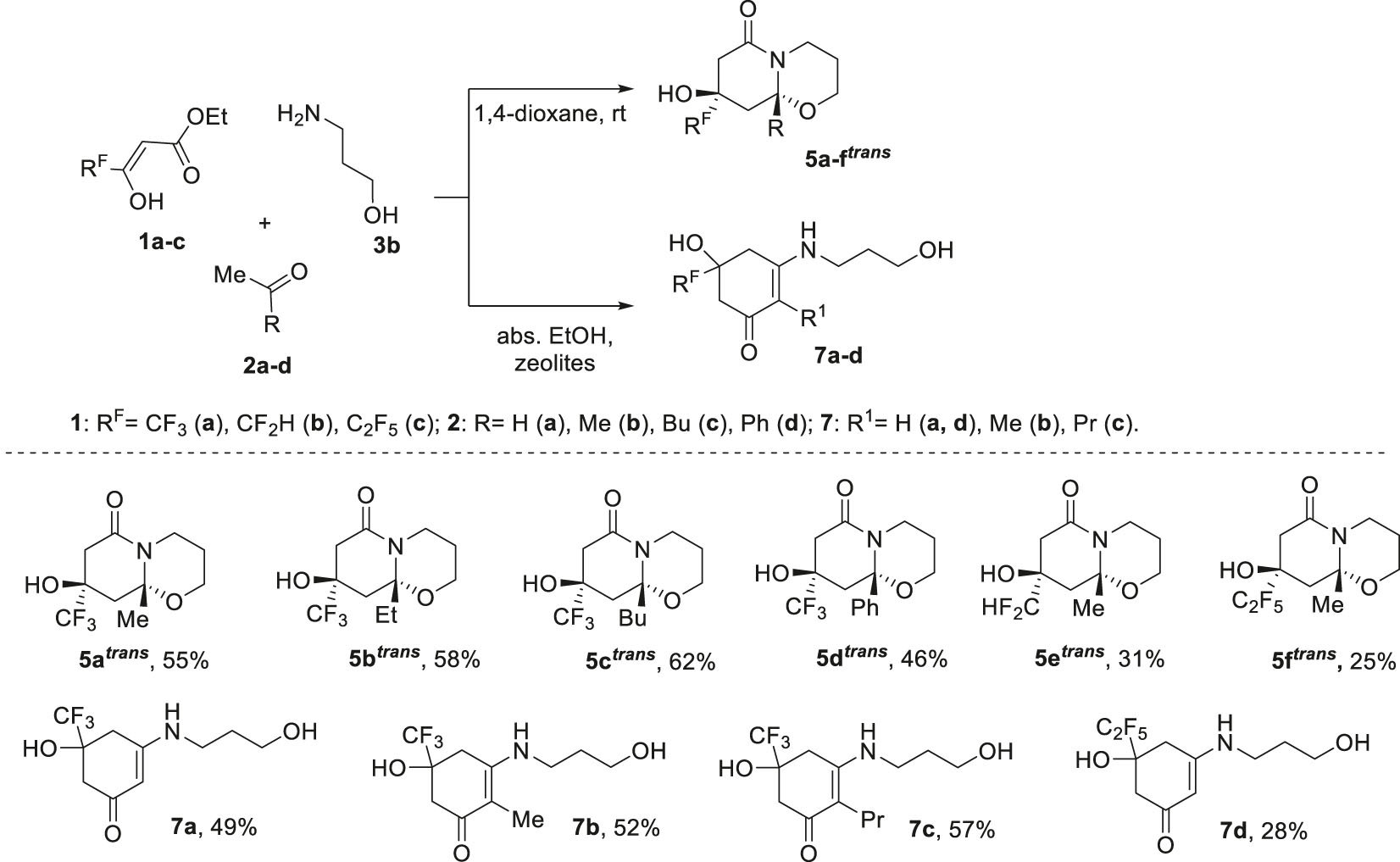
Three-component reaction of polyfluoroalkyl-3-oxo esters 1, methyl ketones 2 and 3-amino-1-propanol 3b.
The use of abs. ethanol with zeolites as a condition for the three-component reaction of trifluoroacetoacetic ester 1a with methyl ketones 2a–c and 3-amino-1-propanol 3b resulted in cyclohexenones 7a–c (Scheme 4). A similar product 7d was obtained in the reaction of pentafluoroethyl substituted ester 1c with acetone 2a and amino propanol 3b under the same conditions.
In the studied three-component reactions, we also introduced amino alcohols 3с–e having various substituents that can affect the result of these transformations. It was found that the use of (+/−)-2-amino-1-propanol 3c in the reaction with ester 1a and acetone 2a in 1,4-dioxane led to cis,cis- and trans,cis-diastereomers of heterocycle 4g (Scheme 5). Obviously, the methyl substituent in the aminoethanol 3a affects the conformation of the resulting bicycle 4g.
