The compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 representing the first ammine/ammonium borate
-
Ingo Widmann
 ,
Birgit Fuchs
,
Birgit Fuchs


Abstract
Orthorhombic crystals of the composition (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 have been synthesized from boron oxide, boron nitride and boric acid in a Walker-type multianvil press under high-pressure/high-temperature conditions of 12.0 GPa and 1,300 °C. The compound crystallizes in the space group Pmn21 (no. 31) with two formula units per unit cell (Z = 2). Its lattice parameters have been determined to be a = 10.7906(4), b = 4.59980(2), and c = 4.2317(2) Å. The structure was elucidated by single-crystal X-ray diffraction, revealing a network of corner-sharing tetrahedra, which form cages containing disordered [NH4] and [BNH3] units. The compound was characterized using powder X-ray diffraction along with vibrational spectroscopy.
1 Introduction
In the last decades, our research group focused its interest on high-pressure chemistry of borates and investigated several borates with the sum formula MB4–x O7 (x = 0 for M = Hg, Ca, Zn, and x = 0.4 for M = La, Pr, Nd). 1 , 2 , 3 , 4 , 5 In a synthesis experiment intended to synthesize an unknown chromium borate, we discovered a borate with the composition (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36. The new structure is homeotypic to compounds crystallizing with the α-SrB4O7 6 , 7 structure type, which include the ambient-pressure phases PbB4O7 7 , 8 , 9 and EuB4O7 10 and the high-pressure phases β-HgB4O7, 1 β-SnB4O7, 11 β-CaB4O7, 2 and LnB3.6O7 (Ln = La, Pr, Nd). 4 , 5 While all the compounds in this structure type, to the best of our knowledge, have a di- or trivalent metal cation, the homeotypic title compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 features [NH4] and [BNH3] units for charge compensation, the latter of which are rare in the literature. While there are several ammonium borates known so far, e. g. HP-(NH4)B3O5, 12 (NH4)B11O16(OH)2, 13 and NH4[B5O6(OH)4]·2H2O, 14 the first and to our knowledge only compound with the structure motif of a [BNH3] unit with boron covalently connected to nitrogen is Cd(NH3)2[B3O5(NH3)]2, which was discovered by Sohr et al. in 2015. 15 In detail, the before-mentioned boron atom is tetrahedrally coordinated by three O and one N atoms forming a BO3(NH3) tetrahedron. Later on, the structural motif of a boron-centered tetrahedron with three oxygen and one nitrogen atom ([BO3N] tetrahedron) could also be observed in the high-pressure oxonitridoborates CrB4O6N 16 and AlB4O6N. 17 In these compounds, the nitrogen atoms are connecting four tetrahedra forming a [N(BO3)4] building unit, with the central nitrogen atom not connected to terminal hydrogen atoms. 16 , 17 In particular, one of these phases, AlB4O6N:Cr3+ showed extraordinary luminescence properties, low thermal expansion, high thermal stability, and hardness suitable for multifunctional applications. 17 , 18
(NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 was synthesized in a high-pressure/high-temperature approach at temperature conditions (1,300 °C/12.0 GPa), at which adducts of ammonia are in general not stable under ambient pressure. For example, (NH4)B3O5 12 disintegrates in a high-temperature-XRD measurement at about 200 °C under normal pressure conditions. 17 This underlines the potential of multianvil experiments to obtain innovative materials, which are not accessible using ambient pressure conditions. Furthermore, to the best of our knowledge this synthesis approach is the first in which a borate structure with an [NH4] unit is obtained from the starting materials h-BN, B2O3 and H3BO3. In the literature, more obvious educts like (NH4)HCO3 or ammonia are reported. 12 , 13 However, a reaction of h-BN with B2O3 and/or H3BO3 is known for the synthesis of other structures, e. g. compounds with the sum formula MB4O6N (with M = Cr, Al). 16 , 17 , 18
In the following, we discuss the high-pressure/high-temperature synthesis, the crystal structure determination, and the vibrational spectroscopic results of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36.
2 Experimental section
2.1 Synthesis
For the synthesis of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36, the starting materials B2O3 (99.9 %, Strem Chemicals, Newburyport, Massachusetts, U.S.A.), h-BN (≥99 %, Strem Chemicals, Newburyport, Massachusetts, U.S.A.), and H3BO3 (≥99.8 %, Carl Roth GmbH & Co. KG, Karlsruhe, Germany) were mixed in a stoichiometric ratio of 1:1:1.36, thoroughly ground in an agate mortar and encapsulated in platinum foil (0.027 mm, 99.9 %, Chem-PUR, Karlsruhe, Germany). The capsule was placed in a crucible composed of h-BN (HeBoSint, P100, Henze Boron Nitride Products AG, Kempten, Germany), closed with a h-BN lid and arranged in an octahedral “14/8” assembly within a tubular graphite resistance heater (FE 254, Schunk Group GmbH, Vienna, Austria). The assembly was placed in the center of eight beveled tungsten carbide cubes (HA-7 %Co, Hawedia, Marklkofen, Germany). The synthesis was performed in a modified Walker-type multianvil device (mavo press LPR 1000–400/50, Max Voggenreiter GmbH, Mainleus, Germany). A more detailed description of the experimental setup can be found in the literature. 19 , 20 , 21 The maximum pressure of 12.0 GPa was reached within 317 min, and the maximum temperature of 1,300 °C within 10 min and then kept constant for 50 min, before it was decreased to room temperature within 60 min. Finally, the sample was decompressed to ambient conditions within 938 min. The reaction product was a colorless solid.
