A mononuclear cobalt(III) 3-(2-pyridyl)-5-phenyl-1,2,4-triazolato complex: hydrothermal synthesis, crystal structure, thermostability, and DFT calculations
-
Huaixian Liu
 and
Qilin Hu
and
Qilin Hu

Abstract
Starting with 1H-3-phenyl-5-(pyridin-2-yl)-1,2,4-triazole (1-Hppt), a Co(III) complex, [Co(ppt)3] (1), has been synthesized by reaction with CoF3 under hydrothermal conditions and characterized by its infrared spectrum and elemental analysis. The structure was determined by single-crystal and powder X-ray diffraction. Density functional theory (DFT) was employed to determine the optimized geometry and preferred conformation of the free ligand. A supramolecular network is formed via π–π stacking interactions. The conformation and geometry of the ligands correspond with the calculated results.
1 Introduction
In recent years, experimental and theoretical efforts have been extensively devoted to crystal engineering of coordination polymers (CPs) from predesigned bridging ligands and metal ions, because such hybrid materials display a variety of regulated architectures and thus many potential functions [1–5]. The interest in polymer architectures arises not only from their potential applications in luminescence, magnetism, sorption, gas adsorption, nonlinear optics, catalytic materials, and medicine [6–10], but also from their intriguing variety of topologies and structural motifs [11–14]. However, it is still a great challenge to rationally design and control the exact structure of CPs. Generally, the structure of target CPs is influenced by several factors, such as counter ions, pH, reaction temperature, and solvent system [15]. In addition to controlling the reaction conditions, using theoretical calculations to predict and guide experimental research has long been a target for scientists [16]. What is even more important is conducting research on the reaction systems based on the combination of theory and experiment, which is conducive to revealing the preferred geometry and constructing a structural model of the compounds [17, 18].
Pyridine-based triazole ligands have been proved as a type of excellent connectors to engender a wide range of CPs resulting from the following advantages: (i) the abundant nitrogen donor atoms in the molecule can be selectively coordinated to metal ions, facilitating the generation of CPs [19] and (ii) the ligand can adopt various coordination modes, such as multidentate or bridging [20–22]. In this work, we have adopted the 3-phenyl-5-(pyridin-2-yl)-1,2,4-triazolato anion (ppt–) as a ligand to Co(III) to assemble a supramolecular architecture.
Based on the above considerations, we have inferred the optimized structure and conformation of the 1H-3-phenyl-5-(pyridin-2-yl)-1,2,4-triazole (1-Hppt) molecule using the density functional theory (DFT) method. The coordination polymer, [Co(ppt)3] (1), has been obtained under the hydrothermal condition, and its structure and thermostability were measured. The theoretical and experimental results are compared and discussed.
2 Results and discussion
2.1 Computational results of the 1-Hppt molecule
The optimized geometries of two different conformations shown in Fig. 1 have been obtained. The calculated bond lengths N1–C2, N1–N2, C2–C3, and N4–C3 are 1.335, 1.351, 1.477, and 1.404 Å, respectively.
The molecular total energies (Etotal), frontier orbital energy levels (EHOMO and ELUMO), and the gaps (ΔE(LUMO–HOMO)) of the two different conformations have been calculated, and the results are listed in Table 1. Compared with the trans-conformation I, the cis-conformation II displays lower total energies, representing the preferred geometry. The surfaces of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for the two conformations are shown in Fig. 2. According MO theory, the HOMO and LUMO are the most important factors that influence the properties of a compound [23].
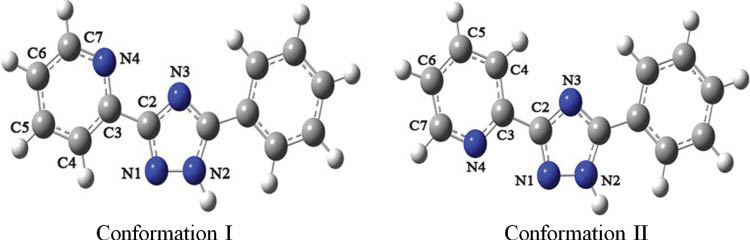
Two different conformations of 1-Hppt.
