Synthesis and characterization of polyvinylmethylsiloxanes by cationic polymerization using a solid green catalyst
-
Djamal Eddine Kherroub
 ,
Mohammed Belbachir
,
Mohammed Belbachir

Abstract
The present work is devoted to the synthesis and characterization of vinylsiloxane polymers produced by the use of an activated natural catalyst known as Maghnite-H+. The cationic ring opening polymerization of pentavinylpentamethylcyclopentasiloxane (V5D5) made it possible to obtain the desired polymeric materials. Through this study, we have adapted a new strategy of synthesis of a siloxane polymer with relatively high molecular mass, using a solid initiator activated by sulfuric acid, which has enabled us to combine the ecological aspect of synthesis and the effectiveness of the catalyst in this kind of reaction. Structural [infrared (IR), proton and carbon nuclear magnetic resonance (1H NMR and 13C NMR)], thermal differential scanning (DSC) and chromatographic (GPC) characterization methods have allowed the products obtained to be identified and their various properties to be focused on. The kinetic study was made to determine the order of the reaction. The proposed reaction mechanism shows the advantages of Maghnite-H+.
1 Introduction
Catalysis has become the indispensable tool for the socio-economic development of industrialized countries, because up to 80% of the manufactured products that surround our daily lives have undergone a catalytic stage during their synthesis. Catalysis greatly increases the production capacity of industrial units by substantially increasing the rate of chemical transformations and improving the selectivity of the reaction to the desired product (1). Catalysis should also make it possible to find ingenious solutions to develop more efficiently the chemical molecules we use (economy of raw materials, energy and investment) (2). Heterogeneous catalysis is the cornerstone of catalysis. In fact, nearly 90% of the processes used by the chemical industry involve heterogeneous catalysis. The development of synthetic materials such as fibers and polymers, which do not naturally exist and have revolutionized the 20th century, has been made possible by the discovery of catalysts for monomer synthesis and polymerization reactions.
Clay minerals are used in many fields. They often serve as catalytic support. The complete determination of their crystalline structure and surface properties is important because they are responsible for the specific properties of the mineral (3).
Biocatalysis is the use of natural catalysts, such as clays in an organic synthesis reaction. The clays are exploited in the formation of amino acids and in their polymerization into polypeptides. Polymerization tests of styrene, methyl methacrylate were carried out on the surfaces of certain clay minerals as attapulgite, kaolin and montmorillonite (4), (5), (6). Polymerization of several heterocyclic and vinyl monomers was carried out within the Laboratory of Polymer Chemistry (University of Oran in Algeria) using the clay of the Maghnia region (Maghnite) activated by sulfuric acid (7), (8), (9).
With this objective, we have designed and realized an alternative method for the synthesis of silicone polymers. This method involves the application of maghnite-H+, an aluminosilicate eco-catalyst developed locally at the Polymer Chemistry Laboratory to initiate the polymerization reaction of pentavinylpentamethylcyclopentasiloxane (V5D5) (Scheme 1A). The reaction was carried out under mild conditions without solvents using a readily separable solid catalyst. The amount of catalyst recovered may be reusable. This study belongs to the purpose of applying the principles of green chemistry and to design cleaner chemical processes not harmful to either human beings or to the environment.

The structure of (A) V5D5 and (B) polyvinylmethylsiloxane.
Silicones are amorphous materials composed of long macromolecular chains characterized by the alternation of silicon and oxygen atoms. The bonds between silicon and oxygen form a very flexible inorganic skeleton with remarkable physico-chemical properties: thermal stability, chemical inertia, etc. Due to their wide variety of presentation, they offer many options for use in a number of cosmetics and healthcare segment application fields. The polyvinylsiloxanes (PVMS) have generally the same macromolecular structure as the silicones. Their chemical formula is: -[(CH2=CH)CH3Si-O]- (Scheme 1B). They are distinguished by the presence of vinyl groups whose opening is at the base of the phenomenon of crosslinking between polymeric chains in the most cases (10), (11). They have evolved considerably: they are very fluid during injection, harden quickly (30–60 s) and have few dimensional variations. They are very rigid (addition of fillers in a proportion of 80% by weight) after polymerization (12), (13). They are used in all processes that take place in a dental laboratory. They are especially suitable for: temporary prostheses, removable prosthesis, ceramic molding, modeling replication, composite molding and implant placement (14), (15), (16), (17), (18), (19). In the case of cyclic monomers, the synthesis reaction of PVMS occurs by ring opening polymerization in the presence of a catalyst. Various types of catalysts have been used previously such as: the strong bases as phosphazene bases (20), (21), (22), (23), strong acids as dodecylbenzenesulfonic acid (24), triflic acid (25), tris(pentafluorophenyl)borane (26) or by gamma radiation (27), etc.
