Synthesis of ring-A serjanic acid derivatives and their cytotoxic evaluation through the brine shrimp lethality assay (BSLA)
-
Elier Galarraga
 ,
Sherezade Gómez
,
Sherezade Gómez

Abstract
Sixteen serjanic acid derivatives were synthesized by modification of the A-ring of the triterpenoid skeleta. Fischer indolization of intermediate 2 with the requisite aromatic hydrazines afforded the indolo-triterpenes 8–13. Also, reaction of 2 with hydroxylamine hydrochloride in pyridine provided the desired C-3 oxime 14, while pyrazine 15 was obtained by condensation of 2 in the presence of ethylenediamine and sulphur in morpholine. Finally, the Claisen-Schmidt condensation of intermediate 2 with corresponding substituted benzaldehydes 16–23, afforded benzylidine ketones 24–31. All compounds were elucidated on the basis of NMR and HR-MS spectroscopic data and evaluated for their in-vitro cytotoxicity to the Brine Shrimp Lethality Assay (BSLA). As a result, the compounds exhibited medium to good cytotoxic potential and this activity was as high as eight times that of serjanic acid (1) on the tested zoophytes.
1 Introduction
The important role that natural products have in drug discovery is reflected in their statistics, with around 60% anti-cancer and 75% anti-infective-approved drugs owing their origin from natural resources [1]. Structural modification of biologically active natural products has led to the development of potentially important bioactive molecules, leads, and drugs [2, 3]. Triterpenoids, on account of their wide distribution in plants, amenable functionality for transformation and promising biological activities have been the target of widespread interest of chemists and biologists.
Oleanane-type triterpenoids are pentacyclic compounds with 30 carbon atoms, biosynthetically derived from the cyclization of squalene [4], [5], [6]. This is a vast class of natural products whose structural diversity includes a wide array of functional groups [7]. Many oleanane-type triterpenoids are reported to have various interesting biological, pharmacological or medicinal activities, such as anti-cancer [8, 9], anti-HIV [10], [11], [12], anti-inflammatory [13, 14] and protection of the liver against toxic injury [15], [16], [17]. In many cases, the potency of these triterpenoids is relatively weak. However, the key functionalities such as C3–OH, Δ12 and C17–COOH embodied in these highly useful natural scaffolds make them suitable starting material for a variety of chemical transformations [18]. At present, most of the research on the chemical modification of pentacyclic triterpenes involves the insertion of heterocyclic rings, either through covalent bonds or by ring fusion. Many biologically active ring-A-modified triterpenoids are of particularly interest, and recently their synthesis has been increasing due to their potential as new drugs.
The brine shrimp lethality assay (BSLA) is an easily performed and cost-effective preliminary standard bioassay based on the use of the simple invertebrate marine organism Artemia salina to detect toxicity of substances. This test has shown positive correlations with some specific cytotoxic assays employing several cancer cell lines such as 9PS, KB and 9K [19], [20], [21] and it also has been suitable in predicting other bioactivities such as pesticidal [22] and trypanocidal [23].
Anticipating novel structures with biological potential, we began the systematic synthesis of serjanic acid (SA) derivatives. SA is a pentacyclic triterpenoid isolated in abundance from the fruits of several Phytolacca species. Despite the structural similarities of SA with other bioactive triterpenes such as oleanolic, ursolic, boswellic and moronic acid, there are only a few studies reporting on the preparation of semi-synthetic derivatives from this acid. Moreover, there are almost no studies on the biological or pharmacological evaluation of these derivatives. Based on the above-mentioned, the present study deals with the synthesis of a series of ring-A-modified derivatives of SA and their in vitro toxicity against brine shrimps (A. salina), with the aim to evaluate them as potential anti-cancer agents.
2 Experimental section
2.1 General procedures
Reagents were obtained from Sigma-Aldrich (Milwaukee, Wisconsin, USA) and were used without any purification. Analytical and preparative thin layer chromatography (TLC) was performed on silica gel (15–40 μm PF254) 0.25 and 0.5 mm thick plates respectively (supplied by Merck), and the spots were visualized by UV-B light or by spraying with AcOH–H2O–H2SO4 (74:16:10) mixture, followed by heating. Column chromatography was performed using silica gel 230–400 mesh. All compounds were fully characterized by IR, HR-EI-MS, 1H NMR and 13C NMR spectroscopy. Infrared spectra were recorded on a Thermo Scientific Serie Nicolet iS10 FTIR spectrophotometer, preparing the samples as potassium bromide disks (1% w/w). EI-HR-MS were recorded on a JEOL JMS-AX505WA double focus mass spectrometer, using EI mode (70 eV) and PFTBA as internal reference, ESI-HR-MS were acquired on a ThermoScientific LTQ-Orbitrap-LC-MS spectrometer. 1H and 13C NMR spectra were recorded on a Bruker Avance 300* spectrometer at 300 and 75 MHz respectively, using CDCl3 as solvent and TMS as internal reference, unless otherwise stated. Melting points were determined using a Krüss-Optronic apparatus and are not corrected.
2.2 Isolation of serjanic acid (1)
Dried and ground fruits of Phytolacca icosandra L., (2.5 kg) were extracted in a Soxhlet apparatus using MeOH as solvent. The methanol extract was concentrated under reduced pressure affording 300 g of a crude brown residue. The residue was submitted to column chromatography on silica gel using solvent mixtures of increasing polarity (CHCl3, EtOAc, MeOH) to afford 15 fractions (A–O). Fraction “K” (34.2 g), was dissolved in MeOH–H2O (7:3 v/v) mixture and concentrated H2SO4 was added until the solution reached pH = 2, then the solution was refluxed for 6 h. The precipitate formed after hydrolysis was filtered off and dried to afford a solid residue (23.5 g) which was submitted to column chromatography on silica gel using CHCl3–(CH3)3CO (9:1 v/v) as eluent. This procedure led to four sub-fractions (A–D). Sub-fractions “B” and “C” of the chromatography process were combined on the basis of their chromatographic profiles, and the combined sub-fractions afforded 4.7 g of serjanic acid (1) (0.2% of dry mass) as a white amorphous solid, m. p. 288–290 °C (lit.: 288–291 °C [24]). All spectroscopic data were in agreement with the literature values [25].
2.3 Synthesis of 3-oxo-serjanic acid (2)
To a solution of compound 1 (2.5 g, 4.99 mmol) in acetone (250 mL) at 0 °C was added Jones reagent (K2Cr2O7–H2SO4) drop wise until yellow-orange colour persisted. The reaction was monitored by TLC till its completion in 2 h. The excess of Jones reagent was eliminated by adding MeOH to the solution and the solution was filtered to remove insoluble residue. The filtrate was concentrated under vacuum to afford a white amorphous solid. The solid was recrystallized from ethanol to afford compound 2 as colourless needles (2.32 g, 93%), m. p. 276–278 °C. All spectroscopic data was in agreement with the literature values [25].
2.4 General procedure for the synthesis of indolo-triterpenes 8–13
To a portion of compound 2 (0.12 mmol) dissolved in glacial AcOH (2 mL) and kept under agitation, a solution of phenyl hydrazines 3–7 (0.12 mmol) dissolved in glacial AcOH (0.5 mL) was added dropwise. The solution was refluxed for 4 h until the end of reaction by TLC monitoring. After cooling, 40 mL of distilled water was added and the resulting solution was extracted with CH2Cl2 (3 × 10 mL). The organic phase was washed with 5% NaOH (2 × 20 mL) and brine (2 × 20 mL), dried with MgSO4 and concentrated under vacuum to obtain the crude reaction product. It was subjected to preparative TLC plates on silica gel (15–40 mm PF254), using hexane-ethyl acetate (2:3 or 1:1 v/v) as solvent mixture to afford the purified product as a solid.
