
CRH receptor antagonist crinecerfont – a promising new treatment option for patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
-
Clemens Kamrath

 und
Hedi L. Claahsen-van der Grinten
und
Hedi L. Claahsen-van der Grinten

Abstract
21-Hydroxylase deficiency (21OHD), the most common form of congenital adrenal hyperplasia (CAH), leads to impaired cortisol synthesis and androgen excess. Current treatments of patients with classic 21OHD with supraphysiological doses of glucocorticoids pose risks such as impaired growth and metabolic complications. We discuss the CRH receptor antagonist as a therapeutic option for children with classic 21OHD. A phase three trial of crinecerfont, a CRH receptor antagonist, offers a promising new treatment option. Crinecerfont helped to reduce glucocorticoid doses and to lower androgen levels. However, the study population may not be fully representative of the general 21OHD population. Successful implementation depends on patient adherence and monitoring to avoid possible complications such as adrenal crises. Overall, crinecerfont represents a valuable development, but further research and careful clinical management are needed to optimize its use in CAH treatment.
Introduction
21-Hydroxylase deficiency (21OHD), the most common form of congenital adrenal hyperplasia (CAH), is a rare autosomal recessive disorder with an incidence of approximately 1 in 15,000 births. 21OHD is caused by mutations in CYP21A2, the gene encoding the adrenal steroid 21-hydroxylase enzyme (CYP21A2). Inefficient cortisol synthesis in patients with CAH leads to adrenocorticotropic hormone (ACTH)-mediated adrenal stimulation, but instead of cortisol, the adrenals produce excess androgen precursors (Figure 1) [1]. Exposure to elevated levels of androgens in childhood is associated with a significant increase in the rate of growth and even more so in the rate of bone maturation, resulting in reduced adult height due to premature epiphyseal closure [2], [3], [4], [5], [6].
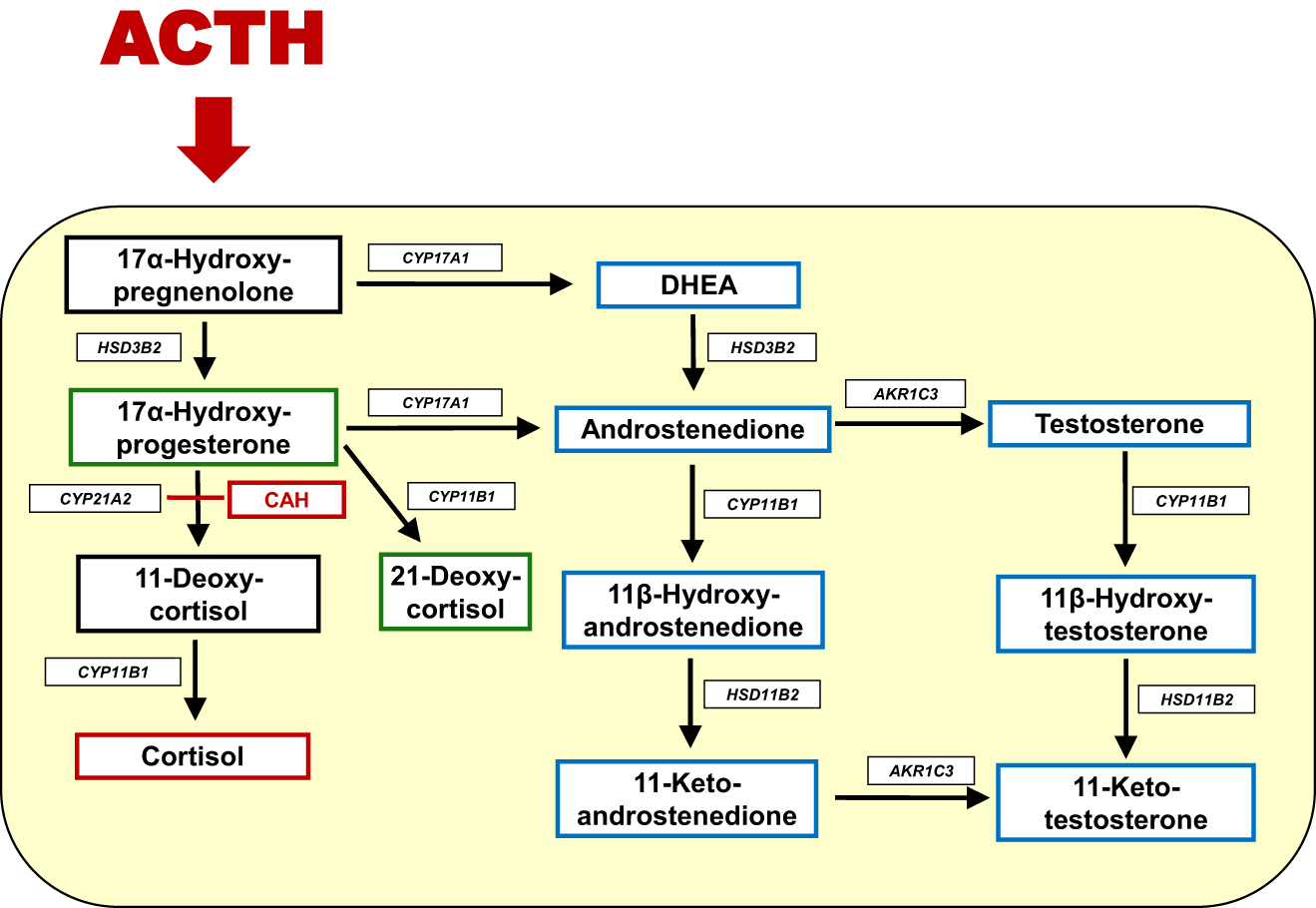
Schematic overview of steroidogenesis in CAH. The enzymatic block in CAH is shown in red. In CAH, ACTH stimulation leads to accumulated 17α-hydroxyprogesterone (17OHP), that is further converted to 21-deoxycortisol through the 11β-hydroxylase activity of CYP11B1·In addition, 17OHP is also converted to the androgen androstenedione. Adrenal androstenedione is converted to testosterone by aldo-keto