Synthesis and characterization of a disordered variant of KB5O7(OH)2
-
Teresa S. Ortner


Abstract
The potassium pentaborate KB5O7(OH)2 reported herein crystallizes in the monoclinic space group P21/c (no. 14) with the lattice parameters a = 904.8(2), b = 753.2(2), c = 1214.9(5) pm, β = 117.16(2)°, and V = 0.73663(5) nm3 (Z = 4). It is a disordered structural variant of an already known compound with the same composition (a = 766.90(3), b = 904.45(3), c = 1223.04(4) pm, β = 119.132(2)°, and V = 0.74101(5) nm3 (Z = 4)) reported by Wu in 2011. The disorder of the potassium cation in the single crystal structure determination of KB5O7(OH)2 presented here leads to a remarkably elongated a axis and a corresponding reduction of the length of the b axis in comparison to the ordered compound. The disordered variant was obtained through a hydrothermal synthesis from KNO3, B2O3, and Ce(NO3)3·6H2O with a molar ratio of 1:1:0.07.
1 Introduction
Borates possess a vast structural flexibility, stemming from the ability of boron atoms to coordinate to three or four oxygen atoms forming trigonal planar or tetrahedral building blocks [1, 2]. The combination of these building blocks can lead to different anionic groups [BxOy]z– forming isolated groups, rings, chains, and layers, with up to three-dimensional networks [3, 4]. A good overview of the structural possibilities is given by Lin and Yang [5], as well as more recently by Bubnova and Filatov [6]. The condensed borate anions are stable host structures for mono-cations and large di-cations. More highly charged cations can form rigid coordination polyhedra promoting more basic (meaning richer in O2–) borate frameworks [7, 8]. The potassium pentaborate presented here is a structural variant of KB5O7(OH)2 reported by Wu in 2011 [9]. Both structures exhibit the same [B5O7(OH)2]– anion consisting of a BO4 tetrahedron linking three planar BO3 groups, in which the terminal oxygen positions are protonated. This structural motive is well known in borates and is present as an isolated group in the minerals santite KB5O8·4H2O [10] and sborgite NaB5O6(OH)4·3H2O [11, 12], or in the synthetic compound K[B5O6(OH)4]·2H2O [13]. The [B5O7(OH)2]nn– chains we report for the non-hydrated, disordered phase KB5O7(OH)2 were first characterized in 1969 by Merlino et al. in the ino-pentaborate larderellite NH4B5O7(OH)2·2H2O [14]. Larderellite also crystallizes monoclinically in the centrosymmetric space group P21/c (no. 14) exhibiting lattice parameters of a = 947.0, b = 763.0, c = 1165.0 pm and β = 97.1° in NH4B5O7(OH)2·2H2O, compared to a = 904.8(2), b = 753.2(2), c = 1214.9(5) pm, and β = 117.16(2)° in the disordered form of KB5O7(OH)2.
In contrast to the ordered phase KB5O7(OH)2 reported by Wu, which was synthesized from a mixture of GaO(OH), H3BO3, KNO3, and additional water, we synthesized the disordered form using hydrate water by employing Ce(NO3)3·6H2O instead of GaO(OH). The hydrothermal approach used to obtain both compounds allowed the growth of high quality crystals, obviating the tendency of borate melts to form glasses rather than crystalline phases [8]. The comparably low reaction temperatures of 513 and 483 K [9] are typical for the formation of a band- or chain-like structure as observed in natural ino-borates [1].
In the following, we describe the synthesis of the disordered potassium pentaborate KB5O7(OH)2. Through its characterization via single-crystal structure analysis as well as vibrational spectroscopy, we have elucidated the remarkable structural novelties resulting from the cation disorder.
2 Experimental section
2.1 Synthesis
The title compound was synthesized hydrothermally by charging a Teflon-lined stainless-steel autoclave (8 mL) with KNO3 (435.3 mg), B2O3 (302.8 mg), and Ce(NO3)3·6H2O (125.9 mg) with a molar ratio of 1:1:0.07. The chemicals were obtained from Carl Roth GmbH (Karlsruhe, Germany, KNO3 and Ce(NO3)3·6H2O), and Strem Chemicals (Newburyport, MA, USA, B2O3) exhibiting at least p.a. quality. Prior to and after the use of the aforementioned chemicals, powder patterns of the educts were collected to control their stability, especially with regard to the water content of Ce(NO3)3·6H2O. The latter nitrate displayed reflections according to pentaaquatrinitratocerium(III) monohydrate Ce(NO3)3(H2O)5·H2O [15]. This compound exhibits five molecules of water in close coordination to the cerium cation, and one water molecule formulated as crystal water confirming the compsition of the educt. The educt mixture was then ground together, heated to and kept at 513 K for 3 days, cooled slowly down to 393 K (1 K h–1), and finally quenched to room temperature. Colorless block-shaped crystals of disordered and ordered KB5O7(OH)2 were obtained alongside traces of another, unknown phase. The product mixture was washed with hot deionized water to remove excess educts.
2.2 Crystal structure analysis
The powder X-ray diffraction pattern of the disordered phase KB5O7(OH)2 was collected with a STOE Stadi P powder diffractometer with transmission geometry and MoKα1 (λ = 70.93 pm) radiation applying a focusing Ge(111) primary beam monochromator and a linear PSD detector. Figure 1 (top) shows the experimental powder pattern of the disordered variant of KB5O7(OH)2 with the according theoretical pattern simulated from the single-crystal data [Fig. 1 (center)]. For comparison, the theoretical pattern stemming from the single crystal data of the ordered structure reported by Wu is also given in Fig. 1 (bottom).

Top: Experimental powder pattern of the hydrothermal synthesis product containing the disordered variant of KB5O7(OH)2; the reflections marked with a cross stem from the ordered variant of KB5O7(OH)2, reflections marked with asterisks result from traces of boric acid, and reflections with a circle stem from an unknown side phase. Center: theoretical powder pattern derived from single-crystal data of the disordered variant of KB5O7(OH)2. Bottom: theoretical powder pattern obtained from single-crystal data of the ordered compound KB5O7(OH)2.
