Rationale Anämieabklärung
-
Jan Hastka
 und
Georgia Metzgeroth
und
Georgia Metzgeroth

Zusammenfassung
Eine Anämie ist definiert als eine Verminderung der Hämoglobinkonzentration unter die alters- und geschlechtsspezifische Norm. Diese beträgt nach der WHO 120 g/L für Frauen und 130 g/L für Männer. Prinzipiell gibt es viele Differentialdiagnosen, die bei der Abklärung einer Anämie ursächlich berücksichtigt werden müssen. Die Diagnose wird zusätzlich dadurch erschwert, dass Anämien häufig nicht nur eine Ursache haben, sondern multifaktoriell bedingt sind. Eine rationale Anämieabklärung sollte immer die epidemiologischen Daten und die individuelle Anamnese berücksichtigen. Eine zentrale diagnostische Rolle spielt nach wie vor die Einteilung der Anämien nach der Größe und dem Hämoglobingehalt der Erythrozyten anhand der Erythrozytenindizes. Die weltweit wichtigste Ursache einer hypochrom-mikrozytären Anämie ist der Eisenmangel, differentialdiagnostisch sind die Anämie der chronischen Erkrankungen (anemia of chronic disorders, ACD) und Thalassämien zu berücksichtigen. Die klinisch wichtigste Ursache einer hyperchrom-makrozytären Anämie sind Störungen des Vitamin-B12- und Folsäurestoffwechsels, bzw. der DNA-Synthese. Die normochrom-normozytäre Gruppe beinhaltet die meisten Anämieformen. Bei deren Abklärung sollte man nicht versuchen alle möglichen Ursachen durch ein allesumfassendes Laborpanel bereits mit der ersten Blutentnahme zu erfassen. Es ist sinnvoller schrittweise vorzugehen und zunächst nur die wichtigsten Ursachen diagnostisch abzudecken. Dies gilt insbesondere für geriatrische und multimorbide Patienten, bei denen der diagnostischen Aufwand nicht nur aus wirtschaftlichen, sondern auch aus ethischen Gründen der individuellen Prognose und den Bedürfnissen des Patienten angepasst werden sollte. Bei ungeklärten Anämien sollte eine Vorstellung bei einem Hämatologen erwogen werden, weil im Zweifelsfall auch eine Knochenmarkpunktion erfolgen muss, um die Hämatopoese genau zu beurteilen und eine hämatologische Grunderkrankung auszuschließen.
Abstract
Anemia is defined as a decrease in the hemoglobin concentration below the age- and sex-matched lower limit (130 g/L in men and 120 g/L in women, WHO). When diagnosing anemia, a number of differential diagnoses must be considered. This is further complicated by the fact that anemias are often multicausal. A rational evaluation of anemia should therefore always take into account the epidemiological data and the individual patient’s history. A central part of the diagnostic process is a classification, based on the size and the hemoglobin content of the red blood cells. For hypochromic microcytic anemia the most important cause worldwide is iron deficiency, with anemia of chronic disorders (ACD) and thalassemia the main differential diagnoses. For hyperchromic macrocytic anemia, the clinically most important causes are disorders of vitamin B12 and folic acid metabolism. Apart from those, most types of anemias fall within the normochromic normocytic group. In these cases, one should not try to investigate all possible causes by carrying out a fully comprehensive laboratory panel on the first blood sample. Instead, it is more appropriate to proceed step-by-step and to evaluate first the most frequent and clinically most important causes. This applies especially to geriatric and multimorbid patients where the diagnostic effort should be adjusted to the individual needs of the patient as well as their prognosis, both from an economical and from ethical point of view. In unexplained anemias, a hematological opinion should be considered because a bone marrow biopsy may be required to accurately evaluate the hematopoiesis and to exclude a hematological disorder.
Rezensierte Publikation:
Nebe C.T.
Einleitung
Eine Anämie ist definiert als eine Verminderung der Hämoglobinkonzentration unter die alters- und geschlechtsspezifische Norm. Dabei verwenden größere Labors meist ihre eigenen Referenzwerte, die aus eigenen, regionalen Erhebungen ermittelt wurden. In den epidemiologischen Studien werden meist die von einer Expertengruppe der WHO im Jahre 1968 festgelegten Kriterien benutzt [1]. Danach ist die Hämoglobinkonzentration alters- und geschlechtsabhängig, die untere Hämoglobingrenze beträgt 120 g/L für Frauen und 130 g/L für Männer (Tabelle 1).
Untere Hämoglobingrenze in Abhängigkeit vom Alter und Geschlecht gemäß der WHO [1].
| Personen | Hämoglobin-Untergrenze, g/L |
|---|---|
| Kinder 6 Monate bis 6 Jahre | 110,0 |
| Kinder 6–14 Jahre | 120,0 |
| Männer | 130,0 |
| Frauen, nicht schwanger | 120,0 |
| Frauen, schwanger | 110,0 |
Natürlich hat die Anämie durch Beeinträchtigung des Herz-Kreislauf-Systems, der zerebralen Leistungsfähigkeit und der Lebensqualität einen Krankheitswert per se. Meistens handelt es sich jedoch nur um ein Symptom einer zugrundeliegenden angeborenen oder erworbenen Erkrankung, bzw. Störung. Dabei gibt es eine breite Differentialdiagnose, die man prinzipiell berücksichtigen muss. Angesichts der hohen Prävalenz der Anämie [2, 3] sollte die Abklärung mit Verstand erfolgen, um den Laboraufwand und die Kosten zu minimieren. Es wäre wünschenswert, die Diagnostik patientenadaptiert zu gestalten, dennoch sollte man nach einem Standardschema vorgehen, das bei Bedarf entsprechend modifiziert und erweitert wird.
Epidemiologie
In der klinischen Praxis beginnt das Standardschema mit der patientenorientierten Auswertung der epidemiologischen Daten, die man im Hinterkopf mit sich trägt. Häufiges ist häufig, Seltenes ist selten. Das gilt nicht nur für die hausärztliche Praxis, sondern auch für Krankenhäuser und mit Ausnahme der Spezialambulanzen sogar für Universitätskliniken.
Im Kindes- und Jugendalter sind Eisenmangelanämie und Thalassämie in Europa die mit Abstand häufigsten Anämieformen, die in ihrer Häufigkeit alle anderen Ursachen um mindestens eine Zehnerpotenz hinter sich lassen [1, 4, 5].
Bei den Erwachsenen kommt die Anämie meist durch eine Beeinträchtigung der Eisenversorgung der Erythropoese zustande. Bei prämenopausalen Frauen und bei Schwangeren ist es in erster Linie ein echter Eisenmangel, bei Personen >65 Jahre ist die Anämie der chronischen Erkrankungen (anemia of chronic disorders, ACD), die pathophysiologisch auf eine Eisenverwertungstörung zurückzuführen ist, mit rund 20% die häufigste Anämieform. Weitere häufige Anämieursachen des geriatrischen Patienten sind Mangelernährung und chronische Niereninsuffizienz [2, 6–11].
Anamnese
Wesentlicher Bestandteil einer rationalen Anämieabklärung ist die Anamnese. Bei Kenntnis der sozialen Situation, der Ernährungsgewohnheiten, der Familiengeschichte und der Vorerkrankungen lässt sich die Anämieursache in der Regel viel effektiver und kostengünstiger abklären. In diesem Zusammenhang ist auch die klinische Einschätzung der Anämiesymptome wichtig. Besteht auch klinisch eine Anämie, oder handelt es sich nur um ein Problem der Normwerte. Das beste Beispiel sind ältere Männer, bei denen physiologischerweise durch den abfallenden Testosteronspiegel das Hämoglobin absinkt, ohne dass es bei den WHO-Normwerten berücksichtigt wird. So ist ein Hämoglobinwert von 120 g/L bei einem 80-jährigen symptomfreien Mann in der Regel kein Grund für eine diagnostische Großaktion.
Labordiagnostik
Die ersten diagnostischen Schritte bei der Abklärung einer Anämie sind abhängig von dem jeweils verwendeten Labor-Routinepanel. In der Regel enthält dieses das komplette Blutbild, bestehend aus Hämoglobin, Erythrozytenzahl, Hämatokrit, den Erythrozytenindizes mittleres korpuskuläres Volumen (MCV) und mittleres korpuskuläres Hämoglobin (MCH), der Anzahl der Thrombozyten und Leukozyten, sowie dem Differentialblutbild. Retikulozyten müssen meist gesondert angefordert werden und liegen in der Regel bei der Erstvorstellung noch nicht vor.
Zunächst sollte man sich das Differentialblutbild anschauen. Bei Nachweis von Blasten liegt eine gravierende hämatologische Grunderkrankung vor, die einer sofortigen Einweisung, bzw. Überweisung zu einem Hämatologen bedarf. Auch bei einer Leukozytenzahl über 25×109/L sollte bei Fehlen anderer offensichtlicher Ursachen eine Überweisung an einen Spezialisten in die Wege geleitet werden, um eine hämatologische Neoplasie auszuschließen. Thrombozytenzahlen über 500×109/L können zwar auch reaktiv, z.B. im Rahmen eines Eisenmangels auftreten, bis zum Beweis des Gegenteils ist jedoch von einer myeloproliferativen Neoplasie auszugehen.
Bei einer isolierten Anämie richten sich die weiteren diagnostischen Maßnahmen nach der Größe und dem Hämoglobingehalt der Erythrozyten. In denjenigen Fällen, in denen bereits zu diesem Zeitpunkt die Retikulozytenzahl vorliegt, kann die Anämie in eine hyperregenerative (Retikulozyten >100/μL) und eine hyporegenerative Form unterteilt werden. Diese Aufteilung ist sehr wertvoll, weil eine hyperregenerative Anämie mit Ausnahme einer therapieassoziierten Regeneration eigentlich nur bei einer subakuten Blutung, bzw. bei einer hämolytischen Anämie vorkommt, die beide umgehend eine weiterführende Diagnostik, bzw. eine intensivere Betreuung benötigen.
Sind die Retikulozyten nicht erhöht, bzw. liegen sie noch nicht vor, so muss man sich zunächst mit den Erythrozytenindizes, dem MCV und MCH begnügen und eine Anämieklassifizierung in hypochrom-mikrozytär (MCH <27 pg; MCV <80 fL), normochrom-normozytär (MCH 27–34 pg; MCV 80–96 fL) und hyperchrom-makrozytär (MCH >34 pg; MCV >96 fL) vornehmen. Diese, seit „immer“ praktizierte Einteilung ist aus klinischer Sicht immer noch sinnvoll, weil sie dafür sorgt, dass man die wichtigsten Anämieformen, bzw. diejenigen Anämieformen, die man wegen deren möglichen Folgen nicht übersehen darf auch nicht übersieht (Abbildung 1). Eine hypochrom-mikrozytäre Anämie ist bis zum Beweis des Gegenteils auf einen Eisenmangel zurückzuführen und gehört entsprechend abgeklärt. Bei einem hyperchrom-makrozytären Blutbild ist zunächst von einem Vitamin-B12-/Folsäuremangel auszugehen und entsprechend zu handeln, um bleibende Schäden vom Patienten abzuwenden.
