Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
-
 ,
,

Abstract
Colchicine derivative bearing substituted bispidine moiety, namely N-{7-(3,7-Di-(tert-butoxycarbonyl)-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-yl)-oxy-7-oxoheptanoyl}-N-deacetylcolchicine, was synthesized and tested for its effect on the net of microtubules (MT) in lung cancer cells A549. The compound induced not only MT depolymerization but stimulated the formation of small tubulin aggregates and long tubulin fibrils localized mainly around nuclei. The assemblies were morphologically different from tubulin clusters induced by structurally related anticancer agent tubuloclustin. The biotests data demonstrate that the depolymerization takes place for both pure tubulin and tubulin in cellulo, while fibrils are formed only in the cells. The research data of structure–activity relationship for several similar colchicine derivatives synthesized in the work give evidence for the proposition that the initial conjugate may interact not only with tubulin and MT in the cells, but also with MT-associated proteins, involved in the process of tubulin polymerization. The ability to affect simultaneously MAP – tubulin interactions opens attractive prospects in the design of novel anticancer agents.
Introduction
Cell dimeric protein α,β-tubulin is a validated molecular target for many anticancer agents [1]. It plays an important role in the formation of cytoskeleton and in the process of cell division, when tubulin polymerizes to microtubules (MTs) [2]. Different types of atypical tubulin assemblies compose under special conditions or in the presence of some metal ions or small molecules [3]. The type of particular assembly induced by tubulin-targeted anticancer drug depends upon its binding site at the protein. Thus, binding of taxol at β-subunit leads to the formation of MT bundles [4]. Ligands of vinca domain at the interface of two neighbor dimers may cause both depolymerization of MTs with the following formation of tubulin paracrystals [5] and assembly to rings sheets etc. (e.g. phomopsin A, dolastatin 10) [6].
The majority of anticancer agents interacting with colchicine binding site (located in β-tubulin at the intradimeric interface of the heterodimer) under normal conditions are causing depolymerization of MTs only [7]. However, some ligands of colchicine domain can in addition stimulate the formation of atypical tubulin assemblies [8], [9], [10], [11], [12], [13], [14], [15], [16]. Many of these ligands represent colchicine (1a, Fig. 1) derivatives, able to provide hydrophobic contacts with α-tubulin subunit [10], [11], [12], [13], [14]. Tubuloclustin (1b) is a typical example of such compound: it initiates MT depolymerization with the following formation of tubulin clusters [10], [14]. Importantly, tubuloclustin is much more cytotoxic to cancer cells than colchicine and has better toxicological profile in vivo [10], [11].
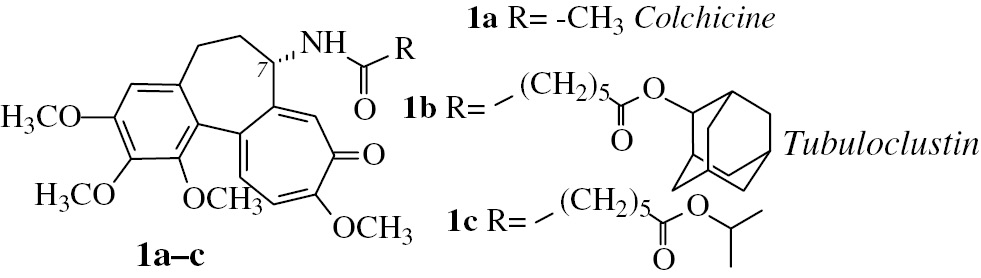
Structures of colchicine (1a) and its derivatives 1b and 1c. Compound 1b (tubuloclustin) stimulates the formation of long tubulin clusters in cancer cells and is more cytotoxic than the parent molecule 1a. Compound 1c with less voluminous than adamantane isopropyl moiety stimulates the formation of “point-like” tubulin clusters and is less cytotoxic than 1b.
Structure – activity relationship in a series of tubuloclustin analogs indicates that the volume of terminal substituent plays a crucial role in clustering [9], [10], [16]. Thus, isopropyl derivative 1c (Fig. 1) stimulates “point-like” clusterization, while conjugates with voluminous and branched moieties, like adamantane and its derivatives stimulate the formation of long “wavy” tubulin clusters [9], [16]. The strength of clustering effect of the compounds correlates with their cytotoxicity to cancer cells and this fact makes interesting additional studies in the field. In the present work we investigated whether an essential enhancement of the bulkiness and branching of the terminal substituent in tubuloclustin would lead to increased clusterization and cytotoxicity. We synthesized and tested novel tubuloclustin analogue – compound 2a (Fig. 2). Unexpectedly, its action on tubulin was qualitatively different: in addition to small tubulin clusters the compound induced the formation of long straight filamentous tubulin structures (Fig. 3, vide infra). This observation stimulated synthesis of compounds 2b-d to determine the structural requirements necessary for manifestation of the unusual effect.
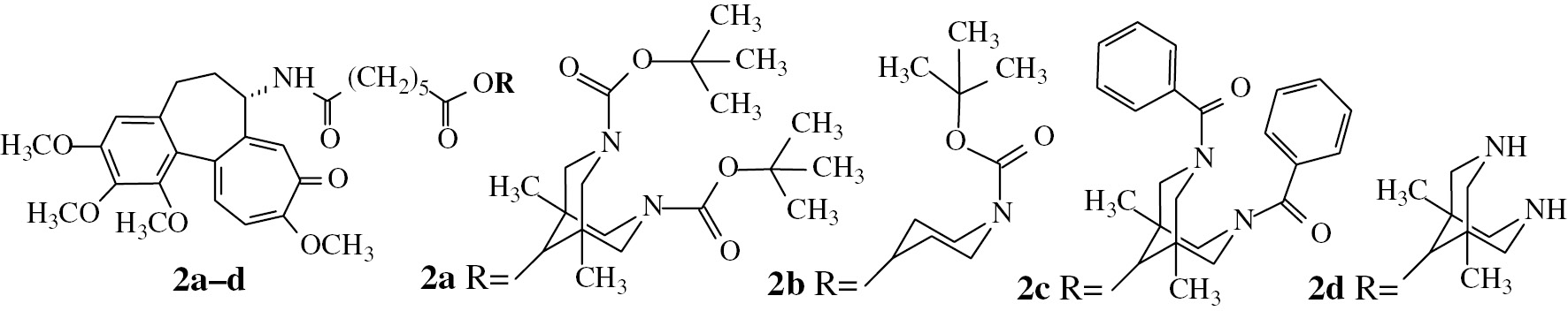
Structures of tubuloclustin analogues synthesized in the present work.
