Investigations of the intrinsic corrosion and hydrogen susceptibility of metals and alloys using density functional theory
-
Christopher D. Taylor
Christopher D. Taylor received his BS from the University of Western Australia, MS from the University of Memphis, and PhD from the University of Virginia. Chris worked at Los Alamos National Laboratory, studying corrosion in oil and gas, materials/environment effects for national security, catalyst design and optimization, and nuclear waste disposition. Chris is currently a Senior Researcher at DNV, and Associate Research Professor at the Ohio State University. Christopher has 100+ publications in science and engineering, with a focus on computational modeling of corrosion.

 und
Huibin Ke
und
Huibin Ke
Huibin Ke received her BS from Harbin Institute of Technology in 2011 and earned her PhD from the University of Wisconsin – Madison in 2017. Since then, she worked as a postdoctoral researcher with Prof. Christopher Taylor at The Ohio State University on ab initio-based modeling of alloy corrosion mechanisms in aqueous solution, after which she worked as a Postdoctoral Research Associate in Pacific Northwest National Laboratory. Her research interest is multi-scale modeling of microstructure evolution of metallic materials.

Abstract
Mechanisms for materials degradation are usually inferred from electrochemical measurements and characterization performed before, during, and after exposure testing and/or failure analysis of service materials. Predicting corrosion and other materials degradation modes, such as hydrogen-assisted cracking, from first-principles has generally been limited to thermodynamic predictions from Pourbaix or Ellingham diagrams and the Galvanic series. Using electronic structure calculations, modern first-principles methods can predict ab initio the key rate-controlling processes for corrosion and hydrogen susceptibility as a function of pH, potential, and solution chemistry, and materials composition and microstructure. Herein we review density functional theory (DFT) approaches for studying the electrochemical reactions occurring on fresh metal and alloy surfaces related to environmentally assisted cracking and localized corrosion/pitting. Predicted changes in surface chemistry as a function of the environment were correlated against experimental crack growth rate data obtained for alloys 718, 725, and pipeline steel under electrochemical control. We also review the application of the method to study the effects of alloying on the chloride susceptibility of stainless steels and Ni–Cr-based corrosion-resistant alloys. Perspectives for improving the model are given, and extending it to future fields of application in corrosion science and engineering.
1 Introduction
Physics-based simulation of materials aging and degradation through the mechanisms of corrosion and hydrogen embrittlement remains a challenging field of research (Barrera et al. 2018; Taylor 2015). Such research is motivated by the necessity for decision-makers in materials engineering and design to develop new materials that are more robust under a variety of environments, to establish fitness for service criteria, and to extend the lifetime of aging structures (Cordeiro et al. 2020; Taylor et al. 2018c). Credible models can help augment and interpret the often-limited set of relevant historical and experimental databases that typically inform such decision-making processes (Melchers 2019). Such models can fall into two generic categories: empirical models, with sometimes (but not always) physically motivated functional forms that are parameterized to laboratory and/or field data (Feliu et al. 1993a, 1993b; Melchers 2019; Mendoza and Corvo 2000; Oesch and Faller 1997); and first-principles models that are physically motivated and parameterized only by fundamental physical constants (Huang et al. 2019; Lin et al. 1981; Sharland 1987; Taylor 2015; Taylor et al. 2018c).
The development of first-principles models for corrosion and hydrogen embrittlement will require a careful re-construction of the key physicochemical processes that contribute to those phenomena (Gunasegaram et al. 2014). These processes include mass-transport from the environment to the near-surface region; chemical and electrochemical reactions at the electrified interface between the material and solution; compositional and microstructural changes in the material itself; interactions of solutes and surfaces with static and dynamic strain fields in the material; and formation and evolution of surface films and scales (Anderko et al. 2008, 2014, 2015; Barrera et al. 2018; Rice and Wang 1989; Shibata 2007; Tehranchi et al. 2020). These processes are dynamic, involve many different species, maybe close to or far from equilibrium, and heavily dependent upon environmental variables such as temperature, pH, electrochemical potential, and the presence of impurities in both the solution and the material itself. Clearly, deriving a completely first-principles approach to corrosion and hydrogen embrittlement phenomena is a challenge for theorists.
In this review, we describe the development of an approach that began with a relatively simple analysis from first principles of the electrode/electrolyte interface (Filhol and Neurock 2006) and moved up to consider the effect of different environmental variables on chemical transformations that take place on activated metal surfaces (Taylor et al. 2007c). The approach was then adapted incrementally to increase in complexity, such that electrochemical reactions on alloy materials in more complex solution chemistry environments can now be estimated entirely from first principles (Ke and Taylor 2020b; Ke et al. 2020b). The method’s assumptions and idealizations are reviewed in detail and potential improvements are suggested that could be made in future developments of the approach. Some new frontiers for the development and application of this methodology are also presented.
2 Density functional theory
Density functional theory (hereafter, DFT) is a method for predicting the structure and properties of matter based on the solution for the ground-state electron density distribution for a given potential field (Jones 2015). In the typical kinds of DFT calculations being performed in contemporary science and engineering, these potential fields often consist of the superposition of individual potential energy fields that are mapped to the nuclei and core electrons of the atoms that constitute solids, liquids, and the molecules and ions in their environment (also known as effective core potentials or pseudopotentials) (Norskov et al. 2010). Additional terms can be added into the electronic structure problems that are solved by DFT to simulate other effects, such as electric fields across materials or interfaces (Hansen et al. 2008; Jacob 2007; Jinnouchi and Anderson 2008b; Karlberg et al. 2007; Lozovoi and Alavi 2007; Ohwaki and Yamashita 2001), the physics of spin–orbit coupling (Boettger and Ray 2002), magnetization and charge-localization constraints (Chen et al. 2014; Mills et al. 2003; Souvi et al. 2013; Scivetti and Persson 2013; Wodtke et al. 2008), self-energy corrections (Mattsson and Jennison 2002), van der Waals interactions (Antony and Grimme 2006; Grimme 2006; Tkatchenko et al. 2010, 2012), and explicit or implicit methods to simulate the effect of solvation on system energies (Marenich et al. 2009; Mathew et al. 2014; Miertuš et al. 1981; Taylor 2009). DFT calculations are time-consuming, even with state-of-the-art computer hardware, and therefore also expensive. For this reason, efforts to reduce the computational burden of DFT calculations, or to approximate their outputs using machine learning and/or coarse-graining, have been popular areas of research for some time (Fellinger et al. 2010; Hegde and Bowen 2017; Martinez et al. 2013, 2020; Schleder et al. 2019).
Because DFT calculations provide information about hypothetical configurations of matter at the level of electronic granularity, a large number of properties can be predicted (Hafner et al. 2006; Ke and Taylor 2019; Parr and Yang 1984; Saal et al. 2013; Yang et al. 1984). Two of the most commonly reported outputs from DFT calculations include ‘optimized geometries’ for atoms and molecules in various states of interaction or compound formation, and the electronic internal energy, which is useful for comparing different states for their relative thermodynamic stabilities and to estimate heats of reaction from the difference in internal energies of product and reactant states (Alfe et al. 2000).
DFT calculations are solved by self-consistent solution of the Kohn–Sham equations (Kohn and Sham 1965), which mirror the concept of ‘molecular orbitals’ in computational quantum chemistry, although the ideas do not exactly correspond (Stowasser and Hoffmann 1999). In the Kohn–Sham equations below, the first term in parentheses is the kinetic energy functional and the second term is the effective potential energy field that is interacting with the occupied one-electron Kohn–Sham orbitals
Whereas the physics of the interaction between charged particles that are fundamental to DFT is formally exact, determining an accurate solution of these equations is limited by (a) the present lack of knowledge of what is called the exchange-correlation functional (but good approximations exist), and (b) the burdensome computational requirements needed to solve this problem which scale as n3, where n is the number of electrons (Korth 2017; Mortensen et al. 2005; Su and Xu 2017). The two effects of exact exchange and electron correlation relate to the quantum mechanical properties of electrons and the difficulty in describing the multi-electron wavefunction in terms of separable electronic orbital functions. A hierarchy of approaches has been developed to incrementally approach the exact energy limit, and increasing computational complexity is required to ascend this hierarchy, known in quantum chemistry as “Jacob’s Ladder” (Car 2016). Whereas this is one source of error in the formalism, which itself is an active field of research (Mardirossian and Head-Gordon 2017), when the method is being applied to practical engineering problems, such as corrosion and hydrogen embrittlement, other sources of error are probably more important and result from the necessity of creating idealized, solvable representations that mimic problems of industrial significance. Even comparison to experiments conducted under well-controlled laboratory conditions often involves many steps of approximation and idealization. For this reason, it is the creation, evolution, and interpretation of DFT models in the context of corrosion science that forms the focus of this review.
A flowchart showing the workflow for DFT calculations is given in Figure 1. A list of useful outputs from first-principles DFT models that can be applied to help interpret corrosion and hydrogen embrittlement is shown in Table 1, adapted from Ke and Taylor (2019).
Workflow of DFT modeling.
Useful outputs from DFT calculations, adapted from Ke and Taylor (2019).
| Output | Application |
|---|---|
| Electron density distribution | Atomic charge analysis Work function Density of states and projected density of states Molecular orbitals and reactivity metrics Electrostatic and electrochemical potentials |
| Potential energy | Evaluate reaction mechanisms Statistical mechanics and thermodynamics Activation energies and reaction kinetics Comparison of different atomic configurations Mechanical properties |
| Normal mode analysis | Vibrational frequency assignment Thermodynamic properties (partition function) |
| Band structure | Photoelectron spectroscopy |
| Equilibrium atomic geometries | Prediction of molecular, crystalline, or defect structure |
3 Using DFT to model the electrode-electrolyte interface
Aqueous corrosion is a redox process involving the dissolution of metal atoms as cations (at the anode), and a counter-electrode reaction (at the cathode) that involves the reduction of species in solution with consumption of the electrons that are produced during anodic dissolution (Tomashov 1961). Some of the fundamental elements that need to be modeled from first principles, therefore, are the structure and properties of the electrochemical interface (Jinnouchi and Anderson 2008b; Lozovoi et al. 2001), and the influence of electrochemical and materials parameters on the reactions that occur there (Anderson and Kang 1998; Jinnouchi and Anderson 2008a; Taylor et al. 2018b). To construct models for materials/environment interfaces, an appropriate function νeff(r) needs to be constructed that creates the potential energy field around which the electronic structure will be solved via the self-consistent field theory. The predominant model used for such scenarios is the “periodic slab” method in which a three-dimensional box is constructed which has a finite number of layers of atoms that minimally represent the material and its surface (i.e., one could use say five layers of metal atoms arranged in a specific crystallographic orientation of interest; often low-index surfaces are used because of their close-packed, stable structure) and either a void or solvated space adjacent and normal to the surface is populated with the environmental molecules of interest (Makov and Payne 1995; Taylor et al. 2006b). This method has been widely used to simulate the catalytic properties of metal surfaces used in industrial catalysis and then later adapted to study systems of interest to corrosion science (Norskov et al. 2010; Skúlason 2015).
Motivated to study the impact of changing electrochemical potentials on the functionality of electrocatalysts and corrosion reactions, Filhol and Neurock (2006), and Taylor et al. (2006b) expanded upon this framework by introducing a virtual potentiostat into the cell (Figure 2). Electrification of the interface to simulate anodic (or cathodic) potentials was effectuated in this method by changing the number of electrons, N in equation (1b), as the constraint to the Kohn–Sham DFT model (Lozovoi and Alavi 2007; Lozovoi et al. 2001). The corresponding variation in electrochemical potential was detected in the simulations by monitoring the difference in the electrostatic potential between the Fermi level of the metal slab system against the potential at the point of the virtual potentiostat (located at the mid-point of the cell, in the environment region); i.e., essentially the work function of the coupled electrode/electrolyte system. This method was used to simulate the electrochemical activation of water, and the impact of changes in potential on the water molecule arrangements near the interface (Desai and Neurock 2003; Filhol and Neurock 2006; Rossmeisl et al. 2006; Taylor et al. 2006a, 2006b, 2007b, 2007c). Of particular interest was to evaluate the effect of surrounding, explicitly modeled water molecules, on the predictions made in the DFT approach. Because the inclusion of water molecules in the slab model adds significant computational complexity, many studies of adsorption exclude “spectator” solvent molecules. The study by Rossmeisl et al. showed that these can be omitted from the calculation with little effect on the chemisorption energies and final results (Rossmeisl et al. 2006). For other calculations, such as metal dissolution, in which solvation of the product state (i.e., a dissolved cation) plays a key role in lowering the system energy, such idealizations would be expected to radically influence the final results.
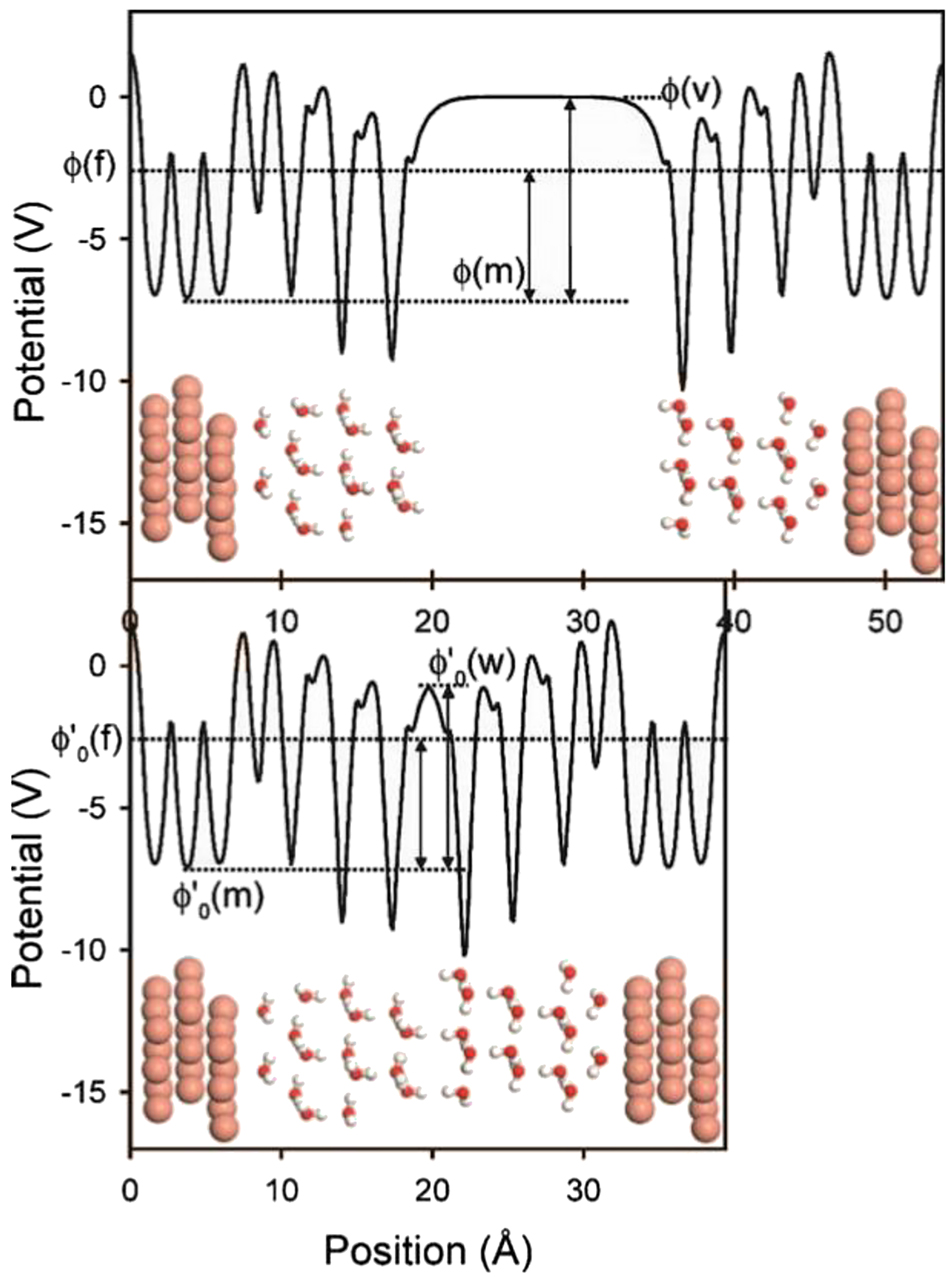
An example of a periodic slab model with virtual potentiostats shown to enable calibration to a vacuum reference state (top), and then electrified reference states (bottom) (Taylor et al. 2006b). Reproduced with permission from the American Physical Society (© 2006). The electrostatic potential profile across the periodic slab is plotted, with the vacuum potential φ(v), the Fermi level φ(m) marked for the top figure, and reference points for the ‘double electrode’ method, φ′0(m), marked and the solution potential φ′0(w). The vacuum potential is used to align the Fermi level of the system relative to vacuum as a reference point for simulations on charged, electrochemically polarized slab systems.
The electrochemical activation of water corresponds to either the cathodic reaction (upon application of a negative potential) or the anodic reaction (for more positive potentials) as follows:
The superscript * denotes that the associated molecule or fragment is adsorbed to the metal surface.
When cathodic potentials are applied, the DFT calculations produce stable geometries for water molecules with the dipole of the water molecule rotating so that the hydrogen atoms become oriented toward the metal electrode (Taylor et al. 2007b). As the potential becomes more cathodic, bonds form between the hydrogen atoms and the metal, breaking the O-H bond in water and producing adsorbed hydrogen and OH−: this is an important step in both the cathodic evolution of hydrogen (Conway and Bockris 1957), which can be a counter electrochemical reaction to anodic dissolution in aqueous corrosion and also in the activation of hydrogen before absorption and embrittlement of the material (DeLuccia 1988). Conversely, when anodic potentials are applied, the DFT model correctly reproduces the opposite behavior: water breaks up to produce OH adsorbed to the surface and liberating H+ which forms H3O+ and H5O2+ moieties in the interslab space, a process that has been observed to occur on metallic surfaces upon polarization in an aqueous environment via in situ surface probes (Maurice and Marcus 2018; Maurice et al. 2000). Figure 3 shows how the DFT energy varies with the electrochemical potential applied by the computational potentiostat model (Figure 2). As the potential varies from cathodic to anodic, the stable phases of the surface, in order, the transition from H adsorbed, to water adsorbed, and hydroxide and oxide, because of the cathodic and then anodic electrochemical activation of water (equations 2a, 2b) and then the subsequent electrochemical activation of OH.

Water activation on Cu(111) using the computational virtual potentiostat method (Taylor et al. 2007b). Reproduced with permission from Taylor and Francis (© 2007). The plot shows the free energy for the Cu(111) system with adsorbed water (filled circles), hydroxide (open circles), oxide (open squares), or hydrogen (open triangles) as obtained from direct DFT calculations of solvated metal slabs.
Of relevance to corrosion was modeling the impact of changing the electrochemical potential of the system not just on the reactivity of water but also on the changes that resulted in the metal slab itself. Using the virtual potentiostat model, a Cu(111) surface was polarized incrementally to progressively more positive surface charge densities, and geometry optimizations performed at each step (Taylor 2012a). After activation of water to OH occurred, a second activation step to adsorbed O resulted from the geometry optimization.
Following this step, a Cu surface atom in the (111) plane was lifted out of the surface and displaced by the adsorbed O, effectively forming a stoichiometric layer of Cu2O on the surface. The results indicated that DFT calculations could provide a powerful method for understanding the electrochemical destabilization of a/the metal surface and could be used in the future to investigate a variety of scenarios relevant to corrosion science and engineering (Figure 4).