2 Experimental
2.1 Materials
Pentavinylpentamethylcyclopentasiloxane (V5D5, 99%) was used as purchased from Aldrich chemical (Algeria), without further purification. Methanol was purified by vacuum distillation. All other products have been used as received. Maghnite was obtained from Algerian company of bentonite (BENTAL), without any pretreatment. All products were obtained from the Aldrich chemical annex in Algeria (Sigma-Aldrich is a known chemical supplier on a global scale).
2.2 Preparation of Maghnite-H+
A mass of 30 g of raw maghnite is combined with 120 ml of distilled water at room temperature, the suspension is left under stirring. After 30 min, 100 ml of a solution of sulfuric acid (0.23 m) is added, the stirring is continued for 48 h. After filtration and subsequent washing, the activated maghnite is dried in an oven for 24 h at a temperature of 105°C. Finally, Maghnite-H+ was crushed, sieved and stored away from air and moisture.
2.3 Polymerization procedure
A total of 0.1 g of Maghnite-H+ was heated before use under vacuum with mechanical stirring for 30 min. The polymerization was carried in bulk. The dried amount of Maghnite-H+ was added to a flask containing 5 g of V5D5, the flask was immersed in an oil bath and brought to a temperature of 60°C under reflux at while being stirred. After 6 h, the reaction was stopped by deactivating the Maghnite-H+ by adding cold water to the reaction mixture. The Maghnite-H+ was recovered by filtration, and the filtrate was precipitated in methanol (non-solvent). The insoluble product was dried at 80°C in vacuum for 6–8 h and weighed as polymer. Excess water was retrieved by evaporation at 105°C, the amount of water necessary to stop the reaction would be then the difference between the initial amount and the recovered amount. It was assumed that the residual material was the remaining monomers and the oligomers formed during the reaction. Regarding the kinetic study, the same procedure described above was repeated by changing the temperature, time and the percentage of the catalyst.
2.4 Characterization methods
2.4.1 X-ray diffraction (XRD)
The XRD patterns of the samples were carried out at room temperature on a Bruker D8 Advance X-Ray diffractometer (40 kV, 30 mA) with a graphite monochromator, using CuKα radiation (λ=0.154 nm) at the rate of 5° min−1 in the range of 2θ=2°–80°.
2.4.2 Infrared spectroscopy (IR)
IR analysis of the polymers obtained was done using a Bruker alpha Fourier transform infrared (FTIR) spectrometer equipped with an ATR accessory.
2.4.3 Nuclear magnetic resonance (NMR)
1H and 13C NMR spectrums were recorded under ambient temperature on Bruker Avance 300 NMR spectrometer, using tetramethylsilane as the internal standard and deuterated chloroform as the solvent.
2.4.4 Differential scanning calorimetry (DSC)
The different thermal characteristics such as Tg of the synthesized polymer were measured by DSC from the corresponding thermal changes in the DSC thermogram using a Setaram 92 DSC apparatus.
2.4.5 Molecular weight measurements
Gel permeation chromatography (GPC) measurements of the samples was performed using a WISP Model 712 (Waters Associates) chromatograph, THF was used as a solvent and the apparatus was calibrated in an initial approximation with polymethyl methacrylate of known molecular weight.
3 Results and discussion
3.1 X-ray diffraction
Figure 1 shows the characterization by XRD analysis of the raw maghnite and maghnite treated with sulfuric acid. It is obvious that the treatment led to the removal of minerals such as calcite and mica, this is confirmed by the decline of the intensity of their peaks compared to the strong peak corresponding to montmorillonite (green area), this elimination is clearer for the quartz, as shown by the reduction of the two peaks at 2θ=21.93° and 26.71° (blue areas). Moreover, the acid treatment caused a shift of the peak of montmorillonite to small values of 2θ from 8.41° to 5.73°, corresponding to an increase of the interlayer distance of montmorillonite (d001) from 10.50 Å to 15.41 Å, this can be explained by the substitution of interlamellar cations of maghnite by the acid protons which have a larger atomic diameter.

XRD patterns of the maghnite before treatment (raw-maghnite) and after treatment (Maghnite-H+).
3.2 Infrared spectroscopy (IR)
Figure 2 provides the IR spectra of the monomer and the obtained products for 2, 4, 6, 8 and 10 h at 60°C. The broad peak seen at 3510 cm−1 for the obtained products is attributed to the OH stretching of the Si-OH end groups in the PVMS chains, the appearance of this peak is due to the linkage between the released proton of Maghnite-H+ and the oxygen atom at the end after the V5D5 ring opening (Scheme 2A), the decrease in its intensity with time of the reaction is clearly noticeable, which may be explained by the increase of polymerization degree leading to smaller number of OH chain ends. The small peak appears at about 3062 cm−1 is attributed to the C-H bond of the vinyl group (CH=C). The band corresponds to the stretch C=C is seen at 1639 cm−1. The decrease in the intensity of this two peaks corresponds to C-H and C=C of the vinyl group for PDVS obtained after long periods of time is perhaps explained by the rupture of the double bond between carbon atoms to establish interchain linkages with CH3 leading to the formation of crosslinked polymers (Scheme 2B). The two bands at 2907 and 2861 cm−1 are, respectively, due to C-H asymmetric/symmetric stretching of CH3. The two peaks seen, respectively, at 2788 and 2734 cm−1 are assigned to the C-H asymmetric/symmetric stretching of CH2. The signal at 1272 cm−1 is assigned to the CH3 symmetric deformation of Si-CH3. Peaks appearing at 1029, 1099 and 469 cm−1 are, respectively, attributed to the stretching vibrations and deformation vibrations of the linear Si-O-Si structures. The signal at 816 cm−1 is due to the Si-C stretching vibrations. The IR spectrum of obtained PVMS using Maghnite-H+ as the catalyst revealed no differences from those obtained by other researchers (28), (29).

