A novel ternary bismuthide, NaMgBi: crystal and electronic structure and electrical properties
-
Takahiro Yamada
 ,
Naoki Matsuo
,
Naoki Matsuo

Abstract
A new ternary sodium magnesium bismuthide, NaMgBi, has been synthesized from the constituent metals, and its crystal structure was determined by single-crystal X-ray diffraction. NaMgBi crystallizes in a tetragonal PbFCl-type structure corresponding to the space group P4/nmm, where Z = 2, a = 4.7123(4) and c = 7.8158(7) Å. The structure is composed of layers formed by edge-sharing Bi tetrahedra centered with Mg stacked in the c-axis direction, and these layers sandwich the Na atoms. First-principles computations based on density functional theory calculations have verified that the most stable atomic configuration is the one in which the Na and Mg atoms occupy the 2a and 2c sites, respectively. The electrical resistivity measured for a sintered polycrystalline sample of NaMgBi with a relative density of 70% was found to gradually decrease from 868 to 26.4 mΩ cm upon increasing the temperature from 297 to 506 K, and the Seebeck coefficient decreased from 273 to 180 μV K−1 upon increasing the temperature from 298 to 496 K. Electronic structure calculations have revealed that NaMgBi must be a semiconductor with a small band gap of ∼0.1 eV.
1 Introduction
Intermetallic compounds composed of multiple elements with large electronegativity differences are known as Zintl phases or polar intermetallics, and they possess both ionic and covalent bonding. This leads to unique crystal structures and a range of electronic structures exhibiting metallic, semi-metallic, and semiconducting behavior [1], [2], [3], [4], [5]. Some metallic compounds show superconductivity [6], [7], [8], [9], while some semimetals and semiconductors exhibit thermoelectric, photovoltaic, and catalytic properties [10], [11], [12], [13], [14], and therefore are useful in the area of materials science. Furthermore, intermetallic compounds with special electronic band structures, in which the conduction and valence bands cross each other near the Fermi level, have attracted growing attention in the area of condensed matter physics as they exhibit topological quantum properties [15], [16], [17]. Therefore, the exploration and characterization of novel Zintl phases and polar intermetallics have become very important active fields from both theoretical and experimental perspectives.
For example, in the Na-Mg-group 14 element (Tt = C, Si, Ge, Sn, or Pb) systems, five intermetallic compounds, namely Na2MgTt, Na2Mg3 Tt 2 (Tt = Sn or Pb), and Na4Mg4Sn3 have been synthesized in the last decade [18], [19], [20]. Compounds of the form Na2MgTt (Tt = Sn or Pb) crystallize in a Li2CuAs-type structure (hexagonal, space group P63/mmc [No. 129]) at T = 293 K, in which honeycomb layers composed of Mg and Tt atoms are stacked alternatively with layers of Na atoms. Na2MgSn is a narrow-gap semiconductor (E g = 0.2–0.4 eV) with a relatively large Seebeck coefficient (+390 μV K−1), while Na2MgPb exhibits metallic electrical properties features two crystal structure phase transitions at higher temperatures. Recently, theoretical calculations have predicted that Na2MgSn has the potential to be a room-temperature thermoelectric material [21], while Na2MgPb is a candidate for Dirac semiconductors derived from its band topology [22].
In terms of the Na-Mg-group 15 element (Pn = N, P, As, Sb, or Bi) systems, only two ternary compounds, i.e., NaMgPn (Pn = As and Sb) have been reported [23], [24], [25]. They crystallize in a PbFCl-type structure (tetragonal, space group P4/nmm [No. 129]), where the layers composed of covalently connected Mg and Pn atoms are separated by double sheets of ionic Na atoms [23], [24], [25], being isostructural with the 111 family of iron-based superconductors, such as LiFePn (Pn = P or As) [26], [27], [28]. However, the electrical properties and electronic structures of compounds of the type NaMgPn have not yet been elucidated. In this study, a new ternary compound, NaMgBi, which contains the heaviest pnictogen, has been synthesized, and its crystal structure and electrical properties were experimentally investigated. Furthermore, the electronic structures of NaMgPn (Pn = As, Sb, and Bi) have been calculated using density functional theory (DFT).
2 Results and discussion
2.1 Chemical characterization and crystal structure determination
In an ingot sample prepared by heating Na, Mg, and Bi metals in an equimolar ratio (Na:Mg:Bi = 1:1:1), many plate-like crystals with sizes greater than several hundred micrometers were found, as shown in Figure 1. Using wavelength-dispersive X-ray (WDX) spectroscopy, the chemical composition of these crystals was determined to be Na, 9.0(2) wt%; Mg, 8.8(1) wt%; and Bi, 84.4(1.3) wt% (total 102(2) wt%), which corresponds to a Na:Mg:Bi molar ratio of 1.01(2):0.94(1):1.05(1); this is close to the ideal composition of NaMgBi (Table 1). The obtained platelet crystals were easily exfoliated and deformed, and a large strain was induced in the crystals when they were divided into sizes suitable for single-crystal X-ray diffraction (XRD) measurements. Therefore, another ingot was prepared using a Na:Mg:Bi ratio of 3:5:2, which is richer in Mg and Na than the previous sample. In this case, platelet single crystals of ∼100 μm in size were obtained. The crystals of NaMgBi were unstable in air, so the crystal used for the XRD measurement was enclosed in a glass capillary.