reductase 1C3 (AKR1C3). Adrenal androstenedione and testosterone are also converted by the 11β-hydroxylase activity of CYP11B1 to the 11-oxygenated androgens 11β-hydroxyandrostenedione and 11β hydroxytestosterone, respectively. The biologic active androgens 11-ketotestosterone and 11-ketodihydrotestosterone that activate the androgen receptor comparable to testosterone and dihydrotestosterone, respectively, are formed in peripheral target tissues of androgen action.
In the treatment of children and adolescents with 21OHD, the challenge is to maintain a good therapeutic balance between iatrogenic glucocorticoid excess and adrenal hyperandrogenaemia [7]. However, the therapeutic glucocorticoid excess that is often required to suppress elevated pituitary ACTH with consequently lowering adrenal androgens carries a risk of long-term complications for the patient, such as impaired final height, increased obesity and long-term complications such as hypertension, insulin resistance and metabolic syndrome [8], [9], [10]. As a new treatment option for these patients, the results of a phase 3 trial of the CRH receptor antagonist crinecerfont have recently been published [11]. CRH antagonists were used as co-administration to current glucocorticoid substitution and aim to reduce ACTH-mediated adrenal hyperandrogenemia in order to move from supraphysiological suppressive glucocorticoid dosing to physiological glucocorticoid replacement therapy.
The CRH receptor antagonist crinecerfont for treatment in patients with CAH
The phase 3 randomized trial published by Sarafoglu et al. included 103 children and adolescents aged 2–17 years (average age 12.1 ± 3.5 years), of whom 69 patients received crinecerfont and 34 placebo for 28 weeks [11]. The mean serum androstenedione concentration of all participants was 431 ng/dL [15.0 nmol/L] (±461 ng/dL) and the mean value for 17α-hydroxyprogesterone (17OHP) was 8,682 ng/dL [263 nmol/L] (±6,847 ng/dL), at a mean glucocorticoid dose of 16.4 mg (±3.9 mg) hydrocortisone equivalent per square metre of body surface area per day. Notably, the mean glucocorticoid dose used was above the range of 10–15 mg/m2 per day recommended by the Endocrine Society [12] as well as hydrocortisone doses reported by registries under real-life conditions in children and adolescent with CAH [13], 14]. In particular, the average physiological production of cortisol by the adrenal gland is about 7 mg/m2 per day [15].