Calculated energies and frontier orbital energies for two conformations of 1-Hppt.
| Conformation I | Conformation II | |
|---|---|---|
| E, hartree | −720.4099 | −720.4109 |
| EHOMO, hartree | −0.22443 | −0.22513 |
| ELUMO, hartree | −0.04197 | −0.04388 |
| ΔE(LUMO–HOMO), eV | 4.97 | 4.93 |

Isodensity surfaces of HOMO and LUMO for 1-Hppt (left: conformation I, right: conformation II).
The natural charges and electron configuration for the conformation II have been calculated by using natural bond orbital (NBO) analysis. As shown in Table 2, Mulliken charges and natural charges of the N1, N2, N3, and N4 atoms are obtained as negative values. The natural charge of N3 (−0.52) is more negative than that of the N1 (−0.36) and N2 (−0.26) atoms in the triazole ring. The natural charge of N4 in the pyridine ring is −0.43. The results demonstrate that all four N atoms provide donor position to coordinate with metal ions.
Natural configurations and natural and Mulliken charges of conformation II of 1-Hppt.
| Atom | Natural electron configuration | Natural charge | Mulliken charge |
|---|---|---|---|
| N1 | [core]2S(1.44)2p(3.84)3p(0.01)3d(0.01) | −0.36286 | −0.49234 |
| N2 | [core]2S(1.44)2p(3.79)3p(0.01)3d(0.01) | −0.25581 | −0.29671 |
| N3 | [core]2S(1.40)2p(4.10)3p(0.01)3d(0.01) | −0.51760 | −0.57613 |
| N4 | [core]2S(1.41)2p(4.01)3p(0.01)3d(0.01) | −0.43053 | −0.49547 |
| C2 | [core]2S(0.82)2p(2.83)3p(0.02)3d(0.01) | 0.31229 | 0.41707 |
| C3 | [core]2S(0.85)2p(2.97)3p(0.02) | 0.16323 | 0.25091 |
| C6 | [core]2S(0.98)2p(3.28)3p(0.01) | −0.27183 | −0.14027 |
| C7 | [core]2S(0.96)2p(3.01)3p(0.01) | 0.01968 | 0.04239 |
2.2 Synthesis of complex 1
The Co(III) complex 1 was synthesized from CoF3 and 1-Hppt in water by hydrothermal reaction at 140 °C for 3 days in the presence of terephthalic acid. The latter was not found to be incorporated in the final product which proved to be [Co(ppt)3] (1) as dark red crystals. The phase purity of bulk 1 was proved by a comparison of the observed and simulated powder X-ray diffraction (PXRD) diagrams as described below.
2.3 Structural description of complex 1
Single-crystal X-ray diffraction analysis reveals that the complex crystallizes in the triclinic system, space group P1̅ with Z = 2 (Table 3). The asymmetric unit of the complex consists of one crystallographically independent Co(III) ion and three [ppt]– anions. The auxiliary ligand (terephthalic acid) did not take part in coordination. As shown in Fig. 3a, the six-coordinated Co(III) center presents a slightly distorted octahedral geometry. The four equatorial positions are occupied by three nitrogen atoms from triazole rings of three different ligands (Co1–N1 1.874(3), Co1–N5 1.900(3), Co1–N9 1.883(3) Å) and one nitrogen atom from a pyridine ring (Co1–N12 1.976(3) Å). Two nitrogen atoms from pyridine rings of two different ligands are located at the axial positions (Co1–N4 1.953(3), Co1–N8 1.954(3) Å) (Table 4). Compared to the previous reports, the Co–N bond lengths of the title complex are similar to those of Co(III) complexes (<2.0 Å [24]), and deviate from the range of Co(II)–N bond lengths (2.037–2.187 Å [25]). For the ligand, the bond lengths of N1–C2, N1–N2, C2–C3, and N4–C3 are 1.332(5), 1.345(4), 1.453(5), and 1.360(5) Å, respectively, in accordance with the calculated values. Adjacent units are connected to each other via two kinds of π−π stacking interactions between benzene and pyridine rings (Fig. 3b). The distance between two benzene rings is 3.909 Å, but 3.720 Å between benzene and pyridine rings, which is close to the π–π interaction distances reported in [26].