SEM image of single crystals of NaMgBi.
Composition of the single crystals of NaMgBi.
| Na | Mg | Bi | Total | |
|---|---|---|---|---|
| wt% | 9.0(2) | 8.8(1) | 84.4(1.3) | 102(2) |
| at% | 33.9(6) | 31.2(4) | 34.9(4) | 100 |
The XRD reflections of the crystal were indexed with tetragonal unit cell parameters, and they were refined to a = 4.7123(4) and c = 7.8158(7) Å. Since the observed extinction conditions were consistent with the space group P4/nmm of the NaMgSb and NaMgAs analogues, the crystal structure was considered to be a stoichiometric compound (i.e., NaMgBi) using a structural model in which the Sb component of NaMgSb (Na, 2c [1/4, 1/4, z(Na) = 0.6384]; Mg, 2a [3/4, 1/4, 0]; Sb, 2c [l/4, 1/4, z(Sb) = 0.2243]) [24] was replaced with Bi. The refined atomic coordinates were z(Na) = 0.6413(7) and z(Bi) = 0.22909(5), and the reliability factors (i.e., R1 and wR) for all data were 2.15 and 4.37%, respectively. When the occupation ratios of the Na and Mg sites were independently refined assuming that the Bi site was fully occupied, they were determined to be 1.0(3) and 1.01(2), respectively. These results are consistent with the composition determined by WDX spectroscopy. Since the distinction between atoms with similar atomic X-ray scattering factors, such as Na and Mg, is difficult by XRD measurements, the occupation of the 2c and 2a sites by Na and Mg atoms, respectively, in the crystal structure of NaMgBi was confirmed by first-principles calculations, as described below.
Details of the XRD data collection and refinement results are listed in Table 2, and the atomic coordinates and atomic displacement parameters are summarized in Table 3. A comparison of the crystal structures, including representative interatomic distances for NaMgPn (Pn = Bi, Sb, or As) is presented in Table 4. The lattice parameters of NaMgBi with the PbFCl-type structure, which contains the heavier pnictogen atom Bi, are significantly larger than those of the other isostructural compounds, namely NaMgSb (tetragonal, a = 4.653, c = 7.640 Å) and NaMgAs (tetragonal, a = 4.422, c = 7.138 Å) [24]. The c/a ratio (=1.658) of NaMgBi was ∼1–2% larger than those of NaMgSb (c/a = 1.642) and NaMgAs (c/a = 1.614).
Crystal data and refinement results for NaMgBi.
| Chemical formula | NaMgBi |
|---|---|
| Formula weight, M r | 256.28 |
| Habit; color | Platelet; silver black |
| Size/mm3 | 0.055 × 0.089 × 0.090 |
| Temperature T/K | 298(2) |
| Crystal system | Tetragonal |
| Space group | P4/nmm (No. 129) |
| Unit cell dimensions | |
| a/Å | 4.7123(4) |
| c/Å | 7.8158(7) |
| Unit cell volume V/Å3 | 173.56(3) |
| Z | 2 |
| Calculated density D cald/g cm−3 | 4.90 |
| Wavelength λ/Å | 0.71073 |
| Absorption correction | Numerical (Sadabs) |
| Absorption coefficient μ/mm−1 | 50.9 |
| Limiting indices hkl | −6 ≤ h ≤ 5 |
| −6 ≤ k ≤ 6 | |
| −10 ≤ l ≤ 10 | |
| F(000)/e | 212 |
| θ range data collection/deg | 2.61–27.48 |
| Reflections collected; unique | 1429; 145 |
| R int | 0.0215 |
| Data; restraints; parameters | 145; 0; 11 |
| R1a; wR2b (I > 2 σ(I)) | 0.0215; 0.0437 |
| R1a; wR2b (all data) | 0.0215; 0.0437 |
| Weight parametersb a; b | 0.0120; 0.1957 |
| Goodness-of-fit S c on F 2 | 1.460 |
| Largest diff. Peak; hole Δρ/e Å−3 | 2.93; −1.89 |
-
a R1 = Σ||F o| − |F c||/Σ|F o|. b wR2 = [Σw(F o 2 − F c 2)2/Σ(wF o 2)2]1/2, w = 1/[σ 2(F o 2) + (aP)2 + bP], where F o is the observed structure factor, F c is the calculated structure factor, σ is the standard deviation of F c 2, and P = (F o 2 + 2F c 2)/3. c S = [Σw(F o 2 − F c 2)2/(n − p)]1/2, where n is the number of reflections and p is the total number of parameters refined.