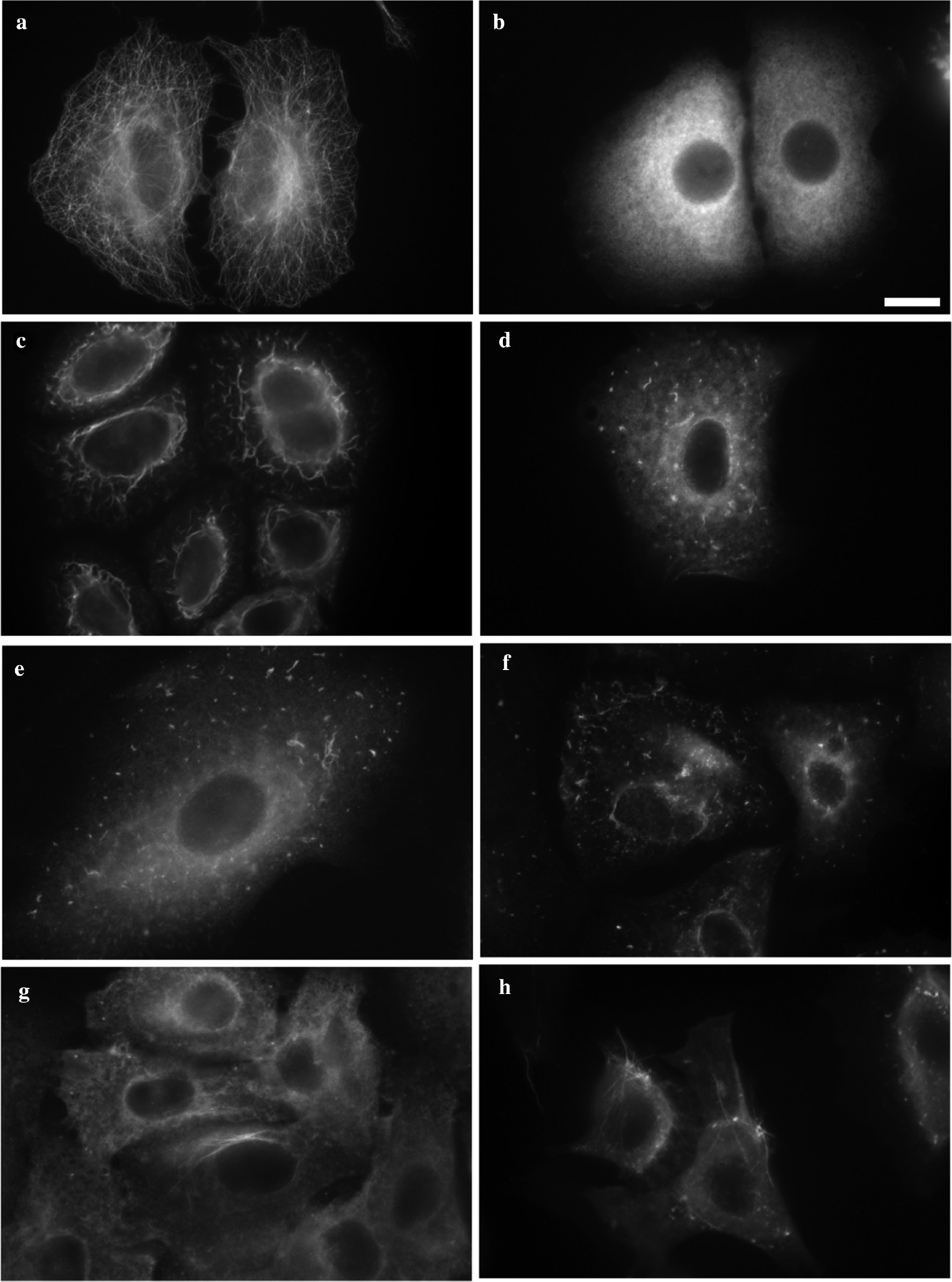
Fluorescence microscopy of MTs and tubulin structures in human lung carcinoma cells A549 treated with: (a) 0.5% DMSO (negative control) – intact microtubules; (b) 1 μM of colchicine (positive control) – depolymerization of MTs, no clustering; (c) 1 μM of tubuloclustin (1) – long wavy clusters; (d) 10 μM 2b – point-like and short wavy clusters; (e) 10 μM 2c – point-like and short wavy clusters; (f) 100 μM 2d – point-like and short wavy clusters; (g) 10 μM 2a – tiny aggregates and long tubulin filaments; (h) 100 μM 2a – small clusters and many long tubulin fibrils. Bar 10 μm.
Target compound 2a was synthesized as depicted on Scheme 1. Initial ketone 3a was obtained using a ring opening reaction in 5,7-dimethyl-1,3-diazaadamantane-6-one by action of Boc2O in the presence of HSO3NH2 and then reduced to the corresponding alcohol 4a [18]. The alcohol 4a was subjected to esterification with polyanhydride of pimelic acid [19] to give ester 5a, and reaction of the latter with N-deacetylcolchicine [20] in the presence of EEDQ led to target conjugate 2a.
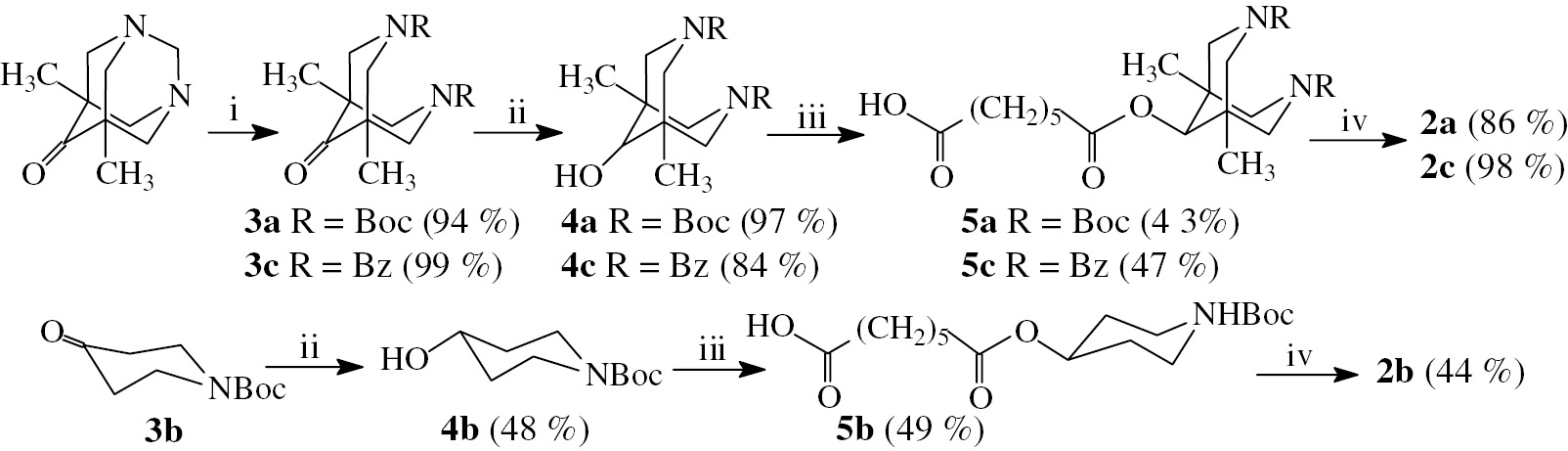
Reagents and conditions: (i) for 3a: Boc2O, HSO3NH2; for 3c: PhCOCl, NaHCO3, H2O–CHCl3; (ii) NaBH4, CH3OH, rt, 12 h; (iii) polyanhydride of pimelic acid, 4-DMAP, CH2Cl2, rt; (iv) N-deacetylcolchicine, EEDQ, CH2Cl2, rt.
