Bis-periazulene: remaining non-alternant isomer of pyrene
-
Akihito Konishi

 ,
Koki Horii
,
Michiyoshi Hirose
,
Koki Horii
,
Michiyoshi Hirose

Abstract
Bis-periazulene (cyclohepta[def]fluorene) is one of the seven possible non-alternant isomers of pyrene. Different from the other isomers, bis-periazulene had remained an uncharacterized hydrocarbon until 2022, since it was first reported in 1955. This paper documents our recent achievements in synthesizing and characterizing bis-periazulene derivatives. Its triaryl derivatives 5c‒e exhibited the superimposed electronic structures of peripheral, polarized, and open-shell π-conjugated systems. Furthermore, in contrast to previous theoretical predictions, bis-periazulene derivatives showed the singlet ground state.
Introduction
π-Conjugated carbocycles containing odd-membered rings are called as non-alternant hydrocarbons. Azulene 1, pentalene 2, and heptalene 3 are the most representative skeletons among non-alternant hydrocarbons. They are composed by the consecutive odd-membered 5–7, 5–5, and 7–7 ring systems, respectively. When one alternately assigns stared (*) and non-stared marks (○) to the sp 2 carbon atoms of these hydrocarbons, the same marks are inevitably next to each other (e.g. red-colored * marks in Fig. 1). On the other hand, in alternant hydrocarbons such as benzene and naphthalene, the alternative assignment of the two marks to the sp 2 carbons is always realized (Fig. 1). The intriguing sp 2-carbon network of non-alternant hydrocarbons has been a major motivation for the research because of their optoelectronic properties, (anti)aromaticity, and magnetisms, which are never shared by alternant hydrocarbons. Non-alternant hydrocarbons in which the naphthalene ring in pyrene 4 is replaced by one and more azulene ring(s) have provided profound insights into the relationship between molecular structure and the main π-conjugated system (Fig. 2) [1], [2], [3], [4], [5]. Recently, the chemistry of non-alternant hydrocarbons has experienced a huge resurgence thanks to the advances in synthetic methodologies and quantum chemical calculations. The small HOMO‒LUMO gaps, ambipolar redox properties and near-infrared absorptions of these π-extended non-alternant systems demonstrate the possibility of their application as functional organic materials. Through pioneering works by Boekelheide [6,7], Reid [8], Gardner [9], Hafner [10], Anderson [11], and Vogel [12], six of the seven possible non-alternant isomers of pyrene, which contain the pentagon and heptagon pair, have been isolated and characterized as stable aromatic molecules, revealing their differences from pyrene in terms of optoelectronic properties.

Examples of non-alternant and alternant hydrocarbons.

Non-alternant isomers of pyrene 4. Blue bold lines represent the embedded pair(s) of pentagon and heptagon.
The only unsynthesized isomer, bis-periazulene [13] (cyclohepta[def]fluorene) 5, had been uncharacterized until 2022, despite many synthetic efforts. This paper summarizes our recent studies on the synthesis and characterization of the triaryl-substituted bis-periazulene derivatives. Our interests are to reveal the π-conjugated network of bis-periazulene and to determine the actual ground state of bis-periazulene [14].
Historical background
In 1955, Reid reported the first synthetic attempt for the unsubstituted 5a [8]. Sutherland followed Reid’s work, and accomplished the synthesis of the tropylium-type cation salt 6a +∙ClO4 − [15,16]. Hafner designed a kinetically protected derivative 5b using three tert-butyl groups and observed the dianion 5b 2− by NMR measurements [17]. Although these synthetic attempts provided useful precursors for 5, the transformations into 5 through deprotonation or two-electron oxidation failed due to the inherently high reactivity of 5 against oxidation and self-polymerization (Fig. 3) [16,17]. Wu reported the synthetic attempt of a π-extended system but it failed to undergo the Lewis-acid mediated cyclization [18]. The theoretical calculations have offered fascinating clues as to the electronic structures of 5 [19]. In 1965, Heilbronner and co-workers reported the theoretical calculations of 5 [19]. Their simplified Pariser–Parr–Pople (PPP) calculations revealed that 5 has a triplet ground state of 3.92 kcal/mol lower than the lowest singlet state. They concluded that the ground state of 5 has either a triplet ground state or a singlet ground state with a low-lying thermally excited triplet state. The recent DFT calculation also predicted that 5 would have an open-shell nature with the triplet ground state approximately 2 kcal/mol lower than the lowest singlet state, despite 5 being drawn as a closed-shell Kekulé structure [13]. Malrieu suggested that the lowest singlet and triplet states of 5 are nearly degenerate in their different equilibrium geometries by using both geometry-dependent Heisenberg Hamiltonian and ab initio methods [20]. These intriguing experimental and theoretical studies demonstrate the value of synthesizing and characterizing 5.