At week 28, the mean glucocorticoid dose could be reduced by 18 %–12.8 mg/m2 per day in the crinecerfont group, with a reduction in mean androstendione levels of 94 ng/dL [3.3 mmol/L] from baseline. In contrast, the mean glucocorticoid dose in the placebo group had to be increased by 6 %–17.0 mg/m2 per day, and the mean androstendione level increased from baseline by 147 ng/dL [5.1 mmol/L] [11]. This shows that this new therapeutic approach using CRH receptor antagonists is a promising adjuvant treatment in CAH and can improve the two main problems in the treatment of CAH patients, namely hyperandrogenemia and supraphysiological doses of hydrocortisone on both sides.
However, some critical aspects should be considered:
The initially poor adrenal suppression of the patients who were included in the crinecerfont trial is striking. Data of the German CAH Registry [16], which provides real-world data on treatment and outcomes, show a mean 17OHP level of 1,567 ng/dL [47.4 nmol/L] (±3,447 ng/dL [104.3 nmol/L], median 327 ng/dL [9.9 nmol/L]) in 475 patients aged 2–17 years (mean age 11.1 ± 5.0 years). The mean androstenedione level was 119 ng/dL [4.2 nmol/L] (±180 ng/dL [6.3 nmol/L], median 40 ng/dL [1.4 nmol/L]) in 318 patients at a mean age of 11.5 ± 4.8 years (the most recent measured values of the patients were presented from children with a clinically documented diagnosis of classic 21OHD; data not published). Data from the international CAH registry reported median 17OHP levels of 958 ng/dL (29 nmol/L) in children below age of 12 years (n=283) and of 1,999 ng/dL (60.5 nmol/L) in adolescents aged 12–18 years. Median levels of androstendione were 0 ng/dL in children below the age of 12 years and 301 ng/dL [10.5 mmol/L] in adolescents ages 12–18 years [13]. These data suggest that the study population of the crinecerfont study may be a selected group of poorly controlled patients. On the other hand, although the cohort studied may not be representative for the entire group of CAH patients, those with poor androgen control despite high doses of glucocorticoids will be the ones who will be the first to be offered the new therapy.
The combination of high glucocorticoid doses and inadequate adrenocortical suppression and the high prevalence of testicular adrenal rest tumours (TART) of 33 % in this cohort is consistent with failure of conventional glucocorticoid treatment to suppress adrenal hyperandrogenism in these patients. Data from studies of 24-h urine steroid metabolite analyses in 109 children with classic CAH showed that treatment failure, defined by insufficiently suppressed adrenal glands despite increased excretion of cortisol metabolites, affected approximately 30 % of the children treated with hydrocortisone [17].
There are several possible reasons why current hydrocortisone treatment fails to effectively suppress the ACTH production that is necessary to reduce adrenal androgen production. First, the current hydrocortisone dose regimen throughout the day does not mimic the circadian rhythm of ACTH production and consequently adrenal androgen production especially during the night [18], 19]. Importantly, modified-released hydrocortisone preparations which are recently introduced are able to reach more stable androgen suppression [20]. The combination treatment of modified-release hydrocortisone together with a CRH receptor antagonist would certainly be a reasonable next therapeutic option.
Second, treatment failure could be due to differences in intra-individual absorption and half-life of oral hydrocortisone [21], 22]. Last, poor hormonal control can be caused by lack of adherence, which affects a large proportion of young patients in particular with chronic illnesses and regular medication use [23], 24]. Nowadays, in modern CAH management with frequent (3–4 monthly) follow-up and monitoring with saliva/serum daily profiles, the majority of patients can be adequately treated with moderate doses of hydrocortisone, the preferred glucocorticoids in paediatric patients, enabling most patients to reach their final height within their target range [25].
Studies in recent years have shown that the androgen excess in children with CAH due to 21OHD is mainly caused by elevated adrenal-derived 11-oxygenated androgens [26], 27]. Adrenal-derived 11β-hydroxyandrostenedione is further metabolized to 11-ketotestosterone and 11-ketodihydrotestosterone, which are biologically highly potent androgens that directly stimulate the androgen receptor comparable with testosterone and dihydrotestosterone, respectively (Figure 1) [28], 29]. Although the superiority of measuring 11-oxygenated androgens over classical androgens for monitoring therapy in patients with CAH has not yet been proven, it would be interesting to study the effect of crinecerfont on this group of androgens in the future.
The role of steroid precursors in CAH
Although the study by Sarafoglu et al. did not show evidence of a particular risk profile of treatment with crinecerfont (adverse event rate 84 % in the crinecerfont group vs. 82 % in the placebo group) [11], we want to highlight the theoretical potential to increased risk of Addisonian crisis due to suppression of ACTH and thereby of adrenal steroid precursors of which some have potential glucocorticoid effects.