The action of compound 2a on the network of MTs in human lung carcinoma A549 cells was studied by immunofluorescence microscopy and gave as it was mentioned above the unexpected results (see Table 1 and Fig. 3). The morphological effect caused by 2a on cellular tubulin was different from that of both tubuloclustin and colchicine (Fig. 3b,c). At 10 μM of 2a the pattern observed represented small aggregates and tubulin filaments localized mainly around nuclei in about 50% of the cells (Fig. 3g). At 100 μM of 2a these long tubulin fibrils were detected in almost 100% of observed cells (Fig. 3h). Cytotoxicity of compound 2a (measured in MTT test [20]) was two orders of magnitude lower than that of tubuloclustin (1), but still in nanomolar range (Table 1).
Cytotoxicity and effect on microtubule network of subject compounds.
| Compound | A549 EC50 (nM)a | Effect on A549 cells MT net (10 μM, 24 h) |
|---|---|---|
| 2a | 430±90 | MT depolymerization |
| Small aggregates and tubulin filaments | ||
| 2b | 54±5 | MT depolymerization |
| Point-like and short wavy clusters | ||
| 2c | 710±90 | MT depolymerization |
| Point-like clusters and short wavy clusters | ||
| 1 | 6 | MT depolymerization |
| Long wavy clusters | ||
| Colchicine | 30 | MT depolymerization |
-
aThe average of three to six experiments.
Literature search for the recent publications concerning mitostatic compounds able to first depolymerize MT and to stimulate the following formation of assemblies from tubulin dimer revealed the data about so called Janus compounds, which bind to colchicine site and in addition to MTs depolymerization stimulate the formation of long filamentous structures of tubulin in cells, sometimes very similar to tubulin fibrils promoted by 2a [21]. However, in contrast to Janus compounds, 2a cannot promote the MT formation from pure tubulin in vitro (data not shown). Moreover, Janus compounds belong to a pyrimido[4,5-b]indole structural type, totally different from that of 2a [21]. Therefore, we studied the structural requirements necessary for the manifestation of the “fibrillation” effect by conjugate 2a.
To check the role of the bridged bicyclic moiety in 2a we synthesized its monocyclic analogue 2b. The synthesis was fulfilled using standard procedure: after reduction of ketone 3b to alcohol 4b [22] the latter was subjected to reaction with polyanhydride of pimelic acid to give monoester 5b (Scheme 1). Its following amidation with N-deacetylcolchicine led to the target conjugate 2b. Compound 2b was one order of magnitude more cytotoxic, than 2a and was as cytotoxic as colchicine (Table 1). Immunofluorescence microscopy study demonstrated that at 10 μM compound 2b caused MT depolymerization in cancer cells and a formation of point-like and short wavy clusters (Fig. 3d) typical to that of less active tubuloclustin analogues [9]. No tubulin filaments were observed for compound 2b, giving evidence that bicyclic moiety in the conjugate 2a was important for unusual action of the latter on MT.
To check whether the filaments could be observed in the presence of analogues of 2a with other substituents in bispidine core, we synthesized N,N-dibenzoyl-conjugate 2c as depicted on Scheme 1. A ring opening reaction in 5,7-dimethyl-1,3-diazaadamantane-6-one was carried out by action of chloroanhydride of benzoic acid in the presence of aqueous solution of NaHCO3 [23]. Obtained ketone 3c was reduced to the corresponding alcohol 4c as described in [17] with slight modifications. The esterification of alcohol 4c by polyanhydride of pimelic acid gave monoester 5c, and amidation of the latter with N-deacetylcolchicine gave target conjugate 2c. Cytotoxicity of compound 2c was close to that of 2a (see Table 1), however it did not cause the formation of tubulin fibrils, but stimulated MT depolymerization and formation of point-like and short wavy tubulin clusters analogously to that of “monocyclic” conjugate 2b and other less active tubuloclustin analogues (Table 1 and Fig. 3e).
The biotesting data gave a reason for the proposition, that unusual action of compound 2a on MT of cancer cells was a result of a cleavage of biodegradable carbamate moiety in the cells. This cleavage should lead to a formation of a conjugate with unsubstituted bispidine (3,7-diazabicyclo[3.3.1]nonane) moiety, able to form complexes with ions of metals [24] and influence tubulin polymerization (sensitive to the presence of divalent cations [25]). However, hydrolysis of compound 2a with trifluoroacetic acid and the following treatment of the A549 cells with the corresponding base 2d indicated that tubulin fibrils still were not formed in these cells. Instead the point-like and short wavy clusters, very similar to that formed in the presence of 2b or 2c were observed (Fig. 3f). This fact indicates that the base 2d penetrates into the cell, but acts differently from the parent molecule 2a.
As though the tert-butoxycarbonyl moiety of 2a seems not to cleave in the cells, the differences in effect might be explained by different binding type of conjugates 2a and 2c with tubulin or by possibility of compound 2a to interact with another biotarget besides tubulin. To check the latter hypothesis, we carried out a study of pure tubulin polymerization in the presence of conjugate 2a under the conditions of MT forming (in the presence of 10% DMSO). The obtained microscopy picture (Fig. 4) showed that 2a prevented MT formation in vitro (only small tubulin aggregates were observed).
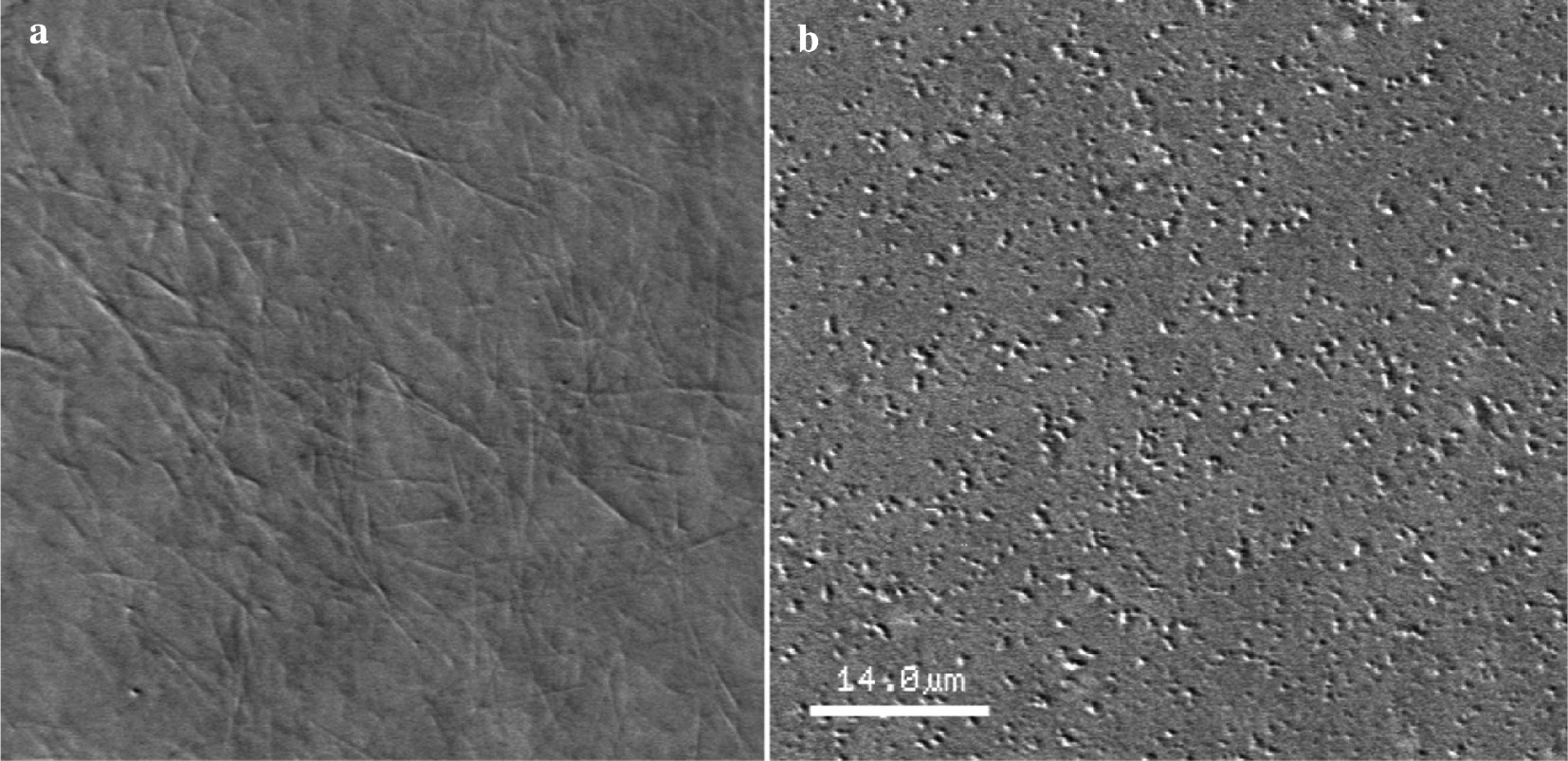
AVEC-DIC microscopy of purified tubulin after induction its polymerization by 10% DMSO: (a) without addition of 2a – MTs and (b) in the presence of 2a at concentration 50 μM – small aggregates.
