Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
-
Yin-Ping Wang


Abstract
For the first time, carborane complexes containing a B(4)–S bond were obtained directly by heating mononuclear Pd-B(4)-bound carborane complexes. A possible mechanism involved in sulfur atom migration is presented in which the leaving group, pyridine, benzyl isocyanide or PPh3, is demonstrated to be the trigger of the reaction process. In this work, efficient routes are developed through one-pot reactions to prepare B(4)-S carborane derivatives.
Introduction
Since the first report of icosahedral carborane clusters in the 1960s, both the synthetic strategies to these compounds and their applications have been extensively developed [1], [2]. Due to its commercial availability, o-carborane (1,2-C2B10H12) has been the most widely investigated by researchers. It is an excellent candidate for synthetic strategies because of its rigid backbone, high thermal stability and aromatic character [3], [4], [5], [6]. Carborane derivatives have been extensively reviewed in relation to their applications in medicine [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], as well as in catalysis [19], [20], [21], [22], polymer chemistry [23], [24], [25], self-assembly [26], [27], [28], [29], [30], and optoelectronic applications [31], [32], [33], [34], [35], [36]. However, their unique structures make selective derivatization difficult, resulting in a limited application scope. Significant research efforts have been focused on the selective functionalization of carboranes [37], [38], [39], [40], [41], [42], [43], [44], [45]. In the work of Xie et al., the selective hydrogenation and functionalization of boron hydrides catalyzed by transition metals has been successfully achieved by introducing a carboxyl group as a directing group on the carborane carbon. A series of methods were developed to synthesize B4-substituted carborane derivatives, including alkenyl, aryl, alkynyl, hydroxyl and amino-substituted carboranes [46], [47], [48], [49], [50], [51]. B–H bonds can also react with strong electrophiles such as halogens and methyl cations, leading to boron-substitution of o-carborane derivatives [52], [53], [54], [55], [56]. Palladium compounds play an important role in a lot of reactions which involve in molecular activation and produce new derivatives through various transformations. In addition, Pd(II) complexes are well known to catalyze the cleavage of carbon-sulfur bonds in fundamental catalytic and environmental aspects [57], [58], [59], [60], [61], [62].
One of the most well-known applications of boron clusters is their use in boron neutron-capture therapy (BNCT) [63], [64], [65], [66], [67]. Because cluster derivatives containing Bcluster–S bonds show affinity for tumours, it seems necessary to develop efficient methods to produce boron clusters containing B–S bonds. Over the past two decades, many boron clusters incorporating sulfur atoms have been reported in the literature [68], [69], [70], [71], [72], [73]. Plešek and Heřmánek [74] obtained the first carborane derivative containing a B–S bond through the B(9,12) atoms via the action of elemental sulfur and carborane in the presence of AlCl3. Despite the range of known cluster derivatives, synthetic methods for their preparation have not been widely developed, particularly for the synthesis of o-carborane derivatives containing B(4)–S bonds.
In our previous work, a series of mononuclear B(4)-borometalate complexes were obtained in high yield via reaction of tetranuclear palladium complex 1 ([Pd4(1-PhN=CS-1,2-C2B10H10)4]) with nucleophilic reagents [75]. Herein, based on our previous work, we report our discovery that by directly heating palladium starting materials containing Pd–B(4) bonds in toluene under the protection of N2, the first examples of carborane compounds containing B(4)–S bonds were obtained. This new strategy provides a novel pathway for B(4)–S bond formation in o-carborane derivatives. By using diferent mononuclear B(4)-borometalate complexes, a plausible mechanism involving sulfur atom migration is discussed.
Results and discussion
Synthesis and characterization of complex 3 ([(Py)2Pd2(4-S-1-PhCN-1,2-C2B10H10)])
Complex 2a ([(Py)2Pd2(4-S-1-PhCN-1,2-C2B10H10)]) was prepared from precursor tetranuclear complex 1 ([Pd4(1-PhCNS-1,2-C2B10H10)4]), which was obtained in high yield (90%) with excellent regioselectivity. Complex 2a was treated in different solvents and at different temperatures to screen the optimal reaction conditions. When complex 2a was heated in toluene at 110°C for 3 min, the solution changed from colorless to yellow. The isolated yield was 70% (Scheme 1).
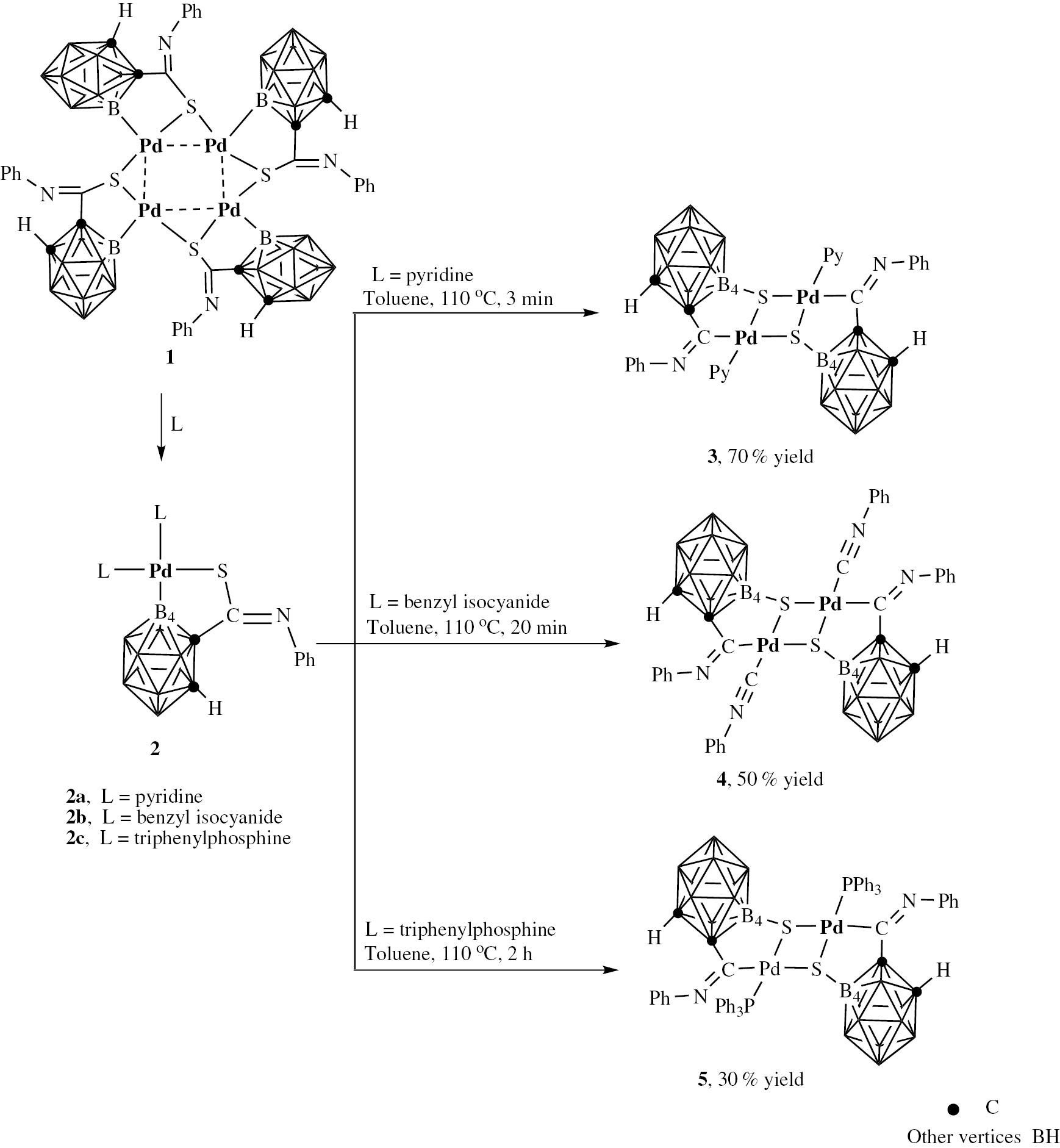
Preparation of B(4)-S dimer complexes.
The isolated product, obtained via column chromatography, was characterized using spectroscopic data and X-ray crystal structure determinations. In the 1H NMR spectrum of complex 3, the signal at 4.29 ppm is characteristic of a Ccage-H resonance, which suggested the presence of a single B–S bond in the dimer structure. In the IR spectrum, the strong band at 2575 cm−1 is characteristic of a B-H stretching mode. In the normal 11B{1H} NMR spectrum, there are three broad signals at –2.2, –11.7 and –18.7 ppm, in a ratio of 4: 14: 2, respectively, indicating the closed structure of the carborane cage. The 11B{1H} NMR spectrum of 3 is significantly different from that of its parent complex 2a [16]. According to this ratio, the shift at –18.66 ppm is obviously a cageB-S resonance, by comparison with its parent complex. The ESI mass spectrum of 3 was consistent with the theoretical isotope distribution and revealed the formation of a dimer complex (See Supplementary Information, Figure 3).
The crystal structure further confirmed complex 3 to be a dimer complex, with S atoms bridging two Pd atoms (Figure 1). In the structure, the S atom has clearly migrated, leading to the formation of a complex with a B(4)–S bond. The coordination mode changed from B(4)–Pd–S–C–Ccage to B(4)–S–Pd–C–Ccage. The C=N distance is 1.270(7) Å in the structure of complex 3, which is almost identical with its parent complex (1.271(3) Å). The length of the Pd–C bond is ca. 2.00 Å and the length of the B–S bond is ca. 1.88 Å. The formation of the B(4)–S–Pd–C–Ccage five-membered ring may be more favourable than forming the B(4)–S–Pd–N–C–Ccage six-membered ring, the former inducing one C atom to bind with the palladium metal center.
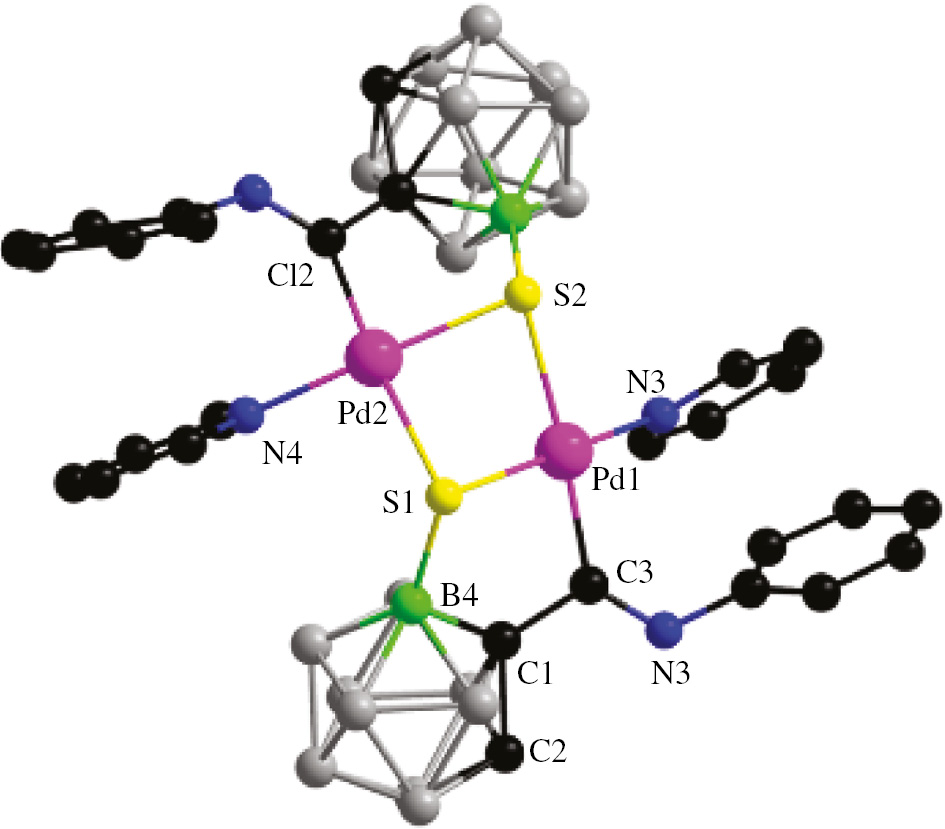
Molecular structure of complex 3: H atoms are omitted for clarity. Color code: C, black; N, blue; Pd, pink; B4, bright green; other B, gray; S, yellow. Selected bond lengths (Å) and angles (deg): Pd(1)–C(3) 1.994(6); Pd(1)–S(1) 2.3402(16); Pd(1)–S(2) 2.4245(15); S(1)–B(4) 1.883(7); C(1)–B(4) 1.708(9); N(1)–C(3) 1.270(7); C(3)–Pd(1)–S(1) 88.63(17); B(4)–S(1)–Pd(1) 96.37(2); Pd(1)–S(1)–Pd(2) 81.09(5); C(1)–B(4)–S(1) 110.3(4).
Synthesis and characterization of complex 4 ([(PhCH2N≡C)2Pd2(4-S-1-PhCN-1,2-C2B10H10)]) and complex 5 ([(PPh3)2Pd2(4-S-1-PhCN-1,2-C2B10H10)])
Based on the simple method for obtaining product 3, the reactivity of similar complexes containing other ligands (L=benzyl isocyanide, PPh3) was also examined similar to that of complex 2a (Scheme 1). The palladium starting materials 2b and 2c were prepared via a similar route to that used to prepare complex 2a, by mixing the ligands with tetranuclear complex 1 in CH2Cl2. The crystal structure of the palladium starting material 2b is shown in the Supplementary Information (Figure 2). The structures of complex 4 and 5 were also confirmed fully by NMR spectroscopy and single-crystal X-ray crystallography. In the 1H NMR spectra of complexes 4 and 5, singlets at δ 3.49 and 4.50 ppm are characteristic of Ccage–H resonances, respectively, which also suggests the formation of single B(4)–S bonds in their dimer complexes. The ESI mass spectrum of complex 5 is also consistent with the theoretical isotope distribution (In Supplementary Information, Figure 4). The molecular structures confirmed that complexes 4 and 5 have the same coordination mode, containing a five-membered ring as in complex 3, in which the S atoms had migrated to the B(4) sites (Figures 2 and 3).
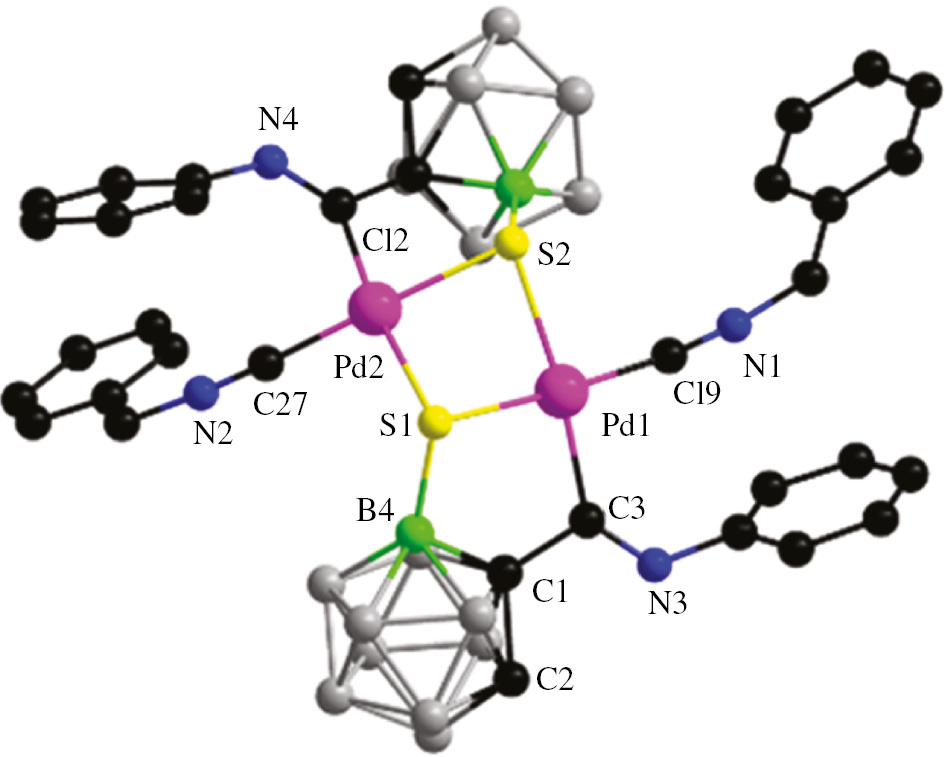
Molecular structure of complex 4: H atoms are omitted for clarity. Color code: C, black; N, blue; Pd, pink; B4, bright green; other B: gray; S, yellow. Selected bond lengths (Å) and angles (deg): Pd(1)–C(19) 1.954(6); Pd(1)–C(3) 2.036(5); Pd(1)–S(1) 2.3630(13); Pd(1)–S(2) 2.4193(12); S(1)–B(4) 1.884(6); N(3)–C(3) 1.255(6); C(1)–B(4) 1.689(8); C(19)–Pd(1)–C(3) 93.9(2); C(3)–Pd(1)–S(1) 90.58(14); B(4)–S(1)–Pd(1); 97.3(2); C(1)–B(4)–S(1) 110.7(4); C(1)–C(3)–Pd(1) 111.4(3).
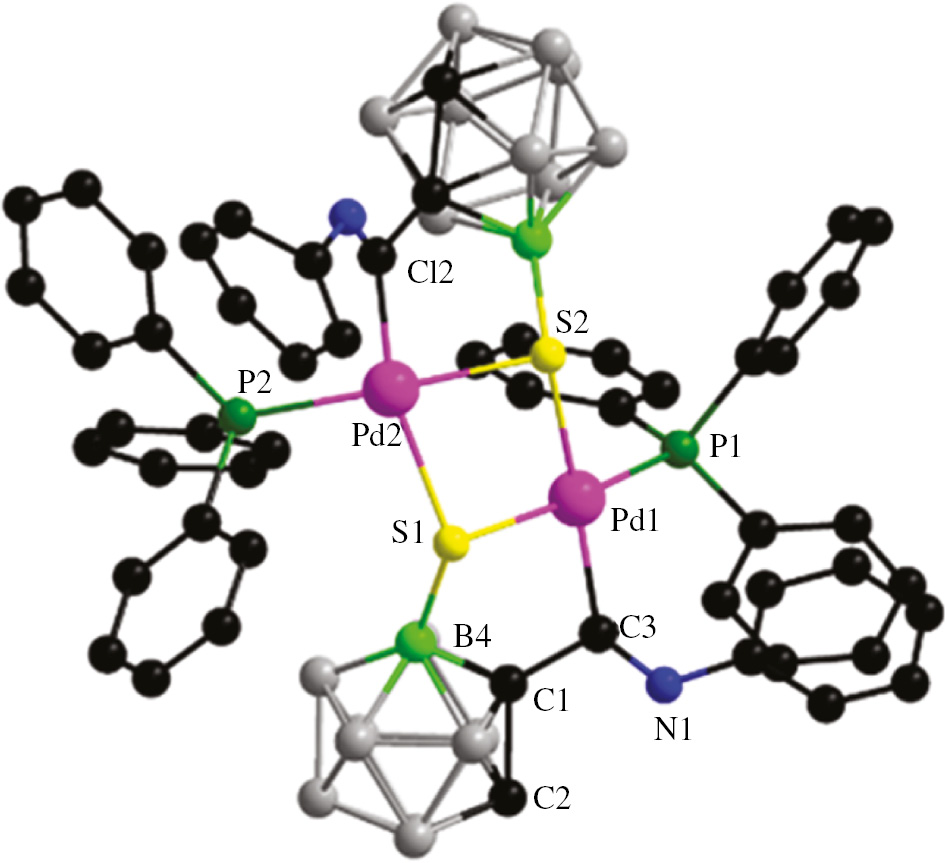
Molecular structure of complex 5: H atoms are omitted for clarity except for those involving hydrogen bonds. Color code: C, black; N, blue; Pd, pink; B4, bright green; other B, gray; P, green; S, yellow. Selected bond lengths (Å) and angles (deg): Pd(1)–C(3) 2.032(5); Pd(1)–S(1) 2.3796(13); Pd(1)–S(2) 2.4166(13); S(1)–B(4) 1.867(6); N(1)–C(3) 1.246(7); B(4)–C(1) 1.735(7); C(3)–Pd(1)–S(1) 86.44(14); B(4)–S(1)–Pd(1) 96.59(18); C(1)–B(4)–S(1) 109.5(4).
A plausible mechanism for the S-atom migration process
To gain insight into the S-atom migration process, Dppe (1, 2-bis(diphenylphosphino)ethane) – a bidentate ligand and poor leaving group for palladium centers – was used to prepare palladium starting material complex 6 (Scheme 2). The molecular structure of complex 6 is shown in the Supplementary Information (Figure 1). The structures of the palladium starting materials indicate that they exhibit coordinated mode paralleling to each other [16]. The complexes differ by the leaving ability of the ligand, which is in the following order: pyridine>benzyl isocyanide>triphenylphosphine>Dppe. However, when complex 6 was treated in the same way as other palladium starting materials, no reaction was observed through monitoring by TLC, despite prolonged reaction time. This suggested that PPh3 or pyridine leaves in the first step, and may trigger the reaction process. Only when there is an unoccupied coordination site at the palladium center can the sulfur atom migration reaction be induced, forming a B(4)–S bond.
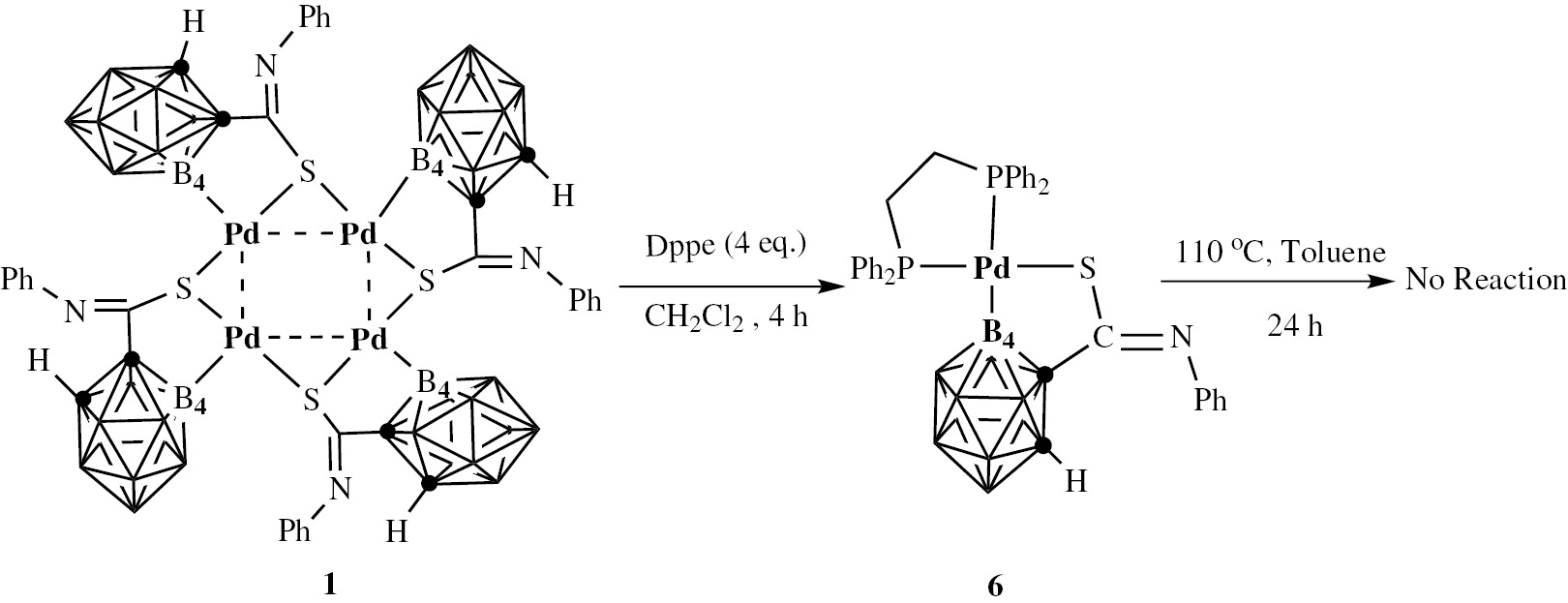
Use of Dppe as ligand to demonstrate the S-atom migration reaction.
The completion time (as monitored by TLC) is in the order: pyridine<benzyl isocyanide<triphenylphosphine<Dppe, which is the inverse of the order of the leaving ability of the ligand. In other words, the easier the ligand can leave, the less time the reaction needs. It seems the leaving ability of the leaving group determines the reaction rate under the heating conditions. All of these sulfur-atom migration reactions involve intramolecular carbon-sulfur bond activation, leading to new derivatives containing B(4)–S bond. The cleavage of carbon-sulfur bonds has been extensively reported in the literature [12].
As shown in Scheme 3, a plausible mechanism has been proposed to illustrate the sulfur atom migration process under optimal heating conditions. The loss of pyridine or PPh3, which is fundamentally important in the reaction, leads to a vacant coordination site. After that, a transition state containing two three-membered rings is formed, in which the Pd center activates the C–S bond. Finally, the rearrangement product is produced. This migration of S atoms to o-carborane B(4) sites, and formation of B(4)-S complexes, is unprecedented.
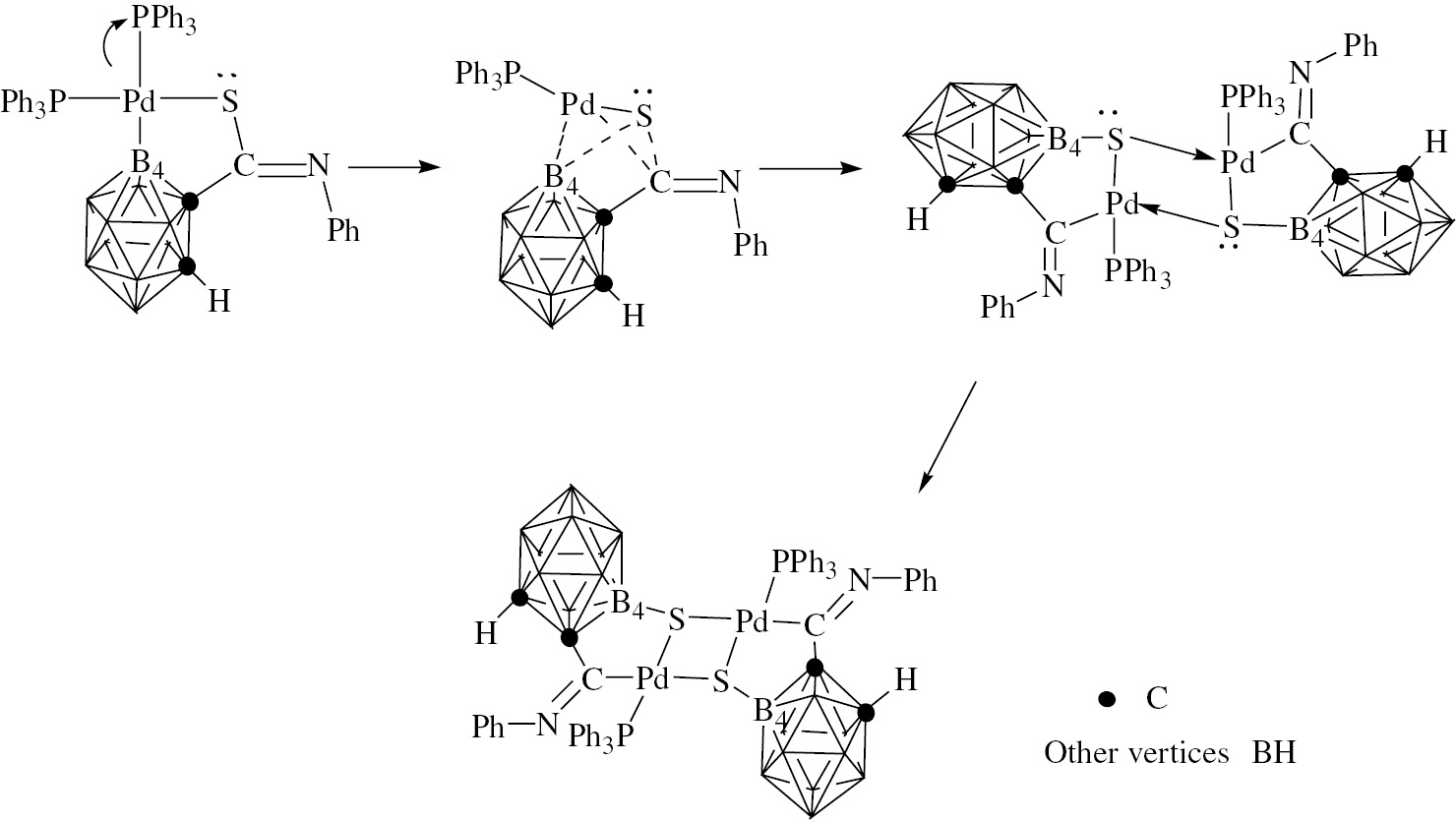
A plausible mechanism of S-atom migration.
Conclusion
Several carborane complexes containing B(4)–S bonds were obtained directly by heating different palladium starting materials containing Pd–B(4) bonds. In these processes, the sulfur atom undergoes an intramolecular migration process. By varying the chelating ligands connected to the palladium center and monitoring the reaction time under the same conditions, a vacant coordination site at palladium was demonstrated to be a crucial prerequisite for the reaction. As for the known methods for the synthesis of o-carboranes containing Bcage–S bonds, this work marks the first example of the preparation of boron cluster complexes containing B(4)–S bonds with the help of transition metal palladium, increasing the scope and generality of these B(4)-S-functionalised derivatives. This connection of sulfur atoms to cage boron atoms can help researchers to prepare new sulfur-containing functional derivatives of carboranes and metallacarboranes for use in drug design, which may have promising biomedical applications and promote the development of multifunctional metallodrugs.
Experimental section
General procedures
All reactions were carried out under an atmosphere of dry, oxygen-free nitrogen using standard Schlenk techniques, with subsequent manipulations under ambient conditions. All solvents were dried and distilled under N2 immediately prior to use. CH2Cl2 was dried over CaH2; toluene and n-hexane were dried over Na. The palladium starting materials 2a and 2c were prepared according to literature procedures [16]. 1H NMR (400 MHz) spectra were measured with an AVANCE III HD spectrometer. 11B NMR (160 MHz) spectra were recorded with a Bruker DMX-500 spectrometer. IR (KBr) spectra were measured with a Nicolet FT-IR spectrophotometer. Elemental analysis was performed on a Vario EL Elemental Analyzer. ESI-MS spectra were recorded on a Bruker micrOTOF11 using electrospray ionization. Diffraction data of all the above were collected on a Bruker Smart APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (λ=0.71073 Å).
X-ray crystal structure determinations
Single crystals of 3, 4, 5, 6, 2b suitable for X-ray diffraction study were obtained at room temperature. X-ray intensity data of 3, 4, 6, 2b were collected at 173 K on a CCD-Bruker SMART APEX system. X-ray intensity data of 5 was collected at 296 K on a CCD-Bruker SMART APEX system. In these data, the disordered solvent molecules which could not be restrained properly were removed using the SQUEEZE route.
In the asymmetric unit of complex 4, 5 ISOR and 5 DFIX instructions were used to restrain ligands and solvent molecules so that there were 35 restraints in the data. The hydrogen atoms of chloroform molecules could not be found and others were placed in calculated positions.
In asymmetric unit of complex 5, there were disordered solvents (one dichloromethane and one diethyl ether molecules) which could not be restrained properly. Therefore, SQUEEZE algorithm was used to omit them. One phenyl group was disordered and it was divided into two parts (69:31). 9 ISOR, 1 FLAT and 8 DFIX instructions were used to restrain the disordered phenyl group so that there were 68 restraints in the data. H2 and H11 were found in difference Fourier map and others were put in calculated positions.
In the asymmetric unit of complex 6, one phenyl group was disordered and it was divided into two parts (81:19). 15 ISOR and 3 DFIX instructions were used to restrain the phenyl groups and carborane cages so that there were 93 restraints in the data.
These structures were solved by direct methods, using Fourier techniques, and refined on F2 by a full-matrix least-squares method. All calculations were carried out with the SHELXTL program. A summary of the crystallographic data and selected experimental information are shown in Supplementary Information Table 1 and Table 2.
CCDC-1559018 (3), 1559019 (4), 1559020 (5), 1559021 (6) and 1559022 (2b) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Synthesis of complex 3 [(Py)2Pd2(4-S-1-PhCN-1,2-C2B10H10)]
The palladium starting material ([(Py)2Pd(1-PhCNS-1,2-C2B10H10)], 54 mg, 0.10 mmol) and distilled toluene (10 mL) were added to a Schlenk flask under nitrogen atmosphere. After the reaction mixture was stirred at 110°C for ca. 3 min, the colorless solution turned bright yellow. The solvent was then removed under vacuum. The crude solid was purified by column chromatography on silica gel. Elution with petroleum ether/CH2Cl2 (1/1) gave the pure complex 3 (64 mg, 70%) as a yellow crystalline solid. 1H NMR (400 MHz, CDCl3): δ/ppm: 1.30–2.75 (m, 18H, B-H); 4.29 (s, 1H, Ccage-H); 8.08, 8.07, 7.01, 6.99, 6.97, 6.96, 6.94 (10H, pyridine H); 7.51, 7.49, 7.47, 6.92, 6.90, 6.85, 6.84 (10H, ArH). 13C NMR (100 MHz, CDCl3): δ/ppm: 52.67, 92.37 (cage C); 181.31 (−N=C−), 152.09, 128.30, 124.62 (pyridine C); 149.81, 137.02, 123.65, 120.56 (ArC). 11B NMR (160 MHz, CDCl3): δ/ppm: –2.24 (4B), –11.71 14B), –18.66 (2B). IR (KBr): ν (cm−1) 1604 (C=N), 2575 (B−H), 1448 (C=N, pyridine). EI-MS: m/z calcd for (3 – 2 pyridine + H)+: 768.20. Found: 768.20. Elemental analysis: calculated for C28H40B20N4Pd2S2: C, 36.32; H, 4.35; N, 6.05. Found: C, 36.42; H, 4.44; N, 6.08.
Synthesis of complex 4 [(PhCH2N≡C)2Pd2(4-S-1-PhCN-1,2-C2B10H10)]
The palladium starting material [(PhCH2N≡C)2Pd(1-PhCNS-1,2-C2B10H10), 63 mg, 0.10 mmol] and distilled toluene (10 mL) were added to a Schlenk flask under nitrogen atmosphere. After the reaction mixture was stirred at 110°C for ca. 20 min, the colorless solution turned yellow green. The solvent was then removed under vacuum. The crude solid was purified by column chromatography on silica gel. Elution with petroleum ether/CH2Cl2 (1/1) gave the pure complex 4 (48 mg, 50%) as a yellow crystalline solid. 1H NMR (400 MHz, CDCl3): δ/ppm: 1.60–3.26 (m, 18H, B-H), 4.29 (s, 2H, Ccage–H); 4.18 (s, 4H, −CH2−); 7.37, 7.36, 7.28, 7.24, 7.10, 7.08, 7.05, 7.04 (20H, ArH). 13C NMR (100 MHz, CDCl3): δ/ppm: 54.86, 94.19 (cage C), 47.38 (−CH2−); 175.43 (−N=C−); 151.71 (−N≡C−); 130.32, 129.12, 128.99, 128.88, 127.36, 125.16, 121.91 (ArC). Elemental analysis: calculated for C32H44B20N4Pd2S2: C, 40.76; H, 4.43; N, 5.59. Found: C, 40.88; H, 4.52; N, 5.61.
Synthesis of complex 5 [(PPh3)2Pd2(4-S-1-PhCN-1,2-C2B10H10)]
The palladium starting material ([(PPh3)2Pd(1-PhCNS-1,2-C2B10H10)], 91 mg, 0.10 mmol) and distilled toluene (10 mL) were added to a Schlenk flask under a nitrogen atmosphere. After the reaction mixture was stirred at 110°C for ca. 2 h, the light yellow solution turned bright yellow. The solvent was then removed under vacuum. The crude solid was purified by column chromatography on silica gel. Elution with petroleum ether/CH2Cl2 (1/1) gave the pure complex 5 (39 mg, 30%) as a yellow crystalline solid. 1H NMR (400 MHz, CDCl3): δ/ppm: 1.26–2.85 (m, 18H, B-H), 3.49 (s, 2H, Ccage–H); 8.44, 7.67, 7.48, 7.46, 7.38, 7.08, 6.84, 6.56 (40H, ArH). 13C NMR (100 MHz, CDCl3): δ/ppm: 51.05, 96.22 (cage C), 175.91 (−N=C−), 148.26, 138.01, 134.77, 134.48, 134.35, 133.29, 132.19, 131.15, 128.75, 128.63, 128.27, 128.05, 125.12, 121.30 (ArC). 11B NMR (160 MHz, CDCl3): δ/ppm: –3.33 (6B), –9.55 (6B), –14.07 (8B). IR (KBr): ν (cm−1) 1608 (C=N), 2589 (B−H). Elemental analysis: calculated for C54H60B20N2P2Pd2S2: C, 50.21; H, 4.70; N, 2.28. Found: C, 50.19; H, 4.68; N, 2.17.
Synthesis of complex 6 [(Dppe)2Pd(1-PhCNS-1,2-C2B10H10)]
Tetranuclear palladium complex 1 (Pd4(1-PhCNS-1,2-C2B10H10)4, 77 mg, 0.05 mmol) was dissolved in 5 mL of CH2Cl2, into which 1,2-bis(diphenylphosphino)ethane (Dppe) (80 mg, 0.2 mmol) was added. After ca. 4 h, the solvent was removed and the residue was washed a few times with n-hexane, producing a white solid. Complex 6 was obtained quantitatively. 1H NMR (400 MHz, CDCl3): δ/ppm: 1.60–3.20 (m, 9H, B-H); 4.22 (s, 1H, Ccage–H); 2.46 (m, 4H, −CH2−); 7.72, 7.70, 7.69, 7.67, 7.62, 7.58, 7.55, 7.51, 7.48, 7.46, 7.41, 7.39, 7.37, 7.35, 7.28, 7.24, 7.08, 7.06, 7.02, 7.01, 6.99 (m, 25H, ArH). 13C NMR (100 MHz, CDCl3): δ/ppm: 62.49, 84.84 (cage C); 150.13 (−N=C−); 134.18, 134.11, 133.99, 133.13, 133.11, 133.00, 132.99, 131.34, 131.24, 130.81, 129,11, 129.07, 129.01, 128.97, 128.71, 128.42 (ArC). IR (KBr): ν (cm−1) 1581 (C=N), 2585 (B−H). Elemental analysis: calculated for C35H39B10NP2PdS: C, 53.80.75; H, 5.15; N, 1.83. Found: C, 53.74; H, 5.03; N, 1.79.
Synthesis of complex 2b [(PhCH2N≡C)2Pd(1-PhCNS-1,2-C2B10H10)]
Tetranuclear palladium complex 1 (Pd4(1-PhCNS-1,2-C2B10H10)4, 77 mg, 0.05 mmol) was dissolved in 5 mL of CH2Cl2, into which benzyl isocyanide (0.05 mL, excess) was added. About 10 min later, the solvent was removed and the residue washed a few times with n-hexane, producing a white solid. Complex 2b was obtained quantitatively. 1H NMR (400 MHz, CDCl3): δ/ppm: 1.59–3.27 (m, 9H, B–H), 4.22 (s, 1H, Ccage–H); 4.83, 4.67 (s, 4H, −CH2−); 7.41, 7.40, 7.37, 7.36, 7.35, 7.32, 7.31, 7.29, 7.24, 7.23 (15H, ArH). 13C NMR (100 MHz, CDCl3): δ/ppm: 62.64, 83.73 (cage C), 150.36 (−N=C−), 171.84 (−N≡C−), 48.24 (−CH2−); 129.33, 129.21 129.16, 128.82, 127.54, 127.02, 123.92, 120.86 (ArC). IR (KBr): ν (cm−1) 1569 (C=N), 2582 (B−H). Elemental analysis: calculated for C25H29B10N4PdS: C, 47.55; H, 4.71; N, 8.90. Found: C, 47.50; H, 4.62; N, 8.86.
Dedicated to
Professor Narayan Hosmane on the occasion of his 70th birthday.
Article note
A collection of invited papers based on presentations at the 16th International Meeting on Boron Chemistry (IMEBORON-16), Hong Kong, 9-13 July 2017.
Acknowledgments
This work was supported by the National Science Foundation of China (21531002, 21374019), the Program for Changjiang Scholars and Innovative Research Team in University (IRT-15R12) and the Shanghai Science and Technology Committee (16ZD2270100).
References
[1] R. N. Grimes, Carboranes, Vol. 9, 2nd ed., in Chap. 9, Icosahedral Carboranes: 1,2-C2B10H12, pp. 301–540, Academic Press, New York, NY (2011).10.1016/B978-0-12-374170-7.00009-4Suche in Google Scholar
[2] P. A. Jelliss, in Boron Science: New Technologies and Applications, in Chap. 15, Photoluminescence from Boron-Based Polyhedral Clusters, N. S. Hosmane (Ed.), pp. 355–384, CRC Press, Boca Raton, FL (2012).Suche in Google Scholar
[3] R. J. Zaldivar, G. S. Rellick, J. Yang. J. Mater. Res.8, 501 (1993).10.1557/JMR.1993.0501Suche in Google Scholar
[4] J. Poater, M. Solà, C. Viñas, F. Teixidor. Chem. Eur. J.22, 7437 (2016).10.1002/chem.201600510Suche in Google Scholar PubMed
[5] A. A. Attia, A. Lupan, R. B. King. Phys. Chem. Chem. Phys.18, 11707 (2016).10.1039/C5CP05708FSuche in Google Scholar PubMed
[6] H. Dai, G. Liu, X. Zhang, H. Yan, C. Lu, Organometallics35, 1488 (2016).10.1021/acs.organomet.6b00082Suche in Google Scholar
[7] A. H. Soloway, W. Tjarks, A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni, J. G. Wilson. Chem. Rev.98, 1515 (1998).Suche in Google Scholar
[8] M. F. Hawthorne, A. Maderna. Chem. Rev.99, 3421 (1999).10.1021/cr980442hSuche in Google Scholar PubMed
[9] J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein, K. A. Stephenson. Coord. Chem. Rev.232, 173 (2002).10.1016/S0010-8545(02)00087-5Suche in Google Scholar
[10] I. B. Sivaev, V. I. Bregadze, N. T. Kuznetsov. Russ. Chem. Bull.51, 1362 (2002).10.1023/A:1020942418765Suche in Google Scholar
[11] M. F. Hawthorne, M. W. Lee. J. Neuro-Oncol.62, 33 (2003).10.1023/A:1023253309343Suche in Google Scholar
[12] Y. Endo, T. Yoshimi, C. Miyaura. Pure Appl. Chem.75, 1197 (2003).10.1351/pac200375091197Suche in Google Scholar
[13] Z. J. Leśnikowski. J. Med. Chem.59, 7738 (2016).10.1021/acs.jmedchem.5b01932Suche in Google Scholar PubMed
[14] S. Xuan, N. Zhao, Z. Zhou, F. R. Fronczek, M. G. H. Vicente. J. Med. Chem.59, 2109 (2016).10.1021/acs.jmedchem.5b01783Suche in Google Scholar PubMed PubMed Central
[15] K. Watanabe, M. Hirata, T. Tominari, C. Matsumoto, Y. Endo, G. Murphy, H. Nagase, M. Inada, C. Miyaura. Biochem. Biophys. Res. Commun.478, 279 (2016).10.1016/j.bbrc.2016.07.027Suche in Google Scholar PubMed
[16] J. Wang, L. Chen, J. Ye, Z. Li, H. Jiang, H. Yan, M. Yu, I. B. Sivaev, V. I. Bregadze, X. M. Wang, Biomacromolecules18, 1466 (2017).10.1021/acs.biomac.6b01845Suche in Google Scholar PubMed
[17] R. Kawasaki, Y. Sasaki, K. Akiyoshi. Biochem. Biophys. Res. Commun.483, 147 (2017).Suche in Google Scholar
[18] G. Calabrese, A. Daou, A. Rova, E. Tseligka, I. S. Vizirianakis, D. G. Fatouros, J. Tsibouklis. Med. Chem. Commun.8, 67 (2017).10.1039/C7MD90004JSuche in Google Scholar PubMed PubMed Central
[19] F. Teixidor, R. Núñez, M. A. Flores, A. Demonceau, C. Viñas. J. Organomet. Chem.614–615, 48 (2000).10.1016/S0022-328X(00)00576-3Suche in Google Scholar
[20] Y. Zhu, K. Vyakaranam, J. A. Maguire, W. Quintana, F. Teixidor, C. Viñas, N. S. Hosmane. Inorg. Chem. Commun.4, 486 (2001).10.1016/S1387-7003(01)00256-8Suche in Google Scholar
[21] J. Zhang, X. Wang, G.-X. Jin. Coord. Chem. Rev.250, 95 (2006).10.1016/j.ccr.2005.06.002Suche in Google Scholar
[22] H. Shen, Z. W. Xie. J. Am. Chem. Soc.132, 11473 (2010).10.1021/ja101796kSuche in Google Scholar PubMed
[23] C. E. Housecroft. J. Organomet. Chem.798, 218 (2015).10.1016/j.jorganchem.2015.04.047Suche in Google Scholar
[24] I. Boldog, P. J. Bereciartua, R. Bulánek, M. Kučeráková, M. Tomandlová, M. Dušek, J. Macháček, D. D. Vos, T. Baše. Cryst. Eng. Comm.18, 2036 (2016).10.1039/C5CE02501JSuche in Google Scholar
[25] M. Y. Tsang, S. Rodríguez-Hermida, K. C. Stylianou, F. Tan, D. Negi, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte, C. Verdugo-Escamilla, M. Guerrero, J. Sort, J. Juanhuix, D. Maspocha, J. G. Planas. Cryst. Growth Des. 17, 846 (2017).10.1021/acs.cgd.6b01682Suche in Google Scholar
[26] H. Lee, C. B. Knobler, M. F. Hawthorne. Chem. Commun.24, 2485 (2000).10.1039/b007353iSuche in Google Scholar
[27] M. A. Fox, A. K. Hughes. Coord. Chem. Rev.248, 457 (2004).10.1016/j.ccr.2003.10.002Suche in Google Scholar
[28] B. H. Northrop, H. B. Yang, P. J. Stang. Chem. Commun.45, 5896 (2008).10.1039/b811712hSuche in Google Scholar PubMed PubMed Central
[29] Z.-J. Yao, W.-B. Yu, Y.-J. Lin, S.-L. Huang, Z.-H. Li, G.-X. Jin. J. Am. Chem. Soc.136, 2825 (2014).10.1021/ja4115665Suche in Google Scholar PubMed
[30] X.-Y. Shen, L. Zhang, Y.-J. Lin, G.-X. Jin. Dalton Trans.43, 17200 (2014).10.1039/C4DT02495HSuche in Google Scholar
[31] J. C. Axtell, K. O. Kirlikovali, P. I. Djurovich, D. Jung, T. V. Nguyen, B. Munekiyo, A. T. Royappa, A. L. Rheingold, A. M. Spokoyny. J. Am. Chem. Soc.138, 15758 (2016).10.1021/jacs.6b10232Suche in Google Scholar PubMed
[32] R. Furue, T. Nishimoto, I. S. Park, J. Lee, T. Yasuda. Angew. Chem. Int. Ed.55, 7171 (2016).10.1002/anie.201603232Suche in Google Scholar PubMed
[33] X. Li, H. Yan, Q. Zhao. Chem. Eur. J.22, 1888 (2016).10.1002/chem.201503456Suche in Google Scholar PubMed
[34] R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani, F. Teixidor. Chem. Rev.116, 14307 (2016).10.1021/acs.chemrev.6b00198Suche in Google Scholar
[35] J. Cabrera-González, C. Viñas, M. Haukka, S. Bhattacharyya, J. Gierschner, R. Núñez. Chem. Eur. J.22, 13588 (2016).10.1002/chem.201601177Suche in Google Scholar
[36] K. Q. Qiu, H. Y. Huang, B. Y. Liu, Y. K. Liu, Z. Y. Huang, Y. Chen, L. N. Ji, H. Chao. ACS Appl. Mater. Interfaces8, 12702 (2016).10.1021/acsami.6b03422Suche in Google Scholar
[37] M. Herberhold, H. Yan, W. Milius, B. Wrackmeyer. Angew. Chem. Int. Ed.38, 3689 (1999).10.1002/(SICI)1521-3773(19991216)38:24<3689::AID-ANIE3689>3.0.CO;2-TSuche in Google Scholar
[38] J.-Q. Wang, C. Zhang, L.-H. Weng, M. Herberhold, G.-X. Jin. Angew. Chem. Int. Ed.44, 259 (2005).10.1002/anie.200461348Suche in Google Scholar
[39] A. M. Prokhorov, P. A. Slepukhin, L. Rusinov, V. N. Kalinin, D. N. Kozhevnikov. Chem. Commun.47, 7713 (2011).10.1039/c1cc12230dSuche in Google Scholar
[40] R. Zhang, L. Zhu, G. F. Liu, H. M. Dai, Z. Z. Lu, J. B. Zhao, H. Yan. J. Am. Chem. Soc.134, 10341 (2012).10.1021/ja303334tSuche in Google Scholar
[41] Z.-J. Yao, G.-X. Jin. Coord. Chem. Rev.257, 2522 (2013).10.1016/j.ccr.2013.02.004Suche in Google Scholar
[42] D. Olid, R. Núñez, C. Viñas, F. Teixidor. Chem. Soc. Rev.42, 3318 (2013).10.1039/c2cs35441aSuche in Google Scholar
[43] J. Estrada, S. E. Lee, S. G. McArthur, A. El-Hellani, F. S. Tham, V. Lavallo. J. Organomet. Chem.798, 214 (2015).10.1016/j.jorganchem.2015.05.008Suche in Google Scholar
[44] Z. Qiu. Tetrahedron Lett.56, 963 (2015).10.1016/j.tetlet.2015.01.038Suche in Google Scholar
[45] R. M. Dziedzic, L. M. Saleh, J. C. Axtell, J. L. Martin, S. L. Stevens, A. T. Royappa, A. M. Spokoyny. J. Am. Chem. Soc.138, 9081 (2016).10.1021/jacs.6b05505Suche in Google Scholar PubMed
[46] Y. J. Quan, Z. W. Xie. J. Am. Chem. Soc.136, 15513 (2014).10.1021/ja509557jSuche in Google Scholar PubMed
[47] H. Lyu, Y. J. Quan, Z. W. Xie. Angew. Chem. Int. Ed.54, 10623 (2015).10.1002/anie.201504481Suche in Google Scholar PubMed
[48] Y. J. Quan, Z. W. Xie. Angew. Chem. Int. Ed.55, 1295 (2016).10.1002/anie.201507697Suche in Google Scholar PubMed
[49] Y. J. Quan, C. Tang, Z. W. Xie. Chem. Sci.7, 5838 (2016).10.1039/C6SC00901HSuche in Google Scholar PubMed PubMed Central
[50] H. Lyu, Y. J. Quan, Z. W. Xie. Angew. Chem. Int. Ed.55, 11840 (2016).10.1002/anie.201605880Suche in Google Scholar PubMed
[51] H. Lyu, Y. J. Quan, Z. W. Xie. J. Am. Chem. Soc.138, 12727 (2016).10.1021/jacs.6b07086Suche in Google Scholar PubMed
[52] Z. P. Zheng, W. Jiang, A. A. Zinn, C. B. Knobler, M. F. Hawthorne. Inorg. Chem.34, 2095 (1995).10.1021/ic00112a023Suche in Google Scholar
[53] L. I. Zakharkin, V. A. Olshevskaya, E. V. Vorontsov, P. V. Petrovsky. Russ. Chem. Bull.45, 2614 (1996).10.1007/BF01431128Suche in Google Scholar
[54] A. Herzog, A. Maderna, G. N. Harakas, C. B. Knobler, M. F. Hawthorne. Chem. Eur. J.5, 1212 (1999).10.1002/(SICI)1521-3765(19990401)5:4<1212::AID-CHEM1212>3.0.CO;2-WSuche in Google Scholar
[55] B. P. Dash, R. Satapathy, J. A. Maguire, N. S. Hosmane. Org. Lett.10, 2247 (2008).10.1021/ol8005248Suche in Google Scholar
[56] Z. Z. Qiu, Y. J. Quan, Z. W. Xie. J. Am. Chem. Soc.135, 12192 (2013).10.1021/ja405808tSuche in Google Scholar
[57] H. Topsøe, B. Hinnemann, J. K. Nørskov, J. V. Lauritsen, F. Besenbacher, P. L. Hansen, G. Hytoft, R. G. Egeberg, K. G. Knudsen. Catal. Today107–108, 12 (2005).10.1016/j.cattod.2005.07.165Suche in Google Scholar
[58] H. X. Li, W. Zhao, H. Y. Li, Z. L. Xu, W. X. Wang, J. P. Lang. Chem. Commun.49, 4259 (2013).Suche in Google Scholar
[59] L. Wang, W. He, Z. Yu. Chem. Soc. Rev.42, 599 (2013).10.1039/C2CS35323GSuche in Google Scholar
[60] W. H. Zhang, Z. G. Ren, J. P. Lang, Chem. Soc. Rev. 45, 4995 (2016).10.1039/C6CS00096GSuche in Google Scholar
[61] Y. Tian, L. Wang, H. Z. Yu. RSC Adv.6, 61996 (2016).10.1039/C6RA12607CSuche in Google Scholar
[62] X. F. Jiang, H. Huang, Y. F. Chai, T. L. Lohr, S. Y. Yu, W. Z. Lai, Y. J. Pan, M. Delferro, T. J. Marks. Nat. Chem. 9, 188 (2017).10.1038/nchem.2637Suche in Google Scholar
[63] A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni, J. G. Wilson. Chem. Rev.98, 1515 (1998).10.1021/cr941195uSuche in Google Scholar
[64] J. A. Todd, P. Turner, E. J. Ziolkowski, L. M. Rendina. Inorg. Chem.44, 6401 (2005).10.1021/ic050265kSuche in Google Scholar
[65] R. Kawasaki, Y. Sasaki, K. Akiyoshi. Biochem. Biophys. Res. Commun. 483, 147 (2017).10.1016/j.bbrc.2016.12.176Suche in Google Scholar PubMed
[66] R. Kawasaki, Y. Sasaki, K. Akiyoshi. Chem. Lett.46, 513 (2017).10.1246/cl.161137Suche in Google Scholar
[67] A. N. Ay, H. Akar, A. Zaulet, C. Viňas, F. Teixidor, B. Zumreoglu-Karan. Dalton. Trans.46, 3303 (2017).10.1039/C7DT00100BSuche in Google Scholar PubMed
[68] J. Wright, A. Kaczmarczyk. Inorg. Chem.12, 1453 (1973).10.1021/ic50124a057Suche in Google Scholar
[69] J. G. Planas, C. Viñas, F. Teixidor, A. Comas-Vives, G. Ujaque, A. Lledós, M. E. Light, M. B. Hursthouse. J. Am. Chem. Soc.127, 15976 (2005).10.1021/ja055210wSuche in Google Scholar PubMed
[70] F. Teixidor, A. Laromaine, R. Kivekäs, R. Sillanpää, C. Viñas, R. Vespalec, H. Horáková. Dalton. Trans.3, 345 (2008).10.1039/B715362GSuche in Google Scholar PubMed
[71] C. Wu, B. Xu, J. Zhao, Q. Jiang, F. Wei, H. Jiang, X. M. Wang, H. Yan. Chem. Eur. J.16, 8914 (2010).10.1002/chem.201000605Suche in Google Scholar PubMed
[72] M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant, C. A. Mirkin. Nat. Chem.3, 590 (2011).10.1038/nchem.1088Suche in Google Scholar PubMed
[73] X. L. Zhang, X. Tang, J. J. Yang, Y. Z. Li, H. Yan, V. I. Bregadze. Organometallics32, 2014 (2013).10.1021/om400097mSuche in Google Scholar
[74] J. Plešek, S. Heřmánek. Collect. Czech. Chem. Commun.46, 687 (1981).10.1135/cccc19810687Suche in Google Scholar
[75] Y. P. Wang, L. Zhang, Y. J. Lin, Z. H. Li, G.-X. Jin. Chem. Eur. J.23, 1814 (2017).10.1002/chem.201603851Suche in Google Scholar PubMed
Supplemental Material
The online version of this article (https://doi.org/10.1515/pac-2017-0609) offers supplementary material, available to authorized users.
©2018 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Artikel in diesem Heft
- Frontmatter
- In this issue
- Preface
- 16th International Meeting on Boron Chemistry (IMEBORON XVI)
- Conference papers
- Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
- When diazo compounds meet with organoboron compounds
- Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
- Novel sulfur containing derivatives of carboranes and metallacarboranes
- Metal–metal bonding in deltahedral dimetallaboranes and trimetallaboranes: a density functional theory study
- Nanostructured boron compounds for cancer therapy
- Heterometallic boride clusters: synthesis and characterization of butterfly and square pyramidal boride clusters*
- Influence of fluorine substituents on the properties of phenylboronic compounds
- Copper-catalyzed asymmetric dearomative borylation: new pathway to optically active heterocyclic compounds
- Borenium and boronium ions of 5,6-dihydro-dibenzo[c,e][1,2]azaborinine and the reaction with non-nucleophilic base: trapping of a dimer and a trimer of BN-phenanthryne by 4,4′-di-tert-butyl-2,2′-bipyridine
- The electrophilic aromatic substitution approach to C–H silylation and C–H borylation
- Recent advances in B–H functionalization of icosahedral carboranes and boranes by transition metal catalysis
- closo-Dodecaborate-conjugated human serum albumins: preparation and in vivo selective boron delivery to tumor
- IUPAC Technical Report
- Risk assessment of effects of cadmium on human health (IUPAC Technical Report)
Artikel in diesem Heft
- Frontmatter
- In this issue
- Preface
- 16th International Meeting on Boron Chemistry (IMEBORON XVI)
- Conference papers
- Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
- When diazo compounds meet with organoboron compounds
- Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
- Novel sulfur containing derivatives of carboranes and metallacarboranes
- Metal–metal bonding in deltahedral dimetallaboranes and trimetallaboranes: a density functional theory study
- Nanostructured boron compounds for cancer therapy
- Heterometallic boride clusters: synthesis and characterization of butterfly and square pyramidal boride clusters*
- Influence of fluorine substituents on the properties of phenylboronic compounds
- Copper-catalyzed asymmetric dearomative borylation: new pathway to optically active heterocyclic compounds
- Borenium and boronium ions of 5,6-dihydro-dibenzo[c,e][1,2]azaborinine and the reaction with non-nucleophilic base: trapping of a dimer and a trimer of BN-phenanthryne by 4,4′-di-tert-butyl-2,2′-bipyridine
- The electrophilic aromatic substitution approach to C–H silylation and C–H borylation
- Recent advances in B–H functionalization of icosahedral carboranes and boranes by transition metal catalysis
- closo-Dodecaborate-conjugated human serum albumins: preparation and in vivo selective boron delivery to tumor
- IUPAC Technical Report
- Risk assessment of effects of cadmium on human health (IUPAC Technical Report)