Main group cyanides: from hydrogen cyanide to cyanido-complexes
-
Axel Schulz studied chemistry at the Humboldt-University and Technical University of Berlin (1987–1992), with a diploma thesis (1993) in inorganic chemistry and a doctoral thesis (1993–1994) in inorganic chemistry (Thomas M. Klapötke, nitrogen chemistry). Thereafter he was a research associate at the university of Glasgow (1995–1997) and a postdoctoral fellow at the ANU in Canberra with Leo Radom (computational chemistry). In 1997, he went to the LMU Munich where he received his second PhD (Dr. Habil.) in 2001 (elementorganic and theoretical chemistry). In 2006 he was appointed full professor for inorganic chemistry at the University of Rostock. Research in the Schulz group largely focuses on fundamental main group element chemistry and the quantum mechanical description of chemical bonding. The group’s more applied interests include the synthesis of new hydrogen acids of pseudohalogens, their stabilization using Lewis acids, the study of their chemical properties and their application in noval reaction pathways.

 and
and
In 2019, Jonas Surkau joined the group of Axel Schulz at the University of Rostock to work on his BSc thesis on the topic of pseudohalogen chemistry. For his MSc (2021) and PhD he set his focus on the stabilization of hydrogen acids by Lewis acids, the influence of adduct formation on the tautomeric equilibrium and their reaction behavior towards hydrocarbons.

Abstract
Homoleptic cyanide compounds exist of almost all main group elements. While the alkali metals and alkaline earth metals form cyanide salts, the cyanides of the lighter main group elements occur mainly as covalent compounds. This review gives an overview of the status quo of main group element cyanides and cyanido complexes. Information about syntheses are included as well as applications, special substance properties, bond lengths, spectroscopic characteristics and computations. Cyanide chemistry is presented mainly from the field of inorganic chemistry, but aspects of chemical biology and astrophysics are also discussed in relation to cyano compounds.
Units of measurement
In this review the units of measurement are used in accordance to the International System of Units (SI). All derived units and their expression in terms of the SI base units are given below.
| Dimension | Unit | Notation | Conversion to SI-units |
|---|---|---|---|
| Energy | H | Hartree | 1 H = 4.3597 × 10−18 J |
| eV | Electronvolt | 1 eV = 1.602 × 10−19 J | |
| Frequency | MHz | Megahertz | 1 MHz = 1 × 106 s−1 |
| Hz | Hertz | 1 Hz = 1 s−1 | |
| Heat quantity | kJ | Kilojoule | 1 kJ = 1 × 103 kg m2 s−2 |
| kcal | Large calorie | 1 kcal = 4.1868 kJ | |
| Length | Å | Ångström | 1 Å = 1 × 10−10 m = 0.1890 B |
| Pressure | Pa | Pascal | 1 Pa = 1 × 10−5 bar |
| Temperature | K | Kelvin | ϑ/°C = T/K − 273.15 |
| Time | h | Hour | 1 h = 3.6 × 103 s |
| min | Minute | 1 min = 60 s | |
| Wave number | cm−1 | Reciprocal centimetre | 1 cm−1 = 100 m−1 |
Introduction
Historical review
Centaurea cyanus is the Latin name for cornflower, which is famous for its intense blue color (Figure 1), but the origin of the color has nothing to do with cyanides. The name was derived from the Greek word for deep blue κυανός (kyanos). However, the accidental discovery of a new blue pigment at the beginning of the eighteenth century can be regarded as the beginning of the cyanide chemistry (Figure 2).

Centaurea cyanus.

Filtered Prussian blue.
As early as 1706 J. Diesbach and J. Dippel accidentally discovered Prussian blue (originally named in German Berliner Blau) (Unknown, 1710; Roth, 2003), a blue dye in the reaction of iron salts (e.g. Eisenvitrol = FeSO4) with lime potash (Kalilauge German for KOH) containing hexa- cyanoferrate(III) salts (Prussiate salts, in German Blutlaugensalze), which were formed in the repeated treatment of Dippel’s Tieröl (oil isolated from a dry distillation of remains of animals, mostly blood) with potash. With an excess of FeSO4 an insoluble intense blue pigment was obtained (eqs. 1, 2, Figure 1, Scheme 1). The first publication appeared in 1710 anonymously in the journal Miscellanea Berolinensia (Kraft, 2011; Unknown, 1710). Ever since Prussian blue became a great economic success as dye (Figure 1) (Andreas, 2017).

Historic reactions that led to the development of CN chemistry.
Only in 1973, A. Ludi et al. succeeded in elucidating the structure of pure Prussian blue: “The results of the chemical analyses clearly demonstrate that all of our samples have a composition close to the ideal formula FeIII4[FeII(CN)6]3 · 15 H2O” (Buser et al., 1977) displaying Fe2+ and Fe3+ bridged by [CN]– ions.
With the recipe for the generation of Prussian blue in hand, many alchemists/chemists studied its properties and chemistry. In 1782, the German-Swedish alchemist Karl Wilhelm Scheele obtained the volatile Berlinerblausäure (= Berlin blue acid, HCN, eq. (3)) during experiments with Prussian blue and yellow Prussiate by treatment with aqueous sulfuric acid. Ever since Scheele’s discovery of HCN, the German name Berlinerblausäure was used, however, often shortened to Blausäure (= blue acid = prussic acid) which is still used for HCN in the German language to date (von Ittner, 1809). It was Joseph Louis Gay-Lussac in 1815 who introduced the French term cyanogène (derived from the Greek word for blue – see above – and γεννάω, I create) for the radical [CN]·, which he figured out to be the central moiety in prussic acid in accordance with C.-L. Berthollet’s work (Berthollet, 1787; Ladenburg, 1907; von Ittner, 1809). Hence, he renamed prussic acid into l’acide hydrocyanique (HCN, hydrogen cyanide; other common names for HCN: hydrocyanic acid, prussic acid). Moreover, J. L. Gay-Lussac observed the formation of gaseous dicyan, NC–CN, on thermal treatment of mercury dicyanide (eq. (4), Scheme 1).
Both HCN and its salts have attracted increasing attention in the past centuries as an important product of the primitive earth atmosphere as well as a reaction intermediate in the formation of important biological molecules such as amino acids, and of course in industry as valuable starting material for a plethora of pharmaceutical and chemical products or for the mining of gold (Abelson, 1966; Loew, 1971; Moffat and Tang, 1976; Park, 2014; Soto-Blanco, 2013).
Cyanide, a classic pseudohalide
With the preparation of HCN by K. W. Scheele in 1782, the first pseudohalogen compounds were isolated, although this term was introduced more than 100 years later (von Ittner, 1809). The pseudohalogen concept was introduced by Lothar Birckenbach and Karl Kellermann in 1925 and further established and justified in a series of experimental reports (Birckenbach and Huttner, 1929; Birckenbach and Kellermann, 1925a; Birckenbach and Linhard, 1929, 1930; Birckenbach et al., 1929a) as well as applied and further developed by others (Brand et al., 2005a, 2007; Hering et al., 2014; Schulz and Klapötke, 1992; Stopenko et al., 1986). Interestingly, the term was introduced by L. Birckenbach and K. Kellermann “to avoid the controversial term radical ” (Birckenbach and Kellermann, 1925a), therefore proposed the term pseudoelement in the free state. Since the [CN]· radical behaves chemically like a halogen, they therefore called it a pseudohalogen . As all pseudohalides, the cyanide ion represents a monovalent resonance stabilized mono-anion. The diatomic [CN]– ion is the simplest of all pseudohalides (such as [N3]–, [OCN]–, [SCN]–, [CNO]–, etc.). Due to the larger number of atoms forming the pseudohalide, usually larger ionic radii and different steric properties are observed compared to the halides. Moreover, unlike the halides, the cyanide with its ambidentate character with respect to bonding can form dative bonds either via the nitrogen or the carbon atom or even via both in a 1,2-bridging coordination mode. The bridging mode is also known from halides (Corain, 1982; Stopenko et al., 1986). Nevertheless, there are a number of chemical properties that are very similar to those of the halogens (Scheme 2). For example, the cyanide fulfills the following criteria with respect to a halogen-like chemical behavior: It forms (i) a strongly bound univalent [CN]∙ radical (eq. 5), (ii) a singly charged anion ([CN]–, eq. 6), (iii) a hydrogen acid (HCN, hydrocyanic acid, eq. 7), (iv) salts of the type M(CN) n with silver, lead and mercuric salts, which exhibit a low solubility (eq. 8), (v) a neutral dipseudohalogen compound (NC–CN, eq. 9), which disproportionates in water (into [CN]– and [OCN]–, eq. 10) and can be added to double bonds (which is not the case for NC–CN but for HCN), and (vi) interpseudohalogen species (Arulsamy et al., 1999; Brand et al., 2005b; Brupbacher et al., 1997; Chow and Britton, 1974; Grundmann and Fulton, 1964; Guo et al., 1996; Hvastijová et al., 1999; Köhler and Lux, 1968; Maier and Teles, 1987a, 1987b; Maier et al., 1996a, 1996b, 1997a, 1997b; McGibbon et al., 1992; Middleton et al., 1958; Pasinszki, 2008; Robles et al., 2011; Schulz and Klapötke, 1996; Vörös et al., 2012, 2015) (e.g. NC–CNO, eq. 11) (Maier and Teles, 1987a) as well as halogen-pseudohalogen compounds (X–CN (Berlinerblau, 1884; Coleman et al., 2007; Corain, 1982; Gail et al., 2004; Geier and Willner, 2008; Hartman and Dreger, 1931; Ho et al., 2006; Kichigina et al., 1998; Mayer, 1969a; Rubo et al., 2006; Slotta, 1934; Wu et al., 1998), X = halogen, eq. 12) (Brand et al., 2005a, 2007; Stopenko et al., 1986). Recently, it was shown that this concept can be extended by the criterion that a pseudohalogen should also form a pseudohalonium ion, e.g. in case of CN of the type [TMS–CN–TMS]+ (eq. 13) (Schulz and Villinger, 2010). Furthermore, the cyano radical is – like the halogen radicals – a strongly electronegative molecule, its electron affinity being measured to be (3.862 ± 0.004) eV (Bradforth et al., 1993).

Overview of cyano-pseudohalogen-chemistry.
Characteristic of cyanides
The term cyanide refers to any compound containing the cyanide ion ([CN]–) with one carbon atom triple bonded to a nitrogen atom. Experimentally, it has been shown that gaseous HCN is partially polymerized at pressures beyond 1.3 GPa (Sinosaki and Hara, 1929). Hydrogen cyanide (HCN) is a colorless gas (TB = 298.8 K) with a faint bitter almond odor, while sodium cyanide (NaCN) and potassium cyanide (KCN) are white crystalline powders. HCN is a weak acid with a pKa of 9.2 (Ambient Water Quality, 1980); therefore, HCN and [CN]– can be transformed into each other depending on pH and temperature. In water at pH = 7, most of the HCN is in the undissociated form. The simple cyanide salts KCN and NaCN are very soluble in water. Besides hydrogen cyanide, cyanides and cyanogen are the most prominent representatives of cyano-compounds. Table 1 summarizes some computed spectroscopic and physical data of these species, while Figure 3 displays the occupied molecular orbitals. The smallest species, the formal mono-pseudohalogen [CN]·, is a radical that is formed when hydrogen cyanide or cyanogen is irradiated (Scheme 2, eq. 5). The [CN]· radical is probably one of the most extensively studied spectroscopic species since it was one of the first detected molecules in the interstellar space (Adams, 1941; Black and van Dishoeck, 1991; Cerny et al., 1978; Cook and Levy, 1973; Forthomme et al., 2014; Lee, 1992; Li et al., 1984; Liszt and Lucas, 2001; Paul and Dalby, 1962; Penzias et al., 1974; Poletto and Rigutti, 1965; Radford and Broida, 1962; Skatrud et al., 1983; Shi et al., 2011; Weinberg et al., 1967; Wootten et al., 1982; Yang et al., 1992). Commonly, [CN]· radicals are detected in mixtures with HCN or NC–CN as it is easily formed by irradiation of these species. The spin density is located mainly at the carbon atom (0.92e vs. 0.08e at the nitrogen atom) (Cochran et al., 1962; Easley and Weltner, 1970; Milligan and Jacox, 1967a, 1967b; Sorensen and England, 2002). So it is not surprising that recombination of two [CN]· radicals form C–C bound cyanogen, NC–CN. Cyanogen (Brotherton and Lynn, 1959) is a linear molecule (D∞h symmetry) that has no dipole moment but a quadrupole moment (Dagg et al., 1986). Due to the delocalization of the CN triple bond also across the C–C bond (hyperconjugation, see Figure 3), the experimental C–C distance of 1.3768(3) Å is very short and thus has partial double bond character (Torrie and Powell, 1989). This also results in a large dissociation energy of 135.14 kcal mol−1 (Feller et al., 2008). Cyanogen polymerizes at temperatures above 573 K to form polycyanogen.
Computed spectroscopic data of some basic CN species (PBE0/aug-cc-pVDZ; dipole moment in Debye, lengths in Å, charges in e, zpe in kcal mol−1, unscaled wave numbers in cm−1) (Schulz, 2021).
| parameter | [CN]⋅ | [CN]– | HCN | NC–CN |
|---|---|---|---|---|
| Ground state | 2Σ+ | 1Σg | 1Σg | 1Σg |
| Dipole | 1.48 | 0.62 | 3.05 | 0.00 |
| r C–N | 1.171 | 1.182 | 1.156 | 1.161 |
| q(C) | 0.43 | −0.21 | 0.09 | 0.19 |
| q(N) | −0.43 | −0.79 | −0.33 | −0.19 |
| λ[a] | 0.80 | 1.01 | 0.82 | 0.78 |
| zpe | 3.11 | 3.06 | 10.25 | 10.35 |
| ν CN | 2173 | 2173 | 2211 | 2467/2282[b] |
-
[a]spλ hybride for the nitrogen lone pair. [b]In-phase (Σg) and out-of-phase (Σu) vibration.
![Figure 3:
Qualitative MO description of [CN]·, [CN]–, HCN, and NC–CN.](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_003.jpg)
Qualitative MO description of [CN]·, [CN]–, HCN, and NC–CN.
Like HCN, cyanogen is a colorless, flammable, and toxic gas. It is formed not only during the thermal decomposition of transition metal cyanides, but also during the blast furnace process according to 2C + N2 → NC–CN + 309 kJ mol−1 (Chase et al., 1974). Formally, cyanogen is the anhydride of ammonium oxalate, [NH4]2[C2O4] = NC–CN ⋅ 4H2O, i.e. it can also be generated by dehydration of ammonium oxalate (Stopenko et al., 1986).
Selected cyanide reactions
In the field of organic as well as inorganic chemistry there are a number of different reactions with HCN or CN sources (mostly metal cyanides or cyanides of weakly coordinating cations (WCC)). Although in the following chapters on the main group cyanides the specific synthetic pathways are discussed for each cyanide class, the most important general cyanide reactions are listed in equations (14)–(24) (Scheme 3). Especially in the field of organic cyanide chemistry, many articles, books and reviews about nitriles (R–CN, organic cyanides) have already been published (Beyer and Walter, 1991; Livinghouse, 1981; Glöcklhofer et al., 2015; Gröger and Asano, 2020; Krüss, 1884; Letts, 1872; Mowry, 1948; Nauth and Opatz, 2019; Plumet and Roscales, 2019; van Epps and Reid, 1916; Yan et al., 2017), so that we have focused here mainly on cyanide reactions in the field of inorganic chemistry (Arlt et al., 2021; Harloff et al., 2019a, 2020a; Stopenko et al., 1986).

Overview of several types of reaction (additional reference to 25: Bläsing et al., 2016a).
HCN sources, toxicology, treatment, and mode of action (Newhouse and Chiu, 2010)
When K. W. Scheele discovered HCN in 1782, he was unaware of its toxicity. The fact that he died of hydrogen cyanide poisoning is probably only a rumor, even if he died relatively early at the age of 44, however after a long illness (Walden, 1943). Nevertheless, the fact that cyanides are highly toxic has been known for a long time and was used as a poison to murder people. Hence, it is not surprising that cyanides are very often even the subject of detective novels such as Agatha Christie’s “Sparkling Cyanide” from 1944 (Christie, 1945).
HCN sources
Cyanides are found both in nature and in everyday life. In nature, HCN – bound in non-toxic cyanogenic glycosides – occurs in some plants. Mostly their fruit kernels contain cyanide. The best-known example are bitter almonds, but small amounts of cyanides are also found in plums, blackthorn, apricots, peaches, etc., in legumes, spurges such as manioc, sweet grasses, flax and columbine. Therefore, the columbine, for example, is avoided by grazing cattle. Cyanogenic glycosides [R1R2C(O–sugar)CN] themselves have no toxic effect, only enzymatic cleavage of the cyanogenic glycoside molecule leads to the release of HCN, which is the actual toxic substance. Cyanides are also released in industry, e.g. agriculture (herbicide), metal industry (metal extraction, leaching of precious metal), chemical industry (waste water). Furthermore, cyanides are also formed during pyrolysis of coal and during fires (of plastics e.g. polyurethanes), so that flue gas mostly contains HCN (up to 1/3) (Newhouse and Chiu, 2010). HCN is also released from biomass burning and volcanoes.
Cyanides are also used in the food industry. For example, cyano-complexes such as alkali ferrocyanide (E 535/E 536, Na4[FeII(CN)6]/K4[FeII(CN)6]) and calcium ferrocyanide (E 538, Ca2[FeII(CN)6]) are used as a food additive. These salts are permitted in small quantities as artificial anti-caking agents, release agents and stabilizers for table salt and table salt substitutes. They are considered harmless, as the HCN cannot be released by the stomach acid.
HCN is also present in cigarette smoke. A smoker ingests 10–400 μg HCN per cigarette through smoke. Even passive smokers still absorb 0.06–108 μg HCN. Compared with a nonsmoker, serum and urine levels of thiocyanate ([SCN]–), the primary metabolite of HCN, are two to five times greater in smokers, clearly demonstrating the markedly increased intake of HCN from cigarette smoke (Newhouse and Chiu, 2010).
Toxicology
Cyanide salts and HCN are very toxic. Inhalation of hydrogen cyanide is more toxic than ingestion of the cyanide salts because they can be detoxified by the first-pass effect in the liver and absorption is slower. Orally ingested alkali cyanide reacts immediately with gastric acid in the stomach to form HCN ([CN]–(aq) + H+(aq) → HCN(aq)). It should be noted that cyanide is also readily absorbed through the skin. In the course of poisoning, headache, dizziness, nausea, feeling of suffocation, metabolic acidosis, hyperventilation, saturation of venous blood (pink skin color), convulsions, unconsciousness, respiratory arrest, and dilated pupils occur. The cause of death is usually central respiratory paralysis due to inhibition of cytochrome c oxidase. The lethal oral dose of KCN and NaCN for humans ranges from 0.5 to 3.5 mg kg−1 body weight (bw) (Newhouse and Chiu, 2010). After inhalation, the distribution of HCN in the body is almost complete after about 5 min. For example, in the bodies of people who died from HCN poisoning, cyanides were found in the blood as well as in the lungs, kidneys, brain, and heart (Gettler and Baine, 1938). The organism has a natural enzymatic detoxification capacity (enzyme rhodanase) of about 0.1 mg kg−1 bw h−1: The enzyme is present in the mitochondria of the liver and kidney and detoxifies cyanide ions by transformation into non-toxic rhodanide ([CN]– + S → [SCN]–). This is the reason why a large part of the cyanide is excreted in the urine as thiocyanates. Only a small part is exhaled as HCN gas. Cyanide binds to Fe3+ ions of the cytochrome c oxidase, whereas binding to Fe2+ ions of hemoglobin is comparatively low. Cyanide binding primarily impairs O2 utilization in the mitochondria. The time course of cyanide poisoning is as follows: unconsciousness sets in after 30 s, respiratory arrest after 3–5 min, and ultimately death after 5–8 min. Even if cardiac arrest does not occur immediately (5–8 min) after inhalation, death can still occur after hours if the clinical picture progresses. A complete cure is only possible with early therapy immediately after poisoning. Depending on the duration of exposure, the level of cyanide concentration ingested, and the type of exposure by inhalation, absorption, or ingestion, the severity of the effect varies. A distinction is made between different degrees of severity of cyanide poisoning: (i) mild form: dizziness, confused state, mild respiratory problems; (ii) acute form: headache with severe dizziness, loss of consciousness, breathing difficulties and convulsions; coma – often with cardiovascular complications such as collapse, pulmonary edema or cardiac arrest; (iii) fulminant form: all of the above symptoms occur very rapidly and can lead to death within minutes. Interestingly, in 75% of all fire deaths (smoke inhalation), pure cyanide poisoning or mixed intoxication with CO is the cause. Even if someone survives cyanide poising, it can lead to long-term neurological damage (Berger, 1997; Gettler and Baine, 1938; Hartwig, 1972; Newhouse and Chiu, 2010).
Treatment of cyanide poisoning
As an antidote in cyanide poisoning after smoke inhalation it is recommended to use hydroxocobalamin (1st choice), in cases of proven and suspected cyanide poisoning, especially in the case of mixed intoxication with CO (e.g. flue gas). Hydroxocobalamin exchanges the [OH]– group for [CN]–, forming the non-toxic cyanocobalamin, or vitamin B12 for short. It is not suitable for oral cyanide poisoning, as the cyanide dose is usually too high. As a 2nd alternative, 4-DMAP (dimethylaminophenol, acts as met-hemoglobin former, Met-Hb) can be utilized when complexing agents such as hydroxocobalamin are not available. This antidote (like NaNO2) oxidizes the divalent iron Fe2+ in hemoglobin to trivalent Fe3+ iron. This then forms a bond with the cyanides. Both agents have similar side effects. First, the hemoglobin in the Met-Hb is no longer available for oxygen transport. On the other hand, both agents cause vasodilation and hypotonias. Finally, sodium thiosulfate, Na2S2O3, which acts as a sulfur donor (sulfur donor: [S2O3]2– → “S” + [SO3]2–), is administered for sequential use with other cyanide antidotes (Anseeuw et al., 2013; Baskin et al., 1992; Bebarta et al., 2012, 2017; Beelitz et al., 2017; Borron et al., 2007; Gracia and Shepherd, 2004; Hall et al., 2007; Henretig et al., 2019; Thompson and Marrs, 2012).
For oral ingestion 4-DMAP should be given, which oxidizes hemoglobin to Met-Hb. Met-Hb in turn binds cyanide. Therapeutic dosage is 3–4 mg 4-DMAP kg−1 bw (about 250 mg). This can bind about 16 mmol cyanide (corresponding to 1000 mg potassium cyanide). Since cyanide is only bound to Met-Hb, but is not eliminated, and there is a risk that it will be released again from the Met-Hb complex, then administration of sodium thiosulfate is useful. If 4-DMAP is not available then immediately infuse sodium nitrite intravenously as countermeasure. Subsequently, Na2[S2O3] solution should be infused. The nitrite like 4-DMAP forms met-hemoglobin, Met-Hb, which reacts with the cyanide in the tissues and renders it harmless. The sodium thiosulfate provides sulfur, which is needed for the natural breakdown of cyanide in the body by enzymes (rhodinase, vide supra). The metabolic inactivation of the cyanide occurs mainly (approx. 80%) via the production of thiocyanate, which is catalyzed by the rhodanese of the liver. Lactacidosis (pH > 7.2) caused by cyanide poisoning requires the administration of NaHCO3 (Anseeuw et al., 2013; Baskin et al., 1992; Bebarta et al., 2012, 2017; Beelitz et al., 2017; Borron et al., 2007; Gracia and Shepherd, 2004; Hall et al., 2007; Henretig et al., 2019; Thompson and Marrs, 2012).
Cyanide poisoning – mechanism of action
Simply put: cyanide blocks cellular respiration in the cells. In the case of poisoning, the cells can no longer utilize the vital oxygen. Therefore, cyanide is considered chemically asphyxiant because it interferes with aerobic metabolism without affecting tissue oxygenation. Since cyanide has a high affinity for iron (III), it results in the formation of a strong bond to tissue cytochrome c oxidase (see above). This in turn leads to inactivation of tissue cytochrome c oxidase. Because cytochrome c oxidase normally takes up oxygen from the blood and functions as an electron acceptor in cellular energy production, this inactivation inhibits cellular respiration (Newhouse and Chiu, 2010). That is, the strong Fe(III)–CN bond inhibits the enzyme and blocks the last step in oxidative phosphorylation. The result is a deficiency of mitochondrial ATP and cell death (Berger, 1997). This leads to an increased anaerobic metabolism. Therefore, blood levels of pyruvic acid, lactic acid, and NADPH increase, while the ATP/ADP ratio consistently decreases. Thus, the first symptoms of acute cyanide poisoning occur in organs with high aerobic energy demands, especially the brain and heart. Inhibition of cellular oxygen consumption results in the presence of oxyhemoglobin in venous blood, which is why the skin turns bright red. All acute effects of cyanide poisoning are closely related to the disruption of aerobic metabolism and the release of secondary neurotransmitters and catecholamines. Acute cyanide poisoning effects include altered respiration, nausea, vomiting, and weakness. Eventually, severe convulsions, coma and death occur. In addition to cytochrome c oxidase, cyanide also binds to other metalloproteins and other cellular molecules, and also contributes to the symptoms of acute cyanide toxicity (Berger, 1997; Gettler and Baine, 1938; Hartwig, 1972; Newhouse and Chiu, 2010).
The deadly HCN probably wrote its worst chapter in the National Socialist concentration camps (extermination camps) during the Second World War. In the Holocaust, millions of Jews were poisoned by the gas “Zyklon B”. “Zyklon B” consisted of liquid HCN, which was dripped onto absorbent carrier materials, e.g. diatomaceous earth, during production. “Zyklon B” was developed to make HCN (boiling point 298.8 K) safe to handle by lowering its vapor pressure.
Safety precautions while working with HCN
Appropriate safety precautions (HCN detector, gas mask, low temperatures, basic solutions, small amounts, pressure and temperatures control, use of powerful fume hoods) should be taken. Note: Some people cannot detect the typical odor (bitter almond oil) of hydrogen cyanide. Therefore, the use of an HCN detector is strongly recommended. Caution! HCN is not only highly toxic but can also decompose explosively under various conditions! Without stabilizer, pure HCN polymerizes very rapidly and exothermically, e.g. under basic conditions (or by autocatalysis) and the resultant HCN polymer subsequently decomposes violently (>373 K).
Scope of this review
Homoleptic cyanide compounds exist of almost all main group elements. While the alkali and alkaline earth metals form cyanide salts, the cyanides of the lighter main group elements occur mainly as covalent compounds. There are already a large number of review articles in the field of organic synthetic chemistry (nitriles) (Beyer and Walter, 1991; Glöcklhofer et al., 2015; Gröger and Asano, 2020; Krüss, 1884; Letts, 1872; Livinghouse, 1981; Mowry, 1948; Nauth and Opatz, 2019; van Epps and Reid, 1916; Yan et al., 2017; Plumet and Roscales, 2019), on E–CN bond formation (Yu et al., 2017), CN-coordination chemistry (Stopenko et al., 1986; Corain, 1982), as well as cyanide salts and compounds (Gail et al., 2004; Rubo et al., 2006). This review presents an overall view of the status quo of main group element cyanides and cyanido complexes. Mainly, only ternary compounds of the formal type E x (CN) y with E = main group element are considered. They can be neutral as well as positively or negatively charged. In some cases, we have also included corresponding CN adducts in the discussion if they show certain properties or stabilize interesting CN fragments. With regard to the alkaline and alkaline earth metals, this review is limited to the pure metal cyanide compounds. Due to the fact that the cyanide anion belongs to the group of pseudohalides, the properties of the cyanide compounds considered are quite similar to those of alkali and earth alkaline halides. Especially at higher temperatures, the cyanide anion starts to rotate and forms a spherical rotor increasing the geometrical similarity to the spherical halides. A significant difference of metal cyanides and metal halides is the existence of various solid-state modifications of the metal cyanides mainly caused by different orientations of the cyanide group at low temperatures and a steric hindrance of its rotation. Due to many similarities of these cyanide compounds, the syntheses and crystal structures of 1st and 2nd main group compounds will be discussed in general. This review also compiles reports on compounds consisting exclusively of one element of the main groups 3–8 paired with at least one cyanide group to provide a summary.
In general, this review focuses on information about syntheses, bond lengths, spectroscopic characteristics, applications, special substance properties and computations. Accordingly, all fields of chemistry, but also biology and astrophysics are engaged. On this way, the report includes experimental findings from the nineteenth century as well as modern quantum mechanistic calculations and sophisticated gas phase experiments.
Hydrogen cyanide
Since HCN occupies a central role in the cyanide chemistry of the main group elements and, moreover, its chemical physical properties are markedly different from those of the alkali cyanides, HCN will be discussed here separately from them. Some important points concerning its history, bonding, and toxicity have been deliberately written into the introduction to emphasize the aspect of its central role.
Similar to hydrogen halides this compound consists of a polar covalent bond between H–CN (see pseudohalide concept in chapter 1.2), which is able to dissociate in aqueous solution to form the rather week acid, hydrocyanic acid (prussic acid), pKa = 9.2 (Ambient Water Quality, 1980; Cotton et al., 1967). Pure hydrogen cyanide is a colorless volatile liquid (TB = 298.75 K) with a characteristic smell of bitter almonds (Cotton et al., 1967). The compound is very poisonous, because it prevents the oxygen uptake in the respiratory chain of the human body (see chapter 1.5.2) (Meredith et al., 1993).
On pure HCN, there are a large number of theoretical (Johansson et al., 1972; Jucks and Miller, 1988; Karpfen, 1996; King and Weinhold, 1995; Kurnig et al., 1990; Moffat and Tang, 1976; Pacansky, 1977; Ramos et al., 1988) and experimental (Al-Azmi et al., 2003; Anex et al., 1988; Beichert et al., 1995; Brown et al., 1981; Earnshawb and Ireland, 1995; Fowler et al., 1994; Giauque and Ruehrwein, 1939; Gutowsky et al., 1992; Jucks and Miller, 1988; Knözinger, 1986; Kurnig et al., 1990; Legon et al., 1977; Meot-Ner and Speller, 1989; Nauta et al., 1999; Wofford et al., 1986; Völker, 1960; Schrems et al., 1987; Sun et al., 2000) studies on its clusters (HCN) n in the gas phase (microwave rotational spectroscopy, IR or matrix IR). These studies show that HCN forms linear H-bonded chains in all its phases. The crystal structure of HCN in the solid phase showed “infinite” parallel linear H-bridged chains (see 2.2 Crystal structure) (Dulmage and Lipscomb, 1951), while a linear structure consisting of three formula units was experimentally derived for the liquid phase (Tyuzyo, 1957). Density and heat capacity measurements of HCN in the gas phase proved the presence of (HCN) n oligomers, especially di- and trimers at 298 K and 1 atm (Felsing and Drake, 1936; Giauque and Ruehrwein, 1939; Sinosaki and Hara, 1929). Using rotational spectroscopy, the HCN dimer and trimer could be characterized without doubt as H-bonded linear species in the gas phase (Brown et al., 1981; Buxton et al., 1981; Ruoff et al., 1988).
It was shown that the formation of very strong hydrogen bonds is mainly responsible for the formation of aggregates in the condensed phase. The energy of the hydrogen bond in gas phase increases from 3.28 to 8.72 kcal mol−1 by addition of a third hydrogen cyanide molecule. Comparing the gaseous and liquid state the energy of the hydrogen bond increases to 4.6 kcal mol−1 due to dimer formation (Pauling, 1968).
Monomeric HCN adducts can be formed with Lewis acids, e.g. HCN–BF3, HCN–NbCl5, or HCN–AsF5) (Burns and Leopold, 1993; Chavant et al., 1975; Davydova et al., 2010; Haiges et al., 2004; Reeve et al., 1993; Saal et al., 2019; Tornieporth-Oetting et al., 1992). The only experimental study of an HCN⋯HCN adduct in the gas phase was described by K. R. Leopold et al. who investigated HCN⋯HCN–SO3 in the gas phase using rotational spectroscopy (Fiacco et al., 2000). A. Schulz and co-workers succeeded in stabilizing an HCN dimer in the solid state by a strong Lewis acid, such as B(C6F5)3, and investigating the influence of different solvents (HCN, CH2Cl2 and aromatic hydrocarbons, such as benzene, toluene) on the crystallization process. Dimer formation was observed (Figure 4, Scheme 4) when HCN or CH2Cl2 were used as solvents, whereas the use of aromatic hydrocarbons led to the formation of monomeric aryl⋯HCN–B(C6F5)3 adducts (Figure 4), which are stabilized via η6-coordination of the aromatic ring system and thus resemble the well-known half-sandwich complexes (Bläsing et al., 2018).

Molecular structure of a Lewis acid stabilized dimer (left) and η6-coordination of the benzene adduct (right) (Bläsing et al., 2018).

Synthesis of an HCN dimer stabilized as B(C6F5)3 adduct (Bläsing et al., 2018).
Hydrogen cyanide is widely used in industrial processes as an intermediate to mainly produce plastics, for example polymethacrylate or polyamides, drugs, insecticides, iron cyanide pigments, organic pigments and also pesticides (Henderson and Cullinan, 2007). A significant amount is also converted into cyanides, which are used in the cyanide process, the surface hardening of metals, in ore dressing and electroplating. Hydrogen cyanide, which is marked by the radioactive carbon isotope H14CN, is used in biochemical research to synthesize traced amino acids (Koschel et al., 1971).
A natural source of hydrocyanic acid is the glycoside amygdaline (C20H27NO11), which is contained in the kernel of apricots, apples and almonds. It is obtained by hydrolysis of the amygdaline and other glycosides in presence of the enzyme β-glycosidase leading to glucose and benzaldehyde (Scheme 5) (Williams, 1915).
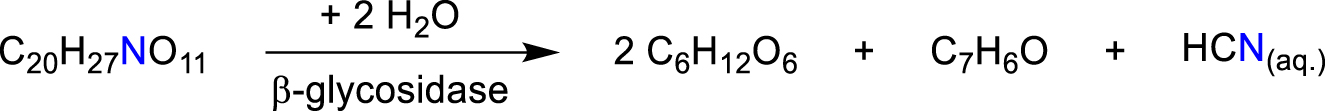
Hydrolysis of the glycoside amygdaline leading to hydrocyanic acid (Williams, 1915).
Synthesis
In 1782 Karl Wilhelm Scheele obtained HCN by treating a solution of dried blood and potash (containing the potassium ferrocyanide, Prussian blue) with dilute acid (Böhland et al., 1978). He also described a synthesis by distillation of ferro cyanide in acid. The observed products of decomposition are merely CO2 and N2. But 30 years later, Joseph Louis Gay-Lussac could provide proof by isolation in a pure state (Williams, 1915).
Today many synthetic routes of hydrogen cyanide are known using different sources of nitrogen (NH3, N2, organic nitrogen compounds) and carbon (coal, CO, CH4). An important industrial process is the catalytical combustion of a gas mixture containing NH3 and CH4, which is an extremely exothermic reaction ΔRH = −949.6 kJ (Scheme 6) (Andrussow, 1926, 1955; Böhland et al., 1978).