Atomic coordinates and anisotropic and equivalent isotropic displacement parameters (U ij and U eq in Å2).
| Atom | Wyckoff site | Occ. | x | y | z | U 11 | U 33 | U eq |
|---|---|---|---|---|---|---|---|---|
| Na | 2c | 1.0 | 1/4 | 1/4 | 0.6413(7) | 0.035(3) | 0.023(3) | 0.0314(18) |
| Mg | 2a | 1.0 | 3/4 | 1/4 | 0 | 0.0093(17) | 0.0183(17) | 0.0123(10) |
| Bi | 2c | 1.0 | 1/4 | 1/4 | 0.22909(5) | 0.0116(3) | 0.0118(3) | 0.0117(2) |
-
U 11 = U 22, U 23 = U 13 = U 12 = 0, U eq = (∑i∑j U ij a i * a j * a i a j)/3.
Comparison of the crystal structures of NaMgPn (Pn = As, Sb, Bi).
| Chemical formula | NaMgBi (this work) |
NaMgSb Ref. [24] |
NaMgAs Ref. [24] |
NaMgAs Ref. [25] |
|---|---|---|---|---|
| Crystal system | Tetragonal | Tetragonal | Tetragonal | Tetragonal |
| Space group | P4/nmm (No. 129) | P4/nmm (No. 129) | P4/nmm (No. 129) | P4/nmm (No. 129) |
| Unit cell dimensions | ||||
| a/Å | 4.7123(4) | 4.653 | 4.422 | 4.417 |
| c/Å | 7.8158(7) | 7.640 | 7.138 | 7.130 |
| c/a | 1.658 | 1.642 | 1.614 | 1.614 |
| Unit cell volume V/Å3 | 173.56(3) | 165.41 | 139.58 | 139.1 |
| Z | 2 | 2 | 2 | 2 |
| Calculated density, D calcd/g cm−3 | 4.90 | 3.40 | 2.91 | 2.92 |
| Atomic positions | ||||
| Na (2c), z | 0.6413(7) | 0.6384 | 0.6539 | 0.6305 |
| Pn (2c), z | 0.22909(5) | 0.2243 | 0.2234 | 0.21441 |
| Atomic distances | ||||
| d(Mg–Pn) | 2.9593(4) | 2.8895 | 2.7260 | 2.6860 |
| d(Na–Pn) | 3.222(6) | 3.164 | 3.073 | 2.967 |
| d(Na–Mg) | 3.662(5) | 3.612 | 3.315 | 3.438 |
In the crystal structure of NaMgBi shown in Figure 2, Bi atoms at the 2c site form regular tetrahedra with one Mg atom in the center (2a site), where the distance between the Mg and Bi atoms [d(Mg–Bi)] of these tetrahedra is 2.9593(3) Å, and ∞[(Mg–Bi)−] layers are formed through edge sharing. The Mg–Bi layers are stacked along the c axis with Na atoms sandwiched between them. The interatomic distance between Na and Bi (3.222(6) Å) is shorter than that between Na and Mg (3.662(5) Å), and these distances have been determined to be 1.4–2.4% larger than the Mg–Sb, Na–Sb, and Na–Mg distances of NaMgSb.

Crystal structure of NaMgBi. Each atom is drawn using displacement ellipsoids at the 99% probability level.
2.2 Electronic properties of the polycrystalline sample
A polycrystalline bulk sample of NaMgBi was prepared at T = 1027 K via a solid-state reaction from a pulverized ingot sample obtained from the three metals in an equimolar ratio. The density estimated from the volume and weight of the bulk sintered sample was 3.40 g cm−3, which is ∼70% of the theoretical density of NaMgBi determined in the previous section. As shown in Figure 3, the powder diffraction peaks exhibited a preferred orientation to the c axis owing to the layered structure, and this can be explained by the PbFCl-type structure determined by single-crystal structure analysis with tetragonal lattice parameters of a = 4.7131(2) and c = 7.8126(3) Å. Although the Na and Mg starting materials have comparatively high vapor pressures, the mass change of the sample in the crucible before and after heating was ≤1%, which indicates that the composition of the synthesized sample was identical to that of the starting material.

XRD pattern of the polycrystalline bulk sample of NaMgBi (a) and the simulated XRD pattern (b).
The variations in the electrical resistivity (ρ) and the Seebeck coefficient (S) of the polycrystalline NaMgBi sample with temperature are shown in Figure 4. More specifically, ρ exhibits semiconducting behavior with values ranging from 868 mΩ cm at 297 K to 26.4 mΩ cm at 509 K. In addition, the S value was determined to be 255 μV K−1 at 298 K. This value decreased gradually with increasing temperature, reaching 180 μV K−1 at 496 K. The positive S value therefore suggests that holes are the majority carriers in NaMgBi.

Variation in the electrical resistivity and Seebeck coefficient of the polycrystalline bulk sample of NaMgBi with temperature.
2.3 Quantum-chemical computations
To confirm the occupation of the Na atom at the 2c site and of the Mg atom at the 2a site in the crystal structure of NaMgBi determined by XRD, first-principles calculations were used to evaluate the formation enthalpies at 0 K for this structure (regular) and for the inverted structure, in which the atomic sites of Na and Mg were exchanged (i.e., Na at the 2a site and Mg at the 2c site) (Figure 5). As indicated, the energy minimum for the regular structure is −40.2 kJ mol−1, which is significantly lower than that of the inverted structure (−23.2 kJ mol−1), thereby indicating that the regular structure is favored. Furthermore, the volume per atom of NaMgBi measured by XRD was 28.9 Å3 atom−1, which is closer to the calculated value for the regular structure.