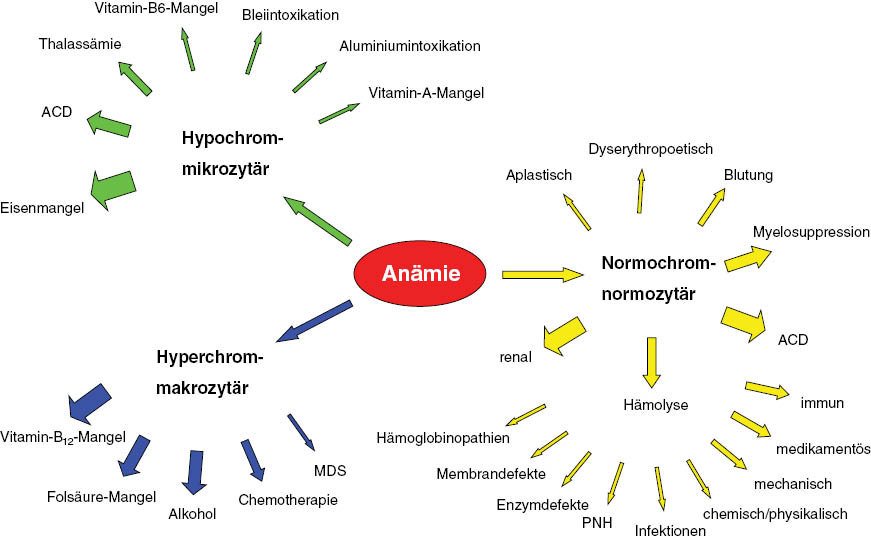
Aufteilung der Anämien nach den Erythrozytenindizes.
Hypochrom-mikrozytäre Anämie
Theoretisch gibt es einige mögliche Ursachen einer hypochrom-mikrozytären Anämie (Abbildung 1). In der klinischen Praxis spielen jedoch nur drei davon eine Rolle: die Eisenmangelanämie, die ACD und die Thalassämien. Was sind die typischen Merkmale dieser Anämieformen und wie kann man sie am einfachsten auseinanderhalten?
Eisenmangelanämie
Eine negative Eisenbilanz führt zunächst zu einem Speichereisenmangel bei noch optimaler Versorgung der Erythropoese. Nach Erschöpfung der Eisenspeicher geht der Eisenmangel in das Stadium der eisendefizitären Erythropoese über, bis schließlich nach Unterschreiten der Hämoglobinnormwerte die Eisenmangelanämie resultiert (Abbildung 2).
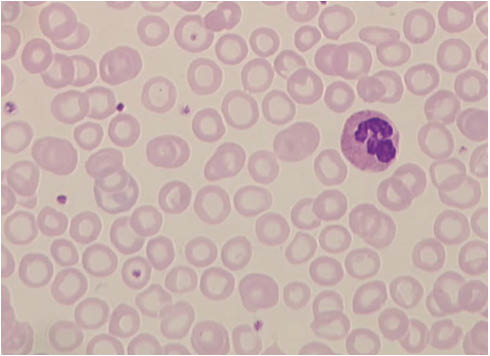
Ausstrich des peripheren Blutes bei einer Eisenmangelanämie.
Die Erythrozyten sind kleiner als normale rote Blutkörperchen. Durch Mangel an Hämoglobin ist die zentrale Aufhellung größer als normal, die meisten Erythrozyten kommen als Ringformen, sogenannte Anulozyten zur Darstellung.
Es gibt zahlreiche Eisenparameter, die zur Beurteilung des Eisenhaushaltes einer Person herangezogen werden können. Dabei ist immer zu berücksichtigen, dass die jeweiligen Parameter nicht den Eisenmangel per se diagnostizieren (Abbildung 3), sondern ein bestimmtes Stadium des Eisenmangels [12]. Ferritin ist ein Spiegel der Eisenspeicher, sagt jedoch nichts über die Eisenversorgung der Erythropoese aus. Dafür müssen andere Parameter eingesetzt werden, die die Eisenversorgung der Erythropoese überwachen. Dazu gehören das Zinkprotoporphyrin (ZPP), die löslichen Transferrinrezeptoren (sTfR), die hypochromen Erythrozyten (HYPO) und das Retikulozytenhämoglobin (CHr). Einen indirekten Hinweis auf eine eisendefizitäre Erythropoese liefert eine erniedrigte Transferrinsättigung von ≤15%. Die Hämoglobinbestimmung wird schließlich benötigt, um den Hämoglobinabfall unter die untere Normgrenze zu beweisen und dadurch den höchsten Schweregrad des Eisenmangels, die Eisenmangelanämie zu diagnostizieren [13].
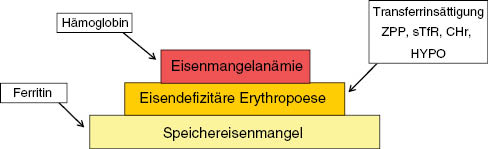
Laborparameter des Eisenstoffwechsels und deren Sensitivität.
Den besten Eisenparameter gibt es nicht. Alle haben ihre Daseinsberechtigung mit individuellen Stärken und Schwächen und müssen deshalb mit Verstand interpretiert werden. Die WHO empfiehlt als die beste Möglichkeit zur Beurteilung des Eisenstatus die Bestimmung von Ferritin im Serum [14]. Das ist von der Theorie her sicher richtig, denn Ferritin ist der einzige Laborparameter, der die Eisenspeicher widerspiegelt und somit den Eisenmangel bereits im Anfangsstadium erfasst. Ein Serumferritin <12 μg/L gilt als beweisend für ein Speichereisenmangel, vermutlich sind jedoch bereits Werte <22 μg/L mit einem klinisch relevanten Speichereisenmangel assoziiert [15]. Dabei gibt es allerdings einen Schönheitsfehler, da das Ferritin auch ein Akut-Phase-Protein ist, das bei entzündlichen Erkrankungen, aber auch bei Lebererkrankungen mitreagiert und deshalb insbesondere bei multimorbiden Patienten nur eingeschränkt verwertbar ist. Vorsicht ist in diesem Zusammenhang auch bei älteren Menschen geboten, weil das Altern mit einem subklinischen inflammatorischen Prozess einhergeht, der den Ferritinspiegel im Serum erhöhen und dadurch einen bestehenden Eisenmangel maskieren kann [16]. So wird generell empfohlen, bei einem normwertigen oder einem erhöhten Ferritin ein zusätzliches Akut-Phase-Protein zu untersuchen, um falsch normale Ferritinwerte auszuschließen [17]. In der Regel wird dabei das C-reaktive Protein (CRP) verwendet. Damit ist das Problem jedoch nicht ganz gelöst, da das Ferritin bei einer Entzündung eine andere Dynamik aufweist als das CRP, so dass auch durch diese Tandembestimmung ein Eisenmangel in etwa 15% nicht erkannt wird. So gibt es inzwischen Empfehlungen, außer dem CRP noch das saure α1-Glykoprotein als ein weiteres Akut-Phase-Protein zu bestimmen, wodurch die Diagnostik jedoch nicht billiger wird [18].
Angesichts der diagnostischen Unsicherheit des Ferritins braucht man insbesondere für multimorbide Patienten eine diagnostische Alternative. Diese wird von den Parametern der eisendefizitären Erythropoese geboten, die die Eisenversorgung der erythrozytären Vorstufen überwachen. Diagnostisch besonders wertvoll ist das ZPP, das bei einem Eisenmangel alternativ zu Häm entsteht, indem Zink statt Eisen in das Protoporphyrin IX eingebaut wird. Dabei werden zwangsläufig alle Störungen des Eisenstoffwechsels erfasst, nicht nur der echte Eisenmangel [19, 20]. Falsch erhöhte Werte werden dagegen nur bei der sehr seltenen kongenitalen erythropoetischen Porphyrie gemessen. Durch die Bestimmung des ZPP bekommt der Kliniker auf die Frage: „Hat die Anämie irgendetwas mit Eisen zu tun?“ eine klare Antwort: ja, bzw. nein. Normale Werte schließen eine Störung des Eisenstoffwechsels (bis auf einen reinen Speichereisenmangel) aus. Erhöhte Werte beweisen eine eisendefizitäre Erythropoese und erlauben auch eine Einschätzung deren klinischen Bedeutung. Eine Anämie tritt in der Regel erst bei einer Verdoppelung des Normwertes bei einem ZPP >80 μmol/mol Häm auf. Bei schwereren Anämien mit einem Hämoglobin <90 g/L liegt das ZPP meist >200 μmol/mol Häm, eine langanhaltende eisendefizitäre Erythropoese kann Werte bis 1000 μmol/mol Häm bewirken.
Um den Eisenmangel nicht nur hinreichend zu diagnostizieren, sondern wirklich zu beweisen, ist generell neben dem Ferritin die Bestimmung eines der Parameter der eisendefizitären Erythropoese zu empfehlen (Tabelle 2). Dabei haben HYPO, CHr und sTfR gegenüber dem ZPP einen wichtigen Vorteil, indem ihre Bestimmung automatisierbar ist. Sie sind jedoch nicht nur vom Eisenstoffwechsel abhängig, so dass bei deren Interpretation auch andere Faktoren berücksichtigt werden müssen. Das sind bei HYPO und CHr generell alle Faktoren, die die erythrozytäre Hämoglobinkonzentration beeinflussen, vor allem jedoch diejenigen, die ein hyperchromes Blutbild verursachen. Bei sTfR ist es insbesondere die Qualität und die Quantität der Erythropoese, die sich neben dem Eisenstoffwechsel in den gemessenen Serumwerten widerspiegelt [21]. Auch eine chronische lymphatische Leukämie (CLL) bewirkt abhängig von der Tumorlast einen Anstieg der sTfR Konzentration, obwohl kein Eisenmangel vorliegt [22].
Typische Konstellation der Eisenparameter in den verschiedenen Stadien des Eisenmangels.
| Stadium | I | II | III |
|---|---|---|---|
| Speichereisenmangel | eisendefizitäre Erythropoese | Eisenmangelanämie | |
| Ferritin | ↓ | ↓ | ↓ |
| Transferrinsättigung | n | ↓ | ↓ |
| CHr | n | ↓ | ↓ |
| HYPO | n | n/↑ | ↑ |
| ZPP | n | ↑ | ↑ |
| sTfR | n | ↑ | ↑ |
| Hämoglobin | n | n | ↓ |
Abhängig von der angestrebten Sensitivität und Spezifität wurden in der Vergangenheit für HYPO und CHr unterschiedliche cut-off Werte vorgeschlagen. Inzwischen werden als Nachweis einer eisendefizitären Erythropoese meist HYPO >5% und CHr <28 pg präferiert [23]. Bei sTfR ist die Situation etwas komplexer. Der Parameter wird mit verschiedenen Methoden gemessen, bei denen Kalibratoren mit unterschiedlicher Transferrinaffinität verwendet werden. Daraus resultieren testabhängige Referenzwerte, die zum Teil erheblich voneinander abweichen (z.B.: 0,4–1,8 mg/L bei Dade Behring und 0,7–4,2 mg/L bei Nichols Institute). Außerdem sind die Referenzwerte offensichtlich nicht immer an Personen ermittelt worden, deren Eisenstoffwechsel sorgfältig untersucht wurde, so dass das „Normalkollektiv“ auch Menschen mit Eisenmangel enthielt. Dies wird dadurch deutlich, dass zum Teil unterschiedliche Referenzwerte für Männer und Frauen angegeben werden, obwohl es bei Personen ohne Eisenmangel keine geschlechtsspezifischen Unterschiede in der sTfR Konzentration gibt. Außerdem wurden die Referenzwerte des Herstellers anhand von Untersuchungen eines Kollektivs mit genau definiertem Eisenstatus zum Teil revidiert [15, 24]. Um das Potential dieses diagnostisch überragenden Parameters voll ausschöpfen zu können ist es deshalb ratsam, die Qualität der angegebenen Referenzwerte anhand der Literatur zu überprüfen, oder sogar eigene Referenzwerte für den verwendeten sTfR-Test zu ermitteln.