Industrial synthesis of HCN by catalytical combustion of CH4 and NH3 (Andrussow, 1926; Andrussow, 1955; Böhland et al., 1978).
The two most famous industrial processes including this reaction are called Andrussow and Degussa process involving a platinum catalyst. The Andrussow process is carried out at temperatures of about 1273 K. The Degussa process requires higher temperatures of about 1473 K and oxygen free conditions (Andrussow, 1926; Andrussow, 1935; Krause, 1965). Yields of 90% can be achieved by fast reaction process and separation of the product (Greenwood et al., 1988).
Another simple approach to isolate hydrogen cyanide is the reaction of cyanide containing salts, for example NaCN, KCN or K4[Fe(CN)6], with acids (Slotta, 1934; Thaulow, 1844; Williams, 1948; Ziegler, 1921). This kind of synthesis is often used in laboratories because of the high stability of the salts and the nearly quantitative conversion. It is also reported a complete reaction of moist CO2 and KCN to hydrogen cyanide (Henninger, 1876).
In the laboratory, if one wishes to produce anhydrous, pure HCN, HCN can be elegantly produced by the solvent-free reaction of NaCN and stearic acid (TM = 342 K) at 353–373 K in vacuo as a gas, which can be immediately condensed into the appropriate reaction vessel (Scheme 7, Figure 5) (Günther et al., 1935; Labbow et al., 2016).
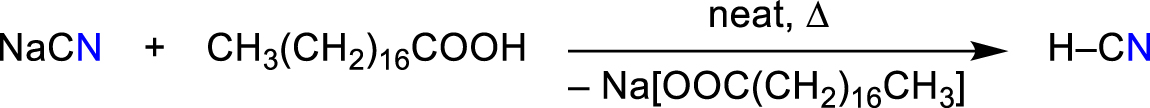
Lab-synthesis of pure dry HCN (Figure 5) (Labbow et al., 2016).

Apparatus for the synthesis of pure HCN with stearic acid and important spectroscopic data: HCN (27.03 g mol−1): T M = 260 K. 1 H NMR (298 K, CD2Cl2, 250.1 MHz): δ = 4.00 (s, 1H, HCN, 1J(1H-13C) = 266 Hz). 13 C{ 1 H} NMR (298 K, CD2Cl2, 62.9 MHz): δ = 110.1 (s, HCN). 14 N{ 1 H} NMR (298 K, CD2Cl2, 36.1 MHz): δ = −125.0 (s, HCN, Δν1/2 = 50 Hz). Raman (632 nm, 6 mW, 20 s, 20 acc., 233 K, cm−1): 819 (1), 1611 (1), 2163 (1), 2197 (10), 3151 (1) (Labbow et al., 2016).
Hydrogen cyanide undergoes fast decomposition, which can be retarded by addition of mineral acids or up to 20% of glycerine (Williams, 1948). Also an adsorption of hydrogen cyanide on appropriate surfaces is possible. A reaction of HCN with NaOH returns to the sodium salt NaCN, which can be stored and converted to hydrogen cyanide easily (Koschel et al., 1971). The decomposition of hydrogen cyanide is mainly a kind of a polymerization reaction. Higher temperatures increase the polymerization and finally lead to the formation of ammonia and ammonium cyanide (Völker, 1960). Polymeric hydrogen cyanide, usually referred to as polymeric hydrocyanic acid or azulminic acid, is formed by anionic polymerization of HCN in aqueous solution. The reaction proceeds via dimeric hydrocyanic acid (iminoacetonitrile). At the same time, ring closure takes place between γ-positioned nitrile groups, which stabilizes the polymer (Marín-Yaseli et al., 2017; Ruiz-Bermejo et al., 2013; Völker, 1960).
As already mentioned, HCN also plays/played a major role in the formation of H/C/N compounds on earth as well as on other celestial bodies, e.g. Titan (Carrasco et al., 2009; Coll et al., 1998; Hörst et al., 2012; Israël et al., 2005; Kaiser and Mebel, 2012; Maillard et al., 2021; Thompson et al., 1994; Rüger et al., 2019; Selliez et al., 2020; Somogyi et al., 2016; Szopa et al., 2006; Vuitton et al., 2010). Titan, the second largest moon in our Solar System, has a dense atmosphere consisting mainly of nitrogen and a few percent methane. Nitrogen radicals are formed in this gaseous environment, for example by the UV radiation of the sun (Kaiser and Mebel, 2012). Their reaction with methane leads to the formation of a dense haze. Because of this formation, the study of this nebula represents a major interest for prebiotic chemistry and planetary sciences (Hörst et al., 2012; Kaiser and Mebel, 2012; Maillard et al., 2021; Sagan and Khare, 1979; Selliez et al., 2020; Somogyi et al., 2016; Rüger et al., 2019). Mainly NH3 and HCN serve as precursors for the formation of more complex nitrogen-containing matter systems. In connection with the atmosphere of Titan, which is often tried to be reproduced in laboratory experiments, the production of so-called tholins (derived from Greek for muddy) plays a major role (Sagan and Khare, 1979). The term coined by C. Sagan describes a mixture of organic substances observed after irradiation of gas mixtures analogous to the atmosphere of Titan. Tholins are thought to be a very complex mixture of different compounds (Sagan and Khare, 1979).
Crystal structure
The calculated molar heat capacities indicate a reversible phase transition of hydrogen cyanide at 170.35 K (Giauque and Ruehrwein, 1939). The experimental determination of two different crystal structures by W. J. Dulmage and W. N. Lipscomb confirmed this result in 1951. Besides an orthorhombic modification, they reported a tetragonal form of hydrogen cyanide for temperatures higher than the temperature of phase transition (Table 2) (Dulmage and Lipscomb, 1951).
Crystallographic data of the tetragonal high temperature modification and orthorhombic low temperature modification of hydrogen cyanide (temperatures in K; lengths in Å; angles in °; volumes in Å3) (Dulmage and Lipscomb, 1951).
| Parameter | HCN tetragonal | HCN orthorhombic |
|---|---|---|
| T | >170 | <170 |
| Space group | I4mm | Imm2 |
| a | 4.63 | 4.13 |
| b | 4.63 | 4.85 |
| c | 4.34 | 4.34 |
| α | 90.00 | 90.00 |
| β | 90.00 | 90.00 |
| γ | 90.00 | 90.00 |
| V | 92.9 | 86.8 |
| Z | 2 | 2 |
Both lattices consist of linear molecule chains parallel to the c-axis of the lattice (Figure 6) and are surrounded by four more chains, which are displaced by half of a lattice parameter to the main chain (Dulmage and Lipscomb, 1951; Langel et al., 1989). Each chain is composed of HCN molecules that are linked by NC–H⋯N hydrogen bonds. The reported structural parameters are identical (rC–N = 1.19 Å; rH–C = 1.07 Å). The hydrogen bond with a computed distance of rNC–H⋯N = 2.08 Å can be considered very strong. For comparison, the gas phase data of HCN are rC–N = 1.15525 Å and rH–C = 1.06666 Å as well as rH1–C1 = 1.06728 Å, rC1–N1 = 1.15361 Å, rN1⋅⋅⋅H2 = 2.21186 Å, rH2–C2 = 1.07300 Å and rC2–N2 = 1.15588 Å for the linear hydrogen bonded dimer H1C1N1⋅⋅⋅H2C2N2 (at the CCSD(T)-F12b/aug-cc-pVQZ level of theory, optimized geometries with C∞v symmetry) (Mihrin et al., 2018).

Left: unit cells of the high-temperature (full lines) and the low-temperature modification (broken lines) of hydrogen cyanide (representation taken from W. J. Dulmage and W. N. Lipscomb), middle: unit cell of the tetragonal and right: of the orthorhombic form (view along a-axis) (Dulmage and Lipscomb, 1951).
In contrast, the low-temperature modification is characterized by an orthorhombic form, which is quite similar to the tetragonal form (Figure 6) (Dulmage and Lipscomb, 1951). Because of parallel molecular and crystal axes a dielectric anisotropy weakens the hydrogen bonds, which leads to slightly stretched intermolecular distances (Baglin et al., 1970).
Due to the polarity of hydrogen cyanide, two models are reported to describe the primitive cell of the low-temperature modification. The polar crystal structure of the first model is based on an arrangement of parallel dipole moments in accordance with W. J. Dumlage and W. N. Lipscomb (space group I4mm). In contrast, the anti-parallel hydrogen-bonded HCN chains and their anti-parallel dipole moments of the second model (space group P4/nmm) leads to a non-polar crystal structure (Figure 7) (Cummins et al., 1988; Panas, 1992).

Polar and non-polar crystal structures of hydrogen cyanide in low-temperature modification (representation taken from Cummins et al., 1988).
Furthermore, more modifications of hydrogen cyanide including different electric conductivities are expected at high pressures of more than 73 GPa. Due to calculations they propose a transition from the orthorhombic to the tetragonal and from the tetragonal to the triclinic modification. The phase transition depends on the pressure and the presence of polar or non-polar phase inside the orthorhombic cell (Khazaei et al., 2011).
HCN/HNC-isomerization
The HNC species is a metastable isomer of hydrogen cyanide, which is mainly detectable in the interstellar matter. Isomeric pairs of small molecular entities such as HCN/HNC are prevalent in the interstellar medium. Previous observations of the HNC/HCN abundance ratio show that the ratio decreases with increasing temperature. It was shown that chemical gas phase reactions affect the HNC/HCN ratio and can thus contribute to the observed dependence, e.g. H + HNC → HCN + H or C + HNC → HCN + C (Graninger et al., 2014). The origin and the mechanism of formation of the isocyanide species is not completely resolved. It is known that the ratio of hydrogen cyanide and hydrogen isocyanide depends on the temperature (Ferus et al., 2011; Wang et al., 2007).
The first infrared spectroscopic (IR) evidence of HNC in laboratory was found by D. E. Milligan and M. E. Jacox during the photolytic decomposition of methyl azide (Milligan and Jacox, 1967a, 1967b). D. E. Milligan could confirm his proposal by studies on the photolysis of HCN in vacuum under ultraviolet light at 14 K (Milligan and Jacox, 1967a, 1967b). The IR data of HNC are presented in Table 3.
IR data of HNC compounds after photolysis of methyl azide in argon matrix at 4 K and after photolysis of HCN in argon matrix at 14 K (wave numbers in cm−1) (Milligan and Jacox, 1967a, 1967b).
| Compound | ν 1 | ν 2 | ν 3 | Compound | ν 1 | ν 2 | ν 3 |
|---|---|---|---|---|---|---|---|
| H14N12C | 2032 | 535 | 3583 | H14N12C | 2029 | 477 | 3620 |
| D14N12C | 1940 | 413 | 2733 | D14N12C | 1940 | 374 | 2769 |
| H15N12C | 2005 | – | 3571 | H15N12C | 2004 | 475 | 3610 |
| D15N12C | 1923 | – | 2710 | – | – | – | – |
| H14N13C | 1990 | – | 3583 | H14N13C | 1987 | 477 | 3620 |
The isocyanide species is an important system of theoretical calculations because its simplicity allows also complex calculations and the results are able to be adopted to more complex systems (Burkholder et al., 1987; Pearson et al., 1975). C. Glidewell and C. Thomson also carried out many calculations using different basis sets to characterize both compounds and their transition state discussing differences of selected methods (Glidewell and Thomson, 1984). The H−C and C−N distances of the T-shaped transition state are quite similar to the distances of the HCN and HNC species, but the N−H distance of the transition state is elongated. An HCN angle of 70°–80° is found for the transition state (HCN → CNH) (Vichietti and Haiduke, 2014). The experimentally observed CN bond in HCN is slightly shorter than in NCH, while the CH bond is slightly longer (Table 4) (Costain, 1958; Okabayashi and Tanimoto, 1993).
Structural data of the isomers HCN and HNC using different basis sets (bond lengths in Å; angles in °) (Gleiter et al., 1995; Pople et al., 1978; Vichietti and Haiduke, 2014).
| Compound | Method + basis set | r A−B | r H−A/B | <(H–A–B) |
|---|---|---|---|---|
| HCN | MP2/6-31G* | 1.177 | 1.069 | 180.0 |
| HF/6-31G* | 1.133 | 1.059 | 180.0 | |
| B3LYP/aug-cc-pVQZ | 1.1451 | 1.0655 | – | |
| MPW1K/aug-cc-pVQZ | 1.1358 | 1.0621 | – | |
| Experimental (Costain, 1958) | 1.154(1) | 1.063(3) | – | |
| HNC | MP2/6-31G* | 1.187 | 1.003 | 180.0 |
| HF/6-31G* | 1.154 | 0.985 | 180.0 | |
| B3LYP/aug-cc-pVQZ | 1.1632 | 0.9964 | – | |
| MPW1K/aug-cc-pVQZ | 1.1543 | 0.9895 | – | |
| Experimental (Okabayashi and Tanimoto, 1993) | 1.16835(2) | 0.996064(3) | – |
P. K. Pearson and H. F. Schaefer reported a transition state structure with HCN/HNC angle of both isomers at 73.7°, so the transition state resembles more the structure of the hydrogen cyanide. The HCN isomer is energetically favoured over the HNC species by ca. 60–80 kJ mol−1 (Pearson et al., 1975; Pople et al., 1978; Redmon et al., 1980; Vazquez and Gouyet, 1981) depending on the used basis sets and method (Komornicki et al., 1977; Loew and Chang, 1971; Pople et al., 1978). W. J. Hehre et al. could confirm the difference of energies between both isomers by experimental measurements ((62.0 ± 8.3) kJ mol−1) whereas A. G. Maki and R. L. Sams reported an energy difference of (43.12 ± 4.61) kJ mol−1 by IR spectroscopic observation (Maki and Sams, 1981; Pau and Hehre, 1982). The activation barriers are computed to lie between 198 and 202 kJ mol−1 for HCN → HNC and 137–141 kJ mol−1 for the reverse process HNC → HCN (Khalouf-Rivera et al., 2019). R. M. Vichietti et al. computed activation enthalpies of 184 kJ mol−1 for the forward and 121 kJ mol−1 for the reverse reaction (Vichietti and Haiduke, 2014).
Spectroscopic data
Vibrational data of solid, liquid and gaseous hydrogen cyanide are listed in Tables 5 and 6. Spectroscopic data of IR and Raman spectra of two different isotopically substituted compounds were discussed in detail by Chadwick and Edwards (1973). Furthermore, Walsh et al. (1978) carried out detailed studies on HCN- and DCN-monomers and dimers in different matrices, e.g. noble gas and N2.
Raman data of HCN at different temperatures (temperatures in K; wave numbers in cm−1).
| Publication | State | T | ν 1 | ν 2 | ν 3 |
|---|---|---|---|---|---|
| Douglas and Sharma (1953) | Gaseous | – | 2095.5 | – | – |
| Pézolet and Savoie (1969) | Liquid | 292 | 2096 | 3215 | 794 |
| 261 | 2097 | 3207 | 798 | ||
| Solid | 183 | 2098, 2104 | 3145, 3180 | 819 | |
| 78 | 2098, 2104 | 3129, 3150 | 827, 840 |
IR data of HCN at different temperatures (temperatures in K; wave numbers in cm−1).
| Publication | State | T | ν 1 | ν 2 | ν 3 |
|---|---|---|---|---|---|
| Hoffmann and Hornig (1949) | Gaseous | – | 2089 | 3312 | 712 |
| Solid | 93 | 2097 | 3132 | 828, 838 | |
| Kozirovski and Folman (1966) | On NaCl | 77 | 2095 | 3145 | 745, 800 |
HCN and adducts of HCN species were studied by means of 1H, 13C, 14N NMR experiments in CD2Cl2 (Table 7) (Alkorta and Elguero, 1998; Arp et al., 2000; Bläsing et al., 2018, 2020; Corain, 1982; Emri and Györi, 1994; Harloff et al., 2019a, 2020b; Klapötke et al., 1996; Mai and Patil, 1986; Mamajanov and Herzfeld, 2009; Nagy et al., 2005; Olsson et al., 1995; Provasi et al., 2005; Saal et al., 2019; Schulz and Klapötke, 1992; Tornieporth-Oetting et al., 1992). It should be noted that in case of adducts such as B(C6F5)3 or its dimer HCN–HCN–B(C6F5)3, all resonances (in the 1H, 13C, 14N and 11B NMR spectra) are very broad but still detectable at 253 K indicating a highly dynamic system (Bläsing et al., 2018). For example, even the nitrogen atoms of HCN–HCN–B(C6F5)3 can be observed in the 14N NMR experiment at −128 and −192 ppm with half widths of 960 and 2250 Hz (cf. HCN: −125 ppm and Δν1/2 = 50 Hz). As expected, upon borane adduct formation (Table 7), the resonance of the proton is shifted to lower field by ca. 2 ppm (cf. HCN 4.00 vs. 6.42 in the dimer adduct) displaying an increase of the acidity. A broad resonance formally assigned to the loosely bound second HCN molecule of HCN–HCN–B(C6F5)3 is detected at 4.02 ppm, indicating dynamic exchange between loosely bound and free HCN species (Bläsing et al., 2018). The 1H NMR spectrum of HC15N shows a sharp doublet at 3.60 ppm, which was directly taken after preparation. The proceeding oligomerization inducing fast intermolecular proton exchanges leads to disappearance of the splitting of the proton resonance and a sharp single signal, while the color turns to yellow (Binsch and Roberts, 1968).
Selected NMR data of HCN, HCN-B(C6F5)3, and HCN-HCN-B(C6F5)3 [δ scale] (Bläsing et al., 2018).
| Compound | 1H | 11 B | 13C{1H} | 14N{1H} |
|---|---|---|---|---|
| HCN | 4.00 | – | 110.1 | −125.0 |
| HCN–B(C6F5)3 | 6.19 | −14.7 | 103.8 | −188.8 |
| HCN–HCN–B(C6F5)3 | 4.02 | −10.9 | 106.9 | −128.1 |
| 6.42 | – | 106.9 | −192.2 |
[HCN]‒ radical anion
After a treatment of cyanide doped alkali halide crystals by γ-irradiation or UV photolysis at low temperatures between 21 and 77 K and followed by tempering to 100 K the [HCN]⋅− radical anion could be detected EPR-spectroscopically. The formation and decomposition of this [HCN]⋅− species has been investigated at different temperatures in detail (Adrian et al., 1969; Beuermann and Hausmann, 1967; Hausmann, 1966; Root et al., 1966). The radical anion could be detected also in potassium cyanide irradiated by UV light at 77 K and in the gas phase during mass spectrometric measurements of methyl and ethyl cyanides (von der Weid et al., 1979; Tsuda et al., 1971).
The stretching mode of the C–N bond is observed in IR spectroscopic measurements at 1758 cm−1 showing partial C≡N bond character (Pacansky et al., 1978). It corresponds to a much weaker C−H bond of lower energy than in the HCN. The EPR measurements support this explanation as they feature a quite high spin density value of ρH = 0.27 for the unpaired electron density on the hydrogen atom (ρC = 0.32 and ρN = 0.41) (Adrian et al., 1969). Furthermore, SCF computations of the orbital containing the unpaired electron indicate a significant hydrogen character and the bond angle decreases from linear structure of HCN to 122°–131° on forming the radical anion. In summary, the [HCN]⋅− radical anion is isoelectronic to the HCO⋅ radical showing quite similar properties (Pacansky et al., 1978; Pedersen, 1970) (Table 8).
Structural data of the [HCN]·− radical anion (bond lengths in Å; angles in °).
| Publication | Exp./calc. | r H−C | r C−N | < (H–C–N) |
|---|---|---|---|---|
| Douglas and Sharma (1953) | Exp. – IR | 1.153 | 1.066 | – |
| Adrian et al. (1969) | Exp. – EPR | – | – | 131 |
| Thomson (1970) | INDO | 1.23 | 1.18 | 133 |
| Pedersen (1970) | INDO | 1.256 | 1.164 | 129 |
| Pacansky et al. (1978) | SCF/CGTO | 1.233 | 1.122 | 121.7 |
[HCN]·+ cation
The formation of the cationic [HCN]⋅+ species is mostly observed during excitation in microwave discharge and in high-energy systems (Forney et al., 1992). The first ionization energy of hydrogen cyanide is determined to about 13.60 eV by photoionization and photoelectron spectroscopy (Dibeler and Liston, 1968; Fridh and Åsbrink, 1975). Furthermore, theoretical calculations indicate that the isocyanide cation species [HNC]⋅+ is more stable than the cyanide cation species [HCN]⋅+, which means a reversion of stability in contrast to the neutral hydrogen cyanide (Murrell and Derzi, 1980). The ground state of [HNC]⋅+ (2Σ+) is decreased by 75–125 kJ mol−1 compared to the ground state of [HCN]⋅+ (2Π) (Forney et al., 1992). G. Frenking and H. Schwarz suggest that the change of electron density causes a reversion of stability (Table 9) (Frenking and Schwarz, 1982). J. N. Murrell and A. A. Derzi suggested that the unpaired electron in [CN]⋅+ is predominantly on the nitrogen atom and hence they expected the 2Σ+ state resulting from interaction with the hydrogen to be more stable for the isomer [HNC]⋅+ as is found (Table 10) (Murrell and Derzi, 1980). Also kinetic studies on a mixture of hydrogen cyanide and hydrogen isocyanide cations indicate a difference of reactivity of both isomers (Petrie et al., 1990).
Total energy, energy of repulsion (ΔErep.) and total energy of electrons (ΔEelec.) of HCN, HNC and their cations (ΔE in kJ mol−1; HF/4-31G) (Frenking and Schwarz, 1982).
| Compound | ΔEtot. | ΔErep. | ΔEelec. |
|---|---|---|---|
| HCN | 0 | 0 | 433.2 |
| HNC | 39.8 | 473.9 | 0 |
| [HCN]⋅+ | 128.1 | 0 | 1702.4 |
| [HNC]⋅+ | 0 | 1574.2 | 0 |
Structural data of cationic hydrogen cyanide species (bond length in Å; calculations using the SCF/cc-pVDZ level of theory) (Murrell and Derzi, 1980).
| Compound | State | r A−B | r H−A |
|---|---|---|---|
| [HCN]⋅+ | 2Π | 1.190 | 1.091 |
| [HCN]⋅+ | 2Σ+ | 1.114 | 1.072 |
| [HNC]⋅+ | 2Π | 1.225 | 1.021 |
| [HNC]⋅+ | 2Σ+ | 1.116 | 1.013 |
[HCNH]+ cation and the [H(HCN) n ]+ cluster ion series
The [HCNH]+ cation, the first representative of the cationic cluster series [H(HCN) n ]+, has been observed in interstellar matter and is a significant part of the ionosphere of Titan, a moon of Saturn (Cravens et al., 2006; Fox and Yelle, 1997; Herbst and Klemperer, 1973; Keller et al., 1998). Because of the cation’s supposed key role in chemistry of interstellar matter, many studies are carried out to determine its structure, fundamentals and transition frequencies (Amano and Tanaka, 1986).
The first synthesis of the [HCNH]+ cation has been carried out 1968. NMR spectroscopic measurements could prove the formation of the cation in a mixture of fluorosulfuric acid HSO3F and antimony pentafluoride SbF5 (1:1) in a solution of HCN and SO2. The first IR measurement of the cation has been published by Altman et al. (1984a). Also in 1968 G. A. Olah and T. E. Kiovsky published another synthetic route of the cation starting with silver cyanide in a solution of HSO3F, SbF5 and SO2 also studied by NMR spectroscopy (Scheme 8, Table 11) (Olah and Kiovsky, 1968).
![Scheme 8:
Synthetic routes of [HCNH]+ (Olah and Kiovsky, 1968).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_008.jpg)
Synthetic routes of [HCNH]+ (Olah and Kiovsky, 1968).
Selected NMR data of [HCNH]+ (chemical shifts in ppm; coupling constants in Hz) (Olah and Kiovsky, 1968; Olah and White, 1969).
| Spectrum | δ | J |
|---|---|---|
| 1H NMR | −7.50 | 8.5 |
| 13C{1H} NMR | −95.7 | 320.0 |
The cation is discussed as possible precursor of HCN and HNC in the interstellar matter. The proposed dissociative recombination reaction leads to the normal hydrogen cyanide and metastable isocyanide in relation of 1:1 (Scheme 9) (Kalescky et al., 2013; Ngassam et al., 2005; Pearson and Schaefer, 1974; Talbi and Ellinger, 1998).

Possible reaction pathways to generate HCN and HNC (Kalescky et al., 2013; Ngassam et al., 2005; Pearson and Schaefer, 1974; Talbi and Ellinger, 1998).
But radio astronomic studies indicate a temperature depending ratio of both isomers and also the position of the isomers inside the molecular cloud is important (Irvine and Schloerb, 1984; Schilke et al., 1992). Further reactions influencing this relation are the equilibrium reaction and ion-neutral reactions, which also occur in interstellar matter (Conrad and Schaefer, 1978; Herbst, 1995; Talbi, 1999; Talbi and Herbst, 1998). Furthermore, the singlet [HCNH]+ in a low-lying vibrational state produces HCN whereas the triplet mostly leads to HNC caused by a smaller bond-dissociation energy of the C−H bond in contrast to the N−H bond in the exited state (Allen et al., 1980; Jursic, 1999). But it will require further studies to permit detailed information about the unexpectedly high ratio of HNC in interstellar matter.
In literature three different singlet isomers of [HCNH]+ are discussed (Figure 8). The linear form, which is the low-lying structure, is isoelectronic to acetylene and two three-bonded forms, in which [H2CN]+ is the energetic most unfavoured one with low isomerization barrier to the linear form (Table 12) (Allen et al., 1980; Conrad and Schaefer, 1978; Jursic, 1999).
![Figure 8:
Structural data of the [HCNH]+ isomers in ground state (bond length in Å; angles in °; DZ SCF method) (Conrad and Schaefer, 1978).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_008.jpg)
Structural data of the [HCNH]+ isomers in ground state (bond length in Å; angles in °; DZ SCF method) (Conrad and Schaefer, 1978).
Relative energies of the [HCNH]+ isomers (in kJ mol−1) calculated with different methods (Conrad and Schaefer, 1978).
| Compound | SCF/DZ | CI/DZ | SCF/DZ + P | CI/DZ + P |
|---|---|---|---|---|
| [HCNH]+ | 0.0 | 0.0 | 0.0 | 0.0 |
| [H2CN]+ | 292.7 | 239.9 | 273.8 | 299.8 |
| [H2NC]+ | 173.3 | 131.5 | 167.5 | 192.6 |
During a period of about 20 years spectroscopic data of the [HCNH]+ cation have been measured several times using different methods, which partially lead to big differences (Table 13).
Spectroscopic data of [HCNH]+ using different methods (wave numbers in cm−1).
| Publication | Exp./calc. | ν NH | ν CH | ν CN | δ HCN | δ HNC |
|---|---|---|---|---|---|---|
| Pearson and Schaefer (1974) | SCF/DZ | 4300 | 3507 | 2354 | 1023 | 922 |
| Lee and Schaefer (1984) | SCF/DZ + P | 3869 | 3501 | 2434 | 937 | 799 |
| CI/DZ + P | 3754 | 3423 | 2283 | 836 | 700 | |
| Altman et al. (1984a) (1984b) | Exp. – IR | 3483 | 3188 | – | – | – |
| DeFrees and McLean (1985) | MP2/6-31G* | 3675 | 3397 | 2164 | 822 | 659 |
| HF/6-31G* | 3465 | 3147 | 2177 | 866 | 699 | |
| Ho et al. (1987) | Exp. – IR | – | – | – | – | 646 |
| Peterson et al. (1995) | HF/4-31G | 3445 | 3176 | 2088 | 901 | 762 |
| HF/6-31G* | 3534 | 3213 | 2100 | 849 | 685 |
Moreover, experimental data of the [H(HCN) n ]+ (n = 1–4) aggregates in the gas phase were published by M. Moet-Ner utilizing a pulsed high-pressure mass spectrometric technique to determine thermochemistry data of these species (Meot-Ner, 1978).
The [CN(HCN) n ]− cluster ion series
M. Moet-Ner, S. Scheiner, and J. F. Liebman reported on thermochemical measurements and computations of HCN-cyanide-aggregates, [CN(HCN) n ]–, in the gas phase utilizing a pulsed high-pressure mass spectrometric technique (Meot-Ner et al., 1988). It was found that the aggregate series [CN(HCN) n ]– (n = 1–7) is formed exothermically for each HCN addition step (ΔH° between −20.7 and −7.6 kcal mol−1). The hydrogen bond energy in [CN(HCN)]– was also determined from ion cyclotron resonance [CN]–/HCN-exchange equilibria measurements (Larson and McMahon, 1984).
The synthesis of salts bearing the [CN(HCN) n ]– cluster ions (Bläsing et al., 2020), was also attempted. For this purpose, pure cyanides of the type [WCC]CN ([WCC]+ = weakly coordinating cation = [Et3NMe]+, [ n Pr3NMe]+, [Ph3PMe]+) (Price et al., 2009), were treated with an excess of liquid HCN (generated from NaCN and stearic acid (TM = 342 K) at 353–373 K under vacuum) (Bläsing et al., 2018; Günther et al., 1935; Labbow et al., 2016). However, this synthetic approach did not work since almost instantaneous polymerization was observed affording a deep brown highly viscous oil. To avoid fast polymerization, [Ph4P]CN and [Ph3PNPPh3]CN (= [PNP]CN), containing bulkier and more symmetrical cations, were used. Indeed, this reaction of [Ph4P]CN/[PNP]CN with liquid HCN (at low temperatures of 263–273 K) finally yielded crystals. X-ray studies revealed unequivocally the presence of hydrogen-bridged linear [CN(HCN)2]– ions in case of the [Ph4P]+ salt, while Y-shaped [CN(HCN)3]– anions were found with the [PNP]+ as counterion (Figures 9 and 10) (Arlt et al., 2021).
![Figure 9:
Ball-and-stick (anions) and wireframe (cations) representations of the molecular structure of [Ph4P][CN(HCN)2] in the crystal. View along [010]. The linear molecular anion is formed by two hydrogen bridges (a and b). A chain is formed when short contacts (c) are considered. A section of such a chain is shown (Pézolet and Savoie, 1969).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_009.jpg)
Ball-and-stick (anions) and wireframe (cations) representations of the molecular structure of [Ph4P][CN(HCN)2] in the crystal. View along [010]. The linear molecular anion is formed by two hydrogen bridges (a and b). A chain is formed when short contacts (c) are considered. A section of such a chain is shown (Pézolet and Savoie, 1969).
![Figure 10:
Ball-and-stick representation of the [CN(HCN)3]– ion in a crystal of [PNP][CN(HCN)3]. The Y-shaped molecular anion is formed by three hydrogen bridges (a–c and d–f). A chain is formed when short contacts (g–j) are considered. A section of such a chain is shown, while the cations are omitted for clarity (Arlt et al., 2021).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_010.jpg)
Ball-and-stick representation of the [CN(HCN)3]– ion in a crystal of [PNP][CN(HCN)3]. The Y-shaped molecular anion is formed by three hydrogen bridges (a–c and d–f). A chain is formed when short contacts (g–j) are considered. A section of such a chain is shown, while the cations are omitted for clarity (Arlt et al., 2021).
The stepwise formation of the HCN solvates [CN(HCN) n ]– were computed to be exothermic and exergonic for n = 1–3, however, the larger n the values for Δ n H°298/Δ n G°298 become less negative (Δ n H°298 = −23.6, −18.7, and −13.0 kcal mol−1; Δ n G°298 = −15.9, −11.1, and −7.0 kcal mol−1; cf. from mass spectroscopy: Δ 1 H°298 = −20.1 ± 1.6 (Chacko et al., 2006) and −20.7 kcal mol−1, as well as Δ 2 H°298 = −16.4 kcal mol−1 and Δ 3 H°298 = −12.6 kcal mol−1) (Meot-Ner et al., 1988).
Owing to the hydrogen bonding within the cluster anions, these species may be regarded as fragments of the solid structure of HCN upon (partial) deprotonation. In accord with computations, linear [CN(HCN)2]– can be regarded as a cyanide ion stabilized by two hydrogen-bonded HCN molecules, while Y-shaped [CN(HCN)3]– is energetically slightly preferred over an analogous linear isomer. There is always only little change of stability when [CN]– and HCN do not form a linear complex, indicating a highly dynamic system at ambient temperatures as proven by computed rather flat rotational potentials either within a linear or Y-shaped HCN framework (Figure 11). Moreover, pseudohalide HCN aggregate ions of the type [N3(HCN)3]−, [OCN(HCN)3]−, [SCN(HCN)2]− and [P(CN·HCN)2]− with similar strong hydrogen bonds and structural motifs have also been isolated and fully characterized (Arlt et al., 2021; Bläsing et al., 2020; Harloff et al., 2020b).
![Figure 11:
Rotational potential for the rotation of a [CN]– ion within the frame of two HCN molecules (Pézolet and Savoie, 1969).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_011.jpg)
Rotational potential for the rotation of a [CN]– ion within the frame of two HCN molecules (Pézolet and Savoie, 1969).
1st Main group – alkali cyanides
The alkali cyanides are extensively investigated especially focusing on the compounds of lithium, potassium and sodium. All alkali cyanides are well water-soluble, sodium cyanide is also soluble in alcoholic solutions. Sodium and potassium cyanide can be received by evaporating their aqueous solution, whereas the synthesis of the lithium, rubidium and cesium compound is more difficult due to their fast decomposition on air. Important properties are listed in Table 14.
Some substance-specific properties of alkali cyanides (temperatures in K, densities in g cm−3).
| Compound | T M | T B | ρ at 298 K | Relative hygroscopicity (DeLong and Rosenberger, 1986) |
|---|---|---|---|---|
| HCN | 259.8 (Henderson and Cullinan, 2007) | 298.8 (Henderson and Cullinan, 2007) | 0.69* (Williams, 1948) | Miscible with water |
| LiCN | 433 (Lely and Bijvoet, 1942) | – | 1.08 (DeLong and Rosenberger, 1986) | Extreme |
| NaCN | 837(1) (DeLong and Rosenberger, 1986) | 1773(10) (Ingold, 1923) | 1.60 (DeLong and Rosenberger, 1986) | Considerable |
| KCN | 907.7 (DeLong and Rosenberger, 1986) | – | 1.52 (DeLong and Rosenberger, 1986) | Little |
| RbCN | 872.2 (Kondo et al., 1979) | – | 2.32 (DeLong and Rosenberger, 1986) | Little |
| CsCN | 801(2) (DeLong and Rosenberger, 1986) | – | 3.41 (DeLong and Rosenberger, 1986) | High |
A review article of A. Rubo et al. also provides further detailed information of properties and also production, uses, e.g. gold mining or electroplating and hardening of metals, or economic aspects (Rubo et al., 2006).
The alkali cyanides are one of the smallest ionic systems having strong rotational-translation coupling in solid phases (Klein et al., 1981). The crystals of alkali cyanides combine properties of ionic and also molecular crystals, so called ionic-molecular crystals, because of the pseudohalogen properties of the cyanide anion (Henriques and von der Weid, 1985). So alkali cyanides are typical alkaline pseudohalogen compounds and their optical, structural and thermic properties are effected by the polyatomic character of the anion. Furthermore, under exclusion from air these compounds are stable against gentle heating and do not decompose.
The spectroscopic data of the alkali cyanides are quite similar, only the ν1 and ν2 frequencies are showing bigger differences in relation to the size of the cation, which pushes the band to smaller energies (Tables 15 and 16) (do Carmo et al., 1981). A. Loupy and J. Corset also reported the independence of the stretching mode ν1 of LiCN, NaCN and KCN from the concentration of the salt. Only the intensities are changing. If the concentration of the salt decreases, the band at lower frequencies increases and that one of higher frequencies declines.
Raman data of different alkali cyanides (wave numbers in cm−1).
| Compound | ν CN | Comments |
|---|---|---|
| LiCN (Loupy and Corset, 1976) | 2079 | In aqueous solution |
| NaCN | 2079 (Loupy and Corset, 1976) | In aqueous solution |
| 2086.7 ± 2.7 (Glockler and Baker, 1942) | – | |
| KCN (Loupy and Corset, 1976) | 2079 | In aqueous solution |
IR data of different alkali cyanides in varying media (wave numbers in cm−1).
| Compound | ν CN | ν 2 CN | ν MC | Comments |
|---|---|---|---|---|
| LiCN | 2080.4 (Ismail et al., 1972) | – | 680.5 (Ismail et al., 1972) | In neon matrix |
| 2084.5 (Ismail et al., 1972) | – | 646.4 (Ismail et al., 1972) | In argon matrix | |
| 2079 (Loupy and Corset, 1976) | – | – | In aqueous solution | |
| 2070, 2086, 2100, 2116 (Loupy and Corset, 1976) | – | – | In DMF | |
| NaCN | 2080 (Miller and Wilkins, 1952) | 3330 (Miller and Wilkins, 1952) | – | – |
| 2090 (do Carmo et al., 1981) | 4156 (do Carmo et al., 1981) | – | – | |
| 2060, 2076 (Loupy and Corset, 1976) | – | – | In DMF | |
| 2060, 2076 (Loupy and Corset, 1976) | – | – | In DMSO | |
| KCN | 2077 (do Carmo et al., 1981) | 4131 (do Carmo et al., 1981) | – | – |
| 2079 (Loupy and Corset, 1976) | – | – | In aqueous solution | |
| 2054, 2064, 2070 (Loupy and Corset, 1976) | – | – | In DMSO | |
| RbCN (do Carmo et al., 1981) | 2071 | 4121 | – | – |
| CsCN (do Carmo et al., 1981) | 2063 | 4103 | – | – |
Synthesis
Until the 1960s, the Castner–Kellner process was the common synthetic route of NaCN using sodium, which was heated by charcoal in an atmosphere of ammonia. The received sodium amide reacts with the charcoal to sodium cyanamide. After a second reaction with one equivalent of charcoal sodium cyanide is obtained (Scheme 10) (Derry and Williams, 1993).