2.4.1 Indolo[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (8)
Pale yellow solid; yield: 74%; m. p. 224–226 °C. – IR (KBr): ν = 3406, 3050, 2949, 1719, 1701, 1550, 1462, 1215, 740 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.71 (s, 1H, NH), 7.41 (dd, J = 1.5, 7.0 Hz, 1H, H-4′), 7.27 (dd, J = 1.2, 7.3 Hz, 1H, H-7′), 7.10 (td, J = 1.5, 7.0 Hz, 1H, H-6′), 7.04 (td, J = 1.2, 7.3 Hz, 1H, H-5′), 5.45 (m, 1H, H-12), 3.70 (s, 3H, OMe), 2.75 (d, J = 14.9 Hz, 1H, H-1a), 2.72 (m, 1H, H-18), 2.19 (d, J = 14.9 Hz, 1H, H-1b), 1.82 (m, 1H, H-9), 1.27, 1.17, 1.16, 1.14, 0.93, 0.79 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 183.9 (C-28), 177.5 (C-30), 143.6 (C-13), 140.9 (C-3), 136.2 (C-8′), 128.3 (C-9′), 123.0 (C-12), 121.0 (C-6′), 118.9 (C-5′), 117.9 (C-4′), 110.4 (C-7′), 106.9 (C-2), 53.3 (C-5), 51.8 (OMe), 46.5 (C-9), 46.2 (C-17), 43.8 (C-20), 42.7 (C-18), 42.5 (C-4), 41.9 (C-14), 39.4 (C-19), 38.1 (C-8), 36.8 (C-10), 34.0 (C-1), 33.9 (C-22), 32.3 (C-7), 31.0 (C-29), 30.4 (C-21), 28.4 (C-27), 27.9 (C-15), 25.7 (C-23), 23.5 (C-24), 23.4 (C-11), 23.3 (C-16), 19.4 (C-6), 17.0 (C-26), 15.6 (C-25). – HRMS (EI, 70 eV): m/z = 571.3932 (calcd. 571.3662 for [C37H49NO4]+).
2.4.2 4′-Bromoindolo[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (9)
Brown solid; yield: 58%; m. p. 211–213 °C. – IR (KBr): ν = 3385, 3060, 2950, 1715, 1595, 1462, 1225, 756 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 8.01 (s, 1H, NH), 7.29 (d, J = 8.0 Hz, 1H, H-5′), 7.20 (d, J = 7.5 Hz, 1H, H-7′), 6.93 (t, J = 7.8 Hz, 1H, H-6′), 5.48 (m, 1H, H-12), 3.75 (s, 3H, OMe), 2.75 (d, J = 14.7 Hz, 1H, H-1a), 2.73 (m, 1H, H-18), 2.20 (d, J = 15.0 Hz, 1H, H-1b), 1.89 (m, 1H, H-9), 1.29, 1.21, 1.20, 1.18, 0.96, 0.85 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 183.6 (C-28), 177.1 (C-30), 142.6 (C-13), 142.2 (C-3), 137.3 (C-8′), 127.2 (C-9′), 123.8 (C-12), 123.1 (C-5′), 122.1 (C-6′), 114.3 (C-4′), 109.6 (C-7′), 107.7 (C-2), 52.6 (C-5), 51.9 (OMe), 46.4 (C-9), 46.1 (C-17), 43.8 (C-20), 42.4 (C-18), 42.1 (C-4), 41.7 (C-14), 39.4 (C-19), 38.9 (C-8), 37.9 (C-10), 33.9 (C-1), 33.6 (C-22), 32.2 (C-7), 30.8 (C-29), 30.4 (C-21), 28.4 (C-27), 27.8 (C-15), 25.8 (C-23), 23.4 (C-11), 23.2 (C-24), 23.1 (C-16), 19.2 (C-6), 16.9 (C-26), 15.7 (C-25). – HRMS (EI, 70 eV): m/z = 649.2251 (calcd. 649.2767 for [C37H48BrNO4]+).
2.4.3 5′-Bromoindolo[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (10)
Brown solid; yield: 72%; m. p. 215–217 °C. – IR (KBr): ν = 3390, 3058, 2959, 1712, 1577, 1462, 1210, 735 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.86 (s, 1H, NH), 7.56 (d, J = 1.5 Hz, 1H, H-4′), 7.20 (dd, J = 1.5, 8.4 Hz, 1H, H-6′), 7.17 (d, J = 8.4 Hz, 1H, H-7′), 5.49 (m, 1H, H-12), 3.75 (s, 3H, OMe), 2.77 (m, 1H, H-18), 2.73 (d, J = 15.0 Hz, 1H, H-1a), 2.17 (d, J = 14.9 Hz, 1H, H-1b), 1.89 (m, 1H, H-9), 1.30, 1.21, 1.19, 1.18, 0.98, 0.85 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 183.1 (C-28), 177.0 (C-30), 142.7 (C-13), 142.3 (C-3), 134.9 (C-8′),130.1 (C-9′), 123.7 (C-6′), 123.6 (C-12), 120.6 (C-4′),112.2 (C-5′), 111.7 (C-7′), 106.8 (C-2), 53.3 (C-5), 51.9 (OMe), 46.4 (C-9), 46.0 (C-17), 43.8 (C-20), 42.4 (C-18), 42.1 (C-4), 41.7 (C-14), 39.4 (C-19), 38.1 (C-8), 36.6 (C-10), 34.0 (C-1), 33.6 (C-22), 32.2 (C-7), 30.9 (C-29), 30.4 (C-21), 28.3 (C-27), 27.8 (C-15), 25.8 (C-23), 23.4 (C-11), 23.3 (C-24), 23.1 (C-16), 19.2 (C-6), 16.8 (C-26), 15.5 (C-25). – HRMS (EI, 70 eV): m/z = 649.2666 (calcd. 649.2767 for [C37H48BrNO4]+).
2.4.4 6′-Bromoindolo[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (11)
Brown solid; yield: 58%; m. p. 205–207 °C. – IR (KBr): ν = 3385, 3060, 2950, 1715, 1595, 1462, 1225, 756 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.97 (s, 1H, NH), 7.44 (d, J = 1.2 Hz, 1H, H-7′), 7.21 (d, J = 8.4 Hz, 1H, H-4′), 7.17 (dd, J = 1.2, 8.4 Hz, 1H, H-5′), 5.48 (m, 1H, H-12), 3.74 (s, 3H, OMe), 3.43 (d, J = 15.6 Hz, 1H, H-1a), 2.77 (d, J = 13 Hz, 1H, H-18), 2.43 (d, J = 15.6 Hz, 1H, H-1b), 1.89 (m, 1H, H-9), 1.29, 1.21, 1.20, 1.18, 0.96, 0.85 (6 × s, 18H, CHMe). – 13C NMR (75 MHz, CDCl3): δ = 183.6 (C-28), 177.1 (C-30), 142.7 (C-13), 141.7 (C-3), 137.0 (C-8′), 126.7 (C-9′), 123.6 (C-12), 121.8 (C-5′), 119.2 (C-4′), 113.8 (C-6′), 113.4 (C-7′), 107.0 (C-2), 53.1 (C-5), 51.8 (OMe), 46.4 (C-9), 46.1 (C-17), 43.8 (C-20), 42.5 (C-18), 42.1 (C-4), 41.7 (C-14), 39.4 (C-19), 38.1 (C-8), 36.6 (C-10), 34.0 (C-1), 33.9 (C-22), 32.2 (C-7), 31.1 (C-29), 30.4 (C-21), 28.4 (C-27), 27.8 (C-15), 25.8 (C-23), 23.4 (C-11), 23.2 (C-24), 23.1 (C-16), 19.2 (C-6), 16.9 (C-26), 15.5 (C-25). – HRMS (EI, 70 eV): m/z = 649.2251 (calcd. 649.2767 for [C37H48BrNO4]+).
