Hämoglobinvarianten – Pathomechanismus, Symptome und Diagnostik
-
Berndt Zur


Zusammenfassung:
Hämoglobinvarianten, die nicht zu den bekannteren Formen der Thalassämien, Sichelzell-, HbC-, HbD-, HbE-Anomalien gezählt werden, stellen häufig eine diagnostische Herausforderung dar. Sehr gute Kenntnisse über die unterschiedliche Symptomatik und der Analytik sind Voraussetzung. Hier ist die laboratoriumsmedizinische Expertise von großer Bedeutung. Das Spektrum der Varianten ist vielfältig und kann bei mangelnder Fachkenntnis zur Fehlinterpretation führen. Hämoglobinvarianten mit niedriger Sauerstoffaffinität können sich durch Zyanose und niedrigen Sauerstoffsättigungswerten bemerkbar machen, welche mit erhöhter Sauerstoffaffinität durch Polyglobulien und die damit zusammenhängenden Komplikationen. Methämoglobinvarianten können besonders in der Pädiatrie ein differentialdiagnostisches Problem darstellen, das eine sorgfältige Begutachtung erfordert. Andere Varianten verursachen durch ihre Instabilität mehr oder weniger stark ausgeprägte Hämolysen oder auch thalassämische Syndrome, die schwere Krankheitsbilder darstellen können. Eine klare Abgrenzung ist nicht immer möglich, da sich die Klinik oft durch mehrere Eigenschaften äußert. Gemeinsam ist vielen Varianten ein autosomal dominanter Erbgang.
Abstract:
The diagnosis of hemoglobin variants that are not any of the better-known forms of thalassemia, sickle cell, HbC, HbD, or HbE anomalies is often challenging and requires detailed knowledge of the difference in symptoms and analysis. Experience in laboratory medicine plays an important role, as the range of variants is extensive and lack of expertise can result in a wrong diagnosis. Hemoglobin variants with low oxygen affinity may present cyanosis and low oxygen saturation levels, whereas variants with increased oxygen affinity show polyglobulia and concomitant complications. Differential diagnosis of methemoglobin variants requires careful assessment, which can be problematic especially in pediatric medicine. Other variants, due to their instability, can cause more or less distinct hemolysis or thalassemia syndromes depicting serious disease patterns. Clear distinction is not always possible, as several symptoms are often present. Many variants are autosomal dominant inherited.
Rezensierte Publikation:
Nebe C.T.
Einleitung
Die häufigsten ursprünglich endemisch vorkommenden Hämoglobindefekte, die Thalassämien und Sichelzellerkrankungen, sind durch Migration schon längst in Mitteleuropa verbreitet und spielen im klinischen Alltag eine zunehmende Rolle. Seltene Hämoglobinanomalien stellen eine diagnostische Herausforderung für den behandelnden und diagnostizierenden Arzt dar und das gesamte Spektrum der Symptome wird zum Teil unterschätzt (Tabelle 1). Medizinische Laboratorien führen als Standarddiagnostik im Allgemeinen Hämoglobinchromatografien oder Hämoglobinelektrophoresen durch. In vielen Fällen reicht dies nicht aus und zur Vermeidung von Fehlinterpretationen sollte die weiterführende Diagnostik in die Hand von Spezialisten gegeben werden. Hier sollte die Laboratoriumsmedizin eine Schlüsselstellung einnehmen. Hämoglobinopathien werden eingeteilt in Thalassämien und Hämoglobin-Strukturanomalien. Die Thalassämien sind durch Imbalance der Globinsynthese gekennzeichnet, die Hämoglobin-Strukturanomalien verursachen die Bildung eines abnormen Hämoglobins, das bekannteste ist das Sichelzellhämoglobin. Strukturdefekte können jedoch auch thalassämische Syndrome verursachen, wie das in Südost-Asien verbreitete Hämoglobin E, daher ist eine Einteilung der Hämoglobindefekte nicht statisch zu begreifen [1]. Hämoglobine, die durch einen Buchstaben gekennzeichnet werden, sind größtenteils endemisch vorkommende Varianten, ebenso das Hb Lepore. Sie werden häufig zusätzlich durch eine Ortsangabe beschrieben (Bsp. Hb D Punjab oder Hb Q Iran). Diese Nomenklatur ist allerdings lückenhaft und ist nach heutigen Erkenntnissen über die Verbreitung einiger Hämoglobinvarianten nicht mehr in allen Fällen richtig. Der Fokus dieser Übersichtsarbeit richtet sich jedoch vorwiegend auf die zahlreichen seltenen Hämoglobin-Strukturanomalien oder Hämoglobinvarianten, die Einfluss auf die Sauerstoffbindung und Stabilität des Hämoglobins ausüben und überwiegend nicht endemisch vorkommen. Die Nomenklatur dieser Varianten richtet sich nach dem Entdeckungsort oder nach der Herkunft des Patienten, allerdings existieren hier keine festen Regeln und die Entdecker haben die Freiheit bei der Namensgebung. Daher ist eine Kategorisierung durch die Benennung nicht möglich. Die Varianten zeichnen sich, im Gegensatz zu den bekannten Anomalien mit autosomal rezessivem Erbgang, häufig durch einen autosomal dominanten Erbgang aus. Da zahlreiche Varianten mehrere Symptome verursachen, ist eine eindeutige Definition nicht in jedem Fall möglich. Instabile Varianten können eine hohe oder niedrige Sauerstoffaffinität haben, Varianten mit Affektion der Sauerstoffaffinität können Hämolysen und Methämoglobinämien auslösen. Sie können auch klinisch unauffällig sein, aber im Kontext anderer Erkrankungen eine Rolle spielen. In dieser Übersichtsarbeit wurde die Einteilung der Varianten aufgrund ihrer Hauptwirkung vorgenommen. Aufgrund der großen Anzahl verschiedener Hämoglobinanomalien werden Varianten vorgestellt, die in ihrer Symptomatik als Beispiel für andere Varianten dienen. Eine Gruppierung innerhalb der Einteilung ist nur selten möglich, da an einem Mutationsort ein Austausch unterschiedlicher Aminosäuren auch unterschiedliche Auswirkung haben kann. Für ein besseres Verständnis werden einige Varianten ausführlicher präsentiert.
Durch Hämoglobinvarianten verursachte Symptome.
| ↓O2-Affinitätsvarianten | ↑O2-Affinitätsvarianten | Methämoglobinvarianten | Instabile und thalassämische Varianten |
|---|---|---|---|
| Zyanose | Erythrozytose | Zyanose | Anämie |
| O2-Sättigung↓ | Hyperviskosität | O2-Sättigung↓ | Hämolyse |
| Anämie | Anämie | Anämie | Ikterus |
| Hämolyse | Hämolyse | Hämolyse | Zyanose |
| Dyspnoe | Thromboembolien | Dyspnoe | O2-Sättigung↓ |
| Methämoglobinämie | Fatigue | Ikterus | ↑O2-Affinität |
| Fatigue | Kopfschmerzen | Hämolyse durch ox. Substanzen | ↓O2-Affinität |
| Nasenbluten | braunes Blut | Dyspnoe | |
| Methämoglobin | |||
| Thromboembolien | |||
| Priapismus | |||
| Splenomegalie | |||
| Hämolyse durch ox. Substanzen | |||
| Pigmenturie |
Grundlagen
Hämoglobin ist ein tetrameres Protein, mit jeweils 2 α- und 2 β-Globinketten, die an hydrophoben Kontaktregionen ionisch, nicht-kovalent gebunden sind und der Hämgruppe, bestehend aus einem Porphyrinring mit zentralem Eisenatom für die Sauerstoffbindung. Die Hämgruppe ist in einer V-förmigen Tasche lokalisiert und zur Stabilisierung in der Tertiärstruktur von hydrophoben Aminosäuren umgeben. Die Konformation des Hämoglobinmoleküls ändert sich durch Oxygenierung, dem R(relaxed)-Zustand und durch Deoxygenierung, dem T(tense)-Zustand. Dieser allosterische Effekt ist für die sigmoidale Sauerstoffbindungskurve des Hämoglobins verantwortlich [2]. Das intraerythrozytäre 2,3 Bis-Phosphoglycerat (2,3-BPG) hat die Funktion als allosterischer Effektor die Sauerstoffaffinität des Hämoglobins zu reduzieren, in dem es an das Desoxyhämoglobin durch Salzbrücken bindet und so die Sauerstoffabgabe erleichtert, es kann sich nicht an Oxyhämoglobin binden. Diese Fähigkeiten sind notwendig, damit das Hämoglobin Sauerstoff in der Lunge aufnehmen und ins Gewebe abgeben kann. Der Bohr-Effekt beschreibt die Sauerstoffsättigungskurve des Hämoglobins in Abhängigkeit des pH-Wertes und des Sauerstoffpartialdruckes und bewirkt eine verstärkte Freisetzung von Kohlendioxid in Gegenwart hoher Sauerstoffkonzentration (Lunge) und eine Freisetzung von Sauerstoff und erleichterte Aufnahme von Kohlendioxid in der Peripherie. Die Hämoglobin-Sauerstoff-Affinität steigt mit sinkender Temperatur, sinkender 2,3-BPG-Konzentration und steigendem pH-Wert [3]. Das Hämoglobin eines Erwachsenen besteht bis zu ca. 97% aus HbA (jeweils 2 α- und 2 β-Globinen), bis zu ca. 3,5% aus HbA2 (2 α- und 2 δ-Globinen) und bis zu 1% aus HbF (2 α- und 2 γ-Globinen). Neugeborene haben einen hohen HbF-Anteil, der sich individuell unterschiedlich schnell auf ca. 1% reduziert. Im gleichen Maße, wie der HbF-Anteil abfällt, steigt der HbA-Anteil, der ca. im sechsten Lebensmonat Erwachsenenwerte erreicht. Klinisch zeigen sich daher Mutationen der γ- und α-Globingene schon bei Neugeborenen, β-Globingen-Mutationen, aufgrund der Umstellung von HbF auf HbA erst im Laufe der ersten Lebensmonate. Auf dem Chromosom 11 codieren 2 Gene das α-Globin, α-1 trägt mit ca. 30%, α-2 zu ca. 70% zur Proteinbiosynthese des α-Globins bei. Auf dem Chromosom 16 codieren jeweils ein Gen für das β-Globin und δ-Globin und 2 Gene für das γ-Globin [4]. In der Sauerstoffdissoziationskurve zeigt sich eine erniedrigte Sauerstoffaffinität durch eine Verschiebung nach links, eine erhöhte Sauerstoffaffinität durch Verschiebung nach rechts [2]. Der p50-Wert beschreibt den Partialdruck von Sauerstoff bei 50%iger Sättigung des Hämoglobins und ist der wichtigste Parameter zur Beurteilung der Sauerstoffaffinität [5]. Die Sauerstoffaffinität des Hämoglobins hat direkten Einfluss auf die Erythropoietinausschüttung (EPO), das überwiegend in den Fibroblasten der peritubulären Zellen der Niere gebildet wird. Der Mechanismus der Erythropoietinbildung ist sehr komplex. Stark vereinfacht gilt nach bisheriger Erkenntnis: Normoxie bewirkt, dass der Transkriptionsfaktor GATA-2 den EPO-Promotor bremst. Die hypoxie-induzierbaren Transskriptionsfaktoren (HIF) werden ebenfalls denaturiert, so dass Erythropoietin nicht gebildet werden kann. Unter Hypoxie wird die Hemmung aufgehoben und die HIF bewirken durch Aktivierung des EPO-Enhancers die Bildung von EPO, das schließlich an spezifische Rezeptoren der Eryhrozytenvorläuferzellen „Colony-Forming Units-Erythroid (CFU-Es)“ bindet. Durch Hemmung der Apoptose und Beschleunigung der Differenzierung und Proliferation von Vorläuferzellen im Knochenmark bewirkt EPO so eine Erhöhung der Erythrozytenzahl. Die Zeitspanne von der Bildung des EPO bis zu einer detektierbaren Erhöhung von Retikulozyten beträgt ca. 3–4 Tage [6]. Eine Mutation mit erhöhter Sauerstoffaffinität, kann zur schlechteren Sauerstoffabgabe führen und eine Erythrozytose bewirken.
