Noted tension headache, anxiety, and depression in a Chinese patient with spinocerebellar ataxia, autosomal recessive 10 caused by a novel anoctamin 10 mutation
-
Xiaoxi Ma


To the editor
Spinocerebellar ataxia, autosomal recessive 10 (SCAR10) is a rare neurogenetic disease due to anoctamin 10 (ANO10) mutations.[1] Herein, we report a Chinese SCAR10 patient with a novel nonsense mutation site in ANO10 gene and highlight several infrequent clinical manifestations.
The proband TYP (IV:2; Figure 1A) was a Chinese woman with a primary school diploma, who was born in a consanguineous family (Figure 1A). At age 33, TYP started to present with gradually aggravating slurred speech and vision, limb weakness, dizziness, and tension headache. Within half a year, she went through progressive gait instability and visible tremor when her hand approached the target, and her handwriting got bigger when writing. She experienced tonic–clonic seizures twice. She also complained of recent memory decline, depression and anxiety since age 34. Her headache worsened after treatment with buspirone.
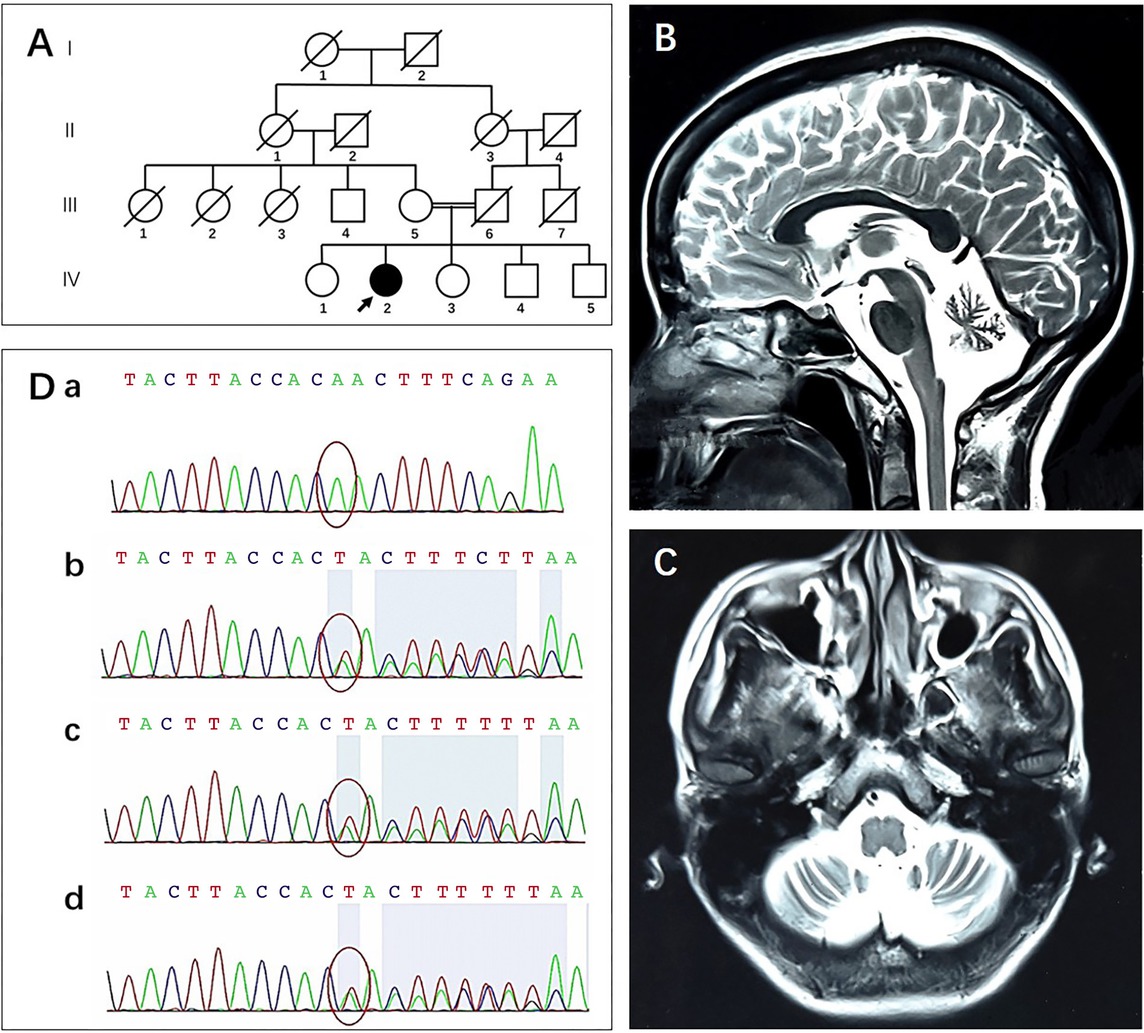
Pedigree, brain MRI, Sanger sequencing. (A) The proband (IV:2, arrow) was born to consanguineous parents (III:5 and III:6). (B and C) Brain MRI shows marked cerebellar atrophy. (D) Red circles indicate the site of mutation. Sanger sequencing confirmed that the proband (IV:2, a) was homozygous for the c.1213_1215delTTA (p. L405del) mutation of ANO10 and her mother (III:5, b), 28-year-old brother (IV:4, c), and 24-year-old brother (IV:5, d) were heterozygous carriers. MRI: magnetic resonance imaging.
At age 35, TYP was seen in the Memory Clinic of Huashan Hospital. A physical examination showed dysarthria, clumsy hands, intention tremor, dysmetria, finger– nose test (+), Romberg’s test (+), drunken gait, binocular horizontal nystagmus, diplopia, bradykinesia, diminished myodynamia (5-), and hyperreflexia. Fasciculation, ankle clonus, pathologic reflex, bowel or bladder dysfunction, pes cavus, and tortuosity of the conjunctival vessels were absent.