Castner–Kellner process to synthesize sodium cyanide (Derry and Williams, 1993).
The quite similar Beilby process was often used to synthesize potassium cyanide, but nowadays this synthetic route has no importance (Scheme 11) (Martin and Barbour, 1915).

Beilby process to synthesize potassium cyanide (Martin and Barbour, 1915).
The most common synthesis of alkali cyanides is the acid base neutralization reaction using hydrogen cyanide and an aqueous solution of alkaline hydroxide (Scheme 12) with yields as high as 98–99% (Guernsey and Sherman, 1926; Meyer, 1921; Reifsig, 1863; Sugisaki et al., 1968).

Neutralization reaction to synthesize alkali cyanides (M = Li+, Na+, K+, Rb+, Cs+) (Guernsey and Sherman, 1926; Meyer, 1921; Reifsig, 1863; Sugisaki et al., 1968).
Because of a fast hydrolysis of alkali cyanides, they are often used as a solution of alcohol and small amounts of water. Especially, the lithium and cesium cyanide are very hygroscopic, which leads to less efficient synthesis of LiCN starting with LiOH and necessitate another synthetic approach. In this context, it is also possible to synthesize most of the alkaline cyanides using dried hydrogen cyanide and the pure alkali metal, which is dissolved in ethanol, ammonia or other non-aqueous solvents (Scheme 13) (DeLong and Rosenberger, 1986; Meyer, 1921). The purification of the salts is often carried out by recrystallization. The solids are suspended in liquid ammonia or ethanol to avoid hydrolysis and less soluble carbonates and cyanates are removed by filtration (Thompson, 1931). Vacuum distillation is reported to be inefficient (Hackspill and Grandadam, 1925).

Synthesis of alkali cyanides using dried HCN and the alkali metal (M = Li+, Na+, K+, Cs+) (DeLong and Rosenberger, 1986; Meyer, 1921).
T. J. Neubert and S. Susman reported another process of purification of NaCN and KCN. After melting the solid in vacuum and increasing the temperature from ambient temperature to 473 K and then to 938 K the alkaline cyanide is zone refined and also filtrated using a frit made of quartz. Single crystals containing 10 ppm Na and very small quantities of [OCN]− can be obtained and are suitable for polarized-light experiments (Neubert and Susman, 1964). LiCN can also be purified by recrystallization in THF. The solvent is removed in vacuum, but one mol percent remains (Rossmanith, 1965). In 1963 I. B. Johns and H. R. DiPietro carried out a reaction of liquid, anhydrous hydrogen cyanide and n-butyllithium in n-hexane under nitrogen atmosphere. The LiCN precipitates immediately in quantitative yield (Scheme 14) (Johns and DiPietro, 1964).

Synthesis of LiCN by Johns and DiPietro (1964).
The decomposition of hydrogen cyanide is one of the main causes of impurities. Several synthetic routes avoiding the use of HCN are published. One of the first is the reaction of alkali metals and silver cyanide in THF and in presence of naphthalene (Scheme 15). The LiCN also crystallizes as its colorless solvate, which is dried in vacuo, and is obtained in yields of 79.6% (Rossmanith, 1965).

Synthesis of LiCN without HCN (Rossmanith, 1965).
In 1997 T. J. Markley et al. reported a metathesis reaction of lithium chloride and sodium cyanide at ambient temperature (Scheme 16). The product could be obtained in a yield of 94.9% after a reaction time of four days. Single crystals were obtained using a mixture of DMF and DMAc (2:1). The solvent can not be removed completely, so the crystals still contain 0.6 mol DMF and 0.4 mol DMAc per equivalent of LiCN (Markley et al., 1997).

Synthesis of LiCN without HCN by Markley et al. (1997).
During researches on the synthesis of Ca(CN)2 in 1912, H. Sulzer carried out a reaction of calcium cyanamide and charcoal, which required high temperatures to melt the mixture minimizing the yield of the reaction because of the preferred reverse reaction. By the addition of fluxing agents, e.g. NaCl or Na2CO3, the reaction temperature could be decreased and H. Sulzer also observed a reaction of the cyanamide and alkali metal salts (Scheme 17) (Sulzer, 1912).

Syntheses of NaCN using CaCN2 as starting material (Sulzer, 1912).
The ratio of anhydrous calcium cyanamide, charcoal and sodium chloride is optimized to 10:2.0–2.5:18–20 and the reaction is carried out in an iron crucible with lid to avoid air. In the case of sodium carbonate, its ratio is decreased to 7.5–9.0 and because of the higher melting point the reaction temperature has to be increased. But this reaction is preferred due to improved separation of alkali cyanide and insoluble alkaline earth carbonates in the melt. In contrast, the use of NaCl leads to soluble alkaline earth chlorides, which can not be separated easily (Sulzer, 1912).
Hydrolysis of alkali cyanides
Hydrolysis of basic alkali metal cyanide begins with dissociation upon addition of water molecules. The autoprotolysis of water provides protons to form hydrogen cyanide. The cyanide ion acts here as a base which grabs the proton. The formation of alkali formate and ammonia is also possible due to hydrolysis of alkali cyanides, while the increasing concentration of ammonia enhances the hydroxide concentration (Scheme 18).

Possible reactions due to hydrolysis of alkali cyanides (M = Na+, K+) (Hinks et al., 1972).
Both types of reaction are prevalently observed at temperatures of more than 288 K, but the formation of hydrogen cyanide dominates at temperatures under 323 K. Higher temperatures of more than 353 K abet the generation of alkaline formate. In the subsequent step, the formed formates can also react with alkali hydroxides to form carbonates (Scheme 19).

Formation of carbonates (Hinks et al., 1972).
Furthermore, cyanates and carbon will be formed, if the cyanides are heated to their melting point. This process is facilitated by an oxygen atmosphere, but an inert atmosphere can not prevent this reaction because the formation of carbonates also permits the generation of cyanates (Scheme 20) (Hinks et al., 1972).

Formation of cyanates due to decomposition of cyanides (Hinks et al., 1972).
Crystal structures
The alkali metal cyanides correspond to the general composition MXY (M = cation of Na, K, Rb; XY = diatomic anion) and are characterized by the pseudocubic NaCl-type structure just below the melting point. They show similar properties as the alkaline halides, e.g. orientational disorder or rapid rotation of the linear [CN]– anion, because of the pseudohalide character of the cyanide anion (see 1.2 Cyanide, a classic pseudohalide) (Atoji, 1971; Kondo et al., 1979).
Lithium cyanide is an exception, since a NaCl-like structure could not be observed, although it should be theoretically possible, and the structures of cesium cyanide are also divergent. However, the difficulty in forming these cyanide compounds makes it difficult to study their crystal structures, resulting in only a few publications that deal with solid state structural studies on these salts. Structural data of various cyanide compounds are listed in Table 17. Further information on crystal data of cyanide compounds and their modifications are given in Table 18.
Structural data of alkali metal cyanides.
Crystallographic data of lithium, sodium, rubidium cyanide and different modifications of potassium cyanide (temperature in K, pressure in MPa, lengths in Å; angles in °; volumes in Å3).
| Parameter | LiCN (Lely and Bijvoet, 1942) orthorhombic | NaCN (Swanson and Tatge, 1953) cubic | NaCN (Swanson and Tatge, 1953) orthorhombic | NaCN (Fontaine, 1975) orthorhombic | NaCN·2H2O (Le Bihan, 1958) monoclinic | RbCN (Yoshimura et al., 1996)cubic | RbCN (Yoshimura et al., 1996) monoclinic | RbCN (Yoshimura et al., 1996)triclinic | RbCN (Rowe et al., 1984) monoclinic |
|---|---|---|---|---|---|---|---|---|---|
| T | – | 299 | 279.6 | 171.9 | – | 295 | 85 | 85 | 4 |
| Space group | Pmcn | P23 | Imm | Pmmn | P2 1 /a | Fm3m | A2/m |
|
Cc |
| a | 3.73 | 5.893 | 3.774 | 3.63 | 6.54(1) | 6.83(1) | 4.81(1) | 9.79(1) | 7.800(1) |
| b | 6.52 | 5.893 | 4.719 | 5.45 | 10.66(1) | 6.83(1) | 4.87(1) | 4.62(1) | 4.876(1) |
| c | 8.73 | 5.893 | 5.640 | 4.85 | 6.08(1) | 6.83(1) | 7.92(1) | 4.88(1) | 9.527(1) |
| α | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 121.1(1) | 90.00 |
| β | 90.00 | 90.00 | 90.00 | 90.00 | 103.0(3) | 90.00 | 122.7(1) | 119.4(1) | 122.25(1) |
| γ | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 90.0(1) | 90.00 |
| V | 212.31 | 204.65 | 100.45 | 95.95 | 413.01 | 318.6 | 156.1 | 154.9 | 306.44 |
| Z | 4 | 4 | 2 | 2 | 4 | 4 | 2 | 2 | 4 |
| Parameter | KCN (Rowe et al., 1983) monoclinic | KCN (Yoshimura, 1989)cubic | KCN[a] (Pople et al., 1978) monoclinic | KCN (Pople et al., 1978) orthorhombic | KCN (Stokes et al., 1993) monoclinic | KCN (Stokes et al., 1993) triclinic | KCN (Decker et al., 1974) monoclinic | KCN (Decker et al., 1974) cubic |
|---|---|---|---|---|---|---|---|---|
| T | 170 | 298 | 163 | 163 | 150 | 110 | 298 | 347 |
| p | 0.101 | 0.101 | 0.101 | 0.101 | 365 | 365 | 2500 | 2200 |
| Space group | Cc | Fm3m | A2/m | Immm | C2/c |
|
Cm | Pm3m |
| a | 7.96(1) | 6.54(1) | 4.61(1) | 4.23(1) | 7.441(1) | 15.882(1) | 5.531(2) | 3.808(3) |
| b | 4.59(1) | 6.54(1) | 4.58(1) | 5.24(1) | 4.595(1) | 4.493(1) | 5.209(1) | 3.808(3) |
| c | 9.18(1) | 6.54(1) | 7.58(1) | 6.15(1) | 9.115(1) | 9.476(1) | 3.743(1) | 3.808(3) |
| α | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 120.55(1) | 90.00 | 90.00 |
| β | 125.3(1) | 90.00 | 122.2(1) | 90.00 | 122.14(1) | 125.25(1) | 85.58 | 90.00 |
| γ | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 | 28.37(3) | 90.00 | 90.00 |
| V | 273.74 | 279.73 | 135.43 | 136.32 | 263.89 | 262.39 | 107.52 | 55.22 |
| Z | 4 | 4 | 2 | 2 | 4 | 4 | 2 | 1 |
-
[a]Metastable compound.
Crystal structure of lithium cyanide
The crystal structure of LiCN has to be discussed separately because its orthorhombic structure (cell dimensions: a = 3.73 Å; b = 6.52 Å; c = 8.73 Å; space group Pmcn) (Lely and Bijvoet, 1942) at ambient temperature and normal pressure is not comparable to the structures of other alkaline cyanides. The unit cell of LiCN contains four molecules. The electron densities of each atom is determined to Li 2.2, C 5.9 and N 7.9 which were derived from the electron map, so the lithium atom is ionized as expected and its missing electron is mainly located to the nitrogen atom (Lely and Bijvoet, 1942). In contrast to the eightfold coordination of CsCN and sixfold coordination of NaCN, KCN and RbCN, the lithium atom is fourfold coordinated (Bijvoet and Lely, 2010; Lely and Bijvoet, 1942; Verweel and Bijvoet, 1939). In detail, one carbon atom and three nitrogen atoms, which are located in the corners of an irregular tetrahedron, surround the lithium atom. J. A. Lely and J. M. Bijvoet report that the transition of the coordination can be described by the different sizes of the involved ions. Furthermore, the special polarizability of the anion due to the lithium ion is also important to the asymmetric surrounding of the cyanide group. The weak coordination of the lithium atom and the asymmetric surrounding also cause holes and channels leading to a small density and an unusual low melting point (Table 14) (Greenwood et al., 1988; Lely and Bijvoet, 1942).
Similarities of crystal structures of MCN (M = Na+, K+, Rb+)
At ambient temperature and normal pressure the crystal structure of sodium, potassium and rubidium cyanide is similar to the pseudocubic unit cell of the NaCl-type structure (type I). The position of the aspherical cyanide ions on the positions of the chloride is described by two theories. The dynamic description was first published by P. A. Cooper, supported by quantum mechanical studies of L. Pauling in 1930 and is predicated on free rotation of the cyanide ions (Pauling, 1930). These ions possess no orientation on their lattice site and because of their high molecular movement they are characterized as a spherical rotor with radius of about r = 1.93 Å (Camargo and von der Weid, 1982; Morris, 1961; Sequeira, 1965). The cyanide anion is consequently very similar to the bromide corresponding to many shared properties of alkali metal halides and alkali metal cyanides. But due to the elliptic shape of the cyanide ion, the influence of temperature and pressure to the rotation of the cyanide ion is stronger and the phase diagram is consequently more complex than those of alkali metal bromides (Heckathorn et al., 1999).
The second model of description was established by R. M. Bozorth in 1922 and further discussed by J. Frenkel in 1935 (Bozorth, 1922; Sequeira, 1965). According to this theory, the cyanide anions are statistically spread on the (111)-axis of the lattice. Therefore, free rotation of the [CN]– seems to be unrealistic due to steric considerations (Durand et al., 1980; Isetti and Neubert, 1957; Yang and Luty, 1989).
In spite of several X-ray structure analyses, neutron diffraction experiments, thermodynamic studies, Raman spectra and NMR spectroscopic measurements a final correlation of the crystal structures to the different models of description is not possible (Atoji, 1971; Bijvoet and Lely, 2010; Coogan and Gutowsky, 1964; Elliott and Hastings, 1961; Messer et al., 1941; Sequeira, 1965; Shimada et al., 1986; Suga et al., 1965; Sugisaki et al., 1968; Verweel and Bijvoet, 1939). M. Atoji carried out several X-ray structure analyses and reported that the cyanide anions are spread on the (111)-axis at temperatures directly above the transition temperature according to the model of J. Frenkel. The deflection turns into the (110)-plane and then into the (100) by increasing temperatures. At the melting point of the compound free rotation is observed, which corresponds to the Pauling model (Atoji, 1971).
The cyanide ion of sodium and potassium cyanide reaches its energetical most favored position on the (100)-axis. Computational studies also indicate a decrease of energy differences as a result of increasing size of the cation, e.g. the energy difference between the [110] and [110] alignment: 22.6 kJ mol−1 (NaCN), 10.5 kJ mol−1 (KCN) and 5.9 kJ mol−1 (RbCN) (LeSar and Gordon, 1982). Theoretical studies of M. Ferrario et al. support the assumption that the degree of [111] orientation raises in correlation to increasing cation radii (Ferrario et al., 1986).
A first order phase transition is observed at temperatures of 288 K (NaCN), 168 K (KCN) and 132 K (RbCN) under normal pressure depending on the size of the cation (Bijvoet and Lely, 2010; Kondo et al., 1979; Shimada et al., 1986; Suga et al., 1965). The cyanides of sodium and potassium are showing [110] orientation in this phase and their dipole moments are still disordered corresponding to changes of entropy during measurements of heat capacities (Matsuo et al., 1968; Suga et al., 1965; Sugisaki et al., 1968). The structural change causes a contraction of one side converting the cubic cell into an orthorhombic cell and ferroelastic order (type A, space group Immm) (Suga et al., 1965; Yang and Luty, 1989).
X-ray structure analysis of RbCN indicates a [111] orientation of the cyanide anions in a monoclinic structure and antiferroelastic order (Bourson et al., 1992; Kondo et al., 1979; Parry, 1962). This structure is also reported in studies of KCN, but in KCN it requires higher pressures or addition of alkali metal halides (Dultz and Krause, 1978; Dultz et al., 1981; Yang and Luty, 1989). In contrast, theoretical studies of R. Le Sar and R. G. Gordon indicates that an orthorhombic structure is energetically preferred by 11.7 kJ mol−1. But due to this small energy difference they do not negate a higher stability of the monoclinic cell at certain temperatures (LeSar and Gordon, 1982). It was shown that a cycle of several heating and cooling of KCN led to a special phenomenon. During such a cycle this compound also shows a monoclinic phase just below the phase transition temperature. Furthermore, this phase occurs in pure form at higher pressures and is similar to the structure of RbCN. But the monoclinic phase can be observed only during the cooling process and not in the heating step (Ortiz-Lopez and Luty, 1988; Parry, 1962; Schmidt et al., 1992). Regarding the first phase transition of NaCN, KCN and RbCN, the phase transition temperature shows linear correlation to the cation size (Scheme 21).

Temperature of first order phase transition of alkali metal cyanides as a function of cation size (representation taken from Kondo et al., 1979).
This underlines the similarity of alkali metal cyanides and halides, which are also characterized by strong correlation of crystalline structure and ratio of ion radii rcation: ranion (Biltz, 1935; Kondo et al., 1979). An increase of this coefficient correlates to an enlarged coordination sphere of an eightfold coordination instead of four in both types of alkali metal salts. NaCN and KCN also undergo a second order phase transformation under normal pressure at 172 K (NaCN) respectively 83 K (KCN) (Table 18). This antiferroelastic structure (type B) is characterized by ordered dipoles of cyanide anions and space group Pmmn (Kondo et al., 1979; Rowe et al., 1977a, 1977b; Suga et al., 1965; Wasylishen and Jeffrey, 1983). The phase transition temperatures are not determined completely, so the correlation to the cation radius is uncertain.
The above Scheme 21 also illustrates the missing disordered structure of LiCN. This structure probably occurs at temperatures of about 400 K expecting a continuously linear correlation. But this temperature is very close to the melting point of about 433 K (Table 14), which is completely different from the melting point of other alkali metal cyanides. But this value perfectly fits to the correlation of the second order phase transition of NaCN and KCN (Kondo et al., 1979; Lely and Bijvoet, 1942). In this context Y. Kondo et al. mentioned that the rotation of the cyanide anions of NaCN, KCN and RbCN during the order-disorder transition is similar to the rotation in liquid phase, but their translation resembles a pseudocubic solid structure. In LiCN, the rotation is similar to the sodium, potassium and rubidium compound, but due to strong coupling to the small lithium cation it also shows translational movement, which destabilizes the pseudocubic structure and leads to melting instead of a phase transition (Mokross and Pirc, 1978; Rehwald et al., 1977).
The changes of the crystal structures of NaCN and KCN are represented in Figure 12. The properties are proved by computational calculations, Raman spectra, NMR spectroscopic measurements and neutron diffraction analyses to study the coupling of translation and rotation (Coogan and Gutowsky, 1964; Decker et al., 1974; Durand et al., 1980; Ehrhardt et al., 1980; Elliott and Hastings, 1961; Kondo et al., 1979; Messer et al., 1941; Loidl et al., 1980a; Wasylishen and Jeffrey, 1983).

Crystal structures of NaCN and KCN in three different phases (representation taken from Buljan et al., 1997).
The neutron diffraction also indicates that the cyanide dipoles of the modification type B (low temperature structure) are ordered parallel in the a,c-plane and are described by alternative inversion in respect to the b-axis (Figure 12) (Rowe et al., 1977b). However, the determination of the carbon and nitrogen positions faces several challenges due to quite similar atomic form factors (in contrast to those of the alkali metal).
Measurements of the heat capacity at temperatures till 14 K and the missing Curie-Weiß behavior reveal no second phase transition of RbCN and CsCN at normal pressure and low temperatures leading to orientation of the cyanide dipoles, which is caused by a “frozen-in state” (Sugisaki et al., 1968). These low temperatures inhibit the collective electric dipole order of induced dipole interaction and fix the orientation of the cyanide groups (Kondo et al., 1979; Sugisaki et al., 1968).
Further solid phases of alkali metal cyanides
In addition to already described solid state phases further modifications of alkali metal cyanides appear as a function of temperature and pressure shown in the following phase diagrams. Especially, the phase diagram of KCN is very complex showing many different phases (Figure 13) (Stokes et al., 1993).

Phase diagram of KCN (p in GPa, T in K, representation taken from Stokes et al., 1993).
The potassium cyanide has a slightly distorted, monoclinic, ferroelectric structure, which is similar to the CsCl-type, with the space group Cm at ambient temperature and high pressures (phase IV) (Decker et al., 1974). The cyanide anions are limited in their rotation and the relative volume is decreased of about 10% causing an increase of the coordination sphere to an eightfold coordination of the cation (Strössner et al., 1985). An additional increase of temperature leads to the phase III characterized by a cubic unit cell and space group
The phase diagram of NaCN also reveals a high-pressure structure IVa, which is quite similar to phase IV of KCN but with space group Pm (Figure 14). The decrease of pressure also leads to an antiferroelectric ordered phase B. The characterization of the coexistent region D between the ferroelectric ordered phase IVa and the phase B is as difficult as in KCN (Strössner et al., 1985).
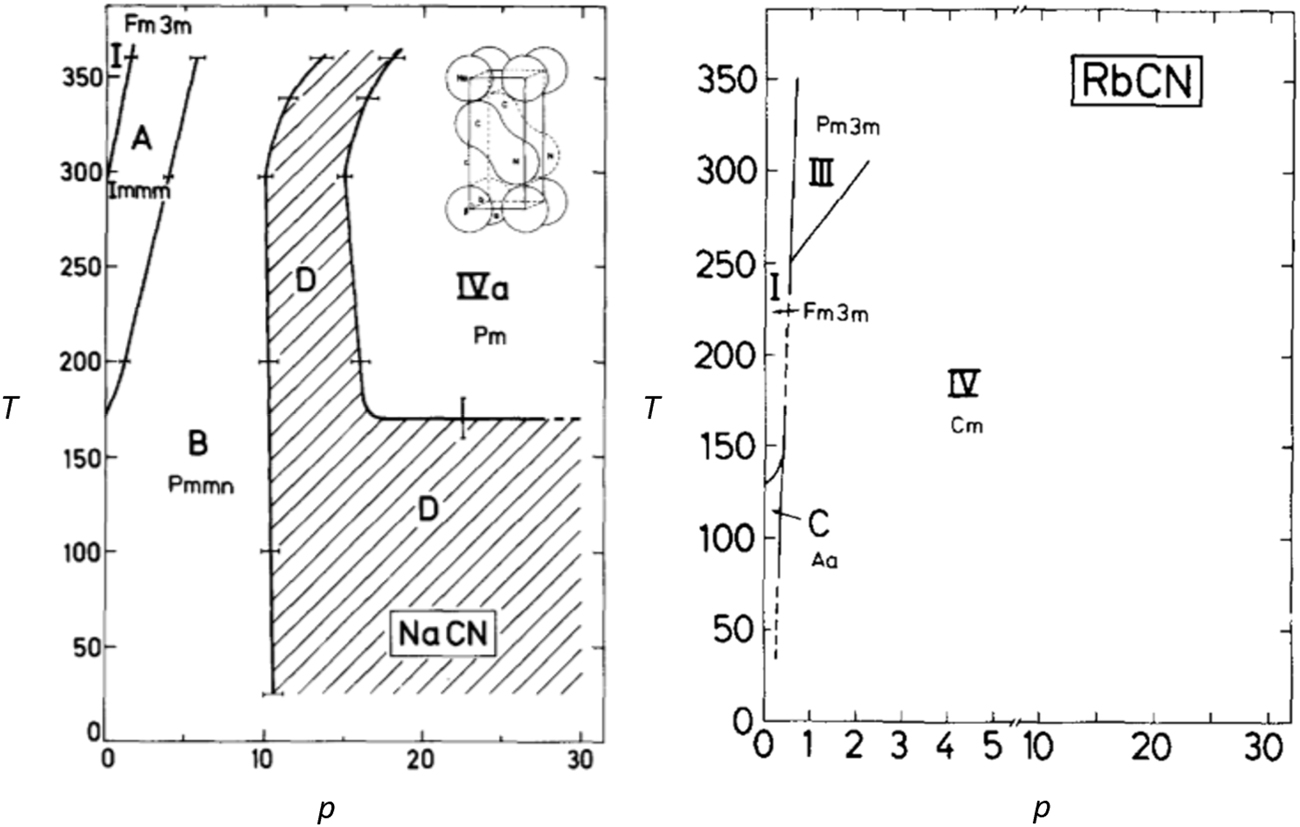
Phase diagram of NaCN and RbCN (T in K; p in GPa; representation taken from Strössner et al., 1985).
The increase of pressure leads to a further phase transition of RbCN from the pseudocubic NaCl structure (phase I) to phase III, which is similar to the CsCl-type, causing a decrease of the volume of about 10% to the eightfold coordination. At pressures of about 1.8 GPa a second phase transition takes place. The observed monoclinic phase IV is quite similar to that phase of KCN (Strössner et al., 1985).
Crystal structure of cesium cyanide
At ambient temperatures cesium cyanide crystallizes in the pseudocubic CsCl-type structure (phase III), which is only formed by other alkali metal cyanides at high pressures (Figure 15). The cyanide anions are orientationally disordered. At about 186 K a phase transition to the rhombohedral structure (phase VII) takes place and each unit cell contains one molecule (Daubert et al., 1976; Knopp et al., 1983; Sugisaki et al., 1968). The axes of the cyanide anions are aligned to the cubic (Borron et al., 2007) direction extending this axis (Loidl et al., 1980b, 1983; Sugisaki et al., 1968). The phase transition from III to VII is not connected to a decrease of the volume and the eightfold coordination is preserved (Strössner et al., 1985).

Phase diagram of CsCN (T in K, p in GPa, representation taken from Strössner et al., 1985).
In summary, the alkali metal cyanides have many similar crystalline phases as a function of temperature and pressure. The phase boundaries differ due to the increasing size of the cation, e.g. the phase boundary is moved to lower pressure by increasing radii. After all, W. Dultz and H. Krause and also K. Strössner et al. provided a phase diagram including all shared phases (Figure 16) (Dultz and Krause, 1978; Strössner et al., 1985).
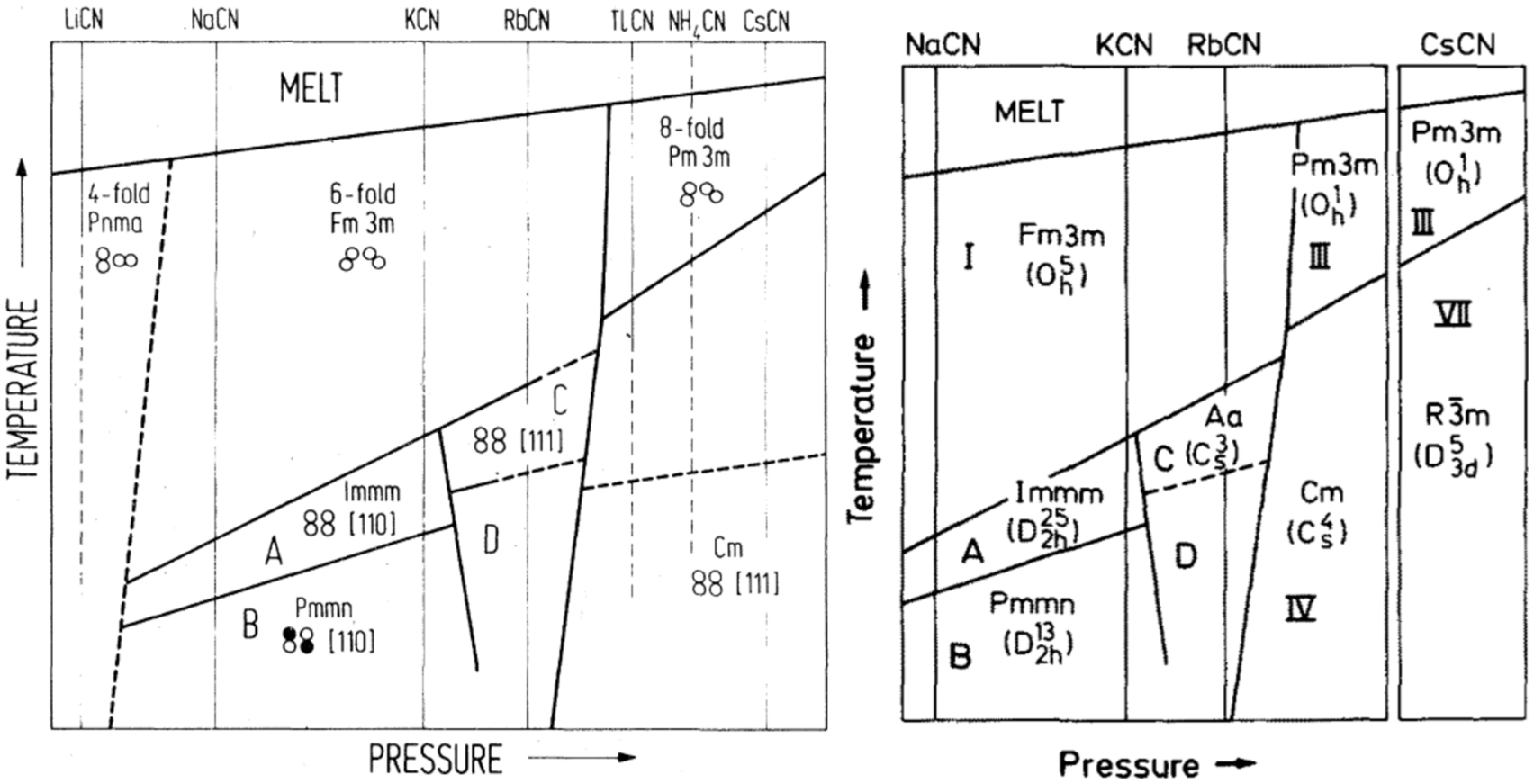
Phase diagrams of alkali metal cyanides (representations taken from Dultz and Krause, 1978; Strössner et al., 1985).
M–CN/M–NC-isomerization – gas phase structure
The alkali metal cyanides are among the best studied metal cyanides. Experimental data and ab initio calculations reveal consistently high ionic character and a low potential energy surface causing simple transformations of these compounds. Overall, three different isomers of metal cyanides are discussed: the linear cyanide structure M−CN, the linear isocyanide structure M−NC and a T-shaped form M[CN] (dihapto, η2). In contrast to the mono-coordinated (monohapto, η1) metal cation of the first two structures, the cation of the T-shaped structure features bi-coordination (Figure 17) (Lee et al., 2007). In the solid state, of course due to a higher coordination number, both M−CN as well as M−NC coordination are found, e.g. in an M–CN–M arrangement, and all isomers were studied in the gas phase.

