Nachweis von schweren angeborenen Immundefekten bei Neugeborenen: Diagnostisches Vorgehen bei Screening, Tracking und Follow-up
-
Stephan Borte
 ,
Michael Borte
,
Michael Borte

Zusammenfassung
In den vergangenen Jahren hat die Messung von T-cell receptor excision circles (TRECs) zum Nachweis von schweren kombinierten T-zellulären Immundefekten (SCID) Einzug in das reguläre Neugeborenenscreening gehalten. Mit der diagnostischen Erweiterung hin zu kappa-deleting recombination excision circles (KRECs) ist dies nun auch für schwere B-zelluläre Immundefekte (primäre Agammaglobulinämien) möglich. Hierbei müssen molekulare Technologien in die Screeninglabore eingeführt werden, die hohe Ansprüche an die Leistungsfähigkeit, das Auflösungsverhalten und die internationale Vergleichbarkeit der Methoden zur Messung von TRECs und KRECs stellen. Angesichts der klinischen Heterogenität der identifizierten Neugeborenen und der diagnostischen Herausforderungen in diesem Altersabschnitt sind zielführende Strategien für das Screening, Tracking und Follow-up erforderlich. In diesem Beitrag diskutieren wir die Möglichkeiten und das Zusammenwirken diagnostischer Verfahren zur zeitnahen Bestätigungsdiagnostik und therapeutischen Klassifikation von Neugeborenen mit schweren angeborenen Immundefekten.
Abstract
Primary immunodeficiencies such as severe combined immunodeficiency and X-linked agammaglobulinemia (Bruton’s disease) are some of the latest additions to population-scale newborn screening campaigns. To identify these diseases at the neonatal stage, molecular technology to detect T-cell receptor excision circles (TRECs) and kappa-deleting recombination excision circles (KRECs) has finally entered newborn screening laboratories, posing considerable demands towards resolution performance and comparability of the TREC and KREC assays used. Moreover, the heterogeneity of patients positively screened by these assays, and the diagnostic challenges in neonates, require comprehensive strategies for screening, tracking and follow-up. In this article, we discuss diagnostic possibilities and render the frame for rapid confirmation and therapeutic classification of neonates with severe primary immunodeficiencies originating from newborn screening programs.
Rezensierte Publikation:
Sack U. Conrad K.
Einleitung
Primäre Immundefekte (PID) sind angeborene Störungen der Immunkompetenz, die sich hauptsächlich durch eine pathologische Infektanfälligkeit äußern. Hierbei herrschen häufig schwere rekurrierende Infektionen mit therapieresistenten, langen Verläufen vor. Zudem finden sich assoziierte Störungen der Immunregulation, die sich in Granulombildung, Autoimmunität, rezidivierendem Fieber, ekzematösen Hauterkrankungen, Lymphoproliferation und chronischen Darmentzündungen manifestieren können [1]. Mehr als 240 Krankheitsentitäten sind bisher als PID definiert worden und ebenso umfangreich stellt sich das Spektrum des klinischen Schweregrades dar [2]. Während die häufigsten angeborenen Immundefekte, wie z.B. die selektive IgA-Defizienz oder der C2-Komplementmangel einen milden Phänotyp aufweisen, der häufig unentdeckt bleibt, sind schwere angeborene Immundefekte mit einer beträchtlichen Mortalität in den ersten Lebensjahren, sowie einer schwerwiegenden Morbidität mit irreversibler Organschädigung behaftet. Dies betrifft insbesondere primäre Immundefekte, die durch das Fehlen oder die funktionelle Anergie von T-Lymphozyten (schwerer kombinierter Immundefekt; SCID) oder B-Lymphozyten (Agammaglobulinämie, z.B. X-linked agammaglobulinemia; XLA) gekennzeichnet sind. Patienten mit solchen schweren angeborenen Immundefekten erscheinen bei Geburt zunächst vollkommen gesund und zeigen erste Manifestationen beim SCID zwischen dem 14. Lebenstag und dem 4. Lebensmonat, und bei XLA-Patienten üblicherweise zwischen dem 8. und 16. Lebensmonat. Obwohl die mütterliche Leihimmunität einen gewissen Abwehrschutz vermittelt, schützt dieser in der Regel auch in den ersten Lebensmonaten nur unzureichend.
Sobald diese passive Antikörperbarriere nachlässt, treten schwerwiegende und zumeist fatal verlaufende Infektionen des respiratorischen und gastrointestinalen Systems auf. Aufgrund der fehlenden eigenen Immunkompetenz der Patienten sind schwere angeborene Immundefekte, insbesondere der SCID, daher immer als pädiatrischer Notfall anzusehen.
Die frühestmögliche Diagnose schwerer angeborener Immundefekte verbessert deren Prognose und Therapieeffizienz erheblich [3–5]. Dies begründet sich vor allem aus der Möglichkeit, frühzeitig Präventionsmaßnahmen zur Vermeidung von Infektionen einzuleiten, sowie iatrogene Schädigungen, z.B. durch die Verabreichung empfohlener Schutzimpfungen, wie z.B. der für immunkompetente Säuglinge ungefährlichen Rotavirus-Impfung (Lebendimpfstoff), abzuwenden [6, 7]. Die Vermeidung solcher Komplikationen verbessert das Gesamtüberleben vor und nach Durchführung einer kurativen hämatopoetischen Stammzelltransplantation, Gentherapie oder supportiven Enzymersatztherapie bzw. Immunglobulin-Substitutionstherapie erheblich [3]. Aufgrund der besonderen Bedeutung einer frühzeitigen präsymptomatischen Diagnose von Patienten mit schweren angeborenen Immundefekten erscheint die Durchführung von Neugeborenenuntersuchungen im Sinne eines Screeningtests sinnvoll [8].
Screeningtest
Zur Rechtfertigung von populationsbezogenen Screeninguntersuchungen sind bereits in den 1960er Jahren Bewertungskriterien von James Wilson und Gunner Jungner aufgestellt worden, die durch entsprechende Zusatzkriterien von Anne Andermann und Michael Petros ergänzt worden sind, um insbesondere Neugeborenenuntersuchungen und genetische Screeningdiagnostik zu evaluieren [8]. Bei Anwendung der Kriterien muss zwischen der Zielerkrankung, die ausgeschlossen werden soll, und dem anzuwendenden Screeningmarker unterschieden werden. Während sich bei einer Reihe metabolischer Erkrankungen der jeweilige Screeningmarker direkt der Pathogenese einer Zielerkrankung zuordnen lässt (z.B. Biotinidasemangel oder klassische Galaktosämie), sind die Anforderungen an Screeningmarker für schwere angeborene Immundefekte anspruchsvoller. Hierbei handelt es sich um eine heterogene Gruppe seltener Erkrankungen, die allerdings mit hinreichender Sensitivität in einem Screeningtest zu erkennen sein muss, um für die Behandlung betroffener Patienten vorteilhaft zu sein. Da schwere angeborene Immundefekte pathogenetisch und immunphänotypisch durch das Fehlen von T- und/oder B-Lymphozyten charakterisiert sind, sollte als Zielerkrankung für das Neugeborenenscreening die „ausgeprägte neonatale Defizienz autologer T- und/oder B-Lymphozyten“ definiert werden. Bei der Evaluation verschiedener Labormethoden zum Nachweis schwerer angeborener Immundefekte bei Neugeborenen hat sich die Messung episomaler Exzisionsprodukte der Lymphozytenrezeptoren (T-cell receptor excision circles, TRECs; und kappa-deleting recombination excision circles, KRECs) durchgesetzt, da diese sowohl hinsichtlich der Testgüte, als auch in der praktischen Durchführbarkeit mit prospektiven Screeninguntersuchungen vereinbar ist [9]. Bei Anwendung der Bewertungskriterien von populationsbezogenen Screeninguntersuchungen für die Messung von TRECs und KRECs zum Nachweis der ausgeprägten neonatalen Defizienz autologer T- und/oder B-Lymphozyten lässt sich ein positives Votum zur Durchführung als Neugeborenenscreening ableiten [8]. Während in den USA in mehreren Bundesstaaten bereits seit 2008 erfolgreich populationsbezogene Screeningprogramme für die ausgeprägte neonatale Defizienz autologer T-Lymphozyten bei Neugeborenen initiiert worden sind, ist nun auch in einigen Ländern der Europäischen Union (Deutschland, Schweden, Frankreich, Großbritannien und Italien) mit Pilotprojekten begonnen worden [10, 11].