2.4.5 7′-Bromoindolo[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (12)
Brown solid; yield: 64%; m. p. 189–191 °C. – IR (KBr): ν = 3390, 3060, 2948, 1701, 1590, 1457, 1195, 741 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.84 (s, 1H, NH), 7.38 (d, J = 7.8 Hz, 1H, H-4′), 7.28 (d, J = 7.8 Hz, 1H, H-6′), 6.96 (t, J = 7.8 Hz, 1H, H-5′), 5.49 (m, 1H, H-12), 3.74 (s, 3H, OMe), 2.77 (d, J = 13 Hz, 1H, H-18), 2.76 (d, J = 14.9 Hz, 1H, H-1a), 2.23 (d, J = 14.9 Hz, 1H, H-1b), 1.86 (m, 1H, H-9), 1.34, 1.23, 1.21, 1.19, 0.97, 0.86 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 183.0 (C-28), 177.0 (C-30), 142.8 (C-13), 141.7 (C-3), 134.8 (C-8′),129.5 (C-9′), 123.7 (C-6′), 123.4 (C-12), 120.2 (C-5′), 117.2 (C-4′), 108.3 (C-7′), 104.2 (C-2), 53.2 (C-5), 51.8 (OMe), 46.4 (C-9), 46.0 (C-17), 43.8 (C-20), 42.4 (C-18), 42.1 (C-4), 41.7 (C-14), 39.4 (C-19), 38.1 (C-8), 36.9 (C-10), 34.1 (C-1), 33.6 (C-22), 32.2 (C-7), 30.9 (C-29), 30.4 (C-21), 28.4 (C-27), 27.8 (C-15), 25.8 (C-23), 23.4 (C-11), 23.3 (C-24), 23.2 (C-16), 19.3 (C-6), 16.8 (C-26), 15.5 (C-25). – HRMS (EI, 70 eV): m/z = 649.2917 (calcd. 649.2767 for [C37H48BrNO4]+).
2.4.6 4′,7′-Dichloroindolo[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (13)
Brown solid; yield: 63%; m. p. 136–138 °C. – IR (KBr): ν = 3337, 3060, 2949, 1696, 1591, 1458, 1210, 757 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.99 (s, 1H, NH), 7.01 (d, J = 8.1 Hz, 1H, H-6′), 6.93 (t, J = 8.1 Hz, 1H, H-5′), 5.47 (t, J = 3.2 Hz, 1H, H-12), 3.73 (s, 3H, OMe), 3.26 (d, J = 15.7 Hz, 1H, H-1a), 2.78 (d, J = 13 Hz, 1H, H-18), 2.41 (d, J = 15.7 Hz, 1H, H-1b), 1.90 (m, 1H, H-9), 1.31, 1.26, 1.21, 1.18, 0.99, 0.84 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 182.6 (C-28), 177.0 (C-30), 142.8 (C-13), 142.7 (C-3), 134.2 (C-8′), 130.4 (C-6′), 126.4 (C-9′), 124.5 (C-4′), 123.6 (C-12), 120.4 (C-5′), 114.6 (C-7′), 108.8 (C-2), 55.3 (C-5), 51.8 (OMe), 46.3 (C-9), 46.0 (C-17), 43.7 (C-20), 42.5 (C-18), 42.1 (C-4), 41.7 (C-14), 39.4 (C-19), 38.4 (C-8), 37.9 (C-10), 34.0 (C-1), 33.5 (C-22), 32.1 (C-7), 301.1 (C-29), 30.4 (C-21), 28.4 (C-27), 27.8 (C-15), 25.7 (C-23), 23.4 (C-11), 23.2 (C-24), 23.1 (C-16), 19.2 (C-6), 16.8 (C-26), 15.6 (C-25). – HRMS (EI, 70 eV): m/z = 639.2730 (calcd. 639.2882 for [C37H47Cl2NO4]+).
2.5 Synthesis of 3-hydroxyimino-olean-12-en-28,30-dioic acid-30-methyl ester (14)
To a solution of 65 mg (0.13 mmol) of ketone 2 dissolved in pyridine (2 mL), hydroxylamine hydrochloride (11.3 mg, 0.16 mmol) was added and the obtained mixture was refluxed for 3 h, cooled and poured into cold water (50 mL) slightly acidified with HCl. The resulted precipitate was filtered, washed with water, dried and recrystallized from absolute ethanol. Colourless needles; yield: 92%; m. p. 262–266 °C. – IR (KBr): ν = 3431, 3055, 2945, 1732, 1709, 1646, 1463, 1211, 769 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 5.36 (m, 1H, H-12), 3.69 (s, 3H, OMe), 3.10 (m, 1H, H-2a), 2.70 (m, 1H, H-18), 2.22 (m, 1H, H-2b), 1.14, 1.12, 1.11, 1.04, 1.01, 0.73 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 182.8 (C-28), 177.0 (C-30), 167.4 (C-3), 143.1 (C-13), 123.1 (C-12), 55.8 (C-5), 51.9 (OMe), 47.2 (C-9), 45.8 (C-17), 43.7 (C-20), 42.4 (C-18), 42.1 (C-19), 41.6 (C-14), 40.3 (C-8), 39.3 (C-4), 38.3 (C-1), 37.0 (C-10), 33.5 (C-22), 32.4 (C-7), 30.4 (C-21), 28.4 (C-29), 27.7 (C-15), 27.3 (C-27), 25.8 (C-23), 23.5 (C-11), 23.4 (C-24), 23.2 (C-16), 19.1 (C-2), 17.6 (C-6), 16.9 (C-26), 14.9 (C-25). – HRMS ((+)-ESI): m/z = 514.3466 (calcd. 514.3454 for C31H48NO5, [M+H]+).
2.6 Synthesis of pyrazine[2,3-b]-olean-12-en-28,30-dioic acid-30-methyl ester (15)
To a solution of 2 (100 mg, 0.2 mmol) in morpholine (6 mL), was added sulphur (72.4 mg, 2.2 mmol) and ethylenediamine (68 μL, 1.01 mmol) at room temperature. The reaction mixture was heated under reflux for 6 h. After cooling, the mixture was poured into water (50 mL) and extracted with ethyl acetate (2 × 15 mL). The organic layer was washed with water and brine, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified over silica gel preparative TLC plates (hexane-ethyl acetate 7:3 v/v) to afford a white solid. Yield: 64%; m.p: 233–235 °C. – IR (KBr): ν = 3418, 2953, 1731, 1693, 1511, 1460, 1208, 755 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 8.41 (d, J = 2.4 Hz, 1H, H-2′), 8.26 (d, J = 2.4 Hz, 1H, H-3′), 5.43 (t, J = 3.1 Hz, 1H, H-12), 3.69 (s, 3H, OMe), 2.98 (d, J = 16.3 Hz, 1H, H-1a), 2.72 (dd, J = 3.5, 13.3 Hz, 1H, H-18), 2.51 (d, J = 16.3 Hz, 1H, H-1b), 1.91 (m, 1H, H-9), 1.29, 1.26, 1.18, 1.15, 0.88, 0.83 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 182.7 (C-28), 176.9 (C-30), 159.7 (C-3), 150.5 (C-2), 142.9 (C-13), 142.4 (C-3′), 141.3 (C-2′), 123.2 (C-12), 53.1 (C-5), 51.8 (OMe), 48.1 (C-1), 45.9 (C-17), 45.8 (C-9), 43.7 (C-20), 42.4 (C-18), 42.1 (C-4), 41.7 (C-14), 39.4 (C-19), 39.2 (C-8), 36.6 (C-10), 33.5 (C-22), 32.0 (C-7), 31.6 (C-29), 30.4 (C-21), 28.4 (C-27), 27.7 (C-15), 25.7 (C-23), 24.2 (C-24), 23.3 (C-11), 23.2 (C-16), 20.1 (C-6), 16.7 (C-26), 15.5 (C-25). – HRMS ((+)-ESI): m/z = 535.3439 (calcd. 535.3458 for C33H47N2O4, [M+H]+).