Different isomers of alkali metal cyanides: Linear cyanide, linear isocyanide and T-shaped structure.
With the exception of LiCN, all other alkali metal cyanides feature the triangular η2-bonded isomer as the lowest lying species in the gas phase (Table 19) (Bak et al., 1970; Clementi et al., 1973; Essers et al., 1982; Lee et al., 2007; Makarewicz and Ha, 1995; Redmon et al., 1980; Schmiedekamp et al., 1980; van Vaals et al., 1983). In the triangular complexes, the M−N distance is much shorter than the M−C distance. The potential barrier connecting the triangular form and the linear cyanide form decreases along the series Li → Fr. As shown in Table 19, these results highly depend on the chosen methods and the basis sets, respectively (Bak et al., 1970; Clementi et al., 1973; Essers et al., 1982; Lee et al., 2007; Makarewicz and Ha, 1995; Redmon et al., 1980; Schmiedekamp et al., 1980; van Vaals et al., 1983). In the crystal structure the lithium favours the coordination to the nitrogen (ratio 3:1) supporting the higher stability of the isocyanide structure. Also experimental rotational and hyperfine spectra from J. J. van Vaals et al. and IR spectra from Z. K. Ismail et al. show the best agreement to the isocyanide structure (Ismail et al., 1972; van Vaals et al., 1983).
Computed relative energies of MCN isomers and their transition states (TS) (in kJ mol−1) (Lee et al., 2007).
| Atom | Method | MCN (linear) | TS1 | M[CN] (η2) | TS2 | MNC (linear) |
|---|---|---|---|---|---|---|
| Li | HF | 25.3 | 39.2 | – | – | 0 |
| CCSD | 10.4 | 22.9 | 0.4 | 3.4 | 0 | |
| CCSD(T) | 8.8 | 20.6 | 0.1 | 2.9 | 0 | |
| MRCI-1 | 4.5 | – | – | – | 0 | |
| MRCI-2 | 1.3 | – | 0 | – | 3.2[a] | |
| MP2-4 | 6.9 | – | 1.2 | – | 0 | |
| Na | HF | 16.8 | 22.5 | 0 | 2.5 | 2.2 |
| CCSD(T) | 10.1 | 15.8 | 0 | 11.2 | 11.1 | |
| MRCI | 6.7 | – | 0 | – | 11.5 | |
| K | HF | 21.6 | 22.7 | 0 | – | 4.1[a] |
| CCSD(T) | 18.3 | 19.3 | 0 | – | 13.0[a] | |
| Rb | HF | 21.6 | 22.2 | 0 | – | 22.2[a] |
| CCSD(T) | 18.3 | 18.8 | 0 | – | 12.5[a] | |
| Cs | HF | 22.0 | 22.1 | 0 | – | 3.9[a] |
| CCSD(T) | 18.6 | 18.8 | 0 | – | 11.3[a] | |
| Fr | HF | 21.4 | 21.5 | 0 | – | 4.1[a] |
| CCSD(T) | 18.5 | 18.5 | 0 | – | 11.9[a] |
-
[a]Saddle point.
Including the effects of correlation in calculations the energy difference of the cyanide and isocyanide structure decreases due to higher stabilisation of the cyanide by correlation (Redmon et al., 1980). While this long-range energy in regard to Coulomb’s law prefers the cyanide structure, the triangular structure is favoured by short range repulsion, which leads to the isocyanide structure as a compromise (Essers et al., 1982). The migration of the lithium cation from the carbon to the nitrogen end of the cyanide anion is correlated to a small energy barrier of about 10.6–14.3 kJ mol−1 due to high electron affinity of the cyanide anion and small ionization potential of the alkali metal leading to a ionic structure with dominating Coulomb force, which is isotropic and facilitates the shift of the lithium (Essers et al., 1982; Rao et al., 1996). This circling motion around the anion is also called “polytopic” (Rao et al., 1996). Due to low energy barriers, the isomerization of all alkali metal cyanides reveals polytopic character (van Vaals et al., 1984b).
In contrast P. v. R. Schleyer et al. prefer a T-shaped structure of LiCN as the most stable based on MP2/6-31G* computations (von Ragué Schleyer et al., 1986). Therefore, B. S. Jursic carried out several studies on suitable basis sets indicating that the T-shaped configuration is slightly more unstable than the isocyanide one, but more stable than the structure of lithium cyanide (Jursic, 1998). The experimental proof of the T-shaped structure is still missing, which could be caused by the small dipole moment of 7.3 D. It should also lead to a hindered detection using rotational and hyperfine spectra. In contrast, the dipole moment of lithium isocyanide is 8.9 D and the dipole moment of lithium cyanide is 9.8 D (von Ragué Schleyer et al., 1986).
In contrast to the lithium compound, the minimum of the T-shaped configuration of NaCN and KCN can be located easily on the potential energy surface due to strong exchange interactions and a negative charge, which is mainly located in an s-type orbital of the cyanide anion (Essers et al., 1982; Greetham and Ellis, 2000; Lee et al., 2007). But the relative energies of different alkali metal cyanide isomers and their transition states also confirm, that the isocyanide structures of potassium and heavier alkali metals are just transition states and that the M−CN bond energy decreases from Li to K (Table 19) (Lee et al., 2006; Wormer and Tennyson, 1981).
Furthermore, the correlation effect leads to an increase of the M−N and C−N bond lengths and a contraction of the M−C bond of triangular isomers (Table 20). On the other hand, it causes an increase of the C−N bond length and a decrease of the M−C bond length of linear MCN isomers (Table 21). One reason might be a decrease of steric repulsion from the nonbonding electron pair of the carbon atom due to the electron correlation (Lee et al., 2007).
Experimental and computational structural data of triangular (η2) M[CN] compounds (bond lengths in Å) (Lee et al., 2007).
| Atom | Method | r M−C | r M−N | r C−N |
|---|---|---|---|---|
| Li | CCSD(T) | 2.13 | 1.85 | 1.18 |
| MRCI-2 | 2.09 | 1.86 | 1.17 | |
| Exp.[a] (van Vaals et al., 1983) | – | 1.760 | 1.168 | |
| Na | CCSD(T) | 2.39 | 2.23 | 1.18 |
| MRCI | 2.72 | 2.26 | 1.17 | |
| Exp. (van Vaals et al., 1984a) | 2.38 | 2.23 | 1.17 | |
| K | CCSD(T) | 2.73 | 2.55 | 1.18 |
| MRCI | 2.86 | 2.59 | 1.17 | |
| Exp. (van Vaals et al., 1984b) | 2.72 | 2.55 | 1.17 | |
| Rb | CCSD(T) | 2.86 | 2.68 | 1.18 |
| Cs | CCSD(T) | 3.03 | 2.82 | 1.18 |
| Fr | CCSD(T) | 3.09 | 2.89 | 1.18 |
-
[a]Supposedly LiNC linear.
Computed structural data of linear (η1) MCN compounds (bond lengths in Å) (Lee et al., 2007).
| Atom | Method | r M−C | r C−N |
|---|---|---|---|
| Li | CCSD(T) | 1.92 | 1.17 |
| MRCI | 1.92 | 1.17 | |
| Na | CCSD(T) | 2.25 | 1.17 |
| MRCI | 2.91 | 1.17 | |
| K | CCSD(T) | 2.60 | 1.17 |
| Rb | CCSD(T) | 2.73 | 1.17 |
| Cs | CCSD(T) | 2.88 | 1.17 |
| Fr | CCSD(T) | 2.94 | 1.17 |
[M(CN)2]− anions
Super(pseudo)halogen anions [M(CN)2]− (M = Na+, Li+)
The term “superhalogen” was established by G. L. Gutsev and A. I. Boldyrev in 1981 and names complexes of the structure [MXk+1]− containing a metal M and electronegative fragments X of normal valence (Gutsev and Boldyrev, 1981). The index k indicates the normal valence of the metal atom, e.g. [BF4]− and [AlF4]−. The electron affinity of these complexes of about 5.0 eV is higher than the electron affinity of chlorine (Behera and Jena, 2012; Sidorov, 1977). Only recently, a superhalogen F@C20(CN)20 with electron affinity of 10.8 eV was designed (Kulsha and Sharapa, 2019). Usually, a superhalogen is a molecule with high electron affinity, which forms a stable anion, and have low proton affinities leading to superacids.
The lithium and sodium compounds of the structure [M(CN)2]− were studied by S. Smuczyńska and P. Skurski using ab initio methods (MP2/6-311 + G(d)). Their main goal was the utilization of pseudohalides to form novel superhalogen anions. In addition to the calculation of the dicyanide anion [M(CN)2]−, they compared this structure to the isocyanide form [M(NC)2]−. All compounds feature D∞h symmetry and the bond length of the carbon and nitrogen atoms are in a range of 1.188–1.192 Å. For all species, a bond between the metal atom and the cyanide respectively isocyanide group was found (Behera and Jena, 2012; Smuczyńska and Skurski, 2009).
Calculations of the vertical electron detachment energy (VDE) also reveal a stronger bond of the lithium to the isocyanide group [Li(NC)2]− (VDE = 7.434 eV) than to the cyanide group (VDE = 7.233 eV), which correlates to the bond length of 1.900 Å for the Li−N bond, respectively 2.043 Å for the Li−C bond. Although, the VDE of the sodium isomers are considerably smaller ([Na(CN)2]−: 7.090 eV, [Na(CN)2]−: 5.871 eV) they are also called superhalogen anions (Smuczyńska and Skurski, 2009).
To continue the studies of the superhalogens with cyanide ligands and their properties, S. Behera and P. Jena carried out calculations of the electron affinity as a function of the number of bonded cyanide ligands and neutral or single and double negative charged complexes, e.g. [Na(CN) n ]x− with n ≤ 3 and x ≤ 2. Focusing on neutral species, they report the NaNC to be the most stable one, while Na(CN)2 and Na(CN)3 tend to form cyanogen (CN)2 by dimerization of two cyanide anions (Figure 18). Due to the fact that the negative charge of superhalogens is mainly located at the electronegative ligand, they named sodium complexes of the type [Na(CN) n ] (with n = 2) super(pseudo)halogens (Behera and Jena, 2012).
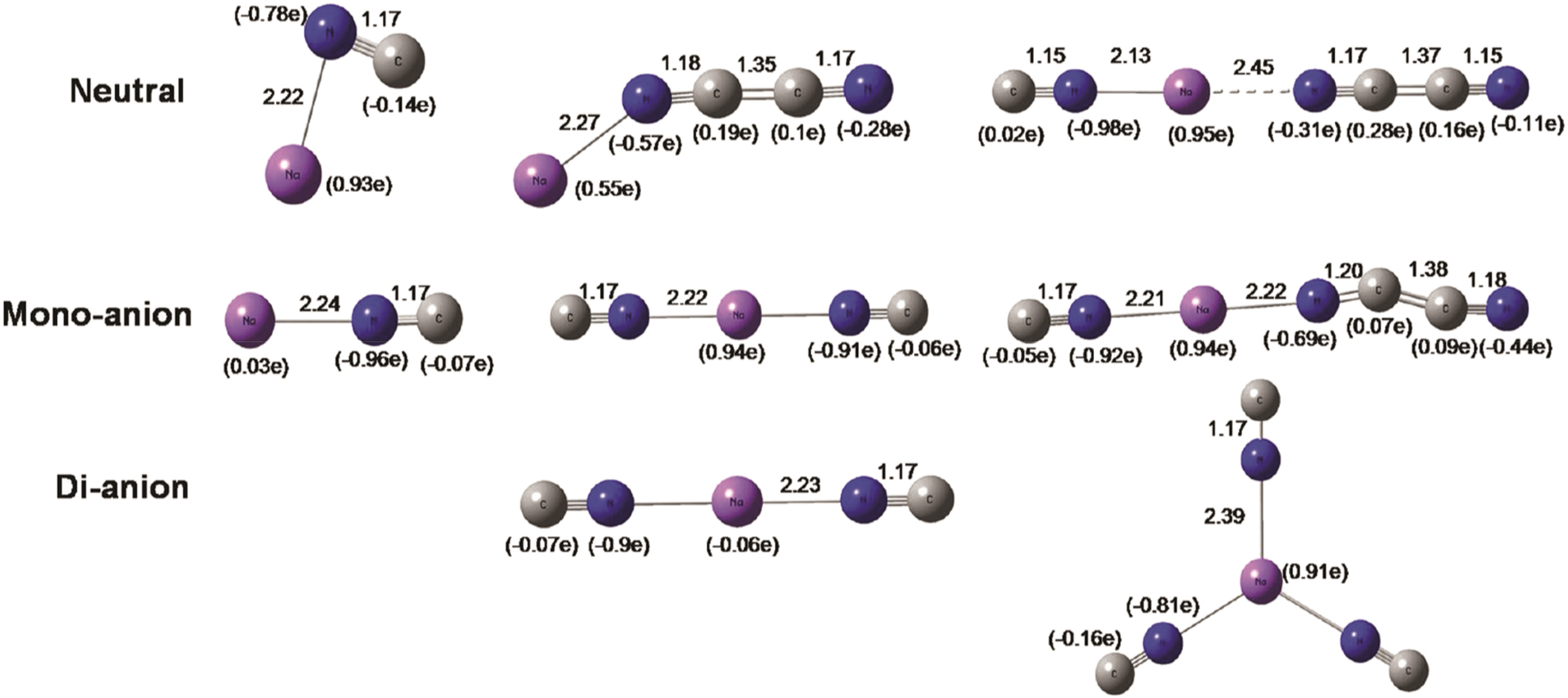
Optimized structures of neutral, single negative and double negative charged Na(CN) n /Na(NC) n complexes (B3LYP/6–311++G(3df); Na – purple; N – blue; C – grey; partial charges in parentheses; representation taken from Behera and Jena, 2012).
2nd Main group
For a long period of time the scientific interest mainly focussed on hydrogen cyanide and the well-known alkali metal cyanides. However, since the observation of alkaline earth cyanides in interstellar matter the increasing interest in these compounds caused several recent studies focusing on computations to analyse experimental spectra. Furthermore, the synthesis of pure alkaline earth cyanides is quite difficult compared to the alkaline mono cyanides, e.g. the crystallization of the barium and strontium compound using an aqueous solution is difficult and the isolation of the magnesium and calcium compound by re-crystallization from heated aqueous solutions is not possible. In contrast to alkali cyanides, which are part of several applications, the salts of alkaline earth cyanides tend to fast decomposition and formation of hydrogen cyanide due to displacement of the weak acid by carbon dioxide or weak organic acids from their solution (Williams, 1915). But they are mostly stable in presence of an inert gas atmosphere or ammonia solution (Franck and Freitag, 1926). As a consequence of these problems (decomposition and difficult isolation), the physical properties of these compounds are still mainly unknown. Only a very broad melting point of calcium cyanide Ca(CN)2 at about (913 ± 30) K by extrapolation and incipient decomposition at temperatures above 623 K are reported (Petersen and Franck, 1938). Moreover, also the number of reported IR spectroscopic data is highly limited (Table 22).
IR data of different alkaline earth dicyanides (wave numbers in cm−1).
| Compound | ν CN | ν MC |
|---|---|---|
| Be(CN)2 (Williams et al., 2001) | 2233 | 756 |
| Mg(CN)2 (Williams et al., 2001) | 2197 | 492 |
| Ca(CN)2 (Loupy and Corset, 1976) | 2068, 2084[a] | – |
| Ba(CN)2 | 2058, 2075, 2088[a] (Loupy and Corset, 1976) | 461, 610, 645 (Miller et al., 1960) |
-
[a]Measurement in DMF.
Alkaline earth monocyanides MCN
The first spectroscopic observation of monocyanides was reported by L. Pasternack and P. J. Dagdigian in 1976. Laser-induced fluorescence measurements of reactions of cyanogen bromide and metals M = Ca2+, Sr2+, and Ba2+ led to very broad bands, which were very similar to the fundamentals of monohalogens and could be allocated to the monocyanides (Pasternack and Dagdigian, 1976). In 1984, additional computations of MCN, where M = Be2+, Mg2+, Ca2+, and Ba2+, carried out by C. W. Bauschlicher and S. R. Langhoff, indicated that L. Pasternack and P. J. Dagdigian observed the isocyanide instead of the cyanide isomers. In contrast to the alkali cyanides, the alkaline earth cyanides mainly prefer a linear isocyanide structure in the 2Σ+ ground state lying 0.3 – 0.5 eV below the energy of the cyanide isomer, which enables a differentiation of both isomers (Table 23). The interconversion barrier from MNC to MCN decreases, e.g. the BeNC and MgNC have a barrier of 0.71 and 0.23 eV, respectively, CaCN and BaCN have no barrier at all, as a consequence of an increased ionic character of the compounds (Bauschlicher et al., 1985; Lanzisera and Andrews, 1997a; Mikhailov et al., 2003).
Computed energies (in eV) of alkaline earth monocyanide isomers (calculations at SCF/TZ2P) (Bauschlicher et al., 1985).
| Compound | MNC | [M(CN)] | MCN | Found |
|---|---|---|---|---|
| BeCN | 4.10 | 3.17 | 3.62 | 4.46 ± 0.29 |
| MgCN | 3.36 | 3.00 | 3.07 | 4.21 ± 0.26 |
| CaCN | 4.12 | 3.88 | 3.78 | 4.46 ± 0.22 |
| BaCN | 4.34 | 4.17 | 4.02 | 4.87 ± 0.26 |
In a reaction of laser-ablated beryllium with hydrogen cyanide, different cyanide and isocyanide compounds of beryllium could be observed and matrix IR spectra were compared to calculated fundamentals using DFT computations (BP86/6-311G*). FTIR spectroscopic measurements of reactions of hydrogen cyanide and laser-ablated beryllium at 6–7 K support this suggestion. In the end, the beryllium isocyanide is the most stable isomer with the cyanide structure lying 15.5 kJ mol−1 higher in energy and no stable T-shaped structure (Lanzisera and Andrews, 1997a).
Further experimental and theoretical studies indicate that not only compounds in the electronic ground state show strong ionic bonding and a linear structure, but also MgNC and CaNC are linear in the energetic minimum of the excited state (Anderson et al., 1994; Steimle et al., 1992, 1994; Whitham et al., 1990). Supplementary molecular orbital calculations (ROHF/TZ2P and SDCI + Q/TZ2P) of the three atoms Mg, C and N were carried out by T. Hirano and co-workers identifying three stationary points: the two linear structures MgCN and MgNC and also a cyclic intermediate with Mg closer to the carbon atom, which is already known from alkali cyanides (Table 24, Figure 19) (Ishii et al., 1993, 1994). These calculations were carried out due to previous observations of the magnesium isocyanide in the circumstellar shell of IRC + 10,216 and were in accordance with experimental data from microwave spectroscopic (MW) measurements (Kawaguchi et al., 1993).
Computed structural data of alkaline earth monocyanide isomers (bond lengths in Å; calculations at SCF/TZ2P, only SrCN at B3LYP/DGDZVP).
| Compound | MCN | MNC | ||
|---|---|---|---|---|
| r M−N | r C−N | r M−C | r C−N | |
| BeCN (Bauschlicher et al., 1985) | 1.529 | 1.159 | 1.693 | 1.132 |
| MgCN (Bauschlicher et al., 1985) | 1.931 | 1.154 | 2.090 | 1.138 |
| CaCN (Bauschlicher et al., 1985) | 2.233 | 1.154 | 2.418 | 1.138 |
| SrCN (Chan and Hamilton, 1998) | 2.350 | 1.188 | 2.512 | 1.175 |
| BaCN (Bauschlicher et al., 1985) | 2.551 | 1.154 | 2.741 | 1.143 |

Computed structural data of the linear isocyanide and cyanide isomer and their intermediate (bond lengths in Å; angle in °, at ROHF/TZ2P) (Ishii et al., 1993).
The properties of the molecular orbitals of the [C≡N]− anion are quite similar to those of the cyano radical (Figure 3) and the SOMO is similarly located at the magnesium atom (Ishii et al., 1993). Overall, the structure is described as an ionic molecule with an oriented Mgδ+ next to a cyano radical [·C≡N:]δ− leading to a very small isomerization barrier of 2170–2230 cm−1 (Ishii et al., 1994).
Removing one single electron from the magnesium leads to the [MgCN]+ cation, which has also a cyclic, T-shaped isomer lying 7.5 kJ mol−1, respectively 12.6 kJ mol−1 below the two linear isomers [MgCN]+, respectively [MgNC]+, displaying similarities to the isoelectronic structure of the NaCN (MP2/6-31G*) (Barrientos and Largo, 1995; Petrie, 1996). The stability of the cyclic structure is a consequence of orbital interaction. Three molecular orbitals are formed of the atomic orbitals of magnesium, carbon and nitrogen and only the lowest molecular orbital is completely occupied. In comparison to the neutral MgCN, the additional half occupied orbital causes an energetic preference of the linear structure (Barrientos and Largo, 1995).
Another T-shaped configuration, which is known from potassium and sodium cyanide, is not known of alkaline earth cyanides due to the strong Coulomb interaction of the metal cation and the cyanide anion, which achieves a maximum for a linear structure (Mikhailov et al., 2003). The same effect influences the structure of SrNC, which was studied using low-resolution laser excitation fluorescence spectroscopy (Douay and Bernath, 1990).
The reaction coordinate of the isomerization of CaNC and CaCN was calculated by T. Taketsugu and co-workers in 2003. Starting with the isocyanide isomer the activation barrier is 25.25 and 7.20 kJ mol−1 from the cyanide side (Figure 20) (Ishii et al., 2003).

Energy profile of the isomerization reaction of CaCN (energy in cm−1, reaction coordinate s in bohr amu1/2) and activation barriers of the isomerization including bond lengths (in Å) and bond angles (in °) of the isomers (representations taken from Ishii et al., 2003).
Alkaline earth dicyanides M(CN)2
Structure of alkaline earth dicyanides
In comparison to the cyanide compounds of the alkaline metals, the alkaline earth dicyanides are less investigated. P. v. R. Schleyer et al. calculated potential energy surfaces of different compounds of the type MC2N2 using the Hartree–Fock method and also the MP-perturbation theory (MP2) to analyse the global minima structures of M(CN)2 with M = Be2+, Mg2+, Ca2+, Sr2+, and Ba2+ extending the calculations of J. Gardner et al. on magnesium species to all alkaline earth metals. In this context, the linear structure of some magnesium species could not be transferred to the later elements (Gardner et al., 1993; Kapp and Schleyer, 1996).
Starting with three different basic linear structures, the metal cation was coordinated by both carbon atoms (CC), by both nitrogen atoms (NN) or by one carbon and one nitrogen atom of the two different cyanide anions (Figure 21).

Considered geometries and connections of structures MC2N2 (representation taken from Kapp and Schleyer, 1996).
The considered linear structures of Ba and Sr are just second-order saddle points, but using HF methods the bent end-on-isomers (CC-2, NN-2, CN-2) are structural minima and using the MP2 level only structure CC-2 is a minimum. In accordance to the other isomers NN-2 and CN-2, the most stable structures are the side-on isomers BB-4 and CB-4, but with small energy differences to the linear ones. P. v. R. Schleyer and J. Kapp summarized that compounds coordinated via nitrogen atoms are more stable than those coordinated via carbon atoms (energies of each favoured N coordination: BaC2N2 – 11.7 kJ mol−1 and SrC2N2 – 9.6 kJ mol−1) and the most stable isomers are showing a side-on coordination of the cyanide anion. Calculating the potential energy surfaces of calcium and magnesium dicyanides using HF method and MP2 method, the minima structures are indeed linear isomers. Although bent structures are not existing (NN-2, CN-2, CC-2) with optimizations leading to the linear isomers, some side-on coordinated structures could be optimized by MP2, for example BB-3 and CB-3 are minimum structures with small energy differences of the side-on isomers. Furthermore, the potential energy surface of the Mg(CN)2 is very flat, so its behavior extremely depends on the basis set and chosen method (Gardner et al., 1993; Petrie, 1999a). For example, using the MP2(full)/6-31G* method the NC–Mg–CN dicyanide is a global minimum structure and the other linear structures are also identified as energetic minima, which are linked by π-complexes (Petrie, 1999a). On that method, these π-complexes are characterized as minima on the energy surface, but calculations on G2-or CBS-Q level suggest that these π-complexes are just transition states upon the potential energy surface and that the dicyanides coordinated via the nitrogen atom (CN–Mg–NC) is the most stable one. The difference of energy between this minimum structure and the highest transition state is only 30 kJ mol−1. The calcium compound is very similar to the Sr and Ba compounds. The energy of the nitrogen dicoordinated isomer is smaller of about 8.8 kJ mol−1 for each cyanide anion than the other two linear isomers, but the structure BB-3 of Ca(CN)2 is most stable by about 18.4 kJ mol−1. The minima of the energy surface of the smallest dicyanide BeC2N2 are only linear structures including the diisocyanide as the most stable one.
Synthesis of alkaline earth dicyanides
Synthesis of M(CN)2 (M = Be2+, Mg2+)
The beryllium dicyanide is formed by dimethylberyllium and an excess of hydrogen cyanide in benzene or other inert solvents. It is also reported that it is insoluble in solvents other than those which cause hydrolysis. The suggestion of a four times coordinated product was disproved by calculations, which were discussed in the chapter above (Coates and Mukherjee, 1963).
In anhydrous conditions, metal chloride and trimethylsilyl cyanide (TMS–CN) are used to synthesize beryllium and magnesium dicyanide. After addition of TMS–CN to the stirred solution of beryllium dichloride in butyl ether at 253 K the resulting solution is stirred at ambient temperatures for 18 h and additional 12 h at 373 K. The solid product is separated by filtration, dried in vacuo and tempered at 773 K for two days. The synthesis of the magnesium compound requires two steps. First, one equivalent of magnesium dichloride is mixed with two equivalents of TMS–CN at ambient temperature and heated under reflux for 18 h. The product is similarly separated by filtration and dried in vacuo. Then two equivalents of TMS–CN are added and the mixture is heated to 473 K for 12 h under nitrogen atmosphere. After tempering the product for 4 h at 648 K, the dicyanide is obtained in a yield of 63% (Williams et al., 2001).
F. Fichter and R. Suter also described a synthesis of magnesium dicyanide using metallic magnesium and a solution of hydrogen cyanide (10–25%). The clear solution rapidly decomposes forming magnesium hydroxide (Fichter and Schölly, 1920; Fichter and Richard, 1922). Therefore, they question the synthesis of O. Schulz (1856a), who reported a high stability of magnesium dicyanide. And the reaction of dry magnesium hydroxide with etheric hydrocyanic acid to form magnesium dicyanide requires small amounts of water, which supports the rapid decomposition of the dicyanide to form the dihydroxide (Fichter and Richard, 1922).
Synthesis of M(CN)2 (M = Ca2+, Sr2+, Ba2+)
The barium and calcium dicyanide are synthesized in a classic neutralization reaction using the metal hydroxide and hydrocyanic acid (Scheme 22) (Williams, 1915).

Syntheses of barium and calcium dicyanide (M = Ba2+, Ca2+) (Williams, 1915).
Although the solid product cannot be isolated from an aqueous solution due to fast decomposition and the solution can only be concentrated up to 35%, which also increases polymerization (Franck and Freitag, 1926). In 1915 H. E. Williams already remarked, that crystals of the magnesium and calcium dicyanides cannot be obtained by removing the solvent of the aqueous solutions (Williams, 1915). And the decomposition of dissolved dicyanides of concentrations higher than 15% is already very fast and depends on the concentration. Oligomers of hydrogen cyanide and ammonium cyanide are formed, which cause the brown color. But solutions of dicyanides are also sensitive to heat and decompose to hydrogen cyanide and metal hydroxide.
The barium dicyanide is obtained as its dihydrate from aqueous solution. By drying at 348 and 373 K the water molecules can be removed (Joannis, 1882a).
In 1926 F. J. Metzger reported another synthesis starting with calcium carbide and addition of hydrogen cyanide under nearly anhydrous conditions. Due to the relatively slow reaction he reported an acceleration of the reaction by addition of 2% water to form the calcium hydroxide in situ, which should directly react with the added hydrogen cyanide. However, it was formed a hydrogen cyanide adduct Ca(CN)2⋅2HCN he could not isolate (Scheme 23) (Metzger, 1926).

Synthesis of the hydrogen cyanide adduct instead of the dicyanide (Metzger, 1926).
H. H. Franck and C. Freitag also reported that the product is a mixture of up to 55% Ca(CN)2 and 25% hydrogen cyanide. Nevertheless, they tried to obtain pure calcium dicyanide from the colorless, powdery di-ammonia complex, which was synthesized using calcium and a solution of ammonium cyanide or salts of calcium, for example Ca(NO3)2, and cyanides as NH4CN or AgCN. The ammonia can be removed by heating up to 453 K in vacuo leading to calcium cyanide in nearly quantitative yields (Scheme 24) (Franck and Freitag, 1926).

Synthesis of calcium dicyanide using ammonia solvate complexes (Franck and Freitag, 1926).
At temperatures higher than 623 K the kinetic stabilized calcium dicyanide decomposes to calcium cyanamide and carbon in an exothermic reaction. If the temperature is strongly increased, the equilibrium will be shifted towards the calcium dicyanide (Scheme 25) (Petersen and Franck, 1938).

Temperature depending equilibrium of calcium dicyanide and calcium cyanamide (Petersen and Franck, 1938).
So the calcium dicyanide can also be obtained by tempering of calcium cyanamide up to 1523 K and fast quenching of the equilibrium to about 573 K (Petersen and Franck, 1938). Such an equilibrium is also known of barium dicyanide, but the temperature range of 500–900 K is much smaller. This equilibrium is part of the reaction of barium carbide and nitrogen and leads to the formation of the cyanamide at lower temperatures of about 773 K and the formation of the dicyanide at higher temperatures (T > 1173 K) in yields of about 82.5% (Scheme 26) (Neubner, 1934; Petersen and Franck, 1938).

Formation of barium cyanamide and dicyanide starting with barium carbide (Neubner, 1934; Petersen and Franck, 1938).
Another synthetic route of alkaline earth dicyanides starts with earth alkaline ferrocyanides. For example, the strontium cyanide is obtained after thermal treatment of strontium ferrocyanide under exclusion of air (Scheme 27) (Schulz, 1856b).

Synthesis of strontium dicyanide starting with strontium ferrocyanide (Schulz, 1856b).
To obtain magnesium, calcium or barium dicyanides, it is also possible to start with an anhydrous double salt mixture of potassium ferrocyanide and calcium chloride or magnesia, which is heated to red heat to form the earth alkaline cyanide and then isolated. The stability of the formed earth alkaline cyanides decreases along Mg(CN)2 → Ba(CN)2 and they cannot be stored in solid state for a long time.
Synthesis of Ra(CN)2
Actually, there is no synthetic route reported to obtain pure radium dicyanide, but the free reaction enthalpy ΔfH°298 K has been estimated to be about (−217 ± 62) kJ mol−1 on the basis of data of the other earth alkaline dicyanides (Wilcox and Bromley, 1963).
Established crystal structures of alkaline earth cyanides
Crystal structures of earth alkaline cyanides are merely known of beryllium and magnesium cyanide. Both structures show a cubic unit cell, further crystallographic data and some atomic distances are given in Table 25. The metal cations are coordinated tetrahedrally by four cyanide anions, which show strong disorder, and the tetrahedra are linked at their vertices (Williams et al., 2001). The literature also offers powder diffraction data of calcium dicyanide of the year 1938, but further analyses of its crystal structure are not published (Hanawalt et al., 1938). The data of strontium dicyanide are also rare. There is only one rudimentary description of its orthorhombic tetrahydrate by A. Joannis from 1882 (Joannis, 1882b).
Crystallographic data of beryllium and magnesium dicyanide (temperatures in K; lengths in Å; angles in °; volumes in Å3, bond lengths in Å) (Williams et al., 2001).
| Parameter | Be(CN)2 cubic | Mg(CN)2 cubic |
|---|---|---|
| T | 298 | 298 |
| Space group |
|
|
| a | 5.339(1) | 6.122(1) |
| α | 90.00 | 90.00 |
| V | 151.2(1) | 229.45(1) |
| Z | 2 | 2 |
| r C−N | 1.187(1) | 1.085(7) |
| r M−CN | 1.718(1) | 2.108(4) |
Further cyanide compounds of the 2nd main group
Calcium monocyanide cation [CaCN]+
The calcium cyanide cation is isoelectronic with potassium cyanide and displays some similar properties. For example, the T-shaped π-complexes T−[MCN]n+ are global minima on the potential energy surface. The linear isomers [CaCN]+ and [CaNC]+ are also minima at the MP2(full)/6-31G* level and identified as second order saddle points by the G2(QCI) procedure. There are also similarities to the T-shaped structures NaCN and MgCN (Petrie, 1999b). The dissociation energy of this cation has been calculated by C. W. Bauschlicher and H. Patridge to be 228.2 kJ mol−1 on SCF(TZ2P) and 268.0 kJ mol−1 on MCPF(ANO) (Bauschlicher and Partridge, 1991).
Superhalides [M(CN)3]− (M = Be2+, Mg2+, Ca2+)
The geometry of superhalides [M(CN)3]− with M = Be2+, Mg2+, Ca2+ is trigonal-planar (D 3h symmetry). The VDEs were determined to be 8.12–8.24 eV of the cyanide and 6.96–8.41 eV of the isocyanide. Overall, the number of coordinated cyanide anions correlates with an increased electronic stability of the compounds due to the strong electron-withdrawing property of the anion (Smuczyńska and Skurski, 2009).
S. Behera and P. Jena also examined the correlation of the VDE and the number of coordinated cyanide anions. Therefore, they had a look on neutral and single or double negatively charged magnesium compounds of the type [Mg(CN) n ]m− with 0 < n < 4 and 0 < m < 2 (Figure 22). The energies of the isocyanide structures are slightly smaller due to the higher electronegativity of the nitrogen and the bond has a high ionic character. Furthermore, the neutral species Mg(CN)3 can also be called a superhalogen (Behera and Jena, 2012).

Optimized geometries (ground states, B3LYP/6–311++G(3df)) of different magnesium cyanide compounds with bond lengths (in Å) and NBO charges (representation taken from Behera and Jena, 2012).
3rd Main group
The general tendencies for compounds of the 3rd main group elements in terms of chemical bonding and the stability of the respective oxidation states are also evident in cyanide chemistry. While boron forms covalent bonds in the energetically favored oxidation state (III), the ionic character of the bonds between aluminum or gallium in the same oxidation state and cyanide units increases significantly. In contrast to these lighter elements, thallium is stabilized in the oxidation state (I) due to the inert pair effect. This results in an alkali metal-like chemical behavior of thallium. TlCN is the only monocyanide that is experimentally known for all elements in this main group. The remaining monocyanides have mainly been studied in theoretical investigations or experimentally by generating these molecules in low amounts in gas phase reactions. Compounds of the constitution E(CN)3 (E = B, Al, In) are known for all elements except thallium. The anionic species [E(CN)4]– are known for boron, gallium and thallium, whereas the anion of indium ([In(CN)4]–) has not been reported so far and regarding aluminium there is just one publication from 1951 reporting the synthesis of Li[Al(CN)4]. The indium cyanide chemistry has not been investigated intensively, presumable reasoned by a missing application for these compounds. The anion [B(CN)4]– is the most frequently applied compound of all reviewed species. There are no significant applications documented for the analogous aluminum or gallium compounds. The thallium monocyanide was utilized in organic syntheses although these procedures did not find a broad application range. As isostructural and isoelectronical to the tricyanomethanide, the tricyanoborate dianion constitutes a unique species in the 3rd main group.
Boron
The chemistry of boron cyanides B(CN) n is largely focused on the tetracyanidoborate anion [B(CN)4]– (Bernhardt et al., 2000; Berkei et al., 2002; Koppe et al., 2007; Küppers et al., 2005). Experimentally known are also the tricyanoborate dianion [B(CN)3]2– and the tricyanoborane B(CN)3 in form of Lewis acid-base complexes (Bernhardt et al., 2011a; Chizmeshya et al., 2007; Williams et al., 2000). The boron monocyanide BCN is well studied by computations and has additionally been observed in gas phase experiments (Lanzisera et al., 1997).
Boron monocyanide BCN
So far, no successful synthesis of stoichiometric boron monocyanide BCN in significant amounts has been published. In 1997, L. Andrews et al. reported results of reactions of laser-ablated boron atoms with hydrogen cyanide while condensing in argon at low temperatures. The generated IR spectra included absorption bands matching the expectations from calculations and predicted absorption shift (Lanzisera et al., 1997). In the following years, several studies described the structure of BCN formed as thin layer on different surfaces (Qin et al., 2012; Shimada et al., 2006; Sugino et al., 2002; Ugarov et al., 1999). While the composition was usually determined by X-ray photoelectron spectroscopy (XPS), the connectivity of the elements was not verified. Furthermore, the specific B–C and C–N absorption bands were not observed in IR spectra.
The first computations of BCN were published by J. B. Moffat (1971). Additional studies suggest that the isocyanide structure BNC is the energetically favoured isomer compared to the BCN molecule. The ground state 1Σ+ energies differ by (39.77 ± 2.09) kJ mol−1 (CASSCF/cc-pVQZ, CCSD(T)/cc-pVQZ) (Martin and Taylor, 1994). The results of different theoretical studies are listed in Table 26.
Structural data of BCN based on computational investigations (bond lengths in Å; wave numbers in cm−1).
| Publication | Method + basis set | r B–C | r C–N | ν CN |
|---|---|---|---|---|
| Moffat (1971) | SCF/GTO | 1.588 | 1.157 | – |
| Glidewell and Thomson (1984) | SCF/6-311G** | 1.585 | 1.133 | – |
| Martin and Taylor (1994) | CASSCF/cc-pVDZ | 1.592 | 1.181 | 2210 |
| Lanzisera et al. (1997) | B3LYP/D95 | 1.60 | 1.17 | 2262.5 |
Noteworthy to Table 26, J. B. Moffat did not optimize the structure using a software algorithm. Instead he varied the B–C distance in constant steps between 1.03 and 2.65 Å and calculated each system in order to find the energetically best structure. Although he stated the C–N distance as 1.157 Å, the results for the optimized structure fit well with later findings. In all cases, the molecule was simulated with a linear structure.
Tricyanoborane B(CN)3
The initial reports regarding tricyanoborane B(CN)3 from 1954 to 1956 have been replicated or verified by others. A reproducible synthesis for Lewis acid-base adduct of B(CN)3 involving trimethylsilyl cyanide TMS–CN as cyanide source and several Lewis bases was published by D. Williams et al. in 2000 (Scheme 28) (Williams et al., 2000).