The typically elevated steroid precursors in patients with 21OHD are 17OHP and 21 deoxycortisol (21DF) (Figure 1) [1], 3], 12]. Especially 21DF, shows clinically significant activity at the human glucocorticoid receptor (hGR), which is relevant in high concentrations such as in untreated or poorly treated 21 OHD patients [30], 31]. In COS7 cells transfected with the human glucocorticoid receptor (hGR), 21DF is able to bind to the hGR leading to translocation and transcription of the hGR into the nucleus [30]. Studies in HEK293 cells showed that 21DF has about half the glucocorticoid potency compared to the classical hGR agonist cortisol [31].
The clinical impact of steroid precursors acting as agonists at the hGR is also known from patients with 17α-hydroxylase deficiency (CYP17A1) [32]. Due to the glucocorticoid activity of accumulating corticosterone, this rare type of CAH usually shows no signs of acute adrenal insufficiency or Addison’s crisis; rather, the affected patients often become noticeable relatively late in adolescence due to a lack of pubertal development or primary amenorrhoea. The in vitro hGR activity of corticosterone is approximately 64 % of that of cortisol [31]. Furthermore, some case reports and smaller cohort studies in patients with classic 21OHD have also been described who, despite lack of treatment, showed no signs of adrenal insufficiency or Addison’s crisis [33]. The most likely reason for this appears to be the high glucocorticoid potency of the precursor steroids. Furthermore, recent studies have shown the impact of free cortisol in classic CAH patients [34]. In a study published by Adriaansen et al. total cortisol levels of classic CAH patients (median 109 nmol/L) were lower compared to levels in non-classic (NC) CAH patients (249 nmol/L) and controls (202 nmol/L), but free cortisol concentrations were similar, indicating comparable availability. After ACTH stimulation, as expected, not total and free cortisol, but total and free 21DF showed a significant increase in CAH patients.
Clinical implications of elevated ACTH
Poorly controlled patients with classic 21OHD, especially those with irregular hydrocortisone intake due to non-adherence, are cortisol deficient but have high levels of ACTH and consequently elevated steroid precursors such as 21DF, leading to some hGR activation that may partly compensate for cortisol deficiency and thus may reduce the risk of Addison’s crisis. This may explain that some classic 21OHD patients survive despite low total cortisol levels [33]. The question therefore arises as to the consequences for a patient whose ACTH-mediated increase in steroid precursors with agonistic glucocorticoid activity is inhibited by a CRH receptor antagonist, while at the same time the hydrocortisone replacement dose is reduced to a lower maintenance level.
Examining another form of CAH, 11β-hydroxylase deficiency (CYP11B1), can provide valuable insights for this assessment. This form of CAH is characterised by an accumulation of steroid precursors, such as 11-deoxycorticosterone, which have mineralocorticoid activity. This leads to an excess of mineralocorticoids and results in hypertension, the opposite of salt wasting. However, when these precursors are suppressed through glucocorticoid treatment, patients may experience clinically significant salt wasting [35], [36], [37].
Young patients who are used to taking medication irregularly are unlikely to change those habits easily when a new drug is added to the treatment plan, making the regimen more complex. Therefore, when suppressing ACTH by adding the CRH receptor antagonist crinecerfont, the increased need for regular hydrocortisone intake especially during stress and illness must be emphasised and the symptoms of glucocorticoid deficiency must be discussed in detail.
The switch from supraphysiological hydrocortisone treatment to physiological replacement therapy in patients treated with crinercerfont should be made carefully and gradually, and only after the clinician has established the patient’s adherence. All measures to prevent Addison’s crisis must be retrained. It is only under these conditions that the new CRH receptor antagonist can become a safe treatment option for children and young people with CAH. To ensure the successful management of the condition at home, greater emphasis should be placed on providing comprehensive education and support to children and their parents, focusing on lifestyle modifications and coping strategies.
Conclusions
The CRH receptor antagonist crinecerfont offers a promising new therapeutic approach for the treatment of patients with classic CAH by reducing the need for high-dose glucocorticoids and better controlling androgen levels. While this represents a significant advance in the treatment of CAH, patient adherence and close monitoring are essential to prevent adrenal insufficiency and other complications. Future research should focus on refining treatment protocols and assessing long-term outcomes to ensure safe and effective use of this therapy in clinical practice.