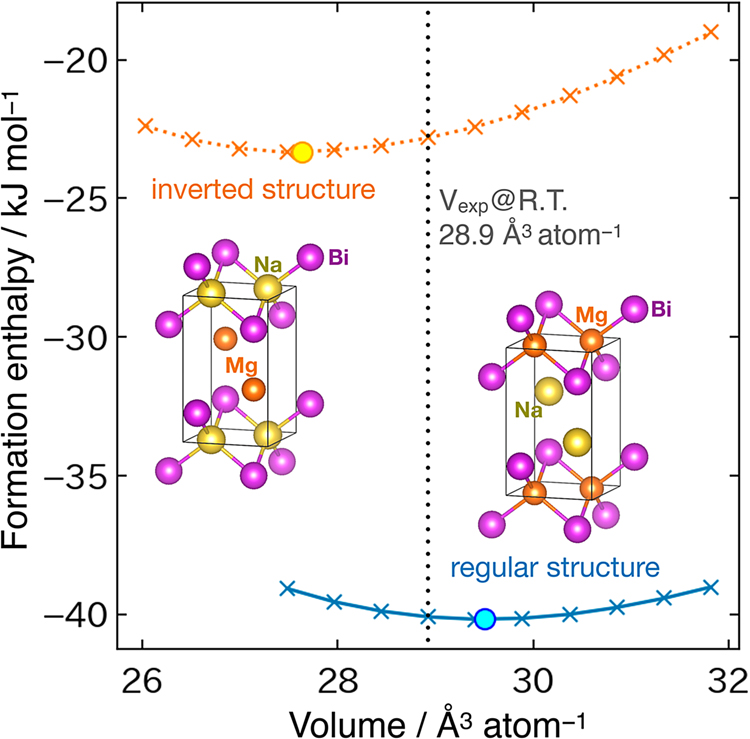
Calculated enthalpy of formation of NaMgBi crystallized in the regular (solid line) and inverse (dotted line) structures as a function of the volume.
The calculated electronic band structures and density of states (DOS) for NaMgBi are shown in Figure 6 together with those of reported isostructural compounds, i.e., NaMgAs and NaMgSb. As indicated, NaMgBi possesses a highly dispersed conduction band that can be attributed mainly to the s orbital of the Bi atoms lying just above the Fermi level at the Γ point, ultimately resulting in a small band gap of 0.09 eV, while NaMgAs and NaMgSb possess semiconducting electronic structures with relatively large band gaps of 0.94 and 1.04 eV, respectively. For all three compounds, large DOS were observed with a steep slope just below the Fermi level (−3.5 up to 0 eV), and these are contributed mainly by the p orbitals of the Pn atoms. This observation is consistent with the fact that the polycrystalline bulk sample of NaMgBi exhibits semiconducting behavior in the measured electrical resistivity, in addition to possessing a relatively large positive Seebeck coefficient. The NaMgBi semiconductor with the smallest band gap among the three NaMgPn compounds could therefore be expected to be a candidate for thermoelectric materials at temperatures close to room temperature.

Calculated band structures and density of states for NaMgPn; Pn = As (a), Sb (b), and Bi (c).
3 Conclusions
A novel intermetallic compound, sodium magnesium bismuthide (NaMgBi), was synthesized by heating stoichiometric amounts of the elemental metals. The crystal structure of NaMgBi (tetragonal, P4/nmm space group, Z = 2, a = 4.7123(4) and c = 7.8158(7) Å) was determined by combining X-ray single-crystal structure analysis with first-principles density functional theory calculations. As a result, a structure based on the PbFCl-type structure was determined, which possesses the following atomic positions: Na at 2c (1/4, 1/4, z) with z = 0.6413(7), Mg at 2a (1/4, 3/4, 0), and Bi at 2c (1/4, 1/4, z) with z = 0.22909(5). NaMgBi was also found to be isostructural with NaMgSb and NaMgAs. Furthermore, the electrical resistivity and Seebeck coefficient of the polycrystalline sample of NaMgBi exhibits negative temperature dependences in the ranges of 868–26.4 mΩ cm at 297–509 K, and 273–180 μV K−1 at 298–496 K. The computed electronic band structure and density of states predict that NaMgBi must be a narrow gap semiconductor with a band gap of 0.09 eV. This band gap is significantly lower than those of NaMgSb (1.04 eV) and NaMgAs (0.94 eV), therefore indicating that this compound could be a candidate for thermoelectric materials at temperatures close to room temperature.