The corresponding reflections of the powder pattern were indexed and refined [16]. The lattice parameters fit well with the parameters obtained from the single-crystal data (see Table 1).
Crystal data and structure refinement of the disordered variant of KB5O7(OH)2 (standard deviations in parentheses).
| Empirical formula | KB5O7(OH)2 (disordered) |
| Molar mass, g mol–1 | 239.21 |
| Crystal system | monoclinic |
| Space group | P21/c (no. 14) |
| Powder data | |
| Powder diffractometer | STOE Stadi P |
| Radiation; wavelength, pm | MoKα1 λ = 70.93 |
| a, pm | 904.8(1) |
| b, pm | 753.0(1) |
| c, pm | 1214.8(2) |
| β, deg | 117.1(1) |
| V, nm3 | 0.7364(1) |
| Single-crystal data | |
| Single-crystal diffractometer | Nonius Kappa-CCD |
| Radiation; wavelength, pm | MoKα; λ = 71.073 (graphite monochromator) |
| a, pm | 904.75(4) |
| b, pm | 753.19(3) |
| c, pm | 1214.90(4) |
| β, deg | 117.156(2) |
| V, nm3 | 0.73663(5) |
| Formula units per cell, Z | 4 |
| Calculated density, g cm–3 | 2.16 |
| Crystal size, mm3 | 0.040 × 0.035 × 0.030 |
| Temperature, K | 296(2) |
| Absorption coefficient, mm–1 | 0.8 |
| F(000), e | 472.0 |
| θ range, deg | 2.5–31.0 |
| Range in hkl | –13 ≤ h ≤ 13, –10 ≤ k ≤ 10, –17 ≤ l ≤ 17 |
| Total no. of reflections | 8929 |
| Independent reflections/Rint | 2337/0.0444 |
| Reflections with I > 2 σ(I) | 1899 |
| Absorption correction | none |
| Data/refined parameters | 2337/154 |
| Goodness-of-fit on Fi2 | 1.069 |
| Final R1/wR2 [I > 2 σ(I)] | 0.0479/0.1135 |
| R1/wR2 (all data) | 0.0641/0.1207 |
| Largest diff. peak/hole, e Å–3 | 0.79/–0.46 |
For the single-crystal structure analysis, irregularly shaped crystals (40 × 35 × 30 μm) of the disordered variant of KB5O7(OH)2 were picked and isolated through polarization contrast microscopy. Collection of the single-crystal data using a Nonius Kappa-CCD diffractometer with graphite-monochromatized MoKα radiation (λ = 71.073 pm) was conducted at room temperature as well as at –50 °C employing liquid nitrogen cooling. No change of the crystal structure was observed while cooling, therefore only data obtained from the room temperature experiment are presented in the following. From the systematic extinctions, the centrosymmetric space group P21/n (no. 14:b2) was derived. For reasons of comparability with the structure variant reported by Wu, a space group transformation to P21/c (no. 14:b1) was performed following a suggested unit cell transformation of (–a, –b, a + c) and an origin shift of (+1/2, +1/2, 0) obtained by the Structure Tidy routine implemented in Platon [17].
All non-hydrogen atoms were refined with anisotropic displacement parameters. The final difference Fourier syntheses did not reveal any significant peaks in the refinement (Shelxl-2013, full-matrix least-squares on F2) [18, 19]. The hydrogen atoms at the hydroxyl groups O7–H1 and O8–H2 were refined isotropically, employing a bond restraint of 83 pm.
All relevant details of the data collection and evaluation are listed in Table 1. Tables 2–6 show the positional parameters, the anisotropic displacement parameters, selected interatomic distances, bond angles, and characteristic data of the hydrogen bond system, respectively.
Atomic coordinates and equivalent isotropic displacement parameters Ueq (Å2) of the disordered variant of KB5O7(OH)2 (space group: P21/c, no. 14) with standard deviations in parentheses.
| Atom | x | y | z | Ueq | S.O.F. |
|---|---|---|---|---|---|
| K1 | 0.6397(2) | 0.0994(2) | 0.2798(2) | 0.0398(3) | 0.46(2) |
| K2 | 0.6693(2) | 0.1878(2) | 0.2898(2) | 0.0462(4) | 0.54(2) |
| O1 | 0.3961(2) | 0.2701(2) | 0.8401(2) | 0.0192(3) | 1 |
| O2 | 0.3500(2) | 0.3963(2) | 0.0025(2) | 0.0185(3) | 1 |
| O3 | 0.1645(2) | 0.4748(2) | 0.7944(2) | 0.0226(3) | 1 |
| O4 | 0.2690(2) | 0.4232(2) | 0.6476(2) | 0.0198(3) | 1 |
| O5 | 0.0242(2) | 0.2896(2) | 0.4887(2) | 0.0186(3) | 1 |
| O6 | 0.1191(2) | 0.1908(2) | 0.6964(2) | 0.0210(3) | 1 |
| O7 | 0.5734(2) | 0.2079(2) | 0.0535(2) | 0.0240(3) | 1 |
| H1 | 0.615(4) | 0.147(4) | 0.019(3) | 0.05(2) | 1 |
| O8 | 0.1930(2) | 0.4785(2) | 0.4379(2) | 0.0214(3) | 1 |
| H2 | 0.122(3) | 0.452(5) | 0.368(2) | 0.05(2) | 1 |
| O9 | 0.1379(2) | 0.9095(2) | 0.4695(2) | 0.0187(3) | 1 |
| B1 | 0.2384(2) | 0.3408(3) | 0.7448(2) | 0.0170(4) | 1 |
| B2 | 0.4387(2) | 0.2886(3) | 0.9611(2) | 0.0175(4) | 1 |
| B3 | 0.2135(2) | 0.4880(3) | 0.9159(2) | 0.0160(3) | 1 |
| B4 | 0.1630(2) | 0.3965(3) | 0.5267(2) | 0.0171(4) | 1 |
| B5 | 0.0039(2) | 0.1904(3) | 0.5771(2) | 0.0161(3) | 1 |
Ueq is defined as one third of the trace of the orthogonalized Uij tensor. All atomic positions comply with a Wyckoff position type 4e.