Acknowledgments
Special thanks to S. Lanzinger and A. Eckert for providing the values for 17OHP and androstenedione from the German CAH register and R. Ranz for support and the development of the documentation software (Institute for Epidemiology and Medical Biometry (CAQM), University of Ulm, Germany). For further information about the German CAH registry, please visit: https://buster.zibmt.uni-ulm.de/projekte/AGS/. We would like to thank all participating centres: Aue Kinderklinik, Augsburg Universitätskinderklinik, Berlin Lichtenberg, Bielefeld Evangelisches Krankenhaus, Bochum Endokrinologikum Ruhr, Bonn Unikinderklinik, Dornbirn Kinderklinik, Dresden Universitäts-Kinderklinik, Erlangen Universitäts-Kinderklinik, Essen Universitätskinderklinik, Frankfurt Universitätskinderklinik, Freiburg Universitätskinderklinik, Greifswald Universitätskinderklinik, Hamburg Altona Kinderkrankenhaus, Hamburg Kinder-MVZ am Wilhelmstift, Heidelberg Prof. Heinrich, Heidelberg Universitäts-Kinderklinik, Homburg Universitäts-Kinderklinik, Innsbruck Universitäts-Kinderklinik, Karlsruhe Kinderklinik, Kiel Universitäts-Kinderklinik, Krefeld Kinderklinik, Köln Unikinderklinik, Magdeburg Universitäts-Kinderklinik, München Haunersche Ki-Klinik, Münster Universitätskinderklinik, Neuburg Donau St. Elisabeth Kinderklinik, Nürnberg Cnopfsche Kinderklinik, Oldenburg Endokrinologisches MVZ Pädiatrie, Paderborn Kinderklinik, Rotenburg Kinderklinik, Tübingen Universitäts-Kinderkliniken, Ulm Endokrinologikum, Ulm Universitäts-Kinderklinik, Wien Universitätskinderklinik.
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Conflict of interest: CK has received consultancy fees from Diurnal for review work. HCvdG was SPRUCE study investigator. CK is the Editor-in-Chief of this journal. However, the manuscript was peer-reviewed by two of the Associate Editors.
-
Research funding: None declared.
-
Data availability: Not applicable.
References
1. Claahsen-van der Grinten, HL, Speiser, PW, Ahmed, SF, Arlt, W, Auchus, RJ, Falhammar, H, et al.. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev 2022;43:91–159. https://doi.org/10.1210/endrev/bnab016.Suche in Google Scholar PubMed PubMed Central
2. Hargitai, G, Sólyom, J, Battelino, T, Lebl, J, Pribilincová, Z, Hauspie, R, et al.. Growth patterns and final height in congenital adrenal hyperplasia due to classical 21-hydroxylase deficiency. Results of a multicenter study. Horm Res 2001;55:161–71. https://doi.org/10.1159/000049990.Suche in Google Scholar PubMed
3. Kamrath, C, Wettstaedt, L, Boettcher, C, Hartmann, MF, Wudy, SA. The urinary steroidome of treated children with classic 21-hydroxylase deficiency. J Steroid Biochem Mol Biol 2017;165:396–406. https://doi.org/10.1016/j.jsbmb.2016.08.006.Suche in Google Scholar PubMed
4. Van der Kamp, HJ, Otten, BJ, Buitenweg, N, De Muinck Keizer-Schrama, SM, Oostdijk, W, Jansen, M, et al.. Longitudinal analysis of growth and puberty in 21-hydroxylase deficiency patients. Arch Dis Child 2002;87:139–44. https://doi.org/10.1136/adc.87.2.139.Suche in Google Scholar PubMed PubMed Central
5. Cordeiro, GV, Silva, IN, Goulart, EM, Chagas, AJ, Kater, CE. Final height in congenital adrenal hyperplasia: the dilemma of hypercortisolism versus hyperandrogenism. Arq Bras Endocrinol Metabol 2013;57:126–31. https://doi.org/10.1590/s0004-27302013000200005.Suche in Google Scholar PubMed
6. Eugster, EA, Dimeglio, LA, Wright, JC, Freidenberg, GR, Seshadri, R, Pescovitz, OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr 2001;138:26–32. https://doi.org/10.1067/mpd.2001.110527.Suche in Google Scholar PubMed
7. Kamrath, C, Wettstaedt, L, Hartmann, MF, Wudy, SA. Height velocity defined metabolic control in children with congenital adrenal hyperplasia using urinary steroid GC-MS analysis. J Clin Endocrinol Metab 2019;104:4214–24. https://doi.org/10.1210/jc.2019-00438.Suche in Google Scholar PubMed