Das Prinzip der Messung episomaler Exzisionsprodukte der Lymphozytenrezeptoren basiert auf der natürlich stattfindenden Rekombination und Affinitätsreifung des T-Zell- und B-Zell-Rezeptors [12]. Hierbei werden aus der Keimbahn-DNA der Immunglobulin-Gene Anteile herausgeschnitten, die nicht am Rekombinationsprozess der Antigenrezeptoren teilnehmen (siehe Abbildung 1A). Während T-Lymphozyten im Thymus zunächst Anteile des δ-Lokus exzidieren, um anschließend den α-Lokus zu rekombinieren, entstehen sogenannte T-cell receptor excision circles (TRECs), welche spontan episomale zirkuläre DNA-Fragmente bilden, die fortan bei Zellteilungen nicht weiter repliziert werden (siehe Abbildung 1B). Nach einem ähnlichen Prinzip findet in B-Lymphozyten im Knochenmark ein Deletionsschritt während des Vk-Jk Rearrangements statt, bei dem kappa-deleting recombination excision circles (KRECs) gebildet werden. Da diese molekularen Prozesse interindividuell einheitlich ablaufen, kann eine uniforme PCR-Strategie angewandt werden um das Vorhandensein und die Kopienzahl von TRECs und KRECs in Trockenblutproben von Neugeborenen zu bestimmen [13]. Da sich bei der Bildung der zirkulären DNA-Fragmente ein einzigartiger Sequenzbereich (sog. signal joint) bildet, kann eine quantitative PCR Methode genutzt werden, um lediglich die bereits aus der Keimbahn-DNA ausgeschnittenen TREC und KREC Fragmente zu detektieren, so dass ein direkter Rückschluss auf die stattgefundene Reifung von T- und B-Lymphozyten möglich ist (Abbildung 1). Da bei diesem Vorgehen keine individuellen Sequenzinformationen gesammelt werden, ist die PCR-Methode am ehesten mit einer virologischen Erregerlastdiagnostik zu vergleichen. Ebenso wie die etablierte Analytik des regulären Neugeborenenscreenings fällt eine solche PCR-Methode jedoch in den Geltungsbereich des Gendiagnostikgesetzes der Bundesrepublik Deutschland (GenDG; §3, Abs. 5 „Genetische Reihenuntersuchungen“), so dass eine schriftliche Aufklärung durch ärztliches Personal mit ausgewiesener Qualifikation zur fachgebundenen genetischen Beratung (FGB) erfolgen muss. Da diese Qualifikation jedoch nicht an allen geburtshilflichen Zentren bzw. Screeninglaboren derzeit vorgehalten werden kann, sind praktikable Lösungen durch den Gesetzgeber zu fordern, um die Einwilligung zum TREC-KREC Screening den bestehenden Screeningprogrammen anzugleichen.
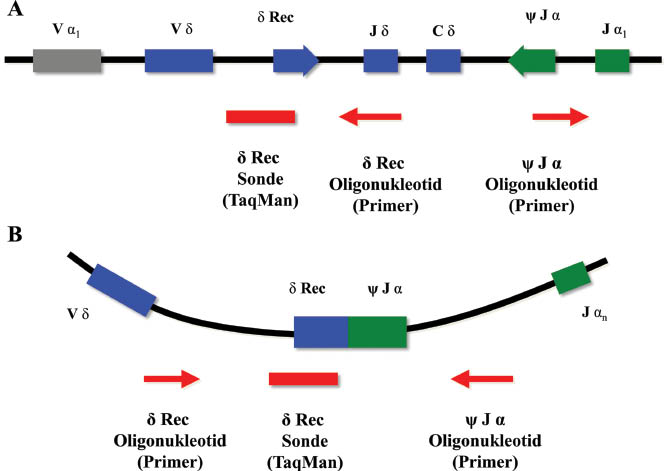
Schematisierte Darstellung der PCR-Strategie zum Nachweis des signal joint von TRECs unter Verwendung von Hydrolysesonden (TaqMan Assay).
(A) Konstitution der Keimbahn-DNA vor Deletion des T-Zell-Rezeptor δ-Lokus. (B) Bildung episomaler zirkulärer DNA-Fragmente (hier TRECs) nach Rekombination des α-Lokus.
Zur Messung von TRECs und KRECs bei Neugeborenen hat sich die Nutzung von 3,2 mm oder 1,5 mm Stanzen aus regulären Trockenblutkarten (z. B. Whatman 903; GE Healthcare, Chalfont St Giles, UK) durchgesetzt, wobei diese im Zeitrahmen der etablierten Logistik des Neugeborenenscreenings gewonnen werden können. Im zuständigen Screeninglabor erfolgt dann der Nachweis von TRECs und KRECs im Probenmaterial, wobei entweder relative Endpunk-PCR- oder absolut quantitative real-time PCR-Methoden angewandt werden können. Weiterhin kann der Screeningtest als Ein-, Zwei- oder Drei-Schritt Protokoll aufgebaut werden (siehe Tabelle 1). Die Verwendung von Sonden (probes) erlaubt hierbei die Erhöhung der Spezifität des nachzuweisenden Amplifikats und erlaubt die zeitgleiche Messung mehrerer verschiedener Amplifikate (multiplexing). Bei der verwendeten Nachweischemie kann zwischen Hydrolysesonden im 5′-Nuklease Verfahren (z. B. TaqMan; LifeTechnologies, Carlsbad, CA, USA), Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET, z. B. PerkinElmer, Waltham, MA, USA) und iso-dG/iso-dC markierten Oligonukleotiden mit Fluoreszenzfarbstoffen und Quenchern (bspw. Plexor; Promega, Fitchburg, WI, USA) gewählt werden. Erfahrungsgemäß erlaubt bereits ein Zwei-Schritt Protokoll die Aufreinigung inhibitorenfreier und konzentrierterer DNA-Template, verglichen mit einer direkten Elution aus getrocknetem Vollblut, so dass besonders im niedrigen Kopienbereich ein besseres Auflösungsverhalten des Screeningtests erzielt werden kann [13]. Dies ist besonders relevant im Hinblick auf die Festlegung eines geeigneten diagnostischen Grenzwertes (cut off), da angeborene T- oder B-Lymphopenien nicht alleinig bei Patienten mit SCID oder XLA beobachtet werden [14]. Ein TREC-KREC Screeningtest, der eine gute Auflösung im niedrigen Kopienbereich bietet, erlaubt die Umsetzung eines durch den Nutzer im Screeninglabor festlegbaren diagnostischen Grenzwertes der die Unterscheidung „typischer“ SCID- und XLA-Patienten von anderen abnormalen Screeningergebnissen ermöglicht (Abbildung 2).
Vergleich der Arbeitsschritte verschiedener Konzepte zur Durchführung eines TREC-KREC Screeningtests.
| Drei-Schritt qPCR Assay | Zwei-Schritt qPCR Assay | Ein-Schritt TR-FRET oder Plexor Assay |
| Filterpapier-Reinigungslösung, pH<5, Entfernen von PCR Inhibitoren | ||
| DNA Elutionslösung, pH>10, Übertragen des Eluats in die qPCR Testplatte | DNA Elutionslösung, pH>10, Übertragen des Eluats in die qPCR Testplatte | |
| 96- oder 384-well triplex qPCR (TREC-KREC-ACTB) | 96- oder 384-well triplex qPCR (TREC-KREC-ACTB) | DNA Elution und TR-FRET TREC oder Plexor TREC-KREC Assay in derselben Testplatte, 96-well Format |

Idealisierter Vergleich der Anwendbarkeit diagnostischer Grenzwerte in Abhängigkeit von der Auflösungsfähigkeit des verwendeten TREC-KREC Screeningtests.