Anisotropic displacement parameters Uij (Å2) of the disordered variant of KB5O7(OH)2 (space group: P21/c, no. 14) with standard deviations in parentheses.
| Atom | U11 | U22 | U33 | U23 | U13 | U12 |
|---|---|---|---|---|---|---|
| K1 | 0.0374(7) | 0.059(2) | 0.0209(5) | 0.0076(7) | 0.0118(5) | 0.0158(7) |
| K2 | 0.0432(7) | 0.069(2) | 0.0212(5) | 0.0059(6) | 0.0097(4) | 0.0000(7) |
| O1 | 0.0147(6) | 0.0226(6) | 0.0187(6) | –0.0031(5) | 0.0062(5) | 0.0033(5) |
| O2 | 0.0157(6) | 0.0207(6) | 0.0165(6) | 0.0010(5) | 0.0052(5) | 0.0059(5) |
| O3 | 0.0194(6) | 0.0296(7) | 0.0150(6) | –0.0030(5) | 0.0045(5) | 0.0088(5) |
| O4 | 0.0186(6) | 0.0224(7) | 0.0183(6) | –0.0029(5) | 0.0084(5) | –0.0068(5) |
| O5 | 0.0176(6) | 0.0211(6) | 0.0160(6) | 0.0010(5) | 0.0066(5) | –0.0052(5) |
| O6 | 0.0200(6) | 0.0257(7) | 0.0148(6) | 0.0002(5) | 0.0058(5) | –0.0069(5) |
| O7 | 0.0201(6) | 0.0280(7) | 0.0212(6) | 0.0024(6) | 0.0071(5) | 0.0117(5) |
| O8 | 0.0244(7) | 0.0231(7) | 0.0193(6) | –0.0003(5) | 0.0123(5) | –0.0076(5) |
| O9 | 0.0164(6) | 0.0213(6) | 0.0165(6) | 0.0012(5) | 0.0060(5) | –0.0056(5) |
| B1 | 0.0153(8) | 0.0192(9) | 0.0143(8) | –0.0031(7) | 0.0047(7) | –0.0008(7) |
| B2 | 0.0143(8) | 0.0166(9) | 0.0207(9) | –0.0014(7) | 0.0072(7) | 0.0004(7) |
| B3 | 0.0134(8) | 0.0156(8) | 0.0167(8) | –0.0015(7) | 0.0048(6) | 0.0015(6) |
| B4 | 0.0165(8) | 0.0171(9) | 0.0196(8) | 0.0006(7) | 0.0101(7) | –0.0007(7) |
| B5 | 0.0159(8) | 0.0157(8) | 0.0173(8) | –0.0004(7) | 0.0081(7) | –0.0018(7) |
Interatomic distances (pm) in the disordered variant of KB5O7(OH)2 (space group: P21/c, no. 14) derived from the single-crystal data (standard deviations in parentheses).
| K1–O7 | 266.1(2) | B1–O1 | 146.7(2) | B4–O4 | 135.3(2) |
| K1–O9 | 278.3(2) | B1–O4 | 146.9(2) | B4–O8 | 137.3(2) |
| K1–O1 | 280.0(2) | B1–O3 | 148.2(2) | B4–O5 | 138.2(2) |
| K1–O4 | 299.0(2) | B1–O6 | 148.6(2) | Ø = 136.9 | |
| K1–O6 | 300.8(2) | Ø = 147.6 | |||
| K1–O2 | 302.0(2) | B5–O6 | 134.4(2) | ||
| K1–O1 | 308.9(2) | B2–O1 | 134.6(2) | B5–O9 | 136.8(2) |
| Ø = 290.7 | B2–O7 | 136.7(2) | B5–O5 | 138.8(2) | |
| B2–O2 | 138.7(2) | Ø = 136.6 | |||
| K2–O7 | 259.7(2) | Ø = 136.6 | |||
| K2–O9 | 273.5(2) | ||||
| K2–O1 | 282.3(2) | B3–O3 | 133.7(2) | ||
| K2–O4 | 301.5(2) | B3–O9 | 137.5(2) | ||
| K2–O5 | 310.7(2) | B3–O2 | 138.7(2) | ||
| K2–O4 | 333.0(2) | Ø = 136.6 | |||
| K2–O3 | 334.2(2) | ||||
| K2–O6 | 339.6(2) | ||||
| K2–O2 | 340.9(2) | ||||
| Ø = 308.4 |
Bond angles (deg) in the disordered variant of KB5O7(OH)2 (space group: P21/c, no. 14) (standard deviations in parentheses).
| O1–B1–O4 | 108.4(2) | O1–B2–O7 | 123.7(2) | O4–B4–O8 | 119.3(2) |
| O1–B1–O3 | 112.5(2) | O1–B2–O2 | 122.2(2) | O4–B4–O5 | 122.4(2) |
| O1–B1–O6 | 107.8(2) | O7–B2–O2 | 114.1(2) | O8–B4–O5 | 118.3(3) |
| O4–B1–O3 | 108.9(2) | Ø = 120.0 | Ø = 120.0 | ||
| O4–B1–O6 | 111.9(2) | ||||
| O3–B1–O6 | 107.4(2) | O3–B3–O9 | 125.3(2) | O6–B5–O9 | 124.5(2) |
| Ø = 109.5 | O3–B3–O2 | 122.1(2) | O6–B5–O5 | 121.5(2) | |
| O9–B3–O2 | 112.6(2) | O9–B5–O5 | 114.0(2) | ||
| Ø = 120.0 | Ø = 120.0 |
Hydrogen bonds in the disordered variant of KB5O7(OH)2 (space group: P21/c, no. 14) obtained from the single-crystal data with standard deviations in parentheses.
| D–H···A | D–H (pm) | H···A (pm) | D···A (pm) | D–H···A (deg) |
|---|---|---|---|---|
| O7–H1···O8i | 83(2) | 201(3) | 269.5(2) | 140(3) |
| O8–H2···O3ii | 83(2) | 250(3) | 319.6(2) | 143(3) |
| O8–H2···O6iii | 83(2) | 233(3) | 298.2(2) | 137(3) |
(i) 1 – x, –0.5 + y, 1.5 – z; (ii) –x, 1 – y, 1 – z; (iii) x, 0.5 – y, –0.5 + z.