She scored 24/30 in Mini-Mental State Examination (MMSE) and 3.7 in Informant Questionnaire on Cognitive Decline in The Elderly, indicating her mild cognitive impairment. Due to poor compliance of the patient, anxiety and depression rating scales were not performed. Electroencephalogram was normal. Magnetic resonance imaging of the brain indicated the presence of cerebellar atrophy (Figure 1B, C).
Sanger sequencing found that she had a homozygous mutation of ANO10, a nonsense mutation of c.1213_1215delTTA (p. L405del) (NM_018075). Her mother (III:5; Figure 1A) and her two brothers (IV:4, 5; Figure 1A) were heterozygous carriers (Figure 1D).
Her symptoms did not progress at the time of her visit at the age of 36. Physical examinations showed binocular horizontal nystagmus, finger–nose test (+, on the left more than on the right), Romberg’s test (+/–), and difficulty walking in a straight line. She scored 24/30 in MMSE and 19/30 in the Montreal Cognitive Assessment-Basic version.
ANO10 spans 2734 bp and consists of 13 exons, 12 of which code 660 amino acid residues. Anoctamin 10, also known as transmembrane protein 16K (TMEM16K), is encoded by ANO10. TMEM16K is a member of TMEM16 protein family, in which all members are composed of eight transmembrane domains. TMEM16 proteins are involved in the process of ion transport, phospholipid scrambling, and regulation of other membrane proteins. Research showed that TMEM16K played a role in the process of inositol 1,4,5-triphate receptor 1(IP3R1)-mediated intracellular Ca2+ release and regulated Ca2+ signaling in Purkinje cells.[2] TMRM16K-knockout mice showed neuromuscular and motor impairment, in line with ataxic phenotypes in SCAR10 patients.[3] c.1213_1215delTTA is a novel deletion variant of ANO10, which could lead to the amino acid change of p. L405del. This mutation might contribute to dysregulation of calcium signaling within cerebellar Purkinje cells, which is the most characteristic pathologic mechanism underlying SCAs.[4] Further functional research is needed to reveal the precise mechanism of ANO10 mutation.