Anämie der chronischen Erkrankungen
Die Anämie der chronischen Erkrankungen (ACD) kommt ebenfalls durch eine eisendefizitäre Erythropoese zustande. Im Unterschied zum echten Eisenmangel ist der Eisenmangel bei der ACD jedoch nur funktionell. Die Pathogenese der ACD ist komplex und multifaktoriell, im Vordergrund steht der Hepcidin-getriggerte Eisenentzug, der den Körper in einen funktionellen Mangelzustand versetzt. Das im Körper reichlich vorhandene Eisen wird im Sinne einer unspezifischen Abwehrmaßnahme im retikuloendothelialen System blockiert und vermindert freigesetzt und damit dem Pathogen bzw. dem entzündlichen Prozess – jedoch leider auch der Erythropoese – entzogen [25–27].
Alle entzündlichen oder malignen Prozesse können eine ACD induzieren. Sie müssen jedoch genügend lange bestehen bleiben, in der Regel mindestens 6–8 Wochen. Besonders erwähnenswert sind hierbei Autoimmunerkrankungen (z.B. Polymyalgia rheumatica, rheumatoide Arthritis, systemischer Lupus erythematodes), chronische Infektionen (z.B. Tuberkulose, Osteomyelitis, Endokarditis) und Tumorerkrankungen. Auch ein Teil der Anämie des Älteren ist eine, durch Zytokindysbalance bedingte ACD. Akute Entzündungen verursachen zwar auch eine Eisenblockade, diese ist jedoch angesichts der kurzen Krankheitsdauer und der langen Lebenszeit der Erythrozyten ohne Bedeutung.
Die ACD spielt in der klinischen Praxis eine wichtige Rolle. Bei hospitalisierten Patienten und bei älteren Menschen gilt sie als die häufigste Anämieform überhaupt [6, 7, 10]. Dabei ist jedoch zu betonen, dass nicht jede Anämie bei einer chronischen Erkrankung einer ACD entspricht, sondern nur solche, die pathophysiologisch auf eine zytokinvermittelte Störung der Eisenverwertung zurückzuführen sind. Deshalb sind die epidemiologischen Erhebungen über die ACD mit Vorsicht zu genießen, da die Diagnose in solchen Fällen meist nur auf einer Ferritin- und/oder CRP-Bestimmung basiert.
Ein eindeutiger Beweis einer ACD ist eigentlich nur durch eine Knochenmarkpunktion mit Eisenfärbung möglich, bei der man sowohl das ausreichend vorhandene Speichereisen, als auch die, durch die eisendefizitäre Erythropoese bedingte Abnahme der eisenhaltigen roten Vorstufen nachweisen kann. Eine Knochenmarkpunktion ist natürlich nicht immer möglich und sie ist auch in den meisten Fällen nicht nötig um eine ACD ausreichend sicher zu diagnostizieren.
Die Schlüsselaufgabe der ACD-Diagnostik besteht im Nachweis der eisendefizitären Erythropoese und Ausschluss eines echten Eisenmangels. Diese Aufgabe kann mit zwei Parametern sehr einfach gelöst werden, mit ZPP und sTfR. Das ZPP erfasst alle Störungen des Eisenmangels, auch diejenige bei der ACD [28]. Die Konzentration der sTfR ist dagegen nur bei einem echten Eisenmangel erhöht, bei einer ACD bleibt sie normwertig [29, 30]. Einen wertvollen Hinweis liefert auch die Höhe des ZPP, so werden Werte >150 μmol/mol Häm nur bei einer sehr schweren ACD beobachtet. Ein ZPP >200 μmol/mol Häm kommt bei einer ACD praktisch nie vor und spricht für eine Eisenmangelanämie. Übersetzt bedeutet es, dass die Minderversorgung der Erythropoese bei einer ACD nicht so ausgeprägt ist, wie bei einer echten Eisenmangelanämie. Dementsprechend ist die ACD in den meisten Fällen normochrom-normozytär. Erst nach einem langen, schweren Verlauf der chronischen Erkrankung entwickelt sich ein hypochrom-mikrozytäres Blutbild. Dabei werden jedoch MCV-Werte <70 fL praktisch nie erreicht.
Man kann natürlich ZPP und sTfR ignorieren und nur mit den klassischen Parametern arbeiten, in diesem Fall ist die Diagnostik jedoch schwieriger und auch nicht ganz eindeutig, insbesondere wenn das Blutbild nur grenzwertig mikrozytär ist. Eine erniedrigte Transferrinsättigung weist in solchen Fällen auf eine eisendefizitäre Erythropoese hin, ein CHr <28 pg beweist eine aktuelle Minderversorgung der Erythropoese und ein Anstieg der HYPO >5% zeigt, dass die Minderversorgung schon länger anhält. Das alles macht jedoch kein Unterschied zwischen einer Eisenmangelanämie und einer ACD. So muss man spätestens jetzt sTfR bestimmen, oder sich auf ein erhöhtes Ferritin verlassen. Das CRP hilft in dieser Situation nicht wirklich weiter. Ein erhöhtes CRP gehört zwar klassischerweise zu einer ACD, es erschwert aber gleichzeitig die Beurteilung des erhöhten Ferritins indem es signalisiert, dass das Ferritin auch falsch normal sein könnte. Oder anders gesagt, eine Eisenmangelanämie hätte bei einer akuten Entzündung genau die gleiche Laborkonstellation. Wenn man also ehrlich ist muss man zugeben, dass die ACD in der klinischen Praxis, bei Kenntnis der chronischen Grunderkrankung, bisher eher angenommen als wirklich eindeutig nachgewiesen wird. Diagnostisch hilfreich wäre sicherlich die Kenntnis des Serumspiegels von Hepcidin, der bei Eisenmangelanämie erniedrigt und bei ACD erhöht ist [31, 32]. Der Parameter wird inzwischen von einigen Labors angeboten und wird zukünftig bei dieser Differentialdiagnose vermutlich eine Schlüsselrolle spielen. Bisher ist er jedoch nicht standardisiert, so dass die ermittelten Werte abhängig von der verwendeten Methode (ELISA, Massenspektrometrie) und dem verwendeten Standard, bzw. von der jeweiligen Fähigkeit, neben dem bioaktiven Hepcidin-25 auch die Isoformen Hepcidin-20 und Hepcidin-22 zu erfassen, variieren [33].
Bei Patienten mit einer ACD und einem koexistierenden Eisenmangel, einer Konstellation wie sie insbesondere bei der rheumatoiden Arthritis nicht selten ist, war die Diagnostik mit den konventionellen Eisenparametern ganz problematisch und eigentlich nur durch eine Knochenmarkpunktion lösbar. Auch in diesen Fällen erwies sich sTfR als sehr hilfreich nachdem gezeigt wurde, dass man mit diesem Parameter einen echten Eisenmangel effektiv und zuverlässig identifizieren kann, auch bei einer chronischen Entzündung. Solange sTfR im Normbereich liegt, muss man bei Patienten mit chronisch entzündlichen Erkrankungen von einer „einfachen“ ACD ausgehen, bei einer erhöhten sTfR-Konzentration liegt dagegen eine ACD mit einem koexistierenden Eisenmangel vor [15, 24]. Diese differentialdiagnostische Bedeutung unterstreicht die Notwendigkeit eines sorgfältig ermittelten cut-off Wertes für eine zuverlässige Anwendung von sTfR bei der Abgrenzung von Patienten mit und ohne Eisenmangel.
Um in komplexen Anämiefällen die diagnostische Sicherheit von sTfR bei Nachweis, bzw. Ausschluß eines Eisenmangels zu steigern, wurde der TfR-F-Index in die Diagnostik eingeführt [15, 24]. Dieser Parameter entspricht einem Quotienten aus der Serumkonzentration von sTfR und dem Zehnerlogarithmus von Ferritin und kann anämische Patienten mit und ohne Eisenmangel unterscheiden, indem bei Auftreten eines Eisenmangels ein cut-off Wert überschritten wird. Wir präferieren weiterhin die individuelle Interpretation der Einzelparameter sTfR und Ferritin, nicht nur weil der Index zwei aussagekräftige Parameter zu einer imaginären Größe degradiert und weil wir in eigenen Untersuchungen die Überlegenheit des TfR-F Index nicht nachvollziehen konnten [22], sondern auch deshalb, weil aus obengenannten Gründen die cut-off Werte des TfR-F-Index testabhängig sind und zum Teil stark voneinander abweichen (R&D Systems: >1,5; Dade Behring: >1,5; Orion Diagnostica: >2,2; Nichols Institute: >3,5; Roche Diagnostics: >3,8), was die Diagnostik nicht gerade erleichtert. Bei den wirklichen differentialdiagnostischen Problemen des sTfR wie Hämolyse oder CLL, bietet der TfR-F-Index gegenüber der sTfR-Einzelbestimmung keinen diagnostischen Vorteil [34].
Um den Anteil der Erythropoese an der gemessenen Konzentration von sTfR einschätzen und differentialdiagnostisch berücksichtigen zu können, wurde in der „Thomas-Tafel“ der TfR-F Index mit CHr als einem zweiten, von der Aktivität der Erythropoese unabhängigen Parameter der eisendefizitären Erythropoese kombiniert [23]. Obwohl auch hier die obengenannten Einwände bezüglich des TfR-F-Index gelten und der cut-off Wert von TfR-F nicht nur testabhängig ist, sondern sich auch abhängig vom CRP verschiebt, erlaubt die 4-Felder-Tafel bei Abklärung einer Anämie eine grobe Orientierung, indem die Anämien ursachenabhängig vier verschiedenen Quadranten zugeordnet werden können. Quadrant 1 (TfR-F-Index <cut-off Wert, CHr ≥28 pg) beinhaltet Anämien mit einem normalen Hämoglobingehalt mit normaler, oder reduzierter Erythropoese (z.B. ACD, renale Anämie, hyporegenerative Anämien, Myelosuppression), während sich im Quadrant 2 (TfR-F-Index >cut-off Wert, CHr ≥28 pg) hyperregenerative Anämien und Eisenmangelanämien unter Substitutionstherapie, bzw. Fälle mit einer leichten eisendefizitären Erythropoese finden. Klassische Eisenmangelanämien landen im Quadrant 3 (TfR-F-Index >cut-off Wert, CHr <28 pg) und Anämien mit einer Eisenverwertungstörung und solche mit einer reduzierten Hämoglobinproduktion (z.B. schwere ACD, ACD kombiniert mit Eisenmangel, Thalassämien) im Quadrant 4 (TfR-F-Index <cut-off Wert, CHr <28 pg).
Thalassämien
Bei den Thalassämien ist das hypochrom-mikrozytäre Blutbild bedingt durch eine hereditäre Minderproduktion einer Hämoglobinkette mit konsekutiver Störung der Hämoglobinsynthese. Da das Hämoglobin ab dem siebten Lebensmonat physiologischerweise in 95%–98% aus HbA1 besteht, das aus zwei α- und zwei β-Ketten zusammengesetzt ist, spielen nur α- und β-Thalassämien in der klinischen Praxis eine Rolle. Dabei können das Krankheitsbild und auch die Laborparameter abhängig von der jeweiligen Genmutation, bzw. von der Anzahl der betroffenen Gene stark variieren.
Die β-Thalassaemia major (Abbildung 4) macht in der Regel keine diagnostischen Probleme. Sie ist gekennzeichnet durch eine schwere, transfusionsbedürftige, meist seit dem frühen Säuglingsalter bestehende hypochrom-mikrozytäre Anämie, die durch eine ausgeprägte Reduktion oder einen kompletten Verlust der β-Ketten zustande kommt.
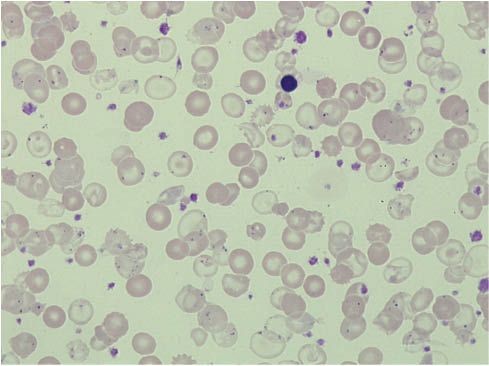
Ausstrich des peripheren Blutes bei einer transfusionsbedürftigen β-Thalassaemia major.