Further details of the crystal structure investigation may be obtained from the Fachinformationszentrum Karlsruhe, D-76344 Eggenstein-Leopoldshafen, Germany (fax: +49-7247-808-666; e-mail: crysdata@fiz-karlsruhe.de, http://www.fiz-karlsruhe.de/request_for_deposited_data.html) on quoting the deposition number CSD-429423.
2.3 Vibrational spectroscopy
The transmission FT-IR spectrum of a single-crystal of the title compound was measured in the spectral range of 600–4000 cm–1 with a Bruker Vertex 70 FT-IR spectrometer (spectral resolution 4 cm–1), equipped with an MCT (Mercury Cadmium Telluride) detector and attached to a Hyperion 3000 microscope. As mid-infrared source, a Globar (silicon carbide) rod was used, whereby 32 scans of the single-crystal placed on a BaF2 sample holder were acquired. Primary data was recorded supported by the spectrometer software Opus[20].
The confocal Raman spectrum of the same single-crystal of the disordered variant of KB5O7(OH)2 was recorded from 100–2000 cm–1 with a HORIBA Jobin Yvon LabRam-HR 800 Raman micro-spectrometer. The sample was excited using a frequency doubled 100 mW Nd-YAG-laser (λ = 532.22 nm) under an Olympus 100× objective lens with a numerical aperture of 0.9. The scattered light was dispersed by an optical grating with 1800 lines mm–1, and collected by a 1024 × 256 open electrode CCD detector. Three ranges with a measurement time of 10 s each were measured with a spectral resolution higher than 2 cm–1 determined by measuring the Rayleigh line. A background correction was applied [21].
2.4 DFT calculations
In addition to the experimental recording of vibrational spectra, quantum-chemical energy minimizations and computations of the harmonic vibrational frequencies were performed. The minimum geometry and the theoretical spectra were calculated using the program Crystal09 [22, 23]. For all atoms, a triple zeta basis set was chosen [24]. Exchange and correlation contributions were calculated with the Pbesolfunctional using a shrinking factor of eight [25]. All calculations were performed on the MACH Supercomputer with 256 processors of the type Intel Westmere EX 2.66 GHz.
3 Results and discussion
3.1 Synthetic conditions
The title compound was obtained through a hydrothermal synthesis as described in the experimental section. To promote single-crystal growth, a low cooling rate of 1 K h–1 was applied. We also varied the educts with respect to the synthesis of the ordered structure variant KB5O7(OH)2 published by Wu substituting boron oxide B2O3 with boric acid H3BO3, whereby the title compound was not formed. A reduction of the reaction temperature from 513 to 483 K as applied by Wu did not lead to product formation either. Furthermore, a reduced reaction time of two days (instead of three) at the maximum temperature of 513 K did not lead to the formation of the desired product. In our experimental approach to yield the disordered variant of KB5O7(OH)2, we employed a small amount of cerium nitrate hexahydrate (0.07 equivalents based on boron), which possibly acted upon the reaction mixture as a mineralizer accelerating or enabling the reaction. Wu reported the addition of GaO(OH) (also 0.07 equivalents relative to boron) to obtain the ordered variant of KB5O7(OH)2. The related compound K[B5O7(OH)2]·H2O reported by Zhang et al., which is not a structural variant but shows great resemblance to the dehydrated KB5O7(OH)2, was, however, obtained by addition of pyridine (14 equivalents based on boron) to the reaction mixture containing boric acid, potassium hydroxide, and water [26].
We were able to isolate single crystals of the disordered variant of KB5O7(OH)2, but only as a minor side phase. As the system is strongly inclined towards formation of the ordered structure variant as reported by Wu, further promotion of the formation of the disordered structure variant appears to be not very promising.
3.2 Crystal structure discussion
The disordered potassium pentaborate KB5O7(OH)2 crystallizes with four formula units (Z = 4) in the monoclinic space group P21/c (no. 14) with the lattice parameters a = 904.8(2), b = 753.2(2), and c = 1214.9(5) pm, β = 117.16(2)° and V = 0.73663(5) nm3. The Figs. 2 and 3 show the crystal structure of the title compound in direct comparison to the ordered structure obtained by Wu along [001] and [100], respectively. The disorder of the cation position results in a remarkable elongation of the crystallographic a axis (766.90(3) → 904.8(2) pm) and a shortening of the b axis [904.45(3) → 753.2(2) pm]. The c axis and the monoclinic angles are similar in both forms: c = 1214.9(5) pm/β = 117.16(2)° and c = 1223.04(4) pm/β = 119.132(2)° for the disordered and the ordered pentaborate, respectively. Interestingly, the volume of the disordered variant [V = 0.73663(5) nm3] is slightly smaller than the volume of the ordered variant (V = 0.74102 nm3).