2.7 General procedure for the synthesis of the benzylidine derivatives 24–31
To a solution of 2 (100 mg, 0.2 mmol) in ethanol (3 mL), was added KOH (0.6 mmol). The mixture was cooled to 0 °C trough ice bath and added drop wise 0.2 mmol of the corresponding benzaldehyde 16–23 dissolved in 1 mL EtOH. The reaction was carried out at 0 °C for the first 5 min and then at room temperature for 24 h. The mixture was poured into water (10 mL) acidified with HCl (three drops) and extracted with ethyl acetate (2 × 15 mL). The organic layer was washed with water and brine, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified over silica gel preparative TLC plates (hexane-chloroform 1:9 v/v) to afford benzylidines 24–31.
2.7.1 2-(p-Nitrobenzylidine)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (24)
Yellow oil; yield: 40%. – IR (KBr): ν = 3500, 2945, 1726, 1697, 1598, 1217, 756 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 8.23 (d, J = 8.7 Hz, 2H, H-3′/5′), 7.52 (d, J = 8.7 Hz, 2H, H-2′/6′), 7.51 (s, 1H, H-1″), 5.38 (m, 1H, H-12), 3.67 (s, 3H, OMe), 2.93 (d, J = 15.0 Hz, 1H, H-1a), 2.68 (dd, J = 3.9, 13.7 Hz, 1H, H-18), 2.36 (d, J = 15.0 Hz, 1H, H-1b), 1.08 (m, H-9), 1.24, 1.18, 1.16, 1.14, 0.86, 0.76 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.3 (C-3), 183.3 (C-28), 176.8 (C-30), 147.2 (C-4′), 143.2 (C-13), 142.4 (C-2), 137.2 (C-1′), 134.6 (C-1″), 130.7 (C-2′/6′), 123.5 (C3′/5′), 122.8 (C-12), 53.1 (C-5), 51.8 (-OMe), 46.0 (C-4), 45.4 (C-17), 45.5 (C-9), 44.1 (C-8), 43.7 (C-20), 42.5 (C-18), 42.0 (C-19), 41.8 (C-14), 39.2 (C-1), 36.4 (C-10), 33.4 (C-22), 31.8 (C-7), 30.3 (C-21), 29.7 (C-23), 28.3 (C-29), 27.6 (C-15), 25.7 (C-27), 23.6 (C-16), 23.1 (C-11), 22.6 (C-24), 20.2 (C-6), 16.5 (C-26), 15.3 (C-25). – HRMS ((+)-ESI): m/z = 632.3501 (calcd. 632.3509 for C38H50NO7, [M+H]+).
2.7.2 2-(o-Chlorobenzylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (25)
Yellow oil; yield: 81%. – IR (KBr): ν = 3500, 2949, 1727, 1698, 1606, 1217, 756 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.63 (s, 1H, H-1″), 7.42 (m, 1H, H-6′), 7.39 (m, 1H, H-3′), 7.26 (m, 1H, H-4′), 7.25 (m, 1H, H-5′), 5.33 (t, J = 3.1 Hz, 1H, H-12), 3.66 (s, 3H, OMe), 2.64 (dd, J = 3.9, 13.7 Hz, 1H, H-18), 2.80 (d, J = 15.0 Hz, 1H, H-1a), 2.11 (d, J = 15.0 Hz, 1H, H-1b), 1.08 (m, H-9), 1.17, 1.15, 1.14, 1.12, 0.86, 0.77 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.5 (C-3), 183.4 (C-28), 176.9 (C-30), 135.2 (C-2), 143.0 (C-13), 134.8 (C-1′), 134.5 (C-1″), 134.4 (C-2′) 130.1 (C-4′), 129.7 (C-3′), 129.4 (C-6′), 126.3 (C-5′), 123.0 (C-12), 53.4 (C-5), 51.8 (OMe), 46.0 (C-4), 45.6 (C-17), 45.6 (C-9), 43.7 (C-20), 43.2 (C-8), 42.4 (C-18), 42.0 (C-19), 41.7 (C-14), 39.2 (C-1), 36.6 (C-10), 33.4 (C-22), 31.9 (C-7), 30.3 (C-21), 29.2 (C-23), 28.3 (C-29), 27.7 (C-15), 25.7 (C-27), 23.5 (C-16), 23.1 (C-11), 22.7 (C-24), 20.2 (C-6), 16.6 (C-26), 15.1 (C-25). – HRMS ((+)-ESI): m/z = 621.3251 (calcd. 621.3269 for C38H50ClO5, [M+H]+).
2.7.3 2-(p-Cyanobenzylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (26)
Yellow oil; yield: 80%. – IR (KBr): ν = 3500, 2932, 2228, 1726, 1698, 1603, 1217, 756 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.67 (d, J = 8.4 Hz, 2H, H-2′/6′), 7.48 (d, J = 8.4 Hz, 2H, H-3′/5′), 7.46 (s, 1H, H-1″), 5.39 (t, J = 3.1 Hz, 1H, H-12), 3.69 (s, 3H, OMe), 2.70 (dd, J = 3.9, 13.7 Hz, 1H, H-18), 2.92 (d, J = 15.0 Hz, 1H, H-1a), 2.28 (d, J = 15.0 Hz, 1H, H-1b), 1.08 (m, H-9), 1.19, 1.15, 1.14, 1.13, 0.85, 0.78 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.3 (C-3), 183.0 (C-28), 176.8 (C-30), 143.2 (C-13), 140.5 (C-1′), 136.7 (C-2), 135.0 (C-1″), 132.1 (C-3′/5′), 130.5 (C2′/6′), 122.8 (C-12), 118.6 (C≡N), 111.7 (C-4′), 53.1 (C-5), 51.8 (OMe), 46.0 (C-4), 45.3 (C-17), 45.3 (C-9), 44.1 (C-8), 43.7 (C-20), 42.5 (C-18), 42.1 (C-19), 41.8 (C-14), 39.2 (C-1), 36.4 (C-10), 33.4 (C-22), 31.8 (C-7), 30.7 (C-21), 29.6 (C-23), 28.3 (C-29), 27.7 (C-15), 25.7 (C-27), 23.6 (C-16), 23.1 (C-11), 22.6 (C-24), 20.2 (C-6), 16.5 (C-26), 15.3 (C-25). – HRMS ((+)-ESI): m/z = 612.3608 (calcd. 612.3611 for C39H50NO5, [M+H]+).
2.7.4 2-(Benzylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (27)
Yellow oil; yield: 80%. – IR (KBr): ν = 3500, 2950, 1726, 1698, 1596, 1217, 756 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.67 (d, J = 8.4 Hz, 2H, H-2′/6′), 7.48 (d, J = 8.4 Hz, 2H, H-3′/5′), 7.46 (s, 1H, H-1″), 5.39 (t, J = 3.1 Hz, 1H, H-12), 3.69 (s, 3H, OMe), 2.70 (dd, 1H, J = 3.9, 13.7 Hz, H-18), 2.92 (d, 1H, J = 15.0 Hz, H-1a), 2.28 (d, 1H, J = 15.0 Hz, H-1b), 1.08 (m, H-9), 1.19, 1.15, 1.14, 1.13, 0.85, 0.78 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.7 (C-3), 183.3 (C-28), 176.9 (C-30), 143.1 (C-13), 137.5 (C-1′), 136.0 (C-2), 133.7 (C-1″), 130.3 (C-3′/5′), 128.4 (C2′/6′), 123.1 (C-12), 53.1 (C-5), 51.8 (OMe), 46.0 (C-4), 45.5 (C-17), 45.2 (C-9), 44.1 (C-8), 43.7 (C-20), 42.5 (C-18), 42.1 (C-19), 41.8 (C-14), 39.2 (C-1), 36.3 (C-10), 33.4 (C-22), 31.9 (C-7), 30.4 (C-21), 29.7 (C-23), 28.3 (C-29), 27.7 (C-15), 25.7 (C-27), 23.6 (C-16), 23.2 (C-11), 22.6 (C-24), 20.3 (C-6), 16.5 (C-26), 15.2 (C-25). – HRMS ((+)-ESI): m/z = 587.3641 (calcd. 587.3658 for C38H51O5, [M+H]+).