4 Experimental section
All manipulations were carried out in an Ar gas-filled glove box (MBraun, O2, H2O < 1 ppm) because of the air sensitivities of the reagents and samples. Na (lump, 99.95%, Nippon Soda Co., Ltd.), Mg (rod: 1.6 mm diameter, 99.95%, The Nilaco Co., Ltd.), and Bi (powder, 99.999%, Sigma-Aldrich Co., LLC.) were weighed at predetermined molar ratios. A sintered BN crucible (inner diameter 6 mm; inner depth 18 mm, Showa Denko, 99.5%) was loaded with the three elements (∼0.5 g) and sealed in a stainless-steel container (SUS316, inner diameter 10.5 mm, length 80 mm). To prepare the polycrystalline bulk NaMgBi material, the mixture was heated at 1023 K for 2 h. Subsequently, the resulting ingot was pulverized in an agate mortar and pressed into a compact specimen (∼14 × 3 × 3 mm3) that was sintered at 1073 K for 10 h in an electrical furnace. Single crystals of NaMgBi were obtained from the ingot sample, in which NaMgBi was the main product.
The powder X-ray diffraction (XRD) pattern of NaMgBi was measured under an Ar atmosphere using CuKα radiation and a diffractometer (D2 PHASER, Bruker AXS). The XRD data of a single NaMgBi crystal sealed in a glass capillary under an Ar atmosphere were collected using a single-crystal X-ray diffractometer (D8 QUEST, Bruker AXS) with MoKα radiation. The Apex3 software package (Bruker AXS) was used to collect the data, refine the lattice parameters, and correct the X-ray absorption effect [29]. The Shelx-2014 program and the WinGX software were used to analyze the crystal structures of the compounds [30, 31]. The VESTA software was used to visualize the crystal structures [32].
Further details of the crystal structure investigation may be obtained from Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: +49-7247-808-666; crysdata@fiz-karlsruhe.de, http://www.fiz-informationsdienste.de/en/DB/icsd/depot_anforderung.html) on quoting the deposition number CSD-2106109.
The electrical resistivities and Seebeck coefficients of the polycrystalline sintered samples were measured under an Ar atmosphere according to the direct current four-probe and temperature gradient methods, respectively, in a temperature range of 297–509 K.
The formation enthalpy of NaMgBi and the electronic structures of NaMgPn (Pn = Bi, Sb, and As) were calculated based on the DFT approach using the Vienna ab initio simulation package [33, 34], in which the ion-electron correlation is described by the projector augmented wave method [35, 36]. The correlation and exchange functions were given by the generalized gradient approximation proposed by Perdew et al. [37]. Pseudo-potentials were employed for Na, Mg, Bi, Sb, and As with 7, 2, 15, 5, and 5 valence electrons, respectively. Furthermore, k-point sampling meshes of 11 × 11 × 7 and a plane-wave energy cutoff of 400 eV were used for the self-consistent calculations.
To evaluate the crystal structure models of NaMgBi, the energies of two structures with different atomic configurations were calculated. In one model, the Na and Mg atoms occupied the same crystallographic sites in the structures of NaMgSb and NaMgAs, while in the inverted structure, the Na and Mg sites were exchanged. The formation enthalpy per mole of atoms (ΔE) of NaMgBi was defined as the difference between the total energy (E) and the concentration weighted total energy of pure BCC-Na, HCP-Mg, and rhombohedral-Bi as follows:
Furthermore, the relationship between the enthalpy and the volume of the cell was examined at several different cell volumes and upon relaxing the atomic positions and cell shapes. For the electronic structure calculations of NaMgPn, non-self-consistent calculations were performed along a highly symmetrical band path using charge densities and wave functions obtained by self-consistent calculations.
Dedicated to: Professor Richard Dronskowski of the RWTH Aachen on the occasion of his 60th birthday.
Funding source: JSPS KAKENHI
Award Identifier / Grant number: 20H02820
Award Identifier / Grant number: 19H02794
Acknowledgments
The authors would like to thank Chikako Nagahama (Tohoku University) for helping with sample preparation, and Takashi Kamaya (Tohoku University) for carrying out the EPMA measurements.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: This research was financially supported by JSPS KAKENHI Grants (20H02820 and 19H02794).