![Fig. 2: Left: Crystal structure of the disordered variant of KB5O7(OH)2 down [001]. K+: large grey spheres; both possible potassium positions are displayed centering the corrugated channels along the b axis; O2–: small blue spheres at the corners of the tetrahedral and of the trigonal-planar groups; B3+: center of tetrahedra and BO3 groups (depicted without polyhedra) as small red spheres; H+: small yellow spheres; Right: ordered structure variant KB5O7(OH)2 by Wu; K+: large white spheres; color scheme of the remaining atoms as aforementioned.](/document/doi/10.1515/znb-2015-0064/asset/graphic/j_znb-2015-0064_fig_002.jpg)
Left: Crystal structure of the disordered variant of KB5O7(OH)2 down [001]. K+: large grey spheres; both possible potassium positions are displayed centering the corrugated channels along the b axis; O2–: small blue spheres at the corners of the tetrahedral and of the trigonal-planar groups; B3+: center of tetrahedra and BO3 groups (depicted without polyhedra) as small red spheres; H+: small yellow spheres; Right: ordered structure variant KB5O7(OH)2 by Wu; K+: large white spheres; color scheme of the remaining atoms as aforementioned.
![Fig. 3: Crystal structure of the disordered variant of KB5O7(OH)2 down [100] (top). Color scheme as mentioned. For comparison, KB5O7(OH)2 by Wu is given below.](/document/doi/10.1515/znb-2015-0064/asset/graphic/j_znb-2015-0064_fig_003.jpg)
Crystal structure of the disordered variant of KB5O7(OH)2 down [100] (top). Color scheme as mentioned. For comparison, KB5O7(OH)2 by Wu is given below.
A more detailed view of the structure of the disordered variant of KB5O7(OH)2 is depicted in the left part of Fig. 4; all relevant positions referred to in the following are highlighted. The disordered phase KB5O7(OH)2 consists of chains of four trigonal-planar BO3 units (Δ), whereby the chain is terminated at both ends by protonated oxygen positions. These chains are interconnected by BO4 tetrahedra (□), of which all four corners are connected to adjacent BO3 units. Thereby, the obtained fundamental building block (FBB) in the disordered variant of KB5O7(OH)2 can be written as <2Δ□>–<2Δ□>–· (after Burns et al. [27]). Fig. 4 (left) shows the representation of the asymmetric unit with displacement ellipsoids (relevant values are given in Tables 2 and 3). For comparison, the ordered phase KB5O7(OH)2reported by Wu is also given in Fig. 4 (right). In the disordered phase KB5O7(OH)2, chains composed of BO3 groups display an angle of 133.5(2)° formed by the positions B3–O9–B5 of adjacent FBBs, resulting in slightly bent chains. The equivalent positions B5–O5–B1 in the ordered variant form a slightly smaller angle of 131.2(2)°. A torsion angle of –7.6(3)° in the Dreierringe [28] (formed by the positions B5–O5–B4–O4) gives rise to a helical wrapping of the disordered structure. In the ordered structure variant, the corresponding torsion angle is very small [–0.7(3)° formed by the positions B1–O3–B2–O1]. At the opposite side of the central tetrahedron, the terminal borate group is connected with a small torsion angle of –0.3(3)° for O1–B2–O2–B3. The ordered structure variant of KB5O7(OH)2, however, shows an angle of 3.6(3)° for the equivalent structural feature (O6–B3–O4–B5) [9].

Asymmetric units in both variants of KB5O7(OH)2. Left: the disordered variant of KB5O7(OH)2; right: KB5O7(OH)2reported by Wu. Displacement ellipsoids are presented at a 50 % probability level.
Bond angles within the trigonal-planar and tetrahedral borate groups (listed in Table 5) concur well with the normal values of 120.0 and 109.4°, respectively [3, 4]. The B–O bond lengths [146.8(3)–148.6(2) pm] are within the normal range commonly reported for BO4 tetrahedra in borates [3]. In the BO3 units, the observed distances range from 133.7(2) to 138.9(2) pm (average value in borates: 137.0 pm [4]).
The terminal oxygen atoms of the BO3 groups are protonated enabling hydrogen bonds as an interconnection between the infinite borate strings. In Table 6, bond lengths, distances, and angles of the three different OH donor and acceptor groups are listed. In the disordered phase KB5O7(OH)2, the O–H distance was restrained to a value of 83(2) pm, which corresponds to the value reported in the ordered structure variant (84 pm). Therefore, also the formed hydrogen bonds (average lengths for the distances H···A and D···A in the disordered form of KB5O7(OH)2 are 228 and 295.8 pm compared to 239 and 313 pm in the ordered form) are comparable [9]. Only the mean hydrogen bond angle of 140° found for the title compound is slightly smaller in comparison to the previously reported average angle of 150° [9] in the ordered phase of KB5O7(OH)2.
In the disordered pentaborate, the positions K1 and K2 are occupied by 46 and 54 %, respectively. Single-crystal diffraction experiments below room temperature (at 213 K) showed no convergence of the cations to a single position. Figure 5 shows the coordination environment of K1 and K2 in direct comparison to the coordination sphere of the K1 position in the ordered structure of KB5O7(OH)2. The K–O coordination distances ranging from 259.6(2) to 340.9(2) pm (average value for K1 and K2 is 300.7 pm) are given in Table 4. The average K1–O distance of 286.9 pm in the ordered form of KB5O7(OH)2 is significantly smaller [9]. The K1 and K2 cations in the title compound exhibit a seven- and nine-fold coordination by oxygen anions, respectively.

Coordination spheres of the two potassium positions K1 and K2 in the disordered variant of KB5O7(OH)2 (left) in comparison to the coordination sphere of the K1 position in the ordered variant of KB5O7(OH)2 (right).
The bond-valence sums of all ions for the disordered variant of KB5O7(OH)2 were calculated from the single-crystal data using the bond-length/bond-strength concept (ΣV) [29, 30]. The results of the calculations (Table 7) correspond well with the expected values for the formal ionic charges of the atoms.