Mutationen
Die genaue Anzahl der Hämoglobinvarianten wird in der Database of Human Hemoglobin Variants and Thalassemias (HbVar) aktuell mit 1212 angegeben, mit den Thalassämie-Mutationen mit 1635. Mutationen im α-Globin führen aufgrund der 2-fachen Codierung des α-Globins seltener zu ausgeprägten Störungen. Die durch Mutation ausgetauschte Aminosäure kann aufgrund unterschiedlicher Eigenschaften (hydrophil, hydrophob, pH-Wert etc.) sehr unterschiedliche Effekte in der Proteinstruktur verursachen. Mutationen der α1β2-Verbindung, der β1α2-Kontaktregion, des c-terminalen Endes, der Hämtasche und das unmittelbar an der Hämbindung beteiligte zentrale und distale Histidin, sind Regionen, die eine Affektion der Sauerstoffbindung und der Stabilität des Hämoglobins bewirken können. Es kann eine Beeinträchtigung des Wechsels von der oxygenierten R-Struktur zur deoxygenierten der T-Struktur oder der Stabilisierung der T-Struktur und R-Struktur des Hämoglobinmoleküls stattfinden [2]. 21 Aminosäuren der Globinkette haben in der quaternären Struktur direkten Hämkontakt (Abbildung 1) [7]. Mutationen hyperinstabiler Varianten verursachen häufig eine Verlängerung oder Verkürzung von Globinketten, die nicht für die Hämoglobinsynthese brauchbar sind und schon in den Vorläuferzellen denaturieren [8]. Posttranslationale Modifikationen, die Diskrepanzen zwischen Gensequenz und Proteinstruktur verursachen sind vereinzelt möglich [9]. Über die Prävalenz der seltenen Hämoglobinvarianten, die nicht zu den „endemische Hämoglobinvarianten“ gezählt werden, gibt es keine verlässlichen Angaben. Verfügbare Zahlen aus Deutschland stammen aus Untersuchungsergebnissen über einen Zeitraum von 4 Jahrzehnten eines spezialisierten universitären Einsenderlabors [10]. Klassische endemische Gebiete für eine geografische-ethnische Disposition, wie bei den Thalassämien, Sichelzellanämien, HbC- und HbE-Anomalien gibt es häufig nicht, mit Ausnahme der hyperinstabilen α-Varianten. Die im Folgenden beschriebenen Hämoglobinvarianten stellen nur eine Auswahl dar, aber sie sind aufgrund ihrer Mutationsorte und klinischen Ausprägung repräsentativ für andere Varianten.
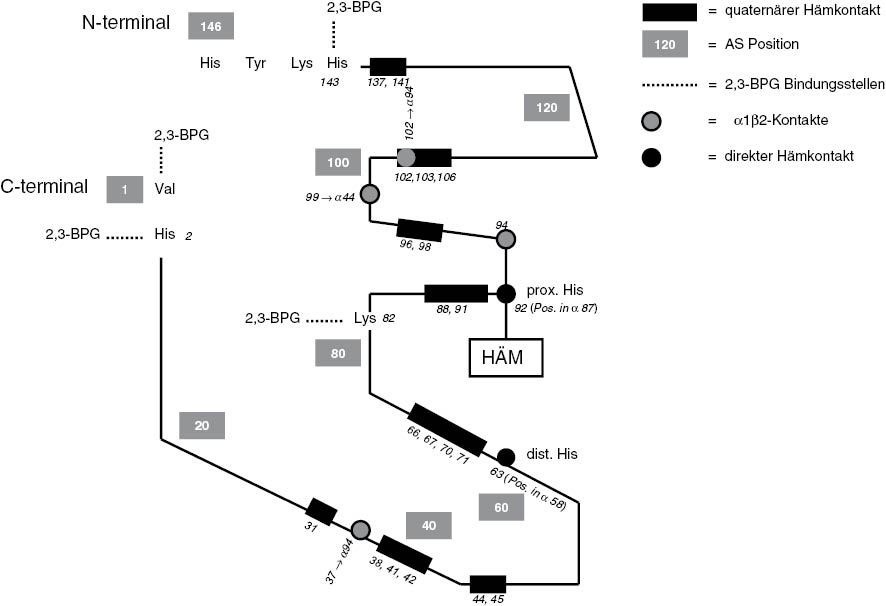
Mutationsorte am Beispiel des Betaglobins, die zu funktionellen Störungen führen können.
Varianten mit Affektion der Sauerstoffaffinität
Varianten erhöhter Sauerstoffaffinität
Hämoglobinvarianten mit erhöhter Sauerstoffaffinität können mit einer Erythrozytose einhergehen und zeichnen sich durch einen erniedrigten p50-Wert aus [11]. Alle bisher bekannten Mutationen, die die Regionen β99 und β146 betreffen, führen zur erhöhten Sauerstoffaffinität des Hämoglobins. Mutationen der 2,3-BPG-Bindungsregionen an Position β143 und β82 führen ebenfalls zur erhöhten Sauerstoffaffinität, wie Mutationen der Bindungsregion am n-terminalen Ende, die nicht zu einer Erythrozytose führen, da in diesem Bereich eine geringere Konzentration von 2,3-BPG beobachtet wird [12]. Das Hb Crete allein verursacht keine spezifische Klinik, jedoch mit kombinierter Mutation im δ-Globingen wird über Polyglobulien, Hämatokriterhöhungen und Splenomegalien berichtet [13]. Hb Montfermeil kann eine leichte Erythrozytose oder auch eine ausgeprägte Polyglobulie mit Hämatokritwerten von 56% und thromboembolischen Ereignissen verursachen [14]. Bei Hb Chesapeake bewirkt die Mutation im α-Globingen nur eine leichte Erythrozytose, in Kombination mit heterozygoter α-Thalassämie eine ausgeprägte Polyglobulie [15]. Homozygot Betroffene mit Hb Saint Nazaire leiden in aller Regel an ausgeprägten Polyglobulien, die regelmäßige Aderlässe erfordern [16]. Eine kombinierte Heterozygotie von β-Thalassämie und Hb Malmö verursacht eine extreme Polyglobulie mit Hämoglobinkonzentrationen bis zu 23 g/dL und Erythrozyten bis zu 10,5 T/L, aber auch ohne kombinierte Thalassämie verursacht der Defekt bei Betroffenen eine Polyglobulie mit Fatigue, Kopfschmerzen und Nasenbluten [17]. Hb Headington kennzeichnet sich durch eine diskrete Erythrozytose, kombiniert mit heterozygoter β-Thalassämie, jedoch durch ausgeprägte Polyglobulie [18]. Es sind einige wenige Varianten bekannt, bei denen die 2,3-BPG-Bindungsstelle des α- oder β-Globins mutiert ist, wie z.B. Hb Helsinki, Hb Providence und Hb Rahere. Betroffene Patienten weisen meist nur eine milde oder keine Erythrozytose auf [19–21]. Die verlängerte β-Globinkette des Hb Saverne verursacht eine Instabilität des Hämoglobins und bewirkt eine hämolytische Anämie [22], die noch stärker beim Hb Köln in Erscheinung tritt [23]. Obwohl beide Varianten eine erhöhte Sauerstoffaffinität besitzen, steht hier die Instabilität im Vordergrund und sie werden als instabile Hämoglobine klassifiziert. Zu den Differentialdiagnosen der Polyglobulie gehören unter anderem auch ein 2,3-BPG-Mutasedefekt, Hämochromatose, Polycythaemia vera, aktivierende Mutation des Erythropoietinrezeptors und sekundäre Erythrozytosen aufgrund von anderen Grunderkrankungen [24]. Physiologisch besitzt das HbF und HbA2 eine erhöhte Sauerstoffaffinität. Ebenso das pathologische β-Tetramer der HbH-Erkrankung, die der α-Thalassämie zuzuordnen ist [25, 26].
Varianten erniedrigter Sauerstoffaffinität
Hämoglobinvarianten mit erniedrigter Sauerstoffaffinität gehen häufig mit einer milden Anämie aufgrund reduzierter Erythropoietinantwort einher und zeichnen sich durch einen erhöhten p50-Wert und erniedrigten Sauerstoffsättigungswert aus. Viele Varianten weisen auch leichte Instabilität und gering erhöhte Methämoglobinkonzentrationen auf. Klinische Zeichen sind Zyanose mit milder Anämie, die jedoch in ihren Ausprägungen sehr unterschiedlich sein können [27]. Die Mutation des Hb Kansas bewirkt eine Begünstigung der T-Struktur des Hämoglobins und verursacht außer einer niedrigen Sauerstoffaffinität eine diskrete Anämie [28]. Hb Beth Israel, eine Mutation an gleicher Position, jedoch anderer Aminosäurensubstitution, bewirkt eine Zyanose und niedrige Sauerstoffsättigungswerte bis zu 63% [29]. Hb Louisville verursacht eine hypochrome, mikrozytäre, hämolytische Anämie und eine Dyspnoe, zumeist ausgelöst durch Infekte oder oxidative Substanzen (Tabelle 2). Familienuntersuchungen ergaben allerdings bei gleicher Genetik eine phänotypisch unterschiedliche Ausprägung [30]. Hb Bassett kennzeichnet sich durch niedrige Sauerstoffsättigungen bis 85%, ohne zusätzliche Auffälligkeiten [31]. Die Mutation des Hb Denver betrifft die Hämbindungsstelle, klinisch wurde eine leichte hämolytische Anämie mit Zyanose beschrieben [32]. Die Mutation des Hb Rothschild destabilisiert die Globinuntereinheiten. Das Hämoglobintetramer besitzt eine niedrige Sauerstoffaffinität, die dissoziierten Dimere eine erhöhte. Klinisch zeigen sich nur erniedrigte Sauerstoffsättigungswerte [33]. Am Beispiel des Hb Venusberg kann man die Problematik der differentialdiagnostischen Einschätzung und der Diagnostik gut veranschaulichen. Hb Venusberg wurde erstmals bei einem 14jährigen Jungen entdeckt, der seit seiner Geburt regelmäßige kardiopulmologische Untersuchungen erhielt, da intermittierend niedrige Sauerstoffsättigungen mit Zyanose nicht erklärt werden konnten. Seine 49jährige Mutter litt seit ihrer Kindheit unter deutlicher Leistungseinschränkung mit intermittierender Lippenzyanose, die regelmäßige diagnostische Klinikaufenthalte nach sich zogen. Durch unauffällige Untersuchungsergebnisse der Hämoglobinelektrophorese wurden bei beiden Patienten eine Hämoglobinvariante nicht mehr vermutet. Schließlich dauerte es 10 Jahre bis die Diagnose gestellt werden konnte [34].
Oxidativ wirkende Medikamente.
| Antibiotika: | Sulfonamide, Nalidixinsäure, Nitrofurantoin, Niridazol |
| Antimalariamittel: | Pamaquin, Primaquin |
| Schmerzmittel: | Phenazon |
| Methb-Therapie: | Methylenblau, Toluidinblau |
| Entzündungshemmer: | Sulfasalazin |
Die Sauerstoffaffinität kann aber auch bei weitverbreiteten Hämoglobinanomalien herabgesetzt sein, wie bei der Sichelzellerkrankung. Patienten weisen mitunter auch erniedrigte Sauerstoffsättigungswerte auf. Dies kann jedoch variieren, je nachdem wie hoch der HbF-Anteil ist, da HbF eine hohe Sauerstoffaffiniät besitzt, wobei dies nur ein Nebenaspekt bei der Betreuung von Sichelzellpatienten darstellt, aber ggf. in der Differentialdiagnose eine Rolle spielen kann [35].
Varianten mit pulsoximetrisch falschen Werten
Die Pulsoximetrie ist ein nicht-invasives photometrisches Verfahren zur kontinuierlichen Messung der arteriellen Sauerstoffsättigung durch einen Fingerclip. Störungen können auftreten durch Bewegungsartefakte, Minderperfusion des Applikationsortes, venöse Pulsation, Dyshämoglobine, optische und elektrische Störstrahlung, intravasal applizierte Farbstoffe, Nagellack und im Einzelfall aufgrund von anatomisch-histologischen Gegebenheiten [36]. Bei einigen Hämoglobinvarianten sind die pulsoximetrisch gemessenen Sauerstoffsättigungswerte falsch niedrig. Das Verhalten der Pulsoximetrie bei Hämoglobinvarianten ist in der Literatur selten beschrieben [37]. Als Beispiel für falsch niedrige Sauerstoffsättigungen in der Pulsoximetrie dient das Hämoglobin Bonn, das bei einem 4-jährigen Jungen und dessen Vater entdeckt wurde. Die diskrepanten blutgasanalytischen und pulsoximetrischen Sättigungswerte zogen zahlreiche kardiopulmologische Untersuchungen nach sich. Beim Vater wurde aufgrund der pulsoximetrischen Werte fälschlicherweise ein Schlaf-Apnoe-Syndrom diagnostiziert. Es vergingen einige Jahre bis die Diagnose gestellt werden konnte. Das Hb Bonn verursacht im in der spektralphotometrischen Messung ein zusätzliches Absorptionsmaximum bei 668 nm des Oxyhämoglobins, was letztendlich zu falsch niedrigen Sauerstoffsättigungswerten in der Pulsoximetrie führte [38].
Methämoglobin-Varianten
Methämoglobin wird auch als Ferrohämoglobin oder Hämiglobin bezeichnet. Es entsteht durch Oxidierung des zentralen Fe2+ des Häms zu Fe3+. MetHb entsteht in vivo zu jeder Zeit, es wird jedoch unmittelbar durch die NADH-abhängige Methämoglobinreduktase zu Hämoglobin reduziert. Normal ist ein MetHb-Anteil unter 1%. Ein Anteil von mehr als 15% macht sich klinisch durch Zyanose bemerkbar, über 30% führen zu Dyspnoe und Verwirrtheit, über 50% zur metabolischen Azidose, Herzrhythmusstörungen, Koma und über 70% zum Tod [39]. Die korrekte Bestimmung des von Hämoglobinvarianten verursachten Methämoglobins (HbM-Methämoglobin) ist problematisch. Die weitverbreiteten Pulsoximeter messen das Hämoglobin-Spektrum bei einer Wellenlänge von 660 nm (Oxyhämoglobin) und 940 nm (Deoxyhämoglobin). Methämoglobine, die nicht durch Hämoglobinvarianten verursacht werden, haben Absorptionsmaximum bei 635 nm, die die durch Hämoglobin M-Varianten verursachten bei 600 nm. Dies verursacht bei der Analyse von HbM-Methämoglobin falsch niedrige Werte. Neuere Multiwellenlängen-Pulsoximeter können die Methämoglobine bei diesen Wellenlängen erfassen [40]. Auch die photometrischen Messmethoden mit Zyanidzusatz führen bei vielen HbM-Varianten, aufgrund langsamer und unvollständiger Reaktion zu falsch niedrigen Werten. Zu den Differentialdiagnosen von Methämoglobinämien zählen NADH-Methämoglobin-Reduktasemangel, Vergiftungen durch Reinigungsmittel und Nitrite u.a. Die Hämoglobinvarianten, die eine MetHb-Bildung verursachen, können therapeutisch nicht durch Redoxfarbstoffe wie Methylenblau behandelt werden, je nach Variante, werden dadurch sogar Hämolysen induziert. Die Mutationen betreffen häufig das proximale Histidin, das unmittelbar kovalent an die Hämgruppe bindet und das nicht-kovalent gebundene, aber unmittelbar zum Häm in Kontakt stehende distale Histidin durch Tyrosin-Austausch. Mutationen an anderen Positionen sind auch möglich. Es besteht in aller Regel ein enger Kontakt zur Hämtasche und dem distalen Histidin. Die substituierten Aminosäuren können eine kovalente Bindung zum Hämeisen eingehen und so eine Stabilisierung des MetHb bewirken. Gemeinsam ist den Hämoglobin M-Varianten eine Instabilität, die sich durch unterschiedlich stark ausgeprägte Hämolyse kennzeichnet. Sichtbares Zeichen einer Methämoglobinämie ist die schokoladenbraune Farbe des Blutes [41, 42]. Die Anteile der Varianten am Gesamthämoglobin betragen zwischen 20 und 40%. Die Mutationen des HbM Saskatoon und HbM Hyde Park betreffen das proximale und das distale Histidin im β-Globin. Klinsch zeigen sich bei beiden Varianten eine Zyanose und eine leichte hämolytische Anämie. HbM Iwate und HbM Boston mit Mutationen des proximalen und distalen Histidins des α-Globingens, sind niedrig-sauerstoffaffine Varianten, die auch schon bei Neugeborenen auftreten können. Es besteht außer einer Zyanose keine andere hämatologische Auffälligkeit. Die MetHb-Konzentration beträgt bei diesen Varianten ca. 10–20%, allerdings sind die Messmethoden und die genauen Konzentrationen aus der Literatur nicht eindeutig [43, 44]. Beim HbM Dothan kommt es zur Affektion des distalen Histidins, was zu einer leichten Instabilität mit Zyanose führt, die MetHb-Konzentration beträgt 20% [45]. Das Hb Chile ist eine instabile hochsauerstoffaffine Variante. Betroffene leiden an einer Zyanose und leichten hämolytischen Anämie, die durch Gabe von Methylenblau und Sulfonamiden verstärkt wird. Das Methämoglobin schwankt zwischen 11 und 18% [46]. Mutationen an gleicher Stelle sind die hochsauerstoffaffinen Hb St. Louis und Hb Genova. Betroffene mit Hb St. Louis leiden ebenfalls an einer MetHb-Zyanose und milder hämolytischer Anämie, mit Hb Genova nur an einer chronischen hämolytischen Anämie [47, 48]. Das Hb Tübingen und Hb Southampton zeichnen sich durch ausgeprägte Hämolysen aus. Durch den Aminosäurenaustausch entstehen hydrophile Reste, die durch Wassereintritt in die Hämtasche die Autooxidation begünstigen. Die Zyanose ist jedoch im Allgemeinen mild ausgeprägt, die Meth-Hb-Konzentration beträgt 13% und werden zu den instabilen Varianten gezählt [49, 50].
HbF-Varianten
Durch Hämoglobin F-Varianten verursachte Methämoglobinämien (Mutationen des γ-Globingens) betreffen nur Neugeborene bis zum 4. Lebensmonat und sind selbstlimitierend, können aber zu unnötigem oder überstürztem ärztlichem Handeln führen. Diese Varianten werden auch als HbFM-Varianten bezeichnet. Nicht alle Mutationen des γ-Globingens verursachen Methämoglobinämien. Die pathophysiologischen Erklärungen sind analog zu den Positionen der β-Globingen-Mutationen zu betrachten, mit dem Unterschied, dass 2 Gene auf Chromosom 16 für das γ-Globin codieren und das HbF generell eine höhere Sauerstoffaffinität besitzt [4]. Das HbFM Fort Ripley, analoge Pathophysiologie zu HbM Hyde Park und das HbFM Osaka, analoge Pathophysiologie zu HbM Saskatoon, machen sich bei betroffenen Neugeborenen durch Methämoglobin verursachte Zyanose und einer hämolytischen Anämie bemerkbar. Die klinische Ausprägung ist individuell sehr unterschiedlich [51, 52]. Beim HbFM Circleville dagegen, gleicher Mutationsort wie HbFM Osaka, aber andere Aminosäurensubstitution wird keine Hämolyse beobachtet [53]. Bei HbF Viseu schwanken die MetHb-Konzentrationen zwischen 16 und 21%, und Neugeborene fallen durch Zyanose auf, ohne weitere Auffälligkeiten [54]. HbF Cincinnati und HbF Sarajevo verursachen bei Neugeborenen eine Zyanose, aber keine Methämoglobinämie. Bei Neugeborenen mit HbF Sarajevo besteht eine ausgeprägte Zyanose. Der analoge Mutationsort des β-Globingens verursacht das Hb Kansas, eine Variante mit niedriger Sauerstoffaffinität. Viele Varianten sind auch hier nicht durch die Standarddiagnostik HPLC oder Elektrophorese detektabel [55, 56].
Instabile Varianten
Diese Varianten zeichnen sich durch die Instabilität des Hämoglobintetramers aus. Einige Mutationen bewirken eine Denaturierung der Hämoglobintetramere schon im Knochenmark und sind als Protein in der Peripherie häufig nicht detektierbar. Die Mutationen, meist De-Novo-Mutationen, betreffen häufig die hydrophoben, wasserabweisenden Gruppen des Hämoglobins. Ein hydrophiler Aminosäurenaustausch führt zu einem Leck und Wassereintritt destabilisiert das Molekül. Ein Aminosäurenaustausch durch Prolin bewirkt eine Instabilität in der Sekundärstruktur durch Beeinflussung der Helixstruktur und den interhelikalen Bindungen. Die Mutationen können auch eine Dissoziation von Globin und Häm bewirken. Das Häm mit zentralem Fe3+ (MetHb) ist aus der Hämtasche herausgelöst und verbindet sich in weiteren Schritten mit denaturierten Globinresten und es entstehen irreversible Hemichrome, die sich mit dem zytosolischen Anteil des Bande-3-Proteins der Erythrozytenmembran verbinden. Diese Verbindung verursacht eine verminderte Verformbarkeit der Erythrozyten und führt zur Entfernung betroffener Erythrozyten in der Milz. Die Präzipitate werden als Heinz’sche Innenkörper bezeichnet und lösen eine Anheftung von IgG-Antikörpern aus, die die Elimination der Erythrozyten zusätzlich triggert. Durch die Anwesenheit des freien Häms und Herauslösung des Eisens wird eine Fenton-Reaktion bewirkt, eine katalysierte Oxidation von Eisensalzen mit Wasserstoffperoxid. Folgen sind eine Verstärkung der Hämoglobindenaturierung, Methämoglobinbildung und des Membranschadens. Daher sind Heinz’sche Innenköper oft erst nach einer Milzentfernung im Blutausstrich detektierbar. Erythrozyten mit Innenkörpern besitzen zudem eine hohe Affinität sich an das Gefäßendothel zu heften. Bei einigen Varianten lösen oxidativ wirkende Substanzen, Medikamente (Tabelle 2) und Infektionen hämolytische Krisen aus. Ein klassisches Zeichen ist die Pigmenturie (brauner Urin), die durch Dipyrrolmethene aus dem Hämabbau verursacht wird. Instabile Varianten mit erhöhter Sauerstoffaffinität zeigen durch die induzierte Erythropoietinausschüttung weniger stark ausgeprägte Anämien. Diagnostisch sind viele Varianten nicht mit den herkömmlichen Methoden HPLC oder Elektrophorese zu detektieren. Bei den hyperinstabilen Varianten finden sich durch den vorzeitigen Untergang in den Vorläuferzellen keine pathologischen Erythrozyten im peripheren Blut [8, 57–60].
Die phänotypische Ausprägung des Hb Köln ist individuell sehr unterschiedlich. Die Krankheit tritt durch die Umstellung von HbF auf HbA bei Kindern frühestens ab dem zweiten bis dritten Lebensmonat mit Ikterus und hämolytischer Anämie in Erscheinung [23]. Pulsoximetrisch gemessene Sauerstoffsättigungswerte sind falsch niedrig [38]. Splenektomien im Kindesalter aufgrund ausgeprägter hämolytischer Anämien und Splenomegalie sind nicht selten indiziert, jedoch wird auch über relativ milde Verläufe mit gelegentlichen hämolytischen Krisen, insbesondere nach Infekten berichtet. Nach Splenektomie neigen Patienten durch die Erhöhung von Innenkörper-Erythrozyten zu thromboembolischen Ereignissen [61, 62]. Die Mutation des Hb Mainz betrifft den gleichen Genort und ist dem Hb Köln in der klinischen Ausprägung ähnlich. Bei einem Betroffenen wurde zusätzlich über eine pulmonale Hypertonie auf dem Boden einer chronischen Hämolyse berichtet [63]. Hb Olmsted verursacht eine schwere hämolytische Anämie, Splenomegalie und Gallensteine im Kindesalter. Bei splenektomierten Patienten wurden thromboembolische Ereignisse und Priapismus beschrieben [62]. Eine Besonderheit ist das hochsauerstoffaffine Hb Zürich. Der Austausch des proximalen Histidins durch Arginin bewirkt eine weite Öffnung der Hämtasche, das Häm wird zu Methämoglobin oxidiert und bewirkt hier im Gegensatz zu den HbM-Varianten eine Destabilisierung des Hämoglobinmoleküls. Durch die weite Öffnung können sich reaktive Komponenten, wie Sulfonamide oder Methylenblau direkt an das Häm binden und zur Methämoglobinbildung beitragen, was zu schweren Hämolysen führt. Die Kohlenmonoxidbindungsfäähigkeit (CO) wird durch die sterischen Verhältnisse im Molekül deutlich verstärkt und stabilisiert durch Verhinderung der Methämoglobinbildung das Hämoglobinmolekül. Daher haben betroffene Raucher mit hohen COHb-Werten deutlich weniger stark ausgeprägte hämolytische Anämien. Die klinische Ausprägung des Hb Zürich variiert, es wurden schwere hämolytische Anämien beschrieben, die sich durch Splenektomie besserten [64, 65]. Das leicht hochsauerstoffaffine Hb Hasharon zeigt bei vielen Betroffenen keine hämatologischen Auffälligkeiten. Es kommt zu einer leicht verminderten α-Globin-Produktion und Imbalance der Globinketten ohne klinisch eine Thalassämie zu verursachen. Allerdings wurde über hämolytische Anämien mit Splenomegalien berichtet. Oxidative Substanzen (Tabelle 2) und Infekte können Hämolysen triggern. Diese Variante wird sehr häufig bei aschkenasischen Juden angetroffen, jedoch kommt sie auch bei anderen Bevölkerungsgruppen vor. Neugeborene mit Hb Hasharon können Hb H (β-Tetramere) und Hb Bart’s (γ-Tetramere) aufweisen, was zu einer Fehldiagnostik führen kann [66–68]. Hb Leiden und Hb Seattle und Hb Evans sind Varianten, bei denen ebenfalls oxidative Substanzen oder Infektionen eine Hämolyse triggern können [69, 70]. Eine nur in den ersten Lebensmonaten klinisch in Erscheinung tretenden instabile γ-Globin-Variante ist das Hb Poole und macht sich durch Ikterus und gering ausgeprägter hämolytischer Anämie bemerkbar [71].
Hyperinstabile und dominante thalassämische Varianten
Hyperinstabile Varianten kennzeichnen sich durch Proteolyse der Globinketten in den Vorläuferzellen der Erythrozytenentwicklung. Die Mutationen verursachen häufig verkürzte oder verlängerte Globinketten, die eine stabile Hämoglobinsynthese verhindern. Ein Überschuss der normalen Globinketten kann eine Globinsynthese-Imbalance bewirken und als Thalassämie in Erscheinung treten [58, 60].
Hyperinstabile α-Globingen-Mutationen
Hyperinstabile α-Globinketten sind klinisch aufgrund der Duplizität der Gene bestenfalls durch grenzwertige hämatologische Veränderungen auffällig. In Verbindung mit einer heterozygoten α-Thalassämie kann eine mehr oder minder schwere HbH-Erkrankung vorherrschen. Einige hyperinstabile α-Varianten kommen gehäuft in Gebieten vor, in denen die α-Thalassämie endemisch ist. Hb Constant Spring bewirkt eine Verlängerung des α-Globins. Eine Instabilität der mRNA bewirkt eine verminderte Translation des Globingens. Durch den hydrophoben Überschuss der α-Kette kommt es zur Präzipitation im Erythrozyten, β-Globinpräzipate verursachen zusätzlich einen Zellschaden [72, 73]. Diese Variante und auch die Varianten Hb Icaria und Hb Koya Dora, verursachen ein α-thalassämisches Krankheitsbild [74, 75]. Hb Taybe ist besonders verbreitet bei Arabern in Israel. In Kombination mit einer α-Thalassämie ist der Phänotyp sehr unterschiedlich [76]. Das Hb Heraklion verursacht in kombinierter Heterozygotie eine atypische, milde HbH-Erkrankung [77]. Hb Agrinio ist extrem instabil und kann nur durch DNA-Sequenzierung detektiert werden. Die Mutation ist in Griechenland relativ häufig und führt in Verbindung mit einer α-Thalassämie zu schweren Formen der HbH-Erkrankung, wie auch das Hb Quong Sze, das hauptsächlich in China vorkommt [78, 79]. Hb Petah Tikva führt hingegen in Kombination zu einer milden Form der HbH-Erkrankung und das Hb Questembert nur zu einer milden chronischen hämolytischen Anämie, die sich unter oxidativem Stress verstärkt [80, 81].
Hyperinstabile β-Globingen-Mutationen
Mutation die zu hyperinstabilen β-Globinketten führen, werden zu den dominanten β-Thalassämien gezählt. Die Missens-, Framshift-, Nonsens-Mutationen führen hier häufig auch zu einer Verkürzung oder Verlängerung des β-Globins. Mutation, die das Exon 3 betreffen, haben im allgemeinen stärkere Auswirkungen, da die veränderte mRNA nicht im Zellkern abgebaut wird und eine Proteinbiosynthese auslöst, bei der Globinfragmente entstehen, die proteolytisch abgebaut werden [58]. Der Phänotyp ist im Gegensatz zu den hyperinstabilen α-Globinen interindividuell sehr ähnlich. Der Phänotyp des Hb Indianapolis in heterozygoter Anlage wird in der Literatur widersprüchlich beschrieben. Einerseits wurde über eine Familie mit Thalassaemia major berichtet mit regelmäßiger Transfusionsbedürftigkeit und Organschäden durch Eisenüberladung. Anderseits haben Betroffene keinerlei hämatologische Veränderungen [82, 83]. Hb Stara Zagora ist eine Variante mit verkürzter Globinkette und löst durch Verhinderung einer Dimerbildung eine unmittelbare Proteolyse aus. Eine transfusionsbedürftige hämolytische Anämie ist die Folge mit dem Krankheitsbild einer Thalassämie intermedia/major [84]. Beim Hb Jambol führt die Globinkettenverlängerung zu dem Krankheitsbild einer Thalassämie intermedia/major ab dem 2. Lebensmonat [85]. Hb Genova ist das Pendant zum Hb Agrinio (s.o.) und verursacht ein ausgeprägtes thalassämisches Syndrom in Kombination mit einer heterozygoten β-Thalassämie eine äußerst schwere Thalassaemia major [48]. Hb Florida bewirkt eine Verlängerung des Globins. Betroffene benötigen gelegentliche Bluttransfusionen und entwickeln eine Hepatosplenomegalie, dem Krankheitsbild einer β-Thalassaemia intermedia [86]. Hb Grand Junction verursacht hingegen nur einen milden thalassämischen Phänotyp [87]. Eine konsequente Einteilung und Klassifizierung der Varianten ist in vielen Fällen nicht sinnvoll. Hämolyse und thalassämisches Krankheitsbild können gleichermaßen von Bedeutung sein.
HbA1c-Analytik
Viele Varianten werden in der Literatur als klinisch unauffällig beschrieben. Bei gleicher Mutation kann sich jedoch eine Variante prinzipiell phänotypisch sehr unterschiedlich ausprägen. Sicher ist, dass einige Varianten die HbA1c-Diagnostik durch ihre Instabilität (verkürzte Erythrozytenüberlebenszeiten) oder die Quantifizierung durch Störung der Analysenmethode beeinträchtigen [88]. Hb Venusberg und Hb Bonn (s.o.) gehören ebenfalls dazu. Als Beispiel dient das Hb UKB, das chromatografisch nicht detektierbar ist, aber elektrophoretisch, es zeigt sich hier mit einer Fraktion von 50,9%. Der HbA1c-Wert liegt in der HPLC bei 5,2%. Eine immunologische Messung allerdings ergab einen HbA1c-Wert von 6,5%. Der 73jährige Patient, bei dem erstmals diese Variante beschrieben wurde, litt seit einigen Jahren an Diabetes mellitus Typ II mit dialysepflichtiger, diabetischer Nephropathie. Möglicherweise haben hier falsch niedrige HbA1c-Werte die Therapie des Diabetes mellitus negativ beeinflusst [89]. Der gleiche Mutationsort des Hb Hafnia verursacht ebenfalls falsch niedrige HbA1c-Werte, wie einige andere sogenannte „Silent-Varianten“ [90, 91].
Diagnostik der Varianten
Die Indikation zur Diagnostik auf Hämoglobinvarianten kann vielfältig sein und setzt das Wissen um das breite Spektrum der klinischen Erscheinungsbilder voraus. Diese können Methämoglobinämien, Zyanosen, Hämolysen, Polyglobulien etc. umfassen (Abbildung 2). Für die Diagnostik von Hämoglobinvarianten ist es daher notwendig, dass die Fragestellung an das Labor kommuniziert wird und ein Austausch mit dem behandelnden Arzt stattfindet. Die Spezialisten in den Laboratorien sollten auch über die klinischen Symptome, Konsequenzen und Therapien der Varianten beraten können. Die Erfahrung des Untersuchers mit bestimmten Untersuchungsverfahren spielt hier eine große Rolle. Es werden häufig eigene verfahrensabhängige Datenbanken für Hämoglobinvarianten erstellt, die sich aus jahrelanger Routine speisen [92]. Einige Hersteller von Analysesystemen stellen auch Sammlungen zur Verfügung, die jedoch nur der Orientierung dienen sollten und nicht zur Diagnosestellung. Hier besteht die Gefahr von Fehldiagnosen, da sich mit jedem Hb-Trennverfahren unterschiedliche Varianten mit gleicher Laufzeit oder in der gleichen Zone darstellen können. Auch unauffällige Hb-Chromatografien oder -Elektrophoresen schließen eine Hb-Variante nicht aus, wenn klinische Erscheinungsbilder (s.o.) durch andere Erkrankungen nicht erklärt werden können. Bei seltenen Varianten sollten zur Diagnosesicherung 2 unterschiedliche Hb-Trennverfahren eingesetzt werden, sinnvoll sind chromatografische und elektrophoretische Verfahren. Einige Varianten zeigen sich nur in einem Verfahren oder es zeigen sich unterschiedliche Varianten-Anteile. Letztendlich muss in den meisten Fällen eine molekulargenetische Analyse durch Globin-Gensequenzierung durchgeführt werden, um eine sichere Diagnose zu stellen. Es ist in jedem Fall notwendig die Untersuchungsverfahren bis zur Diagnosefindung genau aufzulisten. Patienten mit Hämoglobinvarianten haben zum Teil eine lange Anamnese von kostspieligen und belastenden Untersuchungen hinter sich, bis eine Diagnose endgültig gestellt wird.
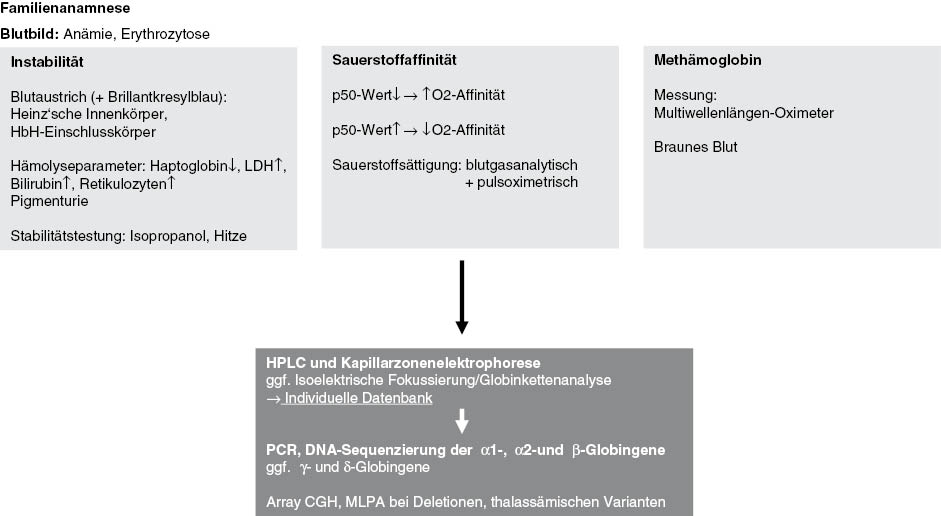
Stufendiagnostik von Hämoglobinvarianten.
Zur kompletten Diagnostik gehören: Blutbild mit Erythrozytenindizes und Retikulozytenzahlen zur Einordnung der Anämie, Serumferritin als Marker für den Eisenspeicher, Serumhaptoglobin als Hämolysemarker. Die meisten Varianten besitzen eine gewisse Instabilität, sie sind häufig grenzwertig normozytär und normochrom bei erhöhten Retikulozytenzahlen und erniedrigter Haptoglobinkonzentration. Bei Varianten mit erhöhter Sauerstoffaffinität können zu den Hämolyseparametern erhöhte Ferritinwerte auf eine Eisenüberladung hinweisen. Eine erweiterte Diagnostik sollte in der Entscheidung des Spezialisten liegen. Wichtige Hinweise können die Hämoglobinchromatografie und/oder Hämoglobinelektrophorese/Hämoglobin-Kapillarzonenelektrophorese liefern, allerdings sind in einigen Fällen die Hämoglobinvarianten nur in einer Untersuchungsmethode oder in gar keiner nachzuweisen. Die Isoelektrische Fokussierung ermöglicht aufgrund der Auftrennung der Hämoglobinfraktionen anhand eines pH-Gradienten noch zusätzliche Identifikationsmöglichkeiten, wenn die HPLC oder Elektrophorese unauffällig ist. Eine Globinkettenanalyse mit HPLC oder Massenspektrometrie kann in einigen Fällen notwendige Hinweise liefern, insbesondere bei einer Globinkettenimbalance. Bei vielen instabilen und hyperinstabilen Varianten ist die Diagnostik generell schwierig. Die Hämoglobinstabilitätstestung ist bestenfalls ein Anhalt, aber keine sichere Analyse, da sich die Varianten in vitro und in vivo unterschiedlich verhalten können. Varianten, die im Knochenmark durch Proteolyse denaturiert werden, können oftmals nur durch genetische Analysen identifiziert werden [92–94]. Blutausstriche sollten in der Pappenheim- und Brilliant-Cresyl-Blau-Färbung für die mikroskopische Beurteilung von Innenkörpern und Einschlusskörpern vorliegen. Die Beurteilung sollte durch einen Fachmann erfolgen. Die Sauerstoffaffinitätsmessung durch Blutgasanalyse ist notwendig um die Hämoglobinvariante zu klassifizieren. Am aussagekräftigsten ist der p50-Wert, der bei Verminderung eine Variante mit erhöhter Sauerstoffaffinität aufdeckt und bei Erhöhung eine Variante mit erniedrigter Sauerstoffaffinität [95]. Man sollte bedenken, dass der p50-Wert jedoch auch bei erhöhtem oder erniedrigtem Kohlendioxid-Partialdruck durch respiratorische, pulmonale und kardiale Grunderkrankungen verändert sein kann. Auch die Pulsoximetrie kann aufgrund erniedrigter Sauerstoffsättigungen zur Diagnostik beitragen. Diskrepante blutgasanalytische und pulsoximetrische Sättigungswerte sind ein deutlicher Hinweis auf eine Hb-Variante, wie beim Hb Bonn (s.o.). Hier kann auch eine spektralphotometrische Hämoglobinanalyse nützlich sein. Bei Verdacht auf Methämoglobin-Varianten, können herkömmliche Messmethoden des Methämoglobins versagen und es sollten Messungen nicht mit herkömmlichen 2-Wellenlängen-Oximetern durchgeführt werden, sondern mit geeigneten Multiwellenlängen-Pulsoximetern. Auch wenn eine Variante eindeutig detektiert wurde, sollte man unter Umständen weitere differentialdiagnostische Untersuchungen durchführen, da nicht nur die Variante die jeweilige Symptomatik alleine verursachen kann.
Therapie
Viele Varianten ziehen keine therapeutischen Konsequenzen nach sich. Allerdings werden zum Teil unnötige medizinische Untersuchungen durchgeführt, die eine Belastung für den Betroffenen darstellen und erhebliche Kosten verursachen können. Bei einigen Varianten kann die Gabe von oxidierenden Substanzen oder durch Infekte hämolytische Krisen ausgelöst werden können (Tabelle 2). Dies betrifft unter anderem die Methämoglobinvarianten, bei denen die Gabe von Methylenblau Hämolysen verursachen kann. Das Methämoglobin kann hier nicht reduziert werden, was jedoch auch differentialdiagnostisch genutzt wird, da sich das Methämoglobin bei Methämoglobinämien anderer Ursachen reduzieren lässt [96]. Niedrigsauerstoffaffinie Varianten können aufgrund niedriger Sauerstoffsättigungswerte zur Überreaktion führen. HbA1c-Bestimmungen können falsch sein, was zu falschen therapeutischen Konsequenzen bei der Behandlung des Diabetes mellitus führen kann [91]. Viele Hämoglobinvarianten benötigen keine gezielte Therapie. Bei Varianten mit niedriger Sauerstoffaffinität ist eine Therapie in aller Regel nicht möglich, ob eine gelegentliche Sauerstofftherapie die Symptome bessert, ist unklar. Varianten mit erhöhter Sauerstoffaffinität und Polyglobulie erfordern zum Teil regelmäßige Aderlässe, um ein Hyperviskositätssyndrom zu vermeiden [97]. Die instabilen Varianten können je nach Ausprägung gelegentliche oder regelmäßige Bluttransfusionen erfordern. Eine Splenektomie ist häufig indiziert, erhöht aber das Risiko von Infektionen und Thromboembolien durch Innenkörper-Erythrozyten. Hydroxyurea-Therapien wurden gelegentlich durchgeführt [98]. Bei thalassämischen Varianten können regelmäßige Bluttransfusionen mit gleichzeitiger Chelat-Therapie notwendig sein [57]. Bei Varianten mit erhöhtem Zellumsatz ist eine regelmäßige Gabe von Folsäure sinnvoll.
Autorenbeteiligung: Alle Autoren tragen Verantwortung für den gesamten Inhalt dieses Artikels und haben der Einreichung des Manuskripts zugestimmt.
Forschungsförderung: Keine.
Interessenkonflikt: Kein Interessenkonflikt.
Literatur
1. Williams TN, Weatherall DJ. World distribution, population genetics, and health Burden of the hemoglobinopathies. Cold Spring Harb Perspect in Med 2012;2:a011692.10.1101/cshperspect.a011692Suche in Google Scholar
2. Perutz MF, Wilkinson AJ, Paoli M, Dodson GG. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev of Biophys Biomol Struct 1998;27:1–34.10.1146/annurev.biophys.27.1.1Suche in Google Scholar
3. Riggs AF. The Bohr effect. Annu Rev Phys1988;50:181–204.10.1146/annurev.ph.50.030188.001145Suche in Google Scholar
4. Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol 2005;33:259–71.10.1016/j.exphem.2004.11.007Suche in Google Scholar
5. Rumi E, Passamonti F, Pagano L, Ammirabile M, Arcaini L, Elena C, et al. Blood p50 evaluation enhances diagnostic definition of isolated erythrocytosis. J Intern Med 2009;265:266–74.10.1111/j.1365-2796.2008.02014.xSuche in Google Scholar
6. Lombardero M, Kovacs K, Scheithauer BW. Erythropoietin: a hormone with multiple functions. Pathobiology 2011;78:41–53.10.1159/000322975Suche in Google Scholar
7. Baldwin J, Chothia C. Haemoglobin: the structural changes related to ligand binding and its allosteric mechanism. J Mol Biol 1979;129:175–220.10.1016/0022-2836(79)90277-8Suche in Google Scholar
8. Ohba Y. Unstable Hemoglobins. Hemoglobin 1990;14:353–88.10.3109/03630269009031998Suche in Google Scholar PubMed
9. Strader MB, Hicks WA, Kassa T, Sigleton E, Soman J, Olsen JS, et al. Post-translational transformation of methionine to aspartate is catalyzed by heme iron and driven by Peroxide a novel subunit-specific mechanism in hemoglobin. J Biol Chem 2014;289:22342–57.10.1074/jbc.M114.568980Suche in Google Scholar PubMed PubMed Central
10. Kohne E, Kleihauer E. Hemoglobinopathies. a longitudinal study over four decades. Dtsch Arztebl Int 2010;107:65–71.10.3238/arztebl.2010.0065Suche in Google Scholar
11. Agarwal N, Mojica-Henshaw MP, Simmons ED, Hussey D, Ou CN, Prchal JT. Familial polycythemia caused by a novel mutation in the beta globin gene: essential role of P50 in evaluation of familial polycythemia. Int J Med Sci 2007;4:232–6.10.7150/ijms.4.232Suche in Google Scholar PubMed PubMed Central
12. David O, Ivaldi G, Rabino-Massa E, Ricco G. Functional studies on nine different haemoglobins with high oxygen affinity. Acta Haematol 2002;108:132–8.10.1159/000064702Suche in Google Scholar PubMed
13. Maniatis A, Bousios T, Nagel RL, Balazs T, Ueda Y, Bookchin RM, et al. Hemoglobin Crete (beta 129 ala leads to pro). a new high-affinity variant interacting with beta 0 -and delta beta 0 -thalassemia. Blood 1979;54:54–63.10.1182/blood.V54.1.54.54Suche in Google Scholar
14. Kister J, Baudin-Creuza V, Kiger L, Préhu C, Papassotiriou I, Riou J, et al. Hb Montfermeil [beta 130(H8) Tyr – Cys]: suggests a key role for the interaction between helix A and H in oxygen affinity of the hemoglobin molecule. Blood Cells Mol Dis 2005;34:166–73.10.1016/j.bcmd.2004.12.003Suche in Google Scholar PubMed
15. Imai K. Hemoglobin Chesapeake (92 alpha, arginine–leucine). Precise measurements and analyses of oxygen equilibrium. J Biol Chem 1974;249:7607–12.10.1016/S0021-9258(19)81281-4Suche in Google Scholar
16. Wajcman H, Kister J, M’Rad A, Promé D, Milipied N, Rapp MJ, et al. Hb Saint Nazaire (beta 103[G5]Phe–Ile): a new example of polycythemia due to a hemoglobin variant with increased oxygen affinity. Am J Hematol 1993;44:16–21.10.1002/ajh.2830440105Suche in Google Scholar PubMed
17. Giordano PC, Harteveld CL, Brand A, Willems LN, Kluin-Nelemans HC, Plug RJ, et al. Hb Malmö [beta-97(FG-4)His–Gln] leading to polycythemia in a Dutch family. Ann Hematol 1996;73:183–8.10.1007/s002770050225Suche in Google Scholar PubMed
18. Rochette J, Barnetson R, Kiger L, Kister J, Littlewood TJ, Webster R, et al. Association of a novel high oxygen affinity haemoglobin variant with delta beta thalassaemia. Br J Haematol 1994;86:118–24.10.1111/j.1365-2141.1994.tb03261.xSuche in Google Scholar PubMed
19. Ikkala E, Koskela J, Pikkarainen P, Rahiala EL, El-Hazmi MA, Nagai K, et al. Hb Helsinki: a variant with a high oxygen affinity and a substitution at a 2,3-DPG binding site (beta82[EF6] Lys replaced by Met). Acta Haematol 1976;56:257–75.10.1159/000207947Suche in Google Scholar PubMed
20. Abraham B, Hicks W, Jia Y, Baek JH, Miller JL, Alayash AI. Isolated Hb Providence β82Asn and β82Asp fractions are more stable than native HbA(0) under oxidative stress conditions. Biochem 2011;50:9752–66.10.1021/bi200876eSuche in Google Scholar PubMed
21. Sugihara J, Imamura T, Nagafuchi S, Bonaventura J, Bonaventura C, Cashon R. Hemoglobin Rahere, a human hemoglobin variant with amino acid substitution at the 2,3-diphosphoglycerate binding site. Functional consequences of the alteration and effects of bezafibrate on the oxygen bindings. J Clin Invest 1985;76:1169–73.10.1172/JCI112072Suche in Google Scholar PubMed PubMed Central
22. Delanoe-Garin J, Blouquit Y, Arous N, Kister J, Poyart C, North ML, et al. Hemoglobin Saverne: a new variant with elongated beta chains: structural and functional properties. Hemoglobin 1988;12:337–52.10.3109/03630268808998034Suche in Google Scholar PubMed
23. Ranney HM, Sharma VS, Noble RW. Unstable hemoglobins and thalassemia: some properties of Hb Köln. Ann N Y Acad Sci 1974;232:293–6.10.1111/j.1749-6632.1974.tb20593.xSuche in Google Scholar
24. Percy MJ, Butt NN, Crotty GM, Drummond MW, Harrison C, Jones GL, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematol 2009;94:1321–2.10.3324/haematol.2009.008037Suche in Google Scholar
25. Raybourne S, Stallings M, Gravely M, Huisman T. Oxygen equilibrium analyses of isolated hemoglobins A2, Lepore-Washington and P-Nilotic. Biochim Biophys Acta 1978;535:78–84.10.1016/0005-2795(78)90034-XSuche in Google Scholar
26. Papassotiriou I, Kister J, Griffon N, Abraham DJ, Kanavakis E, Traeger-Synodinos J, et al. Synthesized allosteric effectors of the hemoglobin molecule: A possible mechanism for improved erythrocyte oxygen release capability in hemoglobinopathy H disease. Exp Hematol 1998;26:922–6.Suche in Google Scholar
27. Stamatoyannopoulos G, Bellingham AJ, Lenfant C, Finch CA. Abnormal hemoglobins with high and low oxygen affinity. Annu Rev Med 1971;22:221–34.10.1146/annurev.me.22.020171.001253Suche in Google Scholar
28. Bonaventura J, Riggs A. Hemoglobin Kansas, a human hemoglobin with a neutral amino acid substitution and an abnormal oxygen equilibrium. J Biol Chem 1968;243:980–91.10.1016/S0021-9258(18)93612-4Suche in Google Scholar
29. Nagel RL, Lynfield J, Johnson J, Landau L, Bookchin RM, Harris MB. Hemoglobin Beth Israel. A mutant causing clinically apparent cyanosis. N Engl J Med 1976;295:125–30.10.1056/NEJM197607152950302Suche in Google Scholar
30. Villegas A, Malcorra JJ, Balda I, Calero F, Porres A, Alvarez-Sala JL, et al. A new Spanish family with Hb Louisville. Am J Med Genet A 1989;32:9–14.10.1002/ajmg.1320320103Suche in Google Scholar
31. Abdulmalik O, Safo MK, Lerner NB, Ochotorena J, Daikhin E, Lakka V, et al. Characterization of hemoglobin bassett (alpha94Asp–Ala), a variant with very low oxygen affinity. Am J Hematol 2004;77:268–76.10.1002/ajh.20184Suche in Google Scholar
32. Srabler SP, Jones RT, Head C, Shih DT, Fairbanks VF. Hemoglobin denver [α2β241 (C7) Phe→Ser]: A Low-O2-Affinity variant associated with chronic cyanosis and anemia. Mayo Clin Proc 1994;69:237–43.10.1016/S0025-6196(12)61062-3Suche in Google Scholar
33. Sharma VS, Newton GL, Ranney HM, Ahmed F, Harris JW, Danish EH. Hemoglobin Rothschild (β37(C3)Trp → Arg): A high/low affinity hemoglobin mutant. J Mol Biol 1980;144:267–80.10.1016/0022-2836(80)90090-XSuche in Google Scholar
34. Zur B, Mayer-Hubner B, Ludwig M, Stoffel-Wagner B. A 14-year-old boy with chronic cyanosis, mild anemia, and limited physical resistance to stress. Clin Chem 2012;58:332–5.10.1373/clinchem.2010.158584Suche in Google Scholar PubMed
35. Abdu A, Gómez-Márquez J, Aldrich TK. The oxygen affinity of sickle hemoglobin. Resp Phys Neurobiol 2008;161:92–4.10.1016/j.resp.2007.12.005Suche in Google Scholar PubMed
36. Chan ED, Chan MM, Chan MM. Pulse oximetry: understanding its basic principles facilitates appreciation of its limitations. Resp Med 2013;107:789–99.10.1016/j.rmed.2013.02.004Suche in Google Scholar PubMed
37. Verhovsek M, Henderson MP, Cox G, Luo H, Steinberg MH, Chui DH. Unexpectedly low pulse oximetry measurements associated with variant hemoglobins. Am J Hematol 2010;85:882–5.10.1002/ajh.21810Suche in Google Scholar PubMed
38. Zur B, Hornung A, Breuer J, Doll U, Bernhardt C, Ludwig M, et al. A novel hemoglobin, Bonn, causes falsely decreased oxygen saturation measurements in pulse oximetry. Clin Chem 2008;54:594–6.10.1373/clinchem.2007.095158Suche in Google Scholar PubMed
39. Percy MJ, McFerran NV, Lappin, Terry RJ. Disorders of oxidised haemoglobin. Blood rev 2005;19:61–8.10.1016/j.blre.2004.02.001Suche in Google Scholar PubMed
40. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg 2012;114:972–8.10.1213/ANE.0b013e318233041aSuche in Google Scholar PubMed
41. Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005;51:434–4.10.1373/clinchem.2004.035154Suche in Google Scholar PubMed
42. Mansouri A, Lurie AA. Concise review – methemoglobinemia. Am J Hematol 1993;42:7–12.10.1002/ajh.2830420104Suche in Google Scholar PubMed
43. Hayashi A, Shimizu A, Yamamura Y, Watari H. Hemoglobins M. Identification of Iwate, Boston, and Saskatoon variants. Science 1966;152:207–8.10.1126/science.152.3719.207Suche in Google Scholar PubMed
44. Upadhye D, Koduri P, Tarakeshwari S, Mehta P, Surve R, Warang P, et al. Hb M Hyde Park and Hb M Boston in two Indian families – a rare cause of methaemoglobinemia. Int J Lab Hematol 2015;37:e40–3.10.1111/ijlh.12281Suche in Google Scholar PubMed
45. Kutlar F, Hilliard LM, Zhuang L, Patel N, Eroglu B, Meiler SE, et al. Hb M Dothan [beta 25/26 (B7/B8)/(GGT/GAG–GAG//Gly/Glu–Glu]; a new mechanism of unstable methemoglobin variant and molecular characteristics. Blood Cells Mol Dis 2009;43:235–8.10.1016/j.bcmd.2009.08.006Suche in Google Scholar PubMed
46. Hojas-Bernal R, McNab-Martin P, Fairbanks VF, Holmes MW, Hoyer JD, McCormick DJ, et al. Hb Chile [beta 28(B10)Leu – Met]: an unstable hemoglobin associated with chronic methemoglobinemia and sulfonamide or methylene blue-induced hemolytic anemia. Hemoglobin 1999;23:125–34.10.3109/03630269908996157Suche in Google Scholar PubMed
47. Wiedermann BF, Indrak K, Wilson JB, Webber BB, Yang KG, Kutlar F, et al. Hb Saint Louis or alpha 2 beta 2(28)(B10)Leu–Gln in a Czechoslovakian male. Hemoglobin 1986;10:673–6.10.3109/03630268609036572Suche in Google Scholar PubMed
48. Badens C, Paolasso C, Fossat C, Wajcman H, Thuret I. Compound heterozygosity for unstable hemoglobin Genova and beta(o)-thalassemia associated with early onset of thalassemia major syndrome. Haematol 2005;90:ECR04.Suche in Google Scholar
49. Philippe M, Larondelle Y, Vaerman JL, Martiat P, Galacteros F, Wajcman H, et al. Hb Tübingen [alpha 2 beta (2)106(G8)Leu–Gln] in a Belgian Family. Hemoglobin 1993;17:373–8.10.3109/03630269308997490Suche in Google Scholar PubMed
50. Eandi Eberle S, Noguera NI, Sciuccati G, Bonduel M, Diaz L, Staciuk R, et al. Hb Southampton [beta106(G8)Leu–Pro, CTG–CCG] in an Argentinean boy. Hemoglobin 2006;30:401–3.10.1080/03630260600755930Suche in Google Scholar PubMed
51. Hain RD, Chitayat D, Cooper R, Bandler E, Eng B, Chui DH, et al. Hb FM-Fort Ripley: confirmation of autosomal dominant inheritance and diagnosis by PCR and direct nucleotide sequencing. Hum Mutat 1994;3:239–42.10.1002/humu.1380030310Suche in Google Scholar PubMed
52. Préhu C, Rhabbour M, Netter JC, Denier M, Riou J, Galactéros F, et al. Hb F-M-Osaka [Ggamma63(E7)His – tyr] in a newborn from southwest France. Hemoglobin 2003;27:27–30.10.1081/HEM-120018433Suche in Google Scholar
53. Dainer E, Shell R, Miller R, Atkin JF, Pastore M, Kutlar A, et al. Neonatal cyanosis due to a novel fetal hemoglobin: Hb F-Circleville [Ggamma63(E7)His–Leu, CATCTT]. Hemoglobin 2008;32:596–600.10.1080/03630260802507915Suche in Google Scholar PubMed
54. Bento C, Magalhães Maia T, Carvalhais I, Moita F, Abreu G, Relvas L, et al. Transient neonatal cyanosis associated with a new Hb F variant: Hb F viseu. J Pediatr Hematol Oncol 2013;35:e77–80.10.1097/MPH.0b013e3182667be3Suche in Google Scholar PubMed
55. Kohli-Kumar M, Zwerdling T, Rucknagel DL. Hemoglobin F-Cincinnati, alpha 2G gamma 2 (C7) Phe–Ser in a newborn with cyanosis. Am J Hematol 1995;49:43–7.10.1002/ajh.2830490108Suche in Google Scholar
56. Zimmermann-Baer U, Capalo R, Dutly F, Saller E, Troxler H, Kohler M, et al. Neonatal cyanosis due to a new (G)γ-globin variant causing low oxygen affinity: Hb F-Sarajevo [(G)γ102(G4)Asn→Thr, AACACC]. Hemoglobin 2012;36:109–13.10.3109/03630269.2012.655872Suche in Google Scholar
57. Efremov GD. Dominantly Inherited beta-Thalassemia. Hemoglobin 2007;31:193–207.10.1080/03630260701290092Suche in Google Scholar
58. Waugh SM, Low PS. Hemichrome binding to band 3: nucleation of Heinz bodies on the erythrocyte membrane. Biochem 1985;24:34–9.10.1021/bi00322a006Suche in Google Scholar
59. Schlüter K, Drenckhahn D. Co-clustering of denatured hemoglobin with band 3: its role in binding of autoantibodies against band 3 to abnormal and aged erythrocytes. Proc Natl Acad Sci USA 1986;83:6137–41.10.1073/pnas.83.16.6137Suche in Google Scholar
60. Williamson D. The unstable haemoglobins. Blood Rev 1993;7:146–63.10.1016/0268-960X(93)90002-LSuche in Google Scholar
61. Eisinger J, Flores J, Tyson JA, Shohet SB. Fluorescent cytoplasm and Heinz bodies of hemoglobin Köln erythrocytes: evidence for intracellular heme catabolism. Blood 1985;65:886–93.10.1182/blood.V65.4.886.886Suche in Google Scholar
62. Phyliky RL, Fairbanks VF. Thromboembolic complication of splenectomy in unstable hemoglobin disorders: Hb Olmsted, Hb Koln. Am J Hematol 1997;55:53.10.1002/(SICI)1096-8652(199705)55:1<53::AID-AJH15>3.0.CO;2-7Suche in Google Scholar
63. Lode HN, Krings G, Schulze-Neick I, Dähmlow S, Schroeder U, Bonnet R, et al. Pulmonary hypertension in a case of Hb-Mainz hemolytic anemia. J Pediatr Hematol Oncol 2007;29:173–7.10.1097/MPH.0b013e318032568cSuche in Google Scholar
64. Tucker PW, Phillips SE, Perutz MF, Houtchens R, Caughey WS. Structure of hemoglobins Zürich [His E7(63)beta replaced by Arg] and Sydney [Val E11(67)beta replaced by Ala] and role of the distal residues in ligand binding. Proc Natl Acad Sci USA 1978;75:1076–80.10.1073/pnas.75.3.1076Suche in Google Scholar
65. Zinkham WH, Houtchens RA, Caughey WS. Relation between variations in the phenotypic expression of an unstable hemoglobin disorder (hemoglobin Zürich) and carboxyhemoglobin levels. Am J Med 1983;74:23–9.10.1016/0002-9343(83)91113-0Suche in Google Scholar
66. Lehmann H, Vella F. Haemoglobin Hasharon. Humangenetik 1974;25:237–40.10.1007/BF00281433Suche in Google Scholar
67. Levine RL, Lincoln, Buchholz WM, Gribble TJ, Schwartz HC. Hemoglobin Hasharon in a Premature-Infant with Hemolytic-Anemia. Pediatr Res 1975;9:7–11.10.1203/00006450-197501000-00002Suche in Google Scholar
68. Zur B, Ludwig M, Stoffel-Wagner B. Hemoglobin Hasharon and hemoglobin NYU in subjects of German origin. Biochem Med 2011;21:321–5.10.11613/BM.2011.043Suche in Google Scholar
69. Bonaventura J, Bonaventura C, Amiconi G, Antonini E, Brunori M. Functional properties of hemoglobin leiden (α2Aβ26 or7 Glu deleted). Arch Biochem Biophys 1974;161:328–32.10.1016/0003-9861(74)90268-9Suche in Google Scholar
70. Huehns ER, Hecht F, Yoshida A, Stamatoyannopoulos G, Hartman J, Motulsky AG. Hemoglobin-Seattle (alpha-2 beta-2 76-Glu): an unstable hemoglobin causing chronic hemolytic anemia. Blood 1970;36:209–18.10.1182/blood.V36.2.209.209Suche in Google Scholar
71. Lee-Potter JP, Deacon-Smith RA, Simpkiss MJ, Kamuzora H, Lehmann H. A new cause of haemolytic anaemia in the newborn. A description of an unstable fetal haemoglobin: F Poole, alpha2-G-gamma2 130 trptophan yeilds glycine. J Clin Pathol 1975;28:317–20.10.1136/jcp.28.4.317Suche in Google Scholar PubMed PubMed Central
72. Wajcman H, Traeger-Synodinos J, Papassotiriou I, Giordano PC, Harteveld CL, Baudin-Creuza V, et al. Unstable and thalassemic alpha chain hemoglobin variants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin 2008;32:327–49.10.1080/03630260802173833Suche in Google Scholar PubMed
73. Hunt DM, Higgs DR, Winichagoon P, Clegg JB, Weatherall DJ. Haemoglobin constant spring has an unstable? chain messenger RNA. Br J Haematol 1982;51:405–13.10.1111/j.1365-2141.1982.tb02796.xSuche in Google Scholar PubMed
74. Efremov GD, Josifovska O, Nikolov N, Codrington JF, Oner C, Gonzalez-Redondo JM, et al. Hb Icaria-Hb H disease: identification of the Hb Icaria mutation through analysis of amplified DNA. Br J Haematol 1990;75:250–3.10.1111/j.1365-2141.1990.tb02658.xSuche in Google Scholar PubMed
75. Brennan SO, Ryken S, Chan T. Hb Koya Dora [alpha142, Term–Ser (TAATCA in alpha2)]: a rare mutation of the alpha2 gene stop codon associated with alpha-thalassemia. Hemoglobin 2010;34:402–5.10.3109/03630269.2010.486344Suche in Google Scholar PubMed
76. Pobedimskaya DD, Molchanova TP, Streichman S, Huisman TH. Compound heterozygosity for two alpha-globin gene defects, Hb Taybe (alpha 1; 38 or 39 minus Thr) and a poly A mutation (alpha 2; AATAAA–>AATAAG), results in a severe hemolytic anemia. Am J Hematol 1994;47:198–202.10.1002/ajh.2830470310Suche in Google Scholar PubMed
77. Douna V, Papassotiriou I, Metaxotou-Mavrommati A, Stamoulakatou A, Liapi D, Kampourakis D, et al. Further identification of the hyperunstable alpha-globin chain variant Hb Heraklion [codons 36/37 (-CCC); Pro–0 (alpha1)] in Greek cases with co-inherited alpha+-thalassemia mutations. Hemoglobin 2008;32:379–85.10.1080/03630260802174021Suche in Google Scholar PubMed
78. Felekis X, Phylactides M, Drousiotou A, Christou S, Kyrri A, Kyriakou K, et al. Hb Agrinio [alpha29(B10)Le–uPro (alpha2)] in combination with – (MED I). Results in a severe form of Hb H disease. Hemoglobin 2008;32:237–46.10.1080/03630260802004103Suche in Google Scholar PubMed
79. Yang Y, Lou J, Liu Y, He Y, Li D. Screening and diagnosis of Hb Quong Sze [HBA2: c.377T>C (or HBA1)] in a prenatal control program for thalassemia. Hemoglobin 2014;38:158–60.10.3109/03630269.2014.910669Suche in Google Scholar PubMed
80. Honig GR, Shamsuddin M, Zaizov R, Steinherz M, Solar I, Kirschmann C. Hemoglobin Petah Tikva (alpha 110 ala replaced by asp): a new unstable variant with alpha-thalassemia-like expression. Blood 1981;57:705–11.10.1182/blood.V57.4.705.705Suche in Google Scholar
81. Wajcman H, Vasseur C, Blouquit Y, Rosa J, Labie D, Najman A, et al. Unstable alpha-chain hemoglobin variants with factitious beta-thalassemia biosynthetic ratio. Am J Hematol 1993;42:367–74.10.1002/ajh.2830420407Suche in Google Scholar PubMed
82. Adams JG, Boxer LA, Baehner RL, Forget BG, Tsistrakis GA, Steinberg MH. Hemoglobin indianapolis (beta 112[G14] arginine). An unstable beta-chain variant producing the phenotype of severe beta-thalassemia. J Clin Invest 1979;63:931–8.10.1172/JCI109393Suche in Google Scholar PubMed PubMed Central
83. Fattori A, Kimura EM, Albuquerque DM, Oliveira DM, Costa FF, Sonati MF. Hb Indianapolis [beta112 (G14) Cys–Arg] as the probable cause of moderate hemolytic anemia and renal damage in a Brazilian patient. Am J Hematol 2007;82:672–5.10.1002/ajh.20860Suche in Google Scholar PubMed
84. Petkov GH, Simjanovska L, Tchakarova P, Efremov GD. Hb Stara Zagora. a new hyper-unstable hemoglobin causing severe hemolytic anemia. Hemoglobin 2005;29:249–56.10.1080/03630260500307766Suche in Google Scholar PubMed
85. Efremov GD, Simjanovska L, Plaseska-Karanfilska D, Stanojevic E, Petkov GH. Hb Jambol: a new hyperunstable hemoglobin causing severe hemolytic anemia. Acta Haematol 2007;117:1–7.10.1159/000096783Suche in Google Scholar PubMed
86. Weinstein BI, Erramouspe B, Albuquerque DM, Olivera DM, Kimura EM, Costa FF, et al. Hb Florida: a novel elongated C-terminal beta-globin variant causing dominant beta-thalassemia phenotype. Am J Hematol 2006;81:358–60.10.1002/ajh.20561Suche in Google Scholar
87. Kent MW, Oliveira JL, Hoyer JD, Swanson KC, Kluge ML, Dawson DB, et al. Hb Grand Junction (HBB: c.348_349delinsG; p.His117IlefsX42): a new hyperunstable hemoglobin variant. Hemoglobin 2014;38:8–12.10.3109/03630269.2013.853672Suche in Google Scholar
88. Schnedl WJ, Krause R, Halwachs-Baumann G, Trinker M, Lipp RW, Krejs GJ. Evaluation of HbA1c determination methods in patients with hemoglobinopathies. Diabetes care 2000;23:339–44.10.2337/diacare.23.3.339Suche in Google Scholar
89. Zur B, Stoffel-Wagner B, Ludwig M. Novel Hemoglobin UKB demonstrates the importance of using different methods of detection. Clin Chim Acta 2014;431:58–9.10.1016/j.cca.2014.01.021Suche in Google Scholar
90. Blanke S, Johnsen A, Wimberley PD, Mortensen HB. Hemoglobin Hafnia: α2(β116 (G18)His→Gln)2; a new hemoglobin variant mistaken for glycated hemoglobin. Biochim Biophys Acta 1988;955:214–9.10.1016/0167-4838(88)90195-1Suche in Google Scholar
91. Lahousen T, Roller RE, Lipp RW, Schnedl WJ. Silent haemoglobin variants and determination of HbA(1c) with the HPLC Bio-Rad Variant II. J Clin Pathol 2002;55:699–703.10.1136/jcp.55.9.699Suche in Google Scholar PubMed PubMed Central
92. Wajcman H, Prehu C, Bardakdjian-Michau J, Promé D, Riou J, Godart C, et al. Abnormal hemoglobins: laboratory methods. Hemoglobin 2001;25:169–81.10.1081/HEM-100104026Suche in Google Scholar PubMed
93. Greene DN, Vaughn CP, Crews BO, Agarwal AM. Advances in detection of hemoglobinopathies. Clin Chim Acta 2015;439:50–7.10.1016/j.cca.2014.10.006Suche in Google Scholar PubMed
94. Ryan K, Bain BJ, Worthington D, James J, Plews D, Mason A, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. Br J Haematol 2010;149:35–49.10.1111/j.1365-2141.2009.08054.xSuche in Google Scholar PubMed
95. Burnett RW. Screening for hemoglobin variants with abnormal oxygen affinity. Clin Chem 2002;48:391.10.1093/clinchem/48.2.391aSuche in Google Scholar
96. Spears F, Banerjee A. Hemoglobin M variant and congenital methemoglobinemia: methylene blue will not be effective in the presence of hemoglobin M. Can J Anaesth 2008;55:391–2.10.1007/BF03021499Suche in Google Scholar PubMed
97. Wajcman H, Galactéros F. Hemoglobins with high oxygen affinity leading to erythrocytosis. New variants and new concepts. Hemoglobin 2005;29:91–106.10.1081/HEM-58571Suche in Google Scholar
98. Rose C, Bauters F. Hydroxyurea therapy in highly unstable hemoglobin carriers. Blood 1996;88:2807–8.10.1182/blood.V88.7.2807.bloodjournal8872807Suche in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Editorial
- Aktuelle Diagnostik in der Hämatologie
- Hämatologie/Hematology
- Stufendiagnostik zur Abklärung von krankhaften Veränderungen der Leukozyten
- Rationale Anämieabklärung
- Diagnostik von myelodysplastischen Syndromen (MDS) und akuten myeloischen Leukämien (AML)
- Differenzialdiagnose BCR-ABL1-negativer myeloproliferativer Neoplasien
- Hämoglobinvarianten – Pathomechanismus, Symptome und Diagnostik
- Molekulargenetische und zytogenetische Diagnostik/Molecular-Genetic and Cytogenetic Diagnostics
- An update of novel identified cytogenetic and molecular biomarkers in the laboratory diagnosis of hematological neoplasia
- Stufendiagnostik der Hämoglobinopathien
- Rational laboratory diagnostics of primary immunodeficiency disorders
Artikel in diesem Heft
- Frontmatter
- Editorial
- Aktuelle Diagnostik in der Hämatologie
- Hämatologie/Hematology
- Stufendiagnostik zur Abklärung von krankhaften Veränderungen der Leukozyten
- Rationale Anämieabklärung
- Diagnostik von myelodysplastischen Syndromen (MDS) und akuten myeloischen Leukämien (AML)
- Differenzialdiagnose BCR-ABL1-negativer myeloproliferativer Neoplasien
- Hämoglobinvarianten – Pathomechanismus, Symptome und Diagnostik
- Molekulargenetische und zytogenetische Diagnostik/Molecular-Genetic and Cytogenetic Diagnostics
- An update of novel identified cytogenetic and molecular biomarkers in the laboratory diagnosis of hematological neoplasia
- Stufendiagnostik der Hämoglobinopathien
- Rational laboratory diagnostics of primary immunodeficiency disorders