In vitro cytotoxicity of hydrazones, pyrazoles, pyrazolo-pyrimidines, and pyrazolo-pyridine synthesized from 6-substituted 3-formylchromones
-
Elier Galarraga
 ,
Neudo Urdaneta
,
Neudo Urdaneta

Abstract
Pyrazoles 4a–f, hydrazones 5a–c and 6a–c, pyrazolo[1,5-a]pyrimidines 7a, b, and pyrazolo[3,4-b]pyridine 8 were prepared in good yields (80–95 %) from the reaction of 6-substituted (H, Me, F) 3-formylchromones 1a–c with N-substituted hydrazines 2a–c and aminopyrazole 3. The cytotoxicity of the synthesized compounds was assessed through the brine shrimp lethality assay. IC50 values were between 80 and 300 μm. Fluorine substitution decreased IC50 values.
1 Introduction
Benzopyrone group-based compounds, such as chromones, are widely recognized as an important class of biological active substances from both natural and synthetic origins [1–3]. Numerous studies show their wide range of activities, such as antioxidant [4], antimicrobial [5, 6], antiviral [7, 8] and antitumor [9, 10]. The chromone nucleus is also found within the chemical structure of flavonoids, an important group of naturally occurring substances that are of current interest because of their cytotoxic activity [11, 12].
Among the functionalized chromones, 3-formylchromone is a highly reactive compound, and is used as a starting material in many reactions due to the presence of three electron-deficient centers at C-2, C-4, and the C-3 formyl group. Reaction of the -CHO group with nitrogen nucleophiles, such as hydrazine and aminopyrazole derivatives, has led to the formation of a variety of molecules that have been studied in detail for being of interest to drug discovery [13–21].
π-Electron-rich compounds like chromone-3-carboxyaldehydes react with aromatic primary hydrazines (1:1 molar ratio) mainly at the formyl group by a straight forward 1,2-addition to form the corresponding hydrazone [22–26]. On prolonged heating, a pyrazole-type structure is produced by a 1,4-addition reaction accompanied by pyrone ring-opening followed by recyclization and proton transfer. Meanwhile, the reaction of 3-formylchromone with equimolar quantities of aminopyrazole derivatives affords only pyrazolo[1,5-a]pyrimidines, which is formed by the abovementioned cyclization process of an imine intermediate [27, 28].
The brine shrimp lethality assay (BSLA) is a preliminary standard bioassay, which is based on the use of a simple zoologic organism, and is used to detect toxicity of substances. BSLA is an easily performed and cost-effective test. It showed positive correlations with specific cytotoxic assays employing KB [29], 9KB [30], and 9PS [31] cancer cell lines and it was also suitable in predicting trypanocidal [32] and pesticidal [33] activities.
The synthesis of nitrogenated derivatives of 3-formylchromone is an important chemical issue; it may also contribute to the identification of new compounds with biological activity. In this work, we present the synthesis and characterization of new pyrazoles 4d–f, hydrazones 5b, 5c, 6a–c and pyrazolo[3,4-b]pyridine 8, along with the known pyrazoles 4a–c, hydrazone 5a and pyrazolo[1,5-a]pyrimidines 7a and 7b. We also report on the in vitro toxicity of the obtained compounds against brine shrimps (Artemia salina).
2 Results and discussion
2.1 Synthesis
The experimental procedure led to the synthesis of pyrazoles 4a–f, hydrazones 5a–c and 6a–c, pyrazolo[1,5-a]pyrimidines 7a, 7b, and pyrazolo[3,4-b]pyridine 8. Derivatives 4d–f, 5b, 5c, 6a–c, and 8 are newly synthesized compounds. The products were obtained from the reaction between equimolar quantities of 3-formylchromone derivatives 1a–c, with the corresponding hydrazines 2a–c or aromatic amine 3 in anhydrous THF. The pyrazolo[3,4-b]pyridine 8 was obtained in two steps: the reaction of 1b with 3 produced a precipitate, which was then refluxed using AcOH as solvent in presence of I2 (Scheme 1). All reactions were monitored by TLC and obtained in good yields (80–95 %). The new compounds were fully characterized using NMR, IR, and HRMS methods.
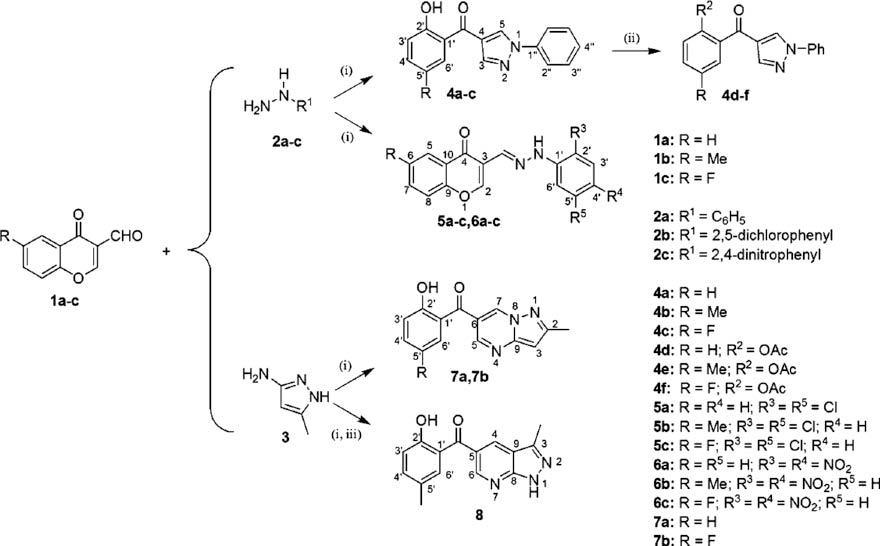
Reagents and conditions: (i) THF, reflux (1–2 h); (ii) Ac2O/H2SO4, reflux (5 h); (iii) AcOH/I2, reflux (3–4 h).