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
1. Schäfer, H., Eisenman, B., Müller, W. Angew. Chem. Int. Ed. Engl. 1973, 12, 694–712; https://doi.org/10.1002/anie.197306941.Search in Google Scholar
2. Corbett, J. D. Inorg. Chem. 2010, 49, 13–28; https://doi.org/10.1021/ic901305g.Search in Google Scholar PubMed
3. Gladisch, F., Steinberg, S. Crystals 2018, 8, 80; https://doi.org/10.3390/cryst8020080.Search in Google Scholar
4. Kauzlarich, S. M., Ed. Chemistry, Structure, and Bonding of Zintl Phases and Ions: Selected Topics and Recent Advances; VCH Publishers: New York, 1996.Search in Google Scholar
5. Fässler, T. F., Ed. Zintl Phases: Principles and Recent Developments; Springer: Heidelberg, Dordrecht, London, New York, 2011.10.1007/978-3-642-21150-8Search in Google Scholar
6. Fässler, T. F., Kronseder, C. Angew. Chem. Int. Ed. Engl. 1997, 36, 2683–2686; https://doi.org/10.1002/anie.199726831.Search in Google Scholar
7. Nagamatsu, J., Nakagawa, N., Muranaka, T., Zenitani, Y., Akimitsu, J. Nature 2001, 410, 63–64; https://doi.org/10.1038/35065039.Search in Google Scholar PubMed
8. Yamada, T., Hirai, D., Hiroi, Z., Yamane, H. Inorg. Chem. 2019, 58, 12911–12917; https://doi.org/10.1021/acs.inorgchem.9b01869.Search in Google Scholar PubMed
9. Yamada, T., Hirai, D., Yamane, H., Hiroi, Z. J. Phys. Soc. Jpn. 2021, 90, 034710; https://doi.org/10.7566/jpsj.90.034710.Search in Google Scholar
10. Kauzlarich, S. M., Brown, S. R., Snyder, G. J. Dalton Trans. 2007, 2099–2107; https://doi.org/10.1039/b702266b.Search in Google Scholar PubMed
11. Yamada, T., Yamane, H., Nagai, H. Adv. Mater. 2015, 27, 4708–4713; https://doi.org/10.1002/adma.201501970.Search in Google Scholar PubMed
12. Asamiya, Y., Yamada, T., Yamane, H. Inorganics 2021, 9, 19; https://doi.org/10.3390/inorganics9030019.Search in Google Scholar
13. Suemasu, T., Usami, N. J. Phys. D Appl. Phys. 2017, 50, 023001; https://doi.org/10.1088/1361-6463/50/2/023001.Search in Google Scholar
14. Hodge, K. L., Goldberger, J. E. J. Am. Chem. Soc. 2019, 141, 19969–19972; https://doi.org/10.1021/jacs.9b09856.Search in Google Scholar PubMed
15. Yang, S.-Y., Yang, H., Derunova, E., Parkin, S. S. P., Yan, B., Ali, M. N. Adv. Phys. X 2018, 3, 1414631; https://doi.org/10.1080/23746149.2017.1414631.Search in Google Scholar
16. Yi, X., Li, W. Q., Li, Z. H., Zhou, P., Ma, Z. S., Sun, L. Z. J. Mater. Chem. C 2019, 7, 15375–15381; https://doi.org/10.1039/c9tc04096j.Search in Google Scholar
17. Xiao, H., Dan, Y., Suo, B. B., Chen, X. J. Phys. Chem. C 2020, 124, 2247–2249; https://doi.org/10.1021/acs.jpcc.9b10295.Search in Google Scholar
18. Yamada, T., Deringer, V. L., Dronskowski, R., Yamane, H. Inorg. Chem. 2012, 51, 4810–4816; https://doi.org/10.1021/ic300184d.Search in Google Scholar PubMed
19. Yamada, T., Ikeda, T., Stoffel, R. P., Deringer, V. L., Dronskowski, R., Yamane, H. Inorg. Chem. 2014, 53, 5253–5259; https://doi.org/10.1021/ic500466w.Search in Google Scholar PubMed
20. Yamada, T., Ishiyama, R., Yamane, H. Jpn. J. Appl. Phys. 2015, 54, 07jc04; https://doi.org/10.7567/jjap.54.07jc04.Search in Google Scholar
21. Wang, C., Chen, Y. B., Yao, S.-H., Zhou, J. Phys. Rev. B 2019, 99, 024310; https://doi.org/10.1103/physrevb.99.024310.Search in Google Scholar
22. Peng, B., Yue, C. M., Zhang, H., Fang, Z., Weng, H. M. npj Comput. Mater. 2018, 4, 68; https://doi.org/10.1038/s41524-018-0124-5.Search in Google Scholar
23. Nowotny, H., Holub, F. Monatsh. Chem. 1960, 91, 877–887; https://doi.org/10.1007/bf00929560.Search in Google Scholar
24. Krenkel, B., Schuster, H.-U. Z. Naturforsch. 1978, 33b, 1080–1082; https://doi.org/10.1515/znb-1978-1005.Search in Google Scholar
25. Somer, M., Carrillo-Cabrera, W., Peters, E.-M., Peters, K., von Schnering, H. G. Z. Kristallogr. 1996, 211, 633; https://doi.org/10.1524/zkri.1996.211.9.633.Search in Google Scholar
26. Deng, Z., Wang, X. C., Liu, Q. Q., Zhang, S. J., Lv, Y. X., Zhu, J. L., Yu, R. C., Jin, C. Q. Physica C 2010, 470, S309–S310; https://doi.org/10.1016/j.physc.2009.11.106.Search in Google Scholar
27. Pitcher, M. J., Parker, D. R., Adamson, P., Herkelrath, S. J. C., Boothroyd, A. T., Ibberson, R. M., Brunelli, M., Clarke, S. J. Chem. Commun. 2008, 5918–5920; https://doi.org/10.1039/b813153h.Search in Google Scholar PubMed
28. Tapp, J. H., Tang, Z. J., Lv, B., Sasmal, K., Lorenz, B., Chu, P. C. W., Guloy, A. M. Phys. Rev. B 2008, 78, 060505; https://doi.org/10.1103/physrevb.78.060505.Search in Google Scholar
29. Adam, M., Hovestreydt, E., Ruf, M., Kaercher, J. Acta Crystallogr. 2015, A71, S194; https://doi.org/10.1107/s2053273315097120.Search in Google Scholar