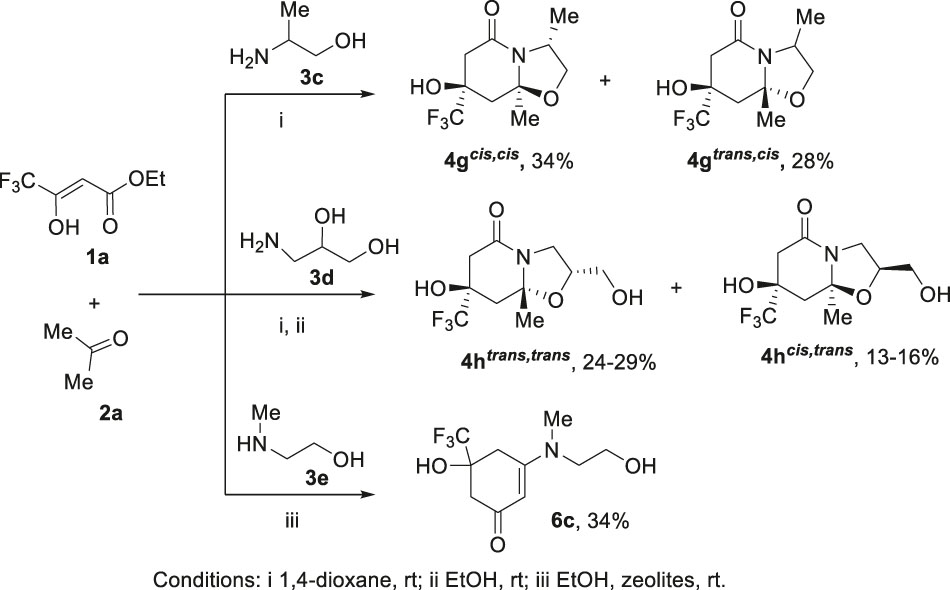
Variation of amino alcohols 3 in the reaction with ester 1a and acetone 2a.
(+/−)-3-Amino-1,2-propanediol 3d can react with ester 1a and acetone 2a as aminoethanol to form hexahydrooxazolo[3,2-a]pyridin-5-one 4h and/or as 3-amino-2-propanol give hexahydropyrido[2,1-b][1,3]oxazin-6-one 5h. However, regardless of the reaction conditions, two trans,trans- and cis,trans-diastereomers of hexahydrooxazolo[3,2-a]pyridin-5-one 4h were obtained. Such reaction course may be preferable because of the thermodynamic benefit of the formation of a five-membered cycle over the formation of a six-membered one.
When 2-(methylamino)ethanol 3e was used as an amino alcohol component in the reaction with ester 1a and acetone 2a, only cyclohexenone 6c could be formed (Scheme 5), because in this case the nitrogen atom cannot act as a bridge centre in the bicycle 4.
The structures of all synthesized compounds were confirmed by IR, 1H, 19F, 13C NMR spectroscopy and high-resolution mass spectrometry. Based on 2D 1H-13C HSQC and HMBC experiments, full correlation of signals in 1H and 13C NMR spectra was carried out. The relative configuration of substituents at stereocenters was determined by homonuclear 2D 1H-1H NOESY experiments. The structures of diastereomers 4 and 5 are shown in schemes 2–4. The relative configurations are given with respect to a hydroxyl group. Diastereomers 4 and 5 are racemates.
A direct confirmation of the trans-position of hydroxyl oxygen and oxazole or oxazine cycle oxygen is the cross-peak in the NOESY spectra between the OH-group proton and the substituent protons at the nodal carbon atom observed for all trans-isomers of type 4 and 5 structures. The spectra of cis-isomers show no cross-peak (OH, R); and the relative configuration is established on the basis of a set of other NOE correlations (see Supplementary).
Some diagnostic features of cis- and trans-configuration were established using the 1Н, 19F NMR spectra analysis of hexahydrooxazolo[3,2-a]pyridine-5-ones 4 isomers. In the 1H spectra, the most striking differences are related to the value of the nonequivalence (Δ6AB) of diastereotope protons at C-6 and to the value of the geminal constant between them (2 J 6AB). Cis-isomers 4 cis is characterized with a significant value Δ6AB = 0.33 – 0.48 ppm, whereas in trans-isomers the value Δ6AB < 0.07 ppm up to the degeneration of AB-system to spin system A2 (Δ6AB = 0). Besides, the constant 2 J 6AB in the transition from cis- to trans-isomers increases in absolute value from 15 to 17.8 Hz.
The presence of long-range spin-spin interaction between the protons of the methylene groups of the pyridinone cycle in compounds 4 and 5 with a characteristic value of the constant 4 J H,H = 1.3 – 3.5 Hz indicates a pseudo-equatorial position of the interacting protons. The identification of axial and equatorial protons was used in the analysis of 2D NOESY spectra and for determining the relative configuration.
In the 19F NMR spectra of compounds 4, 5 having a trifluoromethyl substituent, the 19F signals of trans- diastereomers are observed in a stronger field of δ ∼ 79.8 ppm as compared to cis-diastereomers with δ ∼ 80.4 – 80.8 ppm.
The structures of compounds 4a trans , 4а cis , 4c cis , 4h trans,trans , 5a trans , 7c were confirmed by X-ray diffraction analysis data (Fig. 1 and Fig. 2).