Crystallographic data and data collection and structure refinement details for complex 1.
| Formula | C39H27CoN12 |
| Mr | 722.66 |
| Crystal size, mm3 | 0.17 × 0.15 × 0.10 |
| Crystal system | Triclinic |
| Space group | P1̅ |
| a, Å | 12.4949(12) |
| b, Å | 12.6369(12) |
| c, Å | 13.3796(17) |
| α, deg | 107.975(2) |
| β, deg | 106.025(2) |
| γ, deg | 110.876(3) |
| V, Å3 | 1691.2(3) |
| Z | 2 |
| Dcalcd, g cm−3 | 1.42 |
| μ(CuKα), cm−1 | 43.8 |
| F(000), e | 744 |
| hkl range | ±14, −14→7, ±15 |
| θ range, deg | 3.9–66.2 |
| Tmin/Tmax | 0.523/0.669 |
| Refl. measured/unique/Rint | 10 294/5905/0.058 |
| Param. refined | 469 |
| R(F)/wR(F2) (all reflexions) | 0.0937/0.1444 |
| GoF (F2) | 1.007 |
| Δρfin (max/min), e Å−3 | 0.46/−0.34 |
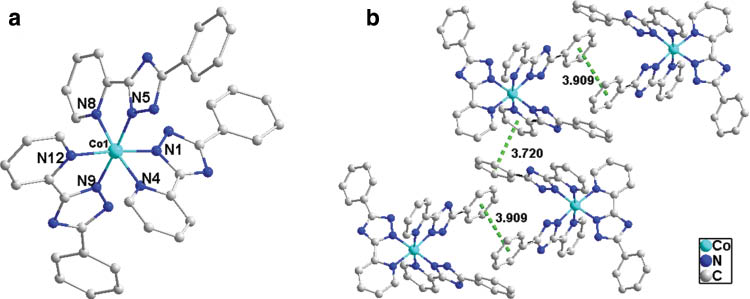
(a) Coordination environment of the Co(III) ion in complex 1; (b) intermolecular π–π stacking in the crystal structure of complex 1.
Selected bond lengths (Å) and bond angles (deg) of complex 1.
| Bond | Bond | Bond | |||
|---|---|---|---|---|---|
| Co(1)–N(1) | 1.874(3) | Co(1)–N(5) | 1.900(3) | Co(1)–N(9) | 1.883(3) |
| Co(1)–N(4) | 1.953(3) | Co(1)–N(8) | 1.954(3) | Co(1)–N(12) | 1.976(3) |
| Angle | Angle | Angle | |||
| N1–Co1–N9 | 92.42(14) | N1–Co1–N5 | 90.11(14) | N9–Co1–N5 | 176.23(14) |
| N1–Co1–N4 | 82.33(14) | N9–Co1–N4 | 88.31(13) | N5–Co1–N4 | 94.80(13) |
| N1–Co1–N8 | 90.78(14) | N9–Co1–N8 | 94.83(14) | N5–Co1–N8 | 82.34(14) |
| N4–Co1–N8 | 172.55(14) | N1–Co1–N12 | 173.69(14) | N9–Co1–N12 | 81.29(14) |
| N5–Co1–N12 | 96.19(14) | N4–Co1–N12 | 96.84(13) | N8–Co1–N12 | 90.32(13) |
It is interesting to note that all [ppt]– ligands exhibit conformation II in the structure of the complex, confirming that the preferred conformation calculated by DFT is correct. The ligands display uniform coordination modes with pyridine N4 and triazole N1 atoms chelating the Co(III) ions. The coordination ability of N3 with the most negative charge is probably restricted by steric hindrance.