2.7.5 2-(m-Fluorobezylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (28)
Yellow oil; yield: 73%. – IR (KBr): ν = 3500, 2932, 1727, 1695, 1588, 1218, 757 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.47 (s, 1H, H-1″), 7.37 (m, 1H, H-3′), 7.20 (m, 1H, H-2′), 7.13 (m, 1H, H-6′), 7.06 (m, 1H, H-4′), 5.42 (t, J = 3.1 Hz, 1H, H-12), 3.71 (s, 3H, OMe), 2.99 (d, J = 16.4 Hz, 1H, H-1b), 2.72 (dd, J = 3.7, 13.5 Hz, 1H, H-18), 2.30 (d, J = 16.4 Hz, 1H, H-1a), 1.08 (m, H-9) 1.21, 1.17, 1.17, 1.15, 0.87, 0.81 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.6 (C-3), 183.3 (C-28), 176.9 (C-30), 164.3/161.0 (C-5′), 143.0 (C-13), 138.0/138.1 (C-1′), 134.9 (C-2), 136.0 (C-1″), 128.9/129.9 (C-3′), 126.1/126.2 (C2′), 123.0 (C-12), 53.1 (C-5), 51.8 (OMe), 46.0 (C-4), 45.2 (C-17), 45.4 (C-9), 44.0 (C-8), 43.7 (C-20), 42.5 (C-18), 42.0 (C-19), 41.8 (C-14), 39.2 (C-1), 36.3 (C-10), 33.4 (C-22), 31.7 (C-7), 30.4 (C-21), 29.6 (C-23), 28.3 (C-29), 27.7 (C-15), 25.6 (C-27), 23.6 (C-16), 23.2 (C-11), 22.6 (C-24), 20.3 (C-6), 16.5 (C-26), 15.2 (C-25). – HRMS ((+)-ESI): m/z = 605.3510 (calcd. 605.3564 for C38H50FO5, [M+H]+).
2.7.6 2-(p-Bromobenzylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (29)
Yellow oil; yield: 73%. – IR (KBr): ν = 3500, 2950, 1728, 1696, 1592, 1216, 757 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.50 (d, J = 8.5 Hz, 2H, H-3′/5′), 7.43 (s, 1H, H-1″), 7.25 (d, J = 8.4 Hz, 2H, H-2′/6′), 5.37 (m, 1H, H-12), 3.66 (s, 3H, OMe), 2.91 (d, 1H, J = 16.4 Hz, H-1a), 2.70 (dd, 1H, J = 4.2, 13.6 Hz, H-18), 2.22 (d, 1H, J = 15.7 Hz, H-1b), 1.08 (m, H-9), 1.16, 1.13, 1.12, 1.10, 0.82, 0.75 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.5 (C-3), 183.3 (C-28), 176.8 (C-30), 143.1 (C-13), 136.1 (C-1″), 134.8 (C-2), 134.3 (C-1′), 131.7 (C-3′/5′), 131.6 (C2′/6′), 122.9 (C-12), 122.7 (C-4′), 53.0 (C-5), 51.8 (OMe), 46.0 (C-4), 45.4 (C-9), 45.2 (C-17), 44.0 (C-8), 43.7 (C-20), 42.4 (C-18), 42.0 (C-19), 41.7 (C-14), 39.2 (C-1), 36.3 (C-10), 33.4 (C-22), 31.8 (C-7), 30.3 (C-21), 29.6 (C-23), 28.3 (C-29), 27.6 (C-15), 25.6 (C-27), 23.6 (C-16), 23.1 (C-11), 22.6 (C-24), 20.2 (C-6), 16.5 (C-26), 15.2 (C-25). – HRMS ((+)-ESI): m/z = 665.2759 (calcd. 665.2763 for C38H50BrO5, [M+H]+).
2.7.7 2-(m-Methoxybenzylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (30)
Yellow oil; yield: 73%. – IR (KBr): ν = 3500, 2949, 1728, 1689, 1597, 1217, 757 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.50 (s, 1H, H-1″), 7.33 (t, J = 7.9 Hz, 1H, H-5′), 7.03 (d, J = 7.7 Hz, 1H, H-4′), 6.96 (m, 1H, H-2′), 6.39 (dd, J = 2.6, 1H, 8.1 Hz, H-6′) 5.40 (m, 1H, H-12), 3.85 (s, 3H, OMe), 3.70 (s, 3H, OMe), 3.04 (d, J = 16.3 Hz, 1H, H-1a), 2.71 (dd, J = 3.5, 9.5 Hz, 1H, H-18), 2.29 (d, J = 16.3 Hz, 1H, H-1b), 1.08 (m, H-9), 1.20, 1.17, 1.15, 1.14, 0.88, 0.80 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.7 (C-3), 183.3 (C-28), 176.9 (C-30), 159.5 (C-3′), 142.9 (C-13), 137.4 (C-2), 137.2 (C-1″), 134.0 (C-1′), 129.3 (C-2′), 123.0 (C-4′), 122.9 (C-12), 122.6 (C-5′), 114.0 (C-6′), 55.3 (OMe), 53.0 (C-5), 51.8 (OMe), 46.0 (C-4), 45.4 (C-9), 45.2 (C-17), 44.0 (C-8), 43.7 (C-20), 42.4 (C-19), 42.0 (C-18), 41.7 (C-14), 39.2 (C-1), 36.3 (C-10), 33.4 (C-22), 31.8 (C-7), 30.3 (C-21), 29.6 (C-23), 28.3 (C-29), 27.7 (C-15), 25.6 (C-27), 23.6 (C-16), 23.1 (C-11), 22.6 (C-24), 20.3 (C-6), 16.5 (C-26), 15.2 (C-25). – HRMS ((+)-ESI): m/z = 617.3766 (calcd. 617.3764 for C39H53O6, [M+H]+).
2.7.8 2-(Pyridin-benzylidine-)-3-oxo-olean-12-en-28,30-dioic acid-30-methyl ester (31)
Yellow oil; yield: 77%. – IR (KBr): ν = 3500, 2949, 1938, 1727, 1684, 1599, 1214, 755 cm–1. – 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 8.64 (d, J = 5.9 Hz, 2H, H-3′/5′), 7.37 (s, 1H, H-1″), 7.29 (d, J = 5.9 Hz, 2H, H-2′/6′), 5.42 (m, 1H, H-12), 3.70 (s, 3H, OMe), 2.96 (d, J = 16.4 Hz, 1H, H-1), 2.75 (d, J = 10.3 Hz, 1H, H-18), 2.30 (d, J = 16.4 Hz, 1H, H-1b), 1.08 (m, H-9), 1.21, 1.16, 1.16, 1.15, 0.87, 0.82 (6 × s, 18H, CMe). – 13C NMR (75 MHz, CDCl3): δ = 207.3 (C-3), 182.1 (C-28), 176.9 (C-30), 149.3 (C-3′/5′), 143.9 (C-1′), 143.3 (C-13), 138.2 (C-2), 133.8 (C-1″), 124.2 (C2′/6′), 122.6 (C-12), 53.1 (C-5), 51.8 (OMe), 45.4 (C-4), 45.4 (C-9), 44.0 (C-17), 43.7 (C-8), 43.7 (C-20), 42.6 (C-18), 42.1 (C-19), 41.8 (C-14), 39.2 (C-1), 36.4 (C-10), 33.4 (C-22), 31.8 (C-7), 30.4 (C-21), 29.5 (C-23), 28.3 (C-29), 27.7 (C-15), 25.7 (C-27), 23.6 (C-16), 23.2 (C-11), 22.6 (C-24), 20.2 (C-6), 16.5 (C-26), 15.3 (C-25). – HRMS ((+)-ESI): m/z = 588.3603 (calcd. 588.3611 for C37H50NO5, [M+H]+).
2.8 Brine shrimp lethality assay (BSLA)
The assay was performed as described previously by Meyer [23] with some minor modifications. Brine shrimp eggs (Gulf Breeze®) were hatched in artificial sea water prepared with commercial salt mixture (Instant Ocean®), illuminated and oxygenated with an aquarium pump. After 48 h incubation at 26 °C, 10 shrimps were transferred with a Pasteur pipette to three sample vials for each of three doses (100, 25, 5 μg mL−1), for a total of nine vials per sample. The compound samples (10 mg) were dissolved in CHCl3 (5 mL). Aliquots of testing solutions 250, 62.5 or 12.5 μL (for 100, 25 and 5 ppm doses) were placed on vials of 5 mL and the solvent was evaporated. The residue was redissolved in 5 μL of Tween 80® and 5 mL of artificial sea water was added. Survivors were counted and the percent deaths at each dose were determined. Control samples were included and assayed simultaneously. IC50 values were calculated from 24 h counts using the probit analysis [26].
3 Results and discussion
3.1 Chemistry
Firstly, SA was isolated from the dried fruits of P. icosandra L. using a procedure detailed in the Experimental Section. The isolated compound was fully characterized by comparison of its spectroscopic data with those previously reported [24, 25] along with its melting point. Subsequently, SA was submitted to a Jone’s oxidation to afford ketone 2 [25]. Compound 2 was then converted into several ring-A indolo-derivatives by its reaction with aromatic hydrazines 3–7 in glacial acetic acid under reflux [27]. Aqueous work-up using NaOH and brine solution followed by simple preparative TLC on silica gel, afforded the purified indolo-triterpenes 8–13 in 58–74% yield. The reaction starting with 3-bromophenylhydrazine yielded a mixture of indoles 9 and 11 in a 2:1 ratio respectively, which could not be separated by sequential preparative silica gel chromatography.
Reaction of ketone 2 with hydroxylamine hydrochloride in pyridine provided the desired C-3 oxime 14 in 92% yield; the compound was precipitated from the solution by adding cold water and filtration in vacuum. Pyrazine 15 was obtained by condensation of ketone 2 in the presence of ethylenediamine and sulphur in morpholine [28]. Aqueous work-up using brine and purification by preparative TLC plates on silica gel afforded 15 in 64% yield.
The synthetic protocol for serjanic acid benzylidine derivatives 24–31 from the intermediate 2 involved a Claisen-Schmidt condensation with the corresponding benzaldehyde 16–23 in the presence of ethanolic KOH at room temperature for 24 h. The work-up was carried out by adding acidified water to precipitate the compounds, extraction with EtOAc and purification by preparative TLC on silica gel. Yields for these compounds were between 40 and 81% (Scheme 1).
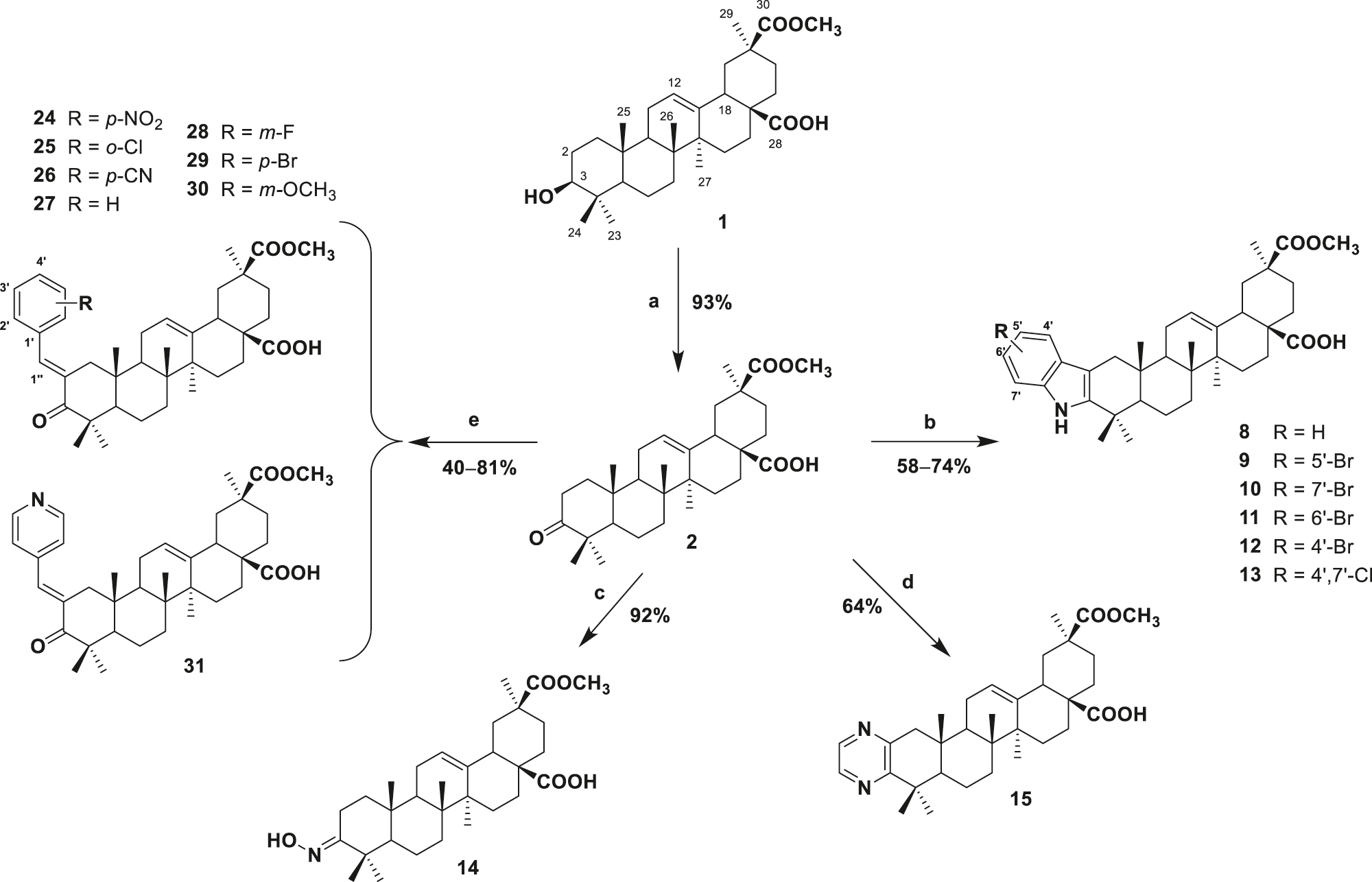
Reagents and conditions: (a) K2Cr2O7–H2SO4, acetone (0 °C); (b) aromatic hydrazine, AcOH, reflux (4 h); (c) NH2OH·HCl, pyridine, reflux (3 h); (d) sulphur, ethylenediamine, morpholine, reflux (6 h); (e) substituted benzaldehyde, ethanolic KOH, r.t. (24 h).
Extensive 1H, 13C NMR, IR and HR-MS spectroscopic analysis confirmed the structures of all serjanic acid derivatives. All compounds exhibited in their IR spectra typical signals expected for the functional groups present in the molecules. For instance, all derivatives showed strong absorption bands ν = 3340–3500 cm−1 (O–H stretching), 3050–3060 cm−1 (=C–H stretching), 1693–1732 cm−1 (C=O stretching) and 1208–1221 cm−1 (C–CO–O stretching). Indolo-triterpene, oxime and pyrazine derivatives presented C=N stretching bands between ν = 1457–1463 cm−1, while aromatic C=C stretching bands between 1550 and 1606 cm−1 were observed for indolo-triterpenes, pyrazine and benzylidines.