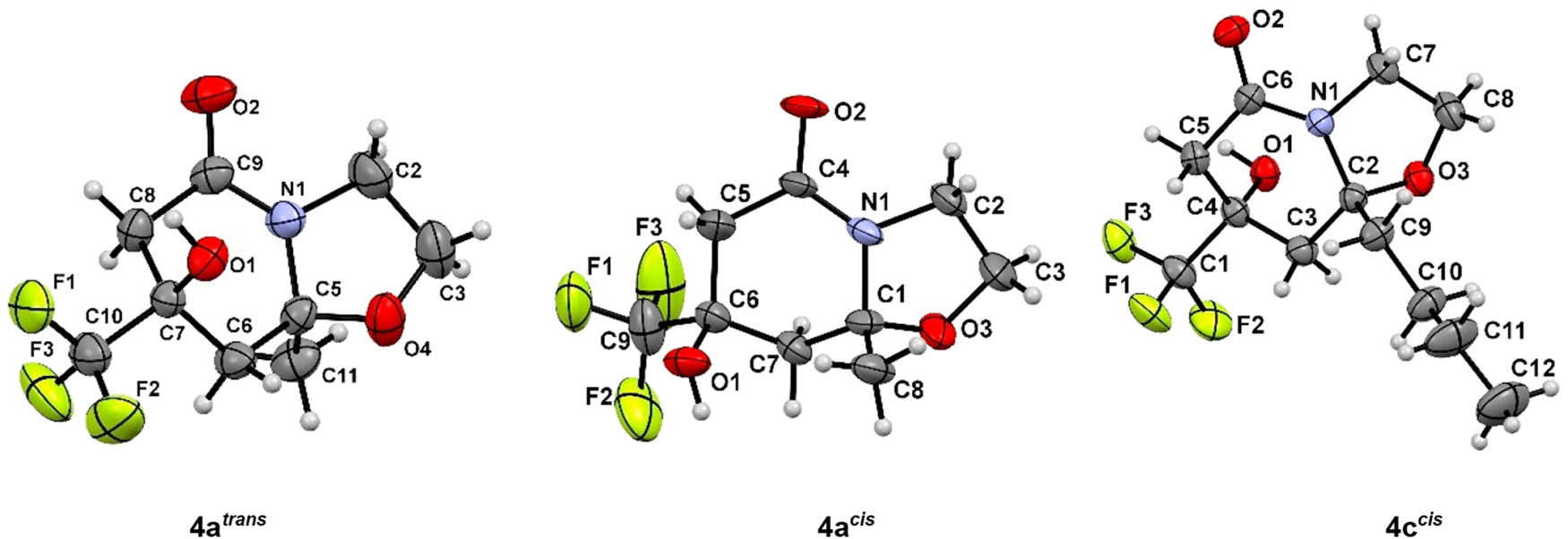
ORTEP diagrams of compounds 4a trans , 4a cis , 4c cis .
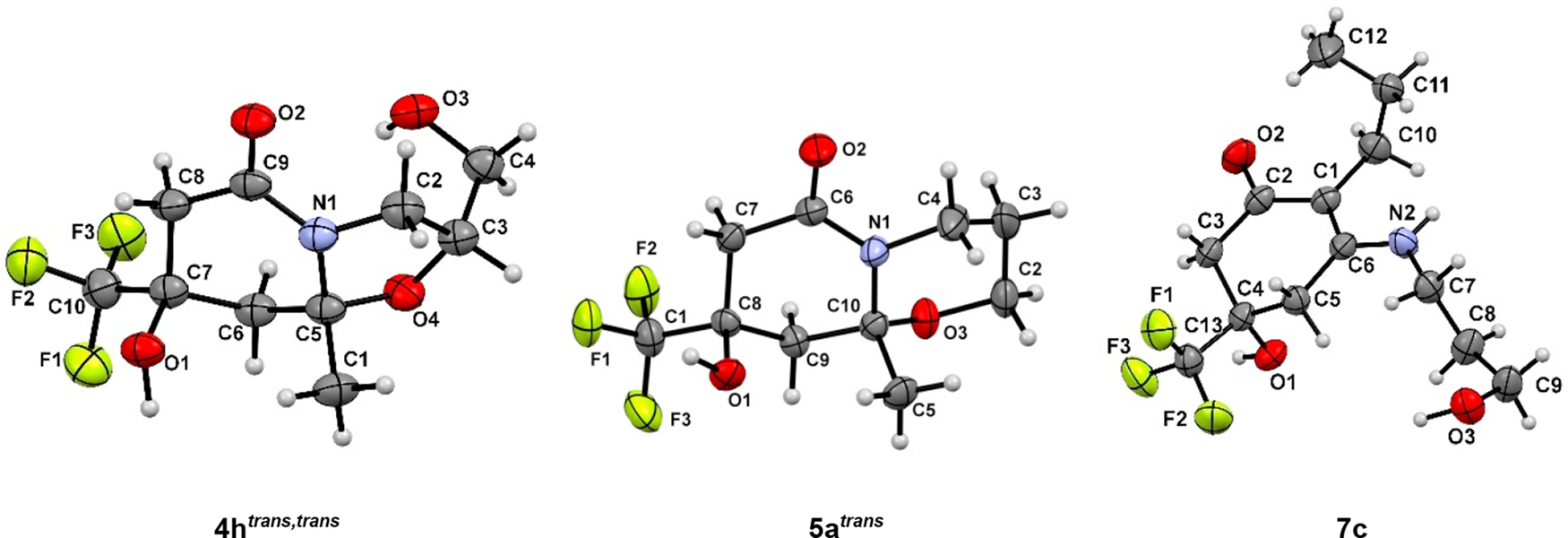
ORTEP diagrams of compounds 4h trans,trans , 5a trans , 7c.