Acylated pyrazoles 4d–f showed IR bands at 3066–3068 (N=CH), 1755–1760 (O–C=O), 1622–1640 (C=O), 1583–1599 (C=C), and 1504–1505 (C=N) cm–1. Their 1H NMR data exhibited the same characteristic signals as already reported for pyrazoles 4a–c [34] and methyl resonances at δ=2.11–2.14 ppm.
Hydrazones 5a–c and 6a–c showed signals in the IR and NMR spectra, which were characteristic of a chromone ring. Compounds 5b and 5c had typical hydrazone IR bands at 3273–2379 (NH), 3060–3064 (N=CH), and 1516–1518 (C=N) cm–1. 1H NMR resonances of 5b were observed at δ=6.76 (dd, J = 2.2/8.4 Hz, 4′-H), 7.18 (d, J = 8.4 Hz, 3′-H), and 7.50 (d, J = 2.2 Hz, 6′-H) ppm corresponding to the 2,5-dichlorophenyl residue, while its 6-methyl group appeared at 2.47 ppm. The presence of the hydrazone moiety was established from signals at 8.12 (s, CH=N) and 8.60 (s, NH) ppm.
Proton signals of 5c were similar to those of 5b. The presence of fluorine at position 6 was evidenced by the multiplicity and chemical shift of the chromone protons at δ=7.89 (dd, J = 3.1/8.2 Hz, 5-H), 7.42 (ddd, J = 3.1/7.6/9.2 Hz, 7-H), and 7.52 (dd, J = 9.2/4.4 Hz, 8-H) ppm. The isomerization of 5b and 5c to pyrazole structures was discarded based on 13C NMR data, which showed the presence of characteristic 1,4-benzopyrone carbonyl carbons between δ=174.9–175.9 (C-4) ppm and vinylic CH=N resonances at 129.9–131.3 ppm.
Hydrazones 6a–c displayed IR bands at approximately 3265–3287 (NH), 3114–3118 (N=CH), 1515–1519 (C=N) and 1584, 1334 (N=O) cm–1. Distinctive 1H NMR signals for the 2,4-dinitrophenyl substituent were located at δ = 8.82–8.85 (H-3′), 8.31–8.33 (H-5′) and 8.08–8.13 (H-6′) ppm, while hydrazone-type protons were detected at 8.85–8.99 (CH=N) and 11.45–11.66 (NH) ppm. The methyl group signal of 6b was located at 2.47 ppm. Characteristic 13C NMR resonances for carbonyl and vinylic CH=N carbons at δ=174.6–175.1 and 125.2–125.8 ppm, excluded a pyrazole structure for 6a–c.
When chromones were substituted with either H (1a) or F (1c) at C-6, the reaction with 3 produced the expected pyrazolo[1,5-a]pyrimidines 7a and 7c. However, the reaction product between 6-methyl substituted chromone 1b with aromatic amine 3 yielded the pyrazolo[3,4-b]pyridine 8 after two steps (see general procedures). IR bands for 8 were present at 3400 (OH), 3203 (NH), 3036 (N=CH), 1634 (C=O), and 1479 (C=N) cm–1. Its 1H NMR spectra revealed signals of a p-hydroxy-methylphenyl residue at δ=2.53 (s, 5′-Me), 6.90 (d, J = 8.4 Hz, 3′-H), 7.22 (d, J = 2.2 Hz, 6′-H), 7.27 (dd, J = 2.2/8.4 Hz, 4′-H), and 10.09 (s, OH) ppm. The formation of the pyrazolo[1,5-a]pyrimidine structure was discarded due to the absence of the characteristic C-3 resonance between δ=93.8–96.9 ppm in the 13C NMR spectra. The presence of 3-methyl-1H-pyrazolo[3,4-b]pyridin-5-yl signals at 2.26 (s, 3-Me), 8.52 (d, J = 1.8 Hz, 4-H), 8.78 (d, J = 1.8 Hz, 6-H) and 13.60 (s, NH) ppm, confirmed the formation of 8.
The formation of regioisomer 8 may arise from an imine intermediary (Scheme 2) that undergoes 1,4-addition at C-2 by attack of C-4′ from the pyrazole instead of the nitrogen atom N-2′. To the best of our knowledge, this is the first report regarding the formation of pyrazolo[3,4-b]pyridines by intramolecular attack of an sp2 carbon atom.