2.4 Thermogravimetric analysis and PXRD of complex 1
The thermal behavior of 1 was studied by thermal gravimetric (TG) measurements under nitrogen atmosphere with a heating rate of 10 °C min−1. In order to confirm the phase purity of the compound, a PXRD experiment was carried out (Fig. 4). The experimental pattern matches with the simulated pattern, indicating the phase purity of the compound within the limits of experimental error. The sample was heated to 800 °C (Fig. 5). The TG curve shows that a continuous weight loss of 92.3 % is observed in the temperature range from 263 to 660 °C, due to the decomposition of the ligands (calcd. 91.84 %). The final residue mass is 7.7 % of the initial mass, which coincides with the calculated value of elemental cobalt (8.2 %), determined by PXRD (PDF 05-0727).

Observed and simulated PXRD diagrams of complex 1.

TGA curve for complex 1.
3 Conclusions
A Co(III) complex with the 3-phenyl-5-(pyridin-2-yl)-1,2,4-triazolato ligand has been determined under hydrothermal conditions. Single-crystal structure analysis has revealed that the title complex features a 0D mononuclear structure, but forms a 3D supramolecular network through π–π stacking. TG analyses showed that the complex possesses high thermostability with decomposition standing at 263 °C. The observed geometric parameters and preferred conformation of the [ppt]– ligand are consistent with the DFT calculated results for the present molecule. The present work is an example of the combination of experiment and theory, providing the guidance for the design and synthesis of coordination polymers.
4 Experimental section
4.1 Materials and instruments
Commercially available solvents, such as 2-cyanopyridine, phenyl hydrazide, absolute methanol, sodium metal, terephthalic acid, and CoF3, were used as received without further purification (Xi’an Haixin Company, China). 1H and 13C nuclear magnetic resonance (NMR; Beijing Oxford Instrument Company, China) spectra were taken with a Varian 400 spectrometer using tetramethylsilane as an internal standard. NMR spectra were obtained from solutions in dimethyl sulfoxide (DMSO). Elemental analyses (C, H, and N) were performed on a Vario EL III analyzer (Nanjing Hua Xin Analysis Instrument Manufacturing Company, China). Infrared (IR) spectra were obtained using KBr pellets on a BEQ VZNDX 550 FTIR (Shimadzu Corporation) instrument within the 400–4000 cm−1 region. The phase purity of the bulk or polycrystalline samples was confirmed by PXRD (Brucker Company, Germany) measurements executed on a Rigaku RU200 diffractometer at 60 kV, 300 mA with CuKα radiation (λ = 1.5406 Å), with a scan speed of 5° min−1 and a step size of 0.02° in 2θ. TG analysis was performed on a SETARAM Setsys16 (Nanjing Research Institute, China) instrument under N2 atmosphere with a heating rate of 10 °C min−1.