For the reactions under consideration, it can be assumed that both bicycles 4, 5 and cyclohexenones 6, 7 are obtained via the enaminoester Y as a key intermediate. We can offer two ways to generate Y: (1) through enamine X formed from ketone 1 and amine 3 (pathway a) by the known enamine mechanism [69] or (2) through aldol 8 (pathway b) (Scheme 6). In the aprotic medium we observed the predominant formation of bicycles 4, 5. Probably, these heterocycles are obtained as a result of intramolecular cyclization of the intermediate Y′ due to nucleophilic addition of the hydroxyl group to the C=N bond (route c) to form the cycle Z, which further underwent intramolecular amidation of the ester fragment to give a bicyclic backbone 4, 5. In ethanol, the reaction can proceed through the alternative pathway d, which becomes the main pathway for 3-aminopropanol 3b. Most likely, in this case hydroxyl reaction centre of the intermediate Y″ is blocked due to its solvation by solvent molecules (ethanol), resulting in intramolecular cyclization of enaminoketone Y″ coming along the path d with the formation of aminocyclohexanones 6, 7 via the condensation ethoxycarbonyl fragment with an activated methylene group. For a spatially distant hydroxypropyl group, more efficient solvation is possible, which is the reason for the preferred formation of cyclohexenones 7. Preparation of hexahydrooxazolopyridinones 4 from aldol 8 in abs. ethanol can be explained by the possible intermediate formation of dihydroxycyclohexenone W (the pathway e) followed by its interaction with aminoethanol 3a. All processes are autocatalysed, because amino alcohols 3a, b act not only as reagents but also as base catalysts.
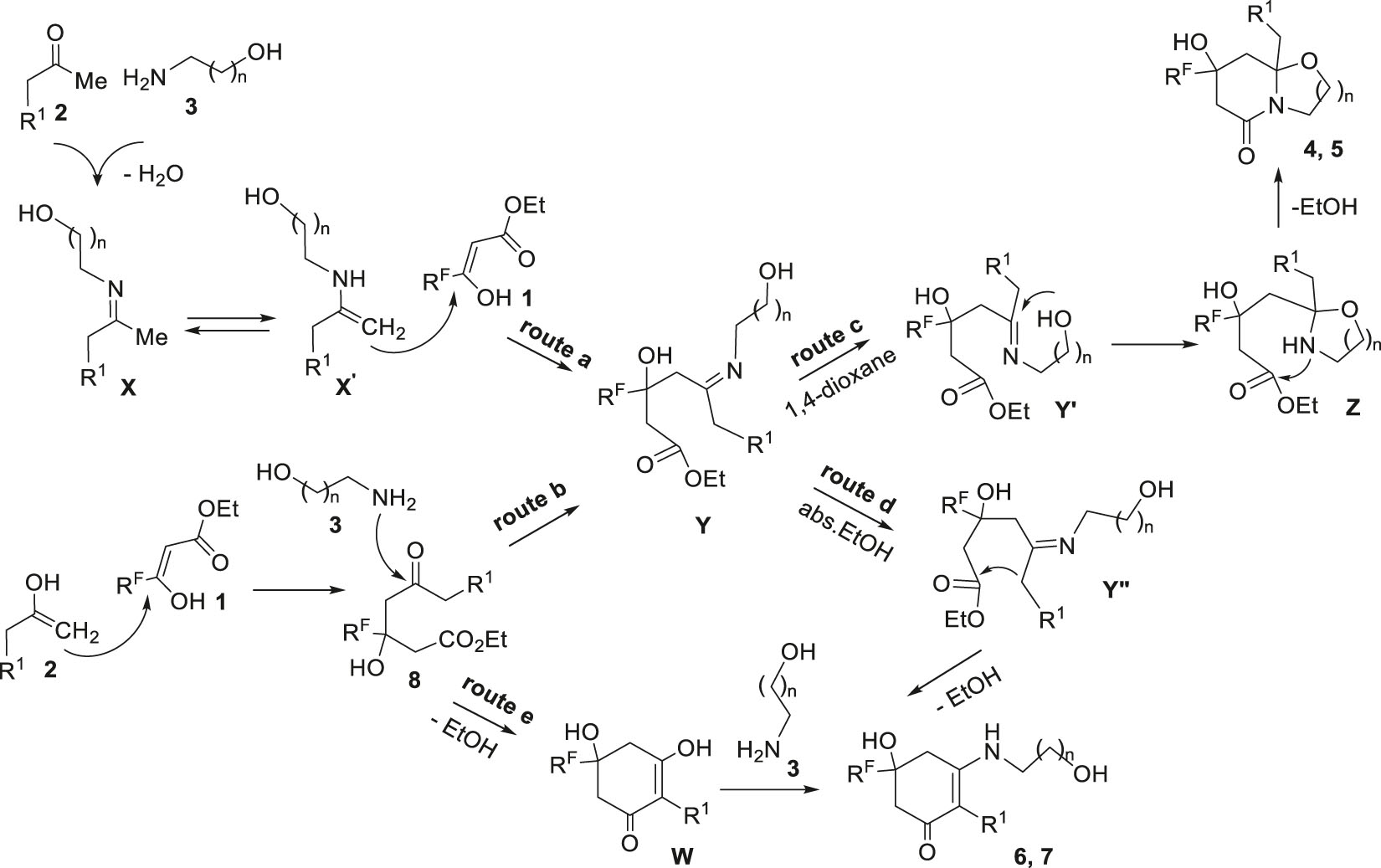
The proposed mechanism for the formation of bicycles 4, 5 and cyclohexenones 6, 7.