![Scheme 2: Formation of pyrazolo[3,4-b]pyridine 8.](/document/doi/10.1515/znb-2014-0236/asset/graphic/znb-2014-0236_scheme2.jpg)
Formation of pyrazolo[3,4-b]pyridine 8.
2.2 Brine shrimp lethality assay
The toxicity of all compounds was assayed against Artemia salina. The dose-dependent assays were carried out between 10 and 500 ppm using a negative control, Tween 80®, at this concentration did not affect this bioassay. The IC50 values of the compounds are presented in Table 1. The study showed that all tested compounds exhibited a toxic effect to the brine shrimp, with derivatives 6c, 7b, 6a and 5c [IC50 = 83.0 ± 16, 94.7 ± 14, 95.3 ± 26 and 98.1 ± 14 μm respectively] among the more active ones. The observed toxicities are slightly lower than the cytotoxicity of Podophyllotoxin, a well known bioactive compound [IC50 = 5.8 μm] [31].
The reasons for this toxic effect are not clear. However, the presence of fluorine increased activity, as could be observed for compounds 4c, 5c, 6c, and 7b. Such results are consistent with the general observation, which states that the presence of aromatic fluorine enhances the overall biological activity of organic compounds on a moderate scale [35, 36]. The presence of the hydroxyl group may also play a role in the toxicity, as evidenced by the increase in the IC50 values for acylated derivatives 4d–f compared with 4a–c. More specific cytotoxic studies that use tumor cell lines MCF-7 (breast) and PC3 (prostate) are currently underway.
Brine shrimp lethality assay of the synthesized compounds.
| Compound | IC50 (μm) | Compound | IC50 (μm) |
|---|---|---|---|
| 4a | 261.8 ± 34 | 5c | 98.1 ± 14 |
| 4b | 202.2 ± 39 | 6a | 95.3 ± 26 |
| 4c | 154.6 ± 49 | 6b | 113.2 ± 16 |
| 4d | 296.5 ± 15 | 6c | 83.0 ± 16 |
| 4e | 319.4 ± 19 | 7a | 101.3 ± 11 |
| 4f | 185.8 ± 24 | 7b | 94.7 ± 14 |
| 5a | 161.2 ± 26 | 8 | 110.4 ± 37 |
| 5b | 170.0 ± 28 |
3 Experimental section
3.1 Chemistry
Melting points were determined using a Krüss-Optronic (San Diego, CA, USA) apparatus, and they were not corrected. 1H NMR, 13C NMR, and DEPT-135 spectra were recorded on a JEOL Eclipse Plus 400 (400 MHz) (Peabody, MA, USA) and Bruker Avance500* (500 MHz) (Billerica, MA, USA) spectrometers, using CDCl3 and [D6]DMSO as solvents. IR spectra were obtained from KBr pellets with Shimadzu IR-408 (Columbia, MD, USA) equipment. The HRMS (70 eV) analyses were conducted in a JEOL JMS-AX505WA (Peabody, MA, USA) double focus mass spectrometer, using the electron impact (EI) method. Analytical thin-layer chromatography was carried out on 0.25 mm layers of silica gel PF254 (Merck) (Kenilworth, NJ, USA).
Reagents 1a–c, 2a–c, and 3 were purchased from Aldrich (Milwaukee, WI, USA) as ‘synthetic grade’ and used without further purification. Compounds 4a–c, 5a, 7a, and 7b were prepared as described in the general procedures. Their physical constants and spectroscopic data were in agreement with those described in the literature [16, 28, 37].
3.2 General procedure for the synthesis of 4d–f
A portion of compound 4a–c (0.4 mmol) was dissolved in an excess of Ac2O (4 mL), after which H2SO4 (18 m, 0.1 mL) was added and refluxed for 5 h. Once finished, 15 mL of iced water was added and extracted with CH2Cl2 (2 × 6 mL). The organic phase was washed with 15 % NaHCO3 (aq.) (4 × 15 mL), and dried over Na2SO4, after which the solvent was evaporated under reduced pressure. The obtained solid was recrystallized from hexane-EtOAc (1:1) solvent mixture.
3.2.1 (2-Acetoxy-phenyl)(1-phenyl-1H-pyrazol-4-yl)methanone (4d)
Colorless crystals. Yield: 85 %. M.p. 108–110 °C. – IR (KBr): v = 3066 (N=CH), 1756 (O–C=O), 1622 (C=O), 1599 (C=C), 1504 (C=N). – 1H NMR (500 MHz, CDCl3): δ = 2.14 (s, 3H, CH3 acetate), 7.18 (d, J = 7.8 Hz,1H, 5′-H), 7.31 (d, J = 7.7 Hz, 1H, 3′-H), 7.33 (d, J=7.3 Hz,1H, 4″-H), 7.43 (dd, J = 7.3/8.2 Hz, 2H, 3″/5″-H), 7.53 (td, J = 1.5/7.7 Hz,1H, 4′-H), 7.67 (d, J = 8.2 Hz, 2H, 2″/6″-H), 7.61 (dd, J = 1.5/7.7 Hz, 1H, 6′-H), 8.06 (s, 1H, 3-H), 8.29 (s, 1H, 5-H) ppm. – 13C NMR (125 MHz, CDCl3): δ = 20.9 (CH3), 119.8 (C-2″/C-6″), 123.6 (C-4), 124.8 (C-1′), 125.9 (C-5′), 127.9 (C-3′), 129.6 (C-4″), 129.7 (C-3″/C-5″), 131.0 (C-6′), 132.3 (C-5), 132.6 (C-4′), 139.3 (C-1″), 142.6 (C-3), 148.3 (C-2′), 169.5 (O–C=O), 187.1 (C=O) ppm. – HRMS: m/z = 306.0998 (calcd. 306.1004 for C18H14N2O3).