Charge distribution in the disordered variant of KB5O7(OH)2 (space group: P21/c, no. 14) calculated with the bond-length/bond-strength concept (ΣV).
| ΣV | ΣV | ||
|---|---|---|---|
| K1 | +0.93 | K2 | +0.94 |
| O1 | –2.08 | O1 | –2.00 |
| O2 | –2.01 | O2 | –1.95 |
| O3 | –1.84 | O3 | –1.87 |
| O4 | –1.92 | O4 | –1.95 |
| O5 | –2.00 | O5 | –2.07 |
| O6 | –1.90 | O6 | –1.84 |
| O7 | –2.13 | O7 | –2.17 |
| O8 | –1.87 | O8 | –1.87 |
| O9 | –2.17 | O9 | –2.19 |
| B1 | +3.01 | B1 | +3.01 |
| B2 | +3.04 | B2 | +3.04 |
| B3 | +3.04 | B3 | +3.04 |
| B4 | +3.02 | B4 | +3.02 |
| B5 | +3.04 | B5 | +3.04 |
| H1 | +0.87 | H1 | +0.87 |
| H2 | +0.87 | H2 | +0.87 |
Left column: values for a fully occupied K1 position; right column: values for a fully occupied K2 position.
3.3 Vibrational spectroscopy
The IR and the Raman spectra of the disordered variant of KB5O7(OH)2are given in Figs. 6 and 7, respectively. The calculations of the irreducible representation of the title compound in the space group P21/c yielded 204 theoretically possible modes: Γ = 51 Au + 51 Bu + 51 Ag + 51 Bg. One Bu and one Au mode are acoustic, the remaining 100 optical u-modes are excitable by IR radiation. The remaining 51 Ag and 51 Bg modes are purely Raman-active.

FT-IR absorbance spectrum of a single-crystal (top curve) and theoretical bands (red lines) of the disordered variant of KB5O7(OH)2 in the range of 600–3500 cm–1.
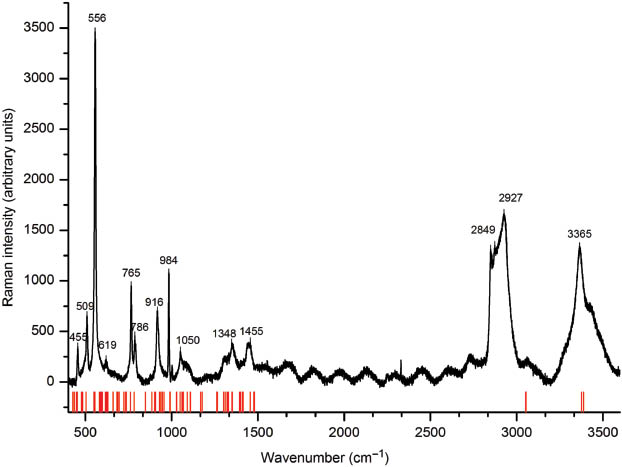
Single-crystal Raman spectrum (top curve) and theoretical bands (red lines) of the disordered variant of KB5O7(OH)2 in the range of 400 to 3600 cm–1.
IR and Raman active bands below 200 cm–1 can be assigned to lattice vibrations involving the cation. Bending and coupled vibrations of BO3 and BO4 units are located between 200 and 900 cm–1 [31]. Due to experimental limitations, an IR characterization below 600 cm–1 was not possible. From 850 to 1600 cm–1, stretching vibrations of B–O units are detected, whereby absorptions of BO4 tetrahedra dominate between 850–1100 cm–1 [32–34]. Absorptions of BO3 units are expected from 1200 to 1450 cm–1 [33–36]. The broad band around 3300 cm–1 can be assigned to O–H groups involved in hydrogen bonding [37, 38]. A more detailed correlation of the observed bands and the according vibrations is given in the following.
3.4 Quantum-mechanical calculations of harmonic vibrational frequencies
For the vibrational band assignment, quantum mechanical calculations were performed using the program Crystal09 [22, 23]. The disordered position of the potassium cation is challenging for quantum chemical calculations. As a consequence, multiple calculations were necessary. Two independent calculations were performed: firstly with a full occupation of the K1 site and secondly with a fully occupied position K2. The similar positions of the vibrational bands in the spectra can be found in the Supplementary Material (Fig. S1). Qualitative band assignments based on the two spectra described above are given in the following. A band-to-band assignment is difficult, as the density of the modes is very high.
The hydrogen stretching bands can clearly be assigned as they represent the only modes showing wavenumbers higher than 3000 cm–1. Around 1600 cm–1, an absorption was found experimentally, which was not obtained in the theoretical work. A possible explanation for this effect could be traces of water on the surface of the measured crystal. The stretching mode of the tetrahedally coordinated boron atoms can theoretically be found between 780 and 1030 cm–1 in the Raman spectrum and between 880 and 1060 cm–1 in the IR spectrum. Stretching frequencies stemming from trigonally coordinated B3+ appear at higher wavenumbers: 1390–1540 and 1300–1480 cm–1 in the IR and Raman spectrum, respectively. Bending modes for the BO4 groups are widespread between 300 and 1000 cm–1. Absorptions resulting from bending of the BO3 groups are located in a more narrow range between 330 and 600 cm–1. In the region between 1050 and 1250 cm–1, H–O–B bending bands are expected. At wavenumbers lower than 250 cm–1, motions in which the potassium cation is involved are dominant, e.g. K–O stretching and O–K–O bending. Table 8 gives all experimentally and theoretically observed frequencies and their assignment. The complete Table of calculated IR and Raman vibrational modes can be obtained as Supporting Information available online (Table S1; see note at the end of the paper for availability).