Cu atom lift-out from a Cu(111) slab with oxygen adsorbed occurs in the DFT simulated system when the electrochemical potentiostat anodic potential is increased to potentials beyond water and hydroxide activation (Taylor 2012a). Reproduced with permission under Creative Commons license, Christopher D. Taylor (© 2012).
4 Modeling adsorption on metal surfaces
The ability of DFT calculations to provide information about reactions and adsorption of molecules and atoms from the environment on to metal surfaces has resulted in their application to the study and comparison of modes of adsorption for chemical corrosion inhibitors (Borck et al. 2007; Cho et al. 1995; Costa and Marcus 2015; Durnie et al. 2005; Jiang and Adams 2003; Jiang et al. 2004; Kokalj et al. 2011; Kovačević and Kokalj 2012, 2013; Kovacevic et al. 2017; Lebrini et al. 2008; Motta et al. 2012; Peljhan and Kokalj 2011a; Sun et al. 2012), to oxygen adsorption as a function of electrochemical potential to form pre-passive monolayer films (Francis and Taylor 2013; German and Efremenko 2004; Hyman and Medlin 2005; Karlberg et al. 2007; Ke and Taylor 2020b; Ke et al. 2020b; Samin and Taylor 2017b, 2018b, 2018c; Taylor 2013; Taylor et al. 2018b; Yeh et al. 2009), and to the influence of interactions between co-adsorbates or alloying elements on processes such as hydrogen uptake (Greeley and Nørskov 2007b; Limmer et al. 2017; Taylor et al. 2011).
Taylor, Kurapati, and Mondal developed a model for molecular adsorption of corrosion inhibitors on to defect sites on passivated metallic surfaces by using a cluster model that calculated the binding energies of the inhibitor molecule to an M(OH)n molecular fragment, building on earlier work by Mondal and Taylor (Mondal and Taylor 2014; Taylor et al. 2018a). The binding energy is computed by the difference inbound (M + A, signifying Metal plus Adsorbate, i.e., the chemical inhibitor molecule) and un-bound (M, A) systems:
Following this equation, more negative binding energies imply stronger chemical bonding because the system M + A has lower potential energy than the isolated parts. Inhibitors can be compared based on their relative binding energy: a more negative binding energy can imply a more effective chemical inhibitor, although other factors such as packing on the surface, availability in mixed-media environments, interactions with corrosion products, etc. may also play a role (Hackerman 1952, 1962; Taylor et al. 2015b). The field of application of quantum chemical methods (including DFT) to the problem of effective corrosion inhibitor selection and design, is proliferous and could entail a review entirely of its own. One approach, developed in the late 60s, involves the regression of computable parameters for molecules based on their electronic structure, against experimentally determined activities-the so-called quantitative structure-activity relationships (Durnie et al. 1999, 2005; Gece 2008; Vosta and Eliasek 1971). This is in effect a semi-empirical way to indirectly link the properties of inhibitor molecules to performance, but does not involve a first-principles mechanistic treatment, and so will not be further described herein.
Oxygen adsorption on metal surfaces is the precursor to the formation of passive films (Francis and Holby 2015; Seyeux et al. 2005). Oxygen binding energies are typically strongly negative values on the order of a few eV, indicating strong binding to metallic surfaces: hence, the propensity for metals to oxidize in the natural environment. Oxygen adsorption is the final step in electrochemical water activation (Taylor et al. 2007b, 2007c), but can also occur directly because of dissociative adsorption of O2 on metal surfaces, a process which is exothermic and therefore spontaneous (Calvo 2014; German and Efremenko 2004; Janik et al. 2009; López-Moreno and Romero 2015; Norskov et al. 2004; Panchenko et al. 2004; Wang et al. 2008). The study of oxygen adsorption on metal surfaces using DFT has seen considerable interest because of the push to develop more efficient oxygen reduction catalysts for fuel cell technologies in the 2000s (German and Efremenko 2004; Hansen et al. 2008; Hyman and Medlin 2005; Janik et al. 2009; Karlberg et al. 2007; Mills et al. 2003; Norskov et al. 2004; Panchenko et al. 2004; Wang et al. 2011, 2012; Yeh et al. 2009). The nature of oxygen binding to metals that are challenging to study experimentally, for example, radioactive elements such as Tc and Pu, has also been studied by Taylor (2013, 2014 to understand more about the corrosion properties of these materials. As oxygen accumulates on close-packed metallic surfaces the adsorption energy changes because of lateral interactions between the adsorbates (Cheng et al. 2014; Tian et al. 2009). When atomic oxygen adsorbs on closest-packed metal surfaces, it typically resides in hollow-sites, bonded to three or four metal atoms, depending on the configuration (Francis and Holby 2015). The adsorption is accompanied by charge transfer. The surface metal atoms that are ‘bonded to’ or first-nearest neighbors to the oxygen atom each develop a partial charge, indicating that the metal surface, even at sub-monolayer coverage of oxygen, already has an oxidized character (Francis and Taylor 2013). As the coverage of oxygen increases, reconstructions can occur in the top layer of metal atoms because of this polarization of the metal atoms, and the repulsions that accumulate between the oxygen atoms.
A study was made of the accumulation of oxygen on the Ni(111) surface to produce monolayer, bilayer, and trilayers of NiO(111) for the purpose of characterizing the changes in valence, coordination, and reactivity that accompany the initial stages of growth of the oxide film (Olatunji-Ojo and Taylor 2013). Table 2 summarizes some of the findings of the paper, showing that, when an appropriately higher level of theory (PBE + U) is used (Dudarev et al. 1998a, 1998b), the so-called “octopolar” reconstruction of the Ni(111) surface to form the first partial monolayer of NiO(111) results from the model, which is a finding that is in agreement with the experimental observations (Barbier et al. 1999). As subsequent layers of the oxide phase form, the energy of oxygen adsorption approaches the energy of formation for bulk NiO (Table 2). The same convergence in energy occurs with the lower level of theory (PBE), but in that case, there is a reduced driving force to form the oxide because PBE underestimates the energy of formation for the oxide phase. Further such studies of these processes will help to elucidate the mechanisms that lead to passive film formation and growth on these metal systems, including identification of the relevant charge transfer processes (Yu et al. 2012), how the reaction energies change with the time evolution of the film structure and thickness, and identification of the role of alloy elements (Castle and Asami 2004; Han et al. 2015; Ke and Taylor 2020b; Ke et al. 2020b; Liu et al. 2015b; Marcus 1994; Seyeux et al. 2013; Yu et al. 2018), solution chemistry (Anderko et al. 2008; Bouzoubaa et al. 2009a, 2009b, 2010; Ke et al. 2020b; Liu et al. 2015a; Samin and Taylor 2018b; Taylor 2012b; Yu et al. 2019), the adhesion at metal/oxide interfaces (Jiang et al. 2008; Wang et al. 2006), and defect states at the metal/oxide and oxide/environment interface that are most relevant for limited and controlled oxide growth as a function of environment and materials composition (Krishnamurthy et al. 2002; Lin et al. 1981; Todorova and Neugebauer 2014, 2015; Samin and Taylor 2018a, 2018d; Seyeux et al. 2013).
Energy of oxide formation on Ni(111) computed per O atom, using DFT at two levels of theory, and charges on oxygen atoms reported for each case (Olatunji-Ojo and Taylor 2013).
| System | E (PBE), Ev | E (PBE/PBE + U) | QO (PBE) | QO (PBE + U) |
|---|---|---|---|---|
| NiO | E f = −1.18 | E f = −2.98 | ||
| Ni(111) ¼ ML OFCC | −2.16 | −2.26 | −0.95 | −1.01 |
| Ni(111) ¼ ML OOCT | +0.32 | −1.26 | −0.77 | −1.21 |
| Ni(111) ¾ ML O | −1.02 | −1.95 (octopolar reconstruction ∼ NiO(111)) | −0.85, −0.84, −0.43 | −1.17 |
| Ni(311) Step | −2.03 (1 O/cell) −1.91 (2 O/cell) −1.51 (4 O/cell) |
−0.98 | ||
| 1 ML NiO(111)/Ni(111) | −1.39 | −2.48 | −1.1 | −1.2 |
| 1.5 ML NiO(111)/NI(111): O-termination | −1.33 | −1.90 | −1.1 (lower), −0.8 (upper) | −1.1 (lower),−0.8 (upper) |
| 2 ML NiO(111)/Ni(222): Ni-termination | −1.10 | −2.98 | −1.1 | −1.2 |
The ability to study the interactions of adsorbates with one another on metal surfaces directly using DFT methods was used to investigate a long-standing question regarding the ability of certain chemical species such as As and S to influence hydrogen uptake kinetics on structural materials (McCright 1977; Protopopoff and Marcus 1987, 1988; Taylor et al. 2011). A Ni(111) surface was studied by Taylor et al. and hydrogen adsorption and absorption energies calculated using DFT (Taylor et al. 2011). Sulfur, a known hydrogen recombination poison, was then adsorbed on the simulated surface and its influence on hydrogen adsorption and absorption energies of nearby sites was determined. Using the energies extracted from the DFT calculations, the hydrogen recombination versus uptake (i.e., absorption into the metal) kinetics were estimated. It was shown that up to certain quantities of sulfur adsorbed on the surface, hydrogen absorption kinetics could be enhanced while recombination was quenched (i.e., the poisoning effect). The kinetics were estimated by raising or lowering the activation energy according to the shift in chemical potential of the adsorbed hydrogen because of its proximity to the sulfur atom. Raising the surface chemical potential by an amount δE was anticipated to lower the activation energy by an equal amount. Beyond a certain sulfur coverage, however, the ability for hydrogen to either adsorb or absorb was entirely quenched as full coverage of sulfur effectively sealed off the surface to further hydrogen adsorption (Figure 5).

DFT calculations of sulfur and hydrogen co-adsorbed show two zones of influence of S on nearby H adsorption energies. Zone A increases the surface chemical potential mildly, lowering the activation energy enough to promote an increase in H absorption into the metal by the uptake enhancement factor, which is dependent upon electrochemical overpotential, h. Zone B increases the surface chemical potential to a much higher extent, creating a field that repulses hydrogen too strongly to allow for either adsorption or absorption to occur. At higher S coverages, there is more zone B than zone A on the surface, thus the uptake effect is coverage dependent (Taylor et al. 2011). Reproduced with permission from IOP Publishing (© 2011).
5 Electrochemisorption and surface Pourbaix diagrams from first-principles
One of the most fundamental tools in the electrochemical corrosion scientist’s toolkit is the Pourbaix diagram (Deltombe et al. 1974). Pourbaix diagrams express in a visual way the most stable phases of an element in the two-dimensional electrochemical phase space of electrochemical potential (usually in units of V NHE) and pH. The Pourbaix diagram is calculated using Gibb’s free energy of the various possible phases and the Nernst equation to adjust the free energies for the conditions of potential and pH. For example, the free energy for the reaction:
is calculated using the Nernst equation as follows:
Where, in this case, m = 2 for the number of H+ exchanged in the reaction and n = 2 for the number of electrons exchanged. RT is the ideal gas constant multiplied by temperature. F is Faraday’s constant, and U is the potential; U0 is the equilibrium potential for the reaction. Owing to the typical linear form of equation (5), Pourbaix diagrams can be solved by linear programming methods and have the appearance of linear divisions of the U-pH two-dimensional space. The Pourbaix diagram, therefore, is a kind of map, where lines mark the transition from one stable phase of the metal/environment system to another. The various regions of the map are sometimes referred to as “corrosion”, “immunity”, and “passivity” depending on the properties of each phase.
Using chemisorption energies one can compute something akin to a Pourbaix diagram for the most stable chemical species that form on a metallic surface: this approach has been extensively developed since the 1980s by Protopopoff and Marcus (Marcus and Protopopoff 1993, 1997; Protopopoff and Marcus 2003, 2005, 2012). For example, water may exist in the undissociated molecular form of H2O on a metallic surface under some conditions of potential and pH, but as the potential becomes anodic or cathodic it may transform according to the reaction sequences shown above (2a–c)a–c(2a–c). The chemisorption energies can be shifted for different conditions of pH and potential using the same rules as the Nernst equation:
Adsorption reactions that include electrochemical transfer can be referred to as electrochemisorption v(Conway et al. 1974).
The electrochemisorption windows of potential for water on a variety of metals were calculated by Taylor, Neurock, and Kelly, as shown in Figure 6 (Taylor et al. 2007c). More noble elements typically have a broad range of electrochemical stability for water, and, in particular Au. Pd and Pt, good hydrogen reduction catalysts, have underpotential regions of stability for hydrogen adsorption from water on the surface. Ni and Mo are electrochemically active toward water under all potentials studied. Mo, Ni, and Cu are also ‘earlier’ oxide formers in the sense that they allow the surface in contact with water to oxidize at lower electrochemical potentials. These first-principles predictions are in broad agreement with the known activities and behaviors of these metals (Henderson 2002; Taylor and Neurock 2005).

Electrochemisorption windows for H (gray), H2O (white), and OH (black) on a series of close-packed metallic surfaces. Standard conditions were assumed (i.e., pH 0, 298 K). A full Pourbaix style treatment would also include an additional axis of pH variation to this kind of phase diagram for each of the surfaces studied, and additional phases (Taylor et al. 2007c). Reproduced with permission from IOP Publishing (© 2007).
Protopopoff and Marcus have created surface phase diagrams for several materials/environment scenarios, including sulfur, hydroxide, thiosulfate, and lead adsorption on various metals and alloys. Their technique primarily used adsorption energies taken from surface science experiments (Marcus and Protopopoff 1993, 1997; Protopopoff and Marcus 1987, 1988, 2003, 2005, 2012). Surface Pourbaix diagrams were created based on DFT calculated adsorption energies for nickel in high purity water under both ambient and relevant temperatures, relevant to the nuclear industry (Taylor et al. 2006a, 2007a). The method has continued to be adopted by others, including Williams et al. (2015) and Yuwono et al. (2019) for Mg, and Kim et al. (2014) for Tc. An example of the Pourbaix diagram for Mg(0001) computed by Yuwono et al. is shown in Figure 7.

A Pourbaix diagram constructed from DFT for the Mg(0001) surface by Yuwono et al. (2019). Reproduced with permission from Elsevier (© 2019). The various θ values correspond to surface coverages of the adsorbates (Mg*O, or Mg*OH, or Mg*H) used for the various colored curves in the plot.
6 Competitive electrochemisorption on metals
Going beyond the adsorption of water and its dissociation products, competitive chemisorption of other species can occur when ions such as chloride compete with water, oxygen, etc. for adsorption sites on the metallic surface (Conway et al. 1974). Chloride ions can adsorb electrochemically to the surface to form a chemisorbed chloride:
This process can compete with the adsorption of oxygen, hydrogen, hydroxide, or molecular H2O. Analysis of the electron density maps produced using DFT shows that the Cl maintains its negative charge on adsorption through polarization of the metal atoms it contacts on adsorption (so the complex Cl–M(surf) could also be written as Cl−–M+ (surf) or even Cl− (M3)+, although Cl adsorption may not always be at threefold hollow sites depending on the surface topology and/or presence of defects) (Andryushechkin et al. 2015; Ke and Taylor 2020b). The change in free energy for each of these reactions can be computed with DFT, and then by using suitable thermodynamic models based on Born-Haber cycles and the knowledge of the reference equilibrium potentials tabulated for hydrogen evolution, chloride reduction potential, etc.(Taylor 2012b) This approach translates the zero Kelvin chemisorption energy (ΔEads, using ads more generally for adsorption-chemical (chemisorption) or physical (physisorption)) into ΔG at realistic temperatures for the reactions relevant to problems in alloy corrosion, crack growth, etc.
The Born–Haber cycle that could be used to compute the free energy for chloride adsorption onto a metallic surface starting with Cl− in the aqueous phase (corresponding to Eq. (9)), ∆Gtarget, is shown in Figure 8. Similar cycles can be derived for hydrogen adsorption, hydrogen sulfide adsorption, water adsorption, etc.

Example Born–Haber cycle for chloride adsorption onto a metallic surface from Cl− in the aqueous phase. ∆Eabinitio is the adsorption energy from the quantum chemical calculation, and the various steps in the cycle refer to the thermodynamic parameters required to get the overall free energy of interest ∆Gtarget (Taylor 2012b). Reproduced with permission from NACE International (© 2012).
Using this data, Taylor computed phase diagrams for the competitive chemisorption of O, OH, H, H2O, N, NH, NH2, NH3, and Cl on Fe(110) surface under conditions relevant to ammonium chloride corrosion, a problem of concern in the refining industry (Akpanyung et al. 2019; Alvisi and de Freitas Cunha Lins 2008; Taylor 2012b). In this failure mode, ammonia and hydrochloric acid condense onto the inner walls of refinery equipment to form the NH4Cl salt, which, being hygroscopic, absorbs water thereby resulting in highly concentrated solutions of Cl− at the saturation concentration around and underneath the salt deposit. Using the DFT adsorption energies, the constructed phase diagram shows that chloride can preferentially chemisorb onto the Fe(110) surface over a certain range of environmental conditions, thereby inhibiting the process of passivation on the metallic surface. Inhibiting the passive film formation can be expected to have implications for the ability of metals in the early stages of localized corrosion to repassivate and hence the transition from metastable to stable pit growth. From experimental investigations, Pletnev et al. (2000), found that chloride accelerates the corrosion of mild steel during active dissolution. Accordingly, Pletnev attributed this to the formation of a surface complex involved adsorbed chloride. Alvarez studied the influence of chloride on pitting potentials and speculated that the specific adsorption of chloride hindered pit repassivation and increased dissolution rate (Alvarez and Galvele 1984). These experimental investigations and interpretations are consistent with the results from these phase diagram studies based on the DFT computations.
7 Quantifying surface coverage: using DFT to generate adsorption isotherms
Whereas the surface Pourbaix and phase diagram approaches allow prediction of the transitions from one dominant surface phase to another, the coverage of a surface can also be mixed with multiple adsorbates having negative free energies of adsorption under any particular condition (Saraby-Reintjes 1985). In such cases, it is of interest to know quantitatively how much of each species is present and in what ratio. The simplest model to predict the surface coverage of a species in the environment onto the surface of a solid is the Langmuir isotherm (Gileadi 1987; Masel 1996). This model assumes that there is no significant interaction between adsorbed atoms or molecules on the solid surface, other than site-blocking (i.e., one molecular species is allowed per site, which can simulate repulsive interactions based upon how one selects the effective size of a ‘site’). For a reaction of the type:
where S represents the solid surface and A the adsorbate, then the equilibrium between surface and environment is given by the Langmuir equation:
here Keq is the equilibrium constant, ΔGad is the free energy of adsorption, R is the ideal gas constant, and T is the absolute temperature. The activities are given by the terms,
The noninteraction assumption inherent in the Langmuir approach may be considered reasonable if one considers that, at the limit where interactions start to be important, the coverage is ‘capped’ by the (usually) repulsive interactions: that is, the fractional surface site coverage term takes this effect implicitly into account through an effective site-blocking, where the ‘size’ of a site is determined by the limit of repulsive interactions between adsorbates (Conway et al. 1974). Although the Langmuir approach may not get the exact shape of the adsorption isotherm, it may be considered approximate enough to start making inferences about corrosion and hydrogen susceptibility.
To quantitatively evaluate this assumption for the case of Cl and O co-adsorption, Samin and Taylor performed some more accurate isotherm calculations using the numerical method of Grand Canonical Monte Carlo (GCMC) (Samin and Taylor 2018b). GCMC is a technique that allows the number of species in an atomistic/molecular simulation, usually on some kind of lattice that represents a crystalline solid or a surface, to vary to meet a given chemical potential (Blum et al. 2005; Byrd et al. 2000; Ceder 1993; Sanchez et al. 1984). Using this technique one can construct an adsorption isotherm without requiring an analytical expression, such as the Langmuir isotherm (Samin and Taylor 2017a; Schmidt et al. 2012). Instead, the surface coverage and arrangement of co-adsorbed species are optimized such that the chemical potential of the surface system obtained by the use of a model Hamiltonian is equal to that of the environment (Blum et al. 2005; Hart et al. 2005; Hegde and Bowen 2017). In the case of chemisorption, this can be achieved by using DFT to develop a database of interaction energies between adsorbed species (known as figures of merit). These interactions can be one-body (i.e., adsorption energy in the limit of no-interaction), two-body (pair-wise interactions), three-body, etc. (see Figure 10). The more interactions that are taken into account in the cluster expansion, the more accurate the isotherm will be.
Samin and Taylor (2018b) demonstrated this technique for Cl and O chemisorption on a Ni(111) surface. Once parameterized, the GCMC algorithm samples from a large ensemble of such configurations that are used to generate the adsorption isotherm as a function of the chemical potential. The many-body interactions produce plateaus in the total surface coverage versus chemical potential plot, indicating that significant chemical potential pressure must be exerted to go beyond a certain nominal coverage (i.e., repulsive interactions between the atoms on the surface effectively prevent them from becoming close neighbors until a high chemical potential is reached through extremely high partial pressures or high concentration solutions): example isotherms are shown in Figure 11. Ultimately the results from Samin and Taylor (2018b) indicated that when chemical potentials remain modest (i.e., typical solution chemistry and ambient temperatures) then the Langmuir approximation can be applied. In such cases, i.e., the left-hand side of Figure 11, the effective surface coverage of ∼0.3 ML (calculated in terms of the ratio of surface adsorbates to the total number of fcc binding sites) is equivalent to 1 ML in the Langmuir definition (100% occupancy of the total accessible surface sites).
8 DFT-informed Langmuir isotherms for Co-adsorption from aqueous solution
When there are multiple species that can interact with the surface, then there may exist a competition among those species for adsorption onto surface sites. In this case, the general form of equation (4) still applies but the total fractional surface coverage must be constrained by an equation of the form:
where j = M (empty site), Cl, O, OH, H2O, etc.
When the adsorbates can vary considerably in size a scale factor may need to be taken into account since one adsorbate may occur >1 equivalent surface sites of one of the smaller species (Conway et al. 1974). Applying this constraint, and a series of equations of form Eq. (11) for each possible adsorption reaction, one can then simultaneously solve for the coverage of each adsorbate with a matrix equation. Alternatively, a Flory–Huggins isotherm may be used (Conway et al. 1974).
Taylor et al. (2018b) applied this Langmuir technique to consider scenarios for a series of commonly used structural base metals. The chemical potentials were determined from the pH and chloride concentration, and the electrochemical potential, and the adsorption equations were solved simultaneously to determine the coverage of each coadsorbate based on the free energy expression. The coverage of Cl, O, and H2O on Ni(111) at various pH, potential coordinates is shown in Figure 12. The coverage is plotted against the electrochemical potential spectrum. The results are of a similar nature to what was seen in Figure 9 for Fe(110): at cathodic potentials water adsorbs (and in fact cathodically dissociates to also form adsorbed hydrogen, although that reaction was not simulated in Figure 12) (Taylor 2012b). As the potential is made more anodic, first the one-electron adsorption of chloride occurs, followed by displacement of the Cl by O. This behavior becomes more pronounced at acidic pH and higher chloride concentrations. The adsorption of chloride becomes less significant or is omitted altogether at higher pH and low chloride concentrations. These observations were interpreted against the scenario that occurs at the bottom of corrosion pits: low pH environments produced by hydrolysis of cations, and high chloride concentrations because of electromigration can lead to conditions which favor chloride adsorption on the exposed metallic surface, thus preventing repassivation (Anderko et al. 2004; Frankel et al. 2017; Sharland 1987). In the next section, the extension of this modeling approach to consider how alloying affects this process will be reviewed.