The data demonstrate that compound 2a stimulates depolymerization of both pure tubulin and tubulin in cellulo, while fibrils are formed (most probably, from depolymerized tubulin) only in the cells. This fact gives evidence for the proposition that conjugate 2a may interact not only with tubulin and MT in the cells, but also with MT-associated proteins, involved in the process of tubulin polymerization. This makes the conjugate under discussion interesting for the additional studies of MAPs binding and its role in altering MT dynamics. The ability of MT-targeting agent to affect simultaneously MAP – tubulin interactions will open attractive prospects in the design of novel agents in anti-cancer therapy.
Experimental part
Chemistry
Liquid column chromatography was performed using silica gel Acros (40–60 μm). Thin-layer chromatography (TLC) was performed on Silufol-UV254 silica gel sheets, and spots were visualized with UV light (λ=254 nm) or stained with iodine vapor or aqueous potassium permanganate solution. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer at 28°C in CDCl3 at 400 and 100 MHz correspondingly. Chemical shifts (δ) are reported in parts per million (ppm) referenced to residual protonated solvent signal (CDCl3, δ=7.26 for 1H NMR) or to carbon signals in the NMR solvent (CDCl3, δ=77.0 for 13C NMR). The following abbreviations were used to designate multiplicities: s=singlet, d=doublet, t=triplet, m=multiplet, br=broad, dd=double-doublet, ddd=double-double-doublet. Electron impact mass spectra (EI-MS) were recorded on a Finnigan MAT INCOSSO spectrometer (electron impact, 70 eV). Matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass-spectra were taken on a Bruker Autoflex II mass spectrometer with 2,5-dihydroxybenzoic acid (DHB). CHN elemental analysis was performed using a Carlo-Erba ER-20 analyser. Infrared spectra (IR) were registered on a Thermo Nicolet IR200 apparatus in the range 900–3500 cm−1 using KBr disks. Melting points were determined using a capillary melting point apparatus and were uncorrected.
Di- tert -butyl 9-oxo-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonane-3,7-dicarboxylate (3a). 5,7-dimethyl-1,3-diazaadamantane-6-one (1.0 g, 5.56 mmol) was mixed with Boc2O (1.42 g, 6.51 mmol) and catalytic amount of sulfamic acid (0.015 ml, 0.33 mmol). After overnight stirring at room temperature ethyl acetate (40 ml) was added and the organic layer was washed with washed with water (3*20 ml) and brine (2*20 ml) and dried over anhydrous Na2SO4. The solvent was distilled off under vacuum, the residue was purified by column chromatography: 10% methanol in CH2Cl2. Yield 1.92 g (94%), white solid, m.p. 200–202°C. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.99 (6H, s, 1,5-CH3), 1.47 (18H, s, tBu), 2.96 (2H, d, J=13.5), 3.00 (2H, d, J=13.5), 4.28 (2H, d, J=12.6), 4.43 (2H, d, J=12.6). 13C NMR (CDCl3, δ, ppm): 16.89 (1,5-CH3), 28.36 (C(CH3)3), 45.96 (C-1,5), 55.97 (CH2), 56.76 (CH2), 80.38 (C(CH3)3), 154.36 (C=O), 213.65 (C 9 =O). Anal. calcd. for, %: C19H32N2O5, %: C 61.93, H 8.75, N 7.60. Found, %: C 61.90, H 8.78, N 7.53.
Di- tert -butyl 9-hydroxy-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonane-3,7-dicarboxylate (4a). To the solution of ketone 3a (1.0 g, 2.72 mmol) in methanol (10 ml) NaBH4 (0.150 g, 3.97 mmol) was added portionwise and the reaction mixture was stirred 12 h at room temperature. The solvent was evaporated at reduced pressure and the residue was purified by column chromatography: 5% methanol in CH2Cl2. Yield 0.980 g (97%), colorless oily liquid. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.90 (6H, s, 1,5-CH3), 1.41 (18H, s, tBu), 2.64 (1H, d, J=13.2), 2.70 (1H, d, J=12.7), 2.94 (1H, d, J=13.0), 3.02 (1H, d, J=12.7), 3.21 (1H, s, H-9), 3.61 (1H, d, J=12.7), 3.73 (1H, d, J=12.7), 3.92 (1H, d, J=13.0), 4.05 (1H, d, J=13.2). 13C NMR (CDCl3, δ, ppm): 20.79 (1,5-CH3), 28.33 (CCH3), 34.88 (C-1,5), 46.75 (CH2), 47.55 (CH2), 53.78 (CH2), 54.80 (CH2), 79.23 (CCH3), 79.56 (C-9), 154.45 (C=O), 154.92 (C=O). Anal. calcd. for, %: C19H34N2O5, %: C 61.60, H 9.25, N 7.56. Found, %: C 61.58, H 9.21, N 7.50.
Tert-butyl 4-hydroxypiperidine-1-carboxylate (4b) was obtained by literature procedure [22].
Polyanhydride of pimelic acid (PPA) was obtained by literature procedure [18].
3,7-Dibenzoyl-1,5-dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane (3c) was synthesized from 5,7-dimethyl-1,3-diazaadamantane-6-one and benzoyl chloride by literature procedure [23].