Comparison and assignment of the experimentally and theoretically observed IR and Raman bands in the disordered variant of KB5O7(OH)2.
| Theoretical | Experimental | Assignment |
|---|---|---|
| IR bands (cm–1) | ||
| 650–696 | 685 | o(H–O–B3) |
| 722–782 | 775 | b(B3–O–B3), b(B3–O–B4) |
| 884–942 | 881 | s(B4–O), o(H–O–K) |
| 990–1029 | 1009 | s(B4–O), b(H–O–B3) |
| 1056–1095 | 1072 | s(B4–O), b(H–O–B3) |
| 1170–1385 | 1225/1292 | s(B4–O), b(H–O–B3) |
| 3400 | 3268 | s(H–O) |
| Raman bands (cm–1) | ||
| 548–554 | 556 | o(H–O–B3) |
| 760 | 765 | b(O–B3–O) |
| 781 | 786 | s(B3–O), b(O–B4–O) |
| 847–955 | 916 | s(B4–O), o(H–O–K) |
| 990 | 984 | s(B4–O), o(H–O–K) |
| 1027–1090 | 1050 | s(B4–O), b(H–O–B3) |
| 1300–1352 | 1348 | s(B3–O) |
| 1390–1476 | 1455 | s(B3–O) |
| 3376–3387 | 3365 | s(H–O) |
s, stretching; b, bending; o, other; pairs of bonded atoms in brackets; B3, trigonal-planar BO3 groups; B4, tetrahedral BO4 groups.
4 Conclusions
The disorder of the potassium cations in the structure variant of KB5O7(OH)2 we were able to present here causes a severe elongation of the crystallographic a axis and a coherent shortening of the b axis of the crystals, distinguishing the title compound from the ordered structure variant KB5O7(OH)2 [a = 766.90(3), b = 904.45(3), c = 1223.04(4) pm, β = 119.132(2)°] reported by Wu in 2011. Vibrational spectroscopic data and their theoretically calculated counterparts support the postulated single-crystal structure solution.
5 Supporting information
A comparison of experimentally observed and theoretical Infrared and Raman bands (Table S1) and a pictorial comparison of theoretical IR and Raman spectra for the two different occupations of the potassium cations K1 and K2 in the disordered phase KB5O7(OH)2 are given as Supporting Information (DOI: 10.1515/znb-2015-0064).
Acknowledgments
We especially thank Dr. G. Heymann for collecting the single-crystal data, and Mag. Dr. C. Hejny for conducting the Raman experiments. Furthermore, we thank MSc. G. Sohr for collecting the FT-IR data, as well as Univ.-Prof. Dr. Roland Stalder (Institute for Mineralogy and Petrography, University of Innsbruck) for the access to the FTIR microscope. The computational work was supported by the Austrian Ministry of Science BMWF as part of the Konjunkturpaket II of the Focal Point Scientific Computing at the University of Innsbruck.
References
[1] E. L. Belokoneva, Crystallogr. Rev. 2005, 11, 151.10.1080/08893110500230792Search in Google Scholar
[2] H. Huppertz, Z. Naturforsch. 2003, 58b, 278.10.1515/znb-2003-0406Search in Google Scholar
[3] E. Zobetz, Z. Kristallogr. 1990, 191, 45.10.1017/S0362152900012733Search in Google Scholar
[4] E. Zobetz, Z. Kristallogr. 1982, 160, 81.10.1524/zkri.1982.160.1-2.81Search in Google Scholar
[5] Z.-E. Lin, G.-Y. Yang, Eur. J. Inorg. Chem. 2011, 2011, 3857.10.1002/ejic.201100270Search in Google Scholar
[6] R. S. Bubnova, S. K. Filatov, Z. Kristallogr. 2013, 228, 395.10.1524/zkri.2013.1646Search in Google Scholar
[7] H. Flood, T. Forland, Acta Chem. Scand. 1947, 1, 592.10.3891/acta.chem.scand.01-0592Search in Google Scholar
[8] N. I. Leonyuk, J. Cryst. Growth 1997, 174, 301.10.1016/S0022-0248(96)01164-5Search in Google Scholar
[9] Q. Wu, Acta Crystallogr. 2011, E67, i67.10.1107/S1600536811043807Search in Google Scholar
[10] S. Merlino, F. Sartori, Contrib. Mineral. Petrol. 1970, 27, 159.10.1007/BF00371981Search in Google Scholar
[11] S. Merlino, F. Sartori, Acta Crystallogr. 1972, B28, 3559.10.1107/S0567740872003498Search in Google Scholar
[12] U. Timper, G. Heller, M. Shakibaie-Moghadam, Z. Naturforsch. 1990, 45b, 1155.Search in Google Scholar
[13] W. H. Zachariasen, H. A. Plettinger, Acta Crystallogr. 1963, 16, 376.10.1107/S0365110X63001006Search in Google Scholar
[14] S. Merlino, F. Sartori, Acta Crystallogr. 1969, B25, 2264.10.1107/S0567740869005553Search in Google Scholar
[15] N. Milinski, B. Ribar, M. Sataric, Cryst. Struct. Commun. 1980, 9, 473.Search in Google Scholar
[16] STOE WinXPOW INDEX, (version 3.0.1.8), Stoe & Cie GmbH, Darmstadt (Germany) 2010.Search in Google Scholar
[17] A. Spek, J. Appl. Crystallogr. 2003, 36, 7.10.1107/S0021889802022112Search in Google Scholar
[18] G. Sheldrick, Acta Crystallogr. 2008, A64, 112.10.1107/S0108767307043930Search in Google Scholar
[19] T. Gruene, H. W. Hahn, A. V. Luebben, F. Meilleur, G. M. Sheldrick, J. Appl. Crystallogr. 2014, 47, 462.10.1107/S1600576713027659Search in Google Scholar
[20] Opus (version 6.5), Bruker Optik GmbH, Ettlingen (Germany) 2008.Search in Google Scholar
[21] LabSpec (version 5), HORIBA Jobin Yvon S.A.S., Villeneuve d’Ascq (France) 2010.Search in Google Scholar
[22] R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders, C. M. Zicovich-Wilson, Z. Kristallogr. 2005, 220, 571.10.1524/zkri.220.5.571.65065Search in Google Scholar