8. Torky, A, Sinaii, N, Jha, S, Desai, J, El-Maouche, D, Mallappa, A, et al.. Cardiovascular disease risk factors and metabolic morbidity in a longitudinal study of congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021;106:e5247–57. https://doi.org/10.1210/clinem/dgab133.Suche in Google Scholar PubMed PubMed Central
9. Balagamage, C, Lawrence, NR, Krone, R, Bacila, IA, Krone, NP. Blood pressure in children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res Paediatr 2024;97:315–25. https://doi.org/10.1159/000533465.Suche in Google Scholar PubMed
10. Barbot, M, Mazzeo, P, Lazzara, M, Ceccato, F, Scaroni, C. Metabolic syndrome and cardiovascular morbidity in patients with congenital adrenal hyperplasia. Front Endocrinol (Lausanne) 2022;13:934675. https://doi.org/10.3389/fendo.2022.934675.Suche in Google Scholar PubMed PubMed Central
11. Sarafoglou, K, Kim, MS, Lodish, M, Felner, EI, Martinerie, L, Nokoff, NJ, et al.. Phase 3 trial of crinecerfont in pediatric congenital adrenal hyperplasia. N Engl J Med 2024;391:493–503. https://doi.org/10.1056/nejmoa2404655.Suche in Google Scholar
12. Speiser, PW, Azziz, R, Baskin, LS, Baskin, LS, Conway, GS, Merke, DP, et al.. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010;95:4133–60. https://doi.org/10.1210/jc.2009-2631.Suche in Google Scholar PubMed PubMed Central
13. Lawrence, N, Bacila, I, Dawson, J, Bryce, J, Ali, SR, van den Akker, ELT, et al.. Analysis of therapy monitoring in the international congenital adrenal hyperplasia registry. Clin Endocrinol 2022;97:551–61. https://doi.org/10.1111/cen.14796.Suche in Google Scholar PubMed PubMed Central
14. Hoyer-Kuhn, H, Huebner, A, Richter-Unruh, A, Bettendorf, M, Rohrer, T, Kapelari, K, et al.. Hydrocortisone dosing in children with classic congenital adrenal hyperplasia: results of the German/Austrian registry. Endocr Connect 2021;10:561–9. https://doi.org/10.1530/ec-21-0023.Suche in Google Scholar
15. Linder, BL, Esteban, NV, Yergey, AL, Winterer, JC, Loriaux, DL, Cassorla, F. Cortisol production rate in childhood and adolescence. J Pediatr 1990;117:892–6. https://doi.org/10.1016/s0022-3476(05)80128-3.Suche in Google Scholar PubMed
16. Hammersen, J, Bettendorf, M, Bonfig, W, Schönau, E, Warncke, K, Eckert, AJ, et al.. Twenty years of newborn screening for congenital adrenal hyperplasia and congenital primary hypothyroidism - experiences from the DGKED/AQUAPE. Med Genet 2022;34:29–40. https://doi.org/10.1515/medgen-2022-2114.Suche in Google Scholar PubMed PubMed Central
17. Kamrath, C, Hartmann, MF, Pons-Kühnemann, J, Wudy, SA. Urinary GC-MS steroid metabotyping in treated children with congenital adrenal hyperplasia. Metabolism 2020;112:154354. https://doi.org/10.1016/j.metabol.2020.154354.Suche in Google Scholar PubMed
18. Frisch, H, Parth, K, Schober, E, Swoboda, W. Circadian patterns of plasma cortisol, 17-hydroxyprogesterone, and testosterone in congenital adrenal hyperplasia. Arch Dis Child 1981;56:208–13. https://doi.org/10.1136/adc.56.3.208.Suche in Google Scholar PubMed PubMed Central
19. Charmandari, E, Matthews, DR, Johnston, A, Brook, CG, Hindmarsh, PC. Serum cortisol and 17-hydroxyprogesterone interrelation in classic 21-hydroxylase deficiency: is current replacement therapy satisfactory? J Clin Endocrinol Metab 2001;86:4679–85. https://doi.org/10.1210/jcem.86.10.7972.Suche in Google Scholar PubMed
20. Merke, DP, Mallappa, A, Arlt, W, Brac de la Perriere, A, Lindén Hirschberg, A, Juul, A, et al.. Modified-release hydrocortisone in congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021;106:e2063–77. https://doi.org/10.1210/clinem/dgab051.Suche in Google Scholar PubMed PubMed Central