4.2 Synthesis of the 1-Hppt ligand
The ligand was synthesized according to [27]. Sodium metal (0.1 g) was added (carefully) to 40 mL absolute methanol followed by the addition of 2-cyanopyridine (2.1 g). The resulting reaction mixture was stirred with a magnetic stirrer for 80 min. Thereafter, it was allowed to cool and phenyl hydrazide (2.1 g) was added. The solution was refluxed for a further 3 h until a yellow solid complex precipitated. The precipitate was filtered off, washed with hot absolute ethanol, and dried in vacuum. The white ligand was precipitated, yield 70 % (based on 2-cyanopyridine). M.p.: 191.8–192.6 °C. – Analysis for C13H10N3: calcd. C 74.97, H 4.85, N, 20.18; found C 74.92, H 4.83, N, 20.12 %. – IR (film): 3404(s), 3213(s), 3057(m), 2361(m), 1670(s), 1612(s), 1589(w), 1446(s), 1313(s), 1180(w), 1053(m), 792(m), 717(m), 696(w), 579(w) cm−1. – 1H NMR (400 MHz, [D6]DMSO, 25 °C, ppm): δ = 6.95 (d, 1H, Ph-H4), 7.49–7.51 (d, 2H, Ph-H2/H6), 7.55–7.57 (dd, 2H, Ph-H3/H5), 8.60–8.61 (dd, 1H, Pz-H3), 7.87–7.93 (d, 1H, Pz-H6), 7.53 (t, 1H, Pz-H4), 8.17–8.19 (t, 1H, Pz-H5), 10.20 (s, 1H, triazole-H). – 13C NMR (75 MHz, [D6]DMSO, 25 °C, ppm): δ = 163.24, 150.75, 148.20, 147.85, 137.04, 134.70, 131.13, 128.31, 127.76, 124.87, 120.81.
4.3 Synthesis of complex 1
A mixture of CoF3 (5.8 mg, 0.05 mmol), terephthalic acid (8.3 mg, 0.05 mmol), 1-Hppt (11.1 mg, 0.05 mmol), and water (6 mL) was sealed in a 10 mL Teflon-lined stainless steal vessel and heated at 140 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. Dark red crystals of complex 1 were collected in a yield of 40 % (based on Co). – Analysis for C39H27CoN12: calcd. C 64.81, H 3.77, N 23.26; found C 64.75, H 3.73, N 23.20 %. – IR (film): 3599(s), 3447(s), 3063(m), 2822(m), 2662(s), 2548(s), 1690(w), 1618(s), 1576(s), 1464(w), 1425(m), 1287(m), 1111(m), 941(w), 785(w), 735(w), 696(w), 529(w).
4.4 X-ray crystallography
All diffraction data were collected on a Bruker Smart Apex CCD diffractometer with graphite-monochromatized CuKα radiation (λ = 1.54178 Å) at 293(2) K. Absorption corrections were applied using Sadabs [28]. The structure was solved by Direct Methods using the Shelxs program of the Shelxtl package and refined with Shelxl-97 [29]. The hydrogen atoms were located geometrically. All non-hydrogen atoms were refined anisotropically. Crystal data and numbers pertinent to data collection and refinement are given in Table 3. Selected bond lengths and bond angles are listed in Tabel 4.
CCDC 1034723 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
4.5 Computational details
Geometry optimization was carried out at the all-electron level using the Gaussian 03 quantum chemistry program package [30] at the B3LYP/6-31G(d) [31, 32] level of theory. Mulliken population analysis, total molecular energies, and NBO calculations were also performed on the B3LYP/6-31G(d) level.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant Nos. 21463020 and 21365016).