IR spectra of PVMS obtained by the polymerization of V5D5 at a temperature of 60°C for different times.
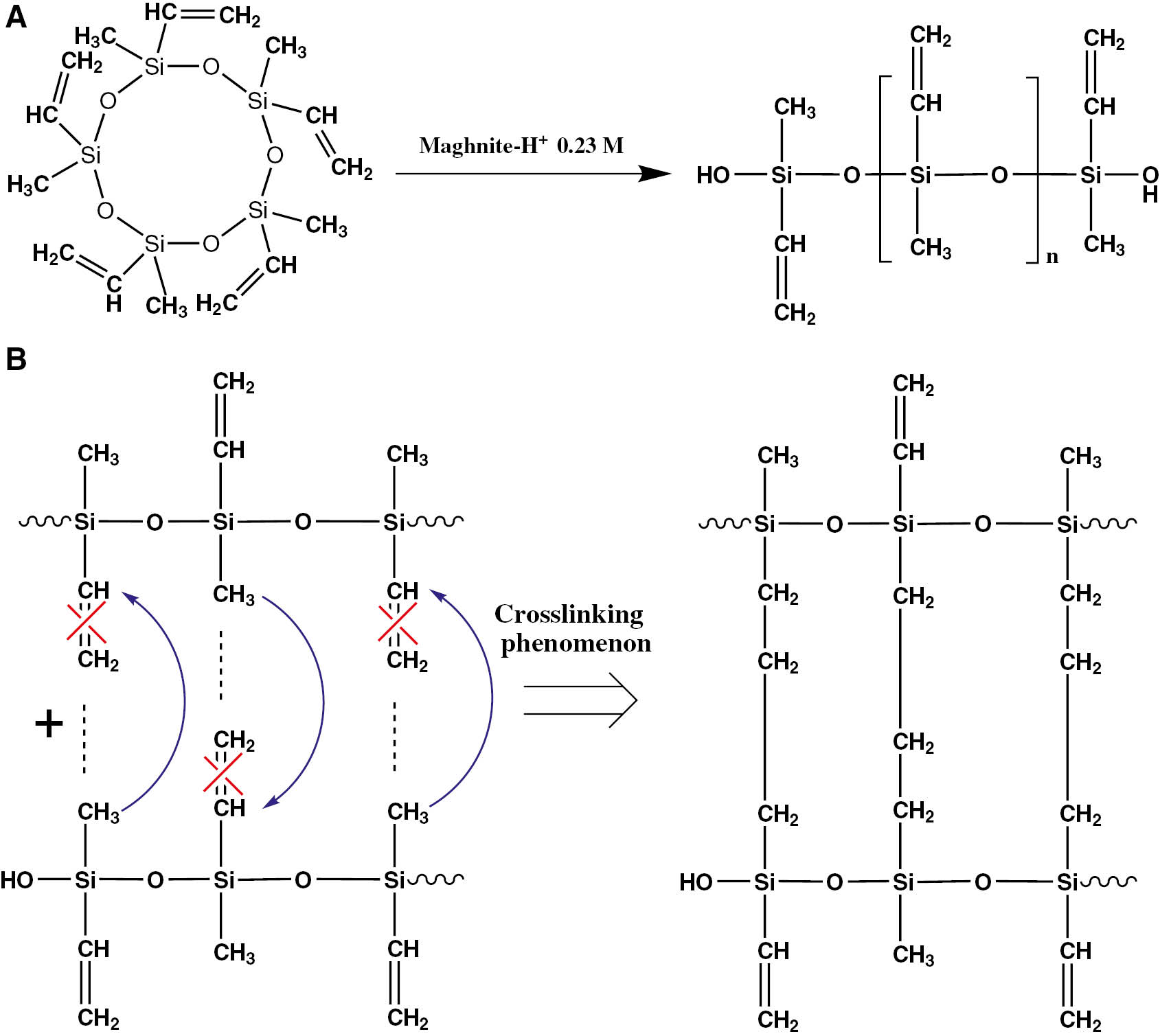
Polymerization of V5D5 by Maghnite-H+.