21. Hindmarsh, PC, Charmandari, E. Variation in absorption and half-life of hydrocortisone influence plasma cortisol concentrations. Clin Endocrinol 2015;82:557–61. https://doi.org/10.1111/cen.12653.Suche in Google Scholar PubMed
22. Charmandari, E, Hindmarsh, PC, Johnston, A, Brook, CG. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: alterations in cortisol pharmacokinetics at puberty. J Clin Endocrinol Metab 2001;86:2701–8. https://doi.org/10.1210/jcem.86.6.7522.Suche in Google Scholar PubMed
23. Kyngäs, HA, Kroll, T, Duffy, ME. Adherence in adolescents with chronic diseases: a review. J Adolesc Health 2000;26:379–88. https://doi.org/10.1016/s1054-139x(99)00042-7.Suche in Google Scholar PubMed
24. McGrady, ME, Hommel, KA. Medication adherence and health care utilization in pediatric chronic illness: a systematic review. Pediatrics 2013;132:730–40. https://doi.org/10.1542/peds.2013-1451.Suche in Google Scholar PubMed PubMed Central
25. Hoyer-Kuhn, H, Eckert, AJ, Binder, G, Bonfig, W, Dübbers, A, Riedl, S, et al.. Impact of newborn screening on adult height in patients with congenital adrenal hyperplasia (CAH). J Clin Endocrinol Metab 2023;108:e1199–204. https://doi.org/10.1210/clinem/dgad307.Suche in Google Scholar PubMed
26. Kamrath, C, Wettstaedt, L, Boettcher, C, Hartmann, MF, Wudy, SA. Androgen excess is due to elevated 11-oxygenated androgens in treated children with congenital adrenal hyperplasia. J Steroid Biochem Mol Biol 2018;178:221–8. https://doi.org/10.1016/j.jsbmb.2017.12.016.Suche in Google Scholar PubMed
27. Turcu, AF, Nanba, AT, Chomic, R, Upadhyay, SK, Giordano, T, Shields, JJ, et al.. Adrenal-derived 11-oxygenated 19-carbon androgens are the dominant androgens 1 in classic 21-hydroxylase deficiency. Eur J Endocrinol 2016;174:601–9. https://doi.org/10.1530/eje-15-1181.Suche in Google Scholar PubMed PubMed Central
28. Swart, AC, Storbeck, KH. 11β-Hydroxyandrostenedione: downstream metabolism by 11βHSD, 17βHSD and SRD5A produces novel substrates in familiar pathways. Mol Cell Endocrinol 2015;408:114–23. https://doi.org/10.1016/j.mce.2014.12.009.Suche in Google Scholar PubMed
29. Storbeck, KH, Bloem, LM, Africander, D, Schloms, L, Swart, P, Swart, AC. 11β-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: a putative role in castration resistant prostate cancer? Mol Cell Endocrinol 2013;377:135–46. https://doi.org/10.1016/j.mce.2013.07.006.Suche in Google Scholar PubMed
30. Pijnenburg-Kleizen, KJ, Engels, M, Mooij, CF, Griffin, A, Krone, N, Span, PN, et al.. Adrenal steroid metabolites accumulating in congenital adrenal hyperplasia lead to transactivation of the glucocorticoid receptor. Endocrinology 2015;156:3504–10. https://doi.org/10.1210/en.2015-1087.Suche in Google Scholar PubMed
31. Engels, M, Pijnenburg-Kleizen, KJ, Utari, A, Faradz, SMH, Oude-Alink, S, van Herwaarden, AE, et al.. Glucocorticoid activity of adrenal steroid precursors in untreated patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2019;104:5065–72. https://doi.org/10.1210/jc.2019-00547.Suche in Google Scholar PubMed
32. Miller, WL, Auchus, RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders [published correction appears in Endocr Rev 2011;32:579]. Endocr Rev 2011;32:81–151. https://doi.org/10.1210/er.2010-0013.Suche in Google Scholar PubMed PubMed Central
33. Adriaansen, BPH, Utari, A, Westra, D, Ariani, MD, Ediati, A, Schröder, MAM, et al.. 46,XX males with congenital adrenal hyperplasia: a clinical and biochemical description. Front Endocrinol (Lausanne) 2024;15:1410122. https://doi.org/10.3389/fendo.2024.1410122.Suche in Google Scholar PubMed PubMed Central
34. Adriaansen, BPH, Utari, A, Olthaar, AJ, van der Steen, RCBM, Pijnenburg-Kleizen, KJ, Berkenbosch, L, et al.. Free cortisol and free 21-deoxycortisol in the clinical evaluation of congenital adrenal hyperplasia. J Clin Endocrinol Metab 2024;24:dgae591. https://doi.org/10.1210/clinem/dgae591.Suche in Google Scholar PubMed