References
[1] H.-L. Sun, Z.-M. Wang, S. Gao, Coord. Chem. Rev. 2010, 254, 1081.10.1016/j.ccr.2010.02.010Search in Google Scholar
[2] Y.-Z. Zheng, Z. Zheng, X.-M. Chen, Coord. Chem. Rev. 2014, 258, 1.10.1016/j.ccr.2013.08.031Search in Google Scholar
[3] E. Pardo, J. Faus, M. Julve, F. Lloret, M. C. Muñoz, J. Cano, R. J. Ruiz-García, J. Am. Chem. Soc. 2003, 125, 10770.10.1021/ja030060fSearch in Google Scholar
[4] X.-Y. Liu, P.-P. Cen, H. Li, H.-S. Ke, S. Zhang, Q. Wei, X. Gang, S.-P. Chen, S.-L. Gao, Inorg. Chem. 2014, 53, 8088.10.1021/ic5010769Search in Google Scholar
[5] B. Moulton, M. J. Zaworotko, Chem. Rev. 2001, 101, 1629.10.1021/cr9900432Search in Google Scholar
[6] S. Leninger, B. Olenyuk, P. J. Stang, Chem. Rev. 2000, 100, 853.10.1021/cr9601324Search in Google Scholar
[7] X.-Y. Liu, S.-P. Chen, T. Grancha, E. Pardo, H.-S. Ke, B. Yin, S.-L. Gao, Dalton Trans. 2014, 43, 15359.10.1039/C4DT02195ASearch in Google Scholar
[8] B. D. Chandler, D. T. Cramb, G. K. H. Shimizu. J. Am. Chem. Soc. 2006, 128, 10403.10.1021/ja060666eSearch in Google Scholar
[9] J. Y. Lee, C.-Y. Chen, H. M. Lee, E. Passaglia, F. Vizza, W. Oberhauser, Cryst. Growth Des. 2011, 11, 1230.10.1021/cg101453mSearch in Google Scholar
[10] D. N. Dybtsev, H. Chun, K. Kim, Angew. Chem. Int. Ed. 2004, 43, 5033.10.1002/anie.200460712Search in Google Scholar
[11] C. N. R. Rao, S. Natarajan, R. Vaidhyanathan, Angew. Chem. Int. Ed. 2004, 43, 1466.10.1002/anie.200300588Search in Google Scholar
[12] W. L. Leong, J. J. Vittal, Chem. Rev. 2010, 111, 688.10.1021/cr100160eSearch in Google Scholar
[13] Y.-G. Huang, F.-L. Jiang, M.-C. Hong, Coord. Chem. Rev. 2009, 253, 2814.10.1016/j.ccr.2009.05.007Search in Google Scholar
[14] G.-P. Yang, L. Hou, X.-J. Luan, B. Wu, Y.-Y. Wang, Chem. Soc. Rev. 2012, 41, 6992.10.1039/c2cs35202hSearch in Google Scholar
[15] X.-Y. Liu, P.-P. Cen, H.-L. Zhou, X.-Y. Jin, Q.-L. Hu, Y.-Q. Ji, Inorg. Chem. Commun. 2015, 53, 11.Search in Google Scholar
[16] F.-P. Huang, J.-B. Lei, Q. Yu, H.-D. Bian, S.-P. Yan, Polyhedron 2012, 34, 129.10.1016/j.poly.2011.12.030Search in Google Scholar
[17] K. B. Lipkowitz, Coord. Chem. Rev. 1998, 98, 1829.10.1021/cr9700179Search in Google Scholar
[18] B. Li, S.-P. Chen, Q. Yang, S.-L. Gao, Polyhedron 2011, 30, 1213.Search in Google Scholar
[19] Y.-P. Tong, S.-L. Zheng, X.-M. Chen, Inorg. Chem. 2005, 44, 4270.10.1021/ic0501059Search in Google Scholar
[20] F.-P. Huang, J.-L. Tian, W. Gu, X. Liu, S.-P. Yan, D.-Z. Liao, P. Cheng, Cryst. Growth Des. 2010, 10, 1145.10.1021/cg901014rSearch in Google Scholar
[21] F.-P. Huang, J.-L. Tian, G.-J. Chen, D.-D. Li, W. Gu, X. Liu, S.-P. Yan, D.-Z. Liao, P. Chen, CrystEngComm. 2010, 12, 1269.10.1039/B915506FSearch in Google Scholar
[22] E. Engel, R. M. Dreizler, J. Comput. Chem. 1999, 20, 31.10.1002/(SICI)1096-987X(19990115)20:1<31::AID-JCC6>3.0.CO;2-PSearch in Google Scholar
[23] J.-P. Zhang, Y.-Y. Lin, X.-C. Huang, X.-M. Chen, J. Am. Chem. Soc. 2005, 127, 5495.10.1021/ja042222tSearch in Google Scholar
[24] A. M. Ritzmann, M. Pavone, A. B. Muñoz-García, J. A. Keith, E. A. Carter, J. Mater. Chem. A. 2014, 2, 8060.10.1039/C4TA00801DSearch in Google Scholar