Man sieht reichlich hypochrome Anulozyten, deren Zellmembran nur leicht mit Hämoglobin beschlagen ist, sowie einzelne Targetzellen. In einigen Erythrozyten kommen als Folge der zeitlich zurückliegenden Splenektomie Howell-Jolly Körperchen zur Darstellung, außerdem reichlich eisenhaltige Pappenheimerkörperchen (Siderozyten), die auf eine Eisenverwertungstörung hindeuten. Zwischen den patienteneigenen Erythrozyten sieht man transfundierte Spendererythrozyten, einen Erythroblasten und zahlreiche Thrombozyten. Labor siehe Tabelle 4, Patient 14.
Wesentlich problematischer ist das Erkennen der β-Thalassaemia minor, bei der die Produktion der β-Ketten nur leicht reduziert ist, so dass häufig gar keine, oder nur eine leicht ausgeprägte Anämie besteht. Dennoch liegt ein hypochrom-mikrozytäres Blutbild vor, das zu dem praktisch normalen Hämoglobin nicht passt und so eine Differenzierung zu Eisenmangelanämie erlaubt. Ein weiterer Unterschied zu Eisenmangelanämie ist das meist normale MCHC (normal 32–36 g/dL) und die minimale Anisozytose (red cell distribution width, RDW <15%). Bestätigt wird die Erkrankung durch die Hämoglobinelektrophorese oder durch HPLC mit dem Nachweis eines erhöhten HbA2-Anteils (normal bis 3,2%). Dabei ist jedoch zu beachten, dass bei einem Eisenmangel das HbA2 selektiv weniger produziert wird, so dass ein Eisenmangel eine β-Thalassaemia minor maskieren kann. In diesem Zusammenhang ist das ZPP hilfreich, weil es eine wesentliche eisendefizitäre Erythropoese ausschließen und den HbA2-Befund untermauern kann. Die Bestimmung des ZPP ist jedoch bei Verdacht auf eine Thalassämie generell sinnvoll. Es ist im Unterschied zu Eisenmangelanämie bei einer Thalassaemia minor normal oder nur leicht erhöht und löst deshalb eigentlich die Differentialdiagnose bereits auf. Ein erhöhtes ZPP spricht bei einer Thalassaemia minor für einen zusätzlichen Eisenmangel, der natürlich auch bei dieser Erkrankung auftreten kann. Ferritin ist differentialdiagnostisch nicht hilfreich, da es zwar die Eisenspeicher überprüft, aber keine Aussage über die Eisenversorgung der Erythropoese erlaubt. Bei einer β-Thalassaemia intermedia, kann ein ZPP bis 100 μmol/mol Häm beobachtet werden. Diese Erhöhung kommt durch die deutlich gesteigerte, ineffektive Erythropoese zustande, indem die Systemkapazität für einen optimalen Eiseneinbau überschritten wird. Ähnliches wird auch bei anderen stark hyperregenerativen Anämien beobachtet.
Bei den α-Thalassämien ist die Situation etwas komplizierter. Als wichtigste Globinkette ist die Existenz der α-Kette durch vier α-Gene gesichert. Der Ausfall aller vier Gene ist mit dem Leben nicht vereinbar und endet mit Hydrops fetalis. Das Fehlen von 3 Genen führt zu einer chronischen hämolytischen Anämie mit einem Hämoglobin von 60–100 g/L, zu einer deutlichen Reduktion von MCV, MCH und MCHC, und zu einer ausgeprägten Anisozytose mit erhöhter RDW. Die Diagnose wird gesichert durch eine Hämoglobinelektrophorese oder durch HPLC mit dem Nachweis des aus vier β-Ketten bestehenden Hämoglobin H (HbH). Bei entsprechendem Verdacht kann man HbH-haltige Erythrozyten in einer Brillantkresylblau Supravitalfärbung durch ihre klassische Golfballmorphologie leicht nachweisen. Die Inkubation mit dem Farbstoff muss jedoch über 3–4 Stunden erfolgen.
Während die schweren Formen der α-Thalassämie kaum übersehen werden, ist der Verlust eines oder zweier α-Gene, ein Zustand der als α-Thalassämie-Merkmal bezeichnet wird, eine echte diagnostische Herausforderung. Die Patienten sind klinisch asymptomatisch und haben normale Hämoglobinwerte. Die Erythrozytenindizes sind jedoch meist erniedrigt, was häufig als Eisenmangel fehlinterpretiert wird. So sollte man grundsätzlich bei allen Personen, die einer entsprechenden ethnischen Gruppe angehören, normwertige Eisenparameter aufweisen und eine normale Hämoglobinelektrophorese und normales HbA2 haben, bei einem hypochrom-mikrozytären Blutbild bis zum Beweis des Gegenteils von einem α-Thalassämie-Merkmal ausgehen. Ein wirklicher Beweis des α-Thalassämie-Merkmal kann nur durch eine DNA-Analyse erfolgen, Hämoglobinelektrophorese und HPLC sind außerhalb der Neonatalperiode dagegen unauffällig.
Konsequenz für die Praxis
Welche Konsequenzen ergeben sich daraus für die klinische Praxis? Ein „All-inklusive-Panel“, das eine hinreichende Abklärung der Hauptursachen einer hypochrom-mikrozytären Anämie erlaubt, würde aus unserer Sicht folgende Parameter enthalten: Retikulozyten, Ferritin, Transferrinsättigung, ZPP, sTfR, CRP, ALAT, Hepcidin und die Hämoglobinelektrophorese. Natürlich ist es sinnvoll, dieses Panel patientengerecht zu adaptieren, bzw. zusammenzustreichen. So ist bei jungen Frauen mit einer hypochrom-mikrozytären Anämie die Bestimmung von Ferritin in der Regel diagnostisch ausreichend, eine Hämoglobinelektrophorese ist generell nur bei einem wirklichen Verdacht auf eine Hämoglobinanomalie, bzw. bei ungeklärten Anämien durchzuführen.
Als Parameter der eisendefizitären Erythropoese werden HYPO und CHr häufig dem ZPP vorgezogen, weil dessen Bestimmung bisher nicht automatisierbar ist. Wir bevorzugen ZPP trotzdem, weil es der einzige Parameter ist, der direkt die Ursache, nämlich die gestörte Eisenversorgung selektiv nachweist und nicht nur deren Folgen.
Hyperchrom-makrozytäre Anämie
Die Gruppe der hyperchrom-makrozytären Anämien umfasst hauptsächlich Anämieformen, die durch eine Beeinträchtigung der Zellteilung, in erster Linie durch Beeinträchtigung der DNA-Synthese zustande kommen. Die klinisch wichtigsten Ursachen sind der Vitamin-B12- und der Folsäuremangel [35–39]. Pathophysiologisch verwandt sind Medikament-assoziierte Anämien mit Störung des Folsäuremetabolismus, bzw. alle Anämien durch Medikamente, die die DNA-Synthese beeinträchtigen wie Zytostatika oder Immunsuppressiva. Bei nur minimaler Makrozytose mit einem MCV knapp über 100 fL muß man auch an eine alkoholtoxisch bedingte Anämie und insbesondere an eine hyperregenerative hämolytische Anämie denken, da die Retikulozyten deutlich größer sind als normale Erythrozyten und deshalb das MCV nach oben verschieben. Bei geriatrischen Patienten stellen die myelodysplastischen Syndrome (MDS) eine wichtige Differentialdiagnose dar. Auch bei diesen Erkrankungen wird häufig eine makrozytäre Anämie beobachtet, die durch eine Störung der DNA-Synthese und der Zellteilung zustande kommt. Vor dem 50. Lebensjahr ist das MDS jedoch eine Rarität.
Vitamin B12 und Folsäure
Die klinisch wichtigste Anämie dieser Gruppe ist auf eine Beeinträchtigung des Folsäure-/Vitamin-B12-Metabolismus zurückzuführen und wird wegen des charakteristischen Knochenmarkbefundes mit Vorherrschen von auffälligen roten Vorstufen, den sogenannten Megaloblasten, unter dem Oberbegriff megaloblastäre Anämie subsummiert. Die gestörte DNA-Synthese ist in diesen Fällen auf eine Beeinträchtigung der Thymidylat-Synthase mit einer verminderten Umwandlung von Desoxyuridinmonophosphat in Desoxythymidinmonophosphat zurückzuführen. Für diesen Reaktionsschritt wird Folsäure benötigt, die nur unter Mitwirkung von Vitamin B12 bereitgestellt werden kann, indem das Vitamin B12 die Folsäure demethyliert und diese dadurch erst funktionsfähig macht. Die abgespaltene Methylgruppe wird auf Homocystein übertragen, das dadurch zu Methionin umgewandelt wird. Die Serumbestimmung von Homocystein bietet somit eine einfache Möglichkeit zur Überprüfung dieses, sehr komplexen Stoffwechselweges. Bei einem Mangel oder einer sonstigen Störung von Vitamin B12 und/oder Folsäure ist das Homocystein im Serum erhöht (Abbildung 5). Bei nur grenzwertig erhöhten Werten muss man jedoch beachten, dass es auch andere Gründe für einen Anstieg von Homocystein gibt, in erster Linie Niereninsuffizienz, Alkoholabusus und insbesondere technische Probleme bei der Verarbeitung der Blutprobe [40]. Als die diagnostisch zuverlässigere Alternative gilt bei der Überprüfung des gesamten Vitamin-B12-Stoffwechsels die Messung der Plasmakonzentration der Methylmalonylsäure (MMA), die Bestimmung der MMA ist sensitiver und spezifischer als die von Homocystein. Sie ist jedoch auch deutlich teurer und nicht überall verfügbar, so dass sie in der klinischen Praxis nur bei diagnostischen Problemfällen zum Einsatz kommt. Wie bei Homocystein, kann auch die Konzentration der MMA bei einer Niereninsuffizienz falsch erhöht sein [40].
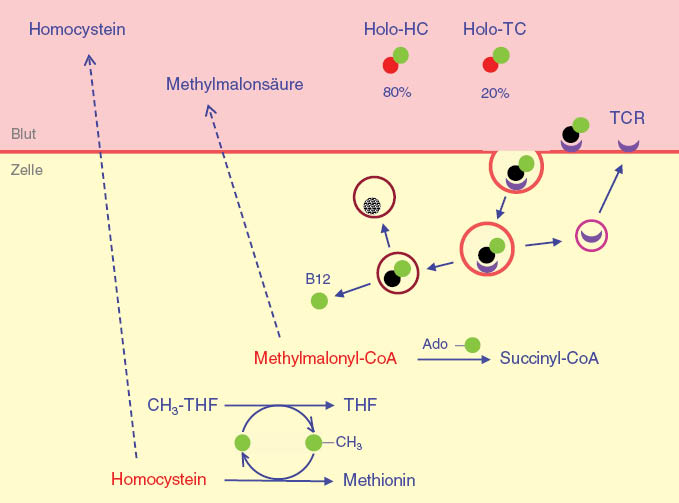
Das Vitamin B12 des peripheren Blutes ist zu etwa 80% an ein Glykoprotein aus der Gruppe der Transcobalamine, das Haptocorrin (früher Transcobalamin I) gebunden.