3,7-Dibenzoyl-9-hydroxy-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonane (4c). To the solution of ketone 3c (0.4 g, 1.06 mmol) in methanol (20 ml) NaBH4 (0.133 g, 3.59 mmol) was added portionwise and the reaction mixture was stirred 12 h at room temperature. The solvent was evaporated at reduced pressure and the residue was dissolved in CH2Cl2 (30 ml), washed by water (3×10 ml), dried over Na2SO4, filtered and evaporated. Yield 0.336 g (84%), white solid. M.p. 238 – 240°C. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.83 (3H, s, CH3), 0.84 (3H, s, CH3), 2.69 (1H, d, J=13.8, CH2), 2.91 (1H, br s, OH), 2.95 (1H, d, J=13.2, CH2), 3.05 (1H, d, J=13.8, CH2), 3.21 (1H, s, C9H), 3.35 (1H, d, J=13.2, CH2), 3.39 (1H, d, J=13.0, CH2), 3.72 (1H, d, J=13.0, CH2), 4.30 (1H, d, J=13.6, CH2), 4.55 (1H, d, J=13.6, CH2), 7.40–7.50 (10H, m, Ph). 13C NMR (CDCl3, δ, ppm): 20.34 (Me), 20.53 (Me), 35.46 (C-Me), 35.40 (C-Me), 45.09 (CH2), 50.89 (CH2), 51.82 (CH2), 57.81 (CH2), 77.92 (C-9), 127.08 (CHPh), 127.18 (CHPh), 128.57 (CHPh), 129.34 (CHPh), 129.52 (CHPh), 135.66 (CPh), 136.09 (CPh), 171.08 (C=O), 170.89 (C=O). IR (cm−1): 3321 br s (O–H), 3058-2844 (C–H), 1635(C=O), 1600 (C=O), 1571 (C–H aryl), 1442, 1429, 1317, 1082, 1020, 916.
General procedure A for the preparation of monoesters of dicarboxylic acids. To a solution of an alcohol in CH2Cl2 (3–5 ml) was added polyanhydride of pimelic acid and catalytic amount (0.01 g) of 4-dimethylaminopyridine (DMAP). The mixture was stirred at room temperature for 48 h and then the solvent was evaporated under reduced pressure. The residue was purified by column chromatography.
7-((3,7-Bis( tert -butoxycarbonyl)-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-yl)oxy)-7-oxo-heptanoic acid (5a) synthesized according to general procedure A from 4a (0.573 g, 1.547 mmol) and PPA (0.506 g, 3.563 mmol). Column chromatography: ethyl acetate/petroleum ether (40–70°C) in a gradient mixture 1:6–1:4. Yield 0.340 g (43%), colorless oily liquid. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.74 (6H, s, 1,5-CH3), 1.37–1.45 (2H, m, γ-CH2), 1.38 (9H, s), 1.39 (9H, s, tBu), 1.60–1.69 (4H, m, β-CH2), 2.32 (2H, t, J=7.4, CH2 CO2CH), 2.37 (2H, t, J=7.4, CH2 CO2H), 2.70 (1H, d, J=13.3, CH2 bicycl.), 2.78 (1H, d, J=13.0, CH2 bicycl.), 2.83 (1H, d, J=13.3, CH2 bicycl.), 2.89 (1H, d, J=13.0, CH2 bicycl.), 3.67 (1H, d, J=12.7, CH2 bicycl.), 3.79 (1H, d, J=12.7, CH2 bicycl.), 3.94 (1H, d, J=13.2, CH2 bicycl.), 4.09 (1H, d, J=13.2, CH2 bicycl.), 4.74 (1H, s, H-9 bicycl.), 8.75–9.08 (1H, br. s., CO2H). 13C NMR (CDCl3, δ, ppm): 20.32 (1,5-CH3), 24.18 (β-CH2), 24.64 (β-CH2), 28.21 (CCH3), 28.28 (CCH3), 28.47 (γ-CH2), 33.61 (α-CH2), 33.94 (α-CH2), 34.24 (C-1,5 bicycl.), 47.53 (CH2 bicycl.), 48.42 (CH2 bicycl.), 53.41 (CH2 bicycl.), 54.50 (CH2 bicycl.), 77.50 (C-9 bicycl.), 79.66 (CCH3), 79.86 (CCH3), 154.22 (tBuOC=O), 154.86 (tBuOC=O), 172.92 (C=O), 178.86 (CO2H). MS (MALDI-TOF), m/z: 512 [M]+, 535 [M+Na]+, 551 [M+K]+. Anal. calcd. for, %: C26H44N2O8, %: C 60.92, H 8.65, N 5.46. Found, %: C 60.96, H 8.59, N 5.42.
7-((1-( Tert -butoxycarbonyl)piperidin-4-yl)oxy)-7-oxoheptanoic acid (5b) synthesized according to general procedure A from 4b (0.500 g, 2.49 mmol) and PPA (0.700 g, 4.929 mmol). Column chromatography: ethyl acetate/petroleum ether (40–70°C) – 1:8. Yield 0.420 g (49%), pale yellow oily liquid. 1H NMR (CDCl3, δ, ppm, J/Hz): 1.35–1.42 (2H, m, γ-CH2), 1.46 (9H, s, tBu), 1.55–1.70 (6H, m, β-CH2+H-3,5 piper.), 1.80–1.87 (2H, m, H-3,5 piper.), 2.32 (2H, t, J=7.4, CH2 CO2CH), 2.36 (2H, t, J=7.0, CH2 CO2H), 3.23 (2H, ddd, J=13.6, 8.6, 3.9, H-2,6 piper.), 3.67–3.72 (2H, m, H-2,6 piper.), 4.92 (1H, tt, J=8.2, 3.9, H-4 piper.). 13C NMR (CDCl3, δ, ppm): 24.24 (β-CH2), 24.55 (β-CH2), 28.36 (CCH3), 28.41 (γ-CH2), 30.52 (CH2CH2N), 33.70 (α-CH2), 34.26 (α-CH2), 40.95 (br s., CH2N), 69.55 (C-4), 79.75 (CCH3), 154.76 (tBuOC=O), 172.87 (C=O), 179.05 (CO2H). MS (MALDI-TOF), m/z: 343 [M]+, 366 [M+Na]+, 382 [M+K]+. Anal. calcd. for, %: C17H29NO6, %: C 59.46; H 8.51. Found, %: C 59.53; H 8.57.