3.3 Proton nuclear magnetic resonance (1H NMR)
In order to identify more precisely the structure of the polymer obtained by the polymerization of V5D5 using the Maghnite-H+ as catalyst, the product was analyzed before and after reaction by NMR analysis by comparing the obtained spectra: that of the monomer and those of the polymers obtained after 6 h and 8 h at 60°C, respectively. The results are shown in Figure 3A–C, which shows the different chemical shifts. In the three spectra, the dominant peak observed at about 0.12 ppm, is attributed to the methyl groups, it is more intense in the spectra of polymers meaning the large number of methyl groups in the polymeric chain. The two peaks at 5.90 and 6.12 ppm are, respectively, assigned to CH2 and CH of the vinyl group. The small peak appearing at 4.86 ppm in the polymer spectra is assigned to the OH groups at the ends of polymer chains. Similar results were obtained by Yang et al. (30). The peak appeared at 1.30 ppm, exclusively for the polymer obtained after 8 h is attributed to the CH2 of the alkane group. These groups can be derived from the breaking of the vinylic double bond by establishing inter-chain propylene bridges.
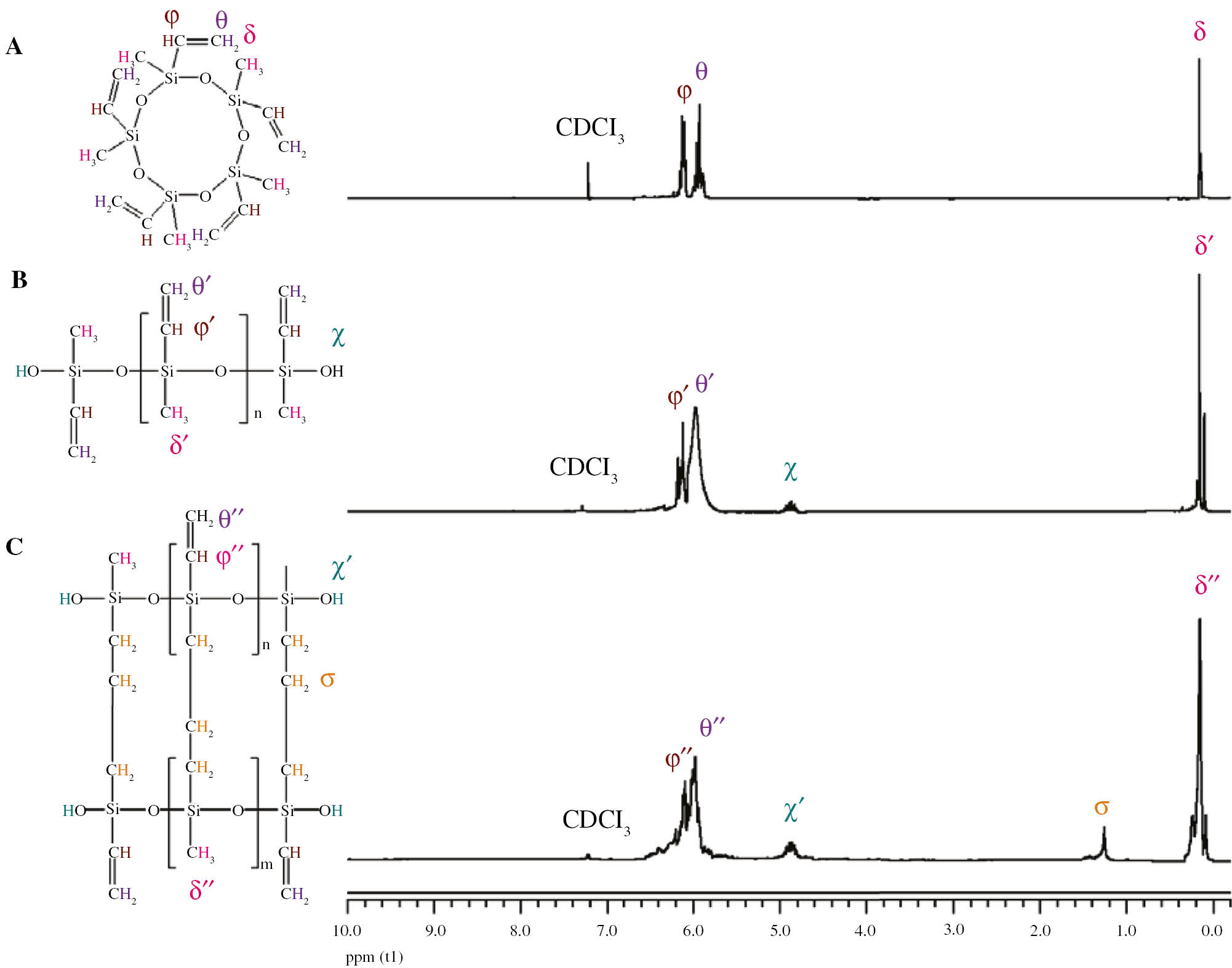
1H NMR spectra of (A) V5D5, (B) polymer obtained after 6 h and (C) polymer obtained after 8 h at a temperature of 60°C.