3.2.2 (2-Acetoxy-5-methylphenyl)(1-phenyl-1H-pyrazol-4-yl)methanone (4e)
Pale yellow crystals. Yield: 87%. M.p. 99–101 °C.– IR (KBr): v = 3065 (N=CH), 1760 (O–C=O), 1640 (C=O), 1598 (C=C), 1505 (C=N). – 1H NMR (500 MHz, CDCl3): δ = 2.11 (s, 3H, CH3 acetate), 2.34 (s, 3H, CH3), 7.06 (d, J = 8.8 Hz, 1H, 3′-H), 7.33 (dd, J = 0.0/8.8 Hz, 1H, 4′-H), 7.40 (d, J = 7.7 Hz, 1H, 4″-H), 7.45 (dd, J = 7.7/8.1 Hz, 2H, 3″/5″-H), 7,67 (d, J=2.0 Hz, 1H, 6′-H), 7,69 (d, J = 8.1 Hz, 2H, 2″/6″-H), 8.06 (s, 1H, 3-H), 8.29 (s, 1H, 5-H) ppm. – 13C NMR (125 MHz, CDCl3): δ = 20.7 (CH3), 20.8 (CH3), 119.7 (C-2″/C-6″), 123.1 (C-1′), 124.8 (C-4), 127.7 (C-3′), 129.7 (C-3″/C-5″), 129.8 (C-4″), 130.8 (C-6′), 132.2 (C-5), 132.7 (C-5′), 135.8 (C-4′), 139.2 (C-1″), 142.5 (C-3), 145.9 (C-2′), 169.5 (O–C=O), 187.1 (C=O) ppm. – HRMS: m/z = 320.1116 (calcd. 320.1161 for C19H16N2O3).
3.2.3 (2-Acetoxy-5-fluorophenyl)(1-phenyl-1H-pyrazol-4-yl)methanone (4f)
Pale yellow crystals. Yield: 91%. M.p. 102–104 °C. – IR (KBr): v=3068 (N=CH), 1755 (O–C=O), 1628 (C=O), 1583 (C=C), 1504 (C=N). – 1H NMR (500 MHz, CDCl3): δ=2.13 (s, 3H, CH3 acetate), 7.14 (dd, J = 4.4/9.2 Hz, 1H, 3′-H), 7.28 (ddd, J=2.9/7.6/9.2 Hz, 1H, 4′-H), 7.36 (d, J = 7.0 Hz, 1H, 4″-H), 7.45 (dd, J = 7.0/8.1 Hz, 2H, 3″/5″-H), 7.66 (dd, J = 2.9/8.8 Hz, 1H, 6′-H), 7.69 (d, J = 8.1 Hz, 2H, 2″/6″-H), 8.05 (s, 1H, 3-H), 8.30 (s, 1H, 5-H) ppm. – 13C NMR (125 MHz, CDCl3): δ = 20.6 (CH3), 115.9/116.2 (C-3′), 118.6/118.9 (C-4′), 119.7 (C-2″/C-6″), 124.2 (C-4), 125.0/125.1 (C-5′), 127.8 (C-4″), 129.6 (C-3″/C-5″), 130.8 (C-5), 133.6/133.7 (C-1′), 139.0 (C-1″), 142.4 (C-3), 143.9 (C-2′), 157.9/161.2 (C-6′), 169.3 (O–C=O), 185.4 (C=O) ppm. – HRMS: m/z = 324.0988 (calcd. 324.0910 for C18H13FN2O3).
3.3 General procedure for the synthesis of 4a–c, 5a–c, and 6a–c
3-Formylchromone derivative 1a–c (1.0 mmol) was dissolved in hot anhydrous THF (4 mL) and a solution of hydrazine 2a–c (1.0 mmol) in THF (2 mL) was added slowly. The mixture was refluxed for 1–2 h; once cooled the solid was filtered, washed with water, and recrystallized from absolute EtOH.
3.3.1 3-[(2,5-Dichlorophenyl)hydrazonomethyl]- 6-methyl-chromen-4-one (5b)
Yellow solid. Yield: 87%. M.p. 221–222 °C. – IR (KBr): v = 3273 (NH), 3060 (N=CH), 1649 (C=O), 1620 (C=C), 1518 (C=N). – 1H NMR (400 MHz, CDCl3): δ = 2.47 (s, 3H, CH3), 6.76 (dd, J = 2.2/8.4 Hz, 1H, 4′-H), 7.18 (d, J = 8.4 Hz, 1H, 3′-H), 7.40 (d, J = 8.8 Hz, 1H, 8-H), 7.48 (d, J = 8.8 Hz, 1H, 7-H), 7.50 (d, J = 2.2 Hz, 1H, 6′-H), 8.05 (bs, 1H, 5-H), 8.12 (s, 1H, CH=N), 8.14 (s, 1H, 2-H), 8.60 (s, 1H, NH) ppm. – 13C NMR (100 MHz, CDCl3): δ = 21.0 (CH3), 113.8 (C-6′), 115.2 (C-3), 118.2 (C-8), 118.9 (C-2′), 119.9 (C-4′), 123.6 (C-10), 125.3 (C-5), 129.9 (CH=N), 132.1 (C-3′), 133.8 (C-5′), 135.2 (C-7), 135.8 (C-6), 141.1 (C-1′), 152.6 (C-2), 154.6 (C-9), 175.9 (C-4) ppm. – HRMS: m/z = 346.0288 (calcd. 346.0276 for C17H12Cl2N2O2).
3.3.2 3-[(2,5-Dichlorophenyl)hydrazonomethyl]- 6-fluoro-chromen-4-one (5c)
Yellow solid. Yield: 85%. M.p. 211–213 °C. – IR (KBr): v = 3279 (NH), 3064 (N=CH), 1646 (C=O), 1620 (C=C), 1516 (C=N). – 1H NMR (400 MHz, CDCl3): δ = 6.77 (dd, J = 2.5/8.5 Hz, 1H, 4′-H), 7.18 (d, J = 8.5 Hz, 1H, 3′-H), 7.42 (ddd, J=3.1/7.6/9.2 Hz, 1H, 7-H), 7.50 (d, J = 2.5 Hz, 1H, 6′-H), 7.52 (dd, J = 4.4/9.2 Hz, 1H, 8-H), 7.89 (dd, J = 3.1/8.2 Hz 1H, 5-H), 8.07 (s, 1H, CH=N), 8.14 (s, 1H, 2-H), 8.60 (s, 1H, NH) ppm. – 13C NMR (100 MHz, CDCl3): δ = 110.2 (C-5), 110.5 (C-8), 114.1 (C-6′), 119.2 (C-2′), 119.9 (C-4′) 122.1 (C-7), 123.2 (C-3), 125.2 (C-10), 131.3 (CH=N), 133.4 (C-3′), 134.2 (C-5′), 142.9 (C-1′), 154.6 (C-2), 158.5/160.9 (C-6), 174.9 (C-4) ppm. – HRMS: m/z = 350.0030 (calcd. 350.0025 for C16H9Cl2FN2O2).
3.3.3 3-[(2,4-Dinitrophenyl)hydrazonomethyl]- chromen-4-one (6a)
Orange solid. Yield: 90%. M.p. 294–295 °C. – IR (KBr): v = 3265 (NH), 3114 (N=CH), 1644 (C=O), 1605 (C=C), 1519 (C=N), 1580, 1334 (N=O). – 1H NMR (400 MHz, [D6]DMSO): δ = 7.56 (dd, J = 7.3/8.1 Hz, 1H, 6-H), 7.72 (d, J = 8.4 Hz, 1H, 8-H), 7.86 (dd, J = 7.3/8.4 Hz, 1H, 7-H), 8.13 (d, J = 9.5 Hz, 1H, 6′-H), 8.16 (d, J = 8.1 Hz, 1H, 5-H), 8.31 (dd, J = 2.2/9.5 Hz, 1H, 5′-H), 8.75 (s, 1H, 2-H), 8.85 (d, J = 2.2 Hz, 1H, 3′-H), 8.97 (s, 1H, CH=N), 11.65 (s, 1H, NH) ppm. – 13C NMR (100 MHz, [D6]DMSO): δ = 117.6 (C-6′), 118.1 (C-3′), 119.1 (C-8), 123.3 (C-5′), 121.5 (C-10), 124.2 (C-5), 125.8 (CH=N), 129.9 (C-3), 130.5 (C-2′), 135.1 (C-7), 126.6 (C-6), 135.8 (C-4′), 142.4 (C-1′), 156.0 (C-9), 159.5 (C-2), 175.1 (C-4) ppm. – HRMS: m/z = 354.0634 (calcd. 354.0600 for C16H10N4O6).
3.3.4 3-[(2,4-Dinitrophenyl)hydrazonomethyl]- 6-methyl-chromen-4-one (6b)
Orange solid. Yield: 87%. M.p. 271–272 °C. – IR (KBr): v = 3270 (NH), 3118 (N=CH), 1644 (C=O), 1595 (C=C), 1515 (C=N), 1584, 1334 (N=O). – 1H NMR (400 MHz, [D6]DMSO): δ = 2.47 (s, 3H, CH3), 7.56 (d, J = 8.4 Hz, 1H, 8-H), 7.65 (d, J = 8.4 Hz, 1H, 7-H), 7,95 (bs, 1H, 5-H), 8.08 (d, J = 9.5 Hz, 1H, 6′-H), 8.32 (dd, J = 2.5/9.5 Hz 1H, 5′-H), 8.67 (s, 1H, 2-H), 8.82 (bs, 1H, 3′-H), 8.85 (s, 1H, CH=N), 11.45 (s, 1H, NH) ppm. – 13C NMR (100 MHz, [D6]DMSO): δ = 20.8 (CH3), 118.1 (C-6′), 118.8 (C-3′), 117.6 (C-8), 121.4 (C-10), 123.1 (C-5′), 125.4 (C-5), 125.2 (CH=N), 129.8 (C-3), 129.9 (C-2′), 135.7 (C-7), 136.0 (C-6), 138.5 (C-4′), 142.6 (C-1′), 154.7 (C-9), 157.8 (C-2), 174.9 (C-4) ppm. – HRMS: m/z = 368.0757 (calcd. 368.0757 for C17H12N4O6).
3.3.5 3-[(2,4-Dinitrophenyl)hydrazonomethyl]- 6-fluor-chromen-4-one (6c)
Orange solid. Yield: 83%. M.p. 261–262 °C. IR (KBr): v = 3287 (NH), 3118 (N=CH), 1640 (C=O), 1605 (C=C), 1519 (C=N), 1584, 1340 (N=O). – 1H NMR (400 MHz, [D6]DMSO): δ = 7.72 (m, 1H, 7-H), 7.81 (m, 1H, 8-H), 7.83 (bs, 1H, 5-H), 8.12 (d, J = 9.5 Hz, 1H, 6′-H), 8.33 (dd, J = 2.5/9.5 Hz 1H, 5′-H), 8.74 (s, 1H, 2-H), 8,85 (d, J = 2.5 Hz, 1H, 3′-H), 8.99 (s, 1H, CH=N), 11,66 (s, 1H, NH) ppm. – 13C NMR (100 MHz, [D6]DMSO): δ = 110.3 (C-5), 110.6 (C-8), 117.7 (C-6′), 118.4 (C-3′), 122.1 (C-10), 123.2 (C-7), 123.4 (C-5′), 125.3 (CH=N), 129.9 (C-3), 130.4 (C-2′), 137.9 (C-4′), 142.1 (C-1′), 152.8 (C-9), 156.5/161.2 (C-6), 158.7 (C-2), 174.6 (C-4) ppm. – HRMS: m/z = 372.0599 (calcd. 372.0506 for C16H9FN4O6).
3.4 General procedure for the synthesis of 7a and 7b
3-Formylchromone derivative 1a or 1c (1.0 mmol) was mixed with aminopyrazole 3 (1.0 mmol) and refluxed in 10 mL of anhydrous THF for 1 h. After cooling, the solid was filtered, washed repeatedly with hot THF, and recrystallized from EtOH to produce TLC pure compounds.
3.5 General procedure for the synthesis of 8
Equimolar quantities of 1b and 3 (1.0 mmol) were refluxed in anhydrous THF for 1–2 h, until the formation of a precipitate. Once separated from the solution, this precipitate was dissolved in AcOH (7 mL) in the presence of I2 (1.0 mmol) and then refluxed for a period of 3–4 h. Upon completion of the TLC reaction, the mixture was poured into crushed ice and treated with NaHCO3 and Na2SO3. The solid was filtered, washed with cold water, and dried.
3.5.1 (2-Hydroxy-5-methylphenyl)(3-methyl- 1H-pyrazolo[3,4-b]pyridin-5-yl)methanone (8)
Pale yellow solid. Yield: 80%. M.p. 198–200 °C. – IR (KBr): v = 3400 (OH), 3203 (NH), 3036 (N=CH), 1634 (C=O), 1597 (C=C), 1479 (C=N). – 1H NMR (400 MHz, [D6]DMSO): δ = 2.26 (s, 3H, CH3 pyrazolopyridine), 2.53 (s, 3H, CH3 2-hydroxy-5- methylphenyl), 6.90 (d, J = 8.4 Hz, 1H, 3′-H), 7.22 (d, J=2.2 Hz, 1H, 6′-H), 7.27 (dd, J = 2.2/8.4 Hz, 1H, 4′-H), 8.52 (d, J = 1.8 Hz, 1H, 4-H), 8.78 (d, J = 1.8 Hz, 1H, 6-H), 10.09 (s, 1H, OH), 13.60 (s, 1H, NH) ppm. – 13C NMR (100 MHz, [D6]DMSO): δ = 12.7 (CH3), 20.5 (CH3), 113.9 (C-5), 117.3 (C-3′), 125.2 (C-1′), 126.9 (C-9), 128.6 (C-4), 131.0 (C-4′), 132.3 (C-6′), 134.6 (C-5′), 143.9 (C-3), 150.8 (C-6), 153.8 (C-8), 155.1 (C-2′), 196.1 (C=O) ppm. – HRMS: m/z = 267.1044 (calcd. 267.1008 for C15H13N3O2).
3.6 BSLA
The assay was performed as described previously by Meyer [31] with some minor modifications. Brine shrimp eggs (Gulf Breeze®) were hatched in artificial sea water prepared with commercial salt mixture (Instant Ocean®), and then illuminated and oxygenated with an aquarium pump. After an incubation period of 48 h at 27 °C, 10 shrimps were transferred with a Pasteur pipette to three sample vials for each of the three doses (500, 100, 10 μg mL–1), for a total of nine vials per sample. The compound samples (10 mg) were dissolved in CHCl3 (5 mL). Aliquotes of testing solutions (1250, 250, or 25 μL for the 500, 100 and 10 ppm doses, respectively) were placed on vials (5 mL) and the solvent was evaporated. The residue was redissolved in 10 μL of Tween 80®, after which artificial sea water (5 mL) was added. Survivors were counted and the percent deaths at each dose were determined. Control samples were included and assayed simultaneously. IC50 values were calculated from 24 h counts using the probit analysis [38].
References
[1] A. M. Edwards, J. B. L. Howell, Clin. Exp. Allergy 2000, 30, 756.10.1046/j.1365-2222.2000.00879.xSuche in Google Scholar
[2] L. H. Hurley, Nat. Rev. Cancer 2002, 2, 188.10.1038/nrc749Suche in Google Scholar
[3] A. K. Mukherjee, S. Basu, N. Sarkar, A. C. Ghosh, Curr. Med. Chem. 2001, 8, 1467.Suche in Google Scholar
[4] W. Samee, P. Nunthanavanit, J. Unwitayatorn, Int. J. Mol. Sci. 2008, 9, 235.Suche in Google Scholar
[5] S. M. Hasan, M. M. Alam, A. Husain, S. Khanna, M. Akhtar, M. S. Zaman, Eur. J. Med. Chem. 2009, 44, 4896.Suche in Google Scholar
[6] R. Amin, B. Krammer, N. Abdel-Kader, T. Verwanger, A. El-Ansary, Eur. J. Med. Chem. 2010, 45, 372.Suche in Google Scholar
[7] J. Ungwitayatorn, W. Samee, J. Pimthon, J. Mol. Struct. 2004, 689, 99.Suche in Google Scholar
[8] P. Nunthanavanit, N. G. Anthony, B. F. Johnston, S. P. Mackay, J. Ungwitayatorn, Arch. Pharm. Chem. Life Sci. 2008, 341, 357.Suche in Google Scholar
[9] W. Huang, M.-Z. Liu, Y. Li, Y. Tan, G.-F. Yang, Bioorg. Med. Chem. 2007, 15, 5191.Suche in Google Scholar
[10] W. Huang, Y. Ding, Y. Miao, M.-Z. Liu, Y. Li, G.-F. Yang, Eur. J. Med. Chem. 2009, 44, 3687.Suche in Google Scholar
[11] W. Ren, Z. Qiao, H. Wang, L. Zhu, L. Zhang, Med. Res. Rev. 2003, 23, 519.Suche in Google Scholar
[12] N. Li, J.-H. Liu, J. Zhang, B.-Y. Yu, J. Agric. Food Chem. 2008, 56, 3876.Suche in Google Scholar
[13] D. W. Engers, A. Y. Frist, C. W. Lindsley, C. C. Hong, C. R. Hopkins, Bioorg. Med. Chem. Lett. 2013, 23, 3248.Suche in Google Scholar
[14] A. Kamal, J. R. Tamboli, V. L. Nayak, S. F. Adil, M. V. P. S. Vishnuvardhan, S. Ramakrishna, Bioorg. Med. Chem. Lett. 2013, 23, 3208.Suche in Google Scholar
[15] Z. Baráth, R. Radics, G. Spengler, I. Ocsovszki, M. Kawase, N. Motohashi, Y. Shirataki, A. Shah, J. Molnár, In vivo 2006, 20, 645.Suche in Google Scholar
[16] A. Lazarenkow, J. Nawrot-Modranka, E. Brzezinka, U. Krajewska, M. Rózalski, Med. Chem. Res. 2012, 21, 1861.Suche in Google Scholar
[17] P. Foltínová, M. Lácová, D. Loos, Il Farmaco 2000, 55, 21.10.1016/S0014-827X(99)00114-7Suche in Google Scholar
[18] H. M. El-Shaaer, P. Foltínová, M. Lácová, J. Chovancová, H. Stankovicova, Il Farmaco 1998, 53, 224.10.1016/S0014-827X(98)00015-9Suche in Google Scholar
[19] B.-d. Wang, Z.-Y. Yang, T.-r. Li, Bioorg. Med. Chem. 2006, 14, 6012.Suche in Google Scholar
[20] Q. Wang, Z.-Y. Yang, G.-F. Qi, D.-D. Qin, Eur. J. Med. Chem. 2009, 44, 2425.Suche in Google Scholar
[21] K. M. Khan, N. Ambreen, S. Hussain, S. Perveen, M. I. Choudhary, Bioorg. Med. Chem. 2009, 17, 2983.Suche in Google Scholar
[22] A. O. Fitton, J. R. Frost, H. Suschitzky, Tetrahedron Lett. 1975, 25, 2099.Suche in Google Scholar
[23] A. O. Fitton, J. R. Frost, H. Suschitzky, J. Chem. Soc., Perkin Trans. 1 1979, 1691.10.1039/p19790001691Suche in Google Scholar
[24] C. K. Ghosh, J. Heterocycl. Chem. 1983, 20, 1437.Suche in Google Scholar
[25] G. Sabitha, Aldrichimica Acta 1996, 29, 15.10.1017/S0278221900601542Suche in Google Scholar
[26] A. S. Plaskon, O. O. Grygorenko, S. V. Ryabukhin, Tetrahedron 2012, 68, 2743.10.1016/j.tet.2012.01.077Suche in Google Scholar
[27] J. Quiroga, D. Mejía, B. Insuasty, R. Abonía, M. Nogueras, A. Sanchez, J. Cobo, J. N. Low, J. Heterocycl. Chem. 2002, 39, 51.Suche in Google Scholar
[28] J. R. Zimmerman, B. J. Myers, S. Bouhall, A. McCarthy, O. Johntony, M. Manpadi, Tetrahedron Lett. 2014, 55, 936.Suche in Google Scholar
[29] P. N. Solis, C. W. Wrigth, M. M. Anderson, M. P. Gupta, J. D. Phillipson, Planta Medica 1993, 59, 250.10.1055/s-2006-959661Suche in Google Scholar
[30] J. E. Anderson, C. M. Goetz, J. M. McLaughlin, Phytochem. Anal. 1991, 2, 107.Suche in Google Scholar
[31] B. N. Meyer, N. R. Ferrigni, J. E. Putnam, L. B. Jacobsen, D. E. Nichols, J. L. McLaughlin, Planta Med. 1982, 45, 31.Suche in Google Scholar
[32] C. L. Zani, P. P. Chavez, R. Queiroz, A. B. de Oliveira, J. E. Cardoso, A. M. Anjos, T. S. Grandi, Phytomedicine 1995, 2, 47.10.1016/S0944-7113(11)80048-6Suche in Google Scholar
[33] L. P. Santos Pimenta, G. B. Pinto, J. A. Takahashi, L. G. F. e Silva, M. A. D. Boaventura, Phytomedicine 2003, 10, 209.10.1078/094471103321659960Suche in Google Scholar PubMed
[34] G. Sabitha, R. S. Babu, J. S. Jadav, Synth. Commun. 1998, 28, 4571.Suche in Google Scholar
[35] A. A. Gakh, M. N. Burnett, J. Fluorine Chem. 2011, 132, 88.Suche in Google Scholar
[36] J.-P. Bégué, D. Bonnet-Delpont in Fluorine and Health, Elsevier, 2008, chapter 13, p. 553.10.1016/B978-0-444-53086-8.00013-8Suche in Google Scholar
[37] G. J. Reddy, D. Latha, C. Thirupathaiah, K. S. Rao, Heterocycl. Commun. 2003, 9, 391.Suche in Google Scholar
[38] D. Finney, Probit Analysis, 3rd Edition, Cambridge University Press, Cambridge, 1971, chapter 3, p. 21.10.2307/2346498Suche in Google Scholar
©2015 by De Gruyter
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Review
- Cerium intermetallics with ZrNiAl-type structure – a review
- Original Communications
- In vitro cytotoxicity of hydrazones, pyrazoles, pyrazolo-pyrimidines, and pyrazolo-pyridine synthesized from 6-substituted 3-formylchromones
- A supramolecular 3-D organic-inorganic hybrid structure based on {PMo12} layers arranged in an alternating mode
- Ionic binuclear ferrocenyl compounds containing 1,1,3,3-tetracyanopropenide anions – synthesis, structural characterization and catalytic effects on thermal decomposition of main components of solid propellants
- Zur Chemie der 1,3,5-Triaza-2-phosphorinan- 4,6-dione. Teil XIV. Darstellung von weiteren P-alkyl- und P-arylsubstituierten 1,3,5-Trimethyl-1,3,5-triaza-2-phosphorinan-4,6-dionen
- Synthesis, biological activity and modeling study of some thiopyrimidine derivatives and their platinum(II) and ruthenium(III) metal complexes
- Synthesis of functionalized benzene using Diels–Alder reaction of activated acetylenes with synthesized phosphoryl-2-oxo-2H-pyran
- Synthesis, structure, and optical nonlinearity of soluble ternary cadmium copper tellurolate complex [Cd(μ-TeTol)4{Cu(PPh3)2}2] (Tol = 4-tolyl)
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Review
- Cerium intermetallics with ZrNiAl-type structure – a review
- Original Communications
- In vitro cytotoxicity of hydrazones, pyrazoles, pyrazolo-pyrimidines, and pyrazolo-pyridine synthesized from 6-substituted 3-formylchromones
- A supramolecular 3-D organic-inorganic hybrid structure based on {PMo12} layers arranged in an alternating mode
- Ionic binuclear ferrocenyl compounds containing 1,1,3,3-tetracyanopropenide anions – synthesis, structural characterization and catalytic effects on thermal decomposition of main components of solid propellants
- Zur Chemie der 1,3,5-Triaza-2-phosphorinan- 4,6-dione. Teil XIV. Darstellung von weiteren P-alkyl- und P-arylsubstituierten 1,3,5-Trimethyl-1,3,5-triaza-2-phosphorinan-4,6-dionen
- Synthesis, biological activity and modeling study of some thiopyrimidine derivatives and their platinum(II) and ruthenium(III) metal complexes
- Synthesis of functionalized benzene using Diels–Alder reaction of activated acetylenes with synthesized phosphoryl-2-oxo-2H-pyran
- Synthesis, structure, and optical nonlinearity of soluble ternary cadmium copper tellurolate complex [Cd(μ-TeTol)4{Cu(PPh3)2}2] (Tol = 4-tolyl)