2.2 X-ray powder diffraction analysis and Rietveld refinement
The reaction product was analyzed with a STOE Stadi P powder diffractometer (STOE & Cie GmbH, Darmstadt, Germany), which was equipped with a Mythen 1 K microstrip detector (Dectris, Baden-Daettwil, Switzerland). The measurement was carried out with Ge(111)-monochromatized MoKα 1 radiation (λ = 0.7093 Å) in transmission geometry with a flat sample holder across a 2θ range of 2.0–60.5°. Figure 1 shows a Rietveld refinement 22 of a sample containing the title compound, performed with the program Topas 4.2. 23 At a 2θ value of about 19° a clear difference can be observed between the measured and calculated curves. The most likely explanation for this discrepancy is the presence of an unidentified side-phase.
![Figure 1:
Rietveld refinement of the powder X-ray diffraction pattern (MoKα
1 radiation) of the sample containing (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 and the additional phases B2O3
24
and NH4[B5O6(OH4)]·2H2O.
25](/document/doi/10.1515/znb-2025-0022/asset/graphic/j_znb-2025-0022_fig_001.jpg)
2.3 Crystal structure analysis
For the single-crystal structure analysis, a Bruker D8 Quest diffractometer (Bruker Corporation, Billerica, Massachusetts, U.S.A.) was used, which was equipped with an Incoatec microfocus X-ray tube (monochromatized MoKα radiation, λ = 0.7107 Å) and a Photon 100 CMOS detector. Data collection and processing was performed with the programs Saint (V8.38A) 26 and Apex III 27 and a multi-scan absorption correction with Sadabs (2016/2). 28 Structure solution and parameter refinement were carried out with Shelxt (2018/2) 29 and Shelxl (2018/3) 30 implemented in the program system Olex2-1.5. 31 The title compound was solved and refined in the orthorhombic space group Pmn21 (no. 31). This non-centrosymmetric space group is supported by failed attempts to solve and refine the structure in the centrosymmetric space group Pmmn (no. 59). (Programs used: Shelxt (2018/2); 29 Direct Methods via Shelxs (2013/1)). 32 , 33 A check with the program Platon (version 170613) 34 clearly confirmed the non-centrosymmetric space group. Furthermore, we excluded a possible inversion twinning because this did not improve the R1 value in the structure refinement. Therefore, the Flack parameter of 0.4(3) (see Table 1) might be caused by the only presence of light elements in the structure. Moreover, the experimental and theoretical transmission coefficients deviate, but since a correction might only have a marginal influence for light elements (B, H, N, O), we retained the structure refinement in its present form.
Single-crystal data and structure refinement of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36. Standard deviations are given in parentheses and refer to the last decimal place.
| Empirical formula | (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 |
| Molar mass, g mol−1 | 177.09 |
| Crystal system | Orthorhombic |
| Space group | Pmn21 (no. 31) |
| Single-crystal diffractometer | Bruker D8 Quest |
| Radiation / wavelength λ, Å | MoKα / 0.71073 |
| a, Å | 10.7902(4) |
| b, Å | 4.5949(2) |
| c, Å | 4.2280(2) |
| V, Å3 | 209.62(2) |
| Formula units per cell Z | 2 |
| Calculated density, g cm−3 | 2.81 |
| Crystal size, mm3 | 0.06 × 0.03 × 0.02 |
| Temperature, K | 213 |
| Absorption coefficient, mm−1 | 0.3 |
| F(000), e | 177 |
| θ range, deg | 3.78–37.67 |
| Range in hkl | ±16; ±6; ±6 |
| Refl. total / independent | 767 / 742 |
| R int / R σ | 0.0316 / 0.0145 |
| Refl. with I > 2 σ(I) | 742 |
| Data / ref. parameters | 767 / 76 |
| T min / T max | 0.715 / 0.747 |
| Absorption correction | Multi-scan |
| Flack parameter | 0.4(3) |
| Final R1 / wR2 (I > 2σ(I)) | 0.0203 / 0.0535 |
| Final R1 / wR2 (all data) | 0.0214 / 0.0539 |
| Goodness-of-fit on F 2 | 1.172 |
| Largest diff. peak / hole, e Å−3 | 0.29 / −0.20 |
With the exception of H, all atoms could be refined anisotropically; the part of the H atoms, which could be localized, had to be refined isotropically. In fact, not all hydrogen atoms were localizable (vide infra). Details of the data collection can be found in the synoptical Table 1. Tables 2 and 3 give the positional and displacement parameters, the Wyckoff positions, and site occupancy factors.
Wyckoff positions, atomic coordinates, isotropic U iso or equivalent isotropic displacement parameters U eq (Å2) and site occupancy factors (s.o.f.) for (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36. U eq is defined as one third of the trace of the orthogonalized U ij tensor (standard deviations in parentheses).
| Atom | Wyckoff position | x | y | z | U eq (U iso for H atoms) | s.o.f. |
|---|---|---|---|---|---|---|
| B1 | 4b | 0.3717(2) | 0.1700(3) | 0.8815(3) | 0.0055(2) | 1 |
| B2 | 4b | 0.2544(2) | 0.3230(3) | 0.3650(3) | 0.0036(2) | 1 |
| B3 | 2a | 1/2 | 0.346(2) | 0.391(2) | 0.0073(9) | 0.36 |
| H1aa | 4b | 0.437(3) | 0.808(8) | 0.461(9) | 0.08(2) | 1 |
| H1ab | 4b | 0.467(4) | 0.74(2) | 0.12(2) | 0.03(2) | 0.5 |
| H1b | 4b | 0.545(7) | 0.52(2) | 0.41(2) | 0.03(2) | 0.32 |
| O1 | 4b | 0.34422(9) | −0.1261(2) | 0.8135(2) | 0.0061(2) | 1 |
| O2 | 2a | 1/2 | 0.2442(3) | 0.7537(3) | 0.0063(3) | 1 |
| O3 | 4b | 0.37089(8) | 0.2394(2) | 0.2192(3) | 0.0071(2) | 1 |
| O4 | 4b | 0.28158(8) | 0.3802(2) | 0.7160(2) | 0.0036(2) | 1 |
| N | 2a | 1/2 | 0.7084(4) | 0.3410(5) | 0.0113(3) | 1 |
Anisotropic displacement parameters U ij (Å2) of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 (standard deviations in parentheses).