30. Sheldrick, G. M. Acta Crystallogr. 2015, C71, 3–8.Search in Google Scholar
31. Farrugia, L. J. Appl. Crystallogr. 1999, 32, 837–838; https://doi.org/10.1107/s0021889899006020.Search in Google Scholar
32. Momma, K., Izumi, F. J. Appl. Crystallogr. 2011, 44, 1272–1276; https://doi.org/10.1107/s0021889811038970.Search in Google Scholar
33. Kresse, G., Furthmüller, J. Phys. Rev. B 1996, 54, 11169–11186; https://doi.org/10.1103/physrevb.54.11169.Search in Google Scholar
34. Kresse, G., Furthmüller, J. Comp. Mater. Sci. 1996, 6, 15–50; https://doi.org/10.1016/0927-0256(96)00008-0.Search in Google Scholar
35. Blöchl, P. E. Phys. Rev. B 1994, 50, 17953–17979; https://doi.org/10.1103/physrevb.50.17953.Search in Google Scholar PubMed
36. Kresse, G., Joubert, D. Phys. Rev. B 1999, 59, 1758–1775; https://doi.org/10.1103/physrevb.59.1758.Search in Google Scholar
37. Perdew, J. P., Burke, K., Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865–3868; https://doi.org/10.1103/physrevlett.77.3865.Search in Google Scholar
© 2021 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this issue
- Laudatio/Preface
- Celebrating the 60th birthday of Richard Dronskowski
- Review
- Orbital-selective electronic excitation in phase-change memory materials: a brief review
- Research Articles
- Solving the puzzle of the dielectric nature of tantalum oxynitride perovskites
- d- and s-orbital populations in the d block: unbound atoms in physical vacuum versus chemical elements in condensed matter. A Dronskowski-population analysis
- Single-crystal structures of A 2SiF6 (A = Tl, Rb, Cs), a better structure model for Tl3[SiF6]F, and its novel tetragonal polymorph
- Na2La4(NH2)14·NH3, a lanthanum-rich intermediate in the ammonothermal synthesis of LaN and the effect of ammonia loss on the crystal structure
- Linarite from Cap Garonne
- Salts of octabismuth(2+) polycations crystallized from Lewis-acidic ionic liquids
- High-temperature diffraction experiments and phase diagram of ZrO2 and ZrSiO4
- Thermal conversion of the hydrous aluminosilicate LiAlSiO3(OH)2 into γ-eucryptite
- Crystal structure of mechanochemically prepared Ag2FeGeS4
- Effect of nanostructured Al2O3 on poly(ethylene oxide)-based solid polymer electrolytes
- Sr7N2Sn3: a layered antiperovskite-type nitride stannide containing zigzag chains of Sn4 polyanions
- Exploring the frontier between polar intermetallics and Zintl phases for the examples of the prolific ALnTnTe3-type alkali metal (A) lanthanide (Ln) late transition metal (Tn) tellurides
- Zwitterion coordination to configurationally flexible d 10 cations: synthesis and characterization of tetrakis(betaine) complexes of divalent Zn, Cd, and Hg
- An approach towards the synthesis of lithium and beryllium diphenylphosphinites
- Synthesis, crystal and electronic structure of CaNi2Al8
- Crystal and electronic structure of the new ternary phosphide Ho5Pd19P12
- Synthesis, structure, and magnetic properties of the quaternary oxysulfides Ln 5V3O7S6 (Ln = La, Ce)
- Synthesis, crystal and electronic structure of BaLi2Cd2Ge2
- Structural variations of trinitrato(terpyridine)lanthanoid complexes
- Preparation of CoGe2-type NiSn2 at 10 GPa
- Controlled exposure of CuO thin films through corrosion-protecting, ALD-deposited TiO2 overlayers
- Experimental and computational investigations of TiIrB: a new ternary boride with Ti1+x Rh2−x+y Ir3−y B3-type structure
- Synthesis and crystal structure of the lanthanum cyanurate complex La[H2N3C3O3]3 · 8.5 H2O
- Cd additive effect on self-flux growth of Cs-intercalated NbS2 superconducting single crystals
- 14N, 13C, and 119Sn solid-state NMR characterization of tin(II) carbodiimide Sn(NCN)
- Superexchange interactions in AgMF4 (M = Co, Ni, Cu) polymorphs
- Copper(I) iodide-based organic–inorganic hybrid compounds as phosphor materials
- On iodido bismuthates, bismuth complexes and polyiodides with bismuth in the system BiI3/18-crown-6/I2
- Synthesis, crystal structure and selected properties of K2[Ni(dien)2]{[Ni(dien)]2Ta6O19}·11 H2O
- First low-spin carbodiimide, Fe2(NCN)3, predicted from first-principles investigations
- A novel ternary bismuthide, NaMgBi: crystal and electronic structure and electrical properties
- Magnetic properties of 1D spin systems with compositional disorder of three-spin structural units
- Amine-based synthesis of Fe3C nanomaterials: mechanism and impact of synthetic conditions
- Enhanced phosphorescence of Pd(II) and Pt(II) complexes adsorbed onto Laponite for optical sensing of triplet molecular dioxygen in water
- Theoretical investigations of hydrogen absorption in the A15 intermetallics Ti3Sb and Ti3Ir