Gibbs’ free energy of adsorbates on Fe(110) at pH 4.45 in a saturated solution of NH4Cl and 298 K (Taylor 2012b). Reproduced with permission from NACE International (© 2012).
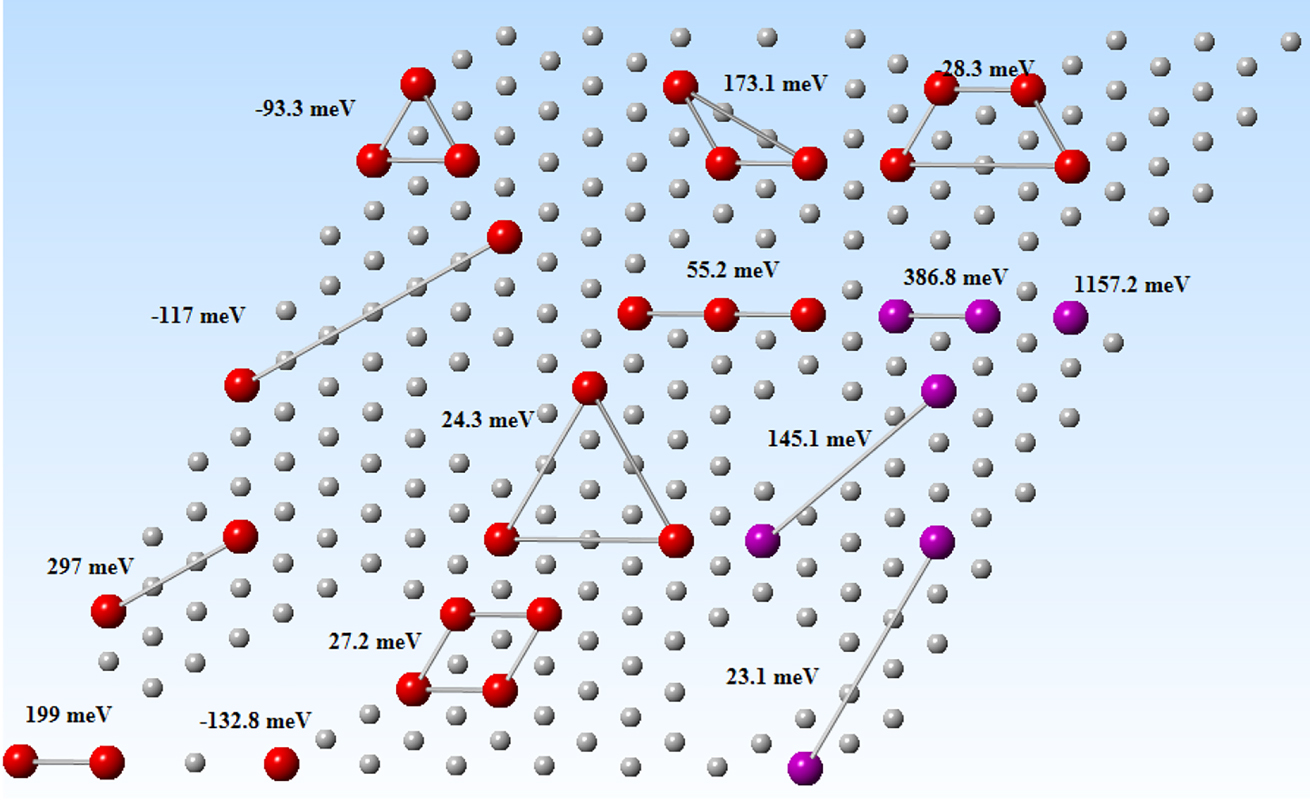
Example of the configurations used to train the cluster expansion model for chloride-oxygen adsorption isotherms on Ni(111). For alloys, the number of configurations needed to train the alloy would need to be significantly expanded (Samin and Taylor 2018b). Reproduced with permission IOP publishing (© 2018).

Isotherms for O and Cl adsorption as a function of the chemical potential of chloride. Typical chemical potentials for chloride ions in solution are from −7 to 0 eV (higher chloride concentration = higher chemical potential). Chemical potentials for oxygen range from −7 to −2 eV, depending on pH (higher pH = higher chemical potential) (Samin and Taylor 2018b). Reproduced with permission from IOP Publishing (© 2018).

Ni(111) surface coverage versus the potential for different Cl molarities (1 mM, 10 mM, 0.1 M, and 1 M, respectively, from left to right) and pH (4, 7, and 10 from top to bottom, respectively) (Taylor et al. 2018b). W stands for water, O for adsorbed oxygen, and Cl for adsorbed chloride. Reproduced with permission from Elsevier (© 2018).
Isotherm models are frequently used in the interpretation of chemical corrosion inhibitor experiments, particularly those that measure inhibitor efficiency versus the concentration of the inhibitor (Christov and Popova 2004; Mondal and Taylor 2014). At the same time, there is some debate about the best way to interpret inhibitor efficiency data and linking it to adsorption phenomena (Kokalj 2010; Kovacevic et al. 2017; Walczak et al. 2019). Taylor et al. published a model for how a first-principles approach to inhibitors could be developed that couples adsorption behavior along with other phenomena that are important such as interaction with corrosion products, partitioning between oil and water phases, speciation, and ion-pair and multilayer formation (Taylor et al. 2015a, 2015c). This remains an area of much interest and such models have yet to see a full development (Kovačević and Kokalj 2013; Kovacevic et al. 2017; Peljhan and Kokalj 2011b). Coupling DFT with other atomistic simulation approaches for inhibitor film formation has seen renewed effort in recent years (Du et al. 2014; Khaled 2003, 2006, 2008, 2009, 2010; Khaled and Sherik 2013; Khaled et al. 2005).
9 Competitive electrochemisorption on alloy surfaces
The development presented so far only treats adsorption on a system where the adsorption energy is homogeneous across the metal surface. When an alloy material is used, however, there will be inhomogeneities across the surface, both at the atomistic level (i.e., in the statistical distribution of elements of the solid-solution alloy matrix) and at the microstructural level (grain-boundaries, secondary phases, intermetallics, etc.). For solid solution alloys, one can use purely statistically distributed (i.e., probabilities of occurrence based on composition) or otherwise informed distributions of the alloying elements and weigh the adsorption isotherm according to the expected distribution of surface sites with distinct adsorption energies. For the case of microstructurally heterogeneous materials, one could also construct adsorption isotherms for each phase and study them in their statistical distribution collectively or individually to understand the unique properties of each microstructural feature and their differentiating surface chemistries that may influence corrosion. For scenarios such as work-hardened zones which have local strain/dislocations, the adsorption energies will shift according to the extent of local strain; these shifts can also be determined from DFT calculations on strained surfaces (Greeley and Mavrikakis 2005; Lei et al. 2013). Francis and Curtin applied DFT to study the impact of localized surface stresses on the adsorption of environmental molecules for their relevance to electrocatalysis (Francis and Curtin 2015). An analogous effort could be applied to infer the impact of local stresses and strains on corrosion-relevant processes at the electrochemical interface.
As an example, consider the case of chloride adsorption on an alloy with unique sites expressed by the term Mi. If there are N unique sites each with their own corresponding adsorption energy and local environment, the possible reactions that could occur on the surfaces can be generally represented as:
For each unique reaction there is a corresponding equilibrium equation:
where there is a coverage term
For most alloy materials, the number of unique surface adsorption sites that would need to be studied with DFT can get extremely large, making it computationally costly to run all combinations. To expedite the determination of adsorption energy at a site i the authors leveraged a machine learning approach by taking the results of ∼150 DFT calculations and then used logistic regression to estimate the adsorption energy from a sum of two-body terms between the metal atoms making up the adsorption site i (Ke and Taylor 2020b; Ke et al. 2020a, 2020b). This approach can be varied based on the geometry of the surface plane and the atomic make-up of the adsorption site. This approach was applied to O and Cl adsorption on close-packed alloy surfaces based on a stainless steel system (Ke et al. 2020a) and a Ni–22Cr system (Ke and Taylor 2020b; Ke et al. 2020b).
Figure 13 shows the performance of the local bonding model, which assumes that the adsorption energy can be obtained only as a sum of nearest-neighbor interactions. The scatter in the parity plot indicates that second-nearest neighbors and the overall matrix composition play a role in influencing the binding energy of both O and Cl to metal alloy surfaces. The order of significance of the elements can be assessed from Figures 13(c) and (d). For oxygen binding, the order from strongest oxygen binding to weakest is: Cr > Mo > W ∼ Mn > Fe > Co > Ru ∼ Ni > Cu. For chloride binding, the order is similar but Mn ∼ Fe > W. Thus, only W looks like it clearly differentiates between Cl and O in terms of binding affinity. Owing to the scatter in the fits, however, it was decided that more detailed analysis was necessary.

Parity plots for the regression against (a) oxygen and (b) chlorine adsorption energies for the series of Ni–Cr alloys and stainless steel alloys. Contributions of alloy elements to (c) oxygen and (d) chloride to adsorption energies (eV) relative to the mean introduced into the Ni–22Cr alloy system.
The first focus of this approach was on studying the role of alloying in influencing the localized corrosion susceptibility of corrosion-resistant alloys (Ke et al. 2020b). The term “corrosion-resistant alloys” refers to the resistance of the alloys to uniform (or general) corrosion because of the presence of robust passive films across the bulk of the material surface. However, microstructural heterogeneities or discrete probabilistic events can create the risk of localized corrosion even in “corrosion-resistant alloys”: hence, many corrosion scientists have favored taking a stochastic approach to modeling localized corrosion (Organ et al. 2005; Stewart and Williams 1992; Riley et al. 1991; Williams et al. 1991). Factors known to enhance the corrosion resistance of an alloy are the adoption of alloying elements such as chromium and molybdenum, and/or the use of nickel-based rather than iron-based alloys (Marcus 1994; Sedriks 1996; Taylor et al. 2018c).
Following the general concept introduced in Figure 12, Ke and Taylor (2020b) sought to quantify the competition between chloride and oxygen for surface adsorption sites as a means to assess a material’s susceptibility versus resistance to localized corrosion within a givenvironment. The adsorption energies of Cl and O were computed for a series of model Ni–22Cr alloy slabs generated using the special quasirandom structure model (Zunger et al. 1990), which is a method for approximating in a periodic model the atomic correlations that exist in truly random solid solutions. The series consisted of the special quasirandom Ni–22Cr slab, generated earlier by Samin and Taylor (Samin and Taylor 2018c), with various elements replacing either Ni or Cr atoms on the surface layer of the slab, at which site the O or Cl atoms were chemisorbed. The chemisorption interaction of Cl and O with the surface is a mixture of covalent and ionic chemical bonds. The possible alloying elements considered were taken from the set: Cr, Mn, Fe, Co, Ni, Cu, Mo, Ru, W.
Because O and Cl both adsorb into three-fold hollow sites on the metallic surface, there are numerous possible combinations of those three binding metal atoms that could coordinate to the O or Cl atom. i.e., for the nickel-chrome binary system, chemically distinct adsorption sites can include Ni–Ni–Ni–O, Ni–Ni–Cr–O, Ni–Cr–Cr–O, etc. It is also likely that second nearest-neighbor effects may play a role in the adsorption environment. The plot of total chlorine versus oxygen adsorption energies for the alloys considered is shown in Figure 14. On the left are all the data points, and on the right are the data points plotted for the case in which the adsorption site has all three of the adsorption sites in common (e.g., Fe–Fe–Fe or Cu–Cu–Cu).
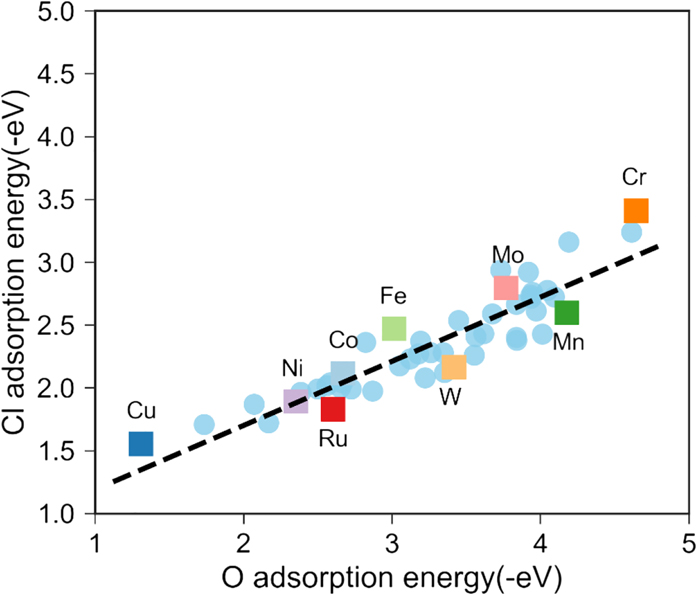
Adsorption energies for Cl versus O on a series of Ni–22Cr alloys with dopant elements added into the surface adsorption sites (Ke and Taylor 2020b; Ke et al. 2020b). The solid squares represent those surface sites which consist of all three elements bonded to Cl or O having the identity as labeled. The remaining data points (blue circles) are composed of mixtures of those elements. Reproduced with permission from IOP Publishing (© 2020).
As seen in Figure 14, generally the chloride and oxide adsorption energies scale with one another. Therefore, from this basis alone it is not clear to determine scenarios in which certain alloying elements or their concentration in the alloy may contribute to corrosion resistance. Instead, the competitive adsorption isotherm model was extended to the alloy surfaces to create surface coverage plots. The coverage plots for a Ni–Cr binary system with water, chloride, and oxygen adsorption as a function of the electrochemical potential (the critical controlling parameter in aqueous corrosion) are shown in Figure 15. The data are shown for an assumed condition of pH 4 (mildly acidic) and high chloride concentration (1 M). Apparent in these plots is the presence of three electrochemically controlled phases on the Ni–Cr binary surface, along with regions of co-adsorption. On the cathodic side, water adsorption is the dominant surface phase (potentially hydrogen adsorption from proton or water reduction as well, although that was not included in this analysis, see the next section). As the potential becomes more anodic, a chloride adsorption phase displaces the water, which we can associate with more active corrosion. At more anodic potentials the oxygen adsorption begins to dominate, as this is a two-electron adsorption process, and will ultimately lead to passivation as multilayer adsorption and formation of the oxide occurs. The multilayer formation of oxide is not able to be addressed within this competitive adsorption framework. Further investigations with multilayer isotherms developed using DFT studies of oxide/metal interfaces like that of Choudhury et al. (2014) would be required in the future. Modeling solute capture in the formation of passive oxide films is also a new frontier that is starting to see interest from surface science measurements and theoretical studies (Sherman et al. 2019; Yu et al. 2018).

Ni–Cr binary alloys of up to 25% Cr composition with the coverage of unique surface phases plotted as a function of the electrochemical potential, which is the critical factor controlling aqueous corrosion. Because of the thermodynamic equilibria, no OH adsorbed phases are present in appreciable quantities for the range of potentials and conditions plotted. The conditions used to solve the Langmuir isotherm model are 300 K, pH 4, and 1 M Cl− concentration (Ke et al. 2020b). Reproduced with permission from IOP Publishing (© 2020).
As seen in Figure 15, the effect of mixing in Cr adds additional structure to the electrochemical phase diagrams. From one perspective, the scale of electrochemical potential can be considered as providing an energy spectrum for the corrosion activity of the alloy surface. Chromium decreases the ability of chloride to totally cover the surface during the electrochemical potentials associated with active corrosion, thus decreasing the corrosion currents possible during localized corrosion. For example, at 25% Cr, there is only a very small range of potentials where Cl is the dominant surface phase, and even in this case the total coverage is relatively small.
For the purposes of understanding the role of additional elements in changing the corrosion susceptibility of an alloy to corrosion, Ke et al. constructed an index that aggregates the information in Figure 15 by integrating the total chloride adsorption across the electrochemical energy spectrum. This quantity was called the chloride susceptibility index (CSI) (Ke et al. 2020b):
Formally, since the surface coverage is a unitless area fraction quantity, the CSI will have units of V.
In Figure 16, Ke et al. (2020b) illustrated the use of the CSI to infer the localized corrosion susceptibility of Ni–Cr–X ternary alloys with substitution of Ni by different elements X. The model shows that Fe and Cu make the alloy more susceptible to chloride interference with passivity (and hence, decreasing the tendency for repassivation), whereas Co, Mn, Ru, Mo, and W, in increasing order of significance, all have positive effects. Of course, there are many other influences these variables can play in affecting corrosion mechanisms; such as the ability to grow sufficiently protective oxide films, their resistance to breakdown, and metallurgical effects that might influence surface segregation, microstructure, and diffusion (Clayton and Olefjord 2011; Grabke et al. 2004; Greeley and Nørskov 2007a; Han et al. 2015; Kirchheim et al. 1989; Liu et al. 2015b; Ma et al. 2017; McCafferty 2000; Punckt et al. 2004; Scully 2009; Shibata 2007; Tang et al. 2014; Qiu et al. 2017).
![Figure 16:
Chloride susceptibility index plotted for a Ni90−xCr10Ax for various potential alloy elements A = [Cu, Fe, Co, Mn, Ru, Mo, W] that could be selected to change the alloy’s net resistance to localized corrosion by integrating the surface coverage of chloride over the relevant electrochemical energy spectrum (Ke et al. 2020b). Reproduced with permission from IOP Publishing (© 2020).](/document/doi/10.1515/corrrev-2020-0094/asset/graphic/j_corrrev-2020-0094_fig_059.jpg)
Chloride susceptibility index plotted for a Ni90−xCr10Ax for various potential alloy elements A = [Cu, Fe, Co, Mn, Ru, Mo, W] that could be selected to change the alloy’s net resistance to localized corrosion by integrating the surface coverage of chloride over the relevant electrochemical energy spectrum (Ke et al. 2020b). Reproduced with permission from IOP Publishing (© 2020).
The CSI was applied to the Ni–Cr and stainless-steel systems and correlated against the repassivation potential obtained from electrochemical measurements (Figure 17) (Ke and Taylor 2020b; Ke et al. 2020a, 2020b). The correlation bears out the general theory proposed by Anderko and Sridhar in which a thermodynamic-kinetic model for repassivation versus salt film formation was proposed as a criterion for localized corrosion pit growth (Anderko et al. 2004, 2006, 2008, 2014). Their model contained adsorption constants and mass-transport data that was fitted to experimental repassivation potential data and polarization curves. The model used in Figure 17, on the other hand, uses only first-principles information but is seen to lead to a similar interpretation and validity relative to the experimental findings.