| Atom | U 11 | U 22 | U 33 | U 12 | U 13 | U 23 |
|---|---|---|---|---|---|---|
| B1 | 0.0087(5) | 0.0037(5) | 0.0040(5) | 0.0001(4) | −0.0025(5) | −0.0003(4) |
| B2 | 0.0043(5) | 0.0039(4) | 0.0026(5) | 0.0005(4) | 0.0001(4) | −0.0001(4) |
| B3 | 0.008(2) | 0.010(2) | 0.004(2) | 0 | 0 | 0.0013(18) |
| O1 | 0.0084(4) | 0.0030(3) | 0.0069(4) | −0.0021(3) | 0.0036(3) | −0.0009(3) |
| O2 | 0.0029(5) | 0.0062(5) | 0.0097(7) | 0 | 0 | −0.0008(4) |
| O3 | 0.0114(4) | 0.0052(4) | 0.0049(4) | 0.0023(3) | 0.0041(4) | 0.0000(3) |
| O4 | 0.0053(4) | 0.0028(3) | 0.0026(3) | 0.0007(3) | −0.0007(3) | −0.0007(3) |
| N | 0.0052(6) | 0.0176(7) | 0.0111(7) | 0 | 0 | −0.0071(7) |
CSD-2410970 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
2.4 IR spectroscopy
A transmission FT-IR spectrum of a single crystal of the title compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36, whose lattice parameters had previously been determined using a single-crystal diffractometer, were measured on a Vertex 70 FT-IR spectrometer in a spectral range of 600–4,000 cm−1 with a spectral resolution of 4 cm−1. The instrument was equipped with a KBr beam splitter, a liquid nitrogen cooled MCT (Mercury Cadmium Telluride) detector and a Hyperion 3000 microscope (Bruker Corporation, Billerica, Massachusetts, U.S.A.). A Globar rod (made of SiC) was used as mid-IR source and a 15 × IR objective as focus. The sample was placed on a BaF2 slide and measured including 300 scans. Atmospheric influences were corrected with the software Opus 7.2. 35
3 Results and discussion
3.1 Crystal structure
The compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 crystallizes in the orthorhombic space group Pmn21 (no. 31) with two formula units in the unit cell and with the lattice parameters a = 10.7902(4), b = 4.5949(2), c = 4.2280(2) Å, and V = 209.62 (2) Å3, exhibiting a slightly modified variant of the α-SrB4O7 structure type. 6 , 7 In fact, (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 is homeotypic to α-SrB4O7, 6 , 7 PbB4O7, 7 , 8 , 9 EuB4O7, 10 β-HgB4O7, 1 β-SnB4O7, 11 β-CaB4O7, 2 and LnB3.6O7 (Ln = La, Pr, Nd). 4 , 5
Similar to the homeotypic compounds, 1 , 2 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 the anionic framework is built up by two crystallographically different boron atoms (B1 and B2), which are tetrahedrally coordinated by oxygen atoms. By alternately connecting these corner-sharing boron tetrahedra, vierer-rings ([B4O7] building blocks) are formed (see Figure 2, one building block is colored in yellow and orange).
![Figure 2:
Visualization of the borate network [B4O7]∞ of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36. The three crystallographically different boron centered tetrahedra are dyed in various shades of blue (light: [B1O4], dark: [B2O4], cyan: [B3O3N]). One single [B4O7] building block is highlighted in yellow and orange. Either [NH4] or [B3NH3] units fill the interstices within the borate framework. All tetrahedra point towards viewing direction [001], which further confirms the non-centrosymmetric space group.](/document/doi/10.1515/znb-2025-0022/asset/graphic/j_znb-2025-0022_fig_002.jpg)
Visualization of the borate network [B4O7]∞ of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36. The three crystallographically different boron centered tetrahedra are dyed in various shades of blue (light: [B1O4], dark: [B2O4], cyan: [B3O3N]). One single [B4O7] building block is highlighted in yellow and orange. Either [NH4] or [B3NH3] units fill the interstices within the borate framework. All tetrahedra point towards viewing direction [001], which further confirms the non-centrosymmetric space group.