Dieser Komplex wird als Holo-Haptocorrin (Holo-HC) bezeichnet und ist biologisch inaktiv. Seine Aufgabe besteht vermutlich im Rücktransport des überschüssigen Vitamin B12 zur Leber. Für die Versorgung der Zellen ist alleine das Holo-Transcobalamin II (Holo-TC) zuständig, das im peripheren Blut nur einen Anteil von etwa 20% aufweist. Holo-TC bindet an einen spezifischen Transcobalamin-II-Rezeptor (TCR) und wird zusammen mit diesem in einem Vesikel in die Zelle aufgenommen. Nach Aufspaltung dieser Verbindung wird der TCR recycled, Transcobalamin II lysosomal abgebaut und das Vitamin B12 der Zelle zur Verfügung gestellt. Bei einem intrazellulären Mangel an funktionsfähigem Vitamin B12 sind die Vitamin-B12-abhängigen Reaktionen beeinträchtigt. Aus diagnostischer Sicht ist der gestörte Abbau von Homocystein und Methylmalonyl-CoA von Bedeutung, weil man durch Bestimmung dieser Parameter im peripheren Blut die Funktionsfähigkeit des gesamten Vitamin-B12-Stoffwechsels überprüfen kann.
In der klinischen Praxis wird die Serumbestimmung der einzelnen Parameter der globalen Überprüfung des gesamten Systems bevorzugt. Bei Vitamin B12 wird traditionell ein cut-off Wert von 200 ng/L verwendet nachdem in mehreren Studien gezeigt werden konnte, dass 60%–80% der Personen mit einem Spiegel <200 ng/L an einem klinisch valenten Vitaminmangel leiden [40]. Höhere Werte von Vitamin-B12 schließen jedoch einen klinisch valenten Vitaminmangel nicht aus. Dies hängt damit zusammen, dass rund 80% des im Blut zirkulierenden Vitamins in einem biologisch inaktiven Komplex, gebunden an Haptocorrin vorliegt. Nur eine kleine, an Transcobalamin II gebundene Fraktion, das sogenannte Holo-Transcobalamin (Holo-TC) ist biologisch aktiv und in der Lage, Zellen mit Vitamin B12 zu versorgen. Die Bestimmung des Holo-TC ist inzwischen kommerziell verfügbar und wird insbesondere für die Frühphase eines Vitamin B12 Mangels angeboten und propagiert. Die Messung ist jedoch auch bei einer ungeklärten hyperchrom-makrozytären Anämie sinnvoll, um die Versorgung der Erythropoese mit Vitamin B12 zu überprüfen. Der Referenzbereich für das Holo-TC beträgt 40–200 pmol/L, bei Werten darunter geht man von einem Mangel an bioaktivem Vitamin B12 aus [41].
Im Fall der Folsäure ist zu beachten, dass die Konzentration der Folsäure im Serum nur die Situation der letzten 1–2 Wochen widerspiegelt und eine, durch Folsäuremangel bedingte Anämie nicht ausschließt. Dazu muss die Folsäure in den Erythrozyten untersucht werden, die entsprechend der Lebensdauer der Erythrozyten eine Beurteilung der letzten 2–3 Monate erlaubt. Wenn man weiß, dass die Folsäure in den Erythrozyten 95% der gesamten Folsäure im Blut ausmacht ist es klar, dass auch eine minimale Hämolyse die Folsäurekonzentration im Serum stark verfälschen kann.
Der morphologische Befund des Vitamin-B12-/Folsäuremangels im Knochenmark mit stark gesteigerter, linksverschobener, ineffektiver Erythropoese spiegelt sich in den Hämolyseparametern wider. Die LDH ist deutlich erhöht und liegt meist um 1000 U/L, das Bilirubin ist leicht erhöht mit Werten um 2 mg/dL. Der Mangel an Vitamin B12 und Folsäure betrifft jedoch nicht nur die Erythropoese, sondern die gesamte Hämatopoese. So gehört auch eine Leukozytopenie mit hypersegmentierten Neutrophilen und eine Thrombozytopenie die auch Werte unter 50×109/L erreichen kann zum Bild eines schweren Vitamin-B12-, bzw. eines Folsäuremangels.
Angesichts des sehr komplexen Stoffwechsels (Abbildung 6) kann ein Vitamin-B12-Mangel viele Ursachen haben. In erster Linie ist eine perniziöse Anämie auszuschließen, eine Autoimmunerkrankung mit Typ-A-Gastritis und Antikörpern gegen Intrinsicfaktor und Parietalzellen. Bei der Perniziosa treten gehäuft Magenkarzinome auf, so dass die Patienten einer engmaschigen gastroenterologischen Überwachung bedürfen. Ein Mangel von Vitamin B12 kann jedoch auch bei anderen Resorptionsstörungen auftreten (atrophische Gastritis, Magenresektionen, exokrine Pankreasinsuffizienz, Darmresektionen, Vitamin-B12-verbrauchende Darmbakterien, Fischbandwurm, Morbus Crohn, Sprue, Zollinger-Ellison-Syndrom, Kalziummangel). Andere Ursachen sind unzureichende Zufuhr (Veganer, Alkoholiker, Ziegenmilch), erhöhter Bedarf (Schwangerschaft, Hämolyse, Neoplasien, Hyperthyreose), Medikamente (Colchicin, Neomycin, Metformin), Inaktivierung durch Lachgas, oder angeborene Probleme (Intrinsicfaktor-Anomalien, Transcobalamin-Mangel, Immerslund-Gräsbeck).
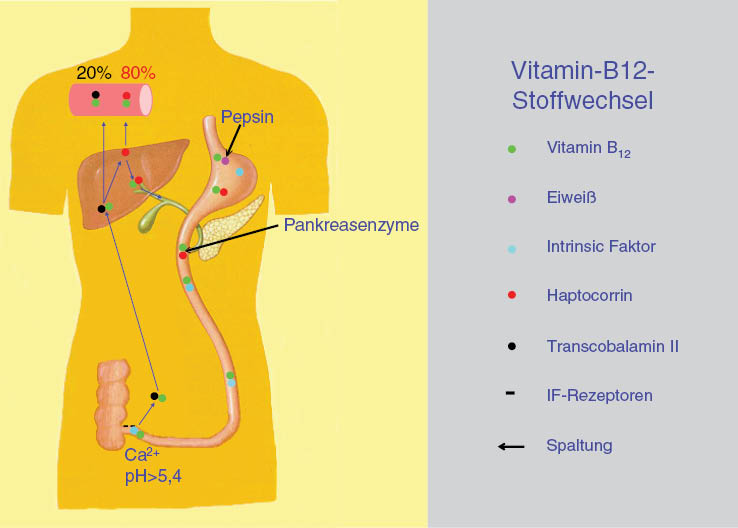
Schematische Darstellung des Vitamin-B12-Stoffwechsels.
Das Vitamin B12 wird mit tierischen Nahrungsmitteln aufgenommen, bei funktionierender Verdauung im Magen freigelegt und an das Haptocorrin gebunden. Im Duodenum wird diese Verbindung durch Pankreasenzyme aufgespalten und das Vitamin B12 auf den Intrinsicfaktor umgeladen, unter dessen Schutz es das terminale Ileum erreicht. Hier erfolgt bei einem pH>5,4 und Anwesenheit von Ca2+ die Bindung an Intrinsicfaktor-Rezeptoren und die Resorption. Das resorbierte Vitamin B12 wird zunächst vom Transcobalamin II aufgenommen. Der Großteil des Vitamins wird jedoch in der Leber auf Haptocorrin umgeladen, mit dem es über die Galle in das Duodenum ausgeschieden wird und einen enterohepatischen Kreislauf begründet. Diesem enterohepatischen Kreislauf ist es zu verdanken, dass der Organismus in der Lage ist, eine komplett fehlende Vitamin-B12-Zufuhr über 3–6 Jahre aus dem geringen physiologischen Speicher von 2–5 mg zu kompensieren.
Bei einem Folsäuremangel steht ursächlich neben einer unzureichenden Zufuhr insbesondere der erhöhter Bedarf im Vordergrund (Schwangerschaft, Hämolyse, Neoplasien, Stress), sowie Medikamente (Barbiturate, Antikonvulsiva, Kontrazeptiva, Sulfasalazin) und insbesondere die therapeutisch eingesetzten Folsäureantagonisten (Methotrexat, Pemetrexed, Sulfonamide, Trimethoprim, Pyrimethamin, Triamteren). Angeborene Ursachen (Dihydrofolatreduktase-Mangel, Formiminotransferase-Mangel) sind dagegen eine Rarität.
Myelodysplastische Syndrome
Die myelodysplastischen Syndrome (MDS) stellen eine heterogene Gruppe von Erkrankungen dar, die vorwiegend bei älteren Menschen diagnostiziert werden [42]. Der Häufigkeitsgipfel liegt zwischen dem 70. und 80. Lebensjahr, vor dem 50. Lebensjahr ist das MDS selten. Das gemeinsame Merkmal dieser Erkrankungen ist eine ineffektive Hämatopoese, die eine oder mehrere Zelllinien betrifft. Das Knochenmark ist meist normo-, oder sogar hyperzellulär, im peripheren Blut findet man dagegen Zytopenien. Das klinische Bild hängt von der betroffenen Zelllinie, bzw. vom Ausmaß der Zytopenie ab. Das Vollbild der Erkrankung ist durch eine ausgeprägte Panzytopenie mit Transfusionsabhängigkeit, Infektanfälligkeit und Blutungsneigung gekennzeichnet, in 20%–30% der Fälle beobachtet man eine Transformation in eine akute myeloische Leukämie.
Die meisten MDS Patienten präsentieren sich mit einer mehr oder weniger ausgeprägten Anämie, diese ist typischerweise normochrom und normo- oder makrozytär. Die Zahl der Retikulozyten kann normal oder vermindert sein. Das gilt auch für sTfR, deren Serumkonzentration trotz der gesteigerten Erythropoese infolge deren schlechten Ausreifung meist normal, oder sogar erniedrigt ist. Eine besondere Entität des MDS ist das sogenannte 5q- Syndrom, das zytogenetisch eine Deletion des kurzen Armes am Chromosom 5 aufweist. Die Erkrankung betrifft vorwiegend ältere Frauen und ist durch eine meist isolierte, makrozytäre Anämie gekennzeichnet.
Konsequenz für die Praxis
Bei Vorliegen einer hyperchrom-makrozytären Anämie sind in der klinischen Praxis zunächst ein Vitamin-B12- und ein Folsäuremangel auszuschließen. Das eingesetzte Laborpanel sollte dabei zumindest Retikulozyten, LDH, Bilirubin, Serumspiegel von Vitamin B12 und die Erythrozyten-Folsäure beinhalten. Um auch komplexere Störungen aufzudecken empfehlen wir, diese Laborparameter um das Homocystein im Serum zu erweitern. Bei einem Vitamin-B12-Mangel muss eine perniziöse Anämie nachgewiesen bzw. ausgeschlossen werden (Gastroskopie, Antikörper). Bleibt die Ursache einer makrozytären Anämie unklar und lässt sie sich auch anamnestisch durch Ernährungsgewohnheiten, Komorbiditäten und Medikation nicht klären, so ist wegen des nötigen, nicht unerheblichen diagnostischen Aufwandes eine Überweisung zum Hämatologen zu empfehlen.
Besondere Beachtung verdient eine makrozytäre Anämie bei Personen >50 Jahre. Lässt sich das Blutbild bei dieser Patientengruppe nicht durch einen Mangel an Vitamin B12 und/oder Folsäure erklären und durch entsprechende Substitution normalisieren, so muss in erster Linie an ein MDS gedacht werden. Auch in diesem Fall sollte eine Überweisung zum Hämatologen erfolgen, da bereits in einem Verdachtsfall eine zeitnahe Knochenmarkpunktion inklusive Zytologie, Histologie, Durchflußzytometrie, Zytogentik und Molekulargenetik angebracht ist.