CSI plotted against the experimentally determined repassivation potentials, Erp: (a) Ni–Cr–Mo alloys (Ke et al. 2020b); and (b) stainless steels (Ke et al. 2020a). Reproduced with permission from IOP Publishing (© 2020).

First-principles Evans diagrams from Luo et al. (Left) Schematic showing the principle of superposition of anodic and cathodic branches, and (right) First-principles polarization curves of pure Mg, Mg–0.3 wt%Ge (labeled as Mg–0.3Ge), and Mg–0.1 wt%Cu (labeled as Mg–0.1Cu) in saturated Mg(OH)2 solution (Luo et al. 2019). Reproduced with permission from IOP Publishing (© 2018).
The chloride susceptibility index can potentially be generalized as a corrosion susceptibility index. On-going work is assessing other potential adsorption phenomena of interest, and the atomistic intermediate states involved in the dissolution of metal atoms as cations to come up with additional features for indexing the susceptibility of a surface to corrosion processes (Ke and Taylor 2020a).
10 Simulation of anodic dissolution and cathodic reduction kinetics from DFT
Anodic dissolution is one of the quintessential processes that come to mind when thinking of aqueous corrosion: the dissipation of metal atoms as ions into solution, along with the formation of oxide scales and rust (Waldman 2015). Discovering opportunities to manipulate materials’ features and composition to mitigate the rate of anodic dissolution using a first-principles approach could have impacts on the integrated computational materials engineering (ICME) of a new generation of corrosion-resistant materials (Taylor et al. 2018c). Following the idea of Marcus, alloying elements can be plotted in a two-dimensional space where the axes are indicative of the metal-metal bond strength (Marcus used cohesive energy) and the metal-oxygen bond strength (Marcus used adsorption energy of oxygen to the surface) (Marcus 1994). The concept is that metals with high metal-metal bond strength will be dissolution moderators, as they will have higher resistance to the reaction which leads to dissolution and/or migration of the metal atoms from the metallic phase into a corrosion product oxide film. At the same time, metals with a high affinity for oxygen will promote the formation of strong robust oxide films. Windl and co-workers recently used DFT data to expand upon this idea by studying the effect of dissimilar elements on metal-metal bond energies (Oberdorfer and Windl 2019). Their results showed that elements with high cohesive energy can, effectively, propagate that high cohesive energy to dissimilar elements when they are alloyed together. In soon-to-be-published work, they have made similar connections for the impact of dissimilar elements on the metal-oxide bonding energy (Chen and Windl 2020). This approach will allow expansion of the Marcus approach, which considers only elements in isolation, to alloy environments.
Simulating the dissolution process using DFT is a challenging undertaking. Greeley et al. evaluated adatom adsorption energies for a series of alloys as a means for estimating dissolution and deposition potentials (Greeley and Nørskov 2007a; Greeley 2010). Taylor examined the metal-metal bond-breaking process that occurs during dissolution by simulating the transition of a metal adatom from a Cu surface into its final state as a solvated ion (Taylor 2009). This work, which is currently being extended to Ni and Ni–Cr alloy systems by Ke and Taylor (2020a), should allow the first-principles estimation of the activation energy for dissolution (i.e., something akin to a Dissolution Resistance Index), and hence allow theorists to go beyond the thermodynamic predictions of the preceding efforts to first-principles models for electrochemical kinetics.
A semi-empirical approach for electrochemical kinetics, which borrows from first-principles calculations but also draws heavily from empirical parameters to scale the model predictions to experiment, has been applied in recent years to the efforts to understand corrosion kinetics on magnesium and magnesium alloys (Ma et al. 2017, 2019, 2020; Liu et al. 2011; Lü et al. 2011). Ma et al. (2017) propose that anodic dissolution kinetics can be derived using the following set of equations:
These equations provide a first-principles basis for deriving the anodic dissolution exchange current density, given a knowledge of the work function of the material, and the energy to create defect states; namely the free surface, vacancies at the surface, and adatoms. In a way, this equation is an analytical, derivable form of the numerical off-lattice kinetic Monte Carlo approach developed by Taylor using the embedded atom method to directly calculate the activation energy of metal atoms at different defect states on a metallic surface (Taylor and Liu 2013), which was in turn inspired from the on-lattice version assumed by Erlebacher and Newman (Artymowicz et al. 2009; Erlebacher 2004). The work function and surface energy may be directly determined from DFT calculations in Ma et al.’s approach (Ma et al. 2017, 2019, 2020), whereas other terms remain fitting parameters because of the challenge in performing DFT calculations sophisticated enough for their accurate determination.
The cathodic reduction kinetics have also been derived based on oxygen reduction or hydrogen reduction, using first-principles data. Taylor investigated the anomalous hydrogen evolution kinetics on Mg (Atrens and Dietzel 2007; Bender et al. 2011; Frankel et al. 2013; Williams et al. 2013) using a variety of DFT calculations performed in the literature and a proposed mechanism that involved Tafel adsorption, site-blocking by OH, and elimination of MgOH+ by dissolution at anodic potentials (Taylor 2016). This last reaction was proposed to account for continued HER at positive potentials by creating new sites for availability for hydrogen adsorption and recombination by removing the OH site “poisons” through an anodic mechanism. Yuwono extended this idea by adding in the Heyrovsky mechanism and parameterizing a full kinetic model using DFT that accounts for much of the observed anomalous hydrogen evolution behavior in Mg and Mg alloy materials (Yuwono et al. 2019).
Corrosion kinetics are traditionally expressed through the mixed potential theory, in which the corrosion current density (i.e., corrosion kinetics, usually on a log scale) is plotted against the electrochemical potential (i.e., the thermodynamic driving force) (Muralidharan 2002). This approach, developed by Evans and Hoar in 1932 and subsequent works (Evans and Hoar 1932, 1934), provides a useful way to consider the contribution of relative cathodic and anodic reactions to the overall current density and corrosion performance of a metal or alloy in an environment, and the effect of various inhibition and corrosion mitigation strategies (Frankel 2016). Reconstruction of these “Evans diagrams” for real and hypothetical Mg alloys using the first-principles basis has been published in recent work (Luo et al. 2019). The approach, similar to the above uses a combination of first-principles DFT evaluated data for work functions and surface adsorption properties, alongside empirically fitted scale factors to try to forecast the effect of different alloying choices on the final electrochemical kinetics of these materials (Luo et al. 2018, 2019). Luo et al. proposed that a semi-empirical implementation of the first-principles electrochemical kinetics using the Butler–Volmer kinetics can be applied to evaluate the relative corrosion behavior of Mg–Ge and Mg–Cu alloys (Luo et al. 2018, 2019). The anodic branch was fitted from the experimental data and first-principles predictions of the anodic equilibrium potential, and the cathodic branch was a combination of fitting parameters and activation energies for HER estimated by evaluating the energy of the intermediate adsorbed hydrogen state for the different alloy combinations and relevant intermetallic phases (Luo et al. 2019).
11 Electrochemisorption effects on hydrogen assisted crack growth
When a crack develops in a material that is exposed to an electrochemical environment, processes similar to those occurring during initiation of localized corrosion can activate (Figure 19): freshly exposed surfaces can either repassivate or remain bare for metal dissolution and hydrogen uptake depending on pH, chemistry, and electrochemical potential (Figure 18). The surface adsorption of oxygen can be a precursor to passive film formation. On the other hand, water or hydrogen adsorption can create a source for surface hydrogen that may then diffuse into the material, thereby potentially accelerating crack growth because of hydrogen modifying the plasticity in the fracture process zone or weakening the atomic planes within the lattice or at grain boundaries, thus lowering the resistance to decohesion (Barrera et al. 2018; Van der Ven and Ceder 2004). Other mechanisms for hydrogen-assisted fracture might include enhanced vacancy formation and coalescence because of H-vacancy complex formation (Fukai 2002), or the formation of brittle hydride phases, depending on the material.

Schematic showing processes that can occur during stress corrosion cracking and/or corrosion fatigue; lowering electrochemical potential from passive to active region, metal dissolution, and water reduction leading to high pH and hydrogen generation which can cause embrittlement. Reproduced with permission from Ramgopal Thodla (2020).
Scully and Moran (1984) and Frankenthal and Milner (Quinlan 1986) conjectured that water reduction reaction on the exposed bare metal surface created on a surface under strain may produce a steady-state concentration of atomic hydrogen on the surface when the reduction reaction occurs via the pathway:
The driving force to create an equilibrium between Hads and Habs will create a boundary condition for the flux of hydrogen into the material, thus providing one impetus for further hydrogen embrittlement of the material via its entry to the fracture process zone.
Environmentally assisted cracking, including stress corrosion cracking and corrosion fatigue, is commonly described as a failure mode that involves the intersection of (a) a susceptible material, with (b) a corrosive environment, and (c) a mechanical driving force (Ford 1984). The flow chart in Figure 20 outlines some of the various kinds of environmentally assisted cracking and the processes and sub-processes that lead to crack initiation and growth in the susceptible material. Although this is not the place for a detailed review of environmentally assisted cracking (see a recent review of hydrogen embrittlement of steels, for example, Barrera et al. (2018)), examination of this flow chart indicates that surface processes may play decisive roles in limiting or enabling environmentally assisted cracking because of the potential significance for mechanisms such as dissolution at the crack tip, water and hydrogen/hydronium adsorption, repassivation, transport, and complexation.

Process and sub-processes that may be active and rate-controlling in corrosion fatigue and stress corrosion. Adapted from Ford (1984).
In a combined theoretical/experimental study, crack growth rate measurements were made under different testing conditions on pipeline steels as a function of the applied cathodic protection potential by Thodla et al. (Thodla et al. 2020b). The crack growth rate trend as a function of potential was compared to the DFT predictions of the potential-dependent surface chemistry. The authors studied the crack growth rate in three grades of pipeline steel that differed in strength and under both static and dynamic loadings. In all three cases, when the potential was scanned from −1050 mV SCE to −800 mV SCE, a drop of between 1–3x orders of magnitude was observed in the crack growth rate, corresponding directly with the potentials at which the coverage of water and atomic hydrogen on the surface drops from 1.0 to 0 monolayers (Figure 21). According to Wei et al. (Lu et al. 1981; Wei 1980; Wei and Gangloff 1989; Wei et al. 1980), there are three possible rate-determining steps for hydrogen-assisted crack growth:
– Transport of species to the crack tip
– Reaction of species at the surface
– Diffusion of hydrogen to the fracture process zone
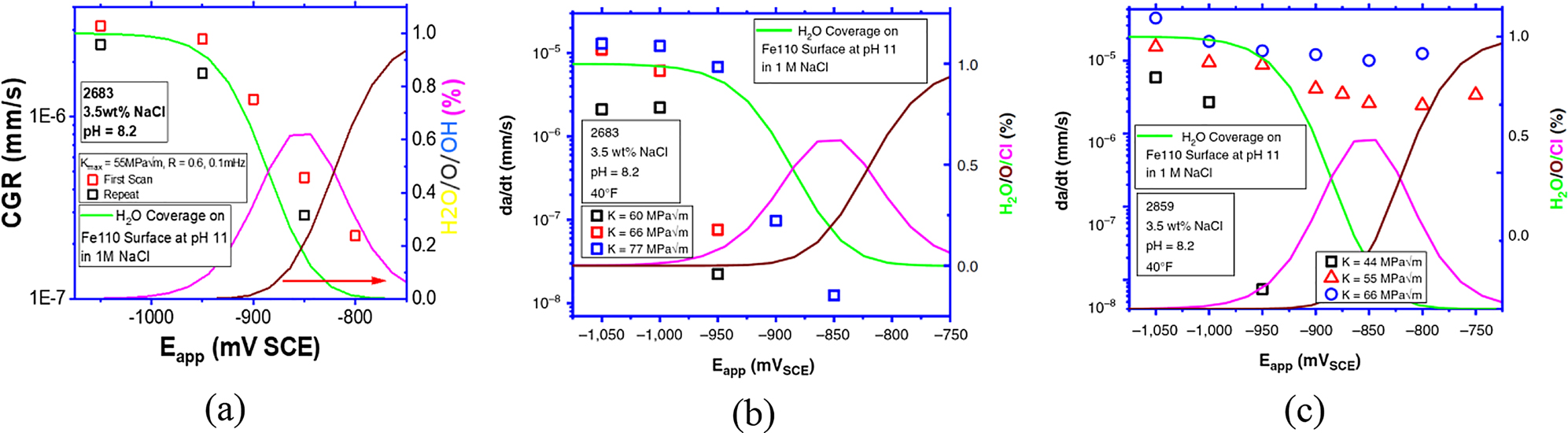
Comparison of the crack growth rate measured at pH 8.2 and water adsorption on Fe110 surface (at pH = 11, which are the expected conditions at the crack tip over a range of cathodic potentials) as a function of applied potential: (a) a 904 MPa strength low alloy steel (sample ID 2683) at 0.1 mHz, ΔK = 22 MPa, and R = 0.6; (b) the 904 MPa strength low alloy steel at various values of K (i.e., limiting static crack growth rate); and (c) an 1166 MPa strength low alloy steel for various values of K (Thodla et al. 2020b).
However, other possible rate-limiting processes may be considered, such as (Thodla et al. 2020b):
– Development of local strains/strain rates high enough to interact with the hydrogen
– Hydrogen interaction with local strain to cause crack propagation
Given the correlation between the predicted change in surface chemistry as a function of electrochemical potential, the reduction in crack growth rate (CGR) with decreasing magnitude of the applied cathodic potential on low alloy steels seems to indicate that surface processes become rate-limiting around −900 mV SCE, and below those potentials, microstructural factors appear to play a more significant role. Figure 21 shows the comparison.
The development of the DFT-based surface coverage model for alloyed materials follows the same approach to be taken for studying the electrochemical potential-dependent behavior of the hydrogen-assisted crack growth for more complex materials, such as corrosion-resistant alloys required for extreme conditions in the oil and gas industry. A study of the hydrogen embrittlement of Inconel alloy 718 under cathodic polarization showed that, such as pipeline steels, hydrogen-assisted crack growth rate can significantly increase once a threshold in the applied cathodic electrochemical potential is crossed (Thodla et al. 2020a). Potentiostatic scratch measurements indicated that the transition from cathodic current transients to anodic transients occurred at the potential associated with the transition from predominantly water-covered metal surface to oxide-covered surface, as predicted by the alloy adsorption isotherm model (Figure 22(a)). Likewise, around this same transition, the crack growth rates associated with hydrogen assisted crack growth increased by several orders of magnitude (Figure 22(b)).

(a) The transitional range for the coverage of water versus oxygen as a function of applied potential to the bare metal surface of alloy Inconel 718 aligns with the transition region between predominantly cathodic transients to anodic transients produced from scratching experiments. (b) Likewise, the transition to higher water coverage/lower oxygen coverage on the Inconel 718 alloy surface induced by cathodic polarization correlates with the increase in crack growth rates on the two heats (120 and 150 ksi) studied for Inconel 718 (Thodla et al. 2020a).
The crack growth characteristics of Inconel alloy 725 under cathodic polarization were studied by Ramgopal et al. (Thodla et al. 2021) and the surface chemisorption model applied to this alloy as a third case study for testing this computational modeling approach. A series of constant K tests on different heats and for different K values showed that crack growth rate associate with hydrogen embrittlement sharply increased by 1–2 orders of magnitude from −850 to −950 mV SCE and then more gradually increased from −900 to −1050 mV SCE. As in the former studies, it was observed in this joint computational/experimental work that the decrease in crack growth rates at less cathodic potentials correlated with the decrease in water coverage that occurs when oxide instead begins to cover the surface sites on the alloy. Based on these insights, a crack-tip strain rate model was developed to account for the changes in the crack growth rate as a function of both the environmental and loading conditions. Figure 23 shows the correlations between CGR and surface coverage for the various heats of 725 studied.

A comparison between the crack growth rate of two heats of 725 (H2 and H4) as a function of water coverage with that of 718 from comparable values of K (∼80 and 100 MPa√m) (Thodla et al. 2021).
In addition to allowing simulations of the potential surface interactions that might occur between a material and the environment that are relevant to hydrogen embrittlement, DFT simulations have also been performed to study some of the details of how hydrogen affects the microstructure. These include the interactions between hydrogen and the metallic lattice (Teus et al. 2007), including diffusion (Chohan et al. 2018), hydrogen, and the decohesion energy of crystallographic planes or grain boundaries (Geng et al. 2005; Tehranchi and Curtin 2017; Tehranchi et al. 2020; Van der Ven and Ceder 2004), hydrogen–vacancy interactions (Lu and Kaxiras 2005; Nazarov et al. 2010; Olsson et al. 2015; Tateyama and Ohno 2003; Taylor et al. 2013, 2014), hydrogen effects at the interface between cementite and the iron matrix (McEniry et al. 2018), formation of hydride phases (Blomqvist et al. 2011; Lillard et al. 2014; Udagawa et al. 2010), and hydrogen interacting with stacking faults and localized plasticity (He et al. 2019; Juan et al. 2002; Lu et al. 2001, 2002; Zamora et al. 2012). These kinds of studies continue to fill out the fundamental knowledge base regarding the influence of hydrogen on materials behavior and will, in time, provide the basis needed for developing fully first-principles informed models of hydrogen-assisted cracking.
12 Passive film breakdown studies
Adsorption reactions on low-index metal surfaces are relatively straightforward calculations compared to those required to study reactions that might take place on the surfaces of oxide materials. This challenge notwithstanding, there remains interested in understanding how oxide films become compromised because of mechanical or chemical interactions, and, in particular, the role of halides in initiating localized corrosion. Efforts that applied DFT in this area in the recent literature include studying the role of chloride in changing the surface stability and vacancy formation mechanisms on passive aluminum oxide surfaces (Marks 2018; Yu et al. 2019), and the displacement of surface OH in hydroxylated nickel oxide surfaces with chloride (Bouzoubaa et al. 2009a, 2010). There still remains to be created, however, an interpretative mechanistic framework that connects these atomistic steps with predictive thermodynamic or kinetic models for passive film breakdown and connection to the electrochemically measured parameters such as the breakdown or repassivation potentials, although one such framework has been initiated that breaks down the various phenomenological contributing factors into kinetic triggers and thermodynamic driving forces (Yu and Marks 2018). We forego a more detailed review of the structure, properties, and reactivity of oxide surfaces surveyed using DFT at this time, and anticipate that advances over the coming years will mature the field toward this direction of integration of DFT insights into phenomenological, electrochemical models for passive film breakdown.
13 Perspectives
As this review indicates, DFT calculations have come a significant way in describing elements of the electrochemical interface relevant to corrosion and hydrogen embrittlement phenomena. There still remains much to be carried out, however, to improve the accuracy and credibility of these models, to expand into new domains of materials corrosion and degradation phenomena, and to develop both larger datasets of relevant parameters computed by first-principles and publicly available algorithms for making those datasets and models usable to the larger corrosion science and engineering community.
13.1 Accuracy and credibility
The physics of DFT continues to be improved with the generation of functionals that are validated against carefully measured experimental data for parameters such as adsorption energies, geometries of molecules and solids, band gaps, vibrational frequencies, etc. Of particular relevance to corrosion are the improvements being made in the electronic structure methods suitable for the study of oxide materials in which self-interaction corrections have been shown to be important (Dudarev et al. 1998b). This is particularly important for oxides in which magnetic moments play a role, such as nickel, chromium, and iron oxides that form the majority of passive films in structural materials (Schmuki 2002). DFT calculations for such systems are notoriously challenging to converge and have had inaccuracies because of the lack of an exact exchange and correlation density functional. As an alternative to improving the physics within the model, machine learning methods can use approximate estimates of parameters from DFT calculations as an input feature that is used in regression models to predict the exact physical parameters with greater accuracy (Hegde and Bowen 2017; Schleder et al. 2019).
Accuracy and credibility can also be improved by including greater physical fidelity in the model construction. For example, much of the work presented in this review focused on data obtained on metals and simulacra of alloy surfaces performed on close-packed, low-index surfaces, usually an fcc (111) plane or a bcc (110) plane. Clearly corrosion phenomena take place on more than this one facet of a polycrystalline material, which is going to be the case in the majority of reported corrosion science experiments and field data gathering reports. Furthermore, other features may be most significant in terms of dominating the kinetics of observable corrosion phenomena (grain boundaries, step-edges, emergent dislocations, and other points of microstructural heterogeneity) such as interfaces between precipitate phases and the matrix (McEniry et al. 2018). As computational resources continue to expand and more researchers enter this field, such features will increasingly be characterized using DFT computation and databases of their properties compiled. Features such as surface roughness that promote micro-crevices can also play a role and are challenging to incorporate into a first-principles model: one way to achieve this is to embed the analysis of the DFT results within a broader model at the macro- and/or meso-scale, for example, phase-field modeling (Wen et al. 2012).
Greater attention to model fidelity can also be placed on the environment side. Much of the adsorption energies reported and used in the works cited in this review were obtained from DFT calculations performed in which the effective potential is constituted by molecules interacting with idealized metallic surfaces in a void. In reality, the molecules and ions will have solvation shells, and the surface will also be solvated. Electric fields of various characters will change the distribution and adsorption energies of these ions, molecules, and solvent species and the electronic structure of the material. The metals and alloys themselves may have a compact of porous, crystalline or amorphous, films of oxides, chlorides, sulfides, or films of mixed composition near the surface that will also change those properties. DFT calculations such as those forming the substance of this review paper are only beginning to explore and characterize the multitudinous variety of contexts in which corrosion of materials may occur. That is why it is pivotal that such expensive and time-consuming calculations be performed within a context informed by high-resolution experimental characterization of corroding interfaces and the existing body of the electrochemical theory of corrosion at both the macroscopic and microscopic scales. DFT models need to be carefully placed within context, and an open mind kept as to practical realities which may cause observed reality to depart from the sparse and hypothetical world in which the model is conceived.
An additional challenge related to accuracy and credibility is the need for validation of the predictions made by DFT to corrosion science and engineering. Most reports of corrosion behavior, whether in the laboratory or in the field, contain a confounding of multiple effects that could be contributing to the observed phenomena. For this reason, new collaborations that involve carefully designed experiments using very clean, optimized materials and high-resolution characterizations alongside DFT analysis need to be formed. The efforts at validation described in this review remain at the level of either supporting inferred hypotheses from experimental and field performance, or approximate comparisons between observed phenomena that may be correlated with surface mechanisms and that calculated for low-index, solid-solution materials (such as the hydrogen assisted crack growth rate comparisons made against the Inconel 718 adsorption model) (Thodla et al. 2020a).
13.2 New domains
Guided by both priorities of government investment and industrial needs, DFT calculations will continue to be applied to both new and long-standing problems in corrosion science and engineering (Perry 2008). Areas of contemporary interest related to corrosion to which DFT is now being applied include design of high entropy and/or multiple principle element alloys for defense and energy applications, environmentally-assisted cracking resistant materials through computationally guided control over microstructural elements, performance of materials in extreme environments including high-pressure high-temperature and sour-service oil and gas recovery, and next-generation nuclear reactor systems operating with molten salt or liquid metal coolants. Resolving materials aging and degradation issues through a combination of first-principles modeling and experiment is also relevant across many industries, including solar power, chemicals production, energy generation, and the maintenance of critical infrastructure. The extent to which DFT calculations will play a role depends upon the perceived critical nature of the problem and the cost-benefit for which creativity in materials approaches may be required. As an example, pipeline corrosion is a well-known and generally well-managed problem but there remains ongoing research to develop new, cheaper, and more environmentally benign chemical corrosion inhibitors to reduce corrosion to more manageable and reliably predictable levels: materials innovation is usually not the focus, however, DFT can inform both pipeline managers and research scientists as to the mechanisms of both corrosion and inhibitor action. On the other hand, the desire to improve the efficiency of power generation through obtaining higher temperatures is requiring a search for new materials and/or multi-material systems that are corrosion resistant under a variety of conditions, and hence, innovation in materials rather than chemical inhibitors is required for this problem set. In sectors such as aerospace and maritime, both materials and coating solutions for corrosion and cracking mitigation remain critical drivers for research that encompasses modeling across multiple scales, including DFT, and improved approaches for experimental characterization and accelerated testing.
13.3 Availability of datasets and models
Up until now the application of DFT to problems in corrosion science and engineering has occurred primarily within research groups, as prototype codes are developed and pieced together with various interfaces to high-performance computing systems, desktop pre-processing and post-processing, and analysis tools such as spreadsheets, python notebooks, and mathematical processing software packages. The task of collecting the datasets that have been generated across numerous projects and activities over the years and making them publicly available, or available in a commercial package, has yet to be performed. Likewise, for the codes that perform the surface Pourbaix analyses, for example, or electrochemical kinetics predictions and corrosion susceptibility indices described in this review. As the techniques become more useful for explaining observed corrosion and hydrogen embrittlement phenomena, these datasets and algorithms should become streamlined and packaged into software platforms for wider adoption. This process can be facilitated through sharing of research codes via code-management websites and the development of an online repository for hosting the relevant fundamentals determined by the costly and time-consuming DFT and similar kinds of computations.
Extending access to DFT databases and codes will have the effect of increasing the experimentation in applying and extending these methods to other problems in electrochemical surface science and corrosion by other research groups in academia and industry. Predictions from DFT computations will start to play a role in the development of new feature sets for machine learning of materials performance as a function of materials microstructural and the environment variables. Likewise, in risk assessment and lifetime prediction models, fundamental materials/environment parameters estimable from first-principles can play a role in defining threshold limits, or classifying compatible and incompatible combinations, in ways similar to how Pourbaix diagrams and electrochemical kinetics are used today. Although it is acknowledged that the materials systems modeled in the idealized DFT calculations can be several times removed from engineering materials, environments, and applications, the translation of the fundamental science into industrial practice can be expected to lead to significant returns.
About the authors

Christopher D. Taylor received his BS from the University of Western Australia, MS from the University of Memphis, and PhD from the University of Virginia. Chris worked at Los Alamos National Laboratory, studying corrosion in oil and gas, materials/environment effects for national security, catalyst design and optimization, and nuclear waste disposition. Chris is currently a Senior Researcher at DNV, and Associate Research Professor at the Ohio State University. Christopher has 100+ publications in science and engineering, with a focus on computational modeling of corrosion.

Huibin Ke received her BS from Harbin Institute of Technology in 2011 and earned her PhD from the University of Wisconsin – Madison in 2017. Since then, she worked as a postdoctoral researcher with Prof. Christopher Taylor at The Ohio State University on ab initio-based modeling of alloy corrosion mechanisms in aqueous solution, after which she worked as a Postdoctoral Research Associate in Pacific Northwest National Laboratory. Her research interest is multi-scale modeling of microstructure evolution of metallic materials.
Acknowledgments
DNV GL USA is acknowledged for supporting the time and effort required to put together this review.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: None declared.
-
Conflicts of interest: The authors declare no conflicts of interest regarding this article.
References
Akpanyung, K.V., Loto, R.T., and Fajobi, M.A. (2019). An overview of ammonium chloride (NH4Cl) Corrosion in the refining unit. J. Phys. Conf. Series 1378: 022089, https://doi.org/10.1088/1742-6596/1378/2/022089.Suche in Google Scholar
Alfe, D., De Wijs, G.A., Kresse, G., and Gillan, M.J. (2000). Recent developments in ab initio thermodynamics. Int. J. Quant. Chem. 77: 871–879.10.1002/(SICI)1097-461X(2000)77:5<871::AID-QUA8>3.0.CO;2-3Suche in Google Scholar
Alvarez, M.G. and Galvele, J.R. (1984). The mechanism of pitting of high purity iron in NaCl solutions. Corrosion Sci. 24: 27–48, https://doi.org/10.1016/0010-938x(84)90133-1.Suche in Google Scholar
Alvaro, A., Thue Jensen, I., Kheradmand, N., Løvvik, O.M., and Olden, V. (2015). Hydrogen embrittlement in nickel, visited by first principles modeling, cohesive zone simulation and nanomechanical testing. Int. J. Hydrogenergy 40: 16892–16900, https://doi.org/10.1016/j.ijhydene.2015.06.069.Suche in Google Scholar
Alvisi, P.P. and de Freitas Cunha Lins, V. (2008). Acid salt corrosion in a hydrotreatment plant of a petroleum refinery. Eng. Fail. Anal. 15: 1035–1041, https://doi.org/10.1016/j.engfailanal.2007.11.014.Suche in Google Scholar
Anderko, A., Sridhar, N., and Dunn, D.S. (2004). A general model for the repassivation potential as a function of multiple aqueous solution species. Corrosion Sci. 46: 1583–1612, https://doi.org/10.1016/j.corsci.2003.10.002.Suche in Google Scholar
Anderko, A., Sridhar, N., Brossia, C.S., and Tormeon, G. (2006). Modeling localized corrosion in complex process environments in the presence of inhibitors, NACE Corrosion. NACE International.10.5006/C2006-06215Suche in Google Scholar
Anderko, A., Sridhar, N., Jakab, M.A., and Tormoen, G. (2008). A general model for the repassivation potential as a function of multiple aqueous species. 2. Effect of oxyanions on localized corrosion of Fe–Ni–Cr–Mo–W–N alloys. Corrosion Sci. 50: 3629–3647, https://doi.org/10.1016/j.corsci.2008.08.046.Suche in Google Scholar
Anderko, A., Gui, F., Cao, L., and Sridhar, N. (2014). Modeling localized corrosion of corrosion-resistant alloys in oil and gas production environments, Corrosion 2014. San Antonio, TX: NACE International.10.5006/C2014-3744Suche in Google Scholar
Anderson, A.B. and Kang, D.B. (1998). Quantum chemical approach to redox reactions including potential dependence: application to a model for hydrogen evolution from diamond. J. Phys. Chem. 102: 5993–5996, https://doi.org/10.1021/jp9810926.Suche in Google Scholar
Andryushechkin, B.V., Zheltov, V.V., Cherkez, V.V., Zhidomirov, G.M., Klimov, A.N., Kierren, B., Fagot-Revurat, Y., Malterre, D., and Eltsov, K.N. (2015). Chlorine adsorption on Cu(111) revisited: LT-STM and DFT study. Surf. Sci. 639: 7–12, https://doi.org/10.1016/j.susc.2015.03.030.Suche in Google Scholar
Antony, J. and Grimme, S. (2006). Density functional theory including dispersion corrections for intermolecular interactions in a large benchmark set of biologically relevant molecules. Phys. Chem. Chem. Phys. 8: 5287–5293, https://doi.org/10.1039/b612585a.Suche in Google Scholar PubMed
Artymowicz, D.M., Erlebacher, J., and Newman, R.C. (2009). Relationship between the parting limit for de-alloying and a particular geometric high-density site percolation threshold. Phil. Mag. 89: 1663–1693, https://doi.org/10.1080/14786430903025708.Suche in Google Scholar
Atrens, A. and Dietzel, W. (2007). The negative difference effect and unipositive Mg+. Adv. Eng. Mater. 9: 292–297, https://doi.org/10.1002/adem.200600275.Suche in Google Scholar
Barbier, A., Renaud, G., Mocuta, C., and Stierle, A. (1999). Structural investigation of the dynamics of the NiO(111) surface by GIXS. Surf. Sci. 433–435: 761–764, https://doi.org/10.1016/s0039-6028(99)00492-6.Suche in Google Scholar
Barrera, O., Bombac, D., Chen, Y., Daff, T.D., Galindo-Nava, E., Gong, P., Haley, D., Horton, R., Katzarov, I., Kermode, J.R., et al.. (2018). Understanding and mitigating hydrogen embrittlement of steels: a review of experimental, modelling and design progress from atomistic to continuum. J. Mater. Sci. 53: 6251–6290, https://doi.org/10.1007/s10853-017-1978-5.Suche in Google Scholar PubMed PubMed Central
Bender, S., Goellner, J., Heyn, A., and Schmigalla, S. (2011). A new theory for the negative difference effect in magnesium corrosion. Mater. Corros. 63: 707–712, https://doi.org/10.1002/maco.201106225.Suche in Google Scholar
Blomqvist, J., Olofsson, J., Alvarez, A.-M. and Bjerken, C. (2011). Structure and thermodynamical properties of zirconium hydrides from first-principle. In: 15th Int. Conf. on Env. Degrad., pp. 671–681.10.1007/978-3-319-48760-1_42Suche in Google Scholar
Blum, V., Hart, G.L.W., Walorski, M.J., and Zunger, A. (2005). Using genetic algorithms to map first-principles results to model Hamiltonians: application to the generalized Ising model for alloys. Phys. Rev. B 72: 165113, https://doi.org/10.1103/physrevb.72.165113.Suche in Google Scholar
Boettger, J.C. and Ray, A.K. (2002). Fully relativistic density functional calculations on hydroxylated actinide oxide surfaces. Int. J. Quant. Chem. 90: 1470–1477, https://doi.org/10.1002/qua.10350.Suche in Google Scholar
Borck, O., Hyldgaard, P., and Schroder, E. (2007). Adsorption of methylamine on alpha-Al2O3(0001) and alpha-Cr2O3(0001): density functional theory. Phys. Rev. B 75: 035403, doi:https://doi.org/10.1103/physrevb.75.035403.Suche in Google Scholar
Bouzoubaa, A., Diawara, B., Maurice, V., Minot, C., and Marcus, P. (2009a). Ab initio modelling of localized corrosion: study of the role of surface steps in the interaction of chlorides with passivated nickel surfaces. Corrosion Sci. 51: 2174–2182, https://doi.org/10.1016/j.corsci.2009.05.048.Suche in Google Scholar
Bouzoubaa, A., Diawara, B., Maurice, V., Minot, C., and Marcus, P. (2009b). Ab initio study of the interaction of chlorides with defect-free hydroxylated NiO surfaces. Corrosion Sci. 51: 941–948, https://doi.org/10.1016/j.corsci.2009.01.028.Suche in Google Scholar
Bouzoubaa, A., Costa, D., Diawara, B., and Marcus, P. (2010). Insight of DFT and atomistic thermodynamics on the adsorption and insertion of halides onto the hydroxylated NiO(111) surface. Corrosion Sci. 52: 2643, https://doi.org/10.1016/j.corsci.2010.04.014.Suche in Google Scholar
Byrd, R.H., Gilbert, J.C., and Nocedal, J. (2000). A trust region method based on interior point techniques for nonlinear programming. Math. Program. 89: 149–185, https://doi.org/10.1007/pl00011391.Suche in Google Scholar
Calvo, E.J. (2014). The kinetics of oxygen electro-reduction: a long way from rust to Li-air batteries. Mater. Corros. 65: 345, https://doi.org/10.1002/maco.201407614.Suche in Google Scholar
Car, R. (2016). Fixing Jacob’s ladder. Nat. Chem. 8: 820–821, https://doi.org/10.1038/nchem.2605.Suche in Google Scholar PubMed
Castle, J.E. and Asami, K. (2004). A more general method for ranking the enrichment of alloying elements in passivation films. Surf. Interface Anal. 36: 220–224, https://doi.org/10.1002/sia.1676.Suche in Google Scholar
Ceder, G. (1993). A derivation of the Ising model for the computation of phase diagrams. Comput. Mater. Sci. 1: 144–150, https://doi.org/10.1016/0927-0256(93)90005-8.Suche in Google Scholar
Chen, J.-h., Long, X.-h., Zhao, C.-h., Kang, D., and Guo, J. (2014). DFT calculation on relaxation and electronic structure of sulfide minerals surfaces in presence of H2O molecule. J. Central South Univ. 21: 3945–3954, https://doi.org/10.1007/s11771-014-2382-9.Suche in Google Scholar
Chen, S.-C. and Windl, W. (2020). Bond synergy model for bond energies in alloy oxides. J. Electrochem. Soc. 167: 141511.10.1149/1945-7111/abc6c7Suche in Google Scholar
Cheng, S.-T., Todorova, M., Freysoldt, C., and Neugebauer, J. (2014). Negatively charged ions on Mg(0001) surfaces: appearance and origin of attractive adsorbate-adsorbate interactions. Phys. Rev. Lett. 113: 136102–136105, https://doi.org/10.1103/physrevlett.113.136102.Suche in Google Scholar
Cho, K., Kishimoto, J., Hashizume, T., Pickering, H.W., and Sakurai, T. (1995). Adsorption and film growth of BTA on clean and oxygen adsorbed Cu(110) surfaces. Appl. Surf. Sci. 87–88: 380–385, https://doi.org/10.1016/0169-4332(94)00506-0.Suche in Google Scholar
Chohan, U.K., Koehler, S.P.K., and Jiminez-Melero, E. (2018). Diffusion of hydrogen into and through gamma-iron by density functional theory. Surf. Sci. 672–673: 56–61, https://doi.org/10.1016/j.susc.2018.02.001.Suche in Google Scholar
Choudhury, S., Morgan, D., and Uberuaga, B.P. (2014). Massive interfacial reconstruction at misfit dislocations in metal/oxide interfaces. Sci. Rep. 4: 6533, https://doi.org/10.1038/srep06533.Suche in Google Scholar PubMed PubMed Central
Christov, M. and Popova, A. (2004). Adsorption characteristics of corrosion inhibitors from corrosion rate measurements. Corrosion Sci. 46: 1613–1620, https://doi.org/10.1016/j.corsci.2003.10.013.Suche in Google Scholar
Clayton, C.R. and Olefjord, l. (2011). Passivity of austenitic stainless steels. Corrosion mechanisms in theory and practice, 3rd ed. CRC Press, New York, NY.Suche in Google Scholar
Conway, B.E. and Bockris, J.O.M. (1957). Electrolytic hydrogen evolution kinetics and its relation to the electronic and adsorption properties of the metal. J. Chem. Phys. 26: 532, https://doi.org/10.1063/1.1743339.Suche in Google Scholar
Conway, B.E., Angerstein-Kozlowska, H., and Dhar, H.P. (1974). On selection of standard states in adsorption isotherms. Electrochim. Acta 19: 455–460, https://doi.org/10.1016/0013-4686(74)87024-6.Suche in Google Scholar
Cordeiro, J.M., Nagay, B.E., Mathew, M.T., and Barao, V.A. (2020). Designing corrosion-resistant alloys. In: Inamuddin, R.B. (Ed.), Alloy materials and their allied applications. Scrivener Publishing, Beverly, MA.10.1002/9781119654919.ch2Suche in Google Scholar
Costa, D. and Marcus, P. (2015). Adsorption of organic inhibitor molecules on metal and oxidized surfaces studied by atomistic theoretical methods. In: Taylor, C.D., and Marcus, P. (Eds.), Molecular modeling of corrosion processes. Hoboken, NJ: ECS-Wiley.10.1002/9781119057666.ch5Suche in Google Scholar
Deltombe, E., de Zoubov, N., and Pourbaix, M. (1974). Atlas of electrochemical equilibria in aqueous solution. Houston, Texas: NACE.Suche in Google Scholar
DeLuccia, J.J. (1988). Electrochemical aspects of hydrogen in metals. In: Raymond, L. (Ed.), Hydrogen embrittlement: prevention and control. Philadelphia: American Society for Testing and Materials.10.1520/STP45284SSuche in Google Scholar
Desai, S.K. and Neurock, M. (2003). First-principles study of the role of solvent in the dissociation of water over a Pt-Ru alloy. Phys. Rev. B 68: 075420, https://doi.org/10.1103/physrevb.68.075420.Suche in Google Scholar
Du, L., Zhao, H., Hu, H., and Yang, J. (2014). Quantum chemical and molecular dynamics studies of imidazoline derivatives as corrosion inhibitor and quantitative structure-activity relationship (QSAR) analysis using the support vector machine (SVM) method. J. Theor. Comput. Chem. 13: 1450012, https://doi.org/10.1142/s0219633614500126.Suche in Google Scholar
Dudarev, S.L., Botton, G.A., Savrasov, S.Y., Humphreys, C.J., and Sutton, A.P. (1998a). Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57: 1505–1509, https://doi.org/10.1103/physrevb.57.1505.Suche in Google Scholar
Dudarev, S.L., Botton, G.A., Savrasov, S.Y., Szotek, Z., Temmerman, W.M., and Sutton, A.P. (1998b). Electronic structure and elastic properties of strongly correlated metal oxides from first principles: LSDA+U, SIC-LSDA, and EELS study of UO2 and NiO. Phys. Status Solidi 166: 429–443, https://doi.org/10.1002/(sici)1521-396x(199803)166:1<429::aid-pssa429>3.0.co;2-f.10.1002/(SICI)1521-396X(199803)166:1<429::AID-PSSA429>3.3.CO;2-6Suche in Google Scholar
Durnie, W., De Marco, R., Jefferson, A., and Kinsella, B. (1999). Development of a structure-activity relationship for oil field corrosion inhibitors. J. Electrochem. Soc. 146: 1751–1756, https://doi.org/10.1149/1.1391837.Suche in Google Scholar
Durnie, W., De Marco, R., Kinsella, B., Jefferson, A., and Pejcic, B. (2005). Predicting the adsorption properties of carbon dioxide corrosion inhibitors using a structure-activity relationship. J. Electrochem. Soc. 152: B1–B11, https://doi.org/10.1149/1.1823991.Suche in Google Scholar
Erlebacher, J. (2004). An atomistic description of dealloying. J. Electrochem. Soc. 151: C614, https://doi.org/10.1149/1.1784820.Suche in Google Scholar
Evans, U.R. and Hoar, T.P. (1932). The velocity of corrosion from the electrochemical standpoint. Part II. Proc. Roy. Soc. A 137: 343.10.1098/rspa.1932.0140Suche in Google Scholar
Evans, U.R. and Hoar, T.P. (1934). The mechanism of metallic corrosion. A view suggested by Whitby’s recent papers. Trans. Faraday Soc. 30: 424–432.10.1039/tf9343000424Suche in Google Scholar
Feliu, S., Morcillo, M., and Feliu, S.Jr. (1993a). The prediction of atmospheric corrosion from meteorological and pollution parameters – II. Long term forecasts. Corrosion Sci. 34: 415–422.10.1016/0010-938X(93)90113-USuche in Google Scholar
Feliu, S., Morcillo, M., and Feliu, S.Jr. (1993b). The prediction of atmospheric corrosion from meteorological and pollution parameters – I. Annual corrosion. Corrosion Sci. 34: 403–313.10.1016/0010-938X(93)90112-TSuche in Google Scholar
Fellinger, M.R., Park, H., and Wilkins, J.W. (2010). Force-matched embedded-atom method potential for niobium. Phys. Rev. B 81: 144119–144133.10.1103/PhysRevB.81.144119Suche in Google Scholar
Filhol, J.S. and Neurock, M. (2006). Elucidation of the electrochemical activation of water over Pd by first principles. Angew Chem. Int. Ed. Engl. 45: 402–6.10.1002/anie.200502540Suche in Google Scholar
Ford, F.P. (1984). Current understanding of the mechanisms of stress corrosion and corrosion fatigue. In: Dean, S.W., Pugh, E.N., and Ugianski, G.M. (Eds.), Environmentally-sensitive fracture: evaluation and comparison of test methods. ASTM STP821. Philadelphia: American Society for Testing and Materials.10.1520/STP34425SSuche in Google Scholar
Francis, M.F. and Curtin, W.A. (2015). Mechanical work makes important contributions to surface chemistry at steps. Nat. Commun. 6: 6261.10.1038/ncomms7261Suche in Google Scholar
Francis, M.F. and Holby, E.F. (2015). Thermodynamics of passive film formation from first principes. In: Taylor, C.D., and Marcus, P. (Eds.), Molecular modeling of corrosion processes: scientific development and engineering applications. John Wiley & Sons, Hoboken, NJ, pp. 157–189.10.1002/9781119057666.ch6Suche in Google Scholar
Francis, M.F. and Taylor, C.D. (2013). First-principles insights into the structure of the incipient magnesium oxide and its instability to decomposition: oxygen chemisorption to Mg(0001) and thermodynamic stability. Phys. Rev. B 87: 075450.10.1103/PhysRevB.87.075450Suche in Google Scholar
Frankel, G., Li, T., and Scully, J.R. (2017). Perspective-localized corrosion: passive film breakdown vs pit growth stability. J. Electrochem. Soc. 164: C180–C181.10.1149/2.1381704jesSuche in Google Scholar
Frankel, G.S. (2016). Fundamentals of corrosion kinetics. In: Hughes, A.E., Mol, J.M.C., Zheludkevich, M.L., and Buchheit, R.G. (Eds.), Active protective coatings. Springer, pp. 17–32.10.1007/978-94-017-7540-3_2Suche in Google Scholar
Frankel, G.S., Samaniego, A., and Birbilis, N. (2013). Evolution of hydrogen at dissolving magnesium surfaces. Corrosion Sci. 70: 104–111.10.1016/j.corsci.2013.01.017Suche in Google Scholar
Fukai, Y. (2002). Superabundant vacancies formed in metal-hydrogen alloys. Phys. Scripta 2003: 11.10.1238/Physica.Topical.103a00011Suche in Google Scholar
Gece, G. (2008). The use of quantum chemical methods in corrosion inhibitor studies. Corrosion Sci. 50: 2981–2992.10.1016/j.corsci.2008.08.043Suche in Google Scholar
Geng, W.-T., Freeman, A.J., Olson, G.B., Tateyama, Y., and Ohno, T. (2005). Hydrogen-promoted grain boundary embrittlement and vacancy activity in metals: insights from Ab initio total energy calculations. Mater. Trans. 46: 756–760.10.2320/matertrans.46.756Suche in Google Scholar
German, E. and Efremenko, I. (2004). Calculation of the activation energies of dissociative oxygen adsorption on the surfaces of rhodium (111), silver (111) and (110), and gold (111). J. Mol. Struct. Theochem 711: 159–165.10.1016/j.theochem.2004.10.001Suche in Google Scholar
Gileadi, E. (1987). The combined adsorption isotherm. Electrochim. Acta 32: 221–229.10.1016/0013-4686(87)85028-4Suche in Google Scholar
Grabke, H.J., Spiegel, M., and Zahs, A. (2004). Role of alloying elements and carbides in the chlorine-induced corrosion of steels and alloys. Mater. Res. 7: 89–95.10.1590/S1516-14392004000100013Suche in Google Scholar
Greeley, J. (2010). Structural effects on trends in the deposition and dissolution of metal-supported metal adstructures. Electrochim. Acta 55: 5545–5550.10.1016/j.electacta.2010.04.055Suche in Google Scholar
Greeley, J. and Mavrikakis, M. (2005). Surface and subsurface hydrogen: adsorption properties on transition metals and near-surface alloys. J. Phys. Chem. B 109: 3460–3471.10.1021/jp046540qSuche in Google Scholar
Greeley, J. and Nørskov, J.K. (2007a). Electrochemical dissolution of surface alloys in acids: thermodynamic trends from first-principles calculations. Electrochim. Acta 52: 5829–5836.10.1016/j.electacta.2007.02.082Suche in Google Scholar
Greeley, J. and Nørskov, J.K. (2007b). Large-scale, density functional theory-based screening of alloys for hydrogen evolution. Surf. Sci. 601: 1590–1598.10.1016/j.susc.2007.01.037Suche in Google Scholar
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27: 1787–1799.10.1002/jcc.20495Suche in Google Scholar
Gunasegaram, D.R., Venkatraman, M.S., and Cole, I.S. (2014). Towards multiscale modelling of localised corrosion. Int. Mater. Rev. 59: 84.10.1179/1743280413Y.0000000024Suche in Google Scholar
Hackerman, N. (1952). Recent advances in understanding of organic inhibitors. Corrosion 18: 332–337.10.5006/0010-9312-18.9.332Suche in Google Scholar
Hackerman, N. (1962). Physical chemical aspects of corrosion inhibition. Corrosion 8: 143–148.10.5006/0010-9312-8.4.143Suche in Google Scholar
Hafner, J., Wolverton, C., and Ceder, G. (2006). Towards computational materials design: the impact of density functional theory on materials research. MRS Bull. 31: 659–668.10.1557/mrs2006.174Suche in Google Scholar
Han, C., Zhang, C., Liu, X., Huang, H., Zhuang, S., Han, P., and Wu, X. (2015). Effects of alloying on oxidation and dissolution corrosion of the surface of gamma-Fe(111): a DFT study. J. Mol. Model. 21: 181.10.1007/s00894-015-2719-9Suche in Google Scholar
Hansen, H.A., Rossmeisl, J., and Norskov, J.K. (2008). Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag, and Ni(111) surfaces studied by DFT. Phys. Chem. Chem. Phys. 10: 3722–3730.10.1039/b803956aSuche in Google Scholar
Hart, G.L.W., Blum, V., Walorski, M.J., and Zunger, A. (2005). Evolutionary approach for determining first-principles Hamiltonians. Nat. Mater. 4: 391–394.10.1038/nmat1374Suche in Google Scholar
He, Y., Li, Y., Zhao, X., and Chen, C. (2019). Effect of hydrogen atom and hydrogen filled vacancies on stacking fault energy in gamma-Fe by first-principles calculations. Int. J. Hydrogenergy 44: 17136–17145.10.1016/j.ijhydene.2019.04.229Suche in Google Scholar
Hegde, G. and Bowen, R.C. (2017). Machine-learned approximations to density functional theory Hamiltonians. Sci. Rep. 7: 42669.10.1038/srep42669Suche in Google Scholar
Henderson, M.A. (2002). The interaction of water with solid surfaces: fundamental aspects revisited. Surf. Sci. Rep. 46: 1–308.10.1016/S0167-5729(01)00020-6Suche in Google Scholar
Huang, L.-F., Scully, J.R., and Rondinelli, J.M. (2019). Modeling corrosion with first-principles electrochemical phase diagrams. Annu. Rev. Mater. Res. 49: 53–77.10.1146/annurev-matsci-070218-010105Suche in Google Scholar
Hyman, M.P. and Medlin, J.W. (2005). Theoretical study of the adsorption and dissociation of oxygen on Pt(111) in the presence of homogeneous electric fields. J. Phys. Chem. B 109: 6304–6310.10.1021/jp045155ySuche in Google Scholar
Jacob, T. (2007). Potential-induced lifting of the Au(100)-surface reconstruction studied with DFT. Electrochim. Acta 52: 2229–2235.10.1016/j.electacta.2006.03.114Suche in Google Scholar
Janik, M.J., Taylor, C.D., and Neurock, M. (2009). First-principles analysis of the initial electroreduction steps of oxygen over Pt(111). J. Electrochem. Soc. 156: B126–B135.10.1149/1.3008005Suche in Google Scholar
Jiang, Y. and Adams, J.B. (2003). First principle calculations of benzotriazole adsorption onto clean Cu(111). Surf. Sci. 529: 428–442.10.1016/S0039-6028(03)00277-2Suche in Google Scholar
Jiang, Y., Adams, J.B., and Sun, D.H. (2004). Benzotriazole adsorption on Cu2O(111) surfaces: a first-principles study. J. Phys. Chem. B 108: 12851–12857.10.1021/jp049457bSuche in Google Scholar
Jiang, Y., Smith, J.R., and Evans, A.G. (2008). First principles assessment of metal/oxide interface adhesion. Appl. Phys. Lett. 92: 141918.10.1063/1.2907339Suche in Google Scholar
Jinnouchi, R. and Anderson, A.B. (2008a). Aqueous and surface redox potentials from self-consistently determined Gibbs energies. J. Phys. Chem. C 112: 8747–8750.10.1021/jp802627sSuche in Google Scholar
Jinnouchi, R. and Anderson, A.B. (2008b). Electronic structure calculations of liquid-solid interfaces: combination of density functional theory and modified Poisson-Boltzmann theory. Phys. Rev. B 77: 245417.10.1103/PhysRevB.77.245417Suche in Google Scholar
Jones, R.O. (2015). Density functional theory: its origins, rise to prominence, and future. Rev. Mod. Phys. 87: 897–923.10.1103/RevModPhys.87.897Suche in Google Scholar
Juan, A., Moro, L., Brizuela, G., and Pronsato, E. (2002). The electronic structure and bonding of an hydrogen pair near a FCC Fe stacking fault. Int. J. Hydrogenergy 27: 333–338.10.1016/S0360-3199(01)00113-6Suche in Google Scholar
Karlberg, G.S., Rossmeisl, J., and Norskov, J.K. (2007). Estimations of electric field effects on the oxygen reduction reaction based on the density functional theory. Phys. Chem. Chem. Phys. 9: 5158–61.10.1039/b705938hSuche in Google Scholar
Ke, H. and Taylor, C.D. (2019). Density functional theory: an essential partner in the integrated computational materials engineering approach to corrosion. Corrosion 75: 708–726.10.5006/3050Suche in Google Scholar
Ke, H. and Taylor, C.D. (2020a). DFT-based calculation of dissolution activation energy and kinetics of Ni–Cr alloys. J. Electrochem. Soc. 167: 131508.10.1149/1945-7111/abbbbdSuche in Google Scholar
Ke, H. and Taylor, C.D. (2020b). First-principles modeling of the repassivation of corrosion resistant alloys: Part I. O and Cl adsorption energy. J. Electrochem. Soc. 167: 111502.10.1149/1945-7111/aba44eSuche in Google Scholar
Ke, H., Taylor, C.D., and Frankel, G.S. (2020a). Application of the chloride susceptibility index (CSI) to study the effect of Ni, Cr, Mn and Mo on the repassivation of stainless steels. J. Electrochem. Soc. 167: 131510.10.1149/1945-7111/abbce2Suche in Google Scholar
Ke, H., Taylor, C.D., Frankel, G.S., Li, T., and Lu, P. (2020b). First-principles modeling of the repassivation of corrosion resistant alloys: Part II. Surface adsorption isotherms for alloys and the chloride susceptibility index. J. Electrochem. Soc. 167: 111501.10.1149/1945-7111/aba3feSuche in Google Scholar
Khaled, K.F. (2003). The inhibition of benzimidazole derivatives on corrosion of iron in 1 M HCl solutions. Electrochim. Acta 48: 2493–2503.10.1016/S0013-4686(03)00291-3Suche in Google Scholar
Khaled, K.F. (2006). Experimental and theoretical study for corrosion inhibition of mild steel in hydrochloric acid solution by some new hydrazine carbodithioic acid derivatives. Appl. Surf. Sci. 252: 4120–4128.10.1016/j.apsusc.2005.06.016Suche in Google Scholar
Khaled, K.F. (2008). Molecular simulation, quantum chemical calculations and electrochemical studies for inhibition of mild steel by triazoles. Electrochim. Acta 53: 3484–3492.10.1016/j.electacta.2007.12.030Suche in Google Scholar
Khaled, K.F. (2009). Monte Carlo simulations of corrosion inhibition of mild steel in 0.5 M sulphuric acid by some green corrosion inhibitors. J. Solid State Electrochem. 13: 1743–1756.10.1007/s10008-009-0845-ySuche in Google Scholar
Khaled, K.F. (2010). Experimental and molecular dynamics study on the inhibition performance of some nitrogen containing compounds for iron corrosion. Mater. Chem. Phys. 124: 760–767.10.1016/j.matchemphys.2010.07.055Suche in Google Scholar
Khaled, K.F. and Sherik, A. (2013). Using neural networks for corrosion inhibition efficiency prediction during corrosion of steel in chloride solutions. Int. J. Electrochem. Sci. 8: 9918–9935.10.1016/S1452-3981(23)13022-7Suche in Google Scholar
Khaled, K.F., Babic-Samardzija, K., and Hackerman, N. (2005). Theoretical study of the structural effects of polymethylene amines on corrosion inhibition of iron in acid solutions. Electrochim. Acta 50: 2515–2520.10.1016/j.electacta.2004.10.079Suche in Google Scholar
Kim, E., Weck, P., Taylor, C.D., Olatunji-Ojo, O., Liu, X.-Y., Mausolf, E., Jarvinen, G.D., and Czerwinski, K.R. (2014). First-principles and kinetic Monte Carlo simulation studies of the reactivity of Tc(0001), MoTc(111) and MoTc(110) surfaces. J. Electrochem. Soc. 161: C83–C88.10.1149/2.097401jesSuche in Google Scholar
Kirchheim, R., Heine, B., Fischmeister, H., Hofmann, S., Knote, K., and Stolz, U. (1989). The passivity of iron–chromium alloys. Corrosion Sci. 29: 899–917.10.1016/0010-938X(89)90060-7Suche in Google Scholar
Kohn, W. and Sham, L.J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140: A1133–A1138.10.1103/PhysRev.140.A1133Suche in Google Scholar
Kokalj, A. (2010). Is the analysis of molecular electronic structure of corrosion inhibitors sufficient to predict the trend of their inhibition performance. Electrochim. Acta 56: 745–755.10.1016/j.electacta.2010.09.065Suche in Google Scholar
Kokalj, A., Kovacevic, N., Peljhan, S., Finsgar, M., Lesar, A., and Milosev, I. (2011). Triazole, benzotriazole, and naphthotriazole as copper corrosion inhibitors. I. Molecular electronic and adsorption properties. ChemPhysChem 12: 3547–3555.10.1002/cphc.201100537Suche in Google Scholar
Korth, M. (2017). Density functional theory: not quite the right answer for the right reason yet. Angew. Chem. Int. Ed. 56: 5396–5398.10.1002/anie.201701894Suche in Google Scholar
Kovačević, N. and Kokalj, A. (2012). Chemistry of the interaction between azole type corrosion inhibitor molecules and metal surfaces. Mater. Chem. Phys. 137: 331–339.10.1016/j.matchemphys.2012.09.030Suche in Google Scholar
Kovačević, N. and Kokalj, A. (2013). The relation between adsorption bonding and corrosion inhibition of azole molecules on copper. Corrosion Sci. 73: 7–17.10.1016/j.corsci.2013.03.016Suche in Google Scholar
Kovacevic, N., Milosev, I., and Kokalj, A. (2017). How relevant is the adsorption bonding of imidazoles and triazoles for their corrosion inhibition of copper? Corrosion Sci. 124: 25–34.10.1016/j.corsci.2017.04.021Suche in Google Scholar
Krishnamurthy, B., White, R.E., and Ploehn, H.J. (2002). Simplified point defect model for growth of anodic passive films on iron. Electrochim. Acta 47: 3375–3381.10.1016/S0013-4686(02)00274-8Suche in Google Scholar
Lebrini, M., Traisnel, M., Lagrenee, M., Mernari, B., and Bentiss, F. (2008). Inhibitive properties, adsorption and a theoretical study of 3,5-bis(n-pyridyl)-4-amino-1,2,4-triazoles as corrosion inhibitors for mild steel in perchloric acid. Corrosion Sci. 50: 473–479.10.1016/j.corsci.2007.05.031Suche in Google Scholar
Lei, H., Wang, C., Yao, Y., Wang, Y., Hupalo, M., McDougall, D., Tringides, M., and Ho, K. (2013). Strain effect on the adsorption, diffusion and molecular dissociation of hydrogen on Mg(0001) surface. J. Chem. Phys. 139: 224702.10.1063/1.4839595Suche in Google Scholar
Lillard, R.S., Taylor, C.D., Wermer, J.R., Mara, N.A., and Cooley, J.C. (2014). A thermal desorption study of the kinetics of uranium hydride decomposition. J. Nucl. Mater. 444: 49–55.10.1016/j.jnucmat.2013.09.032Suche in Google Scholar
Limmer, K.R., Williams, K.S., Labukas, J.P., and Andzelm, J.W. (2017). First principles modeling of cathodic reaction thermodynamics in dilute magnesium alloys. Corrosion 73: 506–517.10.5006/2274Suche in Google Scholar
Lin, L.F., Chao, C.Y., and Macdonald, D.D. (1981). A point defect model for anodic passive films: II. Chemical breakdown and pit initiation. J. Electrochem. Soc. 128: 1194–1198.10.1149/1.2127592Suche in Google Scholar
Liu, J., Zhang, X., Chen, M., Li, L., Zhu, B., Tang, J., and Liu, S. (2011). DFT study on surface properties and dissolution trends of Al(100) surfaces doped with Zn, Ga, In, Sn and Pb. Appl. Surf. Sci. 257: 4004–4009.10.1016/j.apsusc.2010.11.165Suche in Google Scholar
Liu, M., Jin, Y., Zhang, C., Leygraf, C., and Wen, L. (2015a). Density-functional theory investigation of Al pitting corrosion in electrolyte containing chloride ions. Appl. Surf. Sci. 357: 2028–2038.10.1016/j.apsusc.2015.09.180Suche in Google Scholar
Liu, S.Y., Liu, S., Li, D.J., Wang, S., Guo, J., and Shen, Y. (2015b). Ab initio atomistic thermodynamics study on the oxidation mechanism of binary and ternary alloy surfaces. J. Chem. Phys. 142: 064705.10.1063/1.4907718Suche in Google Scholar
López-Moreno, S. and Romero, A.H. (2015). Atomic and molecular oxygen adsorbed on (111) transition metal surfaces: Cu and Ni. J. Chem. Phys. 142: 154702.10.1063/1.4917259Suche in Google Scholar
Lozovoi, A.Y. and Alavi, A. (2007). Vibrational frequencies of CO on Pt(111) in electric field: a periodic DFT study. J. Electroanal. Chem. 607: 140–146.10.1016/j.jelechem.2007.02.030Suche in Google Scholar
Lozovoi, A.Y., Alavi, A., Kohanoff, J., and Lynden-Bell, R.M. (2001). Ab initio simulation of charged slabs at constant chemical potential. J. Chem. Phys. 115: 1661–1669.10.1063/1.1379327Suche in Google Scholar
Lü, B.-L., Chen, G.-Q., Zhou, W.-L., Su, H., and Liu, R. (2011). Ab initio study of influence of 3d alloying elements on corrosion properties of non-passivated nickel-base alloys. J. Nucl. Mater. 418: 286–291.10.1016/j.jnucmat.2011.07.015Suche in Google Scholar
Lu, G. and Kaxiras, E. (2005). Hydrogen embrittlement of aluminum: the crucial role of vacancies. Phys. Rev. Lett. 94: 155501.10.1103/PhysRevLett.94.155501Suche in Google Scholar
Lu, G., Zhang, Q., Kioussis, N., and Kaxiras, E. (2001). Hydrogen-enhanced local plasticity in aluminum: an ab initio study. Phys. Rev. Lett. 87: 095501.10.1103/PhysRevLett.87.095501Suche in Google Scholar
Lu, G., Orlikowski, D., Park, I., Politano, O., and Kaxiras, E. (2002). Energetics of hydrogen impurities in aluminum and their effect on mechanical properties. Phys. Rev. B 65: 064102.10.1103/PhysRevB.65.064102Suche in Google Scholar
Lu, M., Pao, P.S., Weir, T.W., Simmons, G.W., and Wei, R.P. (1981). Rate controlling processes for crack growth in hydrogen sulfide for an alsl 4340 steel. Metall. Trans. A 12: 805–811.10.1007/BF02648345Suche in Google Scholar
Luo, Z., Zhu, H., Ying, T., Li, D., and Zeng, X. (2018). First principles calculations on the influence of solute elements and chlorine adsorption on the anodic corrosion behavior of Mg (0001) surface. Surf. Sci. 672–673: 68–74.10.1016/j.susc.2018.02.002Suche in Google Scholar
Luo, Z., Xu, J., Wang, Y., Xie, T., Zhu, H., Ying, T., Li, D., Wang, L., Zeng, X., Wolverton, C., et al.. (2019). Theoretical analysis of the galvanic corrosion behavior of Mg-Ge binary alloy. J. Electrochem. Soc. 166: C421–C427.10.1149/2.1061913jesSuche in Google Scholar
Ma, H., Chen, X.-Q., Li, R., Wang, S., Dong, J., and Ke, W. (2017). First-principles modeling of anisotropic anodic dissolution of metals and alloys in corrosive environments. Acta Mater. 130: 137–146.10.1016/j.actamat.2017.03.027Suche in Google Scholar
Ma, H., Liu, M., Chen, W., Wang, C., Chen, X.-Q., Dong, J., and Ke, W. (2019). First-principles study on the effects of twin boundaries on anodic dissolution of Mg. Phys. Rev. Mater. 3: 053806.10.1103/PhysRevMaterials.3.053806Suche in Google Scholar
Ma, H., Wu, L., Liu, C., Liu, M., Wang, C., Li, D., Chen, X.-Q., Dong, J., and Ke, W. (2020). First-principles modeling of the hydrogen evolution reaction and its application in electrochemical corrosion of Mg. Acta Mater. 183: 377–389.10.1016/j.actamat.2019.11.025Suche in Google Scholar
Makov, G. and Payne, M.C. (1995). Periodic boundary conditions in ab initio calculations. Phys. Rev. B 51: 4014–4022.10.1103/PhysRevB.51.4014Suche in Google Scholar
Marcus, P. (1994). On some fundamental factors in the effect of alloying elements on passivation of alloys. Corrosion Sci. 36: 2155–2158.10.1016/0010-938X(94)90013-2Suche in Google Scholar
Marcus, P. and Protopopoff, E. (1993). Potential pH diagrams for sulfur and oxygen adsorbed on nickel in water at 25 and 300° C. J. Electrochem. Soc. 140: 1571.10.1149/1.2221604Suche in Google Scholar
Marcus, P. and Protopopoff, E. (1997). Potential-pH diagrams for sulfur and oxygen adsorbed on chromium in water. J. Electrochem. Soc. 144: 1586.10.1149/1.1837645Suche in Google Scholar
Mardirossian, N. and Head-Gordon, M. (2017). Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 115: 2315–2372.10.1080/00268976.2017.1333644Suche in Google Scholar
Marenich, A.V., Cramer, C.J., and Truhlar, D.G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113: 6378–6396.10.1021/jp810292nSuche in Google Scholar
Marks, L. (2018). Competitive chloride chemisorption disrupts hydrogen bonding networks: DFT, crystallography, thermodynamics, and morphological consequences. Corrosion 74: 295–311.10.5006/2555Suche in Google Scholar
Martinez, J.A., Yilmaz, D.E., Liang, T., Sinnott, S.B., and Phillpot, S.R. (2013). Fitting empirical potentials: challenges and methodologies. Curr. Opin. Solid State Mater. Sci. 17: 263–270.10.1016/j.cossms.2013.09.001Suche in Google Scholar
Masel, R.I. (1996). Principles of adsorption and reaction on solid surfaces. Wiley, Hoboken, NJ.Suche in Google Scholar
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T.A., and Hennig, R.G. (2014). Implicit solvation model for rdensity functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140: 084106.10.1063/1.4865107Suche in Google Scholar
Mattsson, A.E. and Jennison, D.R. (2002). Computing accurate surface energies and the importance of electron self-energy in metal/metal-oxide adhesion. Surf. Sci. 520: 1611–1618.10.1016/S0039-6028(02)02209-4Suche in Google Scholar
Maurice, V. and Marcus, P. (2018). Progress in corrosion science at atomic and nanometric scales. Prog. Mater. Sci. 95: 132–171.10.1016/j.pmatsci.2018.03.001Suche in Google Scholar
Maurice, V., Strehblow, H.-H., and Marcus, P. (2000). In situ STM study of the initial stages of oxidation of Cu(111) in aqueous solution. Surf. Sci. 458: 185–194.10.1016/S0039-6028(00)00442-8Suche in Google Scholar
McCafferty, E. (2000). Graph theory and the passivity of iron‐chromium binary alloys. Electrochem. Solid State Lett. 3: 28–30.10.1149/1.1390948Suche in Google Scholar
McCright, R.D. (1977). In: Staehle, R.W. (Ed.), Stress corrosion cracking and hydrogen embrittlement of iron base alloys. France: Unieux-Firminy, pp. 306–325.Suche in Google Scholar
McEniry, E.J., Hickel, T., and Neugebauer, J. (2018). Ab initio simulation of hydrogen-induced decohesion in cementite-containing microstructures. Acta Mater. 150: 53–58.10.1016/j.actamat.2018.03.005Suche in Google Scholar
Melchers, R.E. (2019). Predicting long-term corrosion of metal alloys in physical infrastructure. NPJ Mater. Deg. 3: 4.10.1038/s41529-018-0066-xSuche in Google Scholar
Mendoza, A.R. and Corvo, F. (2000). Outdoor and indoor atmospheric corrosion of non-ferrous metals. Corrosion Sci. 42: 1123–1147.10.1016/S0010-938X(99)00135-3Suche in Google Scholar
Miertuš, S., Scrocco, E., and Tomasi, J. (1981). Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55: 117–129.10.1016/0301-0104(81)85090-2Suche in Google Scholar
Mills, G., Gordon, M.S., and Metiu, H. (2003). Oxygen adsorption on Au clusters and a rough Au(111) surface: the role of surface flatness, electron confinement, excess electrons, and band gap. J. Chem. Phys. 118: 4198.10.1063/1.1542879Suche in Google Scholar
Mondal, S.K. and Taylor, S.R. (2014). The identification and characterization of organic corrosion inhibitors: correlation of a computational model with experimental results. J. Electrochem. Soc. 161: C476–C485.10.1149/2.0191410jesSuche in Google Scholar
Mortensen, J.J., Kaasbjerg, K., Frederiksen, S.L., Norskov, J.K., Sethna, J.P., and Jacobsen, K.W. (2005). Bayesian error estimation in density-functional theory. Phys. Rev. Lett. 95: 216401.10.1103/PhysRevLett.95.216401Suche in Google Scholar
Motta, A., Gaigeot, M.P., Costa, D. (2012). AIMD evidence of inner sphere adsorption of glycine on a stepped (101) boehmite AlOOH surface. J. Phys. Chem. C 116: 23418–23427.10.1021/jp307565pSuche in Google Scholar
Mueller, T., Hernandez, A., and Wang, C. (2020). Machine learning for interatomic potential models. J. Chem. Phys. 152: 050902.10.1063/1.5126336Suche in Google Scholar
Muralidharan, V.S. (2002). An insight into the origin of corrosion potential – a critical review. Bull. Electrochem. 18: 179–186.Suche in Google Scholar
Nazarov, R., Hickel, T., and Neugebauer, J. (2010). First-principles study of the thermodynamics of hydrogen–vacancy interaction in fcc iron. Phys. Rev. B 82.10.1103/PhysRevB.82.224104Suche in Google Scholar
Norskov, J.K., Rossmeisl, J., Logadottir, A., Lindqvist, L., Kitchin, J.R., Bligaard, T., and Jonsson, H. (2004). Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108: 17886–17892.10.1021/jp047349jSuche in Google Scholar
Norskov, J.K., Abild-Pederson, F., Studt, F., and Bligaard, T. (2010). Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 108: 937–943.10.1073/pnas.1006652108Suche in Google Scholar
Oberdorfer, C. and Windl, W. (2019). Bond-order bond energy model for alloys. Acta Mater. 179: 406–413.10.1016/j.actamat.2019.08.048Suche in Google Scholar
Oesch, S. and Faller, M. (1997). Environmental effects on materials: the effect of the air pollutants SO2, NO2, NO and O3 on the corrosion of copper, zinc, and aluminium. A short literature survey and results of laboratory exposures. Corrosion Sci. 39: 1505–1530.10.1016/S0010-938X(97)00047-4Suche in Google Scholar
Ohwaki, T. and Yamashita, K. (2001). A DFT study of electric field effects on proton transfer reactions at H+(H2O)2/Pt(111) and Ag(111). J. Electroanal. Chem. 504: 71–77.10.1016/S0022-0728(01)00430-2Suche in Google Scholar
Olatunji-Ojo, O. and Taylor, C.D. (2013). Changes in valence, coordination and reactivity that occur upon oxidation of fresh metal surfaces. Phil. Mag. 93: 4286–4310.10.1080/14786435.2013.827801Suche in Google Scholar
Olsson, P.A.T., Kese, K., and Holston, A.-M.A. (2015). On the role of hydrogen filled vacancies on the embrittlement of zirconium: an ab initio investigation. J. Nucl. Mater. 467: 311–319.10.1016/j.jnucmat.2015.09.056Suche in Google Scholar
Organ, L., Scully, J.R., Mikhailov, A.S., and Hudson, J.L. (2005). A spatiotemporal model of interactions among metastable pits and the transition to pitting corrosion. Electrochim. Acta 51: 225–241.10.1016/j.electacta.2005.04.018Suche in Google Scholar
Panchenko, A., Koper, M.T.M., Shubina, T.E., Mitchell, S.J., and Roduner, E. (2004). Ab initio calculations of intermediates of oxygen reduction on low-index platinum surfaces. J. Electrochem. Soc. 151: A2016–A2027.10.1149/1.1809586Suche in Google Scholar
Parr, R.G. and Yang, W. (1984). Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 106: 4049–4050.10.1021/ja00326a036Suche in Google Scholar
Peljhan, S. and Kokalj, A. (2011a). DFT study of gas-phase adsorption of benzotriazole on Cu(111), Cu(100), Cu(110), and low coordinated defects thereon. Phys. Chem. Chem. Phys. 13: 20408–20417.Suche in Google Scholar
Peljhan, S. and Kokalj, A. (2011b). DFT study of gas phase adsorption of benzotriazole on Cu(111) Cu(100), Cu(110) and low coordinated defects thereon. Phys. Chem. Chem. Phys. 13: 20408–20417.10.1039/c1cp21873eSuche in Google Scholar
Perry, W. (Ed.). (2008). National Academy of Sciences Grand Challenges for Engineering. National Academy Press.Suche in Google Scholar
Pletnev, M.A., Morozov, S.G., and Alekseev, V.P. (2000). Peculiar effect of chloride ions on the anodic dissolution of iron in solutions of various acidity. Protect. Met. 36: 202–208.10.1007/BF02758391Suche in Google Scholar
Protopopoff, E. and Marcus, P. (1987). Effect of chemisorbed sulfur on the electrochemical hydrogen adsorption and recombination reactions on Pt(111). J. Vac. Sci. Technol. 5: 944.10.1116/1.574303Suche in Google Scholar
Protopopoff, E. and Marcus, P. (1988). Poisoning of the cathodic hydrogen evolution reaction by sulfur chemisorbed on platinum (110). J. Electrochem. Soc. 135: 3073.10.1149/1.2095492Suche in Google Scholar
Protopopoff, E. and Marcus, P. (2003). Potential-pH diagrams for sulfur and hydroxyl adsorbed on copper surfaces in water containing sulfides, sulfites or thiosulfates. Corrosion Sci. 45: 1191–1201.10.1016/S0010-938X(02)00210-XSuche in Google Scholar
Protopopoff, E. and Marcus, P. (2005). Potential–pH diagrams for hydroxyl and hydrogen adsorbed on a copper surface. Electrochim. Acta 51: 408–417.10.1016/j.electacta.2005.04.036Suche in Google Scholar
Protopopoff, E. and Marcus, P. (2012). Potential–pH diagram for sulfur and hydroxyl adsorbed on silver in water containing sulfides. Electrochim. Acta 63: 22–27.10.1016/j.electacta.2011.12.030Suche in Google Scholar
Punckt, C., Bolscher, M., Rotermund, H.H., Mikhailov, A.S., Organ, L., Budiansky, N., Scully, J.R., and Hudson, J.L. (2004). Sudden onset of pitting corrosion on stainless steel as a critical phenomenon. Science 305: 1133–1136.10.1126/science.1101358Suche in Google Scholar
Qiu, Y., Thomas, S., Gibson, M.A., Fraser, H.L., and Birbilis, N. (2017). Corrosion of high entropy alloys. NPJ Mater. Deg. 1: 15.10.1038/s41529-017-0009-ySuche in Google Scholar
Quinlan, J.R. (1986). Induction of decision trees. Mach. Learn. 1: 81–106.10.1007/BF00116251Suche in Google Scholar
Rice, J.R. and Wang, J.-S. (1989). Embrittlement of interfaces by solute segregation. Mater. Sci. Eng. 107: 23–40.10.1016/0921-5093(89)90372-9Suche in Google Scholar
Riley, A.M., Wells, D.B., and Williams, D.E. (1991). Initiation events for pitting corrosion of stainless steel? Corrosion Sci. 32: 1307–133.10.1016/0010-938X(91)90050-YSuche in Google Scholar
Rossmeisl, J., Norskov, J.K., Taylor, C.D., Janik, M.J., and Neurock, M. (2006). Calculated phase diagrams for the electrochemical oxidation and reduction of water over Pt(111). J. Phys. Chem. B 110: 21833–21839.10.1021/jp0631735Suche in Google Scholar
Saal, J.E., Kirklin, S., Aykol, M., Meredig, B., and Wolverton, C. (2013). Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65: 1501–1509.10.1007/s11837-013-0755-4Suche in Google Scholar
Samin, A.J. and Taylor, C.D. (2017a). A computational thermodynamic and kinetic study of chlorine binding to the Zr(0001) surface. Colloids Surf. A 539: 92–100.10.1016/j.colsurfa.2017.11.075Suche in Google Scholar
Samin, A.J. and Taylor, C.D. (2017b). A first principles investigation of the oxygen adsorption on Zr(0001) surface using cluster expansions. Appl. Surf. Sci. 423: 1035–1044.10.1016/j.apsusc.2017.06.210Suche in Google Scholar
Samin, A.J. and Taylor, C.D. (2018a). An analysis of limitng cases for the metal oxide film growth kinetics using an oxygen defects model accounting for transport and interfacial reactions. J. Non-Equil. Thermo. 43: 317–326.10.1515/jnet-2018-0018Suche in Google Scholar
Samin, A.J. and Taylor, C.D. (2018b). A combined density functional theory and Monte Carlo investigation of the competitive adsorption of atomic oxygen and chlorine to the Ni (111) surface. J. Electrochem. Soc. 165: C302–C309.10.1149/2.0031807jesSuche in Google Scholar
Samin, A.J. and Taylor, C.D. (2018c). First-principles investigation of surface properties and adsorption of oxygen on Ni–22Cr and the role of molybdenum. Corrosion Sci. 134: 103–111.10.1016/j.corsci.2018.02.017Suche in Google Scholar
Samin, A.J. and Taylor, C.D. (2018d). A one-dimensional time-dependent model for studying oxide film growth on metal surfaces. J. Appl. Phys. 123: 245303.10.1063/1.5024211Suche in Google Scholar
Sanchez, J.M., Ducastelle, F., and Gratias, D. (1984). Generalized cluster description of multicomponent systems. Physica A Stat. Mech. 128: 334–350.10.1016/0378-4371(84)90096-7Suche in Google Scholar
Saraby-Reintjes, A. (1985). Theory of competitive adsorption and its application to the anodic dissolution of nickel and other iron group metals – 1. Active dissolution in acid solution under steady state conditions. Electrochim. Acta 30: 387–401.10.1016/0013-4686(85)80200-0Suche in Google Scholar
Schleder, G.R., Padilha, A.C.M., Acosta, C.M., Costa, M., and Fazzio, A. (2019). From DFT to machine learning: recent approaches to materials science–a review. J. Phys. Mater. 2: 032001.10.1088/2515-7639/ab084bSuche in Google Scholar
Schmidt, D.J., Chen, W., Wolverton, C., and Schneider, W.F. (2012). Performance of cluster expansions of coverage-dependent adsorption of atomic oxygen on Pt(111). J. Chem. Theor. Comput. 8: 264–273.10.1021/ct200659cSuche in Google Scholar
Schmuki, P. (2002). From Bacon to barriers: a review on the passivity of metals and alloys. J. Solid State Electrochem. 6: 345–364.10.1007/s100080100219Suche in Google Scholar
Scivetti, I. and Persson, M. (2013). The electrostatic interaction of an external charged system with a metal surface: a simplified density functional theory approach. J. Phys. Condens. Matter 25: 355006.10.1088/0953-8984/25/35/355006Suche in Google Scholar
Scully, J.R. (2009). Role of minor alloying elements in the corrosion of selected amorphous alloys. National Science Foundation, Alexandria, VA.Suche in Google Scholar
Scully, J.R. and Moran, P.J. (1984). Influence of strain on the environmental hydrogen-assisted cracking of a high-strength steel in sodium chloride solution. Corrosion 44: 176–185.10.5006/1.3583922Suche in Google Scholar
Sedriks, A.J. (1996). Corrosion of stainless steels. New York, NY: John Wiley & Sons.Suche in Google Scholar
Seyeux, A., Maurice, V., Klein, L.H., and Marcus, P. (2005). In situ scanning tunneling microscopy study of the initial stages of growth and of the structure of the passive film on Ni(111) in 1 mM NaOH(aq). J. Solid State Electrochem. 9: 337–346.10.1007/s10008-004-0627-5Suche in Google Scholar
Seyeux, A., Maurice, V., and Marcus, P. (2013). Oxide film growth kinetics on metals and alloys. J. Electrochem. Soc. 160: C189–C196.10.1149/2.036306jesSuche in Google Scholar
Sharland, S.M. (1987). A review of the theoretical modeling of crevice and pitting corrosion. Corrosion Sci. 27: 289–323.10.1016/0010-938X(87)90024-2Suche in Google Scholar
Sherman, Q.C., Voorhees, P.W., and Marks, L.D. (2019). Thermodynamics of solute capture during the oxidation of multicomponent metals. Acta Mater. 181: 584–594.10.1016/j.actamat.2019.09.043Suche in Google Scholar
Shibata, T. (2007). Passivity breakdown and stress corrosion cracking of stainless steel. Corrosion Sci. 49: 20–30.10.1016/j.corsci.2006.05.031Suche in Google Scholar
Skúlason, E. (2015). Modeling electrochemical reactions at the solid-liquid interface using density functional calculations. Proc. Comput. Sci. 51: 1887–1896.10.1016/j.procs.2015.05.431Suche in Google Scholar
Souvi, S.M.O., Badawi, M., Paul, J.-F., Cristol, S., and Cantrel, L. (2013). A DFT study of the hematite surface state in the presence of H2, H2O and O2. Surf. Sci. 610: 7–15.10.1016/j.susc.2012.12.012Suche in Google Scholar
Stewart, J. and Williams, D.E. (1992). The initiation of pitting corrosion on austenitic stainless steel: on the role and importance of sulphide inclusions. Corrosion Sci. 33: 457–474.10.1016/0010-938X(92)90074-DSuche in Google Scholar
Stowasser, R. and Hoffmann, R. (1999). What do the Kohn−Sham orbitals and eigenvalues mean? J. Am. Chem. Soc. 121: 3414–3420.10.1021/ja9826892Suche in Google Scholar
Su, N.Q. and Xu, X. (2017). Development of new density functional approximations. Annu. Rev. Phys. Chem. 68: 155–82.10.1146/annurev-physchem-052516-044835Suche in Google Scholar
Sun, S., Geng, Y., Tian, L., Chen, S., Yan, Y., and Hu, S. (2012). Density functional theory study of imidazole, benzimidazole and 2-mercaptobenzimidazole adsorption onto clean Cu(111) surface. Corrosion Sci. 63: 140–147.10.1016/j.corsci.2012.05.024Suche in Google Scholar
Tang, Z., Huang, L., He, W., and Liaw, P.K. (2014). Alloying and processing effects on the aqueous corrosion behavior of high-entropy alloys. Entropy 16: 895–911.10.3390/e16020895Suche in Google Scholar
Tateyama, Y. and Ohno, T. (2003). Stability and cauterization of hydrogen-vacancy complexes in α-Fe: an ab initio study. Phys. Rev. B 67: 174105.10.1103/PhysRevB.67.174105Suche in Google Scholar
Taylor, C.D. (2009). The transition from metal-metal bonding to metal-solvent interactions during a dissolution event as assessed from electronic structure. Chem. Phys. Lett. 469: 99–103.10.1016/j.cplett.2008.12.058Suche in Google Scholar
Taylor, C.D. (2012a). Atomistic modeling of corrosion events at the interface between a metal and its environment. Int. J. Corrosion 2012: 1–13, .10.1155/2012/204640Suche in Google Scholar
Taylor, C.D. (2012b). Predictions of surface electrochemistry of saturated and alkaline NH4Cl solutions interacting with Fe(110) from ab initio calculations. Corrosion 68: 591–599.10.5006/0411Suche in Google Scholar
Taylor, C.D. (2013). Oxygen induced transformations of the δ-Pu(111) surface. Surf. Sci. 618: 101–108.10.1016/j.susc.2013.08.016Suche in Google Scholar
Taylor, C.D. (2014). Oxidation of technetium metal as simulated by first principles. J. Phys. Chem. C 118: 10017–10023.10.1021/jp411976sSuche in Google Scholar
Taylor, C.D. (2015). Corrosion informatics: an integrated approach to modelling corrosion. Corrosion Eng. Sci. Technol. 50: 490–508.10.1179/1743278215Y.0000000012Suche in Google Scholar
Taylor, C.D. (2016). A first-principles surface reaction kinetic model for hydrogen evolution under cathodic and anodic conditions on magnesium. J. Electrochem. Soc. 163: C602–C608.10.1149/2.1171609jesSuche in Google Scholar
Taylor, C.D. and Liu, X.-Y. (2013). Investigation of structure and composition control over active dissolution of Fe–Tc binary metallic waste forms by off-lattice kinetic Monte Carlo simulations. J. Nucl. Mater. 434: 382–388.10.1016/j.jnucmat.2012.11.039Suche in Google Scholar
Taylor, C.D. and Neurock, M. (2005). Theoretical insights into the structure and reactivity of the aqueous/metal interface. Curr. Opin. Solid State Mater. Sci. 9: 49–65.10.1016/j.cossms.2006.03.007Suche in Google Scholar
Taylor, C., Kelly, R.G., and Neurock, M. (2006a). First-principles calculations of the electrochemical reactions of water at an immersed Ni(111)/H2O interface. J. Electrochem. Soc. 153: E207.10.1149/1.2357721Suche in Google Scholar
Taylor, C.D., Wasileski, S.A., Filhol, J.-S., and Neurock, M. (2006b). First principles reaction modeling of the electrochemical interface: consideration and calculation of a tunable surface potential from atomic and electronic structure. Phys. Rev. B 73: 165402.10.1103/PhysRevB.73.165402Suche in Google Scholar
Taylor, C., Kelly, R.G., and Neurock, M. (2007a). Theoretical analysis of the nature of hydrogen at the electrochemical interface between water and a Ni(111) single-crystal electrode. J. Electrochem. Soc. 154: F55–F64.10.1149/1.2431326Suche in Google Scholar
Taylor, C.D., Janik, M.J., Neurock, M., and Kelly, R.G. (2007b). Ab initio simulations of the electrochemical activation of water. Mol. Simulat. 33: 429–436.10.1080/08927020601154207Suche in Google Scholar
Taylor, C.D., Kelly, R.G., and Neurock, M. (2007c). First-principles prediction of equilibrium potentials for water activation by a series of metals. J. Electrochem. Soc. 154: F217.10.1149/1.2783780Suche in Google Scholar
Taylor, C.D., Neurock, M., and Scully, J.R. (2011). A first-principles model for hydrogen uptake promoted by sulfur on Ni(111). J. Electrochem. Soc. 158: F36.10.1149/1.3536470Suche in Google Scholar
Taylor, C.D., Hernandez, S.C., Francis, M.F., Schwartz, D.S., and Ray, A.K. (2013). Hydrogen trapping in delta-Pu: insights from electronic structure calculations. J. Phys. Condens. Matter 25: 265001.10.1088/0953-8984/25/26/265001Suche in Google Scholar
Taylor, C.D., Francis, M.F., and Schwartz, D.S. (2014). Thermodynamics, structure and charge state of hydrogen-vacancy complexes in delta-plutonium. Phys. Rev. B 89: 214114.10.1103/PhysRevB.89.214114Suche in Google Scholar
Taylor, C.D., Chandra, A., Vera, J., and Sridhar, N. (2015a). Design and prediction of corrosion inhibitors from quantum chemistry. J. Electrochem. Soc. 162: C347–C353.Suche in Google Scholar
Taylor, C.D., Chandra, A., Vera, J., and Sridhar, N. (2015b). Design and prediction of corrosion inhibitors from quantum chemistry. II. A general framework for prediction of effective oil/water partition coefficients and speciation from quantum chemistry. J. Electrochem. Soc. 162: C347–C353.10.1149/2.0691507jesSuche in Google Scholar
Taylor, C.D., Chandra, A., Vera, J., and Sridhar, N. (2015c). A multiphysics perspective on mechanistic models for chemical corrosion inhibitor performance. J. Electrochem. Soc. 162: C369–C375.10.1149/2.0801507jesSuche in Google Scholar
Taylor, C., Kurapati, Y., and Mondal, S. (2018a). Interaction of model inhibitor compounds with minimalist cluster representations of hydroxyl terminated metal oxide surfaces. Metals 8: 81.10.3390/met8020081Suche in Google Scholar
Taylor, C.D., Li, S., and Samin, A.J. (2018b). Oxidation versus salt-film formation: competitive adsorption on a series of metals from first-principles. Electrochim. Acta 269: 93–101.10.1016/j.electacta.2018.02.150Suche in Google Scholar
Taylor, C.D., Lu, P., Saal, J., Frankel, G.S., and Scully, J.R. (2018c). Integrated computational materials engineering of corrosion resistant alloys. NPJ Mater. Deg. 2: 6.10.1038/s41529-018-0027-4Suche in Google Scholar
Tehranchi, A. and Curtin, W.A. (2017). Atomistic study of hydrogen embrittlement of grain boundaries in nickel: I. Fracture. J. Mech. Phys. Solid. 101: 150–165.10.1016/j.jmps.2017.01.020Suche in Google Scholar
Tehranchi, A., Zhou, X., and Curtin, W.A. (2020). A decohesion pathway for hydrogen embrittlement in nickel: mechanism and quantitative prediction. Acta Mater. 185: 98–109.10.1016/j.actamat.2019.11.062Suche in Google Scholar
Teus, S.M., Shivanyuk, V.N., Shanina, B.D., and Gavriljuk, V.G. (2007). Effect of hydrogen on electronic structure of fcc iron in relation to hydrogen embrittlement of austenitic steels. Phys. Status Solidi 204: 4249–4258.10.1002/pssa.200723249Suche in Google Scholar
Thodla, R., Chandra, A., Li, X., Taylor, C.D., Ke, H., and Sridhar, N. (2020a). Hydrogen embrittlement of 718 under cathodic polarization. Corrosion Sci. 165: 108361.10.1016/j.corsci.2019.108361Suche in Google Scholar
Thodla, R., Sridhar, N., Amaya, H., Fahimi, B., and Taylor, C.D. (2020b). Hydrogen embrittlement of low alloy steels under cathodic polarization. Corrosion 76: 299–311.10.5006/3240Suche in Google Scholar
Thodla, R., Viswanathan, G.B., Amaya, H., Fahimi, B., and Taylor, C.D. (2021). Crack growth behavior of 725 in seawater under cathodic polarization. Mater. Sci. Eng. 2021: 141112. https://doi.org/10.1016/j.msea.2021.141112.Suche in Google Scholar
Tian, F., Jinnouchi, R., and Anderson, A.B. (2009). How potentials of zero charge and potentials for water oxidation to OH(ads) on Pt(111) electrodes vary with coverage. J. Phys. Chem. C 113: 17484–17492.10.1021/jp905377dSuche in Google Scholar
Tkatchenko, A., Romaner, L., Hofmann, O.T., Zojer, E., Ambrosch-Draxl, C., and Scheffler, M. (2010). Van der Waals interactions between organic adsorbates and at organic/inorganic interfaces. MRS Bull. 35: 435–442.10.1557/mrs2010.581Suche in Google Scholar
Tkatchenko, A., DiStasio, R.A., Car, R., and Scheffler, M. (2012). Accurate and efficient method for many-body van der Waals interactions. Phys. Rev. Lett. 108.10.1103/PhysRevLett.108.236402Suche in Google Scholar
Todorova, M. and Neugebauer, J. (2014). Extending the concept of defect chemistry from semiconductor physics to electrochemistry. Phys. Rev. Appl. 1: 014001.10.1103/PhysRevApplied.1.014001Suche in Google Scholar
Todorova, M. and Neugebauer, J. (2015). Identification of bulk oxide defects in an electrochemical environment. Farad. Disc. 180: 97–112.10.1039/C4FD00238ESuche in Google Scholar
Tomashov, N.D. (1961). Development of the electrochemical theory of metallic corrosion. Corrosion 20: 7–14.10.5006/0010-9312-20.1.7tSuche in Google Scholar
Udagawa, Y., Yamaguchi, M., Abe, H., Sekimura, N., and Fuketa, T. (2010). Ab initio study on plane defects in zirconium-hydrogen solid solution and zirconium hydride. Acta Mater. 58: 3927–3938.10.1016/j.actamat.2010.03.034Suche in Google Scholar
Van der Ven, A. and Ceder, G. (2004). The thermodynamics of decohesion. Acta Mater. 52: 1223–1235.10.1016/j.actamat.2003.11.007Suche in Google Scholar
Vosta, J. and Eliasek, J. (1971). Study on corrosion inhibition from aspect of quantum chemistry. Corrosion Sci. 11: 223–229.10.1016/S0010-938X(71)80137-3Suche in Google Scholar
Walczak, M.S., Morales-Gil, P., and Lindsay, R. (2019). Determining Gibbs energies of adsorption from corrosion inhibition efficiencies: is it a reliable approach? Corrosion Sci. 155: 182–185.10.1016/j.corsci.2019.04.040Suche in Google Scholar
Waldman, J. (2015). Rust: the longest war. Simon and Schuster, Delran, NJ.Suche in Google Scholar
Wang, C., Chi, M.F., Wang, G.F., van der Vliet, D., Li, D.G., More, K., Wang, H.H., Schlueter, J.A., Markovic, N.M., and Stamenkovic, V.R. (2011). Correlation between surface chemistry and electrocatalytic properties of monodisperse PtxNi1−x nanoparticles. Adv. Funct. Mater. 21: 147–152.10.1002/adfm.201001138Suche in Google Scholar
Wang, C., Li, D.G., Chi, M.F., Pearson, J., Rankin, R.B., Greeley, J., Duan, Z.Y., Wang, G.F., van der Vliet, D., More, K.L., et al.. (2012). Rational development of ternary alloy electrocatalysts. J. Phys. Chem. Lett. 3: 1668–1673.10.1021/jz300563zSuche in Google Scholar
Wang, F.-H., Liu, S.-Y., Shang, J.-X., Zhou, Y.-S., Li, Z., and Yang, J. (2008). Oxygen adsorption on Zr(0001) surfaces: density functional calculations and a multiple-layer adsorption model. Surf. Sci. 602: 2212–2216.10.1016/j.susc.2008.04.033Suche in Google Scholar
Wang, X.G., Smith, J.R., and Evans, A.G. (2006). Phase diagram for the Ni∕Al2O3 interface and relationships to adhesion. Phys. Rev. B 74: 081403.10.1103/PhysRevB.74.081403Suche in Google Scholar
Wei, R.P. (1980). Rate controlling processes and crack growth response. In: Bernstein, I.M., and Thompson, A.W. (Eds.), Hydrogen effects in metals, p. 677, Warrendale, PA.Suche in Google Scholar
Wei, R.P. and Gangloff, R.P. (1989). Environmentally assisted crack growth in structural alloys: perspectives and new directions. Fracture mechanics: perspectives and directions. West Conshohocken, PA: ASTM.10.1520/STP18827SSuche in Google Scholar
Wei, R.P., Pao, P.S., Hart, R.G., Weir, T.W., and Simmons, G.W. (1980). Fracture mechanics and surface chemistry studies of fatigue crack growth in an aluminum alloy. Metall. Mater. Trans. 11: 151–158.10.1007/BF02700451Suche in Google Scholar
Wen, Y.-H., Chen, L.-Q., and Hawk, J.A. (2012). Phase-field modeling of corrosion kinetics under dual oxidants. Model. Simulat. Mater. Sci. Eng. 20: 035013.10.1088/0965-0393/20/3/035013Suche in Google Scholar
Williams, D.E., Newman, R.C., Song, G., and Kelly, R.G. (1991). Passivity breakdown and pitting corrosion of binary alloys. Nature 350: 216–219.10.1038/350216a0Suche in Google Scholar
Williams, G., Birbilis, N., and McMurray, H.N. (2013). The source of hydrogen evolved from a magnesium anode. Electrochem. Commun. 36: 1–5.10.1016/j.elecom.2013.08.023Suche in Google Scholar
Williams, K.S., Labukas, J.P., Rodriguez-Santiago, V., and Andzelm, J.W. (2015). First principles modeling of water dissociation on Mg(0001) and development of a Mg surface Pourbaix diagram. Corrosion 71: 209–223.10.5006/1322Suche in Google Scholar
Wodtke, A., Matsiev, D., and Auerbach, D. (2008). Energy transfer and chemical dynamics at solid surfaces: the special role of charge transfer. Prog. Surf. Sci. 83: 167–214.10.1016/j.progsurf.2008.02.001Suche in Google Scholar
Yang, W., Parr, R.G., and Pucci, R. (1984). Electron density, Kohn–Sham Frontier orbitals, and Fukui functions. J. Chem. Phys. 81: 2862.10.1063/1.447964Suche in Google Scholar
Yeh, K.-Y., Wasileski, S.A., and Janik, M.J. (2009). Electronic structure models of oxygen adsorption at the solvated, electrified Pt(111) interface. Phys. Chem. Chem. Phys. 11: 10108–10117.10.1039/b909233aSuche in Google Scholar
Yu, J., Rosso, K.M., and Bruemmer, S.M. (2012). Charge and ion transport in NiO and aspects of Ni oxidation from first principles. J. Phys. Chem. C 116: 1948–1954.10.1021/jp208080vSuche in Google Scholar
Yu, X.-X. and Marks, L.D. (2018). Combining the physics of metal/oxide heterostructure, interface dipole, band bending, crystallography, and surface state to understand heterogeneity contrast in oxidation and corrosion. Corrosion 75: 152–166.10.5006/2794Suche in Google Scholar
Yu, X.-x., Gulec, A., Cwalina, K.L., Scully, J.R., and Marks, L.D. (2019). New insights on the role of chloride during the onset of local corrosion: TEM, APT, surface energy, and morphological instability. Corrosion 75: 616–627.10.5006/2991Suche in Google Scholar
Yu, X.X., Gulec, A., Sherman, Q., Cwalina, K.L., Scully, J.R., Perepezko, J.H., Voorhees, P.W., and Marks, L.D. (2018). Nonequilibrium solute capture in passivating oxide films. Phys. Rev. Lett. 121: 145701.10.1103/PhysRevLett.121.145701Suche in Google Scholar
Yuwono, J.A., Birbilis, N., Taylor, C.D., Williams, K.S., Samin, A.J., and Medhekar, N.V. (2019). Aqueous electrochemistry of the magnesium surface: thermodynamic and kinetic profiles. Corrosion Sci. 147: 53–68.10.1016/j.corsci.2018.10.014Suche in Google Scholar
Zamora, R.J., Nair, A.K., Hennig, R.G., and Warner, D.H. (2012). Ab initio prediction of environmental embrittlement at a crack tip in aluminum. Phys. Rev. B 86: 060101.10.1103/PhysRevB.86.060101Suche in Google Scholar
Zunger, A., Wei, S.H., Ferreira, L.G., and Bernard, J.E. (1990). Special quasirandom structures. Phys. Rev. Lett. 65: 353–356.10.1103/PhysRevLett.65.353Suche in Google Scholar
© 2021 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this issue
- Reviews
- Investigations of the intrinsic corrosion and hydrogen susceptibility of metals and alloys using density functional theory
- Integration of green nanotechnology with silica for corrosion inhibition
- Analysis of internal corrosion of supercritical CO2 pipeline
- A review of thermal spray coatings for protection of steels from degradation in coal fired power plants
- Original articles
- The importance of phase composition for corrosion resistance and thermal oxidation behavior of gas-borided layer produced on nickel alloy
- Tribological behavior of stainless steel in sulfuric acid in the presence of Thymus zygis subsp. gracilis essential oil: experimental and quantum chemical studies
Artikel in diesem Heft
- Frontmatter
- In this issue
- Reviews
- Investigations of the intrinsic corrosion and hydrogen susceptibility of metals and alloys using density functional theory
- Integration of green nanotechnology with silica for corrosion inhibition
- Analysis of internal corrosion of supercritical CO2 pipeline
- A review of thermal spray coatings for protection of steels from degradation in coal fired power plants
- Original articles
- The importance of phase composition for corrosion resistance and thermal oxidation behavior of gas-borided layer produced on nickel alloy
- Tribological behavior of stainless steel in sulfuric acid in the presence of Thymus zygis subsp. gracilis essential oil: experimental and quantum chemical studies