SCAs are a large complex spectrum of inherited neurodegenerative disorders characterized by progressive cerebellar ataxia, extrapyramidal signs, peripheral neuropathy, sphincter disturbances, optic atrophy, ophthalmoplegia, cognitive impairment, and epilepsy. SCAs could be divided into autosomal dominant (SCAD) and autosomal recessive (SCAR) forms according to their inheritance mode. The clinical presentation of both SCAD and SCAR is heterogeneous in symptoms, signs, age at onset, and disease worsening, depending on the specific mutation and subtype. According to the specific pathogenesis, SCAR could be divided into different subtypes, including Friedreich’s ataxia, ataxia telangiectasia, and so on. About 40 subtypes of SCAD are further classified into three groups based on their clinical features.[5,6] Unlike its autosomal dominant counterparts, which typically originate from CAG trinucleotide repeat expansions in the respective genes, SCAR10 is caused by single-nucleotide changes in the surrounding exons of ANO10.[7] Overall, genetic screening is recommended for making a definite diagnosis of SCAR10.
SCAR10 is more common in Caucasians than Asians. Based on known cases, the age at onset of SCAR10 patients in China, including this case, ranges from 31 to 39 years. Our patient showed uncommon phenotypes like mild cognitive decline, tonic–clonic seizures, tension headache, depression, and anxiety. Among the other Chinese patients, while most had cognitive impairments, none of them had seizures.[8, 9, 10] What is worth noting is that our patient experienced tension headache at the outset, which aggravated after treatment with buspirone. Minnerop et al.[11] reported a patient with a homozygous c.132dupA mutation, who initially had tension headache and gradually progressed to ataxia. Her tension headache improved after medication with amitriptyline. Tension headache should be considered when diagnosing SCAR10.
In conclusion, we reported here a novel ANO10 mutation in a Chinese SCAR10 patient and highlighted several clinical manifestations that improve our understanding of SCAR10 genetically and clinically.
-
Source of Funding
This work was supported by grants of the Clinical Research Plan of SHDC (grant no. SHDC2020CR4007) and the National Natural Science Foundation of China (82071200).
-
Ethics Approval and Consent to Participate
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Medical Ethics Committee of Huashan Hospital, Fudan University, Shanghai, China (Approval No. 2011-288). Written informed consent was obtained from the proband and her family.
-
Conflict of Interest
None declared.
References
1 Vermeer S, Hoischen A, Meijer RP, Gilissen C, Neveling K, Wieskamp N, et al. Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am J Hum Genet 2010;87:813–9.10.1016/j.ajhg.2010.10.015Search in Google Scholar PubMed PubMed Central
2 Benarroch EE. Anoctamins (TMEM16 proteins): Functions and involvement in neurologic disease. Neurology 2017;89:722–9.10.1212/WNL.0000000000004246Search in Google Scholar PubMed
3 Petkovic M, Oses-Prieto J, Burlingame A, Jan LY, Jan YN. TMEM16K is an interorganelle regulator of endosomal sorting. Nat Commun 2020;11:3298.10.1038/s41467-020-17016-8Search in Google Scholar PubMed PubMed Central
4 Kasumu A, Bezprozvanny I. Deranged Calcium Signaling in Purkinje Cells and Pathogenesis in Spinocerebellar Ataxia 2 (SCA2) and Other Ataxias. Cerebellum 2010;11:630–9.10.1007/s12311-010-0182-9Search in Google Scholar PubMed PubMed Central
5 Soong, Morrison Patrick. Spinocerebellar ataxias. Handb Clin Neurol 2018;155:143–74.10.1016/B978-0-444-64189-2.00010-XSearch in Google Scholar PubMed
6 Sailer A, Houlden H. Recent Advances in the Genetics of Cerebellar Ataxias. Curr Neurol Neurosci Rep 2012;12:227–36.10.1007/s11910-012-0267-6Search in Google Scholar PubMed
7 Maruyama H, Morino H, Miyamoto R, Murakami N, Hamano T, Kawakami H. Exome sequencing reveals a novel ANO10 mutation in a Japanese patient with autosomal recessive spinocerebellar ataxia. Clin Genet 2014;85:296–7.10.1111/cge.12140Search in Google Scholar PubMed