The 1H NMR spectra of all compounds revealed signals corresponding to vinylic, methoxy- and methyl hydrogens appearing between δ = 5.36–5.49, 3.69–3.75 and 0.73–1.34 ppm respectively. All compounds except for 14 exhibited two allylic protons as doublets (J ≈ 15 Hz) around δ = 2.17–3.43 ppm, and were assigned to H-1a and H-1b of the triterpenoid. Signals of aromatic hydrogens were detected between δ = 6.39–8.64 ppm and the –NH proton in compounds 8–13 was detected between 7.71 and 8.01 ppm. A considerable shift to lower field was observed for methyl groups H-23, H-24 and H-25 of indolo-triterpene and pyrazine derivatives, due to the proximity to the heterocyclic ring. Compound 14 displayed two allylic signals at δ = 3.10 and 2.22 ppm, corresponding to the H-2a and H-2b vicinal to the oxime group at C-3 position. Also, a characteristic vinylic proton (H-1″) between δ = 7.23–7.52 ppm was present in derivatives 24–31.
13C NMR signals for all triterpenoidal structures were assigned, and the most relevant signals were: carbonylic carbon atoms between δ = 176.3–183.9 ppm of the acid and ester functionalities, vinylic carbons of the C-12/C-13 double bond between 121.8 and 143.5 ppm, methoxy groups between 51.5 and 52.0 ppm and six methyl carbons around 15.0–31.6 ppm. In derivatives 8–13, carbon atoms C-2 and C-3 suffered a downfield chemical shift due to the aromatic ring formation. Signals for C-2 were located around δ = 106.8–108.8 ppm and C-3 signals around 140.9–142.8 ppm, due the proximity with the nitrogen atom. The rest of the carbon atoms belonging to the indolic nucleus resonates between δ = 108.3–137.3 ppm. Elucidation of the oxime 14 was corroborated by detecting the characteristic hydroxyimine carbon at δ = 167.4 ppm and derivative 15 evidenced a downfield chemical displacement to 48.1 ppm of its C-1 position and exhibited the characteristic pyrazine-ring carbons at δ = 159.7 (C-3), 150.5 (C-2), 142.4 (C-3′) and 141.3 (C-2′) ppm. 13C NMR signals at δ ≈ 207, 142 and 135 ppm for α,β-unsaturated ketones along with aromatic carbons between 114.0 and 164.3 ppm confirmed the product formation of benzylidines 24–31. Some minor signals, product of the “E/Z” isomerization, where found in the NMR spectra of all benzylidines. Formation of the “E” isomer was assumed, due to kinetic and thermodynamic considerations.
A detailed assignment of the 1H and 13C NMR spectral data for all new compounds is given in the Experimental Section.
3.2 Brine shrimp lethality assay (BSLA)
The toxicity of SA and all derivatives was tested against A. salina. The dose-dependent assays were carried out between 10 and 100 ppm using a negative and positive control; Tween 80® at this concentration did not affect this bioassay. IC50 values of the compounds are presented in Table 1.
Brine shrimp lethality assay of the synthesized compounds.
| Compound | IC50 (μm) | Compound | IC50 (μm) |
|---|---|---|---|
| 1 | 85.3 ± 13 | 24 | 39.8 ± 8 |
| 8 | 54.1 ± 4 | 25 | 61.3 ± 33 |
| 9 | 29.3 ± 4 | 26 | 25.9 ± 2 |
| 10 | 28.6 ± 4 | 27 | 60.5 ± 18 |
| 11 | 29.3 ± 4 | 28 | 73.9 ± 12 |
| 12 | 78.8 ± 7 | 29 | 41.6 ± 12 |
| 13 | 49.4 ± 6 | 30 | 162.6 ± 33 |
| 14 | 33.1 ± 6 | 31 | 22.4 ± 4 |
| 15 | 10.3 ± 4 | Podophyllotoxin | 5.8 ± 1 |
The results showed that almost all derivatives presented lower IC50 values with respect to SA, which can be translate into an increase of the cytotoxic potential of these molecules; in contrast, compound 30 showed an increase of almost 50% in the IC50. Concerning indolo-triterpenes, we observe that all derivatives with the exception of 12 presented higher toxicity than 8. The increase in the toxicity with respect to SA was also observed for the majority of benzilydine derivatives, and it is noteworthy to mention that the presence of an electron-withdrawing group in para-position of the phenyl ring resulted in greater bioactivity.
Derivatives 15, 26 and 31 exhibited the lowest IC50 values. Moreover, the observed toxicity of 15 is slightly lower than that of Podophyllotoxin [21], a well-known bioactive natural compound, making 15 a suitable candidate for further biological and pharmacological studies.
4 Conclusions
The obtained results are in accordance with many other studies suggesting that the modification of C-2/C-3 of the triterpene skeleta is of great importance for the biological activity. The study also showed that all tested compounds exhibited a toxic effect to the brine shrimp, with derivatives 15, 26 and 31 being the most effectives. These toxicities were as high as 8 times that of SA.
5 Supporting information
IR, 1H NMR, 13C NMR and HRMS spectra of compounds 1, 2, 8–15 and 24–31 are given as supplementary material available online (https://doi.org/10.1515/znb-2022-0064).
Funding source: USB-DID https://www.did.usb.ve/inicio/coord-de-ccs-básicas
Award Identifier / Grant number: S1-IC-CB-004-15
Acknowledgements
The authors would like to thank USB-DID (project S1-IC-CB-004-15) for financial support. Also, thanks are expressed to Prof. Lourdes Novoa for the support in the structural elucidation of some derivatives and to Prof. Julio C. Herrera for taking and processing HR-MS data.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: This work was funded by USB-DID (project S1-IC-CB-004-15).