7-((N,N’-dibenzoyl-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-yl)oxy)-7-oxoheptanoic acid (5c) synthesized according to general procedure A from 4c (0.201 g, 0.53 mmol) and PPA (0.230 g, 1.62 mmol). Column chromatography: ethyl acetate/petroleum ether (40–70°C) in a gradient mixture 1:5 – 1:1. Yield 0.129 g (47%), white solid, m.p. 93–95°C. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.77 s (3H, CH3), 0.78 s (3H, CH3), 1.40 m (2H, γ-CH2), 1.64–1.73 m (4H, β-CH2), 2.33 t (2H, J=7.3, CH2 CO2H), 2.43 t (2H, J=7.6, CH2 CO2R), 2 bicycl.), 3.27 d (1H, J=12.9, CH2 bicycl. . ), 3.56 d (1H, J=12.9, CH2 bicycl.), 3.81 d (1H, J=13.4, CH2 bicycl.), 4.41 d (1H, J=13.9, CH bicycl.), 4.67 d (1H, J=13.9, CH2 bicycl.), 4.91 s (1H, C9H–O), 7.38–7.54 m (10H, Ph). 13C NMR (CDCl3, δ, ppm): 20.13 (CH3), 20.23 (CH3), 24.20 (β-C), 24.68 (β-C), 28.50 (γ-C), 33.59 (CH2CO2H), 33.94 (CH2-CO2R), 34.82 (CCH3), 34.89 (C CH3), 46.0 (CH2 bicycl.), 51.66 (CH2 bicycl.), 51.82 (CH2 bicycl.), 57.67 (CH2 bicycl.), 77.52 (C9H–O), 127.16 (CH m-Ph), 128.56 (CH o-Ph), 129.51 (CH p-Ph), 129.64 (CH p-Ph), 135.33 (C ipso-Ph), 135.78 (C ipso-Ph), 170.88 (PhC=O), 171.31 (PhC=O), 172.80 (CO2R), 178.16 (CO2H). IR (cm−1): 3199 br (O–H), 3059-2856 (C–H), 1732(C=O), 1641-1595 (C–H aryl), 1454, 1323, 1192, 1024, 937. MS (MALDI-TOF), m/z: 543 [M+Na]+, 559 [M+K]+.
General procedure B for the preparation of conjugates 2a–c. To a solution of compounds 5a–c in CH2Cl2 (5 ml) was added N-deacetylcolchicine and N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ). After stirring for 24 h at room temperature, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (ethyl acetate/petroleum ether (40–70°C) 1:1, then 1% methanol in CH2Cl2).
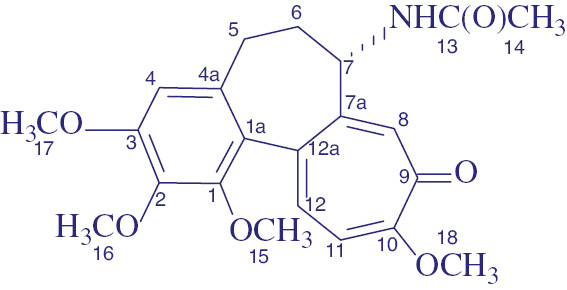
N-{7-(3,7-Di-(tert-butoxycarbonyl)-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-yl)-oxy-7-oxoheptanoyl}-N-deacetylcolchicine (2a) synthesized according to general procedure B from 5a (0.240 г, 0.469 mmol), N-deacetylcolchicine (0.155 g, 0.434 mmol) and EEDQ (0.116 g, 0.470 mmol). Yield 0.317 g (86%), pale yellow oily liquid. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.75 (6H, s, 1,5-CH3 bicycl.), 1.29–1.37 (2H, m, γ-CH2), 1.41 (9H, s, tBu), 1.42 (9H, s, tBu), 1.56–1.66 (4H, m, β-CH2), 1.85 (1H, td, J=12.1, 6.6, H-6 colch.), 2.23 (2H, t, J=7.4, CH2 CONH), 2.27–2.31 (1H, m, J=12.1, 6.5, H-6 colch.), 2.35 (2H, t, J=7.3, CH2CO2), 2.42 (1H, m, H-5 colch.), 2.52 (1H, dd, J=13.1, 6.1, H-5 colch.), 2.72 (1H, d, J=13.3, CH2 bicycl.), 2.79 (1H, d, J=12.6, CH2 bicycl.), 2.83 (1H, d, J=12.6, CH2 bicycl.), 2.90 (1H, d, J=13.3, CH2 bicycl.), 3.65 (3H, s, OCH3), 3.68 (1H, s, J=13.2, CH2 bicycl.), 3.80 (1H, d, J=12.6, CH2 bicycl.), 3.90 (3H, c, OCH3), 3.94 (3H, s, OCH3), 3.99 (1H, d, J=12.6, CH2 bicycl.), 4.00 (3H, s, OCH3), 4.09 (1H, d, J=13.2, CH2 bicycl.), 4.64 (1H, ddd, J=11.1, 7.1, 5.6, H-7 colch.), 4.73 (1H, s, H-9 bicycl.), 6.53 (1H, s, H-4 colch.), 6.86 (1H, d, J=10.6, H-11colch), 7.07 (1H, br.s, NH), 7.34 (1H, d, J=10.6, H-12 colch.), 7.51 (1H, s, H-8 colch.). 13C NMR (CDCl3, δ, ppm): 20.44 (1,5-CH3 bicycl.), 24.79 (β-CH2), 24.87 (β-CH2), 28.30 (CCH3), 28.37 (CCH3), 28.74 (γ-CH2), 29.85 (C-5 colch.), 33.97 (α-CH2), 34.30 (C-1,5 bicycl.), 35.79 (α-CH2), 36.79 (C-6 colch.), 47.61 (CH2 bicycl.), 48.53 (CH2 bicycl.), 52.15 (C-7 colch.), 53.55 (CH2 bicycl.), 54.63 (CH2 bicycl.), 56.05 (OCH3), 56.33 (OCH3), 61.30 (OCH3), 61.51 (OCH3), 77.52 (C-9 bicycl.), 79.47 (CCH3), 79.71 (CCH3), 107.30 (C-4 colch.), 112.66 (C-11 colch.), 125.54, 130.54 (C-8 colch.), 134.12, 135.45, 136.65, 141.63, 151.16, 151.89, 153.47, 154.21 (tBuOC=O), 154.78 (tBuOC=O), 163.95 (C-10 colch.), 172.33 (C=O), 173.02 (C=O), 179.15 (C-9 colch.). MS (MALDI-TOF), m/z: 852 [M]+, 875 [M+Na]+, 891 [M+K]+. Anal. calcd. for, %: C46H65N3O12, %: C 64.84, H 7.69, N 4.93. Found, %: C 64.79, H 7.60, N 4.92. N,N’-di-Boc-derivative 2a (10 mg, 0.011 mmol) dissolved in CH2Cl2 (2 ml) was treated with trifluoroacetic acid (10 μL, 0.130 mmol) and stirred at room temperature for 1 day. The reaction mass was concentrated under reduced pressure at 40°C, diluted with CH2Cl2 (10 ml), washed with saturated solution of NaHCO3 (5 ml), dried over anhydrous Na2SO4. The solvent was distilled off under vacuum to afford compound 6 mg of N-{7-(1,5-Dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-yl)-oxy-7-oxoheptanoyl}-N-deacetylcolchicine (2d).