8 Yang SL, Chen SF, Jiao YQ, Dong ZY, Dong Q, Han X. Autosomal Recessive Spinocerebellar Ataxia Caused by a Novel Homozygous ANO10 Mutation in a Consanguineous Chinese Family. J Clin Neurol 2020;16:333–5.10.3988/jcn.2020.16.2.333Search in Google Scholar PubMed PubMed Central
9 Zhang L, Lyu P, Zhang X, Dan T. Clinical and genetic phenotypes of an autosomal recessive spinocerebellar ataxia-10 patient due to ANO10 gene mutations. Chin J Neurol 2021;54:1256–60.Search in Google Scholar
10 Zhang W, Yuan P, Li X, Yao Y, Wang X, Wang Z, et al. [A case report of ANO10 gene in a family with autosomal recessive cerebellar ataxia]. Bei Jing Yi Xue 2021;43:282–4.(In Chinese)Search in Google Scholar
11 Minnerop M, Bauer P. Autosomal recessive cerebellar ataxia 3 due to homozygote c.132dupA mutation within the ANO10 gene. JAMA Neurol 2015;72:238–9.10.1001/jamaneurol.2014.3918Search in Google Scholar PubMed
© 2022 Xiaoxi Ma, Keliang Chen, Zhenxu Xiao, Qianhua Zhao, published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Articles in the same Issue
- Editorial
- Interpretation of the key issues of expert consensus on immunomodulatory therapies for chronic obstructive pulmonary disease
- Perspective
- Emerging noninvasive neuromodulation methods for functional gastrointestinal diseases
- Gastric electrical stimulation: Overview and summary
- Antibiotic stewardship: Dead bugs do not mutate
- The long and the short of current nanomedicines for treating Alzheimer's disease
- Monkeypox: Clinical issues of concern
- Cortical synaptic mechanism for chronic pain and anxiety in Parkinson’s disease
- Commentary
- Chemical therapy for chronic pancreatitis: An assumption or an alternative?
- Review Article
- Focal liver lesions other than hepatocellular carcinoma in cirrhosis: Diagnostic challenges
- The emergence of travel-related infections in critical care units
- Research progress on N6-adenosylate methylation RNA modification in heart failure remodeling
- Original Article
- Characteristics of the severe acute respiratory syndrome coronavirus 2 omicron BA.2 subvariant in Jilin, China from March to May 2022
- A comprehensive weighted gene co-expression network analysis uncovers potential targets in diabetic kidney disease
- Letter to Editor
- Acute kidney injury associated with severe hypouricemia caused by a novel SLC2A9 mutation: Enlightenment from rare disease to common disease
- Noted tension headache, anxiety, and depression in a Chinese patient with spinocerebellar ataxia, autosomal recessive 10 caused by a novel anoctamin 10 mutation
Articles in the same Issue
- Editorial
- Interpretation of the key issues of expert consensus on immunomodulatory therapies for chronic obstructive pulmonary disease
- Perspective
- Emerging noninvasive neuromodulation methods for functional gastrointestinal diseases
- Gastric electrical stimulation: Overview and summary
- Antibiotic stewardship: Dead bugs do not mutate
- The long and the short of current nanomedicines for treating Alzheimer's disease
- Monkeypox: Clinical issues of concern
- Cortical synaptic mechanism for chronic pain and anxiety in Parkinson’s disease
- Commentary
- Chemical therapy for chronic pancreatitis: An assumption or an alternative?
- Review Article
- Focal liver lesions other than hepatocellular carcinoma in cirrhosis: Diagnostic challenges
- The emergence of travel-related infections in critical care units
- Research progress on N6-adenosylate methylation RNA modification in heart failure remodeling
- Original Article
- Characteristics of the severe acute respiratory syndrome coronavirus 2 omicron BA.2 subvariant in Jilin, China from March to May 2022
- A comprehensive weighted gene co-expression network analysis uncovers potential targets in diabetic kidney disease
- Letter to Editor
- Acute kidney injury associated with severe hypouricemia caused by a novel SLC2A9 mutation: Enlightenment from rare disease to common disease
- Noted tension headache, anxiety, and depression in a Chinese patient with spinocerebellar ataxia, autosomal recessive 10 caused by a novel anoctamin 10 mutation