Bei einem Screeningtest mit geringer (A) bzw. hoher (B) Auflösungfähigkeit im niedrigen Kopienbereich. Die Patienten mit den gesuchten Zielerkrankungen werden jeweils anteilig überlagert durch abnormale Screens, die sich nicht auf einen schweren angeborenen Immundefekt zurückführen lassen.
Zur routinemäßigen Durchführung eines TREC-KREC Screeningtests ist es unerlässlich die Testcharakteristika in der jeweilig zu erwartenden Neugeborenenpopulation des zuständigen Screeninglabors zunächst zu evaluieren und dann fortwährend zu validieren. Üblicherweise sind die Kopienzahlen von TRECs und KRECs in der Gesamtpopulation nicht normal verteilt, so dass 5.000 bis 10.000 Neugeborenenproben evaluiert werden sollten, um geeignete diagnostische Grenzwerte im niedrigen Kopienbereich festlegen zu können und somit auch die Wiederholerrate des Screeningtests in diesem Bereich abzuschätzen (Abbildung 3). Derzeit sind in der Mehrzahl in-house entwickelte Screeningtests in der Anwendung in den USA und Europa, wodurch die Harmonisierung der Methodik und die Übertragbarkeit der diagnostischen Grenzwerte erschwert werden. Die vom Center for Disease Control and Prevention (CDC) für Ringversuche bereitgestellten Trockenblutkarten sind nur hinsichtlich der T-Zellzahlen validiert, so dass deren Nutzung im TREC-KREC Screeningtest unvollständige Ergebnisse liefert. Daher sollten zur fortlaufenden Validierung des TREC-KREC Screeningtests künstliche Kontrollproben hergestellt werden (1: T- B+, 2: T+ B-, 3: T- B-, 4: T+ B+); dies lässt sich durch Depletion von T- und/oder B-Lymphozyten, z.B. in Nabelschnurblut als Spendermaterial erreichen. Alternativ können hierzu auch EBV-Zelllinien verwendet werden, die genomisch stabil integrierte Kopien des TREC oder KREC signal joints tragen (Abbildung 1). Bei Wahl eines absoluten Quantifizierungsmodus ist es vorteilhaft, die gemessenen Kopienzahlen dieser Kontrollproben prospektiv zu dokumentieren, um so die geforderte Qualitätssicherung im Screeninglabor zu komplettieren.
![Abbildung 3 Zusammenstellung der Populationsverteilung von TREC und KREC Kopienzahlen, normalisiert per μL Trockenblut, in einer Kohorte von 1.200 anonym untersuchten Neugeborenenproben [13].](/document/doi/10.1515/labmed-2013-0033/asset/graphic/labmed-2013-0033_fig3.jpg)
Zusammenstellung der Populationsverteilung von TREC und KREC Kopienzahlen, normalisiert per μL Trockenblut, in einer Kohorte von 1.200 anonym untersuchten Neugeborenenproben [13].
In Abhängigkeit von festgelegten diagnostischen Grenzwerten können prospektive TREC-KREC Screeningergebnisse als „normal“ (gleich oder oberhalb der Grenzwerte für TRECs und KRECs) oder als „abnormal“ (unterhalb der Grenzwerte für TRECs und/oder KRECs) gewertet werden. Bei einem abnormalen Testergebnis der ersten Trockenblutstanze wird zunächst eine laborinterne Wiederholungs-Testung (% re-run) mit zwei weiteren Stanzen derselben Trockenblutkarte durchgeführt (Abbildung 4). Nur wenn hierbei in beiden Stanzen weiterhin abnormale Testergebnisse gefunden werden, wird der Trackingprozess initiiert. Der Anteil dieser Neugeborenen an der Gesamtpopulation, bei denen eine separate zweite Trockenblutkarte eingesendet werden muss, wird als % re-call ausgedrückt. Die Differenz zwischen re-run und re-call Prozentsätzen ist vor allem auf präanalytische Einflussfaktoren, wie z. B. die Art der Herstellung der Trockenblutkarte und die Auswahl der Stanzposition auf der Karte zurückzuführen. Die Auswahl geeigneter diagnostischer Grenzwerte sollte sich aber auch an diesen Prozentsätzen orientieren um sicherzustellen, dass der Screeningtest der Gesamtpopulation gegenüber zumutbar ist (Abbildung 5).

Vereinfachtes Flussdiagramm des TREC-KREC Screeningtests mit anschließender Trackingstrategie bei wiederholt „abnormal“ getesteten Trockenblutproben.
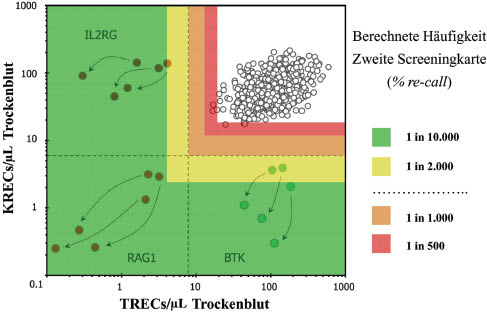
Berechnung der Häufigkeiten wiederholt einzusendender Trockenblutkarten (% re-test) in Abhängigkeit festlegbarer diagnostischer Grenzwerte.
Zu Grunde liegen Screeninguntersuchungen bei 18.500 Neugeborenen (offene Punkte). Die vorteilhaften Auswirkungen eines TREC-KREC Tests mit hoher Auflösungsfähigkeit werden bei SCID Patienten (IL2RG oder RAG1 Mutationen, rote Punkte), oder bei XLA Patienten (BTK Mutationen, grüne Punkte) aufgezeigt.
Trackingprozess
Die Nachverfolgung von Neugeborenen wird bei wiederholt abnormalen Testergebnissen für TREC und/oder KREC Kopienzahlen nach Untersuchung von mindestens 3 unabhängigen Trockenblutstanzen der ersten eingesandten Trockenblutkarte eingeleitet (Abbildung 4). Die Koordination und Dokumentation der Nachverfolgung wird hierbei direkt vom Screeninglabor verantwortet, welches den initialen TREC-KREC Screeningtest durchgeführt hat, kann aber auch an ein spezialisiertes Trackingzentrum übergeben werden.
Zunächst sollten die geburtshilfliche Einrichtung („Arzt-Arzt“) und die Eltern des Neugeborenen („Arzt-Patient“) kontaktiert werden, um zusätzliche Informationen zum Zustand des Kindes in Erfahrung zu bringen. Da wiederholt abnormale TREC und/oder KREC Testergebnisse insbesondere auch bei Frühgeborenen (<32. Schwangerschaftswoche), Kindern mit syndromalen Krankheitsbildern (z.B. Trisomie 21 oder 22q11 Mikrodeletionssyndrom), metabolischen Erkrankungen (z.B. Propionyl-CoA-Carboxylase-Mangel oder Methylmalonazidurie), und schweren konnatalen Infektionen oder Fehlbildungen mit nachfolgender Lymphozyten-Extravasation (z.B. Neugeborenensepsis oder Gastroschisis) auftreten können, ist es wichtig, zusätzliche Untersuchungsbefunde zur Wertung der Verdachtsdiagnose eines schweren angeborenen Immundefekts zusammenzutragen [14]. Wenn in den drei Wiederholungsproben der ersten Trockenblutkarte keinerlei TREC und/oder KREC Kopien sicher nachgewiesen werden können, muss von einem „hochauffälligen“ Befund ausgegangen werden. In diesen seltenen Fällen (ca. 1 auf 20.000 Neugeborene) sollte sofort zu einer klinischen Konsultation unter dem dringenden Verdacht des Vorliegens eines SCID bzw. XLA geraten werden. In jedem anderen Fall wird unverzüglich eine separate zweite Trockenblutkarte zur Nachkontrolle des Neugeborenen angefordert, wobei die Einsendung der zweiten Trockenblutkarte bei Frühgeborenen nicht erst zum errechneten Geburtstermin stattfinden sollte. Im Falle von Bluttransfusionen vor Herstellung der Trockenblutkarte kann ggf. zweimalig getestet werden; sowohl 24 Stunden nach Transfusion, als auch vor Entlassung bzw. innerhalb von 14 Tagen.