N-{7-[1-( tert -butoxycarbonyl)piperidin-4-yloxy]-7-oxoheptanoyl}-N-deacetylcolchicine (2b) synthesized according to general procedure B from 5b (0.055 g, 0.160 mmol), N- deacetylcolchicine (0.050 g, 0.140 mmol) and EEDQ (0.050 г, 0.202 mmol). Yield 0.042 g (44%), pale yellow oily liquid. 1H NMR (CDCl3, δ, ppm, J/Hz): 1.30–1.37 (2H, m, γ-CH2), 1.46 (9H, C, tBu), 1.58–1.65 (6H, m, β-CH2+H-3,5 piper.), 1.78–1.86 (3H, m, H-3,5 piper.+H-6 colch.), 2.23 (2H, t, J=7.5, CH2 CONH), 2.24–2.28 (1H, m, H-6 colch.), 2.30 (2H, t, J=7.3, CH2CO2), 2.42 (1H, m, td, J=13.1, 7.0, H-5 colch.), 2.54 (1H, dd, J=13.1, 6.1, H-5 colch.), 3.22 (2H ddd, J=13.8, 8.5, 3.2, H-2,6 piper.), 3.67 (3H, s, OCH3), 3.68-3.74 (2H, m, H-2,6 piper.), 3.92 (3H, s, OCH3), 3.96 (3H, s, OCH3), 4.01 (3H, s, OCH3), 4.66 (1H, ddd, J=11.7, 7.0, 5.6, H-7 colch.), 4.91 (1H, tt, J=8.1, 4.1, H-4 piper.), 6.35 (1H, br. s, NH), 6.55 (1H, s, H-4 colch.), 6.84 (1H, d, J=10.9, H-11 colch.), 7.32 (1H, d, J=10.9, H-12 colch.), 7.41 (1H, s, H-8 colch.). 13C NMR (CDCl3, δ, ppm): 24.58 (β-CH2), 24.95 (β-CH2), 28.36 (γ-CH2), 28.39 (CCH3), 29.66 (C-3 piper.), 29.87 (C-5 colch.), 30.56 (C-5 piper.), 34.24 (α-CH2), 35.04 (α-CH2), 36.97 (C-6 colch.), 40.24 (br., C-2,6 piper.) 52.04 (C-7 colch.), 56.08 (OCH3), 56.33 (OCH3), 61.37 (OCH3), 61.56 (OCH3), 69.51 (C-4 piper.), 79.62 (CCH3), 107.31 (C-4 colch.), 112.40 (C-11 colch.), 125.60, 130.65 (C-8 colch.), 134.08, 135.24, 136.34, 141.66, 151.22, 151.28, 153.44, 154.71 (tBuOC=O), 163.97 (C-10 colch.), 172.28 (C=O), 172.94 (C=O), 179.34 (C-9 colch.). MS (MALDI-TOF), m/z: 682 [M+], 705 [M+Na+], 721 [M+K+]. Anal. calcd. for, %: C37H50N2O10, %: C 65.08, H 7.38, N 4.10. Found, %: C 65.05, H 7.35, N 4.07.
N-[7-(3,7-dibenzoyl-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-yl)-oxy-7-oxoheptanoyl]-N-deacetylcolchicine (2c) synthesized according to general procedure A from 5c (0.043 g, 0.08 mmol), EEDQ (0.031 g, 0.12 mmol) and 0.038 g N-deacetylcolchicine (0.11 mmol). Yield 0.069 g 4 (98%), pale orange solid, m.p. 174–176°C. 1H NMR (CDCl3, δ, ppm, J/Hz): 0.76 (6H, s, CH3 bicycl.),1.35 (2H, m, γ-CH2), 1.63 (4H, m, β-CH2), 1.84 (1H, m, H-6 colch.),2.24 (2H, t, J=7.1, CH2 CO2H), 2.24–2.27 (1H, m, H-6 colch.), 2.36–2.42 (1H, m, H-5 colch.), 2.41 (2H, t, J=7.5, CH2 CO2R), 2.53 (1H, dd, J=13.1, 5.6, H-5 colch.), 2.84 (1H, d, J=13.9, CH2 bicycl.), 2.95 (1H, d, J=13.9, CH2 bicycl.), 3.13 (1H, d, J=13.1, CH2 bicycl.), 3.26 (1H, d, J=13.1, CH2 bicycl.), 3.53 (1H, d, J=12.6, CH2 bicycl.), 3.66 (3H, s, OCH3), 3.80 (1H, d, J=13.1, CH2 bicycl.), 3.91 (3H, s, OCH3), 3.95 (3H, s, OCH3), 4.00 (3H, s, OCH3), 4.41 (1H, d, J=13.1, CH2 bicycl.), 4.64 – 4.67 (2H, d+m, J=13.1, CH2 bicycl.+H-7 colch.), 4.89 (1H, s, HOC-9 bicycl.), 6.54 (1H, s, H-4 colch.), 6.69 (1H, br. s, NH), 6.85 (1H, d, J=11.2, H-11 colch.), 7.32 (1H, d, J=11.2, H-12 colch.), 7.40–7.51 (11H, m, Ph+H-8 colch.). 13C NMR (CDCl3, δ, ppm): 20.12 (CH3 bicycl.), 20.22 (CH3 bicycl.), 24.81 (β-CH2), 28.69 (β-CH2), 29.82 (γ-CH2), 33.91, 34.79, 34.83, 35.65, 36.63, 45.87 (CH2 bicycl.), 51.81 (CH2 bicycl.), 51.82 (CH2 bicycl.), 52.15 (C-7 colch.), 56.03 (OCH3), 56.32 (OCH3), 57.66 (CH2 bicycl.), 61.31 (OCH3), 61.49 (OCH3), 78.45 (C-9 bicycl.), 107.21 (C-4 colch.), 112.51 (C-11 colch.), 125.50, 127.10, 128.52, 129.38, 129.54, 130.52, 134.09, 135.30, 135.94, 136.49, 141.54, 151.10, 151.78, 153.40, 163.88, 170.74 (PhCONH), 170.98 (PhCONH), 172.31 (C=O), 172.84 (C=O), 179.33 (C-9 colch). IR (cm−1, KBr): 3300 br (N–H), 3059-2858 (C–H), 1738 (C=O), 1.701 (C=O), 1671 (C=O amide), 1637, 1624 (C–H aryl), 1448, 1250, 1140, 1022.MS (MALDI-TOF), m/z: 883 [M+Na]+, 899 [M+K]+.
Biology
MTT cytotoxicity assay
The MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyl-2H-tetrazolium bromide, Roth GmbH, Karlsruhe, Germany) quantitative colorimetric assay was used to measure the cytotoxicity, viability and metabolic activity [20]. A549 human lung epithelial carcinoma cells (CCL-185™) were cultured with Dulbecco´s Modified Eagle medium (DMEM) containing 10% fetal bovine serum and 1% antibiotic penicillin/streptomycin at 37°C under a 5% CO2 humidified atmosphere. The cells were seeded in 96-well plates at a density of 3000 cells per well. Stock solutions of test compounds were prepared in DMSO at concentration 20 mM. Cells were treated for 24 h with selected compounds at 1–2400 nM (8 wells for each concentration). DMSO (0.5%) served as a negative control. Optical density was measured at 550 nm with 690 nm reference filter using EL808 Ultra Microplate Reader (BioTek Instruments, Winooski, VT, USA). Experiments for all compounds were repeated at least 3 times and EC50 values were determined by sigmoid curve fitting using Excel-based software.
Immunofluorescence staining of cellular microtubules and nuclei. For this staining A549 human lung epithelial carcinoma cells were cultured in 12-well plates on small glass coverslips (11 mm diameter) at a density of 20 000 cells per coverslip. Cells were incubated with tested compounds at concentrations of 100–1200 nM for 24 and 48 h. 0.5% DMSO served as a negative control. The cells were fixed and stained as described in [26]. Fixed cells were labelled for tubulin with mouse monoclonal antibody against β-tubulin at a dilution of 1:300 (Sigma, St. Louis, MO, USA), followed by incubation of Alexa Fluor488 labelled goat anti-mouse IgG at a dilution of 1:300 (Invitrogen, Germany). In order to analyze the compound effect on the apoptosis induction effect, the cell nuclei were stained with the Hoechst Nr. 33258 (Sigma, St. Louis, MO, USA) at concentration 5 μg/ml. Images of all samples were acquired with a Nikon Diaphot 300 inverted microscope (Nikon GmbH, Düsseldorf, Germany) equipped with a cooled charge-couple device camera system (SenSys; Photometrics, Munich, Germany).