3.4 Carbon nuclear magnetic resonance (13C NMR)
It was therefore necessary to analyze the products obtained by 13C NMR to provide a complement to the previous study. The results are shown in Figure 4A–C showing the 13C NMR spectra of the monomer, the polymer obtained after 6 h and after 8 h, respectively. The peak located at approximately −0.35 ppm for the monomer and −0.65 ppm for the polymers corresponds to the carbon of CH3. The other two neighboring peaks that appear at 130.02 and 140.95 ppm for the monomer and at 130.39 and 141.01 ppm for the polymers are assigned to CH2 and CH of the vinyl group, respectively. Moreover, there is a creation of two down peaks at 20.10 and 32.87 ppm on the DEPT-135 spectrum of the polymer obtained after 8 h (Figure 4C), which are attributed to the carbon of CH2 of the alkane group, indicating the formation of propylene bridges between linear polymer chains. These results confirm that beyond 6 h of reaction time, the polymer chains can be crosslinked to form organopolysiloxane elastomers.

13C NMR spectra of (A) V5D5, (B) polymer obtained after 6 h and (C) polymer obtained after 8 h at a temperature of 60°C.
3.5 Differential scanning calorimetry (DSC)
DSC was used as a thermal analysis, to identify and confirm at the same time the structure and the purity of the obtained product. Figure 5 shows the DSC thermogram of the polymer obtained after 6 h of reaction time. The thermogram shows a small thermal deformation at about −136.43°C, which corresponds to the glass temperature of the polymer (Tg=−136.43°C), this Tg value is comparable to that of linear PVMS. The result obtained by DSC largely supports the results obtained by IR and NMR for the structure of the polymer obtained after 6 h, it also simulates to a large extent the results obtained in previous research (31), (32).

DSC thermogram of obtained PVMS obtained after 6 h.
3.6 Effect of temperature
In an effort to understand and control more the polymerization reaction of V5D5 catalyzed by Maghnite-H+, we have examined the effect of the temperature of the medium on the reaction that takes place there. Table 1 gives measured values of the monomer conversion and number average molecular mass of the polymers obtained in a temperature range of 10–110°C. The increase in temperature leads to a significant increase in conversion reaching 89% at 60°C, beyond this temperature, this increase becomes negligible until the conversion stabilizes at its maximum at about 93%. On the other hand, the variation in average molecular mass shows two different behaviors, a gradual increase from 10 to 60°C, followed by a reduction after just exceed its highest value at about 60°C, we assume that it is the ceiling temperature, this decrease in average molecular mass can be explained by the fragmentation of the chains suggests the thermal decomposition of PVMS after the breaking of Si-O bonds when approaching the boiling point. The thermal degradation phenomenon reflects a wide divergence between the molecular mass values, resulting in the increase of the polydispersity index, which is found in Table 1.
Effect of reaction temperature on V5D5 polymerizationa.
| T (°C) | Conversion of monomer (wt %) | Mn | Mw/Mn |
|---|---|---|---|
| 10 | 17 | 1821 | 1.05 |
| 30 | 52 | 9210 | 1.05 |
| 50 | 79 | 16,003 | 1.08 |
| 60 | 89 | 21,214 | 1.10 |
| 70 | 92 | 18,547 | 1.22 |
| 90 | 93 | 14,877 | 1.45 |
| 110 | 93 | 10,441 | 1.61 |
aMaghnite-H+/V5D5 weight ration=2%. Reaction time 6 h.
Mn, Number average molecular mass; Mw/Mn: Polydispersity index.
3.7 Effect of Maghnite-H+/monomer weight ratio
In order to study the catalytic action of Maghnite-H+ as a heterogeneous catalyst in the polymerization reaction of V5D5, we performed the reaction with a catalyst content ranging from 0.5 to 4% by weight, so that for each catalyst content, the reaction time varied from 0.5 to 10 h. The results of the influence of the Maghnite-H+ content on the monomer conversion and on the number average molecular mass are shown in Figures 6 and 7, respectively. In all tests, the reaction was carried out in bulk and at a temperature of 60°C. It is clearly noticeable that for all the different Maghnite-H+ contents, the reaction time has an effect proportionally positive on the monomer conversion before 8 h. After this duration, the effect of reaction time has become negative (Figure 6). The reduction in the monomer conversion for large periods may be explained by the occurence of depolymerization phenomenon of polymer chains caused by the active cites of the Maghnite-H+ still remaining in the reaction medium. This result indicates that the Maghnite-H+ can play the opposite role after periods of time sufficiently large. Similar results were previously obtained by several authors (33), (34). On the other hand, the average molecular mass increases with increasing reaction time, the maximum value for the different Maghnite-H+ contents is about 6 h, then it is almost stabilizes for 2 h, so that it begins to decrease. The reduction over time, of the peak corresponding to OH groups exist only at the end of the polymer chains of PVMS synthesized by Maghnite-H+ showing by the IRI analysis (Figure 2), indicates that the chains became longer, resulting in large molecular mass. The temporary stabilization between 6 and 8 h of the average molecular mass and at the same time the increase in the monomer conversion, due to the crosslinking phenomenon leading to branched structures because of the formation of propylene bridges between the linear chains, this explanation is clearly supported by what has been obtained by 13C NMR analysis (Figure 4C). The decrease in the average molecular mass after 8 h can be explained by backbiting degradation in the growing polymer chains, which generates oligomers and cyclic polysiloxanes of varying sizes, thereby increasing the polydispersity index (Table 2).