[25] D. Ghoshal, G. Mostafa, T. K. Maji, E. Zangrando, T. H. Lu, J. Ribas, N. R. Chaudhuri, New J. Chem. 2004, 28, 1204.Search in Google Scholar
[26] J.-B. Lin, J.-P. Zhang, W.-X. Zhang, W. Xue, D.-X. Xue, X.-M. Chen, Inorg. Chem. 2009, 48, 6652.Search in Google Scholar
[27] A. N. Gusev, V. F. Shul’gin, S. B. Meshkova, M. Hasegawa, G. G. Alexandrov, I. L. Eremenko, W. Linert, Inorg. Chim. Acta 2012, 387, 321.10.1016/j.ica.2012.02.016Search in Google Scholar
[28] G. M. Sheldrick, Sadabs, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen (Germany) 2002.Search in Google Scholar
[29] G. M. Sheldrick, Acta Crystallogr. 2008, A64, 112–122.10.1107/S0108767307043930Search in Google Scholar
[30] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian 03 (revision D.01), Gaussian, Inc., Wallingford, CT (USA) 2004.Search in Google Scholar
[31] R. G. Parr, W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989.Search in Google Scholar
[32] A. D. Becke, J. Chem. Phys. 1993, 98, 5648.10.1063/1.464913Search in Google Scholar
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis and structural characterization of a novel copper(II)/lead(II) heterometallic organic–inorganic hybrid
- A new dimeric naphtho-γ-pyrone from an endophytic fungus Aspergillus niger AKRN associated with the roots of Entandrophragma congoënse collected in Cameroon
- A mononuclear cobalt(III) 3-(2-pyridyl)-5-phenyl-1,2,4-triazolato complex: hydrothermal synthesis, crystal structure, thermostability, and DFT calculations
- Short microwave-assisted modular synthesis of naturally occurring oxygenated bibenzyls
- Kristall- und Molekülstruktur von „Urindigo“ (4,4,4′,4′-Tetramethyl-2,2′-bipyrrolidinyliden-3,3′-dion) und die Strukturverwandtschaft zum Indigo
- Synthesis and characterization of a disordered variant of KB5O7(OH)2
- Prenylated 9,10-dihydrophenanthrenes from Macaranga javanica
- The ternary system Tm–Ni–In at 870 K
- Synthesis, crystal structure and magnetism of Eu3Sc2O5Fe2As2
- Novel conformationally constrained pyrazole derivatives as potential anti-cancer agents
- Two new secondary metabolites from the fruits of mangrove Avicennia marina
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis and structural characterization of a novel copper(II)/lead(II) heterometallic organic–inorganic hybrid
- A new dimeric naphtho-γ-pyrone from an endophytic fungus Aspergillus niger AKRN associated with the roots of Entandrophragma congoënse collected in Cameroon
- A mononuclear cobalt(III) 3-(2-pyridyl)-5-phenyl-1,2,4-triazolato complex: hydrothermal synthesis, crystal structure, thermostability, and DFT calculations
- Short microwave-assisted modular synthesis of naturally occurring oxygenated bibenzyls
- Kristall- und Molekülstruktur von „Urindigo“ (4,4,4′,4′-Tetramethyl-2,2′-bipyrrolidinyliden-3,3′-dion) und die Strukturverwandtschaft zum Indigo
- Synthesis and characterization of a disordered variant of KB5O7(OH)2
- Prenylated 9,10-dihydrophenanthrenes from Macaranga javanica
- The ternary system Tm–Ni–In at 870 K
- Synthesis, crystal structure and magnetism of Eu3Sc2O5Fe2As2
- Novel conformationally constrained pyrazole derivatives as potential anti-cancer agents
- Two new secondary metabolites from the fruits of mangrove Avicennia marina