- Assembly of cobalt-p-sulfonatothiacalix[4]arene frameworks with phosphate, phosphite and phenylphosphonate ligands
- Chiral bis(pyrazolyl)methane copper(I) complexes and their application in nitrene transfer reactions
- UoC-6: a first MOF based on a perfluorinated trimesate ligand
- PbCN2 – an elucidation of its modifications and morphologies
- Flux synthesis, crystal structure and electronic properties of the layered rare earth metal boride silicide Er3Si5–x B. An example of a boron/silicon-ordered structure derived from the AlB2 structure type
Articles in the same Issue
- Frontmatter
- In this issue
- Laudatio/Preface
- Celebrating the 60th birthday of Richard Dronskowski
- Review
- Orbital-selective electronic excitation in phase-change memory materials: a brief review
- Research Articles
- Solving the puzzle of the dielectric nature of tantalum oxynitride perovskites
- d- and s-orbital populations in the d block: unbound atoms in physical vacuum versus chemical elements in condensed matter. A Dronskowski-population analysis
- Single-crystal structures of A 2SiF6 (A = Tl, Rb, Cs), a better structure model for Tl3[SiF6]F, and its novel tetragonal polymorph
- Na2La4(NH2)14·NH3, a lanthanum-rich intermediate in the ammonothermal synthesis of LaN and the effect of ammonia loss on the crystal structure
- Linarite from Cap Garonne
- Salts of octabismuth(2+) polycations crystallized from Lewis-acidic ionic liquids
- High-temperature diffraction experiments and phase diagram of ZrO2 and ZrSiO4
- Thermal conversion of the hydrous aluminosilicate LiAlSiO3(OH)2 into γ-eucryptite
- Crystal structure of mechanochemically prepared Ag2FeGeS4
- Effect of nanostructured Al2O3 on poly(ethylene oxide)-based solid polymer electrolytes
- Sr7N2Sn3: a layered antiperovskite-type nitride stannide containing zigzag chains of Sn4 polyanions
- Exploring the frontier between polar intermetallics and Zintl phases for the examples of the prolific ALnTnTe3-type alkali metal (A) lanthanide (Ln) late transition metal (Tn) tellurides
- Zwitterion coordination to configurationally flexible d 10 cations: synthesis and characterization of tetrakis(betaine) complexes of divalent Zn, Cd, and Hg
- An approach towards the synthesis of lithium and beryllium diphenylphosphinites
- Synthesis, crystal and electronic structure of CaNi2Al8
- Crystal and electronic structure of the new ternary phosphide Ho5Pd19P12
- Synthesis, structure, and magnetic properties of the quaternary oxysulfides Ln 5V3O7S6 (Ln = La, Ce)
- Synthesis, crystal and electronic structure of BaLi2Cd2Ge2
- Structural variations of trinitrato(terpyridine)lanthanoid complexes
- Preparation of CoGe2-type NiSn2 at 10 GPa
- Controlled exposure of CuO thin films through corrosion-protecting, ALD-deposited TiO2 overlayers
- Experimental and computational investigations of TiIrB: a new ternary boride with Ti1+x Rh2−x+y Ir3−y B3-type structure
- Synthesis and crystal structure of the lanthanum cyanurate complex La[H2N3C3O3]3 · 8.5 H2O
- Cd additive effect on self-flux growth of Cs-intercalated NbS2 superconducting single crystals
- 14N, 13C, and 119Sn solid-state NMR characterization of tin(II) carbodiimide Sn(NCN)
- Superexchange interactions in AgMF4 (M = Co, Ni, Cu) polymorphs
- Copper(I) iodide-based organic–inorganic hybrid compounds as phosphor materials
- On iodido bismuthates, bismuth complexes and polyiodides with bismuth in the system BiI3/18-crown-6/I2
- Synthesis, crystal structure and selected properties of K2[Ni(dien)2]{[Ni(dien)]2Ta6O19}·11 H2O
- First low-spin carbodiimide, Fe2(NCN)3, predicted from first-principles investigations
- A novel ternary bismuthide, NaMgBi: crystal and electronic structure and electrical properties
- Magnetic properties of 1D spin systems with compositional disorder of three-spin structural units
- Amine-based synthesis of Fe3C nanomaterials: mechanism and impact of synthetic conditions
- Enhanced phosphorescence of Pd(II) and Pt(II) complexes adsorbed onto Laponite for optical sensing of triplet molecular dioxygen in water
- Theoretical investigations of hydrogen absorption in the A15 intermetallics Ti3Sb and Ti3Ir
- Assembly of cobalt-p-sulfonatothiacalix[4]arene frameworks with phosphate, phosphite and phenylphosphonate ligands
- Chiral bis(pyrazolyl)methane copper(I) complexes and their application in nitrene transfer reactions
- UoC-6: a first MOF based on a perfluorinated trimesate ligand
- PbCN2 – an elucidation of its modifications and morphologies
- Flux synthesis, crystal structure and electronic properties of the layered rare earth metal boride silicide Er3Si5–x B. An example of a boron/silicon-ordered structure derived from the AlB2 structure type