Conclusions
In summary, based on the three-component reaction of commercially available reagents, such as polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols, the approaches are offered to obtaining hexahydrooxazolo[3,2-a]pyridin-5-ones, hexahydropyrido[2,1-b][1,3]oxazin-6-ones and cyclohexenones functionalized with amino alcohol fragment. It was found that the cyclization of 3-oxo ester and methyl ketone reagents with 3-aminopropanol in ethanol leads mainly to 3-hydroxypropylaminocyclohex-2-enones, whereas in 1,4-dioxane the main products are hexahydropyrido[2,1-b][1,3]oxazine-6-ones in the form of trans-diastereomers. In contrast to that, similar cyclization with aminoethanol and its analogues, regardless of the reaction conditions, yields hexahydrooxazolo[3,2-a]pyridine-5-ones mainly. In this case, the reaction with alkyl methyl ketones in 1,4-dioxane proceeds stereoselectively to form trans-diastereomers, while a mixture of cis-and trans-diastereomers is generated in ethanol, which allows the cis-isomer to be isolated. In the case that acetophenone is introduced in the cyclization with both amino alcohols, cis-diastereomer heterocycles are the predominant products. The synthesis of 3-hydroxyethylaminocyclohex-2-enones is possible in rare cases only.
Funding source: Russian Foundation for Basic Research
Award Identifier / Grant number: 18-03-00342
Acknowledgements
Analytical studies were carried out using the equipment of the Center for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis of the Russian Academy of Sciences (Ural Branch).
-
Funding: This research was financially supported by Russian Foundation for Basic Research (grant no. 18-03-00342) and the State project АААА-А19-119011790132-7.
References
[1] J. Zhu, H. Bienayme (Ed.), Multicomponent Reactions, Wiley-VCH Weinheim, Germany (2005).Suche in Google Scholar
[2] A. Domling. Chem. Rev. 106, 17 (2006).10.1021/cr0505728Suche in Google Scholar PubMed
[3] B. Ganem. Acc. Chem. Res. 42, 463 (2009).10.1021/ar800214sSuche in Google Scholar PubMed PubMed Central
[4] A. Domling, W. Wang, K. Wang. Chem. Rev. 112, 3083 (2012).10.1021/cr100233rSuche in Google Scholar PubMed PubMed Central
[5] J. Liu, J. Li, L. Zhang, L. Song, M. Zhang, W. Cao, S. Zhu, H. Deng, M. Shao. Tetrahedron Lett. 53, 2469 (2012).10.1016/j.tetlet.2012.03.023Suche in Google Scholar
[6] W. Wang, J. Li, L. Zhang, L. Song, M. Zhang, W. Cao, H. Deng, M. Shao. Synthesis 44, 1686 (2012).10.1055/s-0031-1289761Suche in Google Scholar
[7] K. Karnakar, K. Ramesh, K. H. V. Reddy, B. S. P. Anil Kumar, J. B. Nanubonula, Y. V. D. Nageswar. New J. Chem. 39, 8978 (2015).10.1039/C5NJ01448DSuche in Google Scholar
[8] N. N. Gibadullina, D. R. Latypova, R. A. Novikov, Y. V. Tomilov, V. A. Dokicheva. Arkivoc 222 (2017).10.24820/ark.5550190.p010.003Suche in Google Scholar
[9] L. Zhou, F. Yuan, Y. Zhou, W. Duan, M. Zhang, H. Deng, L. Song. Tetrahedron 3761 (2018).10.1016/j.tet.2018.05.059Suche in Google Scholar
[10] J. D. Bhatt, T. S. Patel, C. J. Chudasama, K. D. Patel. ChemistrySelect 3, 3632 (2018).10.1002/slct.201702285Suche in Google Scholar
[11] C. Dayakar, B. C. Raju, ChemistrySelect 3, 9388 (2018).10.1002/slct.201801430Suche in Google Scholar