These building blocks are linked by corner sharing tetrahedra to a three-dimensional [(B4O7)2–]∞ borate network, showing sechser-, vierer-, and dreier-rings. Hence, cages within the borate framework are formed (see Figure 3), which serve as locations for two different tetrahedral structure motifs, [NH4] and [BNH3] units (see Figures 3 and 4). The [NH4] unit is linked to the borate framework solely by hydrogen bonds to O1, O2, and O3, the [BNH3] unit by hydrogen bonds to O1 and a covalent bond between B3 and an O atom of the [(B4O7)2–]∞ network (see Figure 3 and Table 6). The [BNH3] and [NH4] units are introduced into the [(B4O7)2–]∞ network, leading formally either to a “[B5O7NH3]+” unit or to an arrangement, where the ammonium group is located in a void of the borate network and stabilized via hydrogen bonds in a “[(NH4)+(B4O7)2–]–” unit. Apart from the atoms H1ab, H1b, and B3, all sites are fully occupied and therefore present in both units (see Table 2 and Figure 4). H1ab is also present in both units, but only one of the two positions can be occupied at the same time, which decreases the occupation to 0.5. H1b is solely in the “[(NH4)+(B4O7)2–]–” constellation (see Figure 4a) with a site occupancy factor of 0.32. For H1b, also two positions are possible, of which only one can be occupied simultaneously. Atom B3 is exclusively present in the “[B5O7NH3]+” unit (see Figure 4b) and has an occupancy of 0.36. Therefore, the [NH4] and [BNH3] units both have two atoms of H1aa and one of H1ab and differ by the presence of H1b in [NH4] and B3 in [BNH3]. Based on all the atoms and their site occupancy factors in the structure refinement and their common charges (+3 for B, +1 for H, −3 for N, and −2 for O), the sum formula would be “(B4.36H3.64NO7)0.28−”. Hence, for charge compensation we assume the presence of an additional number of protons distributed over oxygen atoms of the network leading formally to O–H groups, which cannot be detected in the difference Fourier map. Therefore, the structure of the compound has to be taken as tentative, but since there is no evidence of another charge compensation and of an exclusion criterion, we are sure, that our structure model is the most reasonable one. Including these additional protons for charge compensation, the atoms are in the ratio “B4.36H3.92NO7”. However, this does not reflect the actual connectivity of the atoms and so we decided to choose a sum formula that considers the assignment of the hydrogen atoms to specific atoms. Therefore, based on this structural analysis (single-crystal data), the sum formula of this new compound can be written as (NH4)0.64B4O6.72(OH)0.28[B(NH3)]0.36. Thereby, the alternating units [NH4] and [B(NH3)] are in the ratio of 0.64:0.36, which is based on the site occupancy factors of 0.36 of B3, and of 0.32 of H1b (solely in [NH4] unit), which is half of 0.64 because only half of the theoretically H1b sites can be occupied in the entire unit. There are seven O atoms which are partially loaded with protons: “O6.72(OH)0.28”. The sum formula (NH4)0.64B4O6.72(OH)0.28[B(NH3)]0.36 resembles MB4O7, with 0.28 O being connected to H, and M is formally replaced by (NH4)0.64 and [B(NH3)]0.36.
![Figure 3:
Visualization of the “cages”, that are surrounding either (a) the ammonium group [NH4] or (b) the [BO3NH3] units depending on the occupation of the B3 site. The hydrogen bonding situation is marked with dashed lines.](/document/doi/10.1515/znb-2025-0022/asset/graphic/j_znb-2025-0022_fig_003.jpg)
Visualization of the “cages”, that are surrounding either (a) the ammonium group [NH4] or (b) the [BO3NH3] units depending on the occupation of the B3 site. The hydrogen bonding situation is marked with dashed lines.
![Figure 4:
Depiction of the [NH4] (a) and [B3O3NH3] (b) units. Atoms H1ab and H1b are drawn nearly transparent due to the fact that only one of the two positions can be occupied at the same time (according to their site occupancy factors). A mirror plane passes perpendicularly through the N atom in both (a) and (b).](/document/doi/10.1515/znb-2025-0022/asset/graphic/j_znb-2025-0022_fig_004.jpg)
Depiction of the [NH4] (a) and [B3O3NH3] (b) units. Atoms H1ab and H1b are drawn nearly transparent due to the fact that only one of the two positions can be occupied at the same time (according to their site occupancy factors). A mirror plane passes perpendicularly through the N atom in both (a) and (b).
The B1–O and B2–O distances are in the range between 1.414(2) and 1.552(2) Å with average values of 1.49, and 1.46 Å, respectively, which corresponds to the mean value of 1.476(35) Å investigated by Zobetz et al. 36 for B–O distances in [BO4] tetrahedra and to values of the homeotypic compounds α-SrB4O7 6 , 7 and LnB3.6O7 (Ln = La, Pr, Nd). 4 , 5 The B3–O distances are in the range between 1.604(6) and 1.645(3) Å with a mean value of 1.63 Å. This increase may be explained by defective atoms inside the cage and an averaging of the [NH4] and [B–NH3] units in the structure refinement. The B3–N distance has a value of 1.678(6) Å, which is also increased compared to the ammine borate Cd(NH3)2[B3O5(NH3)]2 (1.605(3) Å) 15 and the oxonitridoborate tetrahedra (between 1.553(3) and 1.608(3) Å) 16 , 17 documented in the literature, and may be explained by the defective atoms as well. The N–H distances (hydrogen bond donor) are in the range between 0.96(4) and 1.03(8) Å (see Tables 4 and 6), which agrees well with typical data in the literature (e.g. values between 0.83(2) and 0.99(2) Å in HP-(NH4)B3O5 12 and between 0.84(2) and 0.88(2) Å in [BF3NH3]·3NH3 37 ). The hydrogen bond acceptors have distances between 1.82(4) and 1.99(8) Å, which is in agreement to values between 1.92(2) and 2.13(2) Å in HP-(NH4)B3O5. 12
Interatomic distances (Å) in (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 (standard deviations in parentheses).
| B1– | O1 | 1.422(2) | B3– | O2 | 1.604(6) |
| O2 | 1.525(2) | O3 | 1.645(3) 2 × | ||
| O3 | 1.463(2) | Ø B3–O | 1.63 | ||
| O4 | 1.539(2) | ||||
| Ø B1–O | 1.49 | B3– | N | 1.678(6) | |
| Ø B3–N | 1.68 | ||||
| B2– | O1 | 1.414(2) | |||
| O3 | 1.452(2) | N– | H1aa | 0.96(4) 2 × | |
| O4a | 1.535(2) | H1ab | 1.03(5) | ||
| O4b | 1.552(2) | H1b | 1.03(8) 1 × /0 × | ||
| Ø B2–O | 1.46 | Ø N–H | 1.0 |
-
Mean values are written in bold.