Normochrom-normozytäre Anämie
Im Unterschied zu den hypochrom-mikrozytären und hyperchrom-makrozytären Anämien, deren Abklärung meist relativ einfach ist, stellen die normochrom-normozytären Anämien den Kliniker häufig vor eine wirkliche diagnostische Herausforderung. Das hängt einerseits damit zusammen, dass zu dieser Gruppe die meisten Anämien zählen. Es ist jedoch auch dadurch bedingt, dass Anämien in der Praxis häufig nicht nur eine Ursache haben, sondern in rund 30% multifaktoriell bedingt sind [9]. Durch Überlagerung mehrerer Komponenten können klassische Anämieformen maskiert werden, so dass hinter einem normochrom-normozytären Blutbild auch ein Eisenmangel, bzw. ein Vitamin-B12- oder ein Folsäuremangel stecken können.
Es gibt eine diagnostisch wichtige Frage, die man gleich zu Beginn der Abklärung dieser Anämiegruppe beantworten sollte: Ist die Anämie hyper- oder hyporegenerativ? Klassischerweise wird diese Frage mit den Retikulozyten beantwortet. Hyperregenerative Anämien sind durch Retikulozytenzahlen >100/μL gekennzeichnet, bei den hyporegenerativen Formen liegen die Werte darunter. Als Ursache einer hyperregenerativen Anämie kommen außer einer gesteigerten Erythropoese nach, bzw. unter Therapie (Chemotherapie, Erythropoetinsubstitution, Vitamin-B12- oder Eisensubstitution) nur eine subakute Blutung, sowie eine Hämolyse in Frage. Die subakute Blutung bedarf einer umgehenden internistischen und insbesondere gastroenterologischen Abklärung.
Die Verdachtsdiagnose einer Hämolyse wird untermauert durch eine Erhöhung der LDH und des indirekten Bilirubins, sowie durch ein erniedrigtes Haptoglobin. Bei Vorliegen einer Hämolyse sollte man sich zunächst überlegen und anamnestisch abklären, ob eine angeborene Störung des Erythrozyten (Störung der Hämoglobinsynthese, Enzymdefekt, Membrandefekt) zugrunde liegen könnte (Abbildung 7). In diesem Fall folgt nach Beurteilung des peripheren Blutausstriches die Hämoglobinelektrophorese, die Messung der Erythrozytenenzyme (Glucose-6-Phosphat-Dehydrogenase, Pyruvatkinase), die Bestimmung der osmotischen Resistenz und der EMA-Test (Sphärozytose). Diese Spezialuntersuchungen werden insbesondere wegen des finanziellen Aufwandes in der Regel durch einen Hämatologen veranlasst.
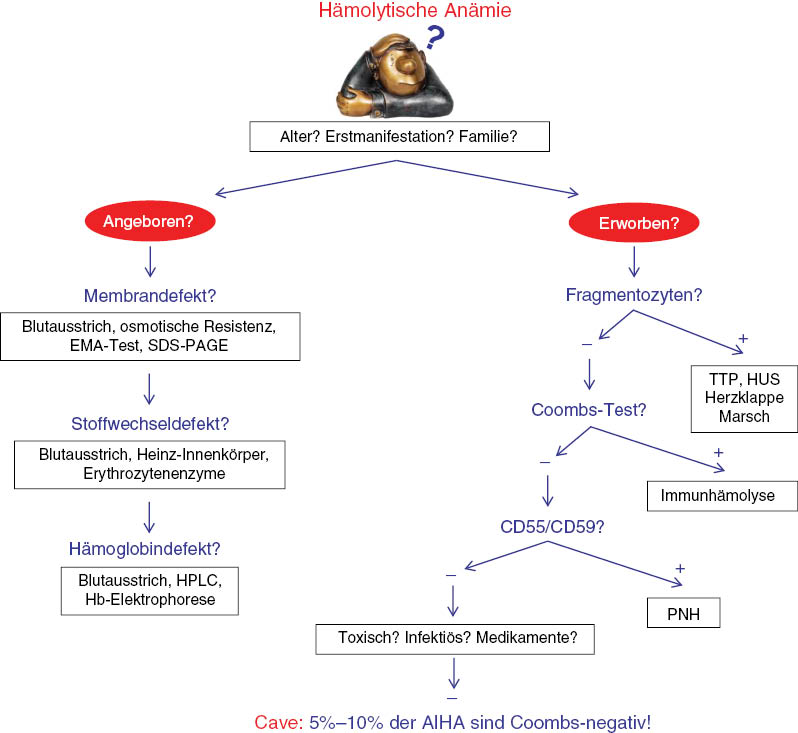
Abklärung einer hämolytischen Anämie.
Ist eine angeborene Anämie als Ursache der Hämolyse unwahrscheinlich, sollten zunächst Fragmentozyten und damit eine mikroangiopathische Anämie bei einer Thrombotisch-thrombozytopenischen Purpura (TTP) oder bei einem Hämolytisch-urämischen Syndrom (HUS) ausgeschlossen werden, weil sie einen therapeutischen Notfall darstellt der sofort durch eine Plasmapherese behandelt werden muss [43, 44]. An diese Diagnose ist insbesondere dann zu denken, wenn gleichzeitig eine Thrombozytopenie vorliegt und wenn es sich um junge Patienten handelt. Bei entsprechendem Verdacht wird die Aktivitätsbestimmung der Metalloprotease ADAMTS13 empfohlen [45].
Nach Ausschluss der mikroangiopathischen Anämie folgt der Coombs-Test zur Abklärung einer Immunhämolyse [46]. Hier sollte man wissen, dass 5%–10% der immunhämolytischen Anämien einen negativen Coombs-Test aufweisen [47]. Dies kann verschiedene Ursachen haben, im Allgemeinen ist es jedoch dadurch bedingt, dass die Autoantikörper in diesen Fällen effektiver sind als das verwendete Coombs-Serum, das erst bei einer Beladung von 500 IgG-Molekülen pro Erythrozyt reagiert. Insbesondere IgG3- und IgA-Autoantikörper verursachen bereits bei deutlich niedrigerer Beladung eine Hämolyse, auch niedrigaffines IgG kann durch Komplementaktivierung eine Hämolyse verursachen, die von einem klassischen Coombs-Test nicht erfasst wird.
Bevor man an ganz seltene Ursachen einer Hämolyse denkt, wie die mechanische Hämolyse (Marschhämolyse, Herzklappe), Hämolyse bei Infektionserkrankungen (Malaria, Gasbrand) oder bei toxischer Belastung (Chemikalien, Medikamente, tierische Gifte), sollte man eine durchflußzytometrische Untersuchung des peripheren Blutes zum Ausschluß einer monoklonalen lymphatischen Population und damit einer hämatologischen Grunderkrankung veranlassen. Auch die Diagnose der einzigen erworbenen korpuskulären hämolytischen Anämie, der Paroxysmalen nächtlichen Hämoglobinurie (PNH), die durch eine anfallsweise auftretende Hämolyse mit Dunkelfärbung des Morgenurin, aber auch durch eine Thrombophilie gekennzeichnet ist, ist inzwischen eine Domäne der Durchflußzytometrie. Der Defekt der Glycosylphosphatidylinositol (GPI)-verankerten Oberflächenmoleküle CD55 und CD59 kann nicht nur an den Erythrozyten, sondern auch an den Neutrophilen nachgewiesen werden, so dass die Diagnose auch nach einer stattgehabten Hämolyse, nachdem der erythrozytäre PNH-Klon weitgehend verschwunden ist, an den Neutrophilen gestellt werden kann [48, 49].
Nach Ausschluss einer hyperregenerativen Anämie bleibt eine breite Differentialdiagnose an hyporegenerativen Formen bestehen, in der kombinierte Anämien, eine nicht ganz lange bestehende ACD, eine Myelosuppression und insbesondere die renale Anämie dominieren. Bei der Abklärung solcher Fälle ist die Bestimmung von sTfR von diagnostischer Bedeutung, insbesondere wenn sie parallel mit ihrem natürlichen diagnostischen Partner ZPP erfolgt [50]. Die Konzentration der sTfR ist abhängig einerseits vom Eisenstoffwechsel, andererseits jedoch von der Quantität und Qualität der Erythropoese. Erhöhte sTfR werden nicht nur bei einem echten Eisenmangel, sondern auch bei einer gesteigerten Erythropoese gemessen. Eine Tandembestimmung von sTfR und ZPP liefert somit wertvolle Hinweise für die Differentialdiagnose einer Anämie, insbesondere wenn sie in Kombination mit der Bestimmung des Erythropoetinspiegels erfolgt. Man kann dadurch nicht nur eine eisendefizitäre Erythropoese nachweisen bzw., ausschließen, sondern auch eine ACD diagnostizieren, die in der Regel normochrom-normozytär beginnt. Bei einem normalen ZPP ist eine Störung des Eisenstoffwechsels ausgeschlossen. In diesen Fällen kann man anhand der Konzentration des sTfR direkt die Quantität und Qualität der Erythropoese ablesen (Tabelle 3). Eine hyperregenerative Anämie bei einer Hämolyse ist mit einem erhöhten sTfR vergesellschaftet, erhöhte Werte werden auch bei den megaloblastären Anämien beobachtet, die noch relativ gut ausreifen. Myelodysplastische Syndrome weisen dagegen trotz einer gesteigerten Erythropoese normale oder erniedrigte Konzentrationen von sTfR auf, weil die Erythropoese stark linksverschoben ist und schlecht ausreift und weil die unreiferen roten Vorstufen deutlich weniger Transferrinrezeptoren tragen als die reiferen Erythroblasten. Hypoplasien und Aplasien der Erythropoese sind an einem deutlich erniedrigten sTfR zu erkennen. Das betrifft therapeutisch bedingte Zustände nach einer Chemotherapie, aber auch die Pure red cell aplasia (PRCA), die durch das praktisch fehlende sTfR bei einem gleichzeitig extrem hohen Erythropoetinspiegel gekennzeichnet ist (Tabelle 4).
Differentialdiagnose der Anämien mit ZPP und sTfR [50].
| ZPP | sTfR | Diagnose |
|---|---|---|
| normal | normal | keine IDE, EP normal |
| ↑ | ↑ | Eisenmangel |
| ↑ | normal | ACD |
| normal | ↑ | keine IDE, EP gesteigert |
| normal | ↓ | keine IDE, EP reduziert |
IDE, eisendefizitäre Erythropoese; EP, Erythropoese; ACD, Anämie der chronischen Erkrankungen.