[12] X.-X. Du, Q.-X. Zi, Y.-M. Wu, Y. Jin, J. Lin, S.-J. Yan, Green Chem. 21, 1505 (2019).10.1039/C8GC03698ESuche in Google Scholar
[13] K. I. Pashkevich, V. I. Saloutin. Russ. Chem. Rev. 54, 1185 (1985).10.1070/RC1985v054n12ABEH003164Suche in Google Scholar
[14] S. Zhu, L. Song, G. Jin, B. Dai, J. Hao. Curr. Org. Chem. 13, 1015 (2009).10.2174/138527209788680772Suche in Google Scholar
[15] Y. Wu, Y. Wang, M. He, X. Tao, J. Li, D. Shan, L. Lv. Mini-Rev. Org. Chem. 14, 350 (2017).10.2174/1570193X14666170511122820Suche in Google Scholar
[16] Y. L. Liu, T. D. Shi, F. Zhou, X. L. Zhao, X. Wang. J. Zhou. Org. Lett. 13, 3826 (2011).10.1021/ol201316zSuche in Google Scholar PubMed
[17] D. O’Hagan, Chem. Soc. Rev. 37, 308 (2008).10.1039/B711844ASuche in Google Scholar
[18] A. Rahman, E. Xie, X. Lin. Org. Biomol. Chem. 16, 1367 (2018).10.1039/C8OB00055GSuche in Google Scholar
[19] B. Jeffries, Z. Wang, J. Graton, S. D. Holland, T. Brind, R. D. R. Greenwood, J.-Y. Le Questel, J. S. Scott, E. Chiarparin, B. Linclau. J. Med. Chem. 61, 10602 (2018).10.1021/acs.jmedchem.8b01222Suche in Google Scholar PubMed
[20] K. Koroniak-Szejn, J. Tomaszewska, J. Grajewski, H. Koroniak. J. Fluorine Chem. 219, 98 (2019).10.1016/j.jfluchem.2019.01.004Suche in Google Scholar
[21] F. Giornal, S. Pazenok, L. Rodefeld, N. Lui, J. P. Vors, F. R. Leroux. J. Fluorine Chem. 152, 2 (2013).10.1016/j.jfluchem.2012.11.008Suche in Google Scholar
[22] B. Romana-Souza, J. Salles dos Santos, L. G. Bandeira, A. Monte-Alto-Costa. Life Sci. 153, 82 (2016).10.1016/j.lfs.2016.04.017Suche in Google Scholar
[23] V. K. Sthalam, A. K. Singh, S. Pabbaraja. Org. Process Res. Dev. 23, 1892 (2019).10.1021/acs.oprd.9b00212Suche in Google Scholar
[24] V. I. Saloutin, Y. V. Burgart, O. G. Kuzueva, C. O. Kappe, O. N. Chupakhin. J. Fluor. Chem. 103, 17 (2000).10.1016/S0022-1139(99)00216-XSuche in Google Scholar
[25] M. V. Pryadeina, O. G. Kuzueva, Y. V. Burgart, V. I. Saloutin, K. A. Lyssenko, M. Yu. Antipin. J. Fluor. Chem. 117, 1 (2002).10.1016/S0022-1139(02)00149-5Suche in Google Scholar
[26] M. V. Pryadeina, Y. V. Burgart, V. I. Saloutin, M. I. Kodess, E. N. Ulomskii, V. L. Rusinov. Russ. J. Org. Chem. 40, 902 (2004).10.1023/B:RUJO.0000044558.47152.65Suche in Google Scholar
[27] M. V. Pryadeina, Y. V. Burgart, V. I. Saloutin, O. N. Chupakhin. Mendeleev Commun. 18, 276 (2008).10.1016/j.mencom.2008.09.017Suche in Google Scholar
[28] M. V. Goryaeva, Ya. V. Burgart, V. I. Saloutin, E. V. Sadchikova, E. N. Ulomskii. Heterocycles 78, 435 (2009).10.3987/COM-08-11524Suche in Google Scholar
[29] M. V. Goryaeva, Ya. V. Burgart, V. I. Saloutin. J. Fluorine Chem. 147, 15 (2013).10.1016/j.jfluchem.2013.01.005Suche in Google Scholar
[30] Y. S. Kudyakova, D. N. Bazhin, M. V. Goryaeva, Ya. V. Burgart, V. I. Saloutin. Russ. Chem. Rev. 83, 120 (2014).10.1070/RC2014v083n02ABEH004388Suche in Google Scholar
[31] M. V. Goryaeva, Y. V. Burgart, M. A. Ezhikova, M. I. Kodess, V. I. Saloutin. Beilstein J. Org. Chem. 11, 385 (2015).10.3762/bjoc.11.44Suche in Google Scholar PubMed PubMed Central
[32] V. I. Saloutin, Y. S. Kudyakova, M. V. Goryaeva, Y. V. Burgart, O. N. Chupakhin. Pure Appl. Chem. 89, 1209 (2017).10.1515/pac-2016-1015Suche in Google Scholar
[33] O. G. Khudina, E. V. Shchegol’kov, Y. V. Burgart, N. P. Boltneva, E. V. Rudakova, G. F. Makhaeva, V. I. Saloutin. J. Fluor. Chem. 210, 117 (2018).10.1016/j.jfluchem.2018.03.007Suche in Google Scholar