All the mean tetrahedral angles are in accordance with the expected regular value of 109.5°. The single angle values for O–B1–O, O–B2–O, O–B3–O, O–B3–N, and B–N–H are also close to the regular value (±6 % deviation). The single angle values for H–N–H are in the range of ±22 % (without consideration of the standard deviation), which is due to a distortion of the [NH4] and [BNH3] tetrahedra (see Table 5). This deviation from C 3ν symmetry is caused by the close proximity of the hydrogen atoms to the mirror plane (see Figure 4) and/or that the common nitrogen atom for [NH4] and [BNH3] does not exactly coincide with the occupancy disorder, which leads to a cigar-shaped deflection of the temperature factor ellipsoids. Additionally, we performed structure refinement attempts in the monoclinic space group Pn, which led to a decrease of the symmetry and an omitting of the mirror plane. In that model, we obtained twice the coordinate set with a fully occupied N atom, and the partial disorder of the H atoms was not present, but the R1 value increased from 0.020 to 0.026 and so the original structure refinement in the space group Pmn21 (no. 31) was retained (Tables 1–6).
Interatomic angles (deg) in (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36. Standard deviations are specified in parentheses. For Ø H–N–H both cases [B3NH3] and [NH4] are distinguished.
| O1–B1–O2 | 109.4(2) | O2–Ba3–N | 114.2(3) |
| O1–B1–O3 | 113.9(2) | O3–B3–N | 103.9(2) 2 × |
| O1–B1–O4 | 112.2(2) | Ø O–B3–N | 107.3 |
| O2–B1–O3 | 107.6(2) | ||
| O2–B1–O4 | 105.8(2) | B3–N–H1aa | 114(2) 2 × |
| O3–B1–O4 | 107.7(2) | B3–N–H1ab | 104(3) |
| Ø O–B1–O | 109.4 | Ø B–N–H | 111 |
| O1–B2–O3 | 114.7(2) | [B–NH 3 ]: | |
| O1–B2–O4a | 113.7(2) | H1aa–N–H1aa | 90(3) |
| O1–B2–O4b | 108.16(9) | H1aa–N–H1ab a | 100(3) |
| O3–B2–O4a | 106.06(9) | H1aa–N–H1ab b | 132(4) |
| O3–B2–O4b | 106.9(2) | Ø H–N–H | 107 |
| O4–B2–O4 | 106.86(8) | ||
| Ø O–B2–O | 109.7 | [NH 4 ]: | |
| H1aa–N–H1aa | 90(3) | ||
| O2–B3–O3 | 109.5(2) 2 × | H1aa–N–H1ab a | 100(3) |
| O3–B3–O3 | 115.7(3) | H1aa–N–H1ab b | 132(4) |
| Ø O–B3–O | 111.6 | H1aa–N–H1b a | 85(5) |
| H1aa–N–H1b b | 125(5) | ||
| H1ab–N–H1b | 102(6) or 123(6) | ||
| Ø H–N–H | 106 or 109 |
Hydrogen bonds (Å, deg) in (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 (standard deviations in parentheses). D stands for donor atom, A for acceptor atom of the corresponding hydrogen bond, and d for distance.
| D–H–A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(D–H–A) |
|---|---|---|---|---|
| N–H1aa⋯O1 | 0.96(4) | 1.82(4) | 2.719(2) | 154(4) |
| N–H1ab⋯O1 | 1.03(5) | 1.94(5) | 2.894(2) | 153(4) |
| N–H1b⋯O2 | 1.03(8) | 1.99(8) | 2.756(2) | 129(6) |
| N–H1b⋯O3 | 1.03(8) | 1.78(8) | 2.617(2) | 135(7) |
3.2 CHARDI and BLBS
The results of the crystal structure refinement were further confirmed by calculations of the bond-length/bond-strength 38 values (BLBS, see ΣV in Table 7), and the charge distribution 39 (CHARDI, see ΣQ in Table 7). The calculated BLBS and CHARDI values for B1, B2 and all the O atoms showed only minimal deviations compared to the regular values of +3 for B, and −2 for O. The atoms inside the cavities showed a deviation of either the BLBS or the CHARDI result compared to the expected values, which are –3 for N, and +1 for H. If we additionally consider the site occupancy factors of 0.36 for B3, 0.5 for H1ab and 0.32 for H1b, all the CHARDI values fit well. The deviating BLBS values (written in italics in Table 7) can be explained by the disorder of atoms inside the cavities.
Charge distribution in (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36, according to the bond-length/bond-strength (ΣV) and CHARDI (ΣQ) concept. Values with a deviation from the expected regular values of more than 12 % are written in italics.