35. Hochberg, Z, Benderly, A, Zadik, Z. Salt loss in congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Arch Dis Child 1984;59:1092–4. https://doi.org/10.1136/adc.59.11.1092.Suche in Google Scholar PubMed PubMed Central
36. Holcombe, JH, Keenan, BS, Nichols, BL, Kirkland, RT, Clayton, GW. Neonatal salt loss in the hypertensive form of congenital adrenal hyperplasia. Pediatrics 1980;65:777–81. https://doi.org/10.1542/peds.65.4.777.Suche in Google Scholar
37. Zadik, Z, Kahana, L, Kaufman, H, Benderli, A, Hochberg, Z. Salt loss in hypertensive form of congenital adrenal hyperplasia (11-beta-hydroxylase deficiency). J Clin Endocrinol Metab 1984;58:384–7. https://doi.org/10.1210/jcem-58-2-384.Suche in Google Scholar PubMed
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Review
- Osteogenesis imperfecta: shifting paradigms in pathophysiology and care in children
- Opinion Paper
- CRH receptor antagonist crinecerfont – a promising new treatment option for patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
- Original Articles
- Age and sex mark clinical differences in the presentation of pediatric type 1 diabetes mellitus
- Geographic information system mapping and predictors of glycemic control in children and youth with type 1 diabetes: a study from Western India
- Body composition assessment measured via bioelectrical impedance analysis in euthyroid children with newly diagnosed Hashimoto’s thyroiditis
- Outcomes of newborns screened for congenital hypothyroidism in Turkey – a single center experience
- High yield of congenital hypothyroidism among infants attending Children Hospital, Nairobi, Kenya. Facility based study in the absence of newborn screening
- Immune checkpoint inhibitors and endocrinopathies in pediatric brain tumor patients
- Assessment of quality of life in families affected by maple syrup urine disease: a cross sectional study
- Case Reports
- Reninoma: an unusual cause of growth failure
- Persistent hypoglycemia in congenital syphilis: hyperinsulinemic hypoglycemia with a focal pancreatic lesion
- Diagnostic challenges in pediatric Cushing’s disease associated with chronic renal failure: a report of three patients
- A novel de novo missense OTC mutation in an Iranian girl: a case report
Artikel in diesem Heft
- Frontmatter
- Review
- Osteogenesis imperfecta: shifting paradigms in pathophysiology and care in children
- Opinion Paper
- CRH receptor antagonist crinecerfont – a promising new treatment option for patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
- Original Articles
- Age and sex mark clinical differences in the presentation of pediatric type 1 diabetes mellitus
- Geographic information system mapping and predictors of glycemic control in children and youth with type 1 diabetes: a study from Western India
- Body composition assessment measured via bioelectrical impedance analysis in euthyroid children with newly diagnosed Hashimoto’s thyroiditis
- Outcomes of newborns screened for congenital hypothyroidism in Turkey – a single center experience
- High yield of congenital hypothyroidism among infants attending Children Hospital, Nairobi, Kenya. Facility based study in the absence of newborn screening
- Immune checkpoint inhibitors and endocrinopathies in pediatric brain tumor patients
- Assessment of quality of life in families affected by maple syrup urine disease: a cross sectional study
- Case Reports
- Reninoma: an unusual cause of growth failure
- Persistent hypoglycemia in congenital syphilis: hyperinsulinemic hypoglycemia with a focal pancreatic lesion
- Diagnostic challenges in pediatric Cushing’s disease associated with chronic renal failure: a report of three patients
- A novel de novo missense OTC mutation in an Iranian girl: a case report