Falls nach Testung von 2 unabhängigen Trockenblutstanzen der zweiten, separat eingesandten, Trockenblutkarte weiterhin abnormale Testergebnisse für TREC und/oder KREC Kopienzahlen nachgewiesen werden, sollte dringend von einer persistenten T- und/oder B-Lymphopenie ausgegangen werden, die zeitnah von einem erfahrenen Pädiater in einem Immundefektzentrum abgeklärt werden sollte (Abbildung 4). Hierzu werden vom Screeninglabor alle zusätzlich verfügbaren Testergebnisse, insbesondere solche des regulären Neugeborenenscreenings die das Vorliegen einer metabolischen Erkrankung andeuten, zusammengetragen. Bei bereits stationär behandlungspflichtigen Patienten (z. B. ausgeprägte Frühgeburtlichkeit <28. Schwangerschaftswoche, schwere konnatale Infektionen, syndromale Fehlbildungen) sollte eine Beratung durch ein Immundefektzentrum angestrebt werden.
Während die Eltern des Neugeborenen nach Untersuchung der zweiten separaten Trockenblutkarte auch bei Normalisierung des Untersuchungsergebnisses unverzüglich informiert werden sollten, findet eine detaillierte Erläuterung der Bedeutung des Untersuchungsbefundes bei erneut abnormalem Testergebnis erst im Immundefektzentrum statt. Hierzu werden die Eltern mit konkreten Kontaktdaten eines wohnortnahen Immundefektzentrums ausgestattet und das Zentrum wird über den zu erwartenden Patienten informiert. Die erfolgreiche Vermittlung des Neugeborenen an ein pädiatrisches Immundefektzentrum ist vom Screeninglabor bzw. dem spezialisierten Trackingzentrum zu überprüfen und zu dokumentieren. Eine Übersicht von pädiatrischen Immundefektzentren in der Bundesrepublik Deutschland kann dem FIND-ID Netzwerk für Angeborene Immundefekte (www.find-id.net) entnommen werden. Die Zentrumsauswahl sollte zunächst wohnortnah erfolgen, da bei Verdacht auf Vorliegen einer schweren T- und/oder B-Lymphopenie intensivierte Hygienemaßnahmen und ggf. eine frühzeitige, strikte Isolierung zur Vermeidung opportunistischer Infektionen unerlässlich sind. Zur Durchführung einer ggf. notwendigen Stammzelltransplantation kann eine Verlegung in ein spezialisiertes Transplantationszentrum notwendig sein, wobei nur solche Zentren ausgewählt werden sollten, die bereits Erfahrungen in der Stammzell- oder Gentherapie bei Patienten mit primären Immundefekten gesammelt haben (optimierte Konditionierungs- und Transplantationsprotokolle).
Follow-up Diagnostik
Die Folgediagnostik von Neugeborenen mit Verdacht auf Vorliegen eines schweren angeborenen Immundefektes muss gleichzeitig mehreren Anforderungen genügen. Einerseits soll das auf die ausgeprägte neonatale Defizienz autologer T- und/oder B-Lymphozyten hinweisende, wiederholt abnormale Screeningergebnis der TREC bzw. KREC Kopienzahlen im Trockenblut durch orientierende (großes Blutbild) und vertiefende zytologische Untersuchungen (Durchflusszytometrie) bestätigt werden und dies auch aus Qualitätssicherungsgründen an das zuständige Screeningzentrum zurückgemeldet werden (Abbildung 4). Andererseits sollen durch immunfunktionelle und molekulargenetische Untersuchungen Rückschlüsse auf den pathogenetischen Hintergrund jedes einzelnen Patienten in Vorbereitung einer individualisierten Therapieplanung gezogen werden. Die Notwendigkeit einer solchen patientenzentrierten Folgediagnostik begründet sich aus der erheblichen molekularen Heterogenität schwerer primärer Immundefekte mit vorwiegend „privaten“ Mutationen und dient ebenso dem internationalen Erfahrungsaustausch der therapeutischen Optionen. Hierbei sollte jegliche Folgediagnostik durch einen erfahrenen pädiatrischen Immunologen an einem Behandlungszentrum für angeborene Immundefekte veranlasst werden. Ebenso sollte die initiale klinische Untersuchung, welche in der neonatalen (präsymptomatischen) Phase schwerer angeborener Immundefekte häufig einen Normalbefund imitiert, sich auch an der AMWF Leitlinie (S2k) zur „Diagnostik von primären Immundefekten“ orientieren [1]. Weiterhin sollte die klinische Untersuchung auch die Wahrscheinlichkeit des Vorliegens solcher Erkrankungen bewerten, die ebenso den Befund einer ausgeprägten neonatalen Defizienz autologer T- und/oder B-Lymphozyten im Neugeborenenscreening „nachahmen“ können, wie beispielsweise angeborene Glykosylierungsdefekte oder Mitochondriopathien [14]. Hierbei kann die Zusammenarbeit mit erfahrenen Neuropädiatern bzw. Stoffwechselexperten sinnvoll sein, da ggf. eine Verlegung in ein dahingehend spezialisiertes Zentrum erforderlich ist.
Durchflusszytometrische Untersuchungen
Schwere angeborene Immundefekte können durch das Fehlen bzw. die funktionelle Anergie von CD3+ T-Lymphozyten oder CD19+ B-Lymphozyten charakterisiert werden. Mit Hilfe eines kombinierten TREC-KREC Screeningtests unter Beachtung populationsadaptierter diagnostischer Grenzwerte werden nur solche Neugeborene identifiziert, die persistent eine primäre oder sekundäre schwere T- und/oder B-Lymphopenie in der Trockenblutpräparation aufweisen (Abbildung 4). Die durchflusszytometrische Lymphozytentypisierung soll nun einerseits eine Bestätigung des Screeningverdachtes durch eine methodisch unabhängige, periphere Vollblutuntersuchung erbringen und gleichzeitig die Zusammensetzung der Lymphozytensubpopulationen, einschließlich Thymusemigranten (recent thymic emigrants, RTEs), beschreiben und ein mütterliches Engraftment mit CD45RO+ T-Zellen ausschließen. Während bei Patienten mit „typischen“ SCID Phänotypen oder Agammaglobulinämie das nahezu vollständige Fehlen von autologen T- und/oder B-Lymphozyten (<300 T-Zellen bzw. <100 B-Zellen pro μL Vollblut) bereits diagnostisch wegweisend ist, können bei klinisch relevanten und durch den TREC-KREC Screeningtest erfassbaren „atypischen“ SCID Phänotypen reduzierte T-Zellzahlen (300-1500 T-Zellen pro μL Vollblut) nachweisbar sein. Allerdings können bei Neugeborenen mit „delayed-onset“ ADA SCID ebenso normale T-Zellzahlen bei stark reduzierten B-Zellzahlen beobachtet werden, so dass bei isoliert erniedrigten KREC-Kopienzahlen auch immer ein SCID-Phänotyp in Betracht gezogen werden muss [15].