| B1 | B2 | B3 | H1aa | H1ab | H1b | O1 | O2 | O3 | O4 | N(a) | N(b) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΣV | +2.95 | +2.95 | +2.05 | +1.21 | +1.00 | +1.00 | −1.76 | −1.85 | −2.06 | −1.89 | −3.99 | −4.42 |
| ΣQ | +3.18 | +3.05 | +1.16 | +0.92 | +0.46 | +0.31 | −2.00 | −1.82 | −1.94 | −1.94 | −3.14 | |
3.3 IR spectroscopy
In Figure 5, an IR spectrum of a single crystal of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 in the spectral region of 600–4,000 cm−1 is depicted. To ensure that the crystal corresponds to the title compound, the lattice parameters of the chosen crystal were determined, which confirmed its identity. The assignment of the vibration bands of the borate network has already been discussed for the homeotypic compounds α-SrB4O7, 40 β-CaB4O7, 2 LaB3.6O7, 4 and PrB3.6O7 4 and was also followed for the title compound. Typically, vibration bands ranging from 850 to 1,100 cm−1 correspond to the stretching modes of [BO4] tetrahedra. 41 The BO3N group of the title compound shows similar absorption bands in this region (e. g. peak at 908 cm−1). At lower wavenumbers, there is an increase in contributions from bending modes and more intricate lattice vibrations. 41 Absorption bands between 1,200 and 1,500 cm−1 (e. g. peaks at 1,248, 1,414 and 1,551 cm−1 in (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36) are commonly associated with the stretching modes of triangular [BO3] units, 42 or with three-fold coordinated oxygen atoms and their corresponding [OB3] units, because of the similar geometry and force constants. 2 , 4 However, in 2013 Kaindl et al. investigated possible band positions of [OB3] units with quantum chemical calculations and showed that vibrations in these ranges are caused by stretching and bending modes of the borate network, whereas peaks of [OB3] were at values below 830 cm−1. 43 As described by Sohr et al. for Cd(NH3)2[B3O5(NH3)]2, 15 stretching of the N–H bonds of ammonia occur at 3,000–3,500 cm−1 and the bending of ammonia at 1,580–1,650 and 1,000–1,050 cm−1. 15 Bending of B–N–H groups occurs at 800–900, 1,100–1,120, and 1,470–900 cm−1. 15 We observed similar bands, which further confirms the structure refinement of [NH4] and [BNH3] groups. In detail, the broad band around 3,200 cm−1 probably corresponds to stretching vibrations of N–H/O–H and the peak at 1,643 cm−1 to bending vibrations of ammonia. However, the N–H/O–H stretching band is more extended in the title compound and is in the range between 2,500 and 3,700 cm−1, which can be explained by the strength of the hydrogen bonds. 44 The hydrogen bonds in the title compound with a distance between donor and acceptor in the range between 2.5 and 3.2 Å (see Table 6) can be assumed as moderately strong, 45 and occur as a broad band in the before mentioned range. 44 Typically, O–H stretching modes occur in the range 2,000–4,000 cm−1, 46 , 47 , 48 but could not be assigned with certainty, because of the large number of ammine/ammonium groups in the structure model which show peaks in the same range. For a more detailed assignment of the vibration modes (e. g. the exact position of stretching of O–H and an explanation for the peak at about 1,712 cm−1) quantum chemical calculations for the title compound would be required, but could not be sufficiently resolved in this complex vibration mode pattern.
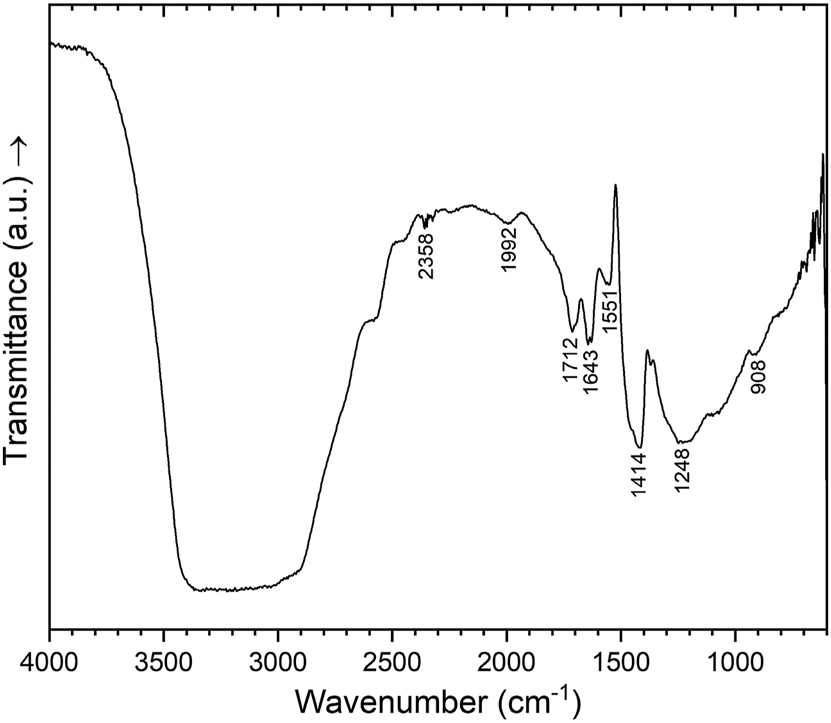
Single crystal IR spectrum of (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 in the spectral region of 600–4,000 cm−1.
4 Conclusions
Orthorhombic crystals of the new compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 have been synthesized in a high-pressure/high-temperature experiment in a multianvil apparatus from the starting materials h-BN, H3BO3, and B2O3, which is to the best of our knowledge the first synthesis approach in which a borate structure with an [NH4] unit is obtained from these starting materials. Its structure could be solved and refined by single-crystal X-ray diffraction, which revealed a three-dimensional [(B4O7)2–]∞ borate network, showing sechser-, vierer-, and dreier-rings. The resulting interstices serve as locations for two different defective tetrahedral structure motifs inside, [NH4] and [B3NH3] units. The title compound is homeotypic to compounds in the α-SrB4O7 6 , 7 structure type. IR data additionally confirm the structure model.
Acknowledgements
The authors thank Assoc.-Prof. Dr. Gunter Heymann for the recording of the single-crystal data, Univ.-Prof. Dr. Roland Stalder for granting us access to the single-crystal IR spectrometer and Dr. Michael Seidl for determining the cell parameters of the crystals for IR spectroscopy. I.W. thanks the Vice Rectorate for Research for the grant of a doctoral fellowship at the University of Innsbruck.
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: The authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: ChatGPT was used to improve language when required.
-
Conflict of interest: The authors state no conflict of interest.
-
Research funding: None declared.