Effect of Maghnite-H+/V5D5 weight ratio on the conversion of monomer.

Effect of Maghnite-H+/V5D5 weight ratio on the average molecular mass.
Kinetic evolution of V5D5 polymerizationa initiated by Maghnite-H+.
| Time (h) | Conversion of monomer (wt %) | Mn | Mw/Mn |
|---|---|---|---|
| 0.5 | 38 | 3278 | 1.06 |
| 1 | 60 | 7958 | 1.07 |
| 2 | 73 | 12,447 | 1.07 |
| 4 | 80 | 18,097 | 1.10 |
| 6 | 89 | 21,214 | 1.10 |
| 8 | 92 | 20,985 | 1.25 |
| 10 | 84 | 17,175 | 1.61 |
aMaghnite-H+/V5D5 weight ration=2%. Reaction temperature 60°C.
3.8 Kinetics and mechanism of the polymerization
In order to study the chemical kinetics of the polymerization reaction of V5D5 catalyzed by Maghnite-H+, we followed the evolution of the concentration of monomer over time, we were interested just for t≤8 h, where there was not the depolymerization phenomenon. The results clearly indicate that the reaction is first-order with respect to monomer (Figure 8). Scheme 3 shows the probable reaction mechanism.
![Figure 8: Representation of Ln ([V5D5]0/[V5D5]t) as a function of time (Maghnite-H+/V5D5 weight ratio=2%, T=60°C).](/document/doi/10.1515/epoly-2017-0039/asset/graphic/j_epoly-2017-0039_fig_008.jpg)
Representation of Ln ([V5D5]0/[V5D5]t) as a function of time (Maghnite-H+/V5D5 weight ratio=2%, T=60°C).

Mechanism of D5V5 polymerization catalyzed by Maghnite-H+.
4 Conclusion
The preparation of polymers based on polyvinylsiloxanes was carried out in bulk and in a single step. The use of Maghnite-H+ as a catalyst for the reaction has made it possible to obtain satisfactory yields at different temperatures. The kinetic study by GPC shows that the yield of the reaction increases with temperature and then stabilizes at its maximum about 93%, regardless of the temperature. The mass content of the Maghnite-H+ also increases the yield, better yield was obtained at 2%, more than this, the Maghnite-H+ becomes a depolymerization agent due to the presence of a sufficiently large number of active sites, this also leads to the decrease in average molecular mass by backbiting phenomenon. The long periods of time cause the reduction of the average molecular mass, the highest value was detected at 6 h. The structure of the polymer was identified by IR, 1H NMR, and 13C NMR. The Tg determined by DSC indicates that the polymer obtained is of linear structure.
The advantage of this technique lies in the fact that the polymerization requires a simpler operating protocol than the protocols used so the catalyst is not toxic, it can be eliminated by simple filtration and recycled. These results open a very important field for the synthesis of silicones by heterogeneous catalysis in accordance with the principles of green chemistry.
References
1. Clark JH, Rhodes CN. Clean synthesis using porous inorganic solid catalysts and supported reagents. 2nd ed. Cambridge: Royal Society of Chemistry; 2000. 10 p.Search in Google Scholar
2. Chen B, Zhan X, Yi L, Chen F. Cationic ring opening polymerization of octamethylcyclotetrasiloxane initiated by acid treated bentonite. Chin J Chem Eng. 2007;15(5):661–5.10.1016/S1004-9541(07)60142-6Search in Google Scholar
3. Belbachir M, Bensaoula A. Composition and method for catalysis using bentonites. US Patent. 2001: No 6, 274,527B1.Search in Google Scholar
4. Kherroub DE, Belbachir M, Lamouri S, Bouhadjar L, Chikh K. Synthesis of polyamide-6/montmorillonite nanocomposites by direct in-situ polymerization catalysed by exchanged clay. Orient J Chem. 2013;29(4):1429–36.10.13005/ojc/290419Search in Google Scholar
5. Kherroub DE, Belbachir M, Lamouri S. Nylon 6/clay nanocomposites prepared with Algerian modified clay (12-maghnite). Res Chem Intermed. 2015;41(8): 5217–28.10.1007/s11164-014-1623-8Search in Google Scholar
6. Kherroub DE, Belbachir M, Lamouri S. Synthesis of poly(furfuryl alcohol)/montmorillonite nanocomposites by direct in-situ polymerization. Bull Mater Sci. 2015;38(1):1–7.10.1007/s12034-014-0818-3Search in Google Scholar
7. Meghabar R, Megherbi A, Belbachir M. Maghnite-H+, an ecocatalyst for cationic polymerization of N-vinyl-2-pyrrolidone. Polymer. 2003;44(15):4097–100.10.1016/S0032-3861(03)00400-2Search in Google Scholar
8. Bouchama A, Ferrahi MI, Belbachir M. Copolymerization of ε-caprolactone with tetrahydrofuran by a solid acid, in the presence of acetic anhydride. J Mater Environ Sci. 2015;6(4):977–82.Search in Google Scholar
9. Kherroub DE, Belbachir M, Lamouri S. Preparation and characterization of organophilic montmorillonite (12-maghnite) using algerian clay. Orient J Chem. 2014;30(4):1647–51.10.13005/ojc/300424Search in Google Scholar
10. Kendrick TC, Parbhoo B, White JW. The silicon-heteroatom bond. Chichester: John Wiley & Sons; 1991. 67 p.10.1002/9780470772447.ch3Search in Google Scholar
11. Chojnowski J, Cypryk M. Silicon-containing polymers. Dordrecht: Kluwer; 2000. 3 p.10.1007/978-94-011-3939-7_1Search in Google Scholar
12. Narins RS, Beer K. Liquid injectable silicone: a review of its history, immunology, technical considerations, complications, and potential. Plast Reconstr Surg. 2006;118(3):77–84.10.1097/01.prs.0000234919.25096.67Search in Google Scholar PubMed
13. Dumitriu S. Polymeric biomaterials. 2nd ed. New York: Marcel Dekker; 2002. 79 p.Search in Google Scholar
14. Friedel C, Crafts JM. Comprehensive organic name reactions and reagents. Compt Rend. 1877;84:1392–6.Search in Google Scholar
15. Crafts JM. Friedel memorial lecture. J Chem Soc. 1900;77:993–8.10.1039/ct9007700993Search in Google Scholar
16. Chee WWL, Donovan TE. Polyvinyl siloxane impression materials: a review of properties and techniques. J Prosthet Dent. 1992;68(5):728–32.10.1016/0022-3913(92)90192-DSearch in Google Scholar
17. Mandikos MN. Polyvinyl siloxane impression materials: an update on clinical use. Aust Dent J. 1998;43(6):428–34.10.1111/j.1834-7819.1998.tb00204.xSearch in Google Scholar
18. Chojnowski J, Cypryk M, Kazmierski K. Cationic polymerization of a model cyclotrisiloxane with mixed siloxane units initiated by a protic acid. Mechanism of polymer chain formation. Macromolecules. 2002;36(27):9904–12.10.1021/ma021060nSearch in Google Scholar
19. Sigwalt P. New developments in cationic polymerization of cyclosiloxanes. Polym J. 1987;19:567–80.10.1295/polymj.19.567Search in Google Scholar
20. Molenberg A, Möller M. A fast catalyst system for the ring-opening polymerization of cyclosiloxanes. Macromol Rapid Commun. 1995;16(6):449–53.10.1002/marc.1995.030160606Search in Google Scholar
21. Pibre G, Chaumont P, Fleury E, Cassagnau P. Ring-opening polymerization of decamethylcyclopentasiloxane initiated by a superbase: kinetics and rheology. Polymer 2008;49(1): 234–40.10.1016/j.polymer.2007.11.017Search in Google Scholar
22. Gee RP. Emulsion polymerization of dimethylcyclosiloxane in cationic emulsion: mechanism study utilizing two phase liquid–liquid reaction kinetics. Colloids Surf A Physicochem Eng Asp. 2015;481:297–306.10.1016/j.colsurfa.2015.05.033Search in Google Scholar
23. Sun CN, Shen MM, Deng LL, Mo JQ, Zhou BW. Kinetics of ring-opening polymerization of octamethylcyclotetrasiloxane in microemulsion. Chin Chem Lett. 2014;25(4):621–6.10.1016/j.cclet.2013.12.021Search in Google Scholar
24. Sijiu J, Teng Q, Xiaoyu L. Kinetic study on the ring-opening polymerization of octamethylcyclotetrasiloxane (D4) in miniemulsion. Polymer 2010;51(18):4087–94.10.1016/j.polymer.2010.06.038Search in Google Scholar