Article note
A collection of invited papers based on presentations at 21st Mendeleev Congress on General and Applied Chemistry (Mendeleev-21), held in Saint Petersburg, Russian Federation, 9–13 September 2019.
Acknowledgements
This work was supported by Russian Foundation for Basic Research (project no. 18-03-00524). Initial reagent 5,7-dimethyl-1,3-diazaadamantane-6-one was synthesized within the State assignment AAAA-A16-116032250004-2. Authors acknowledge the German organization DAAD (German Academic Exchange Service).
References
[1] G. L. Regina, A. Coluccia, V. Naccarato, R. Silvestri. Eur. J. Pharm. Sci. 131, 58 (2019).10.1016/j.ejps.2019.01.028Search in Google Scholar
[2] M. O. Steinmetz1, A. E. Prota. Trends Cel. Biol. 28, 776 (2018).10.1016/j.tcb.2018.05.001Search in Google Scholar
[3] E. Unger, K. J. Bohm, W. Vater. Electron Microsc. Rev. 3, 355 (1990).10.1016/0892-0354(90)90007-FSearch in Google Scholar
[4] D. F. Albertini; J. I. Clark. Cell Biol. Int. Rep. 5, 387 (1981).10.1016/0309-1651(81)90009-6Search in Google Scholar
[5] E. Vassal, C. Barette, X. Fonrose, R. Dupont, E. Sans-Soleilhac, L. Lafanechère. J. Biomol. Screen. 11, 377 (2006).10.1177/1087057106286210Search in Google Scholar PubMed
[6] J. A. Hadfield, S. Ducki, N. Hirst, A. T. McGown. Prog. Cell Cycle Res. 5, 309 (2003).Search in Google Scholar
[7] B. Bhattacharyya, D. Panda, S. Gupta, M. Banerjee. Med. Res. Rev. 28, 155 (2008).10.1002/med.20097Search in Google Scholar PubMed
[8] N. A. Zefirov, A. Kruth, B. Wobith, E. V. Nurieva, S. Riyaz, Ch. V. R. Reddy, S. A. Kuznetsov, O. N. Zefirova. Mendeleev Commun. 28, 475 (2018).10.1016/j.mencom.2018.09.007Search in Google Scholar
[9] N. A. Zefirov, Y. A. Evteeva, B. Wobith, S. A. Kuznetsov, O. N. Zefirova. Struct. Chem. 30, 465 (2019).10.1007/s11224-018-1219-9Search in Google Scholar
[10] O. N. Zefirova, H. Lemcke, M. Lantow, E. V. Nurieva, B. Wobith, G. E. Onishchenko, A. Hoenen, G. Griffiths, N. S. Zefirov, S. A. Kuznetsov. ChemBioChem. 14, 1444 (2013).10.1002/cbic.201300143Search in Google Scholar PubMed
[11] O. N. Zefirova, E. V. Nurieva, Y. S. Glazkova, N. A. Zefirov, A. V. Mamaeva, B. Wobith, V. I. Romanenko, N. A. Lesnaia, H. M. Treshalina, S. A. Kuznetsov. Pharm. Chem. J. 48, 373 (2014).10.1007/s11094-014-1113-8Search in Google Scholar
[12] P. Thomopoulou, J. Sachs, N. Teusch, A. Mariappan, J. Gopalakrishnan, H. G. Schmalz. ACS Med. Chem. Lett. 7, 188 (2015).10.1021/acsmedchemlett.5b00418Search in Google Scholar
[13] C. Vilanova, S. Díaz-Oltra, J. Murga, E. Falomir, M. Carda, J. M. Redondo-Horcajo, J. F. Díaz, I. Barasoain, A. Marco. J. Med. Chem. 57, 10391 (2014).10.1021/jm501112qSearch in Google Scholar
[14] S. K. Kim, S. M. Cho, H. Kim, H. Seok, S. O. Kim, T. K. Kwon, J. S. Chang. Exp. Mol. Med. 45, E19 (2013).10.1038/emm.2013.38Search in Google Scholar
[15] O. N. Zefirova, E. V. Nurieva, D. V. Shishov, I. I. Baskin, F. Fuchs, H. Lemcke, F. Schröder, D. G. Weiss, N. S. Zefirov, S. A. Kuznetsov. Bioorg. Med. Chem. 19, 5529 (2011).10.1016/j.bmc.2011.07.040Search in Google Scholar
[16] O. N. Zefirova, E. V. Nurieva, B. Wobith, V. V. Gogol, N. A. Zefirov, A. V. Ogonkov, D. V. Shishov, N. S. Zefirov, S. A. Kuznetsov. Mol. Divers. 21, 547 (2017).10.1007/s11030-017-9739-6Search in Google Scholar
[17] Ts. Ye. Aghajanian, G. L. Arutyunian, R. K. Shahkhatuni. Chem. Heterocycl. Com. 39, 767 (2003).10.1023/A:1025699129643Search in Google Scholar
[18] J. W. Hill, W. H. Carothers. J. Am. Chem. Soc. 55, 5023 (1933).10.1021/ja01339a054Search in Google Scholar
[19] J. D. Bagnato, A. L. Eilers, R. A. Horton, C. B. Grissom. J. Org. Chem. 69, 8987 (2004).10.1021/jo049953wSearch in Google Scholar
[20] D. Gerlier, N. Thomasset. J. Immunol. Methods, 94, 57 (1986).10.1016/0022-1759(86)90215-2Search in Google Scholar
[21] C. C. Rohena, A. L. Risinger, R. K. V. Devambatla, N. F. Dybdal-Hargreaves, R. Kaul, S. Choudhary, A. Gangjee, S. L. Mooberr. Molecules, 21, 1661 (2016).10.3390/molecules21121661Search in Google Scholar PubMed PubMed Central
[22] P. J. Rayner, P. O’Brien, R. A. J. Horan. J. Am. Chem. Soc. 135, 8071 (2013).10.1021/ja4033956Search in Google Scholar PubMed
[23] Ts. E. Agadzhanyan, A. D. Arutyunyan, G. L. Arutyunyan. Chem. Heterocycl. Com. 28, 772 (1992).10.1007/BF00474490Search in Google Scholar
[24] P. Comba, M. Kerscher, W. Schiek. Prog. Inorg. Chem. 55, 613 (2007).10.1002/nadc.200748645Search in Google Scholar
[25] M. Schliwa, J. Cell Biol. 70, 527 (1976).10.1083/jcb.70.3.527Search in Google Scholar PubMed PubMed Central
[26] A. Al-Haddad, M. A. Shonn, B. Redlich, A. Blocker, J. K. Burkhardt, H. Yu, J. A. Hammer, D. G. Weiss, W. Steffen, G. Griffiths, S. A. Kuznetsov. Mol. Biol. Cell. 12, 2742, (2001).10.1091/mbc.12.9.2742Search in Google Scholar PubMed PubMed Central
©2020 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors