Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
-
Mohammed A. Altahan
 ,
Simon J. Coles
,
Simon J. Coles
 and
Peter N. Horton
and
Peter N. Horton

Abstract
The title compound, [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane) (1), was prepared as a crystalline solid in moderate yield from the reaction of B(OH)3 with [Zn(en)3][OH]2 in aqueous solution (15:1) ratio. The structure contains a neutral bimetallic complex comprised of a unusual dodecaborate(6−) anion ligating two [H3NCH2CH2NH2Zn(en)n]3+ centers in a monodentate (n=1) or tridentate (n=0) manner.
Introduction
Oxidoborates (or more correctly hydroxyoxidoborates) are anionic boron species with boron atoms linked solely to oxygen atoms and such compounds are commonly referred to a ‘borates’ [1]. Polyborate anions can be readily categorized as ‘isolated’ or ‘insular’, containing discrete anionic moieties, or ‘condensed’ with more condensed 2D or 3D polymeric chains, sheets or networks [2], [3], [4]. These anions contain numerous fused B–O rings and cages with the ‘boroxole’ {B3O3} ring system a very common structural motif. Salts containing polyborate anions have attracted recent attention due to their applications as bulk chemicals and possible applications as luminescent, second harmonic generation, ferroelectric, flame retardant and non-linear optical materials [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16].
Polyborates are readily synthesized by solvothermic methods or the addition of B(OH)3 to a basic aqueous solution containing templating cations: polyborate salts containing isolated anions are generally obtained from aqueous solution, whereas solvothermal methods often give the more condensed species [17]. Polyborates crystallize from aqueous solution as cation templated self-assembled solids [18] since in basic aqueous solution B(OH)3 exists as a dynamic combinatorial library (DCL) [19], [20] of numerous polyborate anions [21], [22], [23], [24], [25]. Polyborate salts containing the pentaborate(1−) anion, [B5O6(OH)4]−, are very common and this is because these salts have a strong three-dimensional H-bonded anionic lattice and this lattice is sufficiently flexible to accommodate many medium sized non-metal unicharged cations [26], [27].
We are interested in the synthesis of structurally novel polyborate anions and have adopted a strategy of templating such anions by the use of non-innocent (sterically demanding or multi H-bond donating) and/or more highly charged transition-metal cations. We have recently reported the synthesis of salts containing isolated polyborate anions partnered with inert cobalt(3+) complexes and have described the synthesis and structures of two previously unobserved isolated polyborate anions: heptaborate(3−) and octaborate(2−) [28], [29]. We have also prepared a series of salts and neutral complexes containing more labile copper(2+) centers where pentaborate(1−) or hexaborate(2−) ions are found coordinated to the metal center [30].
We now report an unusual neutral zwitterionic bizinc complex, [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane) (1), derived from a self-assembly in methanolic aqueous solution of B(OH)3 and components of the labile complex salt, [Zn(en)3](OH)2.
Results and discussion
Synthesis and characterization
The title compound, [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (1), was prepared in moderate yield (40%) as a colorless crystalline material by a self-assembly process involving a prolonged (30 days) recrystallization period of an aqueous methanolic reaction mixture originally containing B(OH)3 and [Zn(en)3](OH)2 (eq. 1). The [Zn(en)3](OH)2 was obtained from [Zn(en)3]Cl2 by use of an ion exchange resin.
Compound 1 was characterized primarily by single-crystal XRD study (see below) and its structure is fully consistent with other characterization data that we were able to obtain. Compound 1 is formulated as a neutral molecule although it is zwitterionic with several atoms possessing formal charges and it contains some interesting features (see below). The bulk sample gave satisfactory elemental analysis and its powder XRD pattern was consistent with that derived from the single-crystal XRD analysis, confirming homogeneity. Compound 1 is diamagnetic with a molar magnetic susceptibility of −414.2×10−6 cm3 mol−1 and its IR spectrum shows several strong absorptions in the B–O stretching region which are further assigned as follows: 1355 cm−1 (asymmetric Btrig–O stretch), 1163 cm−1 and 1041 cm−1 (asymmetric Btet–O stretch), 951 cm−1 and 900 cm−1 (symmetric Btrig–O stretch), and 856 cm−1 (symmetric Btet–O) [31]. Compound 1 is decomposed by dissolution in aqueous solution with (1H and 13C) NMR consistent with the ethylenediamine species and 11B NMR showing a single peak (+16.1 ppm) indicative of one signal arising from rapid B(OH)3/OH− exchange equilibria [21], [22], [23], [24], [25].
Solid-state structure
Crystallographic data for compound 1 are given in Table 1, and the structure of 1, together with associated atomic numbering scheme is given in Fig. 1. Its structure is best represented as comprised of a [B12O18(OH)6]6− ligand coordinated to, and bridging two zinc(II) centers.
Crystallographic data for compound 1.
| Formula | C6H48B12N6O32Zn2 |
| D calc./g cm−3 | 1.870 |
| μ/mm−1 | 1.505 |
| Formula weight | 976.96 |
| Color | Colorless |
| Shape | Block |
| Size/mm3 | 0.100×0.090×0.040 |
| T/K | 100(2) |
| Crystal system | Monoclinic |
| Flack parameter | −0.006(7) |
| Hooft parameter | −0.019(7) |
| Space group | Cc |
| a/Å | 17.2990(5) |
| b/Å | 12.5264(3) |
| c/Å | 16.6091(4) |
| α/° | 90 |
| β/° | 105.358(3) |
| γ/° | 90 |
| V/Å3 | 3470.58(16) |
| Z | 4 |
| Z′ | 1 |
| Wavelength/Å | 0.71073 |
| Radiation type | MoKα |
| Θmin/° | 2.436 |
| Θmax/° | 27.484 |
| Measured Refl. | 21551 |
| Independent Refl. | 7310 |
| Reflections used | 7109 |
| R int | 0.0253 |
| Parameters | 536 |
| Restraints | 3 |
| Largest peak | 1.054 |
| Deepest hole | −0.455 |
| GoF | 1.048 |
| wR 2 (all data) | 0.0865 |
| wR 2 | 0.0856 |
| R 1 (all data) | 0.0334 |
| R 1 | 0.0322 |
![Fig. 1:
Diagram showing atomic numbering scheme for [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)] (1).](/document/doi/10.1515/pac-2017-0901/asset/graphic/j_pac-2017-0901_fig_003.jpg)
Diagram showing atomic numbering scheme for [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)] (1).
Compound 1 contains a dodecaborate(6−) anion. This anion was first reported in 1990 in Ag6[B12O18(OH)6]·3H2O [32] and was described as an ‘insular’ polyborate anion. The anion is comprised of six boroxole rings linked together in such a way as to produce a larger central 12-membererd {B6O6} ring (Fig. 2), with each boron atom within this ring being 4-coordinate and carrying a formal negative charge. These six 4-coordinate boron centers (B1–B6) have B–O bond lengths ranging from 1.435(5) to 1.499(6) Å and the O–B–O angles ranging from 106.3(3) to 112.6(3)°. The other six boron centers in the anion, B7–B12, are 3-coordinate and have significantly shorter B–O bonds {1.349(6)–1.389(6) Å} and larger O–B–O angles which range from 116.1(4) to 123.9(4)°. These distances and angles are very similar to distances and angles observed in the previously reported Ag6[B12O18(OH)6]·3H2O [32] and in acyclic polyboroxole ‘chain’ species, [B5O6(OH)4]− [33], [34], [35], [36], [37], [38], [B7O9(OH)5]2− [39], [40], [41], [42], [43], [B9O12(OH)6]3− [44], [45], [46], and related boroxole systems [47], [48], [49] containing both 3- and 4-coordinate boron centers bound to oxygen. The dodecaborate(6−) in 1 is closely related to deprotonated structures Na8[B12O20(OH)4] [50] and Zn6[B12O24] [51] and is an alternative (but not isomeric) to the hydrated dodecaborate found in K4[B12O16(OH)8] [52] which has four 4-coordinate boron atoms and carries a charge of 4–.
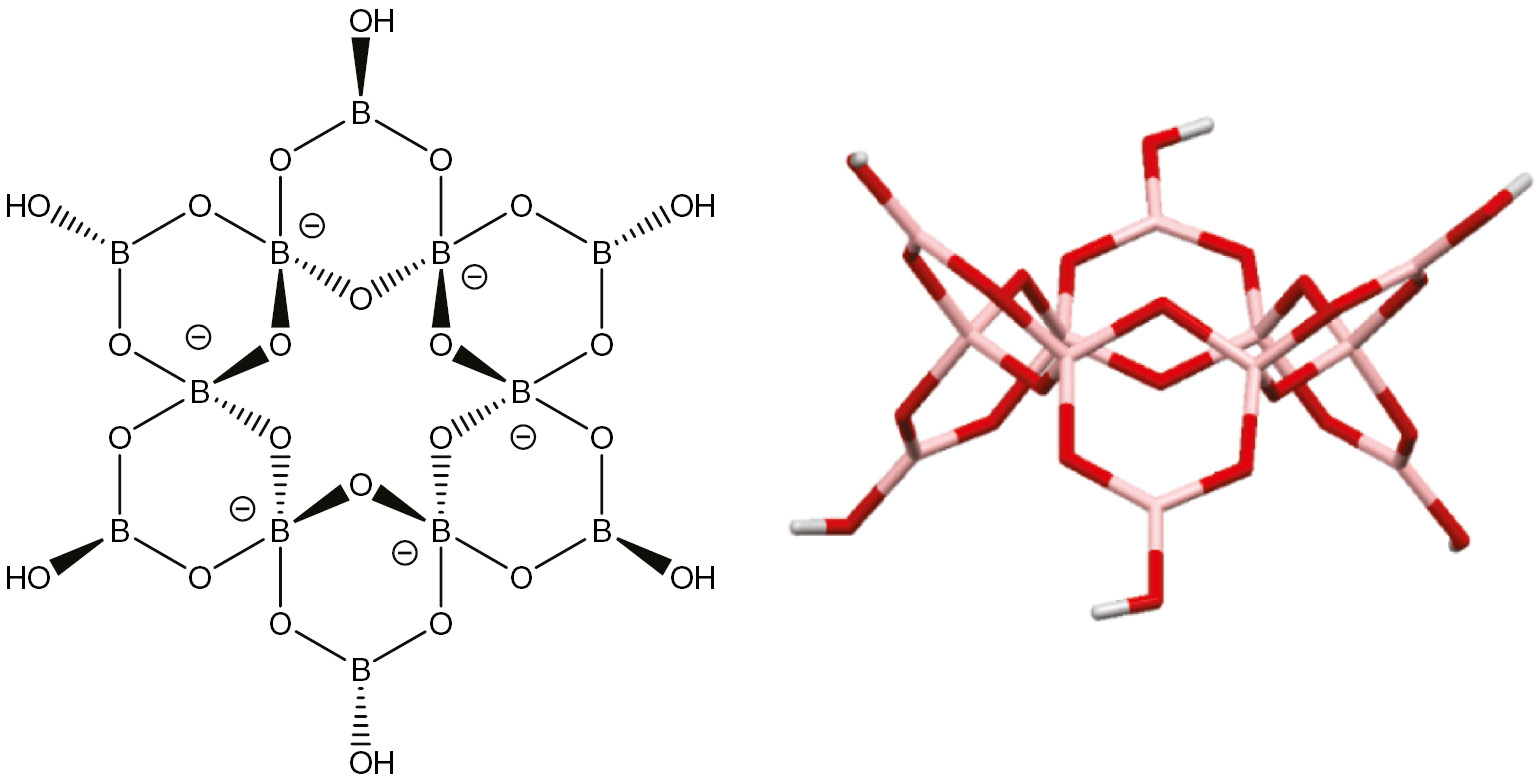
The dodecaborate(6−) ligand as found in 1. The central {B6O6} ring as shown on the left is non-planar with oxygen atoms (red) alternating ‘up’ and ‘down’ as shown in the side-on view (right). Boron atoms are pale pink.
The dodecaborate(6−) anion in 1 utilizes its ‘inner’ ring oxygen atoms to coordinate to the Zn(II) centers and due to their stereochemistry the dodecaborate(6−) anion is ideally set up to bridge metal centers. The dodecaborate(6−) anion has been previously observed coordinated tridentate to metal centers in the following compounds: Na2Cs4Ba2[B12O18(OH)6](OH)4 [53], K7[(BO3)Mn{B12O18(OH)6}]·H2O [54], and K7[(BO3)Zn{B12O18(OH)6}]·H2O [55] with the latter also containing a zinc(II) ion. The coordination modes of the dodecaborate(6−) ligand towards the Zn centers are separated out in Fig. 3. Both Zn atoms in 1 are 4-coordinate and Zn1 is coordinated by the dodecaborate(6−) ligand in a tridentate fashion through oxygen donors O1, O3 and O5 with bond lengths to Zn1 of 1.971(3), 1.976(3) and 1.937(3) Å, respectively. The Zn2 atom is coordinated by the dodecaborate(6−) anion in a mondentate manner solely through O2 with as Zn2–O2 distance of 1.931(3) Å. The coordination number of four is completed on Zn2 by a bidentate en (ethylenediamine) ligand and a monodentate [enH]+ group, whist Zn1 just has one additional monodentate [enH]+ group. The Zn–N distances range from 2.018(5) to 2.043(4) Å, whilst borate O–Zn atom distances (listed above) are significantly shorter and range from 1.937(3) to 1.976(3) Å, and are comparable to those observed elsewhere related zinc-borate species [56], [57], [58], [59], [60], [61], [62], [63] which typically range from 1.919(2) to 2.0099(14) Å. The Zn–N distances are comparable to those observed in related zinc borate complexes with organoamino ligands [56], [57], [58], [59], [60], [61], [62], [63] and ethylenediamine complexes such as [Zn(en)3]Cl2·2H2O [64].
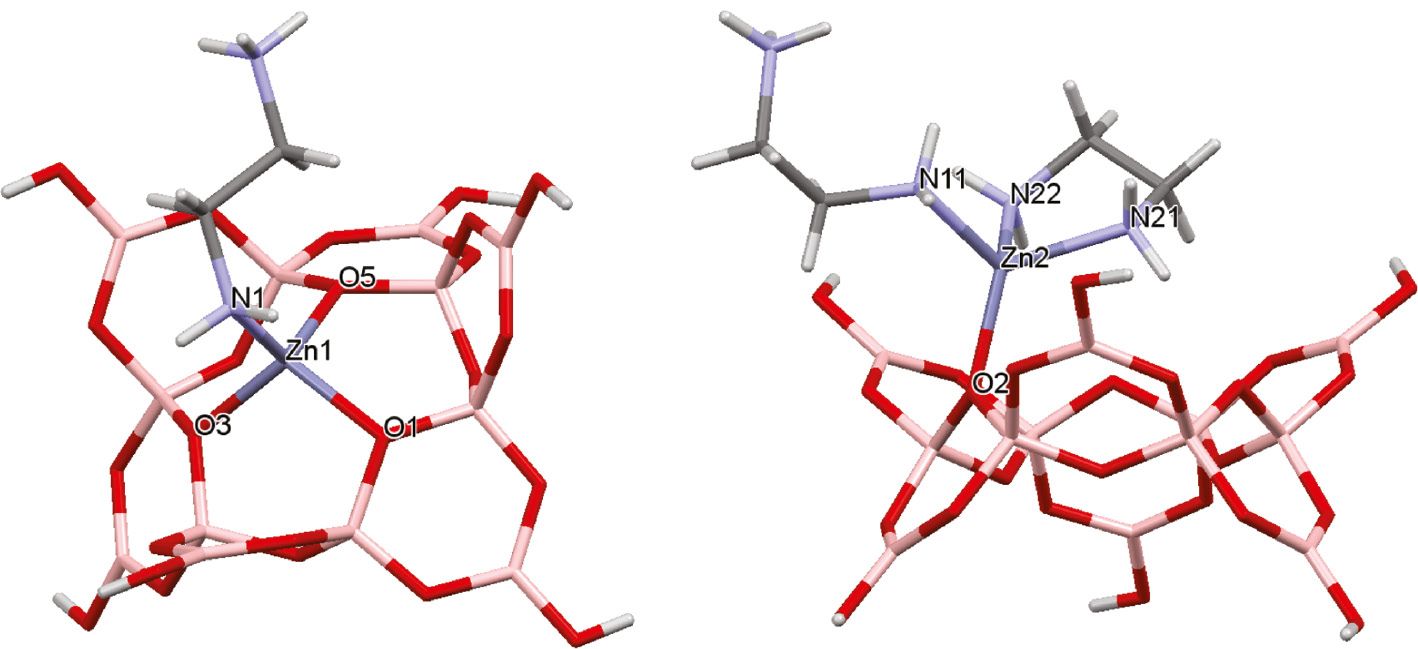
Diagrams showing the two coordination modes of the dodecaborate(6−) ligand. The 4-coordinate Zn1 atom has a tridentate dodecaborate(6−) ligand and a monodentate protonated en ligand (left). The 4-coordinate Zn2 atom a monodentate dodecaborate(6−) ligand, a bidentate en ligand, and a monodentate protonated en ligand coordinated to it.
The hydroxyl hydrogen atoms of the [B12O18(OH)6]6− anion, the amino hydrogen atoms of the en ligands and protonated en ligands, and the waters of crystallization form multiple intramolecular H-bond donor interactions which are presumably responsible for the remarkable self-assembly of 1 from its component parts. In particular, all six hydroxyl groups in the polyborate anion of 1 are involved in energetically favorable R22(8) interactions (Etter nomenclature [65]) with six neighboring molecules of 1, as shown in Fig. 4. Details of these H-bond interactions can be found in the caption to Fig. 4. In addition, the protonated ends of the mondentate H2NCH2CH2NH3 ligands further H-bond to two more molecules of 1 forming a supramolecular chain (Fig. 5) giving an overall ‘coordination number’ of eight for each molecule of 1. The amino hydrogens of the en ligand on Zn2 are involved in an intramolecular H-bond (Fig. 5) and H-bond donation to water molecules, and the waters of crystallization H-bond to each other and dodecaborate anions to further glue the structure together.
![Fig. 4:
The [B12O18(OH)6]6− anion in 1 forms six H-bond donor interactions with six neighboring dodecaborate(6−) units. The interactions involve 8-membered rings R22(8) with D···A oxygen atom distances as follows: O19H19···O14*, 2.779(4) Å; O20H20···O16*, 2.634(4) Å; O21H21···O17*, 2.701(4) Å; O22H22···O8*, 2.899(5) Å; O23H23···O10*, 2.695(4) Å; O24H24···O11*, 2.680(4) Å, where * indicates an acceptor oxygen atom of a neighboring unit. H2O molecules omitted for clarity.](/document/doi/10.1515/pac-2017-0901/asset/graphic/j_pac-2017-0901_fig_011.jpg)
The [B12O18(OH)6]6− anion in 1 forms six H-bond donor interactions with six neighboring dodecaborate(6−) units. The interactions involve 8-membered rings R22(8) with D···A oxygen atom distances as follows: O19H19···O14*, 2.779(4) Å; O20H20···O16*, 2.634(4) Å; O21H21···O17*, 2.701(4) Å; O22H22···O8*, 2.899(5) Å; O23H23···O10*, 2.695(4) Å; O24H24···O11*, 2.680(4) Å, where * indicates an acceptor oxygen atom of a neighboring unit. H2O molecules omitted for clarity.
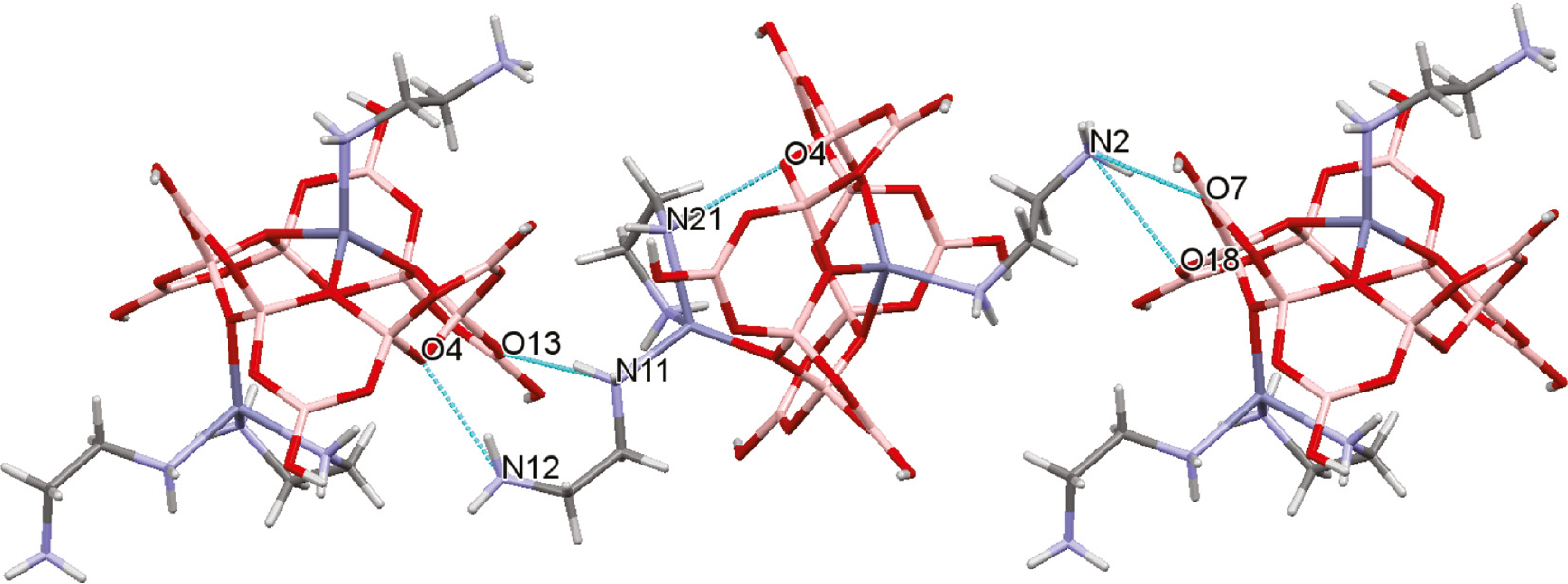
Intermolecular H-bond interactions involving the amino hydrogen atoms link the dodecaborates(6−) anions into an extended chain. The D···A oxygen atom distances as follows: N2H2C···O7*, 2.914(5) Å; N2H2C···O18*, 3.068(5) Å; N11H11B···O13*, 3.053(5) Å; N12H12A···O4*, 2.903(5) Å, where * indicates an acceptor oxygen atom of a neighboring unit. The intramolecular H-bond interaction is also shown: N21H21B···O4, 2.871(5) Å. H2O molecules omitted for clarity.
Conclusion
The zwitterionic bimetallic complex, [(H3NCH2CH2NH2)Zn{κ3O,O′,O″-B12O18(OH)6-κ1O″′}Zn(en)(NH2CH2CH2NH3)]·8H2O has been synthesized in moderate yield from a self-assembly [18] process involving the basic labile zinc(II) complex salt [Zn(en)3](OH)2 and a DCL of polyborate anions [19], [20], [21], [22], [23], [24], [25] derived from B(OH)3. The structure as determined by a single-crystal XRD study reveals that all six borate hydroxyl groups are involved in intermolecular H-bond donor interactions in pairwise R22(8) rings. Intramolecular and intermolecular H-bonds emanating from the amino hydrogen atoms of the ethylenediamine ligands further template the formation of the self-assembled structure.
Experimental
General
NMR spectra were obtained on a Bruker Avance-400 spectrometer in D2O Solution at 128, 400 or 101 MHz for 11B, 1H, and 13C, respectively, and referenced to either BF3·OEt2 (11B) or TMS (1H and 13C). FTIR spectra were obtained as KBr pellets on a Perkin Elmer 100FTIR spectrometer. Powder X-ray diffraction was carried out on a Phillips X’Pert 2040/60 XRD diffractometer with spectra obtained using the Phillips X’Pert Data Collector software. Single-crystal X-ray diffraction was performed at the EPSRC National Crystallographic Service at the University of Southampton. Magnetic susceptibility measurements were performed on a Johnson-Matthey magnetic susceptibility balance. CHN analyses were obtained from OEA Laboratories (Callingham, Cornwall).
Preparation of [Zn(en)3]Cl2·2H2O
Tris(ethylenediamine)zinc(II) chloride dihydrate was prepared as described in the literature [64]. A slight excess of the ethylenediamine (en) (2.10 g, 70%, 24.45 mmol) was added to an aqueous solution of zinc(II) chloride (3.0 g, 22.0 mmol) in distilled water (10 mL). The solution was concentrated by gentle evaporation n a warm water bath before being cooled in an ice bath to yield colorless crystals of tris(ethylenediamine)zinc(II) chloride dihydrate (5.4 g, 70%). M.p.=185–187°C. χm=−211.5×10−6 cm3 mol−1. 1H/ppm: 2.7 (m, 12H, CH2 of en), 4.8 (s, 16 H, NH2, H2O). 13C{1H}/ppm: 39.1. IR (KBr/cm−1): 3383(s), 3261(s), 3154(s), 2949(s), 2933(s), 2885(s), 1651(m), 1587(s), 1458(s), 1331(s), 1275(s), 1147(m), 1023(s), 998(s), 976(m), 645(s), 511(s). [Lit. 3385, 3275, 3150, 2940, 2882, 1653, 1595, 1460, 1335, 1272, 1144, 1022, 996, 977, 644, 515] [66].
Preparation of [Zn2(en)(enH)2{B12O18(OH)6}]·8H2O (1)
A solution of tris(ethylenediamine)zinc(II) chloride dihydrate (1.0 g, 2.8 mmol) in water (10 mL) was added to an aqueous suspension solution of excess activated Dowex 550A (50 g) monosphere anion exchange resin in water (40 mL). The resulting mixture was stirred at room temperature for 24 h. The Dowex 550A resin was separated by filtration with a Buchner funnel and washed four times with a distilled water (4×5 mL). The solution of [Zn(en)3](OH)2 was reduced to a volume of 15 mL using a rotary evaporator. Methanol (15 mL) was added to the concentrated solution, followed by boric acid (2.6 g, 42 mmol). The reaction mixture was gently warmed for 3 h. The solution volume was reduced to 5 mL using a rotary evaporator. The concentrated solution was left for 30 days in NMR tubes for crystallization to yield colorless crystals of [Zn2(en)(enH)2{B12O18(OH)6}]·8H2O (0.55 g, 40%). M.p.300°C. χm=−414.2×10−6 cm3 mol−1. C6H48B12N6O32Zn2. Anal. Calc.: C=7.4%, H=5.0%, N=8.6%. Found: C=6.9%, H=4.7%, N=8.3%. 1H/ppm: 2.75 (m, 12H, CH2 of en), 4.8 (s, 34 H, NH2, H2O, OH). 13C{1H}/ppm: 39.4. 11B/ppm: 16.1. IR (KBr/cm−1): 3433(s), 3373(s), 3268(s), 2986(m), 1637(w), 1550(w), 1355(s), 1163(sh), 1041(s), 951(m), 900(s), 856(m), 758(w), 692(w), 646(w), 616(w), 519(w). p-XRD d-spacing/Å (% rel. int.): 9.40 (68), 8.05 (39), 7.59 (100), 6.78 (75), 4.21 (22), 3.82 (22), 3.14 (28), 2.91 (45).
X-ray crystallography
Crystallographic data for compound 1 is given in Table 1. A suitable crystal (0.10×0.09×0.04) mm3 was selected and mounted on a MITIGEN holder in oil on a Rigaku FRE+ equipped with HF Varimax confocal mirrors and an AFC12 goniometer and HG Saturn 724+ detector diffractometer. The crystal was kept at T=100(2) K during data collection. The data collection was carried out using CrystalClear [67] and cell determination and data reduction was carried out using CrysAlisPro [68]. Using Olex2 [69], the structure was solved and Superflip [70] structure solution program, using the Charge Flipping solution method. The model was refined with version 2014/7 of ShelXL [71] using Least Squares minimisation.
Electronic supplementary information
CCDC1571610 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data request/cif.
Article note
A collection of invited papers based on presentations at the 16th International Meeting on Boron Chemistry (IMEBORON-16), Hong Kong, 9–13 July 2017.
Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for the use of the National Crystallographic Service (NCS).
References
[1] D. M. Schubert. ‘Boron oxide, boric acid and borates’, in Kirk-Othmer Encyclopedia of Chemical Technology (5th Ed), Kirk-Othmer (Ed.), p. 1, J. Wiley and Sons, NY (2011).10.1002/0471238961.0215181519130920.a01.pub3Search in Google Scholar
[2] G. Heller. Top. Curr. Chem.131, 39 (1986).10.1063/1.2814947Search in Google Scholar
[3] C. L. Christ, J. R. Clark. Phys. Chem. Miner.2, 59 (1977).10.1007/BF00307525Search in Google Scholar
[4] E. L. Belokoneva. Crystallogr. Rev.11, 151 (2005).10.1080/08893110500230792Search in Google Scholar
[5] D. M. Schubert. Structure and Bonding105, 1 (2003).Search in Google Scholar
[6] J. M. Simon, R. A. Smith. Glass Tech.41, 169 (2000).10.1057/9780230372443_11Search in Google Scholar
[7] S. Yang, G. Li, S. Tian, F. Liao, J. Lin. Cryst. Growth Des.7, 1246 (2007).10.1021/cg0606794Search in Google Scholar
[8] P. Becker. Adv. Mater.10, 979 (1998).10.1002/(SICI)1521-4095(199809)10:13<979::AID-ADMA979>3.0.CO;2-NSearch in Google Scholar
[9] M. Yang. Z. Zhang, H. Ding, J.-Y. Li. Chem. Res. Chin. Uni.26, 335 (2010).Search in Google Scholar
[10] W. R. Cook, H. Jaffe. Acta Cryst.10, 705 (1957).10.1107/S0365110X57002431Search in Google Scholar
[11] J. Liang, Y.-G. Wang, Y.-X. Wang, F.-H. Liao, J.-H. Lin. J. Solid State Chem.200, 99 (2013).10.1016/j.jssc.2013.01.032Search in Google Scholar
[12] A. K. Paul, K. Sachidananda, S. Natarajan. Cryst. Growth Des.10, 456 (2010).10.1021/cg901161zSearch in Google Scholar
[13] M. A. Beckett, S. J. Coles, P. N. Horton, C. L. Jones, K. Krueger. J. Clust. Sci.28, 2087 (2017).Search in Google Scholar
[14] Y. Wang, S. Pan. Coord. Chem. Rev.323, 15 (2016).10.1016/j.ccr.2015.12.008Search in Google Scholar
[15] P. Becker, P. Held, L. Bohaty. Cryst. Res. Technol.35, 1251 (2000).Search in Google Scholar
[16] N. Jemai, M. Rzaigui, S. Akriche. J. Clust. Sci.26, 2051 (2015).10.1007/s10876-015-0905-7Search in Google Scholar
[17] M. A. Beckett. Coord. Chem. Rev.323, 2 (2016).10.1016/j.ccr.2015.12.012Search in Google Scholar
[18] G. R. Desiraju. Angew. Chem. Int. Ed. Engl.34, 2311 (1995).10.1002/anie.199523111Search in Google Scholar
[19] P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders, S. Otto. Chem. Rev.106, 3652 (2006).10.1021/cr020452pSearch in Google Scholar PubMed
[20] J. Sola, M. Lafuente, J. Archer, I. Alfonso. Chem Commun.50, 4564 (2014).10.1039/C4CC00245HSearch in Google Scholar PubMed
[21] C. G. Salentine. Inorg. Chem.22, 3920 (1983).10.1021/ic00168a019Search in Google Scholar
[22] J. L. Anderson, E. M. Eyring, M. P. Whittaker. J. Phys. Chem.68, 1128 (1964).10.1021/j100787a027Search in Google Scholar
[23] J. B. Farmer. Adv. Inorg. Chem.25, 187 (1982).10.1016/S0898-8838(08)60141-5Search in Google Scholar
[24] H. D. Smith, Jr. R. J. Wiersema. Inorg. Chem.11, 1152 (1972).10.1021/ic50111a056Search in Google Scholar
[25] Y. Zhou, C. Fang, Y. Fang, F. Zhu. Spectrochim. Acta A83, 82 (2011).10.1016/j.saa.2011.07.081Search in Google Scholar PubMed
[26] M. A. Beckett, S. J. Coles, R. A. Davies, P. N. Horton, C. L. Jones. Dalton Trans.44, 7032 (2015).10.1039/C5DT00248FSearch in Google Scholar PubMed
[27] M. A. Beckett, S. J. Coles, P. N. Horton, C. L. Jones. Eur. J. Inorg. Chem.2017, (2017) doi:10.1002/ejic.201700551.10.1002/ejic.201700551Search in Google Scholar
[28] M. A. Altahan, M. A. Beckett, S. J. Coles, P. N. Horton. Inorg. Chem. Commun.59, 95 (2015).Search in Google Scholar
[29] M. A. Altahan, M. A. Beckett, S. J. Coles, P. N. Horton. Inorg. Chem.54, 412 (2015).Search in Google Scholar
[30] M. A. Altahan, M. A. Beckett, S. J. Coles, P. N. Horton. Polyhedron135, 247 (2017).10.1016/j.poly.2017.07.016Search in Google Scholar
[31] J. Li, S. Xia, S. Goa. Spectochem. Acta51A, 519 (1995).10.1142/9789812831545_0009Search in Google Scholar
[32] M. Skakibaie-Moghadam, G. Heller, U. Timper. Zeit fur Kristallogr.190, 85 (1990).10.1524/zkri.1990.190.1-2.85Search in Google Scholar
[33] B. D. Vineyard, H. C. Godt, Inorg. Chem.3, 1144 (1964).10.1021/ic50018a017Search in Google Scholar
[34] C. C. Freyhardt, M. Wiebcke, J. Felsche, G. Englehardt. J. Inclusion. Phenom. Molec. Recognit. Chem.18, 161 (1994).Search in Google Scholar
[35] M. Wiebcke, C. C. Freyhardt, J. Felsche, G. Englehardt. Z. fur. Naturforsch B48, 978 (1993).Search in Google Scholar
[36] M. Z. Visi, C. B. Knobler, J. J. Owen, M. I. Khan, D. M. Schubert. Cryst. Growth Des.6, 538 (2006).10.1021/cg0504915Search in Google Scholar
[37] M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse, K. S. Varma. J. Organometal. Chem.692, 2832 (2007).Search in Google Scholar
[38] M. A. Beckett, P. N. Horton, M. B. Hursthouse, D. A. Knox, J. L. Timmis. Dalton Trans.39, 3944 (2010).10.1039/b927072hSearch in Google Scholar PubMed
[39] Z.-H. Liu, L.-Q. Li, W.-J. Zhang. Inorg. Chem.45, 1430 (2006).Search in Google Scholar
[40] Z.-H. Liu, L.-Q. Li. Cryst. Growth Des.6, 1247 (2006).10.1021/cg0503200Search in Google Scholar
[41] P. Li, L.-Q. Li, H.-S. Huang, Z.-H. Liu. J. Clust. Sci.25, 893 (2014).10.1007/s10876-014-0766-5Search in Google Scholar
[42] C.-Y. Pan, G.-M. Wang, S.-T. Zheng, G.-Y. Yang. Z. Anorg. Allg. Chem.633, 336 (2007).10.1002/zaac.200600264Search in Google Scholar
[43] M. A. Beckett, P. N. Horton, S. J. Coles, D. W. Martin. Inorg. Chem.50, 12215 (2011).Search in Google Scholar
[44] D. M. Schubert, R. A. Smith, M. Z. Visi. Glass Technol.44, 63 (2003).Search in Google Scholar
[45] M. A. Beckett, P. N. Horton, M. B. Hursthouse, J. L. Timmis. Polyhedron77, 96 (2014).10.1016/j.poly.2014.04.019Search in Google Scholar
[46] D. M. Schubert, M. Z. Visi, C. B. Knobler. Inorg. Chem.39, 2250 (2000).10.1021/ic000217uSearch in Google Scholar PubMed
[47] D. M. Schubert, C. B. Knobler, M. Z. Visi. Main Group Chem.11, 69 (2012).10.3233/MGC-2012-0062Search in Google Scholar
[48] M. A. Beckett, S. J. Coles, M. E. Light, L. Fischer, B. M. Stiefvater-Thomas, K. S. Varma. Polyhedron25, 1011 (2006).Search in Google Scholar
[49] M. A. Beckett, Paul Owen, D. E. Hibbs, M. B. Hursthouse, M. K. A. Malik, K. S. Varma. Main Group Chem.2, 251 (1998).10.1080/10241229812331341439Search in Google Scholar
[50] S. Menchetti, C. Sabelli. Acta Cryst.B35, 2488 (1979).10.1107/S0567740879009742Search in Google Scholar
[51] A. Choudury, S. Neeraj, S. Natarajan, C. N. R. Rao. J. Chem. Soc. Dalton Trans. 1535 (2002).10.1039/b108047bSearch in Google Scholar
[52] G.-M. Wang, Y.-Q. Sun, S.-T. Zheng, G.-Y. Yang. Z. Anorg. Allg. Chem.632, 1586 (2006).10.1002/zaac.200600054Search in Google Scholar
[53] T.-J. Zhang, R. Pan, H. He, B.-F. Yang, G.-Y. Yang. J. Clust. Sci.27, 625 (2016).Search in Google Scholar
[54] H.-X. Zhang, J. Zhang, S.-T. Zheng, G.-Y. Yang. Inorg. Chem. Commun.7, 781 (2004).Search in Google Scholar
[55] C. Rong, J. Jiang, Q.-L. Li. Chinese J. Inorg. Chem.28, 2217 (2012).Search in Google Scholar
[56] R. Pan, C.-A. Chen, B.-F. Yang, G.-Y. Yang. J. Clust. Sci.28, 1237 (2017).10.1007/s10876-016-1137-1Search in Google Scholar
[57] H. Jiang, B.-F. Yang, G.-M. Wang, G.-Y. Yang. J. Clust. Sci.28, 1421 (2017).10.1007/s10876-017-1232-ySearch in Google Scholar
[58] L. Wei, A.-H. Sun, Z.-Z. Xue, J. Pan, G.-M. Wang, Y.-X. Wang, Z.-H. Wang. J. Clust. Sci.28, 1453 (2017).10.1007/s10876-017-1158-4Search in Google Scholar
[59] G.-M. Wang, Y.-Q. Sun, G.-Y. Yang. J. Solid State Chem.178, 729 (2005).10.1016/j.jssc.2004.11.016Search in Google Scholar
[60] P. Zhao, L. Cheng, and G.-Y. Yang. Inorg. Chem. Commun.20, 138 (2012).Search in Google Scholar
[61] P. Zhao, Z.-E. Lin, Q. Wei, L. Cheng, G.-Y. Yang. Chem. Commun.50, 3592 (2014).10.1039/C4CC00006DSearch in Google Scholar
[62] D. M. Schubert, F. Alam, M. Z. Visi, C. B. Knobler. Chem. Mater.15, 866 (2003).10.1021/cm020791zSearch in Google Scholar
[63] Y. He, W. Chen, J. Yang, C.-Y. Xi, J.-S. Chen. Chem. Res. Chin. Univ.22, 271 (2006).10.1016/S1005-9040(06)60096-2Search in Google Scholar
[64] C. Muralikrishna, C. Mahadevan, S. Sastry, M. Seshasayee, S. Subramanian. Acta Cryst.C39, 1630 (1983).10.1107/S0108270183009543Search in Google Scholar
[65] M. C. Etter. Acc. Chem. Res.23, 120 (1990).10.1021/ar00172a005Search in Google Scholar
[66] K. Krishnan, R. A. Plane. Inorg. Chem.5, 852 (1966).10.1021/ic50039a031Search in Google Scholar
[67] CrystalClear, Rigaku Corporation, The Woodlands, Texas, USA (2008–2014).Search in Google Scholar
[68] CrysAlisPro Software System, Rigaku Oxford Diffraction, Yarnton, Oxford, UK (2015).Search in Google Scholar
[69] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann. J. Appl. Cryst.42, 339 (2009).10.1107/S0021889808042726Search in Google Scholar
[70] L. Palatinus, G. Chapuis. J. Appl. Cryst.40, 786 (2007).10.1107/S0021889807029238Search in Google Scholar
[71] G. M. Sheldrick. Acta Cryst.C71, 3 (2015).10.1107/S2053229614024218Search in Google Scholar PubMed PubMed Central
Supplemental Material
The online version of this article offers supplementary material (https://doi.org/10.1515/pac-2017-0901).
©2018 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- 16th International Meeting on Boron Chemistry (IMEBORON XVI)
- Conference papers
- Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
- When diazo compounds meet with organoboron compounds
- Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
- Novel sulfur containing derivatives of carboranes and metallacarboranes
- Metal–metal bonding in deltahedral dimetallaboranes and trimetallaboranes: a density functional theory study
- Nanostructured boron compounds for cancer therapy
- Heterometallic boride clusters: synthesis and characterization of butterfly and square pyramidal boride clusters*
- Influence of fluorine substituents on the properties of phenylboronic compounds
- Copper-catalyzed asymmetric dearomative borylation: new pathway to optically active heterocyclic compounds
- Borenium and boronium ions of 5,6-dihydro-dibenzo[c,e][1,2]azaborinine and the reaction with non-nucleophilic base: trapping of a dimer and a trimer of BN-phenanthryne by 4,4′-di-tert-butyl-2,2′-bipyridine
- The electrophilic aromatic substitution approach to C–H silylation and C–H borylation
- Recent advances in B–H functionalization of icosahedral carboranes and boranes by transition metal catalysis
- closo-Dodecaborate-conjugated human serum albumins: preparation and in vivo selective boron delivery to tumor
- IUPAC Technical Report
- Risk assessment of effects of cadmium on human health (IUPAC Technical Report)
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- 16th International Meeting on Boron Chemistry (IMEBORON XVI)
- Conference papers
- Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
- When diazo compounds meet with organoboron compounds
- Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
- Novel sulfur containing derivatives of carboranes and metallacarboranes
- Metal–metal bonding in deltahedral dimetallaboranes and trimetallaboranes: a density functional theory study
- Nanostructured boron compounds for cancer therapy
- Heterometallic boride clusters: synthesis and characterization of butterfly and square pyramidal boride clusters*
- Influence of fluorine substituents on the properties of phenylboronic compounds
- Copper-catalyzed asymmetric dearomative borylation: new pathway to optically active heterocyclic compounds
- Borenium and boronium ions of 5,6-dihydro-dibenzo[c,e][1,2]azaborinine and the reaction with non-nucleophilic base: trapping of a dimer and a trimer of BN-phenanthryne by 4,4′-di-tert-butyl-2,2′-bipyridine
- The electrophilic aromatic substitution approach to C–H silylation and C–H borylation
- Recent advances in B–H functionalization of icosahedral carboranes and boranes by transition metal catalysis
- closo-Dodecaborate-conjugated human serum albumins: preparation and in vivo selective boron delivery to tumor
- IUPAC Technical Report
- Risk assessment of effects of cadmium on human health (IUPAC Technical Report)