[34] L. V. Politanskaya, G. A. Selivanova, E. V. Panteleeva, E. V. Tretyakov, V. E. Platonov, P. V. Nikul’shin, A. S. Vinogradov, Y. V. Zonov, V. M. Karpov, T. V. Mezhenkova, A. V. Vasilyev, A. B. Koldobskii, O. S. Shilova, S. M. Morozova, Y. V. Burgart, E. V. Shchegolkov, V. I. Saloutin, V. B. Sokolov, A. Y. Aksinenko, V. G. Nenajdenko, M. Y. Moskalik, V. V. Astakhova, B. A. Shainyan, A. A. Tabolin, S. L. Ioffe, V. M. Muzalevskiy, E. S. Balenkova, A. V. Shastin, A. A. Tyutyunov, V. E. Boiko, S. M. Igumnov, A. D. Dilman, N. Y. Adonin, V. V. Bardin, S. M. Masoud, D. V. Vorobyeva, S. N. Osipov, E. V. Nosova, G. N. Lipunova, V. N. Charushin, D. O. Prima, A. G. Makarov, A. V. Zibarev, B. A. Trofimov, L. N. Sobenina, K. V. Belyaeva, V. Y. Sosnovskikh, D. L. Obydennov, S. A. Usachev. Russ. Chem. Rev. 88, 425 (2019).10.1070/RCR4871Suche in Google Scholar
[35] S. Tyndall, K. F. Wong, M. A. Van Alstine-Parris. J. Org. Chem. 80, 895 (2015).10.1021/acs.joc.5b01802Suche in Google Scholar
[36] W. Shi, Y. Wang, Y. Zhu, M. Zhang, L. Song, H. Deng. Synthesis 48, 3527 (2016).10.1055/s-0035-1562433Suche in Google Scholar
[37] Y. M. Lin, G. P. Lu, R. K. Wang, W. B. Yi. Org. Lett. 18, 6424 (2016).10.1021/acs.orglett.6b03324Suche in Google Scholar
[38] K. Rajkumar, T. R. Murthy, A. Zehra, P. S. Khursade, S. V. Kalivendi, A. K. Tiwari, R. S. Prakasham, B. C. Raju. ChemistrySelect 3, 13729 (2018).10.1002/slct.201802809Suche in Google Scholar
[39] M. V. Goryaeva, Y. V. Burgart, Y. S. Kudyakova, M. A. Ezhikova, M. I. Kodess, P. A. Slepukhin, V. I. Saloutin. Eur. J. Org. Chem. 6306 (2015).10.1002/ejoc.201500822Suche in Google Scholar
[40] M. V. Goryaeva, Y. V. Burgart, Y. S. Kudyakova, M. A. Ezhikova, M. I. Kodess, V. I. Saloutin. Eur. J. Org. Chem. 3986 (2017).10.1002/ejoc.201700683Suche in Google Scholar
[41] M. V. Goryaeva, S. O. Kushch, O. G. Khudina, Y.V. Burgart, Y. S. Kudyakova, M. A. Ezhikova, M. I. Kodess, P. A. Slepukhin, L. S. Sadretdinova, N. P. Evstigneeva, N. A. Gerasimova, V. I. Saloutin. Org. Biomol. Chem. 17, 4273 (2019).10.1039/C9OB00293FSuche in Google Scholar
[42] T. Someno, S. Kunimoto, H. Nakamura, H. Naganawa, D. Ikeda. J. Antibiot. 58, 56 (2005).10.1038/ja.2005.6Suche in Google Scholar
[43] R. M. Vilar, J. Gil-Longo, A. H. Daranas, M. L. Souto, J. J. Fernandez, S. Peixinho, M. A. Barral, G. Santafé, J. Rodrigueza, C. Jiménez. Bioorg. Med. Chem. 11, 2301 (2003).10.1016/S0968-0896(03)00107-XSuche in Google Scholar
[44] S. Gang, S. Ohta, H. Chiba, O. Jhodo, H. Nomura, Y. Nagamatsu, A. Yoshimoto, J. Antibiot. 54, 304 (2001).10.7164/antibiotics.54.304Suche in Google Scholar PubMed
[45] Y. Asai, N. Nonaka, S.-I. Suzuki, M. Nishio, K. Takahashi, H. Shima, K. Ohmori, T. Onuki, K. Komatsubara, J. Antibiot. 52, 607 (1999).10.7164/antibiotics.52.607Suche in Google Scholar PubMed
[46] E. Martin, A. Moran, M. L. Martin, L. S. Roman, P. Puebla, M. Medarde, E. Caballero, A. San Feliciano. Bioorg. Med. Chem. Lett. 10, 319 (2000).10.1016/S0960-894X(99)00691-5Suche in Google Scholar
[47] M. Amat, M. Pérez, J. Bosch. Synlett. 143 (2011).10.1055/s-0030-1259297Suche in Google Scholar
[48] M. P. Dwyer, J. E. Lamar, A .I. Meyers. Tetrahedron Lett. 40, 8965 (1999).10.1016/S0040-4039(99)01784-0Suche in Google Scholar