Die durchflusszytometrische Lymphozytentypisierung im Probenmaterial von Neugeborenen und Säuglingen stellt spezielle methodische und analytische Anforderungen. Häufig können die zu untersuchenden peripheren Blutproben nur unter schwierigen Bedingungen abgenommen werden und sind somit im Volumen und auch in der Präparationsqualität eingeschränkt. Um dennoch ein weitreichend aussagekräftiges analytisches Ergebnis zu erzielen, sind vorzugsweise polychromatische Panels (z.B. 8-Farb Durchflusszytometrie) anzuwenden. Vorrangig sollten Lymphozytenpopulationen untersucht werden, die sowohl hinsichtlich der Klassifikation schwerer angeborener Immundefekte (T-, B-, NK-Phänotyp), der Einschätzung möglicher spezifischer Therapieanfordeungen und –komplikationen dienen (Engraftment mütterlicher Zellen, GvHD, Prädominanz von TCR γ/δ T-Zellen) und andererseits auch als posttherapeutische Verlaufsparameter genutzt werden können (Thymusemigranten, B-Zell Rekonstitution). Eine mögliche Zusammenstellung solcher durchflusszytometrischer Panels und nachweisbarer Lymphozytenpopulationen ist kürzlich veröffentlicht worden [16]. Bei der Zusammenstellung diagnostisch einzusetzender Oberflächenmarker sollte sowohl bei der Auswahl der Epitopspezifität monoklonaler Antikörper, als auch der konjugierten Fluorochrome, eine Harmonisierung mit umfangreich validierten durchflusszytometrischen Panels (z.B. denen des EuroFlow Konsortiums; www.euroflow.org) oder zumindest auf der Ebene akkreditierter Labore angestrebt werden. Da die Auswertung der Lymphozytentypisierung sowohl relative, als auch absolute Vergleichswerte liefern soll, sind eindeutige Populationsdefinitionen und „Gatingstrategien“ unabdingbar. Da dies bei der Erstellung zytometrischer Daten im ersten Lebensjahr bisher international nur unzureichend beachtet worden ist, können durch Meta-Analysen bisher lediglich vergleichende Normalwerte für CD3+ (CD4+ und CD8+) T-Zellen, CD19+ B-Lymphozyten und CD16/56+ NK-Zellen bereitgestellt werden [17].
Immunfunktionelle Untersuchungen
Die Immunphänotypisierung kann oftmals keine definitive Einschätzung der funktionellen Relevanz eines anzunehmenden schweren Immundefektes liefern, insbesondere wenn die durchflusszytometrische Analyse oder die klinische Untersuchung das Vorliegen eines „atypischen“ Phänotyps nahelegt. Daher sollte zur Komplettierung durchflusszytometrischer Untersuchungen auch (A) die lymphozytäre Proliferationsfähigkeit und Stimulierbarkeit, sowie (B) die Funktionalität der angeborenen Diversität des Immunrepertoires in vitro überprüft werden.
(A) Die klassischen radioaktiven (3H-Thymidin) und nicht-radioaktiven (BrdU) Nachweismethoden der Proliferationsfähigkeit separierter mononukleärer Zellen des peripheren Blutes durch Stimulation mit Mitogenen wie Phytohemagglutinin (PHA) oder Concanavalin A (ConA) bieten nur unzureichende Harmonisierungs- und Vergleichsmöglichkeiten, da die anzuwendenden Separations-, Stimulations- und Auswerteprotokolle erheblich zwischen verschiedenen Laboren differieren und zudem von lokalen Kontrollproben abhängig sind, die zumeist nicht altersgewichtet sind. Dennoch wird erfahrungsgemäß von einer weniger als 10%igen Proliferation PHA-stimulierter Lymphozyten eines „typischen“ SCID-Patienten im Vergleich mit Proben gesunder Probanden ausgegangen, wobei „atypische“ SCID-Patienten bis zu 30% des normalen Proliferationsmesswertes erreichen können. Im Hinblick auf das geringe bereitzustellende Probenvolumen von Neugeborenen mit Verdacht auf einen schweren angeborenen Immundefekt sind separationsfreie Proliferationstests vorzuziehen, insbesondere weil diese mit einzelzellbasierten Analysemethoden wie der Durchflusszytometrie gekoppelt werden können. So sollten moderne, primär nicht-fluoreszierende Proliferationsfarbstoffe (z. B. VPD450; BD Biosciences, Franklin Lakes, NJ, USA) bei der Stimulation von Vollblut mit PHA eingesetzt werden, die dann mit der zellulären Proliferation durch Esterasen-vermittelte Umwandlung in intrazelluläre Fluoreszenzmoleküle korrelieren. Die Auswertung des Stimulationsversuches kann somit in geringen Probenvolumina auch im kinetischen Verlauf (6, 24 und 48 Stunden) erfolgen und erlaubt ebenso eine zelltypspezifische Analyse (CD3+ T-Zellen versus CD19+ B-Zellen), da VPD450 nicht durch Erythrozytenlyse oder Zellfixierung mit Paraformaldehyd beschädigt wird.
Bei zelltyp-unabhängiger Defizienz in der Proliferation von Lymphozyten sollten Defekte der DNA-Reparaturmaschinerie vermutet werden, bei denen eine gesteigerte zelluläre Apoptose durch ionisierende Strahlung, Anhäufung toxischer Metabolite oder durch Behandlung mit Chemotherapeutika ausgelöst wird. SCID-Phänotypen mit „radiosensitiven“ Proliferationsdefekten sind bei Artemis-, DNA Ligase IV-, DNA-PKcs- und Cernunnos-XLF Mangel beschrieben worden, allerdings zeigen auch Patienten mit „Chromosomen-Instabilitätssyndromen“ (z. B. Ataxia teleangiectatica oder Nijmegen-breakage Syndrom) ähnliche Auffälligkeiten [13, 18]. Der Nachweis erhöhter zellulärer Radiosensitivität selbst ist hierbei von Bedeutung für die Auswahl der Konditionierung und Spenderselektion bei einer Stammzelltransplantation der betroffenen Patienten. Die Untersuchung der DNA-Reparaturfähigkeit kann methodisch vielfältig durchgeführt werden; während vormals häufig eine Gamma-Bestrahlung von Lymphozyten, Fibroblasten oder Knochenmarksproben eingesetzt worden ist, sollten heutzutage vorwiegend nicht-radioaktive Stressoren der genomischen Reparaturmechanismen angewandt werden. Inhibitoren der DNA Topoisomerase I (z. B. Camptothecin) können Apoptose-induzierend wirken, wobei Proben von „radiosensitiven“ Patienten im durchflusszytometrischen VPD450-Test eine gesteigerte Proliferationsdefizienz in allen Dosisstufen aufweisen. Alternativ kann auf kommerziell-verfügbare COMET-Tests (Einzelzellgelelektrophorese) zurückgegriffen werden, wobei diese jedoch zunächst mit Patientenproben validiert werden sollten.
Bei Vorliegen grenzwertiger Ergebnisse eines Mitogen-Proliferationstests können andererseits auch Defekte der spezifischen Proliferationsfähigkeit der T- und/oder B-Zellen nach Aktivierung des T-Zell-Rezeptors (TCR) oder B-Zell-Rezeptors (BCR) angenommen werden. Zum Nachweis solcher Defekte können intrazelluläre Calcium-Flux-Messungen auch durchflusszytometrisch durchgeführt werden, wobei eine Aktivierung des TCR mit anti-CD3 Antikörpern und des BCR mit anti-IgM Antikörpern erzielt werden kann. Hierzu werden die zu untersuchenden Lymphozyten zunächst mit einem Calcium-Indikator (z. B. Quest Fluo-8; AAT Bioquest, Sunnyvale, CA, USA) beladen, der nach Aktivierung des TCR bzw. BCR und nachfolgendem Calcium-Einstrom einen >200-fachen Anstieg der Fluoreszenzintensität erfährt. Die Auswertung der zelltypspezifischen Calcium-Flux-Messungen setzt somit die Fluoreszenzintensität des Calcium-Indikators in Abhängigkeit von der Zeit nach Stimulation der TCR bzw. BCR.