Synthesis of the Lewis acid-base adduct by D. Williams et al. (X = SMe, LB = TMS–CN, NMe3, C5H5N) (Chizmeshya et al., 2007; Williams et al., 2000).
Several Lewis acid-base adducts were reported using different Lewis bases and in 2007 A. V. G. Chizmeshya et al. added results for a Lewis adduct with pyridine. All these adducts have one thing in common: they have a nitrogen atom as a donor atom, which forms a dative bond with B(CN)3 via a free electron pair (Figure 23) (Chizmeshya et al., 2007; Williams et al., 2000).
Reported Lewis acid-base adducts (Chizmeshya et al., 2007; Williams et al., 2000).
To achive a quantitative conversion, the first step (as shown in Scheme 28) has to be carried out at higher temperatures of about 333 K. In contrast, the disubstituted product is observed, when the reaction is carried out at ambient temperature. The resulting TMS–CN·B(CN)3 was heated to 523 K to receive B(CN)3 following Scheme 29. Therefore, these Lewis adducts can be utilized as a B(CN)3 precursor.

Reversible formation of non-coordinated B(CN)3 (Williams et al., 2000).
The adduct Me3N·B(CN)3 was also obtained by treating the synthesized B(CN)3 with trimethylamine. Some experimental results from single crystal X-ray diffraction (XRD) experiments and IR measurements are listed in Table 27 (Williams et al., 2000). The tetrahedral structure of the amine adduct exhibits a slight distortion (109.03(13)°–109.6(12)°) and its unit cell is orthorhombic, while the crystals of the pyridine adduct show a monoclinic unit cell (Chizmeshya et al., 2007; Williams et al., 2000).
Experimental data of some reported Lewis adducts of B(CN)3 (bond lengths in Å; wave numbers in cm−1).
| Compound | r B–C | r B–N | r C–N | ν CN |
|---|---|---|---|---|
| TMSCN·B(CN)3 (Williams et al., 2000) | – | – | – | 2310/2232 |
| Me3N·B(CN)3 (Williams et al., 2000) | 1.594(4)/1.586(2) | 1.596(3) | 1.143(3)/1.146(2) | 2225 |
| H5C5N·B(CN)3 (Chizmeshya et al., 2007) | 1.590/1.578/1.582 | 1.571 | 1.133/1.133/1.131 | 2227 |
Tricyanidoborate dianion [B(CN)3]2‒
Recently, E. Bernhardt, H. Willner et al. reported the successful synthesis of the tricyanidoborate dianion [B(CN)3]2– by two different approaches. In case of the use of pure alkali metals like lithium, sodium or potassium, the preferred solvent is ammonia when M[B(CN)4] is treated with elemental alkali metals (Scheme 30).
![Scheme 30:
Synthesis of the tricyanoborate dianion [B(CN)3]2– using alkali metals (M = Li, Na, K) (Bernhardt et al., 2011a).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_030.jpg)
Synthesis of the tricyanoborate dianion [B(CN)3]2– using alkali metals (M = Li, Na, K) (Bernhardt et al., 2011a).
When butyllithium is used, it is feasible to utilize other solvents than ammonia like THF for the synthesis of the lithium salt, but even at higher temperature of 373 K the solvent molecule cannot be removed from Li2[B(CN)3]·THF in vacuum (Scheme 31).

Synthesis of the lithium tricyanoborate using butyllithium (Bernhardt et al., 2011a).
But in both cases, the strong base is necessary to form the product. Even under mild conditions, 233 K and liquid ammonia for example, the solvolysis occurs for all reported salts, proceeds fast for the lithium salt, much slower for the sodium and potassium salts and is kinetically hindered at low temperatures. The solvolysis products like [HB(CN)3]–, [HB(CN)2(C(NH)2)]2– and [H2B(CN)2]– are formed by polarity inversion of protons into hydridic hydrogen in the B–H bond. The more stable sodium and potassium salts start to decompose at 503 K. Some computational data of the tricyanoborate dianion and experimental data of two salts are compiled in Table 28 (Bernhardt et al., 2011a).
Computational data of the tricyanoborate dianion (B3LYP/6–311++G(d,p)) and experimental data of two salts (bond lengths in Å; wave numbers in cm−1) (Bernhardt et al., 2011a).
| Compound | r B–C | r C–N | ν as. BC | ν sym. CN | ν as. CN |
|---|---|---|---|---|---|
| [B(CN)3]2– | 1.526 | 1.181 | 1105[a]/1064[b] | 2121 | 2057 |
| Na2[B(CN)3]·3NH3 | 1.512(4) | 1.161(3) | – | – | – |
| K2[B(CN)3] | 1.513 | 1.165 | 1137[a]/1083[b] | 2098 | 2022 |
-
[a]The first wave number refers to 10B. [b]The second wave number refers to 11B.
The potassium salt K2[B(CN)3] could also by synthesized by J. Landmann et al. starting with K[BH(CN)3] and the non-nucleophilic base K[N(SiMe3)2] leading to the dianion and HN(SiMe3)2 in yields of 97% (Landmann et al., 2017).
The dianion is described as a molecule with D 3h symmetry and a nucleophilic boron atom. It is discussed that the occupied boron p z orbital donates electron density into the anti-bonding orbitals of the cyanide groups. This leads to elongated C–N distances and shortened B–C bond lengths compared to similar structures with an electrophilic boron atom (B(CN)3: ΔrBC = −0.075 Å, ΔrCN = 0.025 Å). Consequently, the negative charge is delocalized over all three cyanide groups illustrated in Figure 24.

Lewis structures of the dianion featuring π-back bonding.
Tetracyanidoborate anion [B(CN)4]‒
The tetracyanidoborate (also known as tetracyanoborate) anion [B(CN)4]– is the most intensively investigated B(CN) n species. The synthesized products, which were described in initial reports from 1951, 1967 and 1977, seem to differ from the expected experimental results for the [B(CN)4]– anion, especially the spectroscopic data (Bessler, 1977; Bessler and Goubeau, 1967; Wittig and Raff, 1951). For example, in 1977, E. Bressler (Bessler, 1977) claimed to have synthesized M[B(CN)4] (M = Cu+, Ag+) starting from BCl3 and MCN, however, it later turned out that the analytical data given in the publication did not correspond to the presumed product, so that it was assumed that the tetraisocyanidoborate anion ([B(NC)4]–) was obtained rather than the tetracyanidoborate. Therefore, the synthesis was presumably achieved for the first time by E. Bernhardt et al. for the tetrabutylammonium, silver and potassium salts in 2000 (Scheme 32) (Bernhardt et al., 2000).

Synthesis of the tetracyanidoborate anion by E. Bernhardt et al. (X = Cl−, Br−) (Bernhardt et al., 2000).
Synthesis. The first reactions described in the literature to prepare [B(CN)4]– date back to 1950 (Grundmann and Beyer, 1954; Wittig and Bille, 1951; Wittig and Raff, 1951). At that time, an attempt was made to react Li[BH4] with hydrocyanic acid. However, this reaction stopped at the Li[BH3(CN)] stage despite a large excess of HCN and temperatures of 373 K. The analogous reaction of Li[AlH4], on the other hand, leads to Li[Al(CN)4]. Around 2000, E. Bernhardt et al. worked intensively on the synthesis of [B(CN)4]– salts and developed several new synthetic routes. The reaction shown in eq. 1 (Scheme 32) can also be understood as the reaction of [ n Bu4N][BX3Br] with CN, where the X stands for Br or Cl, but not for F. In fact, when BF3 was used, no complete conversion could be observed even up to 473 K. Disadvantages of this synthesis route are the long reaction time of 1–3 weeks, the reaction coming to a standstill if the KCN is not always ground up and the column chromatographic purification of the product (Bernhardt et al., 2000). The second synthesis method (Scheme 32, eq. 2) starts from alkali metal tetrafluoridoborates and TMS–CN, but again long reaction times are required for the synthesis of the cyanido(fluorido)borates. For the preparation of M[BF(CN)3], a month is required using KBF4, for LiBF4 1–2 weeks (Bernhardt et al., 2003a). M[B(CN)4] (M = Li+, K+) could not be obtained via this reaction. A more efficient synthesis of the tetracyanidoborates on a larger scale, starting from the commercially available reagents KBF4, LiCl and KCN (Scheme 32, eq. 5), was published in 2003 in connection with investigations on the thermal behavior of the mixed cyanido(fluorido)borates K[BF4–n(CN) n ] (n = 1 – 3) by the same working group (Bernhardt et al., 2003b). The reaction is a solid-state synthesis in which the salt mixture melts already at 280–563 K. After melting of the reactants, the exothermic reaction begins and temperatures of up to 823 K are reached for a short time. After 1 h of reaction time, Li[B(CN)4] is obtained in this way, which can be purified by a rather complex procedure and double re-salting. The yields of this reaction are between 60 and 65%. Simultaneously to our work, a publication by J. A. P. Sprenger et al. appeared in 2015, in which a more efficient synthesis of M[BF(CN)3] (M = Na+, K+) was presented (Sprenger et al., 2015). By addition of 10 mol% TMS–Cl, which seems to catalyze the reaction, the cyanidation with TMS–CN was already successful at 373 K after 6 h of reaction time (yields 80–90%). The silver and potassium salts were synthesized by salt metathesis reaction and the more complex salts listed in Table 30 are accessible via salt metathesis reaction based on the simple alkaline tetracyanidoborates (Bernhardt et al., 2000). Three years after the initial study, E. Bernhardt et al. reported an alternative synthetic route by a sinter process (Scheme 32, eqs. 5 and 6, M = Na+, K+) (Bernhardt et al., 2003b).
Table 29 lists some advantages and disadvantages of the different routes for the synthesis of K[B(CN)4]. The main advantage of E. Bernhardt’s sintering process(Scheme 32, eqs. 5 and 6) is the use of the less expensive KCN as a cyanide source compared to the autoclave and LA-catalyzed reaction (Scheme 33, eqs. 8 and 11), which uses the more expensive TMS–CN. However, E. Bernhardt’s sintering process requires large amounts of LiCl, higher temperatures, more reaction steps and produces a large amount of waste product. In addition, the sintering process is not easy to carry out, as hot spots must be avoided and the purity of the starting materials plays an essential role in obtaining yields of up to 60%, which is, however, difficult to reproduce. The disadvantages of the autoclave reaction compared to the LA catalysis process are the salt metathesis step via silver salts (Scheme 33, eqs. 9 and 10) and the need to use autoclaves. The LA catalysis process represents a rapid, high-yield and easy-to-perform route not only to K[B(CN)4] but to almost all metal tetracyanidoborate salts. Furthermore, the number and amount of impurities (e.g. Cl−, Li+, CN− or Ag+) in the final products is significantly lower compared to the other methods (Bläsing et al., 2016a).
Reaction parameters of different routes to K[B(CN)4].
| Sinter process (Bernhardt et al., 2003b) | Autoclave (Bläsing et al., 2016a) | LA catalysis (Bläsing et al., 2016a) | |
|---|---|---|---|
| Starting material | K[BF4] | [ n Bu4N][BF4] | [ n Pr3NH][BF4] |
| Cyanide source | KCN | TMS–CN | TMS–CN |
| T | 573 K | 473 K | 393 K |
| Reaction time | 1 h | 15 h | 6 h |
| Product | Li[B(CN)4][b] | [ n Bu4N][B(CN)4] | [ n Pr3NH][B(CN)4] |
| Yield | –[b] | 75% | 70–80% |
| Metathese products | [ n Pr3NH][B(CN)4] | Ag[B(CN)4] | None |
| Steps | 3 | 3 | 2 |
| Overall time | 20 h | 25 h | 10 h |
| Waste products | KCN, LiCl, LiF, KCl, n Pr3N | [ n Bu4N]NO3, AgBr | n Pr3N |
| Solvents | CH2Cl2, THF | MeOH, CH3CN | CH2Cl2 |
| Yield K[B(CN)4] | <60%[a] | 60–70% | 60 –80% |
-
[a]It is very hard to achieve 60% yield at all, we never managed to obtain such yields as we only achieved isolated yields between 20 and 35% with the sinter method. However, we do want to stress that the overall yield strongly depends on the purity of the starting materials and avoiding of hot spots during the reaction. [b]The lithium salt is not isolated from the reaction mixture.
(Selected) synthesized salts of [B(CN)4]–.
| Publication | Cation |
|---|---|
| Bernhardt et al. (2000) | [Bu4N]+, Ag+, K+ |
| Berkei et al. (2002) | Hg2+, Hg22+ |
| Bernhardt et al. (2003b) | Li+, Na+, K+, Cs+, Cu+, NH4+, [Et4N]+, [(H2N)3C]+ |
| Küppers et al. (2005) | Rb+, Tl+ |
| Neukirch et al. (2006) | Zn2+, Cu2+ |
| Küppers et al. (2007) | H+, [H3O]+, [H5O2]+ |
| Koppe et al. (2007) | [F5C6Xe]+ |
| Nitschke and Köckerling (2009) | Co2+ |
| Flemming et al. (2010) | [Ph4P]+, [ n Bu4P]+, [EtPh3P]+, [ n BuPh3P]+ |
| Nitschke and Köckerling (2011) | Fe3+, Fe2+ |
| Nitschke et al. (2014) | Mg2+, Ca2+ |
![Scheme 33:
Synthesis of the [B(CN)4]– according to A. Schulz and co-workers (X = Cl−, Br−) (Bläsing et al., 2016a; Ellinger et al., 2014; Ott et al., 2014a, 2014b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_033.jpg)
Synthesis of the [B(CN)4]– according to A. Schulz and co-workers (X = Cl−, Br−) (Bläsing et al., 2016a; Ellinger et al., 2014; Ott et al., 2014a, 2014b).
In 2016, however, the Schulz group reported a very simple synthesis possibility of tetracyanidoborates (Scheme 33) (Bläsing et al., 2016a; Ellinger et al., 2014; Ott et al., 2014a, 2014b). The reaction of [BF4]– salts with TMS–CN was investigated at different temperatures and in the presence of several different Lewis acids, leading to the formation of the known [BF4–n(CN) n ]– (n = 1–4) salts. Depending on the catalyst and the reaction conditions, it is shown that the whole series of [BF4–n(CN) n ]– salts is now accessible under ambient conditions, avoiding long preparation times or excessive production of waste materials. The best synthesis of tetracyanidoborate salts at present is the reaction of [R3NH]BF4 with TMS–CN at 393 K with GaCl3 as Lewis acidic catalyst (Scheme 33, eq. 11). This yields the ammonium salt in very good yields, which can be converted very elegantly with bases to the corresponding metal salts (Scheme 33, eq. 12) (Bläsing et al., 2016a).
Table 30 summarizes several synthesized salts, which can be mainly divided into ionic liquids (ILs), used as additive in dye-sensitized solar cells for example, and common inorganic salts applicable in different fields like fluorine free lithium batteries (Marszalek et al., 2011; Scheers et al., 2014). The tetracyanidoborate anion can also be employed as a starting material of more complex weakly coordinating anions (WCAs) like [B{CN·B(C6F5)3}4]– or for other WCAs like [B(CF3)4]– (Figure 25) (Bernhardt et al., 2001, 2011b; Bernsdorf et al., 2009a).
![Figure 25:
WCA based on [B(CN)4]–⋅4 B(C6F5)3 adduct (= [B{CN·B(C6F5)3}4]–) in comparision to the [B(CF3)]4– ion.](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_025.jpg)
WCA based on [B(CN)4]–⋅4 B(C6F5)3 adduct (= [B{CN·B(C6F5)3}4]–) in comparision to the [B(CF3)]4– ion.
Tetracyanoboronic acids H[B(CN)4]·n H2O (n = 0, 1, and 2) were synthesized by ion exchange (Küppers et al., 2007). These compounds can be again employed to generate several tetracyanidoborate salts by acid-base reaction (Bernsdorf and Köckerling, 2009; Nitschke and Köckerling, 2009, 2011). Furthermore, different magnesia salts are accessible by direct conversion due to the high reductive power of magnesia and the strong acidity of HB(CN)4 (Nitschke et al., 2014). Ionic liquids containing the tetracyanidoborate anion as weakly coordinating anion were synthesized and extensively characterized in several approaches founding a great variety of tetracyanidoborate based ILs (Bläsing et al., 2016a). The tetracyanidoborate salts are stable at ambient temperature and their decomposition is not observed up to 383 K strongly depending on the cation, the alkali salts start to decompose at 773 K for example. Detailed information and additional data of bond lengths and spectroscopic data are represented in Table 31. The solubility also depends on the cation. The metal salts exhibit high hydrolysis stability, when they are exposed to boiling water or concentrated chlorine, and are also stable in dry hydrogen fluoride for over 1 h (Bernhardt et al., 2000).
Experimental data of some [B(CN)4]– salts (bond lengths in Å; wave numbers in cm−1; temperatures in K).
| Cation | r B–C | r C–N | ν CN | ν BC | T M | T Dec. |
|---|---|---|---|---|---|---|
| [H5O2]+ (Küppers et al., 2007) | 1.593(1) | 1.144(2)/1.143(2)/1.143(2) | 2253 | 976/942 | 388 | – |
| Na+ (Bernhardt et al., 2003b; Küppers et al., 2005) | 1.575(2) | 1.130(3) | 2252 | – | 793 | 813 |
| K+ (Bernhardt et al., 2003b; Küppers et al., 2005) | 1.595(1) | 1.142(1) | 2233 | – | 703 | 783 |
| Rb+ (Küppers et al., 2005) | 1.590(1) | 1.136(2) | 2232 | – | 703 | 783 |
| Cs+ (Bernhardt et al., 2003b; Küppers et al., 2005) | 1.582(5) | 1.127(7) | 2230 | – | 693 | 783 |
| Ag+ (Bernhardt et al., 2000, 2003b) | 1.589(2) | 1.131(3) | 2256 | 982/946 | – | 713 |
| Cu+ (Bernhardt et al., 2003b; Küppers et al., 2005) | 1.588(2) | 1.140(3) | 2253 | – | – | 743 |
| NH4+ (Bernhardt et al., 2003b; Küppers et al., 2005) | 1.592(2) | 1.133(2) | 2233 | – | – | 573 |
| [Bu4N]+ (Bernhardt et al., 2000, 2003b) | 1.582(3)/1.590(3) | 1.135(3)/1.141(3) | 2222 | 967/937 | 353 | 623 |
| [Ph4P]+ (Flemming et al., 2010) | 1.596(2)/1.580(2) | 1.141(2)/1.146(2) | 2226 | 985/937 | 466 | 663 |
| Cu2+ (Neukirch et al., 2006) | 1.594(6)/1.591(3)/1.588(8) | 1.131(4)/1.130(3)/1.132(6) | 2234 | 979/946 | – | 623 |
| Zn2+ (Neukirch et al., 2006) | 1.595(2)/1.590(5) | 1.139(2)/1.132(5) | 2234 | 980/946 | – | 723 |
| Hg2+ (Berkei et al., 2002) | 1.60(1)/1.62(2) | 1.12(1)/1.11(3) | 2234 | 976/938 | – | 538 |
| Hg22+ (Berkei et al., 2002) | 1.63/1.57 | 1.12/1.14 | 2225 | 972/938 | – | 439 |
The decomposition of tetracyanoboronic acid was investigated under inert conditions and observed by differential scanning calorimetry (DSC) and IR spectroscopy. The analysis led to a proposed decomposition process illustrated in Scheme 34 (Küppers et al., 2007).

Decomposition process of tetracyanoboronic acid (Küppers et al., 2007).
The single crystal X-ray diffraction revealed a slightly compressed tetrahedral coordination of the boron atom in most cases. The silver(I), copper(I), lithium and sodium salts exhibit perfect tetrahedral symmetry for both the cation and the boron atom. A detailed analysis of the structural parameters and crystal systems was given by the groups of H. Willner and M. Köckerling in 2005 (Küppers et al., 2005). According to the initial report by E. Bernhardt et al., the [Bu4N]+ ion can be described as isolated due to the significant distances to the anion in the crystal structure corresponding to weak interactions between the cation and the anion, which also leads to a small melting point of TM = 353 K (Bernhardt et al., 2000).
Aluminum
The properties of aluminum cyanide compounds Al(CN) n are comparable to those of B(CN) n chemistry even though this class is not as well investigated and applied. The compounds have been mainly investigated by theoretical approaches, while there are only a few experimental studies accessible.
Aluminum monocyanide AlCN
Aluminum monocyanide AlCN has been studied both theoretically and experimentally. The motivation of these investigations originates from a special interest since astrophysicists suspected the molecule AlCN and its isomer AlNC to be the source of some unidentified signals in the MW spectra of the carbon-rich mass-losing star IRC + 10,216 (Fukushima, 1998). Based on this incitement, the first theoretical report was published by B. Ma et al. in 1995. The calculations revealed that the isocyanide is the energetically favored isomer compared to the cyanide (ΔE = 23.0 kJ mol−1, CCSD(T)/TZ2P + f) while both structures are linear molecules (Ma et al., 1995). These findings were confirmed by additional studies and demonstrate a similarity between the monocyanides of boron and aluminum (Jiang et al., 2002; Meloni and Gingerich, 1999; Petrie, 1996; Tokue and Nanbu, 2006, 2011). Furthermore, S. Petrie noted that the isomerization energy of the aluminum species is significantly larger compared to other theoretically investigated monocyanides like NaCN or MgCN (Petrie, 1996). The first experimental work was published by D. V. Lanzisera and L. Andrews operating with the same technique they successfully applied for the detection of BCN (Lanzisera and Andrews, 1997b). In 1999, I. Gerasimov et al. conducted photolysis experiments by laser fluorescence utilizing a supersonic beam containing trimethylaluminum and molecular nitrogen (Gerasimov et al., 1999). Some selected results are shown in Table 32 and a more detailed comparison including numerous results from several publications can be obtained from reports by I. Tokue and S. Nunbu (2011).
Computational data of AlCN and its isomer (bond lengths in Å; wave numbers in cm−1).
| Publication | Method + basis set | Isomer | r Al–E | r C–N | ν CN | ν AlE |
|---|---|---|---|---|---|---|
| Ma et al. (1995) | SCF/TZ2P + f | AlCN | 2.030 | 1.135 | 2479 | 472 |
| AlNC | 1.842 | 1.158 | 2290 | 584 | ||
| Lanzisera and Andrews (1997b) | BP86/6-311G* (LANL2DZ) | AlCN | 2.09 | 1.17 | 2149.7 | – |
| AlNC | 1.94 | 1.19 | 2024.4 | – | ||
| Fukushima (1998) | QCISD(T)/6-311 + G(3df) | AlCN | 2.0215 | 1.1712 | 2222.9 | 469.9 |
| AlNC | 1.8639 | 1.1877 | 2105.2 | 554.2 |
Aluminum tricyanide Al(CN)3
The first hint for a synthesis of aluminum tricyanide was published in 1924 by F. W. Bergstrom when he treated Hg(CN)2 with aluminum metal in liquid ammonia according to 2 Al + 3 Hg(CN)2 → 2 Al(CN)3 + 3 Hg (Bergstrom, 1924). It was found that it is formed Al(CN)3⋅5 NH3. It is assumed that the number of ammonia molecules is much larger in NH3 solution. Al(CN)3⋅5 NH3 is an effloresced white powder. By successive deammonation of the crystals of the composition Al(CN)3⋅n NH3 with n = 13 or 14 at −33 °C the nona- and hexa-ammoniates are obtained as white solids. A substance of the composition AI(CN)3⋅1.5 NH3 can be obtained by heating the penta-ammoniate in vacuum (80 mm Hg). Al(CN)3⋅5 NH3 is decomposed by water, probably to form aluminum hydroxide, ammonia and ammonium cyanide. This is also the reason why Al(CN)3 cannot be obtained from aqueous solution, as it immediately starts to hydrolyse.
The synthesis of pure Al(CN)3 as the framework compound was achieved by a metathesis reaction according to Scheme 35 (Williams et al., 2001). Single crystal XRD experiments indicate that Al(CN)3 crystallizes in a prussian blue-like octahedral structure. As mentioned before, strong absorption bands at 2220 and 530 cm−1 observed in the IR spectra indicate evidence of cyanide groups. The recorded 27Al NMR spectrum exhibits seven signals. The authors assigned each signal to one of seven possible aluminum environments with varying ratios of surrounding carbon and nitrogen atoms due to C, N static disorder (Williams et al., 2001). G. Wittig and H. Bille described the formation of Al(CN)3 in the reaction of AlH3 with HCN, which crystallizes from the etheric solution as Al(CN)3⋅O(C2H5)2 (Scheme 36) (Wittig and Bille, 1951).

Synthesis of Al(CN)3 by J. Kouvetakis et al. (2001).
![Scheme 36:
Synthesis of Al(CN)3 and Li[Al(CN)4] according to G. Wittig and H. Bille (Wittig and Bille, 1951).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_036.jpg)
Synthesis of Al(CN)3 and Li[Al(CN)4] according to G. Wittig and H. Bille (Wittig and Bille, 1951).
Ternary Al–C–N compounds were observed in a thin film deposited using inductively coupled plasma (ICP) assisted DC magnetron sputtering (Jiang et al., 2002).
A comparison of calculated and observed data for the Al(CN)3 framework was given by A. V. G. Chizmeshya et al. in 2007. The report included additional data on gallium and indium frameworks. The values of C–N bond lengths are reported as 1.164 Å for experimental and 1.158 Å for computational investigations (Chizmeshya et al., 2007).
Although the number of publications regarding the aluminum tricyanide Al(CN)3 is small, this compound has been studied theoretically as a gas phase molecule and experimentally as a framework compound. In gas phase Al(CN)3 is reported with a Al–C distance of 1.914 Å (SCF/LANL2DZP) and 1.912 Å (B3LYP/LANL2DZP) while the C–N bond length of the cyanide groups varies between 1.143 Å (SCF/LANL2DZP) and 1.170 Å (B3LYP/LANL2DZP), respectively. This constitutes the significant difference in calculated IR absorption bands of the C–N stretching mode of 2525 cm−1 (SCF/LANL2DZP) and 2276 cm−1 (B3LYP/LANL2DZP), respectively. A. Y. Timoshkin and H. F. Schaefer summarized that the results indicate the importance of the inclusion of electron correlation for such systems whereby the HF method is inadequate for a more precise description (Timoshkin and Schaefer, 2000). This is confirmed by IR measurements of the framework compound synthesized by A. V. G. Chizmeshya et al. which exhibits an absorption band at 2220 cm−1 assigned to the C–N stretching mode and therefore agrees more accurately with the results of the DFT calculations (Mikhailov et al., 2003).
Cyanidoaluminate anions [Al(CN)6–n](3–n)‒ (n = 0, 1, 2)
Interestingly, there is only one publication on the synthesis of cyanidoaluminates from 1951 by G. Wittig and H. Bille (Scheme 36) (Wittig and Bille, 1951). It describes the reaction of Li[AlH4] with hydrocyanic acid, in the course of which hydrogen and Li[Al(CN)4] are formed. This complex salt precipitates from the ethereal solution as a colorless powder and was shown to be Li[Al(CN)4]. It is decomposable even under exclusion of moisture and oxygen, as can be seen from the yellow coloration shortly after preparation and the decrease of the cyanide content. In water it decomposes to form aluminum hydroxide, lithium cyanide and prussic and hydrocyanic acid (Wittig and Bille, 1951).
The anionic species of aluminum cyanide compounds shown in Figure 26 have been recently studied by theoretical approaches. The initial work of S. Smuczyńska and P. Skurski on the [AlCN]– anion reported an Al–C distance of 1.975 Å and a C–N bond length of the cyanide group of 1.182 Å (MP2/6-311 + G(d)) (Smuczyńska and Skurski, 2009). Additional results by T. Sommerfeld and B. Bhattarai reported insignificantly longer bond lengths (ΔrAl–C = + 0.01 Å) in regard to the anion and an increase of the bond length from the anion to the dianion (ΔrAl–C = + 0.11 Å) and to the trianion (ΔrAl–C = + 0.17 Å) while the C–N bond length of the cyanide groups is insignificantly elongated (ΔrC–N = + 0.005 Å and ΔrC–N = + 0.008 Å, MP2/aug-cc-pVDZ). The authors added that the trianion [Al(CN)6]3– is stable considering the VDEs (Sommerfeld and Bhattarai, 2011).

Anion, dianion and trianion of aluminum cyanide compounds.
Generally, both structure optimizations used MP2 methods and were focused on the question of stability of the anions. S. Smuczyńska and P. Skurski noted that only one minimum energy structure was found on the potential energy surface (PES) of [Al(CN)4]–. The authors also investigated the VDEs of various [M(CN) n ]– species with M = Li+, Na+, Be2+, Mg2+, Ca2+, B3+ and Al3+ and concluded that the electronic stability of the anion increases with an increasing number of cyanide ligands resulting in the highest VDE for [Al(CN)4]– (Smuczyńska and Skurski, 2009). T. Sommerfeld et al. calculated two isomers of the aluminum cyanide trianion, [Al(CN)6]3– and [Al(NC)6]3–, using ab initio methods (Sommerfeld and Bhattarai, 2011). These two isomers are predicted to be electronically stable and show significant barriers with respect to the dissociation of [CN] – ions.
Gallium
The cyanide-substituted gallium species are not as well investigated as the corresponding boron or aluminum species. Although computational studies are accessible as well, the part of experimental studies has increased compared to the first two elements of this main group, but the overall number of publications regarding gallium cyanide chemistry is smaller compared to boron and aluminum. Especially, the Ga(CN)3 has been studied most intensively of all gallium cyanide species. Similarities of gallium and aluminum are existing in the tendency to form framework structures and in regard to the small number of studies investigating the anionic species of these elements.
Gallium monocyanide GaCN
Similar to the boron and aluminum analogues, the first successful synthesis of gallium monocyanide GaCN was reported by D. V. Lanzisera and L. Andrews in their analysis of the reaction of laser-ablated elements with hydrogen cyanide in 1997 (Lanzisera and Andrews, 1997b). Two years later this investigation was followed by an additional theoretical and experimental work of K. A. Walker et al. (2001). The results of these reports are listed comparatively in Table 33.
Computational data of GaCN (bond lengths in Å; wave number in cm−1).
| Publication | Method + basis set | Isomer | r Ga–E | r C–N | ν CN | ν GaE |
|---|---|---|---|---|---|---|
| Lanzisera and Andrews (1997b) | BP86/6-311G* (LANL2DZ) | GaCN | 2.12 | 1.17 | 2147.0 | 339.3 |
| GaNC | 1.97 | 1.19 | 2023.0 | 388.2 | ||
| Petrie (1999a) | MP2(full)/6-31G* | GaCN | 2.073 | 1.191 | – | – |
| GaNC | 1.962 | 1.201 | – | – | ||
| Walker et al. (2001) | B3LYP/aug-cc-pVTZ | GaCN | 2.098 | 1.158 | – | – |
| GaNC | 1.965 | 1.176 | – | – |
The question regarding the energetically preferred isomer depends on the employed computational method. The isocyanide isomer is the stable isomer at the DFT level calculated by D. V. Lanzisera and L. Andrews (1997b). At the G2 level, the gallium cyanide represents the global minimum on the PES and is therefore the favored isomer but not at G2[thaw(QCI)]. Due to the similar ground state energies and small isomerization barrier, S. Petrie concluded that the interaction between the gallium atom and the cyanide unit possesses mainly ionic character (Petrie, 1999a).
In 2010 K. Jacobs et al. suggested the intermediate formation of GaCN in a process resulting in GaN. The authors postulated an interaction of ammonia and graphite forming hydrogen cyanide, which reacts with gallium to form volatile GaCN in the second step. This mechanism is based on mass spectrometric findings and thermodynamic calculations (Jacobs et al., 2010).
Gallium tricyanide Ga(CN)3
Gallium tricyanide, Ga(CN)3, was initially reported by L. C. Brousseau et al. in 1997 as an anhydrous Prussian blue-like crystallizing compound (Figure 27). The synthesis was achieved by a reaction of azidodichlorogallane and trimethylsilyl cyanide TMS–CN (Scheme 37).

Ga(CN)3 as a molecule, part of a Lewis acid-base adduct and part of a framework.

Initial synthesis of Ga(CN)3 (Brousseau et al., 1997).
The compound is unstable in presence of air or at temperatures above 723 K. The IR spectrum revealed a strong absorption band at 2215 cm−1 for the CN group and 440 cm−1 assigned to the Ga–C stretching mode which is similar to related compounds like GaMe2CN (νCN = 2207 cm−1, νGaC = 429 cm−1). The bond lengths of rGa–C = 2.072 Å and rC–N = 1.148 Å were determined by powder XRD and Rietveld analysis. The authors suggested that the high building tendency and stability of Ga(CN)3 in contrast to otherwise substituted gallium species could originate from a metal ligand π-back bonding. A second synthetic route, starting from GaCl3 and TMS–CN, was published to gain access to highly pure and crystalline Ga(CN)3 (Scheme 38) (Brousseau et al., 1997).