Beispiele von individuellen Patienten mit verschiedenen Anämieformen mit den jeweiligen Laborparametern. Differentialdiagnostisch interessante Partner sind insbesondere das Zinkprotoporphyrin (ZPP), die löslichen Transferrinrezeptoren (sTfR) und das Erythropoetin im Serum (EPO), weil sie eine Beurteilung des Eisenstoffwechsels, sowie der erythropoetischen Aktivität im Knochenmark erlauben.
| Nr. | Diagnose | Hb g/L | MCV 80–98 fL | Reti 25–102 ×109/L | CHr 28–35 pg | Ferritin 16–252 μg/L | ZPP <40 μmol/molHäm | STfR 0,8–1,8 mg/L | EPO 5–26 U/L | Sonstiges |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Speichereisenmangel | 134 | 83 | 60 | 32 | 12 | 39 | 1,4 | 12 | Blutverlust bei Hämorrhoidalblutung |
| 2 | Eisendefizitäre EP | 121 | 79 | 31 | 28 | 7 | 125 | 2,1 | 9 | Hypermenorrhoe |
| 3 | Eisenmangelanämie | 71 | 78 | 21 | 23 | 5 | 297 | 4,2 | 11 | Hypermenorrhoe |
| 4 | Eisenmangelanämie, therapiert | 142 | 92 | 78 | – | 1 | 29 | 1,5 | – | seit 3 Monaten Fe2+ p.o., noch Speichereisenmangel |
| 5 | Eisenmangelanämie, hereditär | 50 | 54 | 35 | 11 | – | 1650 | – | – | Polymorphismus im Transferringen |
| 6 | ACD bei Polymyalgia rheumatica | 98 | 84 | 56 | 27 | 657 | 156 | 1,2 | 11 | Hb hat sich unter Cortison-Therapie normalisiert |
| 7 | ACD+Eisenmangel | 76 | 75 | 42 | – | 105 | 189 | 2,5 | – | Rheumatoide Arthritis |
| 8 | Vitamin B12-Mangel | 78 | 106 | 20 | 39 | 697 | 82 | 10,0 | 94 | LDH 1773 U/L; B12 32 ng/L |
| 9 | AIHA vom Wärmetyp | 88 | 105 | 706 | 34 | – | – | 5,5 | – | Bilirubin 5,5 mg/dL; Haptoglobin <0,07 g/L |
| 10 | AIHA vom Kältetyp | 112 | 103 | 178 | 40 | 78 | 60 | 5,1 | 37 | Bilirubin 5,1 mg/dL; Haptoglobin <0,07 g/L |
| 11 | AIHA, Coombs-negativ | 99 | 88 | 118 | 36 | 231 | 78 | 3,7 | 40 | Hb hat sich unter Cortisontherapie normalisiert |
| 12 | α-Thalassämie-Merkmal | 119 | 70 | 49 | 24 | 261 | 47 | 1,5 | 12 | Patientin asymptomatisch |
| 13 | α-Thalassämie, HbH-Krankheit | 83 | 81 | 415 | 22 | 315 | 85 | 9,0 | 97 | Bilirubin 2,4 mg/dL; LDH 690 U/L |
| 14 | β-Thalassaemia major | 81 | 67 | 392 | 18 | 4311 | 105 | 13,0 | 115 | Splenektomie; polytransfundiert; Patient Abb. 4 |
| 15 | β-Thalassaemia minor | 115 | 62 | 49 | 20 | 420 | 47 | 2,1 | 15 | keine Transfusionen |
| 16 | β-Thalassämie+Eisenmangel | 132 | 65 | 49 | – | 15 | 38 | 1,89 | – | Thalassaemia minor+Speichereisenmangel |
| 17 | Pyruvatkinase-Mangel, hereditär | 107 | 114 | 630 | 40 | 2213 | 36 | 4,0 | 21 | Splenektomie; keine Transfusionen;Bilirubin 4,8mg/dL |
| 18 | Renale Anämie | 88 | 87 | 44 | 29 | 466 | 55 | 0,47 | 8 | Kreatinin 1,9 mg/dL, GFR 36 mL/min |
| 19 | Aplastische Anämie | 37 | 119 | 5 | 42 | 521 | 85 | 0,48 | 6039 | Thrombozyten 6×109/L, Leukozyten 3,4×109/L |
| 20 | Pure red cell aplasia | 51 | 94 | 6 | – | 238 | 36 | 0,28 | 1227 | Hb hat sich unter Cortisontherapie normalisiert |
| 21 | MDS, 5q- Syndrom | 96 | 84 | 19 | – | 979 | 38 | 0,34 | 1788 | polytransfundiert |
| 22 | MDS, 5q- Syndrom | 143 | 87 | 69 | 34 | 1182 | 42 | 0,99 | 19 | Patientin Nr. 21 unter Lenalidomid |
| 23 | MDS, Typ RA | 86 | 97 | 52 | 39 | 736 | 68 | 0,81 | 70 | Labor zum Zeitpunkt der Diagnosestellung |
| 24 | MDS, Typ RA | 79 | 86 | 40 | 40 | 3887 | 51 | 0,33 | 144 | Patient Nr. 23 nach 30 Erythrozyten-Konzentraten |
| 25 | Multiples Myelom | 82 | 100 | 12 | – | 238 | 57 | 0,54 | – | praktisch 100% Knochenmarkinfiltration |
| 26 | Akute myeloische Leukämie | 68 | 88 | 7 | – | 385 | 43 | 0,18 | – | dichte Knochenmarkinfiltration |
| 27 | NHL nach Hochdosistherapie | 79 | 85 | 6 | – | 132 | 36 | 0,29 | – | nach autologer Stammzelltransplantation |
| 28 | PMF, polytransfundiert | 53 | 88 | 8 | – | 1124 | 44 | 0,36 | – | Knochenmark: ausgeprägte Fibrosierung |
Referenzwerte: Laktatdehydrogenase, LDH [109–250 U/L]; Vitamin B12 [182–625 ng/L]; Bilirubin [0,1–1,2 mg/dL]; Haptoglobin [0,3–2,0 g/L]; Abkürzungen: Hb, Hämoglobin; MCV, mittleres korpuskuläres Volumen; Reti, Retikulozyten; CHr, Retikulozytenhämoglobin; EP, Erythropoese; NHL, Non-Hodgkin-Lymphom; PMF, Primäre Myelofibrose; GFR, glomeruläre Filtrationsrate; AIHA, autoimmunhämolytische Anämie.
Eine besondere Erwähnung verdient die renale Anämie, die insbesondere bei älteren Personen von großer klinischer Bedeutung ist und in manchen Studien sogar als die häufigste Anämie des geriatrischen Patienten gehandelt wird [9]. Bedingt durch den Verlust an funktionsfähigem Nierengewebe ist im Alter nicht nur die Ausscheidungsfunktion der Niere, sondern auch die Produktion von Erythropoetin eingeschränkt. Bei Einschätzung der Nierenfunktion alleine anhand des Kreatininwertes wird die Nierenfunktion des älteren Patienten meist zu positiv bewertet und die renale Komponente der Anämie häufig nicht erkannt. Normwertiges Kreatinin und normwertiges Erythropoetin schließen eine renale Anämie nicht aus. Das Kreatinin ist infolge der reduzierten Muskelmasse häufig normal, so dass die Beurteilung der Nierenfunktion nach der Kreatininclearance erfolgen sollte. Eine Cockroft-Clearance ist dabei völlig ausreichend. Eine renale Anämie kann bereits bei einer Kreatinin-Clearance von 50 mL/min auftreten, bei Werten <30 mL/min gehört sie praktisch dazu [51, 52]. Entsprechend der reduzierten Erythropoese liegen die sTfR-Werte bei einer renalen Anämie im unteren Normbereich. Das Erythropoetin ist normal oder leicht erhöht, die Erhöhung ist jedoch nicht adäquat zum Hämoglobinwert (Tabelle 4).
Eine Myelosuppression bei Patienten nach Chemotherapie, oder Patienten mit einer Knochenmarkinfiltration durch eine hämatologische Neoplasie oder durch ein Karzinom ist ebenfalls sehr häufig mit einer Anämie vergesellschaftet, die in der Regel normochrom-normozytär ist. In der hausärztlichen Praxis soll es sich sogar um die zweithäufigste Anämieursache handeln [9]. In diesem Zusammenhang ist es sinnvoll zunächst zu überprüfen ob rote Vorstufen, sogenannte Erythroblasten in das periphere Blut ausgeschwemmt werden, weil deren Vorkommen auf einen schweren Knochenmarkschaden im Rahmen einer Knochenmarkkarzinose, Knochenmarkfibrosierung oder Infiltration durch eine hämatologische Grunderkrankung hinweist. Ergänzend zu dem selbstverständlichen Differentialblutbild ist bei ungeklärten Fällen auch eine durchflusszytometrische Untersuchung des peripheren Blutes durchzuführen, um ggf. eine monoklonale lymphatische Population nachzuweisen.
Konsequenz für die Praxis
Bei der breiten Differentialdiagnose einer normochrom-normozytären Anämie ist es vernünftiger, nicht den Ehrgeiz zu entwickeln, alle möglichen Ursachen bereits durch die erste Blutentnahme zu erfassen. Es ist sinnvoller, bei der Abklärung schrittweise vorzugehen und zunächst nur die wichtigsten Ursachen diagnostisch abzudecken. Die erste Laboruntersuchung sollte Retikulozyten, LDH, Kreatinin im Serum, Kreatinin-Clearance, Ferritin, Transferrinsättigung, Bilirubin, ALAT und das CRP beinhalten, durch die Bestimmung von sTfR, ZPP und Erythropoetin wird die Auflösung der Differentialdiagnose sicher erleichtert. Das weitere diagnostische Vorgehen hängt von der Befundkonstellation dieser Laborparameter ab. Dabei sollte man nicht allzu enttäuscht sein, wenn die ersten Ergebnisse zu keinem eindeutigen Ergebnis führen. Normochrom-normozytäre Anämien sind eben sehr häufig nicht monokausal, sondern multifaktoriell, insbesondere wenn es sich um ältere, multimorbide Patienten handelt. In solchen Fällen geht es nicht darum, alle Komponenten herauszufinden, die zu der festgestellten Anämie beigetragen haben. Primär geht es darum, die Hauptursache zu finden. Außerdem müssen alle diejenigen Komponenten nachgewiesen, bzw. ausgeschlossen werden die man leicht behandeln kann, wie Substrat- oder Erythropoetinmangel.
Manche halten ein manuelles Differentialblutbild für einen Anachronismus. In unseren Augen ist es die Basis einer jeden Anämieabklärung, die durch das Labor-Routinepanel nicht aufgeklärt wurde. Bezüglich der immunhämolytischen Anämie ist zu betonen, dass diese nicht selten im Rahmen einer hämatologischen Neoplasie auftreten und deshalb eine Vorstellung dieser Fälle bei einem Hämatologen sicher kein Fehler ist. Dies gilt auch für andere ungeklärte Anämien, weil im Zweifelsfall auch eine Knochenmarkpunktion erfolgen muss, um die Hämatopoese genau zu beurteilen und um eine hämatologische Grunderkrankung sicher auszuschließen.
Autorenbeteiligung: Alle Autoren tragen Verantwortung für den gesamten Inhalt dieses Artikels und haben der Einreichung des Manuskripts zugestimmt.
Forschungsförderung: Keine.
Interessenkonflikt: Kein Interessenkonflikt.