[49] C. Escolano, M. Amat, J. Bosch. Chem. – Eur. J. 12, 8198 (2006).10.1002/chem.200600813Suche in Google Scholar
[50] M. D. Groaning, A. I. Meyers. Tetrahedron 56, 9843 (2000).10.1016/S0040-4020(00)00926-1Suche in Google Scholar
[51] M. Amat, N. Llor, R. Griera, M. Pérez, J. Bosch. Nat. Prod. Commun. 6, 515 (2011).Suche in Google Scholar
[52] P. Aeberli, W. J. Houlihan. J. Org. Chem. 34, 165 (1969).10.1021/jo00838a036Suche in Google Scholar
[53] P. Aeberli, J. H. Gogerty, W. J. Houlihan, L. C. Iorio. J. Med. Chem. 19, 436 (1976).10.1021/jm00225a023Suche in Google Scholar
[54] A. I. Meyers, M. A. Sturgess. Tetrahedron Lett. 30, 1741 (1989).10.1016/S0040-4039(00)99568-6Suche in Google Scholar
[55] M. Jida, R. Deprez-Poulain, S. Malaquin, P. Roussel, F. Agbossou-Niedercorn, B. Depreza, G. Laconde. Green Chem., 12, 961 (2010).10.1039/b924111fSuche in Google Scholar
[56] M. Amat, N. Llor, R. Griera, M. Pérez, J. Bosch. Nat. Prod. Commun. 6, 515 (2011).10.1177/1934578X1100600412Suche in Google Scholar
[57] S. Malaquin, M. Jida, J. Courtin, G. Laconde, N. Willand, B. Deprez, R. Deprez-Poulain. Tetrahedron Lett. 54, 562 (2013).10.1016/j.tetlet.2012.11.082Suche in Google Scholar
[58] S. M. Allin, C. I. Thomas, J. E. Allard, M. Duncton, M. R. J. Elsegooda, M. Edgar. Tetrahedron Lett. 44, 2335 (2003).10.1016/S0040-4039(03)00250-8Suche in Google Scholar
[59] S. M. Allin, L. J. Duffy, P. C. Bulman Page, V. McKeea, M. J. McKenzie. Tetrahedron Lett. 48, 4711 (2007).10.1016/j.tetlet.2007.05.030Suche in Google Scholar
[60] O. Bassas, N. Llor, M. M. M. Santos, R. Griera, E. Molins, M. Amat, J. Bosch. Org. Lett. 7, 2817 (2005).10.1021/ol0505609Suche in Google Scholar
[61] M. Amat, M. M. M. Santos, O. Bassas, N. Llor, C. Escolano, A. Gomez-Esque, E. Molins, S. M. Allin, V. McKee, J. Bosch. J. Org. Chem. 72, 5193 (2007).10.1021/jo070539gSuche in Google Scholar
[62] J. C. Hubert, J. B. P. A. Wijnberg, W. N. Speckamp. Tetrahedron 31, 1437 (1975).10.1016/0040-4020(75)87076-1Suche in Google Scholar
[63] M. Amat, N. Llor, J. Bosch. Tetrahedron Lett. 35, 2223 (1994).10.1016/S0040-4039(00)76803-1Suche in Google Scholar
[64] E. Airiau, N. Girard, A. Mann, J. Salvadori, M. Taddei. Org. Lett. 11, 5314 (2009).10.1021/ol902279mSuche in Google Scholar PubMed
[65] J. Salvadori, E. Airiau, N. Girard, A. Mann, M. Taddei. Tetrahedron 66, 3749 (2010).10.1016/j.tet.2010.03.064Suche in Google Scholar
[66] M. A. Estiarte, M. Rubiralta, A. Diez, M. Thormann, E. Giralt. J. Org. Chem. 65, 6992 (2000).10.1021/jo000416vSuche in Google Scholar PubMed
[67] M. Weber, S. Jautze, W. Frey, R. Peters. J. Am. Chem. Soc. 132, 12222 (2010).10.1021/ja106088vSuche in Google Scholar PubMed
[68] J. Jiang, R J. DeVita, G. A. Doss, M. T. Goulet, M. J. Wyvratt. J. Am. Chem. Soc. 121, 593 (1999).10.1021/ja983389nSuche in Google Scholar
[69] W. Notz, F. Tanaka, C. F. Barbas. Acc. Chem. Res. 37, 580 (2004).10.1021/ar0300468Suche in Google Scholar PubMed
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/pac-2019-1216).
© 2020 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Artikel in diesem Heft
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors
Artikel in diesem Heft
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors