Acute kidney injury associated with severe hypouricemia caused by a novel SLC2A9 mutation: Enlightenment from rare disease to common disease
-
Sanxi Ai


To the editor
Hypouricemia is defined as serum uric acid (UA) <2.0 mg/dL. It is an uncommon condition with a prevalence ranging from 0.19% to 0.58%.[1] The mechanisms of hypouricemia include decreased production of UA and increased UA excretion via the kidneys. Hereditary renal hypouricemia (RHUC) is a rare autosomal hereditary disorder caused by recessive mutations in genes encoding UA transporters in the proximal renal tubule. Patients with RHUC are often asymptomatic, but some patients can develop exercise-induced acute renal failure (EIAKI). We present a young Chinese man who presented with EIAKI associated with RHUC and report a novel mutation.
A 28-year-old Chinese man was admitted to our hospital with muscular soreness, weakness, and decreased urine output for 2 days, which were experienced after intense exercise. Four months before admission, he was diagnosed with hypertension, obesity, and proteinuria (0.47 g/d), and he had been on valsartan with well-controlled blood pressure and normal serum creatinine for the past 3 months. On physical examination, his body mass index was 28 kg/m2 and blood pressure was 150/90 mmHg without other abnormal signs. The laboratory tests showed serum creatinine at 2.0 mg/dL, serum UA at 0.35 mg/dL, and creatine kinase at 117 U/L. Urinalysis showed no hematuria, epithelial cells, or casts. Urine protein excretion was 0.26 g/d and urine protein electrophoresis suggested glomerular proteinuria. Diabetes mellitus, infection, systemic autoimmune diseases, malignancy, and postrenal obstruction were excluded by extensive studies. A family history investigation revealed that the patient’s parents had a consanguineous marriage.
Hydration and alkalization with normal saline were initiated, and valsartan was discontinued. The patient’s kidney function completely recovered after 2 weeks, with a serum creatinine level of 1.1 mg/dL. Repeated laboratory tests before discharge showed a very low serum UA level (0.17 mg/dL). Renal hypouricemia was suspected and subsequent examinations revealed high (143.5%) renal fractional excretion of UA (FEUA) and impaired responses to benzbromarone and pyrazinamide inhibition tests. The patient’s parents and two siblings had normal blood pressure and normal serum creatinine measurements. The patient’s sister had a low UA level (1.8 mg/dL) but no symptoms. The UA levels of his parents and brother were normal.
Genomic DNA was isolated from the peripheral blood of the patient. All of the exons and intron flanking regions of the SLC22A12 gene (NM_144585.2) and of both SLC2A9 isoforms (GLUT9L, NM_020041; GLUT9S, NM_001001290) were analyzed via direct sequencing.
In SLC2A9, a novel homozygous nonsense mutation (NM_020041: c.944G > A [p. W315X]/NM_001001290: c.857G > A [p. W286X]) was identified (Figure 1), affecting both SLC2A9 isoforms. According to the ACMG classification criterion, the mutation was pathogenic. The genetic data of his family members were not available.
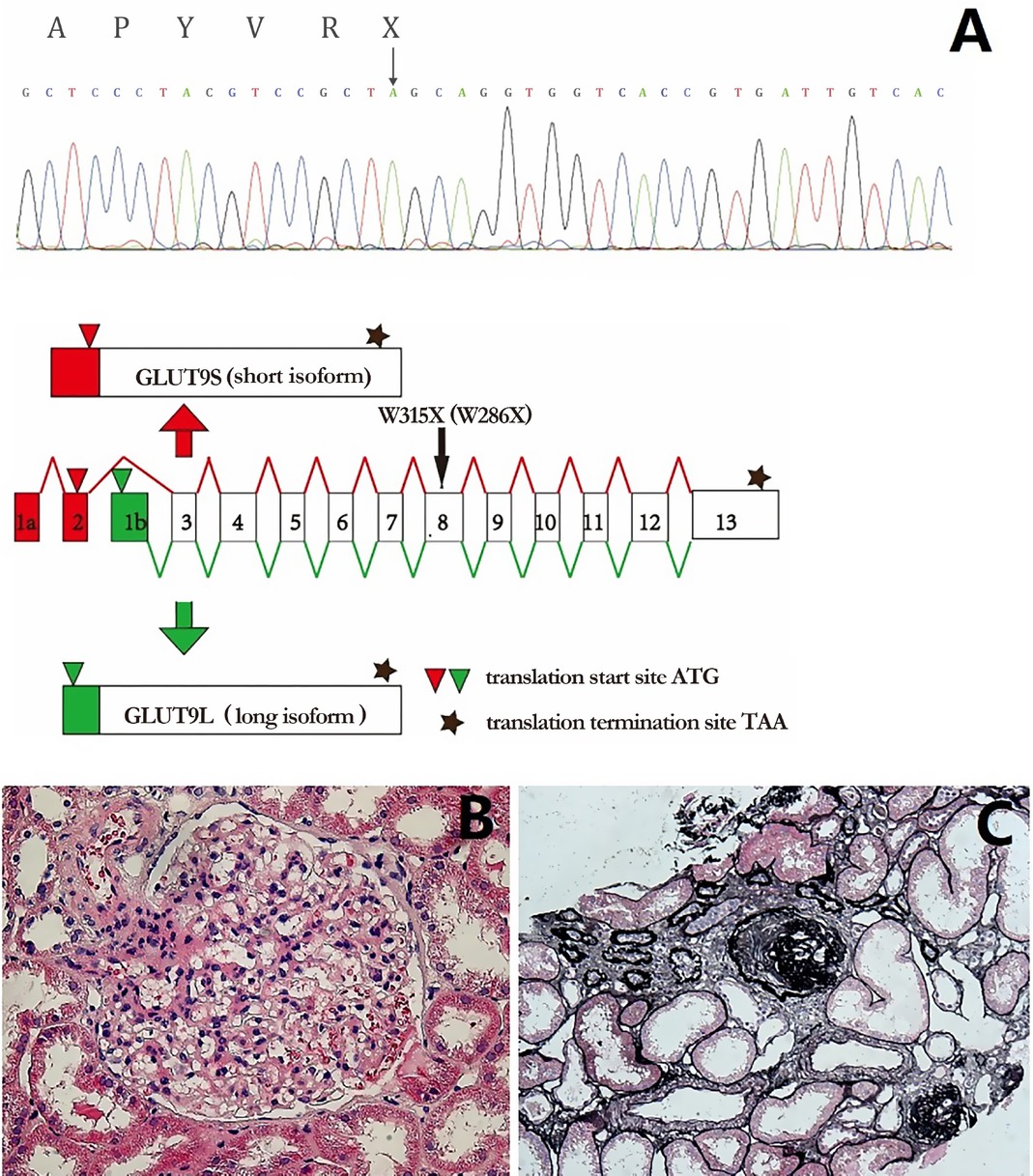
Molecular characterization and renal histology of the patient. A. Homozygous nonsense mutation c.944G > A (NM_020041, GLUT9L)/c.857G > A (NM_001001290, GLUT9S), illustrated by black arrow, led to truncated protein p. W315X (NP_064425.2, GLUT9L)/p. W286X (NP_001001290.1, GLUT9S). B. Marked glomerular hypertrophy with a mean diameter of 240 μm. C. Global sclerosis (9.7%) with focal tubular atrophy and interstitial fibrosis (PASM, ×100).
Ten months after the first admission, he was rehospitalized because of increased urine protein excretion (0.7–1.7 g/d) with no hematuria. His serum creatinine and blood pressure were normal, and his UA was still low (0.17 mg/ dL). A renal biopsy was performed and light microscopy revealed glomerular hypertrophy. Globally, sclerotic glomeruli (9.7%) were observed with focal tubular atrophy and interstitial fibrosis (Figure 1).
RHUC is clinically characterized by hypouricemia and high FEUA (>10%). According to the mutated genes, RHUC is classified into two types: (1) RHUC1 caused by mutations in the SLC22A12 gene and (2) RHUC2 caused by mutations in the SLC2A9 gene. The SLC22A12 gene encodes UA transporter 1, which is specifically expressed on the apical membrane of the proximal tubules. Dozens of mutations in SLC22A12 have been described.[2] The human SLC2A9 gene encodes two isoforms of facilitative glucose transporter 9 (GLUT9), GLUT9L (540 bp) and GLUT9S (512 bp), through the use of alternative promoters (Figure 1A), which are expressed on the basolateral and apical membrane, respectively. Before this report, only a total of 18 mutations in SLC2A9 have been identified.[3,4]
Patients with homozygous or compound heterozygous loss-of-function mutations in the SLC2A9 gene had more severe manifestations than those with heterozygous mutations, presenting with markedly low serum UA levels and notably high FEUA (>150% most times).[5] We identified a novel homozygous nonsense mutation p. W315X (GLUT9L)/p. W286X (GLUT9S) in the SLC2A9 gene, thus confirming the diagnosis of RHUC2. Our patient’s extremely low serum UA level and markedly high FEUA were consistent with previously reported cases. The mutation results in a truncated protein, and we assume that it leads to the loss of function of GLUT9.
Patients with RHUC are often asymptomatic, but some patients can develop EIAKI and nephrolithiasis. Our patient experienced an AKI onset after exercise, which was considered as EIAKI associated with renal hypouricemia. The exact mechanism of EIAKI associated with RHUC remains unclear. Two hypotheses have been previously proposed. The first hypothesis is acute UA nephropathy, which is caused by an elevation in UA production during severe exercise, thus leading to increased urinary UA excretion and renal UA precipitation.[6] However, in many patients with EIAKI, renal biopsies did not show tubular UA crystallization.[7] The second hypothesis is ischemic kidney injury due to renal vasoconstriction which is mediated by oxygen free radicals accumulation during exercise. UA is a powerful antioxidant in humans, and thus, it is speculated that exercise-induced oxygen free radicals accumulation and ischemic renal injury are potentiated by a shortage of UA in patients with RHUC. This hypothesis is supported by biopsy findings of acute tubular necrosis in many patients with EIAKI.[7] However, EIAKI has rarely been described in patients with hypouricemia due to xanthinuria (hypouricemia due to decreased production of UA), suggesting that the antioxidant activity of UA may not be the main factor for EIAKI in patients with RHUC. The present case refused a renal biopsy during the EIAKI episode; therefore, we were unable to identify the pathological features during EIAKI. Obesity-related glomerulopathy was considered the main cause of his isolated nonnephrotic proteinuria.
In summary, this report described a young man with EIAKI associated with RHUC, and we identified a novel homozygous nonsense mutation (p. W315X [GLUT9L]/p. W286X [GLUT9S]) in the SLC2A9 gene. Our report may provide new insights into the clinical and molecular findings of RHUC and provide therapeutic targets for hyperuricemia and gout.
Ethics Approval and Consent to Participate
This study was approved by the Institutional Review Board of Peking Union Medical College Hospital (S-K1511). The patient provided consent to participate in the study.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and for any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Availability of Data and Material
The datasets that were used or analyzed during the current study are available from the corresponding author upon reasonable request.
-
Conflict of Interest
The authors declare that they have no competing interests.
-
Source of Funding
This study was funded by the Chinese Academy of Medical Science (CAMS) Initiative for Innovative Medicine (2017-I2M-2-001) for manuscript writing and publication.
-
Authors’ Contributions
Xu L performed data analysis and manuscript preparation. Ai S performed data analysis and wrote the manuscript. Ai S and Xu L contributed equally to this work. Zheng K contributed to the modification of the manuscript and the analysis of the data.
Acknowledgment
The authors acknowledged Hua Zheng and Yulin Mai for their contribution in data collection, Wenling Ye for her help in pathological studies, and Limeng Chen for her help in data analysis.
References
1 Kuwabara M, Niwa K, Ohtahara A, Hamada T, Miyazaki S, Mizuta E, et al. Prevalence and complications of hypouricemia in a general population: a large-scale cross-sectional study in Japan. PLoS One 2017;12:e0176055.10.1371/journal.pone.0176055Search in Google Scholar PubMed PubMed Central
2 Zhou Z, Ma L, Zhou J, Song Z, Zhang J, Wang K, et al. Renal hypouricemia caused by novel compound heterozygous mutations in the SLC22A12 gene: a case report with literature review. BMC Med Genet 2018;19:142.10.1186/s12881-018-0595-8Search in Google Scholar PubMed PubMed Central
3 Wang C, Wang J, Liu S, Liang X, Song Y, Feng L, et al. Idiopathic renal hypouricemia: A case report and literature review. Mol Med Rep 2019;20:5118-24.10.3892/mmr.2019.10726Search in Google Scholar PubMed PubMed Central
4 Shen H, Feng C, Jin X, Mao J, Fu H, Gu W, et al. Recurrent exercise-induced acute kidney injury by idiopathic renal hypouricemia with a novel mutation in the SLC2A9 gene and literature review. BMC Pediatr 2014;14:73.10.1186/1471-2431-14-73Search in Google Scholar PubMed PubMed Central
5 Dinour D, Gray NK, Campbell S, Shu X, Sawyer L, Richardson W, et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J Am Soc Nephrol 2010;21:64-72.10.1681/ASN.2009040406Search in Google Scholar PubMed PubMed Central
6 Sperling O. Hereditary renal hypouricemia. Mol Genet Metab 2006;89:14-8.10.1016/j.ymgme.2006.03.015Search in Google Scholar PubMed
7 Ohta T, Sakano T, Igarashi T, Itami N, Ogawa T, ARF Associated with Renal Hypouricemia Research Group. Exercise-induced acute renal failure associated with renal hypouricemia: Results of a questionnaire-based survey in Japan. Nephrol Dial Transplant 2004;19:1447–53.10.1093/ndt/gfh094Search in Google Scholar PubMed
© 2022 Sanxi Ai, Lubin Xu, Ke Zheng published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Articles in the same Issue
- Editorial
- Interpretation of the key issues of expert consensus on immunomodulatory therapies for chronic obstructive pulmonary disease
- Perspective
- Emerging noninvasive neuromodulation methods for functional gastrointestinal diseases
- Gastric electrical stimulation: Overview and summary
- Antibiotic stewardship: Dead bugs do not mutate
- The long and the short of current nanomedicines for treating Alzheimer's disease
- Monkeypox: Clinical issues of concern
- Cortical synaptic mechanism for chronic pain and anxiety in Parkinson’s disease
- Commentary
- Chemical therapy for chronic pancreatitis: An assumption or an alternative?
- Review Article
- Focal liver lesions other than hepatocellular carcinoma in cirrhosis: Diagnostic challenges
- The emergence of travel-related infections in critical care units
- Research progress on N6-adenosylate methylation RNA modification in heart failure remodeling
- Original Article
- Characteristics of the severe acute respiratory syndrome coronavirus 2 omicron BA.2 subvariant in Jilin, China from March to May 2022
- A comprehensive weighted gene co-expression network analysis uncovers potential targets in diabetic kidney disease
- Letter to Editor
- Acute kidney injury associated with severe hypouricemia caused by a novel SLC2A9 mutation: Enlightenment from rare disease to common disease
- Noted tension headache, anxiety, and depression in a Chinese patient with spinocerebellar ataxia, autosomal recessive 10 caused by a novel anoctamin 10 mutation
Articles in the same Issue
- Editorial
- Interpretation of the key issues of expert consensus on immunomodulatory therapies for chronic obstructive pulmonary disease
- Perspective
- Emerging noninvasive neuromodulation methods for functional gastrointestinal diseases
- Gastric electrical stimulation: Overview and summary
- Antibiotic stewardship: Dead bugs do not mutate
- The long and the short of current nanomedicines for treating Alzheimer's disease
- Monkeypox: Clinical issues of concern
- Cortical synaptic mechanism for chronic pain and anxiety in Parkinson’s disease
- Commentary
- Chemical therapy for chronic pancreatitis: An assumption or an alternative?
- Review Article
- Focal liver lesions other than hepatocellular carcinoma in cirrhosis: Diagnostic challenges
- The emergence of travel-related infections in critical care units
- Research progress on N6-adenosylate methylation RNA modification in heart failure remodeling
- Original Article
- Characteristics of the severe acute respiratory syndrome coronavirus 2 omicron BA.2 subvariant in Jilin, China from March to May 2022
- A comprehensive weighted gene co-expression network analysis uncovers potential targets in diabetic kidney disease
- Letter to Editor
- Acute kidney injury associated with severe hypouricemia caused by a novel SLC2A9 mutation: Enlightenment from rare disease to common disease
- Noted tension headache, anxiety, and depression in a Chinese patient with spinocerebellar ataxia, autosomal recessive 10 caused by a novel anoctamin 10 mutation