Synthesis of highly pure Ga(CN)3 (Brousseau et al., 1997).
Molecular Ga(CN)3 was studied theoretically by A. Y. Timoshkin and H. F. Schaefer in 2000. Compared to values derived from HF calculations (rC–N = 1.142 Å at SCF/LANL2DZP), the results from approaches utilizing DFT methods indicate an elongated C–N bond length (rC–N = 1.169 Å at B3LYP/LANL2DZP) while both methods lead to similar Ga–C distances of rGa–C = 1.922 Å. The calculated IR absorption bands using DFT methods are in reasonable agreement with the observed bands from the Ga(CN)3 framework (νCN = 2282 cm−1 at B3LYP/LANL2DZP, νCN = 2318 cm−1 at B3LYP/DZP), but HF methods would lead to significantly increased wave numbers (νCN = 2533 cm−1 at SCF/LANL2DZP). Questioning the energetic stability of both isomers, the results do not provide a final answer since different computational methods led to opposing data regarding the relative energies of the gallium cyanide and gallium isocyanide (Timoshkin and Schaefer, 2000).
Very little is known about molecular Lewis acid-base adducts of Ga(CN)3. In 2007, A. V. G. Chizmeshya et al. analyzed Ga(CN)3·2 NC5H5 revealing that the addition of Ga(CN)3 (as coordination polymer) to pyridine leads to the formation of an adduct complex with two pyridine molecules (Ga(CN)3·2 NC5H5) directly bound to the gallium center in contrast to the boron analogue where only one solvent molecule was attached. The average Ga–C bond length was documented as rGa–C = 2.027 Å and the average C–N bond length was determined to be rC–N = 1.065 Å while the calculated bond lengths differed significantly (rGa–C = 1.946 Å and rC–N = 1.161 Å at LDA). The difference of Ga–C distances was mainly caused by a computational method called local density approximation (LDA) which is known for its tendency to underestimate bond lengths. Concerning the experimental data, the authors supposed that refinement artifacts in context of small scattering factors of carbon and nitrogen caused the disagreement. The recorded IR spectrum exhibited a strong absorption at 2180 and 436 cm−1 (Table 34) (Chizmeshya et al., 2007).
Experimental and computational data of different Ga(CN)3 species (bond lengths in Å; wave numbers in cm−1).
| Compound | Exp./calc. | r Ga–C | r C–N | ν CN | ν GaC |
|---|---|---|---|---|---|
| Ga(CN)3 (molecular) (Timoshkin and Schaefer, 2000) | SCF/LANL2DZP | 1.922 | 1.142 | 2533 | 451 |
| B3LYP/LANL2DZP | 1.922 | 1.169 | 2282 | 431 | |
| Ga(CN)3 (framework) (Brousseau et al., 1997) | Exp. – XRD, IR | 2.072 | 1.148 | 2215 | 440 |
| Ga(CN)3·2 NC5H5 (Chizmeshya et al., 2007) | Exp. – XRD, IR | 2.027 | 1.065 | 2180 | 436 |
| LDA | 1.946 | 1.161 | – | – |
Tetracyanido gallide anion [Ga(CN)4]‒
In their report on the Ga(CN)3 coordination polymer, L. C. Brousseau et al. synthesized and analyzed lithium and copper tetracyanido gallide, Li[Ga(CN)4] and Cu[Ga(CN)4], as well. The substances are accessible by direct addition of the metal cyanide salt MCN to Ga(CN)3 (Scheme 39, M = Li+, Cu+).

Synthesis of salts containing the tetracyanido gallide anion (Brousseau et al., 1997).
The cubic crystal system consists of MN4 and GaC4 tetrahedrons. Since it is not possible to distinguish between carbon and nitrogen, a general disorder could not be excluded. The Ga–E bond length is determined as rGa–E = 2.00(1) Å, the bond length of the cyanide group is reported as rC–N = 1.07(1) Å and the cyanide stretching mode was found at νCN = 2227 cm−1 for the lithium salt. The compound Zn(CN)2 crystallizes in the same structural type with a systematical disorder and shows an absorption band for the cyanide closely related to the lithium salt at 2218 cm−1 (Brousseau et al., 1997).
Indium
As discussed before, the tricyanido species of group 13 elements seem to be little studied in contrast to the corresponding halides. Indium tricyanide was reported a long time ago. In 1869, R. E. Mayer already described the reaction of indium acetate with aqueous hydrogen cyanide solution, which shows no precipitation, whereas the reaction with KCN leads to a white precipitate, which dissolves again in the excess of KCN (Meyer, 1869). The dissolving white precipitate was probably K[In(CN)]4. On boiling, indium hydroxide is formed.
The indium cyanide chemistry is similar to the gallium chemistry, but has not been investigated intensively. In particular, the indium monocyanide is not as well theoretically studied as the gallium or aluminum analogue. Similar to other E(CN)3 compounds with E = 3rd main group element, the framework structure has been analyzed in the late 1990s. Syntheses of defined In(CN)3 Lewis base adducts failed in first attempts due to the formation of undefined mixtures (Chizmeshya et al., 2007). Reports of anionic species are not published so far.
Indium monocyanide InCN
Indium monocyanide InCN has been obtained experimentally by ablation of indium and the reaction of these ablated gas phase atoms with hydrogen cyanide (Lanzisera and Andrews, 1997b). Furthermore, computational calculations were conducted for comparison. The studies focused on vibration and rotation spectra (Chizmeshya et al., 2007; Walker et al., 2001). The DFT calculations by D. V. Lanzisera and L. Andrews resulted in an In–C distance of rIn–C = 2.28 Å and a C–N distance of rC–N = 1.17 Å (BP86/6-311G*(LANL2DZ)) and the computational value of the cyanide absorption band is νCN = 2142 cm−1 compared to the experimental value of νCN = 2132.4 cm–1 (Lanzisera and Andrews, 1997b). These properties were also gained by applying the rigid bender model resulting in rIn–C = 2.268 Å and rC–N = 1.143 Å, respectively (Walker et al., 2001).
Furthermore, D. V. Lanzisera and L. Andrews noted that the energy difference between both indium species InCN and InNC is small and interconversion between both isomers is facile reporting bond length of rIn–N = 2.14 Å and rC–N = 1.19 Å (BP86/6-311G*(LANL2DZ)) in regard to the isocyanide (Lanzisera and Andrews, 1997b).
Indium tricyanide In(CN)3
In attempts to prepare indium oxycyanide by the action of cyanogen on indium oxyiodide a small amount of material of the analytical composition In(CN)3 was obtained. This compound was extremely deliquescent, unstable in water and causes considerable doubt about the validity of the earlier reports (Goggin et al., 1966a). In contrast to earlier reports, P. L. Goggin et al. could not report a reaction in aqueous solution due to solvolysis. Furthermore, they reported that indium metal reacts with mercury cyanide in liquid ammonia solution, but not in ethanol or liquid hydrogen cyanide solutions. After evaporating the solvent and heating in vacuo at 373 K, the product found was In(CN)3·n NH3. However, pure In(CN)3 could be obtained as sublimate by heating the ammoniate at 573 K in a nitrogen stream. The reaction of HCN gas with indium metal or with indium hydroxide at 623 K also leads to the isolation of small amounts of In(CN)3 (Goggin et al., 1966b).
The initial synthesis of indium tricyanide In(CN)3, as reported by P. L. Goggin et al., was an inefficient procedure that has not been adopted in additional studies (Goggin et al., 1966b). In 1998, D. Williams et al. reported that In(CN)3 can be prepared by treating freshly sublimed InCl3 with an excess of TMS–CN in dry ether/hexane solutions. After extraction with warm hexane, which led to nanoporous cubic In(CN)3 (Scheme 40) (Williams et al., 1998). The crystal structure is similar to those of Al(CN)3 and Ga(CN)3 which have been described earlier. It was also shown that In(CN)3 can be combined with Ga(CN)3 to form solid solutions in which the lattice parameter varies smoothly with concentration across the entire compositional range.

Synthesis of pure In(CN)3 (Williams et al., 1998).
The structural characteristics of In(CN)3 are determined as rIn–C = 2.251(1) Å and rC–N = 1.125(1) Å. The cyanide stretching mode was observed at νCN = 2200 cm−1 and the band at ν = 415 cm−1 was assigned to the In–CN stretching mode. These structures have zeolitic properties which have been described earlier for Prussian blue-like structures. In this context the authors noted the incorporation of solvent molecules or krypton atoms in the zeolitic structure of In(CN)3 which was verified by powder XRD (Williams et al., 1998). For example, the compound reversibly incorporates krypton atoms into the empty cavities to form In(CN)3⋅Kr.
The molecular structure of gas phase In(CN)3 was computed by A. Y. Timoshkin and H. F. Schaefer (Timoshkin and Schaefer, 2000). The results differ from the solid state structure of In(CN)3 which forms a coordination polymer (framework). The In–C bond length was calculated as rIn–C = 2.080 Å while the bond length of the cyanide group is reported as rC–N = 1.169 Å (B3LYP/LANL2DZP). The result of the cyanide absorption band of νCN = 2274 cm−1 is consistent to other E(CN)3 molecules (Timoshkin and Schaefer, 2000).
Thallium
The significant differences in the chemical properties of thallium and indium additionally influence the reaction behavior and substance properties of their compounds. In the last 90 years, various investigations have been carried out and frequently reported on thallium(I) cyanide, especially in aqueous mixtures. As far as the 3rd main group is concerned, thallium is the only element that forms the distinct EICN compound (E = B, Al, Ga, In, and Tl) (Bassett and Corbet, 1924; Lanzisera and Andrews, 1997b; Penneman and Staritzky, 1958). Many studies of thallium(III) species focused on complexes formed in aqueous solutions and their characteristics (Bányai et al., 2001; Batta et al., 1993; Blixt et al., 1989).
Thallium monocyanide TlCN
In contrast to the compounds of other elements of the 3rd main group, thallium monocyanide, TlCN, has been studied intensively. As a result of the inert pair effect, the oxidation state + I is more stable compared to the lighter group 13 elements. It should be noted that the chemical behavior of thallium(I) is similar to that of an alkali metal. Consequently, TlCN is known for more than 90 years, when H. Bassett and A. S. Corbet studied different aqueous mixtures of TlCN and KCN in 1924 (Bassett and Corbet, 1924). Even though, later findings questioned the existence of K[Tl(CN)2] announced in their work, it underlines that TlCN was discussed even before the 1930s. In addition to the synthesis of TlCN by reaction of thallium nitrate and potassium cyanide, R. A. Penneman and E. Staritzky described the formation of TlCN by cation exchange using an aqueous solution of potassium cyanide (Penneman and Staritzky, 1958). The IR absorption band of the cyanide stretching mode at νCN = 2048 cm−1 reported in this paper was confirmed by other reports, which included results for the application of thallium cyanide in organic synthesis (Penneman and Staritzky, 1958; Taylor et al., 1978).
E. C. Taylor et al. described a convenient and quantitative preparation of anhydrous thallium(I) cyanide under non-aqueous conditions by the reaction of dry hydrogen cyanide with thallium(I) phenoxide, together with some results on the use of TlCN for the preparation of ketonitriles, cyanoformates and trimethylcyanosilane (Taylor et al., 1978).
Thallium dicyanide Tl(CN)2
A framework compound with the empirical formula Tl(CN)2 was synthesized by D. Williams et al. However, this formal Tl(CN)2 is composed of a thallium mono-cation Tl+ and a thallium tri-cation Tl3+ resulting in a formula of TlITlIII(CN)4 = TlCN⋅Tl(CN)3 (Scheme 41) (Williams et al., 2001).

Synthesis of Tl(CN)2 by D. Williams et al. (Williams et al., 2001).
The structure was described as closely related to disordered Cd(CN)2 with
Thallium tricyanide Tl(CN)3
Due to the redox potentials of Tl+ and Tl3+ the formation of thallium tricyanide Tl(CN)3 was disregarded for a long time, but in the late 1980s, J. Glaser and several co-workers started systematic studies on thallium cyanide complexes in aqueous solution using manifold NMR techniques and could confirm the formation of Tl(CN)3 in solution. The suggestion of E. Penna-Franca and R. W. Dodson that Tl(CN)3 could be existing was verified (Batta et al., 1993; Blixt et al., 1989; Penna-Franca and Dodson, 1955). Furthermore, J. Glaser, M. Sandström and co-workers published structural data of thallium cyanide complexes of the structure [Tl(CN) n ](3–n)+ for n = 2, 3, 4 using X-ray and vibrational spectroscopy techniques in 1995 (Blixt et al., 1995). In the following years equilibrium dynamics including different ligand exchange processes were investigated (Bányai et al., 1997, 2001). A synthesis of Tl(CN)3 using thallium trichloride and trimethylsilyl cyanide by D. Williams et al. was not successful due to the formation of a material Tl2(CN)4 containing Tl+ and Tl3+, but four years later results of single crystal XRD experiments on the unstable adduct Tl(CN)3·OH2 (Nagy et al., 2005; Williams et al., 2001).
M. Maliarik, I. Tóth and co-workers dissolved Tl2O3 in water in the presence of strongly complexing cyanide ions. By this procedure they were able to identify the formation of Tl(CN)3(aq) by means of 205Tl NMR studies (Nagy et al., 2005). The dominant cyano complex of thallium(III) obtained in this way is Tl(CN)3(aq), which is in equilibrium with bicyano and tetracyano species (Scheme 42). When aqueous solutions of the MCN (M = Na+, K+) salts are used to dissolve thallium(III) oxide, the equilibrium in the liquid phase shifts completely to the [Tl(CN)4]– complex, which dominates at higher pH values and CN/Tl2O3 ratios (Nagy et al., 2005). The Tl(CN)3 can be selectively extracted from aqueous solution containing Tl(CN)3(aq) with diethyl ether. Depending on the water content in the ether phase, Tl(CN)3⋅H2O or TlI[TlIII(CN)4] crystals can be produced by slow evaporation of the solvent. In the crystal structure of Tl(CN)3⋅H2O, the thallium(III)-ion has a trigonal-bipyramidal coordination with three cyanide ions in the equatorial plane, while an oxygen atom of the water molecule and a nitrogen atom from a cyanide ligand bonded to an adjacent thallium complex form a linear O–Tl–N fragment. Cyanide ligands act as bridging ligands connecting the thallium units in an infinite zigzag chain structure. Besides the formation of the TlIII–CN complexes, notable redox reactions take place in the Tl2O3/HCN/H2O system. The redox reaction is complete when all thallium is in the form of TlI (reduction product) and the most stable cyano-complex of thallium(III), [TlIII(CN)4]–, which yields TlI[TlIII(CN)4] upon crystallization (see above) (Nagy et al., 2005).
– fromTl2O3 in aqueous HCN (Nagy et al., 2005).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_042.jpg)
Synthesis of Tl(CN)3(aq) and [Tl(CN)4](aq)– fromTl2O3 in aqueous HCN (Nagy et al., 2005).
Tetracyanidothalliate anion [Tl(CN)4]‒
The tetracyanidothalliate anion [Tl(CN)4]– has been prepared in aqueous solutions for the purpose of studying the different resulting species like Tl(CN)3(aq.), [Tl(CN)2]+(aq.) or [TlCN]2+(aq.). The determination of different characteristics like complex stability and electrochemical properties was achieved in these studies (Batta et al., 1993; Bányai et al., 1997, 2001; Blixt et al., 1989, 1995; Nagy et al., 2005). In this context, salts like K[Tl(CN)4] have been synthesized and crystallized by J. Glaser et al. in 1995 (and M. Maliarik, I. Tóth and co-workers – see above) by converting Tl(ClO4)3 into K[Tl(CN)4] according to Scheme 43 (Blixt et al., 1989).
![Scheme 43:
Synthesis of the potassium salt of [Tl(CN)4]– (Blixt et al., 1989).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_043.jpg)
Synthesis of the potassium salt of [Tl(CN)4]– (Blixt et al., 1989).
The salts K[Tl(CN)4] and TlI[TlIII(CN)4] are isostructural and derivatives of the Scheelite-type structure. The thallium–carbon distance was determined as rTl–C = 2.175(10) Å which agrees with the overall tendency of decreasing bond lengths with an increasing number of cyanides. The C–N bond length of the cyanide group was determined as rC–N = 1.153(14) Å (Nagy et al., 2005). A strong absorption band of the cyanide stretching mode was observed at νCN = 2180 cm−1 and the Tl–C band at νTlC = 358 cm−1 (Blixt et al., 1995). It should be noted that the crystal data of D. Williams et al. (Williams et al., 2001) of K[Tl(CN)4] and TlI[TlIII(CN)4] differ markedly from the data for the compound of the same composition, reported by M. Maliarik, I. Tóth and co-workers (Nagy et al., 2005).
4th Main group
While the cyanide compounds of the heavier 4th main group elements are not well investigated, the carbon cyanides have been studied by several approaches and the compounds have been analyzed for their characteristics. The inert pair effect also plays a major role in this group. For example, dicyanocarbene, C(CN)2, is a highly reactive covalent molecular substance, while lead(II) dicyanide, Pb(CN)2, is a classic, poorly soluble pseudohalogen salt. The C(CN)4, in turn, can be generated, while the Pb(CN)4 is unknown.
Of practical relevance are the cyanogen (CN)2 and the tricyanomethanide anion [C(CN)3]–. While the acute toxicity of (CN)2 constitutes a difficult aspect for research facilities, the reactivity and reaction behavior enable unique applications in synthetic chemistry. The [C(CN)3]– anion possesses weakly coordinating characteristics due to delocalization effects within the anion and forms ILs with appropriate cations which can be applied in different fields. There are no significant applications known for Si, Ge, Sn or Pb cyanides and the interests in these compounds are so far based on fundamental research.
Carbon
The carbon cyanide compounds are by far the best investigated element cyanide species of all 4th main group compounds. The first reports about cyanogen (CN)2 have been published in 1815 (Figure 28) (Gay-Lussac, 1815).
![Figure 28:
Structures of the mainly spectroscopically studied carbon monocyanide CCN, (CN)2 known since 1815, the highly reactive dicyanocarbene C(CN)2, the [C(CN)3]– and tetracyanomethane C(CN)4.](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_028.jpg)
Structures of the mainly spectroscopically studied carbon monocyanide CCN, (CN)2 known since 1815, the highly reactive dicyanocarbene C(CN)2, the [C(CN)3]– and tetracyanomethane C(CN)4.
Cyanogen thus represents one of the earliest known cyanide compounds of all substances studied besides HCN and cyanido ferrate salts (see introduction). Similar to the tetracyanidoborate anion, tricyanomethanide [C(CN)3]– has proven to be a WCA for ILs and has been intensively investigated with regard to its properties and possible applications (Brand et al., 2006). While carbon monocyanide CCN has mainly been studied due to spectroscopic interests motivated by observations of interstellar media, dicyanocarbene C(CN)2 has been investigated regarding the appearance as highly reactive intermediate in several reactions (Kakimoto and Kasuya, 1982; Swenson and Renaud, 1965). Tetracyanomethane C(CN)4 was successfully synthesized and characterized in 1969 (Mayer, 1969b).
Carbon monocyanide CCN
Speculations about the existence of the carbon monocyanide cation [CCN]+ in interstellar media constituted the motive for the initial investigations. The first reports were published in the early 1980s. The studies covered experimental reactions of the cation with different small molecules like H2O, H2S, HCN, hydrocarbons and other as well as computations of the molecule discussing geometrical characteristics and mechanistic considerations (Knight et al., 1988; Kraemer et al., 1984; Largo-Cabrerizo and Barrientos, 1988; McEwan et al., 1983; Raksit and Bohme, 1985; Tao et al., 2002a, 2002b; Wu et al., 2000). The ion can be generated by laser ablation of graphite and an ensuing reaction with gaseous hydrogen cyanide, similar to the procedure for 3rd main group monocyanides (Parent, 1990). The computational studies found a C–C bond length of rC–C = 1.383 Å and a C–N bond length of rC–N = 1.149 Å (HF/6-31G* and HF/6-31G(d)). The characteristic cyanide stretching mode was calculated to appear between νCN = 2536 cm−1 and νCN = 2257 cm−1 depending on basis set (Knight et al., 1988; Largo-Cabrerizo and Barrientos, 1988). Another study confirmed the band at νCN = 2529 cm−1 by SCF calculations but documented additional values at νCN = 2342 cm−1 (CI–SD) and νCN = 2249 cm−1 (CI-SDQ) (Kraemer et al., 1984). A conclusion derived from the theoretical approaches is that the cyanide isomer is unstable compared to the [CNC]+ cation. Based on these findings, C. G. Freeman, M. J. McEwan and co-workers speculate that the [CCN]+ cation is unlikely to be present in interstellar media (Knight et al., 1988).
The linear CCN molecule and its isomer CNC has initially been studied by A. J. Merer and D. N. Travis in 1965 and 1966, respectively (Merer and Travis, 1965, 1966). Since these first studies, different investigations have been conducted concerning the IR and MW characteristics while both, experimental and computational results were published (Belbruno et al., 2001; Brazier et al., 1987; Georgieva and Velcheva, 2006; Grant Hill et al., 2011; Hakuta and Uehara, 1983; Kakimoto and Kasuya, 1982; Kawaguchi et al., 1984; Ohshima and Endo, 1995). The total number of publications specifically on the rotational spectra and the associated rotational constants is high despite the synthetic irrelevance of this molecule (Brazier et al., 1987; Kohguchi et al., 1997; Oliphant et al., 1990). The studies closer to a synthetic application investigated the kinetic constants of CCN radical reactions (Wang et al., 2006; Zhu et al., 2003). The computational results of bond distances compared in Table 35 vary insignificantly for the C–C bond length and C–N distance, independent on the level of theory and basis sets.
Experimental and computational data of CCN (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r C–C | r C–N | ν CN |
|---|---|---|---|---|
| Hakuta and Uehara (1983) | Exp. – LIF | – | – | 1916.2 |
| Oliphant et al. (1990) | Exp. – EES | – | – | 1930.7 |
| Martin et al. (1994) | CASSCF/pVDZ | 1.4152 | 1.1993 | 1975.2 |
| CSSD(T)/TZ2P | 1.3908 | 1.1811 | 1995.8 | |
| Belbruno et al. (2001)[a] | B3LYP/6-311G* | 1.372 | 1.198 | – |
| B3LYP/cc-pVTZ | 1.3680 | 1.1795 | – | |
| B3LYP/cc-pVQZ | 1.3669 | 1.1780 | – | |
| Georgieva and Velcheva (2006) | B3LYP/6–31 + G(d,p) | 1.372 | 1.189 | 2033 |
| Hill et al. (2011) | CSSD(T)/VDZ-F12 | 1.3825 | 1.1857 | 1923.4 |
| CSSD(T)/VTZ-F12 | 1.3808 | 1.1849 | 1930.3 | |
| CSSD(T)/VDZ-F12 | 1.3805 | 1.1846 | 1932.3 |
-
[a]Calculations of CCN cluster.
The carbon monocyanide CCN molecule can be generated by a reaction of acetonitrile and microwave-discharge products of sulfur hexafluoride, flash photolysis of diazoacetonitrile or photolysis of cyanotrichloromethane (Hakuta and Uehara, 1983; Merer and Travis, 1965; Zhu et al., 2003).
The anionic [CCN]– molecule was investigated by several groups mainly for spectroscopic characteristics as well. Among the theoretical approaches, aspects concerning the geometry of cluster molecules [CnN]– with n > 2 were debated and whether the energetically favored structure would be linear or bent, especially calculations on higher levels of theory indicate a linear structure (Pascoli and Lavendy, 1999; Zhan and Iwata, 1996). The structural data are shown in Table 36.
Experimental and computational data of [CCN]– (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r C–C | r C–N | ν CN |
|---|---|---|---|---|
| Wang et al. (1995) | RHF/3-21G | 1.3443 | 1.1945 | – |
| Zhan, Iwata (1996) | MP2(full)/6-31G(d) | 1.357 | 1.224 | 1940.4 |
| MP2(full)/6-311G(d) | 1.360 | 1.217 | 1920.4 | |
| MP2(full)/6-311G(df) | 1.354 | 1.213 | 1940.4 | |
| Pascoli, Lavendy (1999) | B3LYP/aug-cc-pVTZ | 1.344 | 1.207 | 1758 |
| B3LYP/6-311G* | 1.350 | 1.210 | – | |
| Couturier-Tamburelli et al. (2003), Guennoun et al. (2003) | B3LYP/6-31G** | – | – | 1860 |
| Exp. – IR | – | – | 1779 |
Calculations of spectroscopic data fit well with the observations of νCN = 1779 cm−1 applying DFT methods and additional scaling results in a further decreased value of νCN = 1785 cm−1 as reported by I. Couturier-Tamburelli et al. (Guennoun et al., 2003). The synthesis of this anion was achieved by different techniques. It was usually prepared with other molecules of the [C n N]– constitution and is accessible by laser ablation of different inorganic substances like K3[Fe(CN)6], graphite/KCN or graphite/N2 as well as by UV photoisomerization of cyanoacetylene or dicyanoacetylene and by electron impact on gaseous acetonitrile (Garand et al., 2009; Guennoun et al., 2003; Wang et al., 1995).
The according [CCN]3– anion has so far been calculated twice by P. Pyykkö and Y. Zhao and by M. K. Georgieva et al. and was reported with a characteristic cyanide absorption band at νCN = 1756 cm−1 (HF/6-31G*) or νCN = 1728 cm−1 (B3LYP/6–31 + G(d,p)), respectively (Georgieva and Velcheva, 2006). The C–C and C–N distances were also documented by P. Pyykkö and Y. Zhao (rC–C = 1.315 Å, rC–N = 1.310 Å, HF/6-31G*) (Pyykko and Zhao, 1990). In 1989, G. Boche et al. also reported the synthesis of a lithium dianion containing [TMS–CCN]2–, which could be crystallized, and features several independent molecules with different structural parameters in the unit cell. The synthesis begins with a deprotonation of trimethylsilylacetonitrile with either two n-butyllithium or lithium diisopropylamide in ether/hexane that leads to the “dianion” Li2(TMS–CCN), which crystallizes from this solution as [(Li2(TMS–CCN))12(Et2O)6(C6H14)]. Twelve “dianions” form an aggregate with a crystallographic inversion center. There are three different “dianion” moieties, which differ in the number of their N–Li and C–Li contacts. The formal dianion, [TMS–CCN]2–, can also be considered as an [CCN]3– trianion (with TMS+ + 2Li+ as counter ions) with bond lengths of rC–C = 1.319 Å, rC–N = 1.235 Å in average (Zarges et al., 1989).
Cyanogen (CN)2
Already in 1959, T. K. Brotherton and J. W. Lynn published a detailed review about cyanogen, (CN)2, a compound that was first successfully synthesized by L. J. Gay-Lussac in 1815, when he thermally decomposed silver cyanide (Brotherton and Lynn, 1959). While it is not settled with certainty that the substance had been prepared earlier than 1815, it is well known that the first preparation of oxalic acid was accomplished by F. Wöhler in 1825 by hydrolysis of cyanogen (Wöhler, 1825a). In the following, we will only briefly describe the chemistry of (CN)2, otherwise we refer to the primary literature as well as the review articles.
Cyanogen is a highly flammable gas at ambient temperature and normal pressure and exothermically combusts in presence of oxygen, whereas a mixture of cyanogen and 14 vol% oxygen is explosive. Its toxicity results from the disproportion with water into hydrogen cyanide and isocyanic acid HNCO (Haas, 1978). Some additional characteristics are given in Table 37 (see also 1.5 HCN sources, toxicology, treatment, and mode of action (Newhouse and Chiu, 2010)).
Some experimental data of cyanogen (bond lengths in Å; wave number in cm−1; temperatures in K).
| Publication | r C–C [a] | r C–N [a] | ν as CN, exp. [b] | T M | T B | T C |
|---|---|---|---|---|---|---|
| Perry and Bardwell (1925) | – | – | – | 245.25 | 251.98 | – |
| Cook and Robinson (1935) | – | – | – | 245.3 | 251.98 | – |
| Pauling et al. (1939) | 1.37(2) | 1.16(2) | – | – | – | – |
| Haas (1978) | – | – | 2156 | 245.3 | 252.0 | 299.8 |
-
[a]Bond lengths determined by electron diffraction. [b]Wave numbers determined by IR spectroscopy.
Cyanogen can be synthesized by several different approaches. In laboratory scales, the decomposition of metal cyanides in an atmosphere of inert gas commonly provides cyanogen in sufficient amounts (Scheme 44). The presence of oxidizing agents can decrease the reaction temperature and increase the yield.

Cyanogen received by decomposition of metal cyanides (Haas, 1978).
It is better to thermolyze silver cyanide to generate (CN)2 than to use mercury dicyanide, which is easy to sublimate without releasing cyanogen (Haas, 1978). But in general, the decomposition of metal cyanides to obtain cyanogen is used less often in the meantime due to its inefficiency, hazardous nature and the danger of explosions caused by impurities like silver fulminate or mercury fulminate (Brotherton and Lynn, 1959).
The reaction of copper sulfate pentahydrate and potassium cyanide also leads to the formation of cyanogen in laboratory scales and the copper cyanide can be regenerated by addition of iron trichloride causing additional release of cyanogen (Scheme 45) (Haas, 1978).

Alternative formation of cyanogen in laboratory scales (Haas, 1978).
Another synthetic route is the pyrolysis of diacetylglyoxime, which also avoids the formation of additional toxic by-products and the reaction scale can be adjusted easily (Scheme 46) (Park et al., 1990).

Cyanogen received by pyrolysis of diacetylglyoxime (Park et al., 1990).
Industrial processes are based on the oxidation of hydrogen cyanide with oxygen. Cyanoformamide and oxamide are accessible on this way as well (Scheme 47). The kind of product, which is preferentially formed, depends on the reaction conditions due to the co-catalytic effect of copper nitrate and the solvent. The use of acetonitrile leads to the formation of cyanogen, aqueous acetic acid increases the formation of cyanoformamide at 303–323 K, while the increase of temperature to 333–353 K favors oxamide (Riemenschneider, 1978).

Formation of cyanogen, cyanoformamide and oxamide by oxidation of hydrogen cyanide (Riemenschneider, 1978).
As a highly reactive compound, cyanogen is an appropriate building block in organic chemistry. The reactions with amines, hydrazine, semicarbazide, aminoguanidine, hydroxylamine, ethene, dienes or magnesium organic compounds are reviewed in detail by T. K. Brotherton and J. W. Lynn (1959).
Cyanogen also polymerizes to black paracyanogen under the influence of heat or light, which has been known since the beginning of the nineteenth century (Delbrück, 1847). The amorphous structure of this polymer consists mostly of carbon atoms being sp2-hybridized and bridging nitrogen (Figure 29). Heating the polymer to 873–1073 K leads to the release of cyanogen. This structure motiv is supported by a strong IR absorption band at 1500 cm−1 and a significantly weaker one at 2200 cm−1. These absorption bands represent the C–N double bond vibration and the terminal C–N triple bond vibration, respectively. Moreover, 13C solid state NMR and conductivity experiments also support the structure of paracyanogen as illustrated in Figure 29 (Maya, 1993).
Possible representation of the structure of paracyanogen (representation taken from L. Maya (Maya, 1993) and based on a proposed structure by Brotherton and Lynn (1959).
Dicyanocarbene C(CN)2
The first description of dicyanocarbene C(CN)2, which is also called dicyanomethylene, was published in 1965. The initial report by J. S. Swenson and D. J. Renaud regarded the formation of C(CN)2 as an intermediate. The authors reported that the formation of 1,1-dicyanotetramethylcyclopropane due to in situ generated C(CN)2 in presence of tetramethylethylene (TME) proves the existence of the carbene in the reaction mixture as a highly reactive intermediate (Scheme 48) (Swenson and Renaud, 1965).
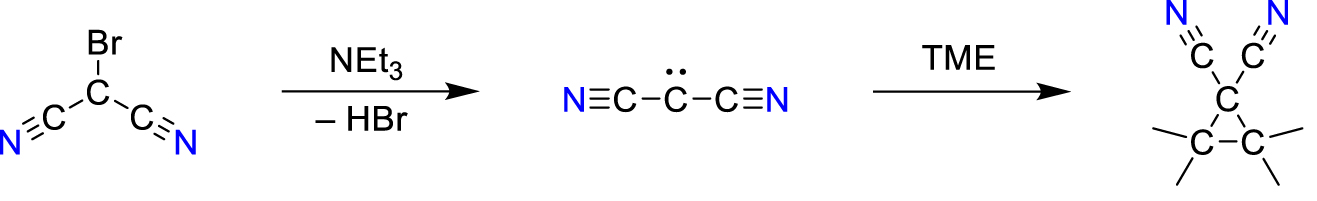
Formation of 1,1-dicyanotetramethylcyclopropane due to the reaction of in situ generated C(CN)2 with tetramethylethylene (TME) (Swenson and Renaud, 1965).
Ensuing this observation, E. Wasserman et al. published results including electron paramagnetic resonance (EPR) measurements of dicyanocarbene resulting from a photolysis of diazomalononitrile (Scheme 49).

C(CN)2 formed by photolysis of diazomalononitrile (Wasserman et al., 1965).
Based on their results, E. Wasserman and co-workers interpreted the structure of dicyanocarbene as linear in a fluorolobe matrix but slightly bent at the carbon center in a hexafluorobenzene matrix (Wasserman et al., 1965). This second observation was supported a few years later by R. R. Lucchese and H. F. Schaefer and their calculations using SCF methods (Lucchese and Schaefer, 1977). More recent calculations predict a linear structure of the ground state molecule in contrast to the first theoretical report, but bent structures of excited states have also been calculated (Blanksby et al., 2000; Chaudhuri and Krishnamachari, 2007; Hajgató et al., 2002; Maier et al., 2003). R. Hoffmann and co-workers concluded based on their theoretical approaches that there should be no doubt that the ground state of these molecules, including dicyanocarbene, would be a linear triplet (Hoffmann et al., 1968). Further reports dealt with the experimental and theoretical values for the IR active stretching mode of the cyanide groups and the C–C bond length (Table 38).
Some computational data of C(CN)2 (bond lengths in Å; angles in °; wave number in cm−1).
| Publication | Method + basis set | state | r C–C | r C–N | < (C–C–C) | ν CN |
|---|---|---|---|---|---|---|
| Lucchese and Schaefer (1977) | SCF/D95V | 3B1 | 1.407 | 1.154 | 132.5 | – |
| 1A1 | 1.421 | 1.160 | 114.9 | – | ||
| Blanksby et al. (2000) | B3LYP/6-31 + G(d) | 3Σg | 1.3176 | 1.1941 | 180.0 | – |
| 1A1 | 1.3694 | 1.1811 | 123.6 | – | ||
| Hajgató et al. (2002) | B3LYP/67–311 + G(3df) | 3B1 | 1.310 | 1.183 | 180.0 | – |
| 1A1 | 1.359 | 1.170 | 125.2 | – | ||
| Maier et al. (2003) | B3LYP/6-311 + G* | 3Σg | 1.312 | 1.186 | 180.0 | 2023.4 |
| Chaudhuri and Krishnamachari (2007) | CASSCF/ANO | 3Σg | 1.301 | 1.197 | 180.0 | 1892.4 |
| 1A1 | 1.400 | 1.153 | 113.2 | 2348.9 |
Furthermore, the report of R. R. Lucchese and H. F. Schaefer from 1977 noted an interesting inconsistency between the experiment and expectations regarding the bond lengths in the molecule. The C–C distance was calculated as rC–C = 1.41 Å, which is slightly closer to a double than a single bond (rcov(C–C) = 1.50 Å, rcov(C=C) = 1.34 Å) (Lucchese and Schaefer, 1977). This issue can be explained by the resonance structures shown in Figure 30.