Literatur
1. World health organisation. Nutritional anemia: report of a WHO scientific group. Geneva, Switzerland: WHO Press, 1968.Suche in Google Scholar
2. Kassebaum NJ, Jasrasaria R, Naghavi M, Wulf SK, Johns N, Lozano R, et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood 2014;123:615–24.10.1182/blood-2013-06-508325Suche in Google Scholar PubMed PubMed Central
3. Shander A, Goodnough LT, Javidroozi M, Auerbach M, Carson J, Ershler WB, et al. Iron deficiency anemia–bridging the knowledge and practice gap. Transfu Med Rev 2014;28:156–66.10.1016/j.tmrv.2014.05.001Suche in Google Scholar PubMed
4. Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Deut Arzteblatt Int 2011;108:532–40.10.3238/arztebl.2011.0532Suche in Google Scholar
5. Eber S, Dickerhoff R. Anemia and hemoglobin diseases in patients with migration background. Deut Med Wochenschr 2014;139:434–40.10.1055/s-0034-1369813Suche in Google Scholar
6. Joosten E, Lioen P. Iron deficiency anemia and anemia of chronic disease in geriatric hospitalized patients: how frequent are comorbidities as an additional explanation for the anemia? Geriatr Gerontol Int 2014; doi: 10.1111/ggi.12371. [Epub ahead of print].10.1111/ggi.12371Suche in Google Scholar PubMed
7. Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood 2004;104:2263–8.10.1182/blood-2004-05-1812Suche in Google Scholar PubMed
8. Guralnik JM, Ershler WB, Schrier SL, Picozzi VJ. Anemia in the elderly: a public health crisis in hematology. Hematology Am Soc Hematol Educ Program 2005:528–32.10.1182/asheducation-2005.1.528Suche in Google Scholar PubMed
9. Merlo Ch M, Wuillemin WA. Prevalence and causes of anemia in a city general practice. Praxis 2008;97:713–8.10.1024/1661-8157.97.13.713Suche in Google Scholar
10. Migone De Amicis M, Poggiali E, Motta I, Minonzio F, Fabio G, Hu C, et al. Anemia in elderly hospitalized patients: prevalence and clinical impact. Int Emergen Med 2015;10:581–6.10.1007/s11739-015-1197-5Suche in Google Scholar PubMed
11. Tettamanti M, Lucca U, Gandini F, Recchia A, Mosconi P, Apolone G, et al. Prevalence, incidence and types of mild anemia in the elderly: the “Health and Anemia” population-based study. Haematologica 2010;95:1849–56.10.3324/haematol.2010.023101Suche in Google Scholar PubMed PubMed Central
12. Hastka J, Lasserre JJ, Schwarzbeck A, Reiter A, Hehlmann R. Laboratory tests of iron status: correlation or common sense? Clin Chem 1996;42:718–24.10.1093/clinchem/42.5.718Suche in Google Scholar
13. Metzgeroth G, Hastka J. Diagnostic work-up of iron deficiency Laboratoriums Medizin 2004;28:391–9.10.1515/LabMed.2004.054Suche in Google Scholar
14. World health organisation, centers for disease control and prevention. Assessing the iron status of population. Geneva, Switzerland: WHO Press, 2004.Suche in Google Scholar
15. Suominen P, Punnonen K, Rajamaki A, Irjala K. Serum transferrin receptor and transferrin receptor-ferritin index identify healthy subjects with subclinical iron deficits. Blood 1998;92:2934–9.10.1182/blood.V92.8.2934.420k07_2934_2939Suche in Google Scholar
16. Cankurtaran M, Yavuz B, Halil M, Ulger Z, Haznedaroglu I, Ariogul S. Increased ferritin levels are critical due to age-associated inflammation and may obscure underlying iron deficiency in the geriatric population. Eur Geriatr Med 2012;3:277–80.10.1016/j.eurger.2012.06.005Suche in Google Scholar
17. World health organisation, centers for disease control and prevention. Assessing the iron status of population: Geneva, Switzerland: WHO Press, 2007.Suche in Google Scholar
18. Thurnham DI, McCabe LD, Haldar S, Wieringa FT, Northrop-Clewes CA, McCabe GP. Adjusting plasma ferritin concentrations to remove the effects of subclinical inflammation in the assessment of iron deficiency: a meta-analysis. Am J Clin Nutr 2010;92:546–55.10.3945/ajcn.2010.29284Suche in Google Scholar PubMed
19. Labbe RF, Finch CA, Smith NJ, Doan RN, Sood SK, Madan N. Erythrocyte protoporphyrin/heme ratio in the assessment of iron status. Clin Chem 1979;25:87–92.10.1093/clinchem/25.1.87Suche in Google Scholar
20. Lamola AA, Eisinger J, Blumberg WE. Erythrocyte protoporphyrin/heme ratio by hematofluorometry. Clin Chem 1980;26:677–8.10.1093/clinchem/26.5.677Suche in Google Scholar
21. Kohgo Y, Niitsu Y, Condo H, Kato J, Tsushima N, Sasaki K, et al. Serum transferrin receptor as a new index of erythropoiesis. Blood 1987;70:1955–8.10.1182/blood.V70.6.1955.1955Suche in Google Scholar
22. Metzgeroth G, Schultheis B, Kuhn C, Dorn-Beineke A, LaRosee P, Hehlmann R, et al. The soluble transferrin receptor reflects tumor load in chronic lymphocytic leukemia. Clin Chem Lab Med 2007;45:1313–8.10.1515/CCLM.2007.287Suche in Google Scholar PubMed
23. Thomas C, Thomas L. Biochemical markers and hematologic indices in the diagnosis of functional iron deficiency. Clin Chem 2002;48:1066–76.10.1093/clinchem/48.7.1066Suche in Google Scholar
24. Punnonen K, Irjala K, Rajamaki A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood 1997;89:1052–7.10.1182/blood.V89.3.1052Suche in Google Scholar
25. Weiss G, Goodnough LT. Anemia of chronic disease. New Engl J Med 2005;352:1011–23.10.1056/NEJMra041809Suche in Google Scholar PubMed
26. Cartwright GE, Lee GR. The anaemia of chronic disorders. Br J Haematol 1971;21:147–52.10.1111/j.1365-2141.1971.tb03424.xSuche in Google Scholar PubMed
27. Weiss G. Iron metabolism in the anemia of chronic disease. Biochim Bioph acta 2009;1790:682–93.10.1016/j.bbagen.2008.08.006Suche in Google Scholar PubMed
28. Hastka J, Lasserre JJ, Schwarzbeck A, Strauch M, Hehlmann R. Zinc protoporphyrin in anemia of chronic disorders. Blood 1993;81:1200–4.10.1182/blood.V81.5.1200.1200Suche in Google Scholar
29. Ferguson BJ, Skikne BS, Simpson KM, Baynes RD, Cook JD. Serum transferrin receptor distinguishes the anemia of chronic disease from iron deficiency anemia. J Lab Clin Med 1992;119:385–90.Suche in Google Scholar
30. Skikne B, Flowers CH, Cook JD. Serum transferrin receptor: a quantitative measure of tissue iron deficiency. Blood 1990;75:1870–6.10.1182/blood.V75.9.1870.1870Suche in Google Scholar
31. Nemeth E. Hepcidin biology and therapeutic applications. Expert Rev Hematol 2010;3:153–5.10.1586/ehm.10.1Suche in Google Scholar PubMed
32. Kemna EH, Tjalsma H, Willems HL, Swinkels DW. Hepcidin: from discovery to differential diagnosis. Haematologica 2008;93:90–7.10.3324/haematol.11705Suche in Google Scholar PubMed
33. D’Angelo G. Role of hepcidin in the pathophysiology and diagnosis of anemia. Blood Res 2013;48:10–5.10.5045/br.2013.48.1.10Suche in Google Scholar PubMed PubMed Central
34. Metzgeroth G, Kripp M, Muller N, Schultheis B, Bonatz K, Walz C, et al. The soluble transferrin receptor (TfR)-F-Index is not applicable as a test for iron status in patients with chronic lymphocytic leukemia. Clin Chem Lab Med 2009;47:1291–5.10.1515/CCLM.2009.273Suche in Google Scholar PubMed
35. Whitehead VM. Acquired and inherited disorders of cobalamin and folate in children. Br J Haematol 2006;134:125–36.10.1111/j.1365-2141.2006.06133.xSuche in Google Scholar PubMed
36. Hoffbrand AV, Weir DG. The history of folic acid. Br J Haematol 2001;113:579–89.10.1046/j.1365-2141.2001.02822.xSuche in Google Scholar PubMed
37. Stabler SP. Vitamin B12 deficiency. N Engl J Med 2013;368: 2041–2.10.1056/NEJMcp1113996Suche in Google Scholar PubMed
38. Quadros EV. Advances in the understanding of cobalamin assimilation and metabolism. Br J Haematol 2010;148:195–204.10.1111/j.1365-2141.2009.07937.xSuche in Google Scholar
39. Allen LH. Causes of vitamin B12 and folate deficiency. Food Nutr Bull 2008;29(2 Suppl):S20–34; discussion S5–7.10.1177/15648265080292S105Suche in Google Scholar
40. Carmel R, Green R, Rosenblatt DS, Watkins D. Update on cobalamin, folate, and homocysteine. Hematology Am Soc Hematol Educ Program 2003;1:62–81.10.1182/asheducation-2003.1.62Suche in Google Scholar
41. Nexo E, Hoffmann-Lücke E. Holotranscobalamin, a marker of vitamin B-12 status: analytical aspects and clinical utility. Am J Clin Nutr 2011;94:3595.10.3945/ajcn.111.013458Suche in Google Scholar
42. Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943–64.10.1182/blood-2013-03-492884Suche in Google Scholar
43. Scully M. Trends in the diagnosis and management of TTP: European perspective. Transfus Apher Sci 2014;51:11–4.10.1016/j.transci.2014.08.001Suche in Google Scholar
44. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005;365:1073–86.10.1016/S0140-6736(05)71144-2Suche in Google Scholar
45. Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001;413:488–94.10.1038/35097008Suche in Google Scholar PubMed
46. Dacie SJ. The immune haemolytic anaemias: a century of exciting progress in understanding. Br J Haematol 2001;114:770–85.10.1046/j.1365-2141.2001.02945.xSuche in Google Scholar PubMed
47. Segel GB, Lichtman MA. Direct antiglobulin (“Coombs”) test-negative autoimmune hemolytic anemia: a review. Blood Cell Mol Dis 2014;52:152–60.10.1016/j.bcmd.2013.12.003Suche in Google Scholar PubMed
48. Hochsmann B, Rojewski M, Schrezenmeier H. Paroxysmal nocturnal hemoglobinuria (PNH): higher sensitivity and validity in diagnosis and serial monitoring by flow cytometric analysis of reticulocytes. Ann Hematol 2011;90:887–99.10.1007/s00277-011-1177-4Suche in Google Scholar PubMed PubMed Central
49. Nebe T, Schubert J, Gutensohn K, Schrezenmeier H. Flow cytometric analysis of GPI-deficient cells for the diagnosis of paroxysmal nocturnal hemoglobinuria. Laboratoriums Medizin 2003;27:257–65.10.1515/LabMed.2003.038Suche in Google Scholar
50. Metzgeroth G, Adelberger V, Dorn-Beineke A, Kuhn C, Schatz M, Maywald O, et al. Soluble transferrin receptor and zinc protoporphyrin–competitors or efficient partners? Eur J Haematol 2005;75:309–17.10.1111/j.1600-0609.2005.00515.xSuche in Google Scholar PubMed
51. Hsu CY, Bates DW, Kuperman GJ, Curhan GC. Relationship between hematocrit and renal function in men and women. Kidney Int 2001;59:725–31.10.1046/j.1523-1755.2001.059002725.xSuche in Google Scholar PubMed
52. Ble A, Fink JC, Woodman RC, Klausner MA, Windham BG, Guralnik JM, et al. Renal function, erythropoietin, and anemia of older persons: the InCHIANTI study. Arch Int Med 2005;165:2222–7.10.1001/archinte.165.19.2222Suche in Google Scholar PubMed
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Editorial
- Aktuelle Diagnostik in der Hämatologie
- Hämatologie/Hematology
- Stufendiagnostik zur Abklärung von krankhaften Veränderungen der Leukozyten
- Rationale Anämieabklärung
- Diagnostik von myelodysplastischen Syndromen (MDS) und akuten myeloischen Leukämien (AML)
- Differenzialdiagnose BCR-ABL1-negativer myeloproliferativer Neoplasien
- Hämoglobinvarianten – Pathomechanismus, Symptome und Diagnostik
- Molekulargenetische und zytogenetische Diagnostik/Molecular-Genetic and Cytogenetic Diagnostics
- An update of novel identified cytogenetic and molecular biomarkers in the laboratory diagnosis of hematological neoplasia
- Stufendiagnostik der Hämoglobinopathien
- Rational laboratory diagnostics of primary immunodeficiency disorders
Artikel in diesem Heft
- Frontmatter
- Editorial
- Aktuelle Diagnostik in der Hämatologie
- Hämatologie/Hematology
- Stufendiagnostik zur Abklärung von krankhaften Veränderungen der Leukozyten
- Rationale Anämieabklärung
- Diagnostik von myelodysplastischen Syndromen (MDS) und akuten myeloischen Leukämien (AML)
- Differenzialdiagnose BCR-ABL1-negativer myeloproliferativer Neoplasien
- Hämoglobinvarianten – Pathomechanismus, Symptome und Diagnostik
- Molekulargenetische und zytogenetische Diagnostik/Molecular-Genetic and Cytogenetic Diagnostics
- An update of novel identified cytogenetic and molecular biomarkers in the laboratory diagnosis of hematological neoplasia
- Stufendiagnostik der Hämoglobinopathien
- Rational laboratory diagnostics of primary immunodeficiency disorders