Selected examples of synthetic attempts of bis-periazulenes 5.
Resonance structures of bis-periazulene
The double peri-benzoannulation on an azulene characterizes the electronic features of 5 (Fig. 4a). In the closed-shell Kekulé structure (5a-i), the hexagons within 5 adopt o-quinoidal forms, which should peripherally delocalize the 14π-system via an equivalent resonance/interconversion [21,22]. When the quinoidal forms are transformed into the 6π-benzenoid forms, two possible structures are described. One is the charge-separated zwitterionic structure (5a-ii), which is reminiscent of azulene-like properties [23], [24], [25], [26]. The other is the open-shell diradical structure (5a-iii), where an m-quinodimethane subunit, a typical non-disjoint non-Kekulé diradical [27], [28], [29], engages in the stabilization of the triplet state. These considerations strongly prompted us to synthesize 5 and characterize its electronic structures, including the actual ground state.

(a) Resonance structures of 5a. Bold lines indicate the peripherally conjugated circuit. (b) Target kinetically protected derivatives of 5c‒e.
Synthesis of triaryl-substituted bis-periazulene
To improve stability, we planned to introduce ortho-disubstituted aromatic groups into the three reactive sites of 5a (Fig. 4b and Scheme 1). 2,4,6-Trimethylphenyl (Mes) (c), 2,4,6-triisopropylphenyl (Trip) (d), and 2,6-dichlorophenyl (Dcp) (e) groups were introduced into the pentagon of 9-aryl-9H-fluorene 7. Formylation of 7 followed by an introduction of (trimethylsilyl)acetylene into 8 afforded fluorene derivative 9. Treating 9 with In(OTf)3 directly gave 10 as the indium(III) center would activate the carbonyl and facilitate cyclization. Nucleophilic attack on enone 10 with mesitylmagnesium bromide afforded 1,4-adduct 11 and oxidation by 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) gave mesityl-substituted enone 12. Re-treatment of 12 with mesitylmagnesium bromide afforded alcohol 13 as a 1,2-adduct to the carbonyl. Treating alcohol 13 with HBF4·Et2O immediately produced the tropylium-type cation salt 6 +·BF4 − [30]. For the deprotonation of 6 + into 5, the choice of a suitable base was important. When we applied NaH to the deprotonation of 6d + [31,32], an over-reduced radical anion 5d ·‒ was produced.

Synthesis of triaryl bis-periazulene 5.
Alternatively, changing the base into LiH successfully transformed 6 + into the desired molecule 5. The difference in solvation of THF to alkali ions may be responsible for this result. A solution of 5c‒e in THF gradually decomposed upon exposure to air at room temperature (half-lives: 2.4 h (5c), 19 h (5d), and 73 h (5e)).
Molecular geometry: peripheral 14π-character
The bond lengths of bis-periazulene were determined from the X-ray crystal structure of 5c‒e. The main core exhibits a slight bond length alternation (BLA) but assumes an approximate C 2v symmetry. In Fig. 5, an ORTEP drawing of 5d is shown as a representative example. The fundamental geometrical characteristics of 5d are mostly identical to those of 5c and 5e. To evaluate the canonical structure contributing to the observed geometry, we focused on the bridging bond a between the pentagon and heptagon (Fig. 5a). The bond length of the a bond in 5d is 1.391(2) Å, which is considerably shorter than that of azulene (1.489 Å) [33], but longer than the corresponding bonds of Anderson’s (1.333 Å) [34] and Vogel’s (1.354 Å) [35] non-alternant isomers of pyrene isomers that have strong peripheral 14π-character. Some double bond characteristic of the a bond implies that a quinoidal form (5a-i in Fig. 4) emerges as one of the dominant resonance structures. The BLA of the two hexagons is not small according to the harmonic oscillator model of aromaticity (HOMA) [36,37] analysis (ca. 0.6), which supports the quinoidal character (Fig. 5b). Contrarily, the HOMA value indicates a larger value for the perimeter of the main core of 5d (0.88), suggesting that peripheral 14π-electron delocalization is induced by the interconversion between equivalent two quinoidal forms (5a-i).