Resonance structures of dicyanocarbene.
Hence, the C–N distance would be expected to have a significantly stronger double bond character. Instead, the calculated value of rC–N = 1.15 Å agrees with the expectations of a C–N triple bond (rcov(C≡N) = 1.14 Å) (Lucchese and Schaefer, 1977; Pyykkö and Atsumi, 2009). Additional studies led to similar results with slightly longer C–N distances of rC–N = 1.183 Å or rC–N = 1.1941 Å, but significantly shorter C–C bond lengths of rC–C = 1.3176 Å or rC–C = 1.310 Å indicating a triple bond character (Blanksby et al., 2000; Hajgató et al., 2002). Some studies also investigated the MS spectra of different dicyanocarbene precursors and detected the [C(CN)2]+ cation in addition (Blanksby et al., 2000; Hajgató et al., 2002; Smith-Gicklhorn et al., 2002).
Tricyanomethanide [C(CN)3]‒
Due to the high reactivity and strong acidity the synthesis and following isolation of tricyanomethane HC(CN)3, which is also known as cyanoform in regard to the haloform compounds HCX3 with X = Cl−, Br− and I−, was difficult. However, in 1977 B. Bak and H. Svanholt could identify cyanoform by MW spectroscopy (Bak and Svanholt, 1977). An excellent review on different reported approaches to generate pure cyanoform was given by L. B. McCusker, J. D. Dunitz and co-workers in 2010 (Šišak et al., 2010). Seven years later K. Banert et al. published another report dealing with the formation of HC(CN)3. Their isolation is based on a reaction of the potassium tricyanomethanide and a strong acid, sulfuric acid for example, and following sublimation (Scheme 50) (Banert et al., 2017).

Synthesis of tricyanomethane by K. Banert, H. Beckers et al. (n = 1, 2) (Banert et al., 2017).
With an approximate pKa of −5, cyanoform is classified as a super acid. Studies on the crystal structure also led to the discovery of the dicyanoketenimine as a tautomeric species (Šišak et al., 2010).
In 1896 Schmidtmann reported the synthesis of cyanoform, but actually he isolated and identified salts containing the tricyanomethanide anion. His early work included the simple silver and sodium tricyanomethanide salts and he already noticed analogies between the silver tricyanomethanide and silver halides (Brand et al., 2008). His findings were confirmed and pursued three years later by A. Hantzsch and G. Osswald who especially mentioned the similar characteristics of tricyanomethane and trinitromethane and their anions, respectively (Hantzsch and Osswald, 1899). Four years after the initial report of the series of pseudohalogens, L. Birckenbach and K. Huttner devoted a chapter of their essay entitled “Über Pseudohalogene” (About pseudohalogens) to the tricyanomethanide (see 1.2 Cyanide, a classic pseudohalide) (Birckenbach and Kellermann, 1925a, 1930; Birckenbach and Kolb, 1933; Birckenbach and Linhard, 1929; Brand et al., 2007; Jäger et al., 1992). The authors concluded that the reaction behavior and the general characteristics of the tricyanomethanide prove that the concept of pseudohalogens is not limited to inorganic chemistry but can also be applied to organic molecules and that the halogen-like chemical behavior results from the electronic configuration of the valence sphere (Birckenbach et al., 1929b; Brand et al., 2008). In 1922, W. Madelung and E. Kern already described the analogies of tricyanomethanide, dicyanamide and thiocyanate concerning characteristics and reaction behavior (Madelung and Kern, 1922a).
There are various successful synthetic routes to tricyanomethanide salts. The most common methods utilize malononitrile as starting material. The two main procedures are illustrated in a generalized modality in Scheme 51 (Schmidtmann, 1896; Trofimenko et al., 1962).

Generalized synthetic routes to obtain tricyanomethanide salts (Schmidtmann, 1896; Trofimenko et al., 1962).
Aside of the interests in the fundamental characteristics of cyanoform and its anion, tricyanomethanides were investigated for possible applications as ILs and building blocks in coordinative polymers (Batten and Murray, 2003; Bernsdorf et al., 2009a; Brand et al., 2006; Cioslowski et al., 1991; Hipps and Aplin, 1985; Weidinger et al., 2012; Zhang et al., 2003). Starting in the second half of the nineteenth century, various tricyanomethanide salts were synthesized and characterized by different groups and a detailed review on metal containing tricyanomethanide salts (especially Cu(II), Ni(II) and Co(II)) was given by J. Kohout et al. (2000). Several studies on ILs containing [C(CN)3]– as WCA have been published in the early years of this century and different synthetic routes towards various carbon based ILs were reported by A. Schulz and co-workers in 2006. The resulting room temperature ionic liquids (RTILs) are distinguished by low melting points and relatively high decomposition points ([EMIM][C(CN)3]: TM = 262 K, TDec. = 513 K; [BMIM][C(CN)3]: TM = 225 K, TDec. = 543 K) (Brand et al., 2006). Three years later, A. Schulz and co-workers expanded the methodology and synthesized advanced WCAs. The concept included the formation of Lewis acid-base adducts between nitrogen and bulky substituted boron atoms leading to a different group of carbon based ILs (Bernsdorf et al., 2009a). A rather unusual but nevertheless interesting application was investigated in 2009 when Z. Shou and J. M. Shreeve and co-workers described hypergolic ILs based on different WCAs including tricyanomethanide (Gao et al., 2009). Another possible application are alkylmethylimidazolium cation/tricyanomethanide anion based membranes that exhibit CO2 selectivity and permeability (Tzialla et al., 2013).
The molecular characteristics of the tricyanomethanide anion were elaborated by different groups and different approaches. The C–C and C–N bond lengths were determined by various computational levels of theory and by XRD (Andersen and Klewe, 1963; Brand et al., 2006; Dixon et al., 1986). Similar to the dicyanocarbene, the C–C distance is shorter than the expected value of a C–C single bond and thereby indicates a significant double bond character which can be explained by the resonance structure shown in Figure 31. Due to the strong delocalization (122.8 kcal mol−1 resonance energy) (Brand et al., 2008) within the [C(CN)3]– ion, it is planar in contrast to [H2CCN]–. Note [HC(CN)2]– is also planar. A CN group attached to a methanide C atom forms always a linear or nearly linear [< (C–C–N) = 175°] CCN moiety. Upon further CN substitution the C–CN bond lengths increases (CM: 1.380, DCM: 1.391, TCM: 1.406 Å) while the CN bond lengths decreases (CM 1.179, DCM: 1.169, TCM: 1.162 Å). Due to the very good delocalization of the π-electrons, the planar [C(CN)3]– anion is only very weakly basic, which conversely means that HC(CN)3 is a very strong acid. There are two almost energy-equal tautomers of the acid, namely non-planar cyanoform HC(CN)3 and a planar ketenimine tautomer HNC(CN)2 (Figure 31) (Banert et al., 2017).
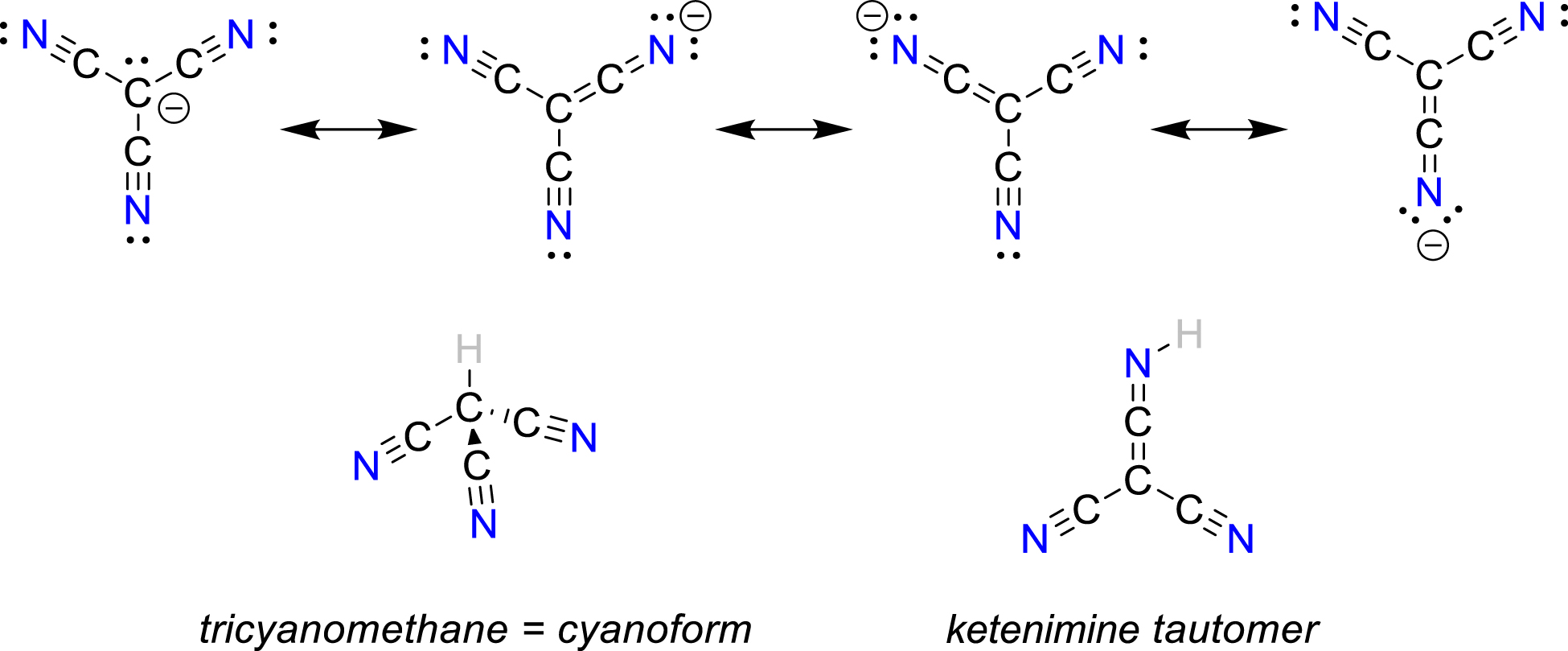
Top: Possible resonance structures of the tricyanomethanide anion and bottom: Tautomers of HC(CN)3.
Based on computational results of geometrical properties, frequency analyses have been carried out to predict vibration spectra and to compare them with experimental results (Beaumont et al., 1984; Long et al., 1962). Early calculations of the asymmetric C–N stretching mode from 1986 using a 3-21G basis set predicted the appearance around νCN = 2471 cm−1 (Dixon et al., 1986). These results disagree with experimental values by Δν = 300 cm−1. With the advancements in computational chemistry and higher levels of theory, this variance was reduced to Δν = 60 cm−1 in 2012 (Weidinger et al., 2012). Some results from different references are compiled in Table 39.
Experimental and computational data of the [C(CN)3]– anion (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | Cation | r C–C | r C–N | ν as. CN |
|---|---|---|---|---|---|
| Long et al. (1962) | Exp. – IR | K+ | – | – | 2175 |
| Andersen and Klewe (1963) | Exp. – XRD | K+ | 1.40 | 1.15 | – |
| Desiderato and Sass (1965) | Exp. – XRD | [H4N]+ | 1.40(1) | 1.15(1) | – |
| Konnert and Britton (1966) | Exp. – XRD | Ag+ | 1.54(9) | 1.10(9) | – |
| Köhler and Seifert (1968) | Exp. – IR | [SnMe2]2+ | – | – | 2185 |
| [SnPh2]2+ | – | – | 2175–90 | ||
| Beaumont et al. (1984) | Exp. – IR | K+ | – | – | 2170 |
| Dixon et al. (1986) | Exp. – XRD, IR | [Fe(Cp)2]+ | 1.402 | 1.159 | 2162 |
| [Bu4N]+ | – | – | 2158 | ||
| HF/3-21G | – | 1.399 | 1.149 | 2471 | |
| Cioslowski et al. (1991) | HF/6-31G* | – | 1.412 | 1.145 | – |
| Dua et al. (2000) | B3LYP/6-31 + G* | – | 1.4126 | 1.1732 | – |
| Brand et al. (2006) | Exp. – Raman | [EMIm]+ | – | – | 2178 |
| Exp. – Raman | [BMIm]+ | – | – | 2166 | |
| B3LYP/aug-cc-pVTZ | – | 1.406 | 1.162 | 2292 | |
| Bernsdorf et al. (2009a) | Exp. – IR | K+ | – | – | 2171 |
| exp. – IR | Ag+ | – | – | 2174 | |
| Weidinger et al. (2012) | Exp. – IR | K+ | – | – | 2170 |
| MP2/aug-cc-pVDZ | – | – | – | 2146.7 | |
| B3LYP/aug-cc-pVDZ | – | – | – | 2230.9 | |
| B3LYP/aug-cc-pVTZ | – | – | – | 2238.1 |
Regarding the computational approaches, the tricyanomethanide anion is computed with D 3h symmetry. This assumption was generally confirmed by XRD of different salts. However, observations indicating a C 2v distortion depending on the specific salt need to be mentioned (Dixon et al., 1986).
Tetracyanomethane C(CN)4
The synthesis of tetracyanomethane C(CN)4 was achieved by E. Mayer in 1969 by metathesis of cyanogen chloride and silver tricyanomethanide (Scheme 52).

Synthesis of C(CN)4 by E. Mayer (1969b).
Mayer also recorded IR spectra of the product to determine whether he obtained tetracyanomethane or tricyano-isocyano-methane. According to his interpretation, the spectra confirmed a molecule exhibiting T d symmetry and consequently the formation of C(CN)4 instead of its isomer and the T d symmetry was confirmed by ensuing studies (Hester et al., 1970; Mayer, 1969b; Oberhammer, 1971). Furthermore, Mayer documented the high reactivity of C(CN)4 by conducting several experiments. He proved that the highly reactive compound readily undergoes hydrolysis in acidic and basic aqueous solutions (Scheme 53).

Hydrolysis of C(CN)4 in acidic and basic aqueous solution (Mayer, 1969b).
Also, the reverse reaction to form tricyanomethanide salts starting with the C(CN)4 occurs with different salts. The conversion with lithium chloride gave lithium tricyanomethanide and cyanogen chloride in quantitative yields (Scheme 54). Additionally, the author mentioned that the formation of the [C(CN)3]– anion was partially observed in the KBr pellet which was prepared for IR spectroscopic measurements (Mayer, 1969b).
![Scheme 54:
Reverse reaction of C(CN)4 to form salts containing [C(CN)3]– (Mayer, 1969b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_054.jpg)
Reverse reaction of C(CN)4 to form salts containing [C(CN)3]– (Mayer, 1969b).
Other reports investigated structural aspects by electron diffraction in gas phase, single crystal XRD and computations (Britton, 1974; Oberhammer, 1971; Salzner and von Raguè Schleyer, 1992; Wiberg and Rablen, 1993). IR measurements generally confirmed the data obtained by E. Mayer in 1969 (Hester et al., 1970). Due to E. Mayer’s findings, a number of studies dealing with C(CN)4 were conducted, but since the report on the crystal structure has been published by D. Britton in 1974, no ensuing experimental studies have been published (Britton, 1974). A compilation of published results is given in Table 40.
Experimental and computational data of C(CN)4 (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r C–C | r C–N | ν CC | ν CN |
|---|---|---|---|---|---|
| Mayer (1969b) | Exp. – IR | – | – | 1058 | 2273 |
| Hester et al. (1970) | Exp. – IR – solid | – | – | 1056 | 2276 |
| Exp. – IR – vapor | – | – | 1061 | 2270 | |
| Oberhammer et al. (1971) | Exp. – GED | 1.484(5) | 1.161(3) | – | – |
| Britton (1974) | Exp. – XRD | 1.488 | 1.168 | – | – |
| Salzner and von Rahué Schleyer (1992) | HF/6-31G** | 1.484 | 1.131 | – | – |
| Wiberg and Rablen (1993) | RHF/6-31G* | 1.484 | 1.131 | – | – |
| MP2/6-31G* | 1.479 | 1.181 | – | – |
Two studies in the early 1990s supplementary discussed the varying properties of carbon tetrahalides. Both studies referred to the destabilizing character of the cyanide being part of the pseudohalogens in contrast to the stabilizing effect of fluorine substituents in compounds of a central carbon (Salzner and von Raguè Schleyer, 1992). According to K. B. Wiberg and P. R. Rablen, the destabilization is based on the strong π-acceptor properties of the cyanide group and the resulting accumulation of positive charge at the carbon center, which leads to an increasing coulomb repulsion (Wiberg and Rablen, 1993).
Graphitic carbon nitride (g-C3N4)
Graphitic carbon nitride (g-C3N4) is a class of binary CN layer compounds (often with non-zero hydrogen contents) based on heptazine and poly(triazine imide) units that exhibit different degrees of condensation, properties and reactivities depending on the reaction conditions (T, p, solvent, starting materials, etc., Scheme 55). Graphitic carbon nitride can be produced by polymerization of cyanamide, dicyandiamide or melamine. There are also methods of synthesizing graphitic carbon nitrides by heating a mixture of melamine and uric acid in the presence of alumina or by condensation of melamine and cyanuric chloride with triethylamine as solvent under supercritical conditions. Pyrolysis of melamine (in the presence of hydrazine) also yields g-C3N4. Graphitic carbon nitride (g-C3N4) has attracted much attention because of its high activity and efficient absorption of visible light. It is a binary CN layer compound with high chemical and thermal stability due to the strong covalent bond between the carbon and nitrogen atoms in the conjugated layer structure. Graphitic carbon nitride has a moderate band gap energy of 2.7 eV (460 nm), which can harvest visible light, and a suitable CB and VB edge position for both water reduction and oxidation. For this reason, g-C3N4 has quickly become a modern area of research as a potential “green” photocatalyst and has already expanded the field of photocatalysis. There is an abundance of review articles (Chen and Bai, 2020; Darkwah and Ao, 2018; Liu et al., 2020; Ong et al., 2016; Rhimi et al., 2020; Wen et al., 2017; Zhang et al., 2019) as well as articles dealing with its synthesis, characterization and application, which we do not wish to repeat here and therefore refer to these publications.

Synthesis of g-C3N4 (Chen and Bai, 2020; Darkwah and Ao, 2018; Liu et al., 2020; Ong et al., 2016; Rhimi et al., 2020; Wen et al., 2017; Zhang et al., 2019).
Silicon
Silicon cyanide compounds (cyanosilanes) have not been studied as intensively as carbon cyanides. The silicon monocyanide SiCN (Apponi et al., 2000; Flores, 2005; Guélin et al., 1986; Jin-chai et al., 2001; Largo-Cabrerizo, 1988; Maier et al., 1998; McCarthy et al., 2001; Ng et al., 2006; Pham et al., 2006; Richardson et al., 2003; Yamamoto and Okabe, 2011) has primarily been studied while the number of reports on Si(CN)2 (Maier and Reisenauer, 2005; Sakai and Inagaki, 1990) and Si(CN)4 is comparatively small. Salts bearing the octahedral hexacyanidosilicate, [Si(CN)6]2–, have been reported recently (Harloff et al., 2020a, 2019b; Smallwood et al., 2019). About neutral binary Si–CN compounds (e.g. SiCN and Si(CN)2) (Fukushima and Ishiwata, 2013; Guélin et al., 1986; Maier and Reisenauer, 2005; Maier et al., 1998; Mandal and Roesky, 2010; McCarthy et al., 2001; Richardson et al., 2003), which have been studied in an argon matrix or observed in the envelope of a star (Guélin et al., 1986, 2004; Mandal and Roesky, 2010), little is known, although cyanide-containing silanes, such as cyanotrimethylsilane (TMS–CN), are well known in organic chemistry for their versatile use as cyanosilylation reagents in combination with Lewis acids or bases (Bither et al., 1958; Booth and Frankiss, 1968; Groutas, 2001; Kalikhman et al., 2007; North et al., 2012). The more highly substituted dicyanodimethylsilane can be used as a protecting group (Mai and Patil, 1986; Ryu, 2001; Ryu et al., 1978), while the tricyanomethylsilane has not yet been isolated but is believed to be formed in situ in a reaction of MeSiCl3 with KCN (Duboudin et al., 1983).
Silicon monocyanide SiCN and its substituted cation
The term “SiCN” is also commonly used in material sciences for amorphous solids and nanoparticles containing mainly silicon, carbon and nitrogen and should not be confused with the silicon monocyanide molecule which has so far not been synthesized in significant amounts (Jin-chai et al., 2001; Ng et al., 2006; Pham et al., 2006).
The motivation behind the investigation of the silicon monocyanide radical [SiCN]⋅ corresponds with those of many other lighter element monocyanide compounds. An initial report about a new molecule observed in the envelope of the star IRC + 10216 in 1986 was followed by theoretical studies regarding the characteristics of this compound (Guélin et al., 1986). According to these calculations, the cyanide and its isomer [SiNC]⋅ both presumably appear linear in the 2Π ground state with the cyanide being the more stable isomer at several MP levels, but not at UHF level being the isocyanide the favored isomer (Largo-Cabrerizo, 1988). A more extensive study was conducted by G. Maier et al. in 1998 including additional computations and IR measurements of prepared [SiCN]⋅ molecules trapped in an argon matrix (Maier et al., 1998).
Both studies reported findings on the isomerization barrier, which is reported to be ΔE = 75 kJ mol−1 and isomerization occurring at a wave length of 366 nm. This aspect was additionally highlighted in a detailed report by N. A. Richardson et al. including calculations using numerous levels of theory. The results matched the predictions by A. Largo-Cabrerizo from 1988 well, even though the levels of theory explicitly differ (Largo-Cabrerizo, 1988; Richardson et al., 2003). Although some levels of theory indicate that the isocyanide is the stable isomer, both authors concluded that the cyanide species is predicted to be more stable by about ΔE ≈ 9 kJ mol−1 and that the barrier of isomerization accounts for about ΔE(TS) ≈ 95 kJ mol−1 (Table 41). The detailed report by N. A. Richardson et al. illustrates the significance of the employed computational method (Richardson et al., 2003). Due to the interest of astronomers and astrophysicists, P. Thaddeus and co-workers recorded and analyzed radio and rotational spectra of the molecule (Apponi et al., 2000; McCarthy et al., 2001).
Some computational data of [SiCN]⋅ (bond lengths in Å; wave numbers in cm−1; energies in kJ mol−1).
| Publication | Method + basis set | r Si–C | r Si–N | ν CN | ΔE(iso.) | ΔE(TS) |
|---|---|---|---|---|---|---|
| Largo-Cabrerizo (1988) | UHF/6-31G(d) | 1.866 | 1.746 | 2135 | −10.0 | – |
| MP2/6-31G(d) | – | – | – | 9.6 | 85.8 | |
| MP3/6-31G(d) | – | – | – | 3.8 | – | |
| Maier et al. (1998) | B3LYP/6-311G** | 1.857 | 1.752 | 2046.1 | 10.5 | – |
| Richardson et al. (2003) | SCF/TZ2P | 1.8812 | 1.7471 | 2458 | −16.66 | 101.07/91.27 |
| SCF/cc-pVTZ | 1.8826 | 1.7461 | 2459 | −20.72 | 101.57/89.51 | |
| CCSD/TZ2P | 1.8717 | 1.7577 | 2185 | 9.29 | 95.33/99.65 | |
| CCSD/cc-pVTZ | 1.8663 | 1.7502 | 2194 | 6.32 | 94.50/95.75 | |
| CCSD(T)/TZ2P | 1.8697 | 1.7594 | 2102 | 12.02 | 91.82/99.56 | |
| CCSD(T)/cc-pVTZ | 1.8635 | 1.7514 | 2109 | 9.46 | 90.85/95.63 | |
| Flores (2005) | B3LYP/6-311G* | 1.851 | 1.743 | – | 8.0 | 87.50 |
| QCISD/6-311G* | 1.863 | 1.751 | – | – | – | |
| MRCISD/cc-pVDZ | 1.879 | 1.785 | – | – | – | |
| Yamamoto and Okabe (2011) | B3LYP/6–311 ++ G(3df,3pd) | 1.846 | 1.733 | – | 1.993 | – |
By treatment of TMS–CN with [TMS–H–TMS]WCA ([WCA]– = [B(C6F5)4]–), it was possible to prepare the first example of a bissilylated [TMS–CN–TMS]+ ion, a so-called pseudohalonium cation, in high yields. It is thermally stable up to 461 K and the Si–CN–Si moiety is almost linear (<(C1–N–Si1) = 176.8(2)° and < (N–C1–Si2) = 178.5(2)°) with rather long Si–C (1.890(2) Å) and Si–N (1.888(2) Å) bond lengths (Schulz and Villinger, 2010).
Dicyanosilylene Si(CN)2
Studies on dicyanosilylene are rare but the accessible reports consist of detailed theoretical calculations and even experimental results from gas phase reactions of silicon with cyanogen. The initial report was published by S. Sakai and S. Inagaki in 1990 and additional information were contributed by G. Maier and H. P. Reisenauer in 2005, especially, the formation of three different open-chain silylenes, dicyanosilylene, cyano(isocyano)silylene and di(isocyano)-silylene, originating from a cyclic intermediate (Maier and Reisenauer, 2005; Sakai and Inagaki, 1990). In the following year, M. Z. Kassaee et al. also examined different silylenes including dicyanosilylene in a theoretical approach (Kassaee et al., 2006). Regarding the energetic difference between dicyanosilylene and di(isocyano)silylene, the calculations by G. Maier and H. P. Reisenauer indicate a lower energy of Si(NC)2. Due to the singlet ground state, an angled geometry results from the quantum mechanical calculations with < (C–Si–C) = 94.3° and a nearly linear Si–C–N angle of about < (Si–C–N) ≈ 170.4° (B3LYP/6-311 + G(3df,3pd)) (Maier and Reisenauer, 2005). The characteristics of Si(CN)2 are given in Table 42.
Computational data of Si(CN)2 and its isomers (bond lengths in Å; angles in °; wave numbers in cm−1; energies in kJ mol−1).
| Publication | Method + basis set | Compound | r Si–X | r C–N | < (A–Si–A) | < (Si–A–B) | ν as CN, calc. | ΔErel. |
|---|---|---|---|---|---|---|---|---|
| Maier and Reisenauer (2005) | B3LYP/6-311 + G(3df,3pd) | Si(CN)2 | 1.872 | 1.157 | 94.3 | 170.4 | 2252 | 0.0 |
| NCSiNC | 1.886/1.751 | 1.156/1.180 | 95.8 | 166.5/169.8 | 2261/2095 | −0.8 | ||
| Si(NC)2 | 1.758 | 1.179 | 97.0 | 165.0 | 2089 | −8.0 | ||
| Kassaee et al. (2006) | B3LYP/6–311++G** | Si(CN)2 | 1.88 | 1.16 | 94.2 | 170.0 | – | – |
| MP2/6-311G** | Si(CN)2 | 1.87 | 1.18 | 93.8 | 172.1 | – | – |
Silicon tetracyanide Si(CN)4
Silicon tetracyanide has been studied once by C. Glidewell in 1981 who determined the Si–C bond length of the tetrahedral molecule as rSi–C = 1.756 Å and the bond length of the cyanide group as rC–N = 1.163 Å in a theoretical study (MNDO) (Glidewell, 1981). A. Schulz and co-workers describe the formation of Si(CN)4 in the thermal decomposition of [ n Pr3NH]2[Si(CN)6] (Harloff et al., 2019b).
Hexacyanidosilicates [Si(CN)6]2–
Starting from fluoridosilicate precursors in pure cyanotrimethylsilane, TMS–CN, a series of different ammonium salts [R3NMe]+ (R = Et, n Pr, n Bu) were synthesized with [SiF(CN)5]2– and [Si(CN)6]2– dianions in simple, temperature-controlled F−/[CN]–-exchange reactions (Figure 32, Schemes 56 and 57). With decomposable, non-innocent cations like [R3NH]+, metal salts of the type M2[Si(CN)6] (M = Li+, K+) could be prepared by neutralization reactions with corresponding metal hydroxides. The ionic liquid [BMIm]2[Si(CN)6] (TM = 345 K, [BMIm]+ = 1-butyl-3-methyl-imidazolium) was obtained by a salt metathesis reaction (Harloff et al., 2019b).
![Figure 32:
Ball-and-stick representation of the molecular anion structure in the crystal: Left: [Ph4P]2[SiCl0.78(CN)5.22] · 4 CH3CN. Middle: [
n
Pr3NH]2[SiF(CN)5]. Right: [
n
Pr3NH]2[Si(CN)6]. Cations and solvent molecules are omitted for clarity (Harloff et al., 2019b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_032.jpg)
Ball-and-stick representation of the molecular anion structure in the crystal: Left: [Ph4P]2[SiCl0.78(CN)5.22] · 4 CH3CN. Middle: [ n Pr3NH]2[SiF(CN)5]. Right: [ n Pr3NH]2[Si(CN)6]. Cations and solvent molecules are omitted for clarity (Harloff et al., 2019b).
![Scheme 56:
Synthesis of salts bearing the [Si(CN)
x
Cl
y
]2– (x + y = 6) ion (Campbell et al., 2015).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_056.jpg)
Synthesis of salts bearing the [Si(CN) x Cl y ]2– (x + y = 6) ion (Campbell et al., 2015).
![Scheme 57:
Synthesis of salts bearing the [Si(CN)6]2– ion (Finger and Sundermeyer, 2016; Glasnov et al., 2012; Holbrey et al., 2010; Jost et al., 2016; Thielemann and Spange, 2017; Zheng et al., 2007).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_057.jpg)
Synthesis of salts bearing the [Si(CN)6]2– ion (Finger and Sundermeyer, 2016; Glasnov et al., 2012; Holbrey et al., 2010; Jost et al., 2016; Thielemann and Spange, 2017; Zheng et al., 2007).
Starting from chloride-containing precursors to synthesize salts with the [Si(CN)6]2– dianion does not work well (e.g. SiCl4 with two equivalents of [WCC]CN and four equivalents of AgCN; Scheme 56) (Campbell et al., 2015). All attempts at a full hexa-substitution failed, even when using a large excess of both cyanide sources.
P. Portius and co-workers also reported this problem and attempted to further convert the respective isolated compounds (PPN)2[Si(CN) x Cl y ] (x + y = 6) to the corresponding hexacyanidosilicate by multiple isolation/reaction with PPN[CN] ([WCC]+ = [PPN]+ = [(Ph3P)2N]+) (Smallwood et al., 2019). However, it can be assumed that a small chloride content always remains, as A. Schulz and co-workers were able to show (Harloff et al., 2019b). Therefore, the two synthesis routes A and B in Scheme 57 are more suitable for synthesizing highly pure, chloride-free [Si(CN)6]2– salts. As shown in Scheme 57, amines NR3 (R = Et, n Pr) can be reacted directly with aqueous hexafluorosilicic acid, H2SiF6, leading to the formation of the corresponding [R3NH]2[SiF6] salts in good yields. Secondly, ammonium methyl carbonates, ionic liquids with a decomposable anion (Finger and Sundermeyer, 2016; Glasnov et al., 2012; Holbrey et al., 2010; Jost et al., 2016; Thielemann and Spange, 2017; Zheng et al., 2007), were treated with aqueous H2SiF6, resulting in the preparation of tetraalkylated ammonium salts [R3NMe]2[SiF6] (R = Et, n Pr and n Bu) in very good yields. With the hexafluoridosilicates in hand, reaction with a large excess of TMS–CN, as a cyanidation reagent, led to the formation of salts containing the [SiF(CN)5]2– ion (after 2 h at 298 K). However, when the temperature was raised to 373 K, the reaction led exclusively to the formation of [Si(CN)6]2– salts (reaction time of 2 h). With small amounts of GaCl3 as Lewis acid catalyst added to the reaction mixture, the reaction time could be shortened (Bläsing et al., 2016a, 2016b).
The great advantage of [R3NH]2[Si(CN)6] salts over [R3NMe]2[Si(CN)6] salts is that the former have a decomposable, non-innocent cation (Bläsing et al., 2016a; Bresien et al., 2015a). Therefore, these salts are particularly suitable for the synthesis of metal salts, as they can be reacted with corresponding metal bases (Scheme 58, eq. 1). The potassium salt can be converted into the BMIm salt in a mixture of H2O/CH2Cl2 at 298 K. Thermal treatment of [R3NH]2[Si(CN)6] results in the liberation of HCN, free amine and Si(CN)4 (Scheme 58, eq. 3) (Harloff et al., 2019b).
![Scheme 58:
Synthesis of salts bearing the [Si(CN)6]2– ion (Harloff et al., 2019b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_058.jpg)
Synthesis of salts bearing the [Si(CN)6]2– ion (Harloff et al., 2019b).
Functionalized imidazolium cations were combined with the hexacyanidosilicate anion by salt metathesis reactions with K2[Si(CN)6], whereby ionic compounds of the general formula [R-Ph( n Bu)Im]2[Si(CN)6] (R = 2-Me, 4-Me, 2,4,6-Me = Mes, 2-MeO, 2,4-F, 4-Br; [Im]+ = imidazolium). All synthesized imidazolium hexacyanidosilicates decompose when thermally treated above 368 K (369–437 K). Furthermore, the hexa-borane adduct [Mes( n Bu)Im]2{Si[CN·B(C6F5)3]6}·6 CH2Cl2 (Figure 33), which is thermally stable up to 488 K, is obtained from the reaction of [Mes( n Bu)Im]2[Si(CN)6] with Lewis acid B(C6F5)3 (Harloff et al., 2020a).
![Figure 33:
Molecular structure of the {Si[CN·B(C6F5)3]6}2– anion (Harloff et al., 2020a).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_033.jpg)
Molecular structure of the {Si[CN·B(C6F5)3]6}2– anion (Harloff et al., 2020a).
Germanium
While germanium monocyanide and its ions have exclusively been studied theoretically (Petrie, 1999b; Wang et al., 2005), germanium(II) and germanium(IV) cyanide have been prepared successfully (Bither et al., 1958; Bundhun et al., 2012; Menzer, 1958; Onyszchuk et al., 1986). Recently, salts containing the [Ge(CN)6]2– anion were prepared (Smallwood et al., 2019).
Germanium monocyanide GeCN
S. Petrie examined trends of the main group elements of the third row and noted that GeCN is linear and the stable isomer in contrast to the lighter third row element monocyanides. Additionally, the barrier of isomerization appears to be lower compared to the corresponding potassium, calcium and gallium compounds.
Furthermore, they calculated the germanium cyanide cation and documented that its characteristics are comparable to those of isoelectronic GaCN in terms of the molecular geometry and the more stable isocyanide isomer (Petrie, 1999b). These results were confirmed by Q. Wang et al. in a detailed study concerning exclusively GeCN and its ions in 2005. According to their results, the cyanide species is the favored isomer of the anionic compound. A triplet state is documented for the anion while the singlet state of the isocyanide species is energetically favored by the cation (Wang et al., 2005). Selected calculated values are given in Table 43.
Computational data of GeCN, its isomer and the cationic and anionic species (bond lengths in Å; wave numbers in cm−1; energies in kJ mol−1).
| Publication | Method + basis set | Compound | r Ge–X | r C–N | ν CN | ΔErel. |
|---|---|---|---|---|---|---|
| Petrie (1999b) | MP2(full)/6-31G* | GeCN | 1.996 | 1.148 | – | – |
| GeNC | 1.774 | 1.189 | – | – | ||
| [GeCN]+ | 1.909 | 1.199 | – | – | ||
| [GeNC]+ | 1.798 | 1.219 | – | – | ||
| Wang et al. (2005) | B3LYP/6-311G(d) | GeCN | 1.9593 | 1.1648 | 2178 | 0.00 |
| GeNC | 1.8773 | 1.1857 | 2039 | 21.48 | ||
| 1[GeCN]+ | 1.9211 | 1.1658 | 2227 | 800.9 | ||
| 3[GeCN]+ | 1.9590 | 1.1682 | 2123 | 1115.28 | ||
| 1[GeNC]+ | 1.7949 | 1.2060 | 1975 | 780.21 | ||
| 3[GeNC]+ | 2.0411 | 1.1630 | 2091 | 1050.09 | ||
| 1[GeCN]– | 1.9843 | 1.1733 | 2089 | −100.06 | ||
| 3[GeCN]– | 2.0063 | 1.1703 | 2113 | −182.50 | ||
| 1[GeNC]– | 1.9629 | 1.1808 | 2037 | −49.95 | ||
| 3[GeNC]– | 1.9925 | 1.1771 | 2077 | −137.75 | ||
| QCISD/6-311G(d) | GeCN | 1.9738 | 1.1732 | 2140 | – | |
| GeNC | 1.8846 | 1.1907 | 2045 | – | ||
| 1[GeCN]+ | 1.9203 | 1.1734 | 2194 | – | ||
| 3[GeCN]+ | 2.0147 | 1.1778 | 2072 | – | ||
| 1[GeNC]+ | 1.7882 | 1.2102 | 1978 | – | ||
| 3[GeNC]+ | 2.3799 | 1.2534 | 1745 | – | ||
| 1[GeCN]– | 2.0185 | 1.1771 | 2111 | – | ||
| 3[GeCN]– | 2.0362 | 1.1771 | 2094 | – | ||
| 1[GeNC]– | 2.0016 | 1.1839 | 2072 | – | ||
| 3[GeNC]– | 2.0218 | 1.1829 | 2075 | – |
Germanium dicyanide Ge(CN)2
J. Satge and co-workers reported the successful synthesis of germanium dicyanide Ge(CN)2 among other germanium(II) pseudohalogens in 1986 by different synthetic routes following Scheme 59 (Onyszchuk et al., 1986).