-
Data availability: The raw data can be obtained on request from the corresponding authors. CSD-2410970 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
References
1. Emme, H.; Weil, M.; Huppertz, H. Z. Naturforsch. 2005, 60b, 815–820.10.1515/znb-2005-0801Suche in Google Scholar
2. Huppertz, H. Z. Naturforsch. 2003, 58b, 257–265.10.1515/znb-2003-0403Suche in Google Scholar
3. Huppertz, H.; Heymann, G. Solid State Sci. 2003, 5, 281–289; https://doi.org/10.1016/s1293-2558(02)00057-2.Suche in Google Scholar
4. Teichtmeister, T. A.; Widmann, I.; Bayarjargal, L.; Tribus, M.; Heymann, G.; Wurst, K.; Huppertz, H. Eur. J. Inorg. Chem. 2024, 27, e202300555 (10 pages). https://doi.org/10.1002/ejic.202300555.Suche in Google Scholar
5. Teichtmeister, T. A.; Bernhart, A. H.; Huppertz, H. Z. Kristallogr. New Cryst. Struct. 2024, 239, 531–532; https://doi.org/10.1515/ncrs-2024-0074.Suche in Google Scholar
6. Krogh-Moe, J.; Magnéli, A.; Seip, R.; Seip, H. M. Acta Chem. Scand. 1964, 18, 2055–2060; https://doi.org/10.3891/acta.chem.scand.18-2055.Suche in Google Scholar
7. Perloff, A.; Block, S. Acta Crystallogr. 1966, 20, 274–279; https://doi.org/10.1107/s0365110x66000525.Suche in Google Scholar
8. Corker, D. L.; Glazer, A. M. Acta Crystallogr. 1996, B52, 260–265.10.1107/S0108768195013310Suche in Google Scholar
9. Stein, W. D.; Liebertz, J.; Becker, P.; Bohatý, L.; Braden, M. Eur. Phys. J. B 2012, 85, 236 (5 pages). https://doi.org/10.1140/epjb/e2012-21062-y.Suche in Google Scholar
10. Machida, K.-I.; Adachi, G.-Y.; Shiokawa, J. Acta Crystallogr.B 1980, B36, 2008–2011.10.1107/S0567740880007820Suche in Google Scholar
11. Knyrim, J. S.; Schappacher, F. M.; Pöttgen, R.; Schmedt auf der Günne, J.; Johrendt, D.; Huppertz, H. Chem. Mater. 2007, 19, 254–262.10.1021/cm061946wSuche in Google Scholar
12. Sohr, G.; Wilhelm, D.; Vitzthum, D.; Schmitt, M. K.; Huppertz, H. Z. Anorg. Allg. Chem. 2014, 640, 2753–2758; https://doi.org/10.1002/zaac.201400312.Suche in Google Scholar
13. Huang, C.; Zhang, F.; Zhang, B.; Yang, Z.; Pan, S. New J. Chem. 2018, 42, 12091–12097; https://doi.org/10.1039/c8nj02359j.Suche in Google Scholar
14. Domenech, V.; Solans, J.; Solans, X. Acta Crystallogr. 1981, B37, 643–645.10.1107/S0567740881003750Suche in Google Scholar
15. Sohr, G.; Ciaghi, N.; Schauperl, M.; Wurst, K.; Liedl, K. R.; Huppertz, H. Angew. Chem. Int. Ed. 2015, 54, 6360–6363; https://doi.org/10.1002/anie.201500706.Suche in Google Scholar PubMed
16. Fuchs, B.; Johrendt, D.; Bayarjargal, L.; Huppertz, H. Angew. Chem. Int. Ed. 2021, 60, 21801–21806; https://doi.org/10.1002/anie.202110582.Suche in Google Scholar PubMed PubMed Central
17. Widmann, I.; Kinik, G.; Jähnig, M.; Glaum, R.; Schwarz, M.; Wüstefeld, C.; Johrendt, D.; Tribus, M.; Hejny, C.; Bayarjargal, L.; Dubrovinsky, L.; Heymann, G.; Suta, M.; Huppertz, H. Adv. Funct. Mater. 2024, 34, 2400054 (9 p.); https://doi.org/10.1002/adfm.202400054.Suche in Google Scholar
18. Widmann, I.; Dubrovinsky, L.; Huppertz, H. Z. Naturforsch. 2025, 80b, 277–283.10.1515/znb-2025-0024Suche in Google Scholar
19. Huppertz, H. Z. Kristallogr. 2004, 219, 330–338; https://doi.org/10.1524/zkri.219.6.330.34633.Suche in Google Scholar
20. Walker, D.; Carpenter, M. A.; Hitch, C. M. Am. Mineral. 1990, 75, 1020–1028.Suche in Google Scholar
21. Walker, D. Am. Mineral. 1991, 76, 1092–1100.10.1007/978-1-4615-3968-1_10Suche in Google Scholar
22. Rietveld, H. J. Appl. Crystallogr. 1969, 2, 65–71; https://doi.org/10.1107/s0021889869006558.Suche in Google Scholar
23. Topas 4.2, Bruker AXS Inc.: Madison, Wisconsin (USA), 2009.Suche in Google Scholar
24. Prewitt, C. T.; Shannon, R. D. Acta Crystallogr. 1968, B24, 869–874.10.1107/S0567740868003304Suche in Google Scholar
25. Möhlen, M.; Neumüller, B.; Dashti-Mommertz, A.; Müller, C.; Massa, W.; Dehnicke, K. Z. Anorg. Allg. Chem. 1999, 625, 1631–1637.10.1002/(SICI)1521-3749(199906)625:6<954::AID-ZAAC954>3.0.CO;2-BSuche in Google Scholar
26. Saint ; Bruker AXS Inc.: Madison, Wisconsin (USA), 2018.Suche in Google Scholar
27. Apex3; Bruker AXS Inc.: Madison, Wisconsin (USA), 2016.Suche in Google Scholar
28. Sadabs ; Bruker AXS Inc.: Madison, Wisconsin (USA), 2016.Suche in Google Scholar
29. Sheldrick, G. M. Acta Crystallogr. 2015, A71, 3–8.10.1107/S2053273314026370Suche in Google Scholar
30. Sheldrick, G. M. Acta Crystallogr. 2015, C71, 3–8.Suche in Google Scholar
31. Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341; https://doi.org/10.1107/s0021889808042726.Suche in Google Scholar
32. Sheldrick, G. M. Shelxs 2013/1. Program for the Solution of Crystal Structures; University of Göttingen: Göttingen (Germany), 2013.Suche in Google Scholar
33. Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112–122.10.1107/S0108767307043930Suche in Google Scholar
34. Spek, A. J. Appl. Crystallogr. 2003, 36, 7–13; https://doi.org/10.1107/s0021889802022112.Suche in Google Scholar
35. Opus . Bruker Corporation: Billerica, Massachusetts (USA), 2012.Suche in Google Scholar
36. Zobetz, E. Z. Kristallogr. 1990, 191, 45–57; https://doi.org/10.1524/zkri.1990.191.1-2.45.Suche in Google Scholar
37. Kraus, F.; Baer, S. A. Z. Anorg. Allg. Chem. 2010, 636, 414–422; https://doi.org/10.1002/zaac.200900392.Suche in Google Scholar
38. Brown, I. D.; Altermatt, D. Acta Crystallogr. 1985, B41, 244–247.10.1107/S0108768185002063Suche in Google Scholar
39. Hoppe, R. Z. Kristallogr. 1979, 150, 23–52; https://doi.org/10.1524/zkri.1979.150.14.23.Suche in Google Scholar
40. Weir, C. E.; Schroeder, R. J. Res. Natl. Bur. Stand., Sect. A 1964, 68, 465–487.10.6028/jres.068A.045Suche in Google Scholar PubMed PubMed Central
41. Ross, S. D. Spectrochim. Acta, Part A 1972, 28, 1555–1561; https://doi.org/10.1016/0584-8539(72)80126-0.Suche in Google Scholar
42. Laperches, J. P.; Tarte, P. Spectrochim. Acta 1966, 22, 1201–1210; https://doi.org/10.1016/0371-1951(66)80023-1.Suche in Google Scholar
43. Kaindl, R.; Sohr, G.; Huppertz, H. Spectrochim. Acta, Part A 2013, 116, 408–417; https://doi.org/10.1016/j.saa.2013.07.072.Suche in Google Scholar PubMed
44. Pazé, C.; Bordiga, S.; Lamberti, C.; Salvalaggio, M.; Zecchina, A.; Bellussi, G. J. Phys. Chem. B 1997, 101, 4740–4751; https://doi.org/10.1021/jp970649z.Suche in Google Scholar
45. Steiner, T. Angew. Chem. Int. Ed. 2002, 41, 48–76; https://doi.org/10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u.10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-USuche in Google Scholar
46. Jun, L.; Shuping, X.; Shiyang, G. Spectrochim. Acta, Part A 1995, 51, 519–532; https://doi.org/10.1016/0584-8539(94)00183-c.Suche in Google Scholar
47. Nakamoto, K.; Margoshes, M.; Rundle, R. E. J. Am. Chem. Soc. 1955, 77, 6480–6486; https://doi.org/10.1021/ja01629a013.Suche in Google Scholar
48. Hammer, V. M. F.; Libowitzky, E.; Rossman, G. R. Am. Mineral. 1998, 83, 569–576; https://doi.org/10.2138/am-1998-5-617.Suche in Google Scholar
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- In this issue
- Research Articles
- A MEMS-based gas sensor for H2S detection with enhanced performance using a Ni–In2O3/ZnO nano-composite
- Ultra-sensitive and rapid response H2S microsensor based on Co–ZnO with ppm level detection
- Crystal chemistry and structural phase transition in CaLa2Zn1–x Ca x Ti2O9 (x = 0.00, 0.15, 0.30, 0.45, 0.90 and 1.00)
- RE 10Co3In10 (RE = Y, Gd, Tb, Dy, Ho, Er, Tm) – a new intergrowth structure with CsCl- and AlB2-type slabs
- Crystal structures of phases from the solid solutions DyNiIn1−xSn x
- Tricyanomethanides of the scandium group obtained from aqueous solution: syntheses, crystal structures and Raman spectra of Sc[C(CN)3]3(H2O)3, Y[C(CN)3]3(H2O)2 and La[C(CN)3]3(H2O)4
- Three pnictides with Sm6Rh30Si19-type structure: Sm6Rh30P19, Lu6Rh30P19 and Eu6Rh30Sb19
- The compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 representing the first ammine/ammonium borate
- Extended investigations on the pressure stability of AlB4O6N:Cr3+
- Synthesis and crystal structures of new phases in the system Hf–Ta–O–N
Artikel in diesem Heft
- Frontmatter
- In this issue
- Research Articles
- A MEMS-based gas sensor for H2S detection with enhanced performance using a Ni–In2O3/ZnO nano-composite
- Ultra-sensitive and rapid response H2S microsensor based on Co–ZnO with ppm level detection
- Crystal chemistry and structural phase transition in CaLa2Zn1–x Ca x Ti2O9 (x = 0.00, 0.15, 0.30, 0.45, 0.90 and 1.00)
- RE 10Co3In10 (RE = Y, Gd, Tb, Dy, Ho, Er, Tm) – a new intergrowth structure with CsCl- and AlB2-type slabs
- Crystal structures of phases from the solid solutions DyNiIn1−xSn x
- Tricyanomethanides of the scandium group obtained from aqueous solution: syntheses, crystal structures and Raman spectra of Sc[C(CN)3]3(H2O)3, Y[C(CN)3]3(H2O)2 and La[C(CN)3]3(H2O)4
- Three pnictides with Sm6Rh30Si19-type structure: Sm6Rh30P19, Lu6Rh30P19 and Eu6Rh30Sb19
- The compound (NH4)0.64B4.36O6.72(OH)0.28(NH3)0.36 representing the first ammine/ammonium borate
- Extended investigations on the pressure stability of AlB4O6N:Cr3+
- Synthesis and crystal structures of new phases in the system Hf–Ta–O–N