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
1. Gupta, R., Gabrielsen, B., Ferguson, S. M. Curr. Drug Discov. Technol. 2005, 2, 203–219; https://doi.org/10.2174/157016305775202937.Suche in Google Scholar
2. Grabley, S., Sattler, I. Natural products for lead identification: nature is a valuable resource for providing tools. In Modern Methods of Drug Discovery; Hillisch, A., Hilgenfeld, R., Eds. Birkhäuser-Verlag: Basel, 2003; Chapter 5; pp. 87–107.10.1007/978-3-0348-7997-2_5Suche in Google Scholar
3. Lee, K. H. J. Nat. Prod. 2010, 73, 500–516; https://doi.org/10.1021/np900821e.Suche in Google Scholar
4. Dewick, P. M. Medicinal Natural Products, 2nd ed.; Wiley & Sons: England, 2002; Chapter 5; pp 212–219.Suche in Google Scholar
5. Connolly, J. D., Overton, K. H. The triterpenoids. In Chemistry of Terpenes and Terpenoids; Newman, A. A., Ed. Academic Press: New York, 1972; pp. 207–287.Suche in Google Scholar
6. Nakanishi, K., Ito, S. Biosynthesis of oleanene and ursene triterpenes in tissue culture of Isodon japonicus. In Natural Products Chemistry, Vol. 3; Nakanishi, K., Goto, T., Ito, S., Natori, S., Nozoe, S., Eds. Kodansha: Tokyo, 1983; pp. 185–187.Suche in Google Scholar
7. Devon, T. K., Scott, A. I. The terpenes. In Handbook of Naturally Occurring Compounds, Vol. 2; Academic Press: New York, 1972; pp. 281–384.10.1016/B978-0-12-213602-3.50011-2Suche in Google Scholar
8. Meng, Y. Q., Liu, D., Cai, L. L., Chen, H., Cao, B., Wang, Y. Z. Bioorg. Med. Chem. 2009, 17, 848–854; https://doi.org/10.1016/j.bmc.2008.11.036.Suche in Google Scholar
9. Liby, K. T., Sporn, M. B. Pharmacol. Rev. 2012, 64, 972–1003; https://doi.org/10.1124/pr.111.004846.Suche in Google Scholar
10. Ma, C., Nakamura, N., Miyashiro, H., Hattori, M., Shimotohno, K. Phytother. Res. 1998, 12, 138–142; https://doi.org/10.1002/(sici)1099-1573(1998)12:1+<s138::aid-ptr276>3.0.co;2-5.10.1002/(SICI)1099-1573(199803)12:2<138::AID-PTR200>3.0.CO;2-CSuche in Google Scholar
11. Cos, P., Maes, L., Berghe, D. V., Hermans, N., Pieters, L., Vlietinck, A. J. Nat. Prod. 2004, 67, 284–293; https://doi.org/10.1021/np034016p.Suche in Google Scholar
12. Yu, D., Sakurai, Y., Chen, C. H., Chang, F. R., Huang, L., Kashiwada, Y., Lee, H. J. Med. Chem. 2006, 49, 5462–5469; https://doi.org/10.1021/jm0601912.Suche in Google Scholar
13. Kwon, T. H., Lee, B. M., Chung, S. H., Kim, D. H., Lee, Y. S. Bull. Kor. Chem. Soc. 2009, 30, 119–123.10.5012/bkcs.2009.30.1.119Suche in Google Scholar
14. Ikeda, Y., Murakami, A., Ohigashi, H. Mol. Nutr. Food Res. 2008, 52, 26–42; https://doi.org/10.1002/mnfr.200700389.Suche in Google Scholar
15. Liu, J. J. Ethnopharmacol. 2005, 100, 92–94; https://doi.org/10.1016/j.jep.2005.05.024.Suche in Google Scholar
16. Tian, Z., Lin, G., Zheng, R. X., Huang, F., Yang, M. S., Xiao, P. G. World J. Gastroenterol. 2006, 12, 874–879; https://doi.org/10.3748/wjg.v12.i6.874.Suche in Google Scholar
17. Shyu, M. H., Kao, T. C., Yen, G. C. J. Agric. Food Chem. 2010, 58, 6110–6118; https://doi.org/10.1021/jf100574j.Suche in Google Scholar
18. Pathak, A., Singh, S. K., Biabani Farooq, M. A., Kulshreshtha, D. K., Puri, S. K., Srivastava, S., Kundu, B. Comb. Chem. High Throughput Screening 2002, 5, 241–248; https://doi.org/10.2174/1386207024607275.Suche in Google Scholar
19. Solis, P. N., Wrigth, C. W., Anderson, M. M., Gupta, M. P., Phillipson, J. D. Planta Med. 1993, 59, 250–252; https://doi.org/10.1055/s-2006-959661.Suche in Google Scholar
20. Anderson, J. E., Goetz, C. M., McLaughlin, J. M. Phytochem. Anal. 1991, 2, 107–111; https://doi.org/10.1002/pca.2800020303.Suche in Google Scholar
21. Meyer, B. N., Ferrigni, N. R., Putnam, J. E., Jacobsen, L. B., Nichols, D. E., McLaughlin, J. L. Planta Med. 1982, 45, 31–34; https://doi.org/10.1055/s-2007-971236.Suche in Google Scholar
22. Zani, C. L., Chaves, P. P., Queiroz, R., De Oliveira, A. B., Cardoso, J. E., Anjos, A. M., Grandi, T. S. Phytomedicine 1995, 2, 47–50; https://doi.org/10.1016/s0944-7113(11)80048-6.Suche in Google Scholar
23. Santos Pimenta, L. P., Pinto, G. B., Takahashi, J. A., e Silva, L. G., Boaventura, M. A. Phytomedicine 2003, 10, 209–212; https://doi.org/10.1078/094471103321659960.Suche in Google Scholar PubMed
24. Morales, M. A. Rev. Latinoam. Quim. 1977, 9, 94–95.Suche in Google Scholar
25. Alvarado, M., Moreno, M., Rodriguez, V. M. Phytochemistry 1981, 20, 2436–2438; https://doi.org/10.1016/s0031-9422(00)82687-4.Suche in Google Scholar
26. Finney, D. Probit Analysis, 3rd ed.; Cambridge University Press: Cambridge, 1971; Chapter 3; pp. 21–49.10.2307/2346498Suche in Google Scholar
27. Finlay, H. J., Honda, T., Gribble, W. Arkivoc 2002, 12, 38–46.10.3998/ark.5550190.0003.c05Suche in Google Scholar
28. Xu, J., Li, Z. X., Luo, J., Yang, F., Liu, T., Liu, M. Y., Qiu, W. W., Tang, J. J. Med. Chem. 2012, 55, 3122–3134; https://doi.org/10.1021/jm201540h.Suche in Google Scholar PubMed
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/znb-2022-0064).
© 2022 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this issue
- Research Articles
- Crystal structures of sildenafil compounds with nitrate and di(citrato)zinc counterions
- Synthesis, crystal structure, and properties of three lead(II) complexes based on the 1,10-phenanthroline ligand
- A highly selective and sensitive fluorescent sensor based on a 1,8-naphthalimide with a Schiff base function for Hg2+ in aqueous media
- Co-crystallization of dimethyl N-cyanodithioiminocarbonate and bis[(aqua)-µ2-hydroxy-n-butyldichlorotin(IV)]
- Synthesis of ring-A serjanic acid derivatives and their cytotoxic evaluation through the brine shrimp lethality assay (BSLA)
- Electron density of the 1:2 complex of valinomycin with calcium triflate observed in crystals of the composition (valinomycin)Ca2(OTf)4(THF)5(H2O)4
- Two zinc and cadmium coordination polymers constructed with bis(4-(1H-imidazol-1-yl)phenyl)methanone and naphthalene-1,4-dicarboxylate ligands: synthesis and structural characterization
- Characterization of hydrophilic carbon nanohorns prepared by the arc-in-water method
- Crystal structure determination and characterization of Sm3SiO5F3
- A four-fold three-dimensional zinc(II) coordination polymer based on 4,4′-bis(2-methyl-imidazolyl)biphenyl and 5-sulfoisophthalate ligands: synthesis, structure and properties
- Synthesis, structures, and photophysical properties of two Cu(I) complexes supported by N-heterocyclic carbene and phosphine ligands
- Synthesis of chiral binaphthol-based bishydroxylamines
Artikel in diesem Heft
- Frontmatter
- In this issue
- Research Articles
- Crystal structures of sildenafil compounds with nitrate and di(citrato)zinc counterions
- Synthesis, crystal structure, and properties of three lead(II) complexes based on the 1,10-phenanthroline ligand
- A highly selective and sensitive fluorescent sensor based on a 1,8-naphthalimide with a Schiff base function for Hg2+ in aqueous media
- Co-crystallization of dimethyl N-cyanodithioiminocarbonate and bis[(aqua)-µ2-hydroxy-n-butyldichlorotin(IV)]
- Synthesis of ring-A serjanic acid derivatives and their cytotoxic evaluation through the brine shrimp lethality assay (BSLA)
- Electron density of the 1:2 complex of valinomycin with calcium triflate observed in crystals of the composition (valinomycin)Ca2(OTf)4(THF)5(H2O)4
- Two zinc and cadmium coordination polymers constructed with bis(4-(1H-imidazol-1-yl)phenyl)methanone and naphthalene-1,4-dicarboxylate ligands: synthesis and structural characterization
- Characterization of hydrophilic carbon nanohorns prepared by the arc-in-water method
- Crystal structure determination and characterization of Sm3SiO5F3
- A four-fold three-dimensional zinc(II) coordination polymer based on 4,4′-bis(2-methyl-imidazolyl)biphenyl and 5-sulfoisophthalate ligands: synthesis, structure and properties
- Synthesis, structures, and photophysical properties of two Cu(I) complexes supported by N-heterocyclic carbene and phosphine ligands
- Synthesis of chiral binaphthol-based bishydroxylamines