(B) Bei nachweisbar niedrigen oder normalen T-Zellzahlen und einer reduzierten lymphozytären Proliferationsfähigkeit sollte eine oligoklonale Expansion von T-Zellpopulationen in Betracht gezogen werden. Dies wird zumeist bei „atypischen“ SCID Phänotypen mit hypomorpher Restfunktion der betroffenen Krankheitsgene beobachtet. Da das Vorliegen oligoklonaler T-Zellpopulationen Einfluss auf das Konditionierungs- und Transplantationsschema haben kann, sollte die Verteilung der Vbeta-Familien vorhandener T-Zellen untersucht werden. Diese Klonalitätsanalyse kann wahlweise durch DNA-Amplifikation der CDR3 Region der beta-Kette des TCR mit nachfolgender PAGE-Fragmentlängenanalyse durchgeführt werden (Spectratyping), oder aber durch eine RNA-Expressionsanalyse der Vbeta-Transkripte mittels real-time PCR [19]. Im methodischen Vergleich erlaubt die RNA-transkriptbasierte Klonalitätsuntersuchung vorwiegend Rückschlüsse auf den augenblicklichen funktionellen Zustand der T-Zell Repertoires.
Die Untersuchung des Funktionsumfanges der B-Zellen ist sowohl bei Verdacht auf Vorliegen einer schweren B-, als auch isolierten oder kombinierten T-Lymphopenie sinnvoll. Hierbei sollten zunächst die Serum-Immunglobuline (IgM, IgG, IgA und IgE) bestimmt werden, wobei die diaplazentare Übertragung von IgG und IgA im Rahmen der mütterlichen Leihimmunität zu beachten ist [20]. Daher sind diese serologischen Messwerte vor allem als Ausgangspunkt für Verlaufsuntersuchungen zu werten, können aber sehr hilfreich sein, den Zeitpunkt einer nötigen Therapieinitiierung bzw. -intensivierung zu definieren. Erhöhte Spiegel an Serum-IgE sind insbesondere bei Patienten mit SCID Omenn-Phänotyp beschrieben worden und können eine entsprechende Diagnose komplettieren [21].
Aufgrund der Unzulänglichkeiten serologischer Methoden in der Einschätzung der körpereigenen Produktion von Immunglobulinen in der Neonatal- und Säuglingsperiode werden zusätzliche Untersuchungstechniken benötigt. So können die RNA-Transkripte der Immunglobulin-Isotypen durch eine real-time PCR gemessen werden. Diese Transkripte können aufgrund alternativer Prozessierung sowohl als sekretierte Form als auch in membranständiger Form vorliegen und korrelieren mit der körpereigenen B-zellulären Antikörperproduktion bzw. der Oberflächenexpression auf B-Zellen [22, 23]. Als Probenmaterial kann <250 μL peripheres, stabilisiertes Vollblut (z. B. PAXgene RNA System; PreAnalytiX, Hombrechtikon, Schweiz) eingesetzt werden. Da die gemessenen Immunglobulin-RNA-Transkripte nicht von der Mutter übertragen werden können, erlaubt diese Untersuchung die Einschätzung der eigenen Antikörperproduktion des Neugeborenen auch unmittelbar innerhalb der ersten Lebensmonate.
Alternativ kann die B-zelluläre Immunglobulinproduktion auch ex vivo nach Stimulation von separierten mononukleären Zellen des peripheren Blutes (PBMC) in einem ELISPOT Testverfahren gemessen werden [24, 25]. Hierzu werden die PBMC zunächst für 3-5 Tage in Gegenwart von Zytokinen (z. B. Interleukin-4, Interleukin-10, oder Interleukin-21) und spezifischen Rezeptorliganden (z. B. CD40-Ligand, oder TLR 7/8 Agonist R848) kultiviert und anschließend für 48 Stunden auf der Membran einer ELISPOT Testplatte aufgebracht, die mit Isotyp-spezifischen Detektionsantikörpern beschichtet ist. Die Auswertung erfolgt einzelzellbasiert auf einem automatischen Lesesystem und gibt die Menge freigesetzter Immunglobuline pro B-Lymphozyt im Verhältnis zur eingesetzten Gesamtzellzahl an. Aufgrund des zeitlichen Aufwands und der benötigten Probemengen zur Zellseparation findet dieses Untersuchungsverfahren allerdings nur bedingt Anwendung im klinischen Kontext.
Immunfunktionelle Untersuchungen umfassen ebenso den Nachweis krankheitsassoziierter Proteine einschließlich Enzyme, sofern andere Untersuchungstechniken, wie zum Beispiel molekulargenetische Verfahren, hierbei Nachteile bergen. Dies ist beispielsweise der Fall beim differentialdiagnostischen Vorgehen bei Verdacht auf Ataxia teleangiectatica (AT; Louis-Bar-Syndrom). Neugeborene mit AT können deutlich erniedrigte T- und B-Zellzahlen bei Geburt aufweisen und werden teilweise durch den TREC-KREC Screeningtest miterfasst [13, 26]. Da die genomische Textur des AT zugrundeliegenden ATM Gens mit über 59 Exons für klassische Sanger-Sequenzierstrategien eine zeit- und kostenineffiziente Hürde darstellt, sollte in einem ersten Untersuchungsschritt vorzugsweise die Anwesenheit des ATM Proteins nachgewiesen werden [27]. Dies kann klassischerweise durch einen Westernblot mit Vollblutlysat oder durchflusszytometrisch mittels intrazellulärer Proteinfärbung realisiert werden. Analog zu diesem Vorgehen können auch weitere differentialdiagnostische Proteinanalysen nach einem wiederholt abnormalen TREC-KREC Screeningtest durchgeführt werden. Ebenso können durch Messung der Aktivität der Adenosin-Desaminase (ADA) und der Purinnukleosid-Phosphorylase (PNP) entsprechende SCID-Phänoytpen diagnostiziert werden.
Molekulargenetische Untersuchungen
Derzeit sind mehr als 18 monogenetische Defekte beschrieben worden, die in einem Phänotyp schwerer kombinierter Immundefekte resultieren [28]. Ebenso können primäre Agammaglobulinämien mit isoliert fehlenden B-Lymphozyten auf mehr als 6 Gendefekte zurückgeführt werden [12]. Pathogenetisch können die zugrundeliegenden Defekte für Reifungsstörungen vor allem der lymphatischen Reihe im Knochenmark, sowie in primären und sekundären lymphatischen Geweben verantwortlich gemacht werden. Durchflusszytometrische Untersuchungen des peripheren Blutes erlauben die Einteilung von SCID-Patienten entsprechend des T- B+/- NK+/- Systems, das Rückschlüsse auf zugrunde liegende Gendefekte erlaubt [27]. Die molekulare und zellfunktionelle Heterogenität schwerer kombinierter Immundefekte zeigt sich ebenso in den klinischen Therapieerfahrungen, die zur Anwendung „gen-spezifischer“ Protokolle bei der Konditionierung und Stammzelltransplantation oder Gentherapie bei SCID Patienten führte [29, 30].
In Vorbereitung einer etwaigen Stammzelltransplantation sollte frühzeitig eine HLA-Typisierung initiiert werden. Hierbei können sich analytische Schwierigkeiten bei Kontamination des peripheren Blutes des Patienten mit übertragenen mütterlichen T-Zellen ergeben. Im Falle einer haploidentischen Transplantation mit der Mutter als Spender würde dies eine untergeordnete Rolle aufgrund der anzunehmenden Empfängertoleranz spielen, allerdings sollte der haploide Genotyp des Patienten vorausschauend auch aus anderem Material (z. B. Wangenabstrich von der Mundschleimhaut oder Fibroblasten der Haut) bestimmt werden.