[23] R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, E. Pascale, B. Civalleri, K. Doll, N. M. I. J. Bush, P. D’Arco, M. Llunell, Crystal09-User’s Manual, University of Torino, Torino (Italy) 2009.Search in Google Scholar
[24] M. F. Peintinger, D. V. Oliveira, T. Bredow, J. Comput. Chem. 2013, 34, 451.10.1002/jcc.23153Search in Google Scholar
[25] J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou, K. Burke, Phys. Rev. Lett. 2008, 100, 136406-1–136406-4. Erratum: J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou, K. Burke, Phys. Rev. Lett. 2009, 102, 039902-1.10.1103/PhysRevLett.102.039902Search in Google Scholar
[26] H.-X. Zhang, J. Zhang, S.-T. Zheng, G.-Y. Yang, Cryst. Growth Des. 2005, 5, 157.Search in Google Scholar
[27] P. C. Burns, J. D. Grice, F. C. Hawthorne, Can. Mineral. 1995, 33, 1131.Search in Google Scholar
[28] F. Liebau, Structural Chemistry of Silicates, Springer-Verlag, Berlin, 1985. The term “Dreierring” was initially coined by F. Liebau in his book Structural Chemistry of Silicates (Springer, Berlin, 1985). It is derived from the German word “Drei”, which means three. However, a “Dreierring” is not a three-membered ring, but a six-membered ring comprising three tetrahedral (or, as in borate structures also possible, trigonal-planar) centers. Similar terms exist for rings made up of two, four, five, and six centers, namely “Zweier-”, “Vierer-”, “Fünfer-”, and “Sechserringe”, respectively.10.1007/978-3-642-50076-3Search in Google Scholar
[29] I. D. Brown, D. Altermatt, Acta Crystallogr. 1985, B41, 244.10.1107/S0108768185002051Search in Google Scholar
[30] N. E. Brese, M. O’Keeffe, Acta Crystallogr. 1991, B47, 192.10.1107/S0108768190011041Search in Google Scholar
[31] H. R. Xia, L. X. Li, B. Teng, W. Q. Zheng, G. W. Lu, H. D. Jiang, J. Y. Wang, J. Raman Spectrosc. 2002, 33, 278.10.1002/jrs.847Search in Google Scholar
[32] S. D. Ross, Spectrochim. Acta 1972, 28A, 1555.10.1016/0584-8539(72)80126-0Search in Google Scholar
[33] J. P. Laperches, P. Tarte, Spectrochim. Acta 1966, 22, 1201.10.1016/0371-1951(66)80023-1Search in Google Scholar
[34] K. Machida, H. Hata, K. Okuno, G. Adachi, J. Shiokawa, J. Inorg. Nucl. Chem. 1979, 41, 1425.Search in Google Scholar
[35] W. C. Steele, J. C. Decius, J. Chem. Phys. 1956, 25, 1184.10.1063/1.1743175Search in Google Scholar
[36] G. D. Chryssikos, J. Raman Spectrosc. 1991, 22, 645.10.1002/jrs.1250221109Search in Google Scholar
[37] W. Baur, Acta Crystallogr. 1972, B28, 1456.10.1107/S0567740872004455Search in Google Scholar
[38] K. Nakamoto, M. Margoshes, R. E. Rundle, J. Am. Chem. Soc. 1955, 77, 6480.10.1021/ja01629a013Search in Google Scholar
Supplemental Material:
The online version of this article (DOI: 10.1515/znb-2015-0064) offers supplementary material, available to authorized users.
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis and structural characterization of a novel copper(II)/lead(II) heterometallic organic–inorganic hybrid
- A new dimeric naphtho-γ-pyrone from an endophytic fungus Aspergillus niger AKRN associated with the roots of Entandrophragma congoënse collected in Cameroon
- A mononuclear cobalt(III) 3-(2-pyridyl)-5-phenyl-1,2,4-triazolato complex: hydrothermal synthesis, crystal structure, thermostability, and DFT calculations
- Short microwave-assisted modular synthesis of naturally occurring oxygenated bibenzyls
- Kristall- und Molekülstruktur von „Urindigo“ (4,4,4′,4′-Tetramethyl-2,2′-bipyrrolidinyliden-3,3′-dion) und die Strukturverwandtschaft zum Indigo
- Synthesis and characterization of a disordered variant of KB5O7(OH)2
- Prenylated 9,10-dihydrophenanthrenes from Macaranga javanica
- The ternary system Tm–Ni–In at 870 K
- Synthesis, crystal structure and magnetism of Eu3Sc2O5Fe2As2
- Novel conformationally constrained pyrazole derivatives as potential anti-cancer agents
- Two new secondary metabolites from the fruits of mangrove Avicennia marina
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis and structural characterization of a novel copper(II)/lead(II) heterometallic organic–inorganic hybrid
- A new dimeric naphtho-γ-pyrone from an endophytic fungus Aspergillus niger AKRN associated with the roots of Entandrophragma congoënse collected in Cameroon
- A mononuclear cobalt(III) 3-(2-pyridyl)-5-phenyl-1,2,4-triazolato complex: hydrothermal synthesis, crystal structure, thermostability, and DFT calculations
- Short microwave-assisted modular synthesis of naturally occurring oxygenated bibenzyls
- Kristall- und Molekülstruktur von „Urindigo“ (4,4,4′,4′-Tetramethyl-2,2′-bipyrrolidinyliden-3,3′-dion) und die Strukturverwandtschaft zum Indigo
- Synthesis and characterization of a disordered variant of KB5O7(OH)2
- Prenylated 9,10-dihydrophenanthrenes from Macaranga javanica
- The ternary system Tm–Ni–In at 870 K
- Synthesis, crystal structure and magnetism of Eu3Sc2O5Fe2As2
- Novel conformationally constrained pyrazole derivatives as potential anti-cancer agents
- Two new secondary metabolites from the fruits of mangrove Avicennia marina