25. Conan JT, William PW, Guoping C. Acid and base catalyzed ring-opening polymerization of 2,2,4,4,6,6-hexamethyl-8,8-diphenylcyclotetrasiloxane. Polymer 2003;44(15):4149–55.10.1016/S0032-3861(03)00373-2Search in Google Scholar
26. Chojnowski J, Rubinsztajn S, Fortuniak W, Kurjata J. Oligomer and polymer formation in hexamethylcyclotrisiloxane (D3)–hydrosilane systems under catalysis by tris(pentafluorophenyl)borane. J Inorg Orgamet Polym. 2007;17(1):173–87.10.1007/s10904-006-9083-2Search in Google Scholar
27. Alexandrova L. Bulk polymerization of 1,3,5,7-tetravinyltetramethylcyclotetrasiloxane induced by gamma radiation. Radiat Phys Chem. 1998;51(1):101–6.10.1016/S0969-806X(97)00106-0Search in Google Scholar
28. Hao L, Xu W, An Q. Synthesis, film morphology and performance of novel crosslinked polysiloxane with end-capped epoxy groups on cotton substrates. Fiber Polym. 2014;15(8):1567–74.10.1007/s12221-014-1567-zSearch in Google Scholar
29. Efimenko K, Crowe JA, Manias E, Schwark DW, Fischer DA, Genzer J. Rapid formation of soft hydrophilic silicone elastomer surfaces. Polymer 2005;46(22):9329–41.10.1016/j.polymer.2005.07.046Search in Google Scholar
30. Yang L, Cao K, Huang Y, Chang G, Zhu F, Yang. Synthesis and properties of cross-linkable polysiloxane via incorporating benzocyclobutene. High Perform Polym. 2010;26(4):463–9.10.1177/0954008313518567Search in Google Scholar
31. Julie A, Crowe W, Jan G. Formation and properties of responsive siloxane-based polymeric surfaces with tunable surface reconstruction kinetics. Adv Funct Mater. 2009;19(3):460–9.10.1002/adfm.200800622Search in Google Scholar
32. Crowe-Willoughby JA, Stevens DR, Genzer J, Clarke LI. Investigating the molecular origins of responsiveness in functional silicone elastomer networks. Macromolecules. 2010;43(11):5043–51.10.1021/ma100470wSearch in Google Scholar
33. Kherroub DE, Belbachir M, Lamouri S. Cationic ring opening polymerization of ε-caprolactam by a montmorillonite clay catalyst. BCREC 2014;9(1):74–80.10.9767/bcrec.9.1.5555.74-80Search in Google Scholar
34. Kherroub DE, Belbachir M, Lamouri S. Study and optimization of the polymerization parameter of furfuryl alcohol by algerian modified clay. Arab J Sci Eng. 2015;40(1):143–50.10.1007/s13369-014-1512-xSearch in Google Scholar
©2017 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this Issue
- Elastic recovery of polyamide 6 matrix nanocomposites and their basalt fiber co-reinforced hybrids
- Bio-based chitosan/PVdF-HFP polymer-blend for quasi-solid state electrolyte dye-sensitized solar cells
- Synergistic effects of retinoic acid and graphene oxide on the physicochemical and in-vitro properties of electrospun polyurethane scaffolds for bone tissue engineering
- Non-isothermal crystallization and thermal degradation kinetics of MXene/linear low-density polyethylene nanocomposites
- Extraction and characterization of chitin and chitosan: applications of chitosan nanoparticles in the adsorption of copper in an aqueous environment
- Systematic evaluation of pH and thermoresponsive poly(n-isopropylacrylamide-chitosan-fluorescein) microgel
- Plasticizer effect on melt blending of polylactide stereocomplex
- Polypropylene/basalt thick film composites: structural, mechanical and dielectric properties
- Performance properties of rigid polyurethane-polyisocyanurate/brewers’ spent grain foamed composites as function of isocyanate index
- Synthesis and characterization of polyvinylmethylsiloxanes by cationic polymerization using a solid green catalyst
Articles in the same Issue
- Frontmatter
- In this Issue
- Elastic recovery of polyamide 6 matrix nanocomposites and their basalt fiber co-reinforced hybrids
- Bio-based chitosan/PVdF-HFP polymer-blend for quasi-solid state electrolyte dye-sensitized solar cells
- Synergistic effects of retinoic acid and graphene oxide on the physicochemical and in-vitro properties of electrospun polyurethane scaffolds for bone tissue engineering
- Non-isothermal crystallization and thermal degradation kinetics of MXene/linear low-density polyethylene nanocomposites
- Extraction and characterization of chitin and chitosan: applications of chitosan nanoparticles in the adsorption of copper in an aqueous environment
- Systematic evaluation of pH and thermoresponsive poly(n-isopropylacrylamide-chitosan-fluorescein) microgel
- Plasticizer effect on melt blending of polylactide stereocomplex
- Polypropylene/basalt thick film composites: structural, mechanical and dielectric properties
- Performance properties of rigid polyurethane-polyisocyanurate/brewers’ spent grain foamed composites as function of isocyanate index
- Synthesis and characterization of polyvinylmethylsiloxanes by cationic polymerization using a solid green catalyst