Während die Entscheidung zur Stammzelltransplantation bisher hauptsächlich aufgrund des klinischen Verlaufs, sowie zellulärer und immunfunktioneller Untersuchungen getroffen worden ist, stellt die präsymptomatische Früherkennung von Neugeborenen mit schweren angeborenen Immundefekten neue Anforderungen an die Algorithmen zur Therapieentscheidung. Insbesondere für die immunologischen Funktionsuntersuchungen bei Säuglingen stehen nur unzureichende Referenzbereiche zur Verfügung, da altersgewichtete Kontrollproben nicht regulär verfügbar sind. Daher kommt molekulargenetischen Untersuchungen bei Neugeborenen mit Vorliegen einer ausgeprägten neonatalen Defizienz autologer T-Lymphozyten (Verdacht SCID Phänotyp) eine besondere Bedeutung zu, wobei die Initiierung einer kausalen Therapie weder durch den erforderlichen Zeitumfang molekularer Untersuchungen, noch durch das Ausbleiben einer finalen genetischen Diagnose verzögert werden darf. Bei Neugeborenen mit ausgeprägter neonataler Defizienz autologer B-Lymphozyten (Verdacht Agammaglobulinämie) erlaubt die übertragene mütterliche Leihimmunität, die in der Regel bis zum 6. Lebensmonat anhält, zumeist die Komplettierung der genetischen Diagnostik vor Therapiebeginn [31]. In diesem Sinne erscheint es aber auch sinnvoll, eine molekulargenetische Untersuchung bereits bei der ersten kinderärztlichen Untersuchung wiederholt abnormal getesteter Neugeborener einzuleiten. Da bei der Mehrzahl der Kinder nicht-konsanguiner Eltern die krankheitsverursachenden Mutationen spontan auftreten, sind Familienuntersuchungen initial nur bedingt hilfreich.
Molekulargenetische Untersuchungen in einem klinischen Kontext werden derzeit vorwiegend durch Auswahl von annotierten Kandidatengenen für den vermuteten angeborenen Immundefekt begonnen. Hierbei sind häufig mehrere Gene unterschiedlicher Komplexität zu untersuchen, so dass ein erheblicher Aufwand bei der Amplifikation und Analyse der gewünschten Sequenzbereiche bei Anwendung klassischer Sanger-Methoden besteht. Zudem stellt die Einschränkung auf Kandidatengene im Hinblick auf die molekulare Heterogenität und die bekannten Überlappungseffekte im klinischen Phänotyp des Patientenklientels einen Nachteil dar, insbesondere bei sequentiellem Vorgehen („Gen-für-Gen“). Neuere molekulargenetische Methoden können diese Nachteile teilweise bzw. vollständig umgehen, wobei insbesondere sequenzanalytische Herausforderungen bei stetig ansteigendem Datenumfang derzeit noch die vorrangige Limitation der zeitnahen Ergebnisübermittlung darstellt. Einerseits können durch gezielte Exon-Anreicherung (z. B. HaloPlex Technologie) individualisierte Diagnostikpanels für derzeit bis zu 250 Kandidatengene auf einem einzigen Mikrofluidic-Genchip untergebracht werden, andererseits verspricht die dritte Generation von Sequenziergeräten eine kosten- und zeiteffiziente Analyse ganzer Transkriptome oder Exome [32]. Der Vorteil transkriptom- bzw. exom-weiter Untersuchungen liegt in der Möglichkeit, gleichzeitig bekannte Kandidatengene umfassend sequenziell zu überprüfen und neue krankheitsassoziierte Gene schwerer angeborener Immundefekte zu erkennen. Im Hinblick auf die evidenzbasierte Begründung lebenslanger supportiver oder invasiver kausaler Therapien, wie einer hämatopoetischen Stammzelltransplantation, wird dies zunehmend an Bedeutung gewinnen [33].
Zusammenfassung
Während das Konzept des Neugeborenenscreenings für schwere angeborene Immundefekte auf der Basis von TRECs und KRECs international noch eine Neuerung darstellt, sind die beim Tracking und der Folgediagnostik anwendbaren Strategien bereits vielfach etabliert und validiert. Allerdings erfordern die präanalytischen Herausforderungen der Untersuchung von Neugeborenenproben die Anpassung bestehender Testverfahren und diagnostischer Algorithmen. Angesichts der Notwendigkeit einer schnellstmöglich umfassenden Abklärung hinsichtlich therapeutischer Maßnahmen, erfordert die Folgediagnostik von im Screening auffälligen Neugeborenen mit Verdacht auf Vorliegen eines schweren angeborenen Immundefektes ein komplexes interdisziplinäres Zusammenwirken von Screeningzentren, immundiagnostischen und humangenetischen Laboren im Kontext mit spezialisierten Behandlungszentren für primäre Immundefekte im Sinne einer individualisierten Medizin.
Interessenkonflikt und Dank
M.B., A.P. und U.S. deklarieren keinerlei Interessenkonflikte. S.B. erhielt Unterstützung von PerkinElmer Inc. zur Durchführung von Forschungs- und Entwicklungsdienstleistungen. Die Autoren danken weiterhin für die Unterstützung durch die Jeffrey Modell Foundation, dem Sächsischen Staatsministerium für Soziales und Verbraucherschutz sowie dem Bundesministerium für Bildung und Forschung (BMBF 1315883).
Literatur
1. Farmand S, Baumann U, von Bernuth H, Borte M, Förster-waldl E, Habermehl P, et al. Leitlinie „Diagnostik von primären Immundefekten“. Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) 2011. Available at: http://www.awmf.org/uploads/tx_szleitlinien/027-050l_S2k_Diagnostik_Primäre_Immundefekte_2011-12.pdf Accessed: 16 March 2013).Suche in Google Scholar
2. Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2011;2:54.Suche in Google Scholar
3. Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood 2011;117:3243–6.10.1182/blood-2010-08-300384Suche in Google Scholar PubMed
4. Puck JM; SCID Newborn Screening Working Group. Population-based newborn screening for severe combined immunodeficiency: steps toward implementation. J Allergy Clin Immunol 2007;120:760–8.10.1016/j.jaci.2007.08.043Suche in Google Scholar PubMed
5. Plebani A, Soresina A, Rondelli R, Amato GM, Azzari C, Cardinale F, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol 2002;104: 221–30.10.1006/clim.2002.5241Suche in Google Scholar PubMed
6. Bakare N, Menschik D, Tiernan R, Hua W, Martin D. Severe combined immunodeficiency (SCID) and rotavirus vaccination: reports to the Vaccine Adverse Events Reporting System (VAERS). Vaccine 2010;28:6609–12.10.1016/j.vaccine.2010.07.039Suche in Google Scholar PubMed
7. Patel NC, Hertel PM, Estes MK, de la Morena M, Petru AM, Noroski LM, et al. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. N Engl J Med 2010;362:314–9.10.1056/NEJMoa0904485Suche in Google Scholar PubMed PubMed Central
8. Borte S, von Döbeln U, Hammarström L. Guidelines for newborn screening of primary immunodeficiency diseases. Curr Opin Hematol 2013;20:48–54.10.1097/MOH.0b013e32835a9130Suche in Google Scholar PubMed
9. Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol 2012;129:607–16.10.1016/j.jaci.2012.01.032Suche in Google Scholar PubMed PubMed Central
10. Verbsky J, Thakar M, Routes J. The Wisconsin approach to newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2012;129:622–7.10.1016/j.jaci.2011.12.004Suche in Google Scholar PubMed
11. Puck JM. The case for newborn screening for severe combined immunodeficiency and related disorders. Ann NY Acad Sci 2011;1246:108–17.10.1111/j.1749-6632.2011.06346.xSuche in Google Scholar PubMed PubMed Central