(a) ORTEP drawing of 5d with at the 50 % probability level. (b) Selected bond lengths with HOMA values (italic) of 5d.
Aromaticity: charge separated zwitterionic character
The magnetic criteria for aromaticity illustrate another aspect of the π-conjugation of 5d (Fig. 6). The NICS(1) calculations of 5a′ (The substituents of the X-ray geometry of 5d were replaced by hydrogen atoms.) demonstrate that the negative values of −14.7, −16.4, and −18.5 ppm are induced on the fluorene moiety, whereas weakly aromatic character of −3.1 ppm appears on the heptagon. According to the anisotropy of the induced current density (ACID) [38,39] calculation, the induced ring currents clock-wisely flow on the 6–5–6 tricyclic system. The electrostatic potential (ESP) map of 5a′ indicates that the pentagon and the adjacent hexagons have a negative charge, whereas the heptagon has a relatively positive charge. Judging from this magnetic/electrostatic evaluation, 5 should be described by the combination of a fluorenyl anion and an allyl cation, suggesting that the charge-separated zwitterionic structure is a non-negligible resonance structure (5a-ii in Fig. 4). Experimentally, 5 behaves as a basic hydrocarbon, similar to azulene-based hydrocarbons [25,40–45]. 5c was readily protonated by CF3SO3H to a tropylium-type cation salt 5c +·OSO2CF3 −.

(a) ACID plots with NICS(1) values of 5a′. (b) ESP map of 5a′.
Magnetic properties: open-shell singlet character
The determined physical properties of 5c–e demonstrated an open-shell character, which has not been found in other pyrene’s non-alternant isomers. From the Superconducting quantum interference device (SQUID) measurements, the singlet‒triplet energy gap (ΔE S‒T) of 5d was determined to be −454 K (−0.90 kcal/mol) (Fig. 7a). A weak open-shell character of 5d (y 0: 0.23) was predicted by the PUHF/def2-SVP method. The ground state of 5d is determined to be singlet, contrary to previous predictions that 5a should be a triple ground state. Due to the small ΔE S‒T, no NMR signal of the main core of 5d in THF-d 8 was observed even at −100 °C. Owing to the small ΔE S‒T, the electron spin resonance (ESR) measurements of 5c–e clearly displayed signals typical of the thermally accessible triplet species (zero field splitting parameters of 5d: |D| = 0.01956 cm−1 and |E| = 0.00231 cm−1).
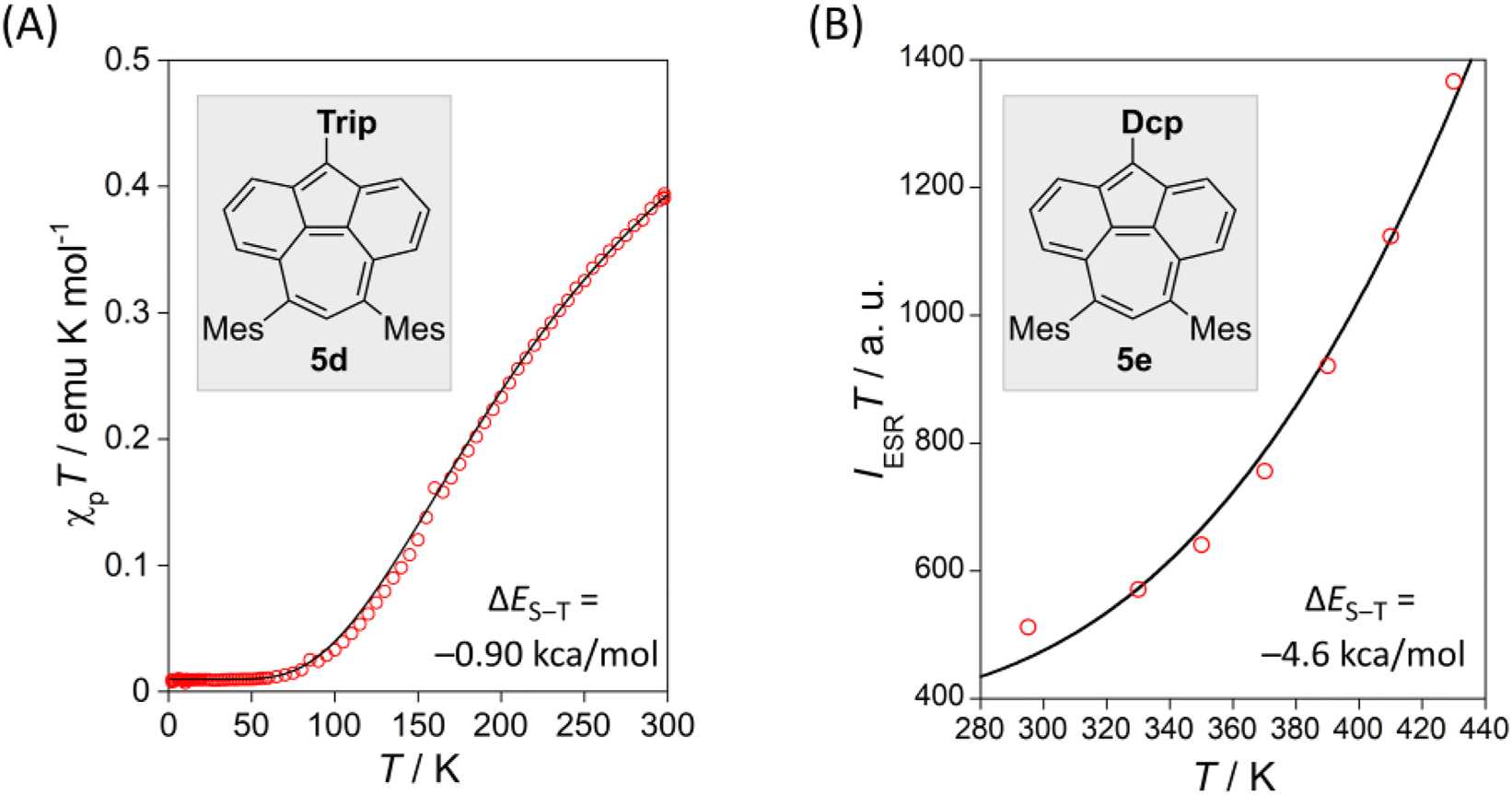
(a) χp T–T plot of the microcrystalline sample of (a) 5d (from SQUID) and (b) 5e (from ESR). The measured data (red circles) and Bleaney–Bowers fit (−).
The introduced aryl groups may affect the energy gap between the lowest singlet and triplet states. The SQUID and ESR measurements of 5e, in which a Dcp group is introduced instead of the Trip group of 5d, showed an expanded ΔE S‒T of approximately −2300 K (−4.6 kcal/mol) (Fig. 7b), five times larger than that of 5d, due to the electron-withdrawing character of Dcp. The inductive effect of the Dcp group should stabilize the polarized negative charge on the pentagon of 5, lowering the singlet energy level and strengthening the pairing of two unpaired electrons. The two impacts can qualitatively explain the expansion of the ΔE S‒T of 5e.
Optical property
Figure 8a shows the electronic absorption spectrum of 5c in THF. The longest absorption band of 5c was a weak and broad band centered at around 1700 nm, reaching over 2000 nm. No solvatochromism of 5c was observed among THF, CH2Cl2, and benzene, presumably because the polarized negative charge distributes on the fluorenyl moiety and the dipole moment is not large. The DFT calculations of singlet 5c at the UCAM-B3LYP/def2-SVP demonstrates that the HOMO (ψ 149) and LUMO (ψ 150), regardless of the spin direction, is mainly distributed on the fluorenyl and dibenzo[a,c]cycloheptenyl moiety, respectively (Fig. 8b). The time-dependent (TD)-DFT calculations of 5c reveal that the lowest transition, approximately 1700 nm, is ascribed to the HOMO→LUMO transition across the pentagon to the heptagon. The observed HOMO→LUMO transition energy of 5c (∼0.7 eV) is much smaller than those of pyrene (3.6 eV) and other non-alternant isomers (1.5–2.6 eV). The molecular orbital characteristics originating from the non-alternant character of 5, as well as the appearance of the open-shell character, resulted in the unusual electronic absorption spectrum.

(a) Electronic absorption spectra of 5c in THF (red), benzene (blue) and CH2Cl2 (green) in the region of visible to near-IR. The background signals at 1700 nm and over 2200 nm arose from an overtone of the C−H vibrations of the solvent. (b) Frontier molecular orbitals of 5c in the singlet state calculated at the UCAMB3LYP/def2-SVP//RCAM-B3LYP-D3/def2-SVP level with the lowest energy transition estimated at the TD-UCAM-B3LYP/def2-SVP//RCAM-B3LYP-D3/def2-SVP level.
Conclusion
We demonstrate the synthesis and characterization of the persistent bis-periazulene derivatives 5c‒e, the remaining non-alternant isomer of pyrene. Triaryl-substituted 5c‒e exhibit singlet ground states, contrary to previous theoretical predictions for 5a. Peri-benzoannulation into an azulene core characterizes 5 as an unprecedented hydrocarbon that contains three aspects of π-conjugation: peripheral, charge-separated, and open-shell π-conjugations. Further studies, such as determining the actual ground state of 5a and the substituent or π-extension [46] effect on the ΔE S‒T of 5 will help us advance in the understanding of this exciting hydrocarbon.
Funding source: Japan Society for the Promotion of Science
Award Identifier / Grant number: 20J10273
Award Identifier / Grant number: 20K05464
Funding source: Ministry of Education, Culture, Sports, Science and Technology
Award Identifier / Grant number: 21H05212
Funding source: Core Research for Evolutional Science and Technology
Award Identifier / Grant number: JPMJCR20R3
Acknowledgement
We thank the financial support that funded our research on bis-periazulene chemistry (JSPS KAKENHI (20K05464 [A.K.]), MEXT Grant-in-Aid for Transformative Research Areas (A) “Digitalization-driven Transformative Organic Synthesis (Digi-TOS)” (21H05212 [M.Y.]), JST CREST, Grant Number JPMJCR20R3, Japan and JSPS Fellowship for Young Scientists (20J10273 [K.H.])). In addition, we gratefully acknowledge the critical contributions of our collaborators, Assoc. Prof. R. Kishi and the late Prof. M. Nakano (Osaka University) for the quantum chemical calculations and Assoc. Prof. D. Shiomi, Prof. K. Sato, and Prof. T. Takui (Osaka Metropolitan University) for the magnetic measurements.
References
[1] K. Hafner, K. H. Häfner, C. König, M. Kreuder, G. Ploss, G. Schulz, E. Sturm, K. H. Vöpel. Angew. Chem. Int. Ed. Engl. 2, 123 (1963), https://doi.org/10.1002/anie.196301231.Search in Google Scholar
[2] K. Hafner. Angew. Chem. Int. Ed. Engl. 3, 165 (1964), https://doi.org/10.1002/anie.196401651.Search in Google Scholar
[3] K. Hafner. Pure Appl. Chem. 28, 153 (1971), https://doi.org/10.1351/pac197128020153.Search in Google Scholar
[4] K. Hafner. Pure Appl. Chem. 54, 939 (1982), https://doi.org/10.1351/pac198254050939.Search in Google Scholar
[5] Y. Tobe. Chem. Rec. 15, 86 (2015), https://doi.org/10.1002/tcr.201402077.Search in Google Scholar PubMed
[6] V. Boekelheide, W. E. Langeland, C.-T. Liu. J. Am. Chem. Soc. 73, 2432 (1951), https://doi.org/10.1021/ja01150a007.Search in Google Scholar
[7] V. Boekelheide, G. K. Vick. J. Am. Chem. Soc. 78, 653 (1956), https://doi.org/10.1021/ja01584a036.Search in Google Scholar
[8] D. H. Reid, W. H. Stafford, J. P. Ward. J. Chem. Soc. 84, 1193 (1955), https://doi.org/10.1039/jr9550001193.Search in Google Scholar
[9] P. D. Gardner, C. E. Wulfman, C. L. Osborn. J. Am. Chem. Soc. 80, 143 (1958), https://doi.org/10.1021/ja01534a039.Search in Google Scholar
[10] K. Hafner, R. Fleischer, K. Fritz. Angew. Chem., Int. Ed. Engl. 4, 69 (1965), https://doi.org/10.1002/anie.196500692.Search in Google Scholar
[11] A. G. Anderson, A. A. MacDonald, A. F. Montana. J. Am. Chem. Soc. 90, 2993 (1968), https://doi.org/10.1021/ja01013a065.Search in Google Scholar
[12] H. Reel, E. Vogel. Angew. Chem., Int. Ed. Engl. 11, 1013 (1972), https://doi.org/10.1002/anie.197210131.Search in Google Scholar
[13] M. Nendel, B. Goldfuss, K. N. Houk, U. Grieser, K. Hafner. Theor. Chem. Acc. 102, 397 (1999), https://doi.org/10.1007/s002140050511.Search in Google Scholar
[14] K. Horii, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, A. Konishi, M. Yasuda. J. Am. Chem. Soc. 144, 3370 (2022), https://doi.org/10.1021/jacs.2c00476.Search in Google Scholar PubMed
[15] R. Munday, I. O. Sutherland. Chem. Commun. 569 (1967), https://doi.org/10.1039/C19670000569.Search in Google Scholar
[16] R. Munday, I. O. Sutherland. J. Chem. Soc. C 1427 (1969), https://doi.org/10.1039/J39690001427.Search in Google Scholar
[17] U. Grieser, K. Hafner. Chem. Ber. 127, 2307 (1994), https://doi.org/10.1002/cber.1491271132.Search in Google Scholar
[18] S. Das, J. Wu. Org. Lett. 17, 5854 (2015), https://doi.org/10.1021/acs.orglett.5b03028.Search in Google Scholar PubMed
[19] P. Baumgartner, E. Weltin, G. Wagnière, E. Heilbronner. Helv. Chim. Acta 48, 751 (1965), https://doi.org/10.1002/hlca.19650480410.Search in Google Scholar
[20] N. Guihery, D. Maynau, A. Jean-Paul Malrieu. New J. Chem. 22, 281 (1998), https://doi.org/10.1039/a708330k.Search in Google Scholar
[21] A. Konishi, Y. Okada, R. Kishi, M. Nakano, M. Yasuda. J. Am. Chem. Soc. 141, 560 (2019), https://doi.org/10.1021/jacs.8b11530.Search in Google Scholar PubMed
[22] A. Konishi, K. Horii, D. Shiomi, K. Sato, T. Takui, M. Yasuda. J. Am. Chem. Soc. 141, 10165 (2019), https://doi.org/10.1021/jacs.9b04080.Search in Google Scholar PubMed
[23] P. Liu, X. Chen, J. Cao, L. Ruppenthal, J. M. Gottfried, K. Müllen, X. Wang. J. Am. Chem. Soc. 143, 5314 (2021), https://doi.org/10.1021/jacs.1c01826.Search in Google Scholar PubMed
[24] X. Yang, X. Shi, N. Aratani, T. P. Gonçalves, K.-W. Huang, H. Yamada, C. Chi, Q. Miao. Chem. Sci. 7, 6176 (2016), https://doi.org/10.1039/c6sc01795a.Search in Google Scholar PubMed PubMed Central
[25] X. Fu, H. Han, D. Zhang, H. Yu, Q. He, D. Zhao. Chem. Sci. 11, 5565 (2020), https://doi.org/10.1039/d0sc00770f.Search in Google Scholar PubMed PubMed Central
[26] C. Zhen, S. Lu, M. Lin, J. Wu, I. Chao, C. Lin. Chem. Eur J. 27, 16682 (2021), https://doi.org/10.1002/chem.202102781.Search in Google Scholar PubMed
[27] W. T. Borden, Diradicals, W. T. Borden (Ed.), Wiley-Interscience: New York (1982).Search in Google Scholar
[28] A. Rajca. Chem. Rev. 94, 871 (1994), https://doi.org/10.1021/cr00028a002.Search in Google Scholar
[29] M. Abe. Chem. Rev. 113, 7011 (2013), https://doi.org/10.1021/cr400056a.Search in Google Scholar PubMed
[30] M. Hagel, J. Liu, O. Muth, H. J. Estevez Rivera, E. Schwake, L. Sripanom, G. Henkel, G. Dyker. Eur. J. Org. Chem. 3573 (2007), https://doi.org/10.1002/ejoc.20070017.Search in Google Scholar
[31] S. Arikawa, A. Shimizu, R. Shintani. Angew. Chem. Int. Ed. 58, 6415 (2019), https://doi.org/10.1002/anie.201902006.Search in Google Scholar PubMed
[32] K. Katayama, A. Konishi, K. Horii, M. Yasuda, C. Kitamura, J. Nishida, T. Kawase. Commun. Chem. 2, 136 (2019), https://doi.org/10.1038/s42004-019-0236-y.Search in Google Scholar
[33] B. Dittrich, F. P. A. Fabbiani, J. Henn, M. U. Schmidt, P. Macchi, K. Meindl, M. A. Spackman. Acta Crystallogr. B 74, 416 (2018), https://doi.org/10.1107/s2052520618010120.Search in Google Scholar
[34] A. G. Anderson Jnr, S. C. Critchlow, L. C. Andrews, R. D. Haddock. Acta Crystallogr. C 46, 439 (1990), https://doi.org/10.1107/s0108270189007857.Search in Google Scholar
[35] E. Vogel, H. Wieland, L. Schmalstieg, J. Lex. Angew. Chem. Int. Ed. Engl. 23, 717 (1984), https://doi.org/10.1002/anie.198407171.Search in Google Scholar
[36] J. Kruszewski, T. M. Krygowski. Tetrahedron Lett. 13, 3839 (1972), https://doi.org/10.1016/s0040-4039(01)94175-9.Search in Google Scholar
[37] T. M. Krygowski. J. Chem. Inf. Comput. Sci. 33, 70 (1993), https://doi.org/10.1021/ci00011a011.Search in Google Scholar
[38] R. Herges, D. Geuenich. J. Phys. Chem. A 105, 3214 (2001), https://doi.org/10.1021/jp0034426.Search in Google Scholar
[39] D. Geuenich, K. Hess, F. Köhler, R. Herges. Chem. Rev. 105, 3758 (2005), https://doi.org/10.1021/cr0300901.Search in Google Scholar PubMed
[40] K. Hafner, G. Hafner-Schneider, F. Bauer. Angew. Chem., Int. Ed. Engl. 7, 808 (1968), https://doi.org/10.1002/anie.196808081.Search in Google Scholar
[41] K. Nakasuji, E. Todo, I. Murata. Angew. Chem. Int. Ed. Engl. 16, 784 (1977), https://doi.org/10.1002/anie.197707841.Search in Google Scholar
[42] E. Todo, K. Yamamoto, I. Murata. Chem. Lett. 8, 537 (1979), https://doi.org/10.1246/cl.1979.537.Search in Google Scholar
[43] Z. Yoshida, M. Shibata, E. Ogino, T. Sugimoto. Tetrahedron Lett. 25, 3343 (1984), https://doi.org/10.1016/s0040-4039(01)81380-0.Search in Google Scholar
[44] K. Yamane, H. Yamamoto, M. Nitta. J. Org. Chem. 67, 8114 (2002), https://doi.org/10.1021/jo020136a.Search in Google Scholar PubMed
[45] A. Konishi, A. Morinaga, M. Yasuda. Chem. Eur J. 24, 8548 (2018), https://doi.org/10.1002/chem.201801915.Search in Google Scholar PubMed
[46] F. Wu, J. Ma, F. Lombardi, Y. Fu, F. Liu, Z. Huang, R. Liu, H. Komber, D. I. Alexandropoulos, E. Dmitrieva, T. G. Lohr, N. Israel, A. A. Popov, J. Liu, L. Bogani, X. Feng. Angew. Chem. Int. Ed. 61, e202202170 (2022), https://doi.org/10.1002/anie.202202170.Search in Google Scholar PubMed PubMed Central
© 2023 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- Preface for special issue of ICPOC-25 in Hiroshima
- Conference papers
- Control of chirality inversion kinetics of triple-helical metallocryptands
- Cooperativity in molecular recognition of feet-to-feet-connected biscavitands
- Bis-periazulene: remaining non-alternant isomer of pyrene
- Closed-shell and open-shell dual nature of singlet diradical compounds
- Anticancer activity and DNA interaction of bis(pyridyl)allene-derived metal complexes
- Reactivity of electrophilic cyclopropanes
- m-Quinodimethane-based fused-ring triplet hydrocarbons
- White light emission from an upconverted emission based on triplet-triplet annihilation with rose bengal as the sensitizer
- Fluorosumanenes as building blocks for organic crystalline dielectrics
- Recent advances in developing tetrathiafulvalene analogs of electrode materials: discovery of an in-cell polymerization technique
- The current landscape of author guidelines in chemistry through the lens of research data sharing
- Role of fiber density of amine functionalized dendritic fibrous nanosilica on CO2 capture capacity and kinetics