12. van Zelm MC, van der Burg M, Langerak AW, van Dongen JJ. PID comes full circle: applications of V(D)J recombination excision circles in research, diagnostics and newborn screening of primary immunodeficiency disorders. Front Immunol 2011;2:12.10.3389/fimmu.2011.00012Suche in Google Scholar PubMed PubMed Central
13. Borte S, von Döbeln U, Fasth A, Wang N, Janzi M, Winiarski J, et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012;119:2552–5.10.1182/blood-2011-08-371021Suche in Google Scholar PubMed
14. Borte S, Wang N, Oskarsdóttir S, von Dobeln U, Hammarstrom L. Newborn screening for primary immunodeficiencies: beyond SCID and XLA. Ann NY Acad Sci 2011;1246:118–30.10.1111/j.1749-6632.2011.06350.xSuche in Google Scholar PubMed
15. Speckmann C, Neumann C, Borte S, la Marca G, Sass JO, Wiech E, et al. Delayed-onset adenosine deaminase deficiency: strategies for an early diagnosis. J Allergy Clin Immunol 2012;130:991–4.10.1016/j.jaci.2012.04.004Suche in Google Scholar PubMed
16. Boldt A, Borte S, Fricke S, Kentouche K, Emmrich F, Borte M, et al. Eight-color immunophenotyping of T-, B-, and NK-cell subpopulations for characterization of chronic immunodeficiencies. Cytometry B Clin Cytom 2014;86:191–206.10.1002/cytob.21162Suche in Google Scholar PubMed
17. Sack U, Gerling F, Tárnok A. Age-Related Lymphocyte Subset Changes in the Peripheral Blood of Healthy Children – a Meta-Study. Transfus Med Hemother 2007;34:176–81.10.1159/000101357Suche in Google Scholar
18. Dvorak CC, Cowan MJ. Radiosensitive severe combined immunodeficiency disease. Immunol Allergy Clin North Am 2010;30: 125–42.10.1016/j.iac.2009.10.004Suche in Google Scholar PubMed PubMed Central
19. Ochsenreither S, Fusi A, Busse A, Nagorsen D, Schrama D, Becker J, et al. Relative quantification of TCR Vbeta-chain families by real time PCR for identification of clonal T-cell populations. J Transl Med 2008;6:34.10.1186/1479-5876-6-34Suche in Google Scholar PubMed PubMed Central
20. Borte S, Janzi M, Pan-Hammarström Q, von Dobeln U, Nordvall L, Winiarski J, et al. Placental transfer of maternally-derived IgA precludes the use of guthrie card eluates as a screening tool for primary immunodeficiency diseases. PLoS One 2012;7:e43419.10.1371/journal.pone.0043419Suche in Google Scholar PubMed PubMed Central
21. Ozcan E, Notarangelo LD, Geha RS. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol 2008;122:1054–62.10.1016/j.jaci.2008.10.023Suche in Google Scholar PubMed
22. Peterson ML. Mechanisms controlling production of membrane and secreted immunoglobulin during B cell development. Immunol Res 2007;37:33–46.10.1007/BF02686094Suche in Google Scholar PubMed
23. Terada T, Kaneko H, Fukao T, Teramoto T, Asano T, Li AL, et al. Semiquantitative evaluation of mRNAs for the membranous form of immunoglobulin heavy chain is useful for investigating the etiology in CVID. Scand J Immunol 2003;58: 649–54.10.1111/j.1365-3083.2003.01350.xSuche in Google Scholar PubMed
24. Borte S, Pan-Hammarström Q, Liu C, Sack U, Borte M, Wagner U, et al. Interleukin-21 restores immunoglobulin production ex vivo in patients with common variable immunodeficiency and selective IgA deficiency. Blood 2009;114: 4089–98.10.1182/blood-2009-02-207423Suche in Google Scholar PubMed
25. Borte S, Lanig H, Borte M, Fasshauer M, Sack U. Therapeutic implications of the IL-21: IL-4 receptor system in children with common variable immunodeficiency syndrome. Klin Padiatr 2010;222:362–7.10.1055/s-0030-1265207Suche in Google Scholar PubMed
26. Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, et al. Newborn Screening for SCID Identifies Patients with Ataxia Telangiectasia. J Clin Immunol 2013;33:540–9.10.1007/s10875-012-9846-1Suche in Google Scholar PubMed PubMed Central
27. Becker-Catania SG, Chen G, Hwang MJ, Wang Z, Sun X, Sanal O, et al. Ataxia-telangiectasia: phenotype/genotype studies of ATM protein expression, mutations, and radiosensitivity. Mol Genet Metab 2000;70:122–33.10.1006/mgme.2000.2998Suche in Google Scholar PubMed
28. van der Burg M, Gennery AR. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Pediatr 2011;170:561–71.10.1007/s00431-011-1452-3Suche in Google Scholar PubMed PubMed Central
29. Hönig M, Schulz A, Friedrich W. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Klin Padiatr 2011;223:320–5.10.1055/s-0031-1287826Suche in Google Scholar PubMed
30. Cavazzana-Calvo M, Fischer A, Hacein-Bey-Abina S, Aiuti A. Gene therapy for primary immunodeficiencies: Part 1. Curr Opin Immunol 2012;24:580–4.10.1016/j.coi.2012.08.008Suche in Google Scholar PubMed
31. Okocha IU, Hanson CG, Chinen J, Shearer WT. Decline of antibodies in XLA infant: when to start IVIG. Allergy 2011;66:434–5.10.1111/j.1398-9995.2010.02481.xSuche in Google Scholar PubMed PubMed Central
32. Ghosh S, Krux F, Binder V, Gombert M, Niehues T, Feyen O, et al. Array-based sequence capture and next-generation sequencing for the identification of primary immunodeficiencies. Scand J Immunol 2012;75:350–4.10.1111/j.1365-3083.2011.02658.xSuche in Google Scholar PubMed
33. Chou J, Ohsumi TK, Geha RS. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiencies. Curr Opin Allergy Clin Immunol 2012;12:623–8.10.1097/ACI.0b013e3283588ca6Suche in Google Scholar PubMed
©2014 by Walter de Gruyter Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Allergie und Autoimmunität/Allergy and Autoimmunity
- Nachweis von schweren angeborenen Immundefekten bei Neugeborenen: Diagnostisches Vorgehen bei Screening, Tracking und Follow-up
- Mediators in pleural effusions of different origin: a two-step diagnostic study
- Übersichtsarbeit/Review
- Die Umsetzung der Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (RiliBÄK) aus Sicht der überwachenden Länderbehörden
- Klinische Chemie und Stoffwechsel/Clinical Chemistry and Metabolism
- NMR-Spektroskopie – ein modernes Werkzeug zur Serum-Analytik von Lipoproteinen und Metaboliten
- Molekulargenetische und zytogenetische Diagnostik/Molecular-Genetic and Cytogenetic Diagnostics
- 13. Jahrestagung der Sektion Molekulare Diagnostik der DGKL am 15. und 16. Mai 2014 in der Evangelischen Akademie Tutzing
- Kongressbericht/Congress Report
- Analytica Conference 2014, München 02. April 2014
Artikel in diesem Heft
- Frontmatter
- Allergie und Autoimmunität/Allergy and Autoimmunity
- Nachweis von schweren angeborenen Immundefekten bei Neugeborenen: Diagnostisches Vorgehen bei Screening, Tracking und Follow-up
- Mediators in pleural effusions of different origin: a two-step diagnostic study
- Übersichtsarbeit/Review
- Die Umsetzung der Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (RiliBÄK) aus Sicht der überwachenden Länderbehörden
- Klinische Chemie und Stoffwechsel/Clinical Chemistry and Metabolism
- NMR-Spektroskopie – ein modernes Werkzeug zur Serum-Analytik von Lipoproteinen und Metaboliten
- Molekulargenetische und zytogenetische Diagnostik/Molecular-Genetic and Cytogenetic Diagnostics
- 13. Jahrestagung der Sektion Molekulare Diagnostik der DGKL am 15. und 16. Mai 2014 in der Evangelischen Akademie Tutzing
- Kongressbericht/Congress Report
- Analytica Conference 2014, München 02. April 2014