Ge(CN)2 received by silver cyanide or exchange reaction using trimethylsilyl cyanide (Onyszchuk et al., 1986).
In contrast to the exchange reaction using trimethylsilyl cyanide TMS–CN, the reaction of germanium diiodide and silver cyanide leads to the highest yields (80%), but also the use of mercury(II) dicyanide is possible. After removal of the solvent the product is obtained as a viscous liquid being very sensitive to moisture and showing a dominant absorption band at νCN = 2090 cm−1 assigned to the cyanide stretching mode. The authors also reported further experiments carried out on Ge(CN)2 including cycloadditions, insertion reactions or formation of Lewis acid-base complexes being so far the only experimental study on the preparation and chemical behavior of Ge(CN)2 (Onyszchuk et al., 1986). A theoretical study by H. F. Schaefer and co-workers highlighted germanium dicyanide and investigated isomers and electronic states which led to the perception that the dicyanide isomer in the singlet ground state is energetically favored. The Ge–C bond length was calculated as rGe–C = 1.970–2.005 Å (MP2, BLYP) and the bond length of the cyanide group as rC–N = 1.156–1.196 Å (BHLYP, MP2). Additionally, the cation (doublet ground state, rGe–C = 1.890 Å, rC–N = 1.176 Å, B3LYP) and anion (doublet ground state, rGe–C = 2.022 Å, rC–N = 1.178 Å, B3LYP) of Ge(CN)2 were calculated (Bundhun et al., 2012).
Germanium tetracyanide Ge(CN)4
In 1958, two independent reports were published describing the preparation of germanium tetracyanide Ge(CN)4. W. Menzer converted the germanium tetraiodide with silver cyanide while T. A. Bither et al. utilized germanium chloride and trimethylsilyl (iso)cyanide (Scheme 60) (Bither et al., 1958; Menzer, 1958).

Synthesis of Ge(CN)4using germanium tetrahalides (Bither et al., 1958; Menzer, 1958).
The products were described as hygroscopic and insoluble in several solvents. The decomposition occurred by heating as well as immediately at air forming hydrogen cyanide (Bither et al., 1958; Menzer, 1958). With the exception of an elemental analysis reported by T. A. Bither and co-workers, no analytical identification of the molecule has been performed and no further studies have been published since these early reports.
Hexacyanidogermanate [Ge(CN)6]2–
The [PPN]+ salt with [Ge(CN)6]2– counterion has been synthesized, isolated, and fully characterized in the reaction of (PPN)CN and GeCl4 (Smallwood et al., 2019). However, more forcing conditions are required for the final reaction step [Ge(CN)5Cl]2− + CN− → [Ge(CN)6]2– + Cl−. In order to complete the Cl−/[CN]–-exchange, the chloride-containing product was further treated with NaCN in MeCN to give [PPN]2[Ge(CN)6] after re-crystallization. [PPN]2[Ge(CN)6] is moderately air sensitive and dissolves readily in polar aprotic solvents without noticeable dissociation.
Tin
Although tin halides are well known and organotin compounds are applied in organic syntheses, the cyanide compounds have been object of only a few studies. Whereas tin dicyanide Sn(CN)2 was prepared and analyzed by Mössbauer spectroscopy, tin tetracyanide Sn(CN)4 was applied as a starting material but not characterized (Harrison, 1972; Harrison and Stobart, 1973; Jung et al., 2010; Renz et al., 2009a). Recently, salts containing the [Sn(CN)6]2– anion were prepared (Smallwood et al., 2019).
Tin dicyanide Sn(CN)2
Whereas tin(II) halides are well known, tin(II) cyanide, Sn(CN)2, has not been studied intensively. So far, only two reports by P. G. Harrison from 1972 to 1973 are available (Harrison, 1972; Harrison and Stobart, 1973). The synthesis was achieved by protolysis of dicyclopentadienyl tin(II) with hydrogen cyanide to give a colorless, air sensitive solid with absorption bands at νCN = 2168 cm−1 and νCN = 2179 cm−1 in the IR spectrum (Scheme 61) (Harrison, 1972; Harrison and Stobart, 1973). It should be noted that the carbodiimide Sn(CN2) has been isolated from the solid-state metathesis reaction between equimolar amounts of Li2(CN2) and SnCl2 (Löber et al., 2019).

Synthesis of Sn(CN)2 by protolysis (Harrison, 1972; Harrison and Stobart, 1973).
Tin tetracyanide Sn(CN)4
Similar to the tin(II) compound, tin(IV) cyanide Sn(CN)4 has not been studied intensively as well. F. Renz et al. published two reports on the investigation of pentanuclear clusters with tin tetracyanide as bridging unit between iron complexes. The Sn(CN)4 was reported to be accessible by conversion of tin(IV) chloride and potassium cyanide (Scheme 62) (Jung et al., 2010; Renz et al., 2009a).

Synthesis of Sn(CN)4 by F. Renz et al. (Jung et al., 2010; Renz et al., 2009a).
An adduct of Sn(CN)4 was obtained in the reaction of SnF4 with an excess of TMS–CN resulting in the formation of Sn(CN)4⋅2 MeCN (Smallwood et al., 2019).
Hexacyanidostannate(IV) [Sn(CN)6]2–
As early as 1987, K. B. Dillon et al. studied the reaction of SnX4 (X = Cl−, Br−) with [ n Bu4N]CN but failed to isolate [ n Bu4N][Sn(CN)6] as they found always cyanohalogenostannates(IV) of the type [ n Bu4N]2[SnX m (CN) n ] (m + n = 6). However, on the basis of 119Sn NMR studies a resonance at ca. −917 ppm was assigned to the [Sn(CN)6]2– ion in solution (Dillon and Marshall, 1987). In 2019, P. Portius and co-workers described the synthesis of [PPN]2[Sn(CN)6] from Sn(CN)4⋅2 MeCN (see above) and an excess of [PPN]CN (Scheme 63). The νCN stretching modes of the slightly distorted octahedral anion (rSn−C = 2.06(1) – 2.10(1) Å) were found at 2157, 2153, 2146 cm−1 in the solid and at 2157.3 cm−1 in solution (Smallwood et al., 2019).
![Scheme 63:
Synthesis of [PPN]2[Sn(CN)6] (Smallwood et al., 2019).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_063.jpg)
Synthesis of [PPN]2[Sn(CN)6] (Smallwood et al., 2019).
Lead
Except for the observations by R. M. Kroeker and J. Pacansky of Pb(CN)2 formed by treating a magnesium layer, which has a monolayer of cyanoacetic acid and is coated with lead, with an electron beam, there is just one article reporting the quantitative formation of lead dicyanide by treatment of lead dihydroxide with an excess of potassium cyanide (Herz and Neukirch, 1923; Kroeker and Pacansky, 1982). But a retry of this reaction in an aqueous system led to a mixture of Pb(CN)2 having two distinct signals in a Raman spectrum (ν = 2057 cm−1 and ν = 2085 cm−1) and a lead compound containing nitrate and hydrogenoxide, where the isolation of lead dicyanide in quantitative amounts was not possible (Schulz et al., 2021). In a US patent from 1954 the synthesis of Pb(CN)2 is described: Pb(CN)2 is prepared by heating a dialkyllead dicyanide, such as diethyllead dicyanide, at 393–533 K. After sublimation a solid material, Pb(CN)2, was isolated (Shapiro et al., 1954).
Astonishingly, also in the CCSD data base there is no entry of Pb(CN)2. The only known and structurally characterized ternary Pb–C–N compounds are lead tricyanomethanides Pb[C(CN)3]2 (Deflon et al., 2006), lead dicyanamide Pb[N(CN)2]2 (Jürgens et al., 2002), and lead cyanamide Pb[NCN] (Zhao et al., 2018), which were synthesized from salt metathesis reactions of PbCl2 with Ag[C(CN)3], Na[N(CN)2] or Ag2[NCN].
5th Main group
The 5th main group is dominated by trisubstituted homoleptic cyanides due to the characteristic oxidation states (III) and (V) of the later elements.
The most important pnictogen monocyanides are nitrogen and phosphorus monocyanide, which are triplet biradicals. Die P–C bond is elongated and smaller in energy compared to the N–C bond, which leads to no formation of the nitrene configuration and the cyanide group is characterized by a triple bond. This is confirmed by shortened bond lengths and increased wave numbers of the stretching mode.
The pnictogen dicyanides are also dominated by nitrogen and phosphorus dicyanide. The differences of these two compounds mainly base on the various electronegativities. Hence, the central nitrogen atom of the dicyanamide anion has a significant negative charge, while the phosphorus atom has a positive charge. On the other hand, the dicyanamide anion is characterized by an angle of 120° indicating sp2-hybridization while the phosphorus species shows an angle of about 90° pointing to a square planar structure.
In regard to the pnictogen tricyanide compounds, the increase of the period of the central atom leads to a decrease of hybridization, to an increasing wave number of the stretching mode of the cyanide groups and to a decreasing angle between the cyanide groups. The diminishment of the angle is analogous to the less coordinated pnictogen cyanide compounds, while the increase of the wave number is contrary to the trends of the pnictogen mono- and dicyanides.
R. Dronskowski et al. investigated in detail the interactions in [E(CN)3] n (E = N–Sb, n = 1–3 and ∞) by means of computations. They found a significant preference for chain-like structures over molecular units (such as monomers, dimers or trimers) the heavier the pnictogen E. In addition, evidence of cooperativity in chains of pnictogen-linked E(CN)3 species is discussed. It is assumed that the pnictogen bonds are strengthened by such a cooperative effect (George et al., 2014).
Nitrogen
Cyanonitrene NCN and its ions
Many publications of cyanonitrene were published in the middle of the twentieth century (Milligan et al., 1965). The synthesis can be carried out as a photolysis of cyanogen azide using UV light at wavelengths of 280–300 nm or as a thermal decomposition (Scheme 64) (Dammeier and Friedrichs, 2010).

Synthesis of cyanonitrene by photolysis or thermal decomposition (Dammeier and Friedrichs, 2010).
On that way H. Okabe and A. Mele could determine, that the bond dissociation energy of the N−CN3 bond is Δ(NCN3) = (414.5 ± 20.9) kJ mol−1 and the standard formation enthalpy is ΔfH°(NCN) = (481.5 ± 20.9) kJ mol−1 (Okabe and Mele, 1969). In 1997, G. B. Ellison and co-workers published another synthetic route of cyanonitrene in terms of a radiation of cyanamine using oxide ion chemistry. The formed cyanonitrene anion is separated and photodetached by light of a CW Ar III ion laser with a wavelength of λ = 351.1 nm. The removed photoelectrons are focused and detected by passing them through an energy analyzer. The electron affinity of the radical is calculated from its photoelectron spectra and the determined thermochemistry compared to computational results (Clifford et al., 1997).
The supposed occurrence in interstellar matter and the radical structure of the cyanonitrene led to many theoretical studies of the molecular structure (Armstrong et al., 2000; Blanksby et al., 2000; Jiang et al., 2003; Schiavon et al., 2012; Yang et al., 2011). A. G. Anastassiou and H. E. Simmons reported a triplet ground state of cyanonitrene, which is lower in energy in contrast to three possible singlet states due to repulsion considerations and described by three mesomeric structures (Figure 34). Furthermore, they argued, the two unpaired electrons should be distributed among two degenerated, orthogonal molecular orbitals (Anastassiou and Simmons, 1967). In addition, experimental and computational values of the cyanonitrene and its isomers are compared in Table 44.

Lewis representation of the triplet ground state of cyanonitrene (Anastassiou and Simmons, 1967).
Experimental and computational values of cyanonitrene (bond lengths in Å; wave numbers in cm−1; electron affinity (EA) in eV).
| Compound | Exp./calc. | r C−N | ν CN | E A |
|---|---|---|---|---|
| [NCN]− | B3LYP/6-31 + G(d) | 1.2404 (Blanksby et al., 2000) | – | – |
| NCN | Exp. – LES, IR, NIPS | 1.230944(14) (Beaton et al., 1996) | 1197 (Milligan and Jacox, 1966) | 2.484 (Clifford et al., 1997) |
| B3LYP/6-31 + G(d) | 1.2341 (Blanksby et al., 2000) | – | 2.50 (Blanksby et al., 2000) | |
| [NCN]+ | B3LYP/6-31 + G(d) | 1.2377 (Blanksby et al., 2000) | – | – |
Dicyanamide [N(CN)2]‒
In 1922 W. Madelung and E. Kern published their studies on the synthesis and properties of dicyanamides (Madelung and Kern, 1922a; Madelung and Kern, 1922b). The consideration of properties was based on the diagonal relationship of the periodic table of elements and could be confirmed by them. Sodium dicyanamide can be technically prepared by introducing gaseous cyanogen chloride into a mixture of sodium hydrogencyanamide and sodium hydroxide solution, according to a patent by F. Thalhammer and H. Trautz in 2000 (Scheme 65) (Thalhammer and Trautz, 2000).
![Scheme 65:
Synthesis of Na[N(CN)2] (Thalhammer and Trautz, 2000).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_065.jpg)
Synthesis of Na[N(CN)2] (Thalhammer and Trautz, 2000).
As early as 1922, W. Madelung and E. Kern recognized that sodium dicyanamide at “dark red heat” trimerizes to trisodium tricyanmelaminate (Scheme 66). In 1997, the group of A. P. Purdy studied the preparation and properties of lithium dicyanamide. In the cause of this study they discovered a trimerization reaction, which was investigated thermoanalytically for the first time (Purdy et al., 1997). A. P. Purdy and co-workers were able to show, among other things, with DSC measurements that the sodium dicyanamide trimerizes in the temperature range of 553–673 K and the lithium dicyanamide trimerizes in the temperature range of 533–653 K. In a series of papers, W. Schnick and co-workers confirmed the assumptions of W. Madelung and E. Kern and the data of A. P. Purdy et al. on the trimerization of sodium dicyanamide (Irran et al., 2001, 2002; Jürgens et al., 1998, 2000, 2001, 2002, 2005; Lotsch et al., 2004).
![Scheme 66:
Trimerization of M[N(CN)2] (Purdy et al., 1997).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_066.jpg)
Trimerization of M[N(CN)2] (Purdy et al., 1997).
Furthermore, this work of W. Schnick and co-workers provided many interesting results on sodium dicyanamide (including IR data, DSC measurements on a reversible phase transition and powder diffractograms of the both phases), on its trimerization (including DSC measurements on the irreversibility of this solid reaction) and on the trisodium tricyanmelaminate (including a powder diffractogram of the anhydrous compound, a powder diffractogram, and a single crystal X-ray structural analysis of the trihydrate). Another 2001 publication of the group of W. Schnick on the preparation and properties of potassium dicyanamide and rubidium dicyanamide was concerned with the trimerization to tripotassium tricyanmelaminate and to trirubidium tricyanmelaminate, respectively (Scheme 66) (Irran et al., 2001). W. Schnick and co-workers described, among other things, that the potassium dicyanamide melts at 505 K, in the temperature range of 583–703 K its anions trimerize, and the melt solidifies during the formation of the tripotassium tricyanamide. For rubidium dicyanamide W. Schnick and co-workers found a melting point of 463 K and a temperature range of 533–643 K for the trimerization of its anions, although here, too, the melt solidifies. The cesium dicyanamide was already characterized by P. Starynowicz in 1991 by a single crystal X-ray structure analysis (Starynowicz, 1991). With the strong Lewis acid B(C6F5)3, both dicyanamide, [N(CN)2]–, and its trimer tricyanomelaminate, [C3N3(NCN)3]3– form stable adduct-anions, {N[CN⋅B(C6F5)3]2}– and {C3N3[NCN⋅B(C6F5)3]3}3– (Bernsdorf et al., 2009b; Voss et al., 2011).
Lab synthesis of dicyanamides usually begins with commerically available disodium cyanamide, Na2[NCN], which is treated with cyanogen bromide in an aqueous solution and the solvent is then removed by distillation (Scheme 67). It should be noted that the reaction of cyanogen bromide and cyanamide (H2NCN, monomer of melamine) does not lead to the expected product. The cyanamide is stable in aqueous solution, tends to oligomerize and acts as acid (Madelung and Kern, 1922a).

Synthesis of the sodium salt of dicyanamide (Madelung and Kern, 1922a).
In 1964, H. Köhler analyzed IR spectra of the sodium dicyanamide and different dicyanamide complexes and found for Na[N(CN)2] a stretching mode for νCN = 2179 cm−1 (Köhler, 1964). Further studies on the dicyanamide anion in different salts, its symmetry and behavior in IR light were published by M. Kuhn and R. Mecke (Table 45) (Kuhn and Mecke, 1961a). On the basis of the IR active modes they could confirm a bent structure of C 2v symmetry with a partial double bond character within the N−CN unit indicating the importance of mesomeric structures in the Lewis picture (Figure 35).
IR data of different metal dicyanamide salts (wave numbers in cm−1) (Kuhn and Mecke, 1961a).
| Compound | Na+ | Ag+ | Cu+ | Cu2+ | Hg2+ | Pb2+ |
|---|---|---|---|---|---|---|
| ν sym. CN | 2179 | 2183 | 2247 | 2203 | 2165 | 2125/60 |
| ν as. CN | 2232 | 2227 | 2283 | 2273 | 2251 | 2217 |

Mesomeric structures of the dicyanamide anion.
H. Köhler et al. studied potassium dicyanamide and determined its structure experimentally (Table 46) (Köhler et al., 1977).
Experimental and computational bond length (in Å) and bond angles (in°) of the dicyanoamide radical, anion and cation.
| Compound | Exp./calc. | r C−N | r N−CN | < (C–N–C) | < (N–C–N) |
|---|---|---|---|---|---|
| [N(CN)2]– | Exp. – XRD[a] (Köhler et al., 1977) | 1.155/1.145 | 1.304/1.317 | 120.8 | 172.6/172.9 |
| [N(CN)2]– | B3LYP/6–311++G** (Schalley et al., 1998) | 1.180 | 1.315 | 122.6 | 173.4 |
| [N(CN)2]∙ | B3LYP/6–311++G** (Schalley et al., 1998) | 1.182 | 1.302 | 124.1 | 173.1 |
| [N(CN)2]+ | B3LYP/6–311++G** (Schalley et al., 1998) | 1.198 | 1.280 | 130.9 | 171.8 |
-
[a]As potassium salt.
Computed data (B3LYP/6–311++G**) (Schalley et al., 1998) of the dicyanamide anion, [N(CN)2]–, the neutral radical, [N(CN)2]⋅, and the cation, [N(CN)2]+, are summarized in Table 47 indicating that the loss of electrons increases the enthalpy of formation while the zero-point vibrational energy (ZPVE) decreases.
Total energy E tot , ZPVE (in Hartree) and formation enthalpy (ΔfH in kJ mol−1) of the dicyanamide anion, radical and cation (E tot calculated using B3LYP/6–311 + G**) (Schalley et al., 1998).
| Compound | E tot. | ZPE | ΔfH |
|---|---|---|---|
| [N(CN)2]− | −240.4649 | 0.0211 | 23.0 |
| [N(CN)2]∙ | −240.3366 | 0.0192 | 360.1 |
| [N(CN)2]+ | −239.9035 | 0.0189 | 1498.0 |
The experimental and computational vibrational spectra of sodium dicyanamide were analyzed by M K. Georgieva in 2005 showing good agreement to calculated data (Table 48). As illustrated by a population analysis, the negative net charges are found at all nitrogen atoms with a slightly more negative charge on the central N atom (−0.72e), while a positive charge of + 0.94e sits on both carbon atoms (Figure 36) (Brand et al., 2009).
Experimental and computational IR data of Na[N(CN)2] (wave numbers in cm−1).
| Publication | Exp./calc. | ν sym. CN | ν as. CN |
|---|---|---|---|
| Georgieva and Binev (2005) | Exp. – IR | 2179 | 2232 |
| MP2/6-31G* | 2183 | 2199 | |
| B3LYP/6-31G* | 2209 | 2186 |

Negative partial charge distributation of the dicyanamide ion (Brand et al., 2009).
In 2009, N. Carrasco et al. repeated the formation of Titan’s (moon of the Saturn) tholins (see 2.7 [HCNH]+ cation and the [H(HCN) n ]+ cluster ion series), which is a polymeric mixture of high-nitrogen carbon compounds discovered on Titan for the first time (Sagan and Khare, 1979), when they could detect dicyanamide. In that experiment plasma radio frequency discharges are initialized in an atmosphere of nitrogen and methane. The formed products were investigated spectroscopically using IR spectroscopy, mass spectrometry and scanning electron microscopy. Characteristic mass-charge ratios are m/z = 66.01 of the dicyanamide ion as well as m/z = 40.01 and m/z = 26.00 of the fragments [NCN]– and [CN]–, respectively (Carrasco et al., 2009).
In addition, D. Ergöçmen and J. M. Goicoechea prepared the heavier homologue of the dicyanamide, the cyapho-cyanamide anion [N(CN)(CP)]–. Sodium phosphanide was added to the N-precursor dimethyl-N-cyanocarbonimidate leading to the sodium salt Na[N(CN)(CP)] and two equivalents of methanol (Scheme 68) (Ergöçmen and Goicoechea, 2021).
![Scheme 68:
Synthesis of the cyapho-cyanamide anion [N(CN)(CP)]– (Ergöçmen and Goicoechea, 2021).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_068.jpg)
Synthesis of the cyapho-cyanamide anion [N(CN)(CP)]– (Ergöçmen and Goicoechea, 2021).
Nitrogen tricyanide N(CN)3
Although N(CN)3 is a very simple molecule, it is unknown. On the one hand, this is due to the lack of suitable precursors, and on the other hand to the well-known isomeric equilibrium with the dicyano-carbodiimide (Scheme 69), which is very unstable with regard to oligomerization or polymerization. While tricyanamine has so far been elusive and has only been investigated theoretically (Riggs and Radom, 1985; Williams and Damrauer, 1971), there are experimental, i.e. spectroscopic, indications of the existence of dicyano-carbodiimide at very low temperatures (Ar matrix). However, it must be pointed out that the existence could not be confirmed in subsequent experiments (Sato et al., 2003, 2004). Although tricyanamine was mentioned by E. C. Franklin in his book “The Nitrogen System of Compounds” (Franklin, 1935), tricyanamine or its structural isomer dicyano-carbodiimide have only be observed as short-lived intermediates and all published procedures so far are of little preparative use.

Isomerism between tricyanamine and dicyano-carbodiimide.
In 2007, P. Politzer and co-workers published calculations dealing with directed, covalent bonds of molecules RX3 including nucleophiles and central atoms of the 5th main group, which led to the σ-hole concept. The geometry and electrostatic potential of N(CN)3 was optimized using DFT calculations. The ability for induction and resonance leads to a strong electron-withdrawing character of the cyanide group resulting in a planar structure of N(CN)3. The length of the carbon-nitrogen bond was found of 1.365 Å (B3PW91/6-31G**) indicating a distinct double bond character, which was confirmed by NBO analysis. The hybridization of the central nitrogen atom is sp2 and it possesses a 2p orbital, which is orthogonally orientated to the molecular plane (Figure 37) (Murray et al., 2007a).

Lewis representations of the nitrogen tricyanide (Murray et al., 2007a).
Polymerization of N(CN)3 formally leads to (C3N4)x. As previously explained for graphitic g-(C3N4) (see 6.1.6 Graphitic carbon nitride (g-C3N4)), the single-source concept is probably the most promising for attempts to produce a metastable carbonitride. This means that a reactive molecular precursor would be used, which can be condensed into a three-dimensional composite via the polymer intermediate stage. The simplest molecule with the right composition is the hypothetical tricyanamine N(CN)3, which is in equilibrium with dicyano-carbodiimide NC–N═C═N–CN via the well-known amine-imide structural isomerism (Scheme 69).
Phosphorus
Based on typical oxidation states of phosphorus, especially compounds with three and five substituents, such as PCl3 and PCl5, are known. Accordingly, the phosphorus tricyanide is known, which is also probably the best-known phosphorus cyanide compound, while the phosphorus pentacyanide P(CN)5 has not yet been studied in as much detail as P(CN)3. The reason for this is the redox instability of P(CN)5 with regard to the release of cyanogen, (CN)2. It is known that CN ligands have a comparatively low oxidation potential, which is reflected in the low thermal stability of P(CN)5, which easily releases cyanogen at room temperature to form P(CN)3. Also, the related [P(CN)6]– ion, on the other hand, is presumably stabilized by hyperconjugation. However, the synthesis of the [P(CN)6]– ion using conventional methods from (fluoro)phosphorus precursors is complicated by an affinity of P(CN)5 for F− (590 kJ mol−1), which is far greater than that for [CN]– (198 kJ mol−1) and could not be realized so far (Bläsing et al., 2016b; Bresien et al., 2015b; Smallwood et al., 2019).
Cyanophosphide PCN
The synthetic route often bases on an ac discharge in a mixture of gaseous starting materials, for example a dilute mixture of red phosphorus vapor and cyanogen (p = 1.33 Pa) in presence of argon gas (p = 4.67 Pa). But the use of phosphorus trichloride or phosphine in flash photolysis as phosphorus precursors in an atmosphere of nitrogen is also possible (Scheme 70) (Basco and Yee, 1968a; Halfen et al., 2012).

Flash photolysis of PCN using phosphine, cyanogen and nitrogen gas (Basco and Yee, 1968a).
The existence of small phosphoric molecules in interstellar matter inspired A. Largo et al. to carry out theoretical studies on PCN and its isomer PNC and also their ions. The ground state of both linear isomers is 3Σ−, the C–N bond length is not affected and very similar to the cyanide radical and also the P–N bond has a typical length of a single bond. In contrast to the isovalent CN2, the bond lengths of PCN are less affected by mesomeric effects. The two unpaired electrons are also mainly located at the phosphorus atom. Furthermore, a nitrene isomer with a P–C triple and a C–N single bond is not located as a minimum structure, which is a consequence of the high bond energy of the C–N triple bond (ΔBH = 888 kJ mol−1) in contrast to a C–P triple bond (ΔBH = 515 kJ mol−1). The proton affinity of the phosphorus atom is 541.8 kJ mol−1, the proton affinity of the nitrogen atom of PCN is 752.8 kJ mol−1 (Largo and Barrientos, 1991; Pham-Tran et al., 2006) (Table 49).
Experimental data of PCN and computational data of PCN and its cation (bond lengths in Å; dipole moments μ in D; wave numbers in cm−1).
| Compound | Exp./calc. | r P−C | r C−N | μ | ν sym. CN |
|---|---|---|---|---|---|
| PCN[a] | Exp. – MW (Halfen et al., 2012) | 1.732(2) | 1.167(2) | – | – |
| B3LYP/cc-pVTZ (El-Yazal et al., 1997) | 1.7237 | 1.1697 | 2.68 | 2037 | |
| UHF/6-31G* (Largo and Barrientos, 1991) | 1.724 | 1.174 | 2.42 | 1771 | |
| [PCN]+ | B3LYP/cc-pVTZ (El-Yazal et al., 1997) | 1.6901 | 1.1783 | 3.67 | 2045 |
| UHF/6-31G* (Largo and Barrientos, 1991) | 1.675 | 1.193 | 3.85 | 2030 |
-
[a]Ground state 3Σ– of PCN.
Dicyanophosphide [P(CN)2]–
Dicyanophosphide, [P(CN)2]–, is well described (Harloff et al., 2020b; Schmidpeter and Zwaschka, 1977a, 1977b; Schmidpeter et al., 1985a; Sheldrick et al., 1979). A. Schmidpeter and F. Zwaschka first reported on the synthesis and properties of the ion in 1977 (Schmidpeter and Zwaschka, 1977a). [P(CN)2]– is stable in the form of the crown ether sodium salt both as a solid and in solution. The preparation is carried out by reacting P(CN)3 with diethyl phosphites, Na[PO(OEt)2], and [18]-crown-6 in THF under cooling to 253 K. In this case the product crystallizes with crown ether and THF in ratio of 1:1:1 (Scheme 71). It is also possible to use other alkali metal salts, for example KF, to transform the P(CN)3 into its phosphide anion in presence of crown ether. In all cases the crown ether is important to stabilize the salt as solid or in solution (Schmidpeter and Zwaschka, 1977a, 1977b; Schmidpeter et al., 1985a; Sheldrick et al., 1979).

Synthesis of dicyanophosphide using an alkaline diethylphosphite salt in presence of crown ether (Schmidpeter and Zwaschka, 1977a, 1977b; Schmidpeter et al., 1985a; Sheldrick et al., 1979).
Another synthetic route is based on the degradation of white phosphorus, P4, by the cyanide ion (as MCN salt). The [CN]– ion as a strong anionic nucleophile is able to generate different PCN-ions in the presence of acetonitrile and stoichiometric amounts of [18]-crown-6, but mainly the [P(CN)2]– can be isolated (Scheme 72). The observed dicyanophosphide salt [18]-crown-6-K][P(CN)2] has an orthorhombic crystal lattice with the space group Pna21 (Schmidpeter et al., 1985a).
![Scheme 72:
Synthesis of the dicyanophosphide anion (M = [18]-crown-6-Na, [18]-crown-6-K, NEt4, NBu4, N(PPh3)2) (Schmidpeter et al., 1985a).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_072.jpg)
Synthesis of the dicyanophosphide anion (M = [18]-crown-6-Na, [18]-crown-6-K, NEt4, NBu4, N(PPh3)2) (Schmidpeter et al., 1985a).
The crown ether is essential to increase the solubility of the cyanide salt. The increasing dark-red color of the reaction mixture indicates the reaction process due to the formation of polymeric phosphides. Though the ratio n/2 of phosphorus and cyanide determines the formation of the polymeric phosphide, but the largest observed phosphide has the index n = 16 which corresponds with the best stoichiometry (Scheme 73). A higher ratio leads to free cyanide anions and a smaller ratio means unreacted phosphorus. Furthermore, the formation of different phosphides (n ≤ 16) hinders the isolation of the pure salt M[P(CN)2] (Schmidpeter et al., 1985a). It is also possible to influence the equilibrium between phosphorus and dicyanophosphide on one side and phosphorus tricyanide/polyphosphides on the other side to increase the amount of the dicyanophosphide anion (Scheme 73). In this context, A. Schmidpeter et al. reported a strong influence of different nucleophiles on the equilibrium. For example, X = [CN]– led to a shift to the left side, but in case of X = H− a shift to the right side was observed (Schmidpeter et al., 1985a).
![Scheme 73:
Equilibrium of dicyanophosphide [P(CN)2]– (Schmidpeter et al., 1985a).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_073.jpg)
Equilibrium of dicyanophosphide [P(CN)2]– (Schmidpeter et al., 1985a).
In 2018, L. B. Macdonald and co-workers published a new approach to the dicyanopnictides [E(CN)2]– (E = P, As). Similar to an approach reported by A. Schmidpeter the phosphino ligand bis(diphenylphosphino)ethane (dppe) in [dppeE][BPh4] (E = P, As) was displaced by cyanide ligands using tetraphenylphosphonium cyanide [PPh4]CN as cyanide source (Binder et al., 2018).
In the crystal the nitrogen atoms of the cyanides are aligned towards the cation, but the phosphorus atom is not, which leads to bent cyanide groups as shown in Table 50. Dicyanophosphide is angled, in crystalline [[18]-crown-6-K][P(CN)2], the nitrogen of the CN group coordinates to the K+ ion so that zigzag P–C–N⋅⋅⋅K⋅⋅⋅N–C–P chains are present in the crystal (Schmidpeter et al., 1985a). The calculated negative net charge (C: −0.16; N: −0.36) is delocalized on the cyanide group corresponding to the shortened P–C bond and widened C–P–C angle in contrast to the P(CN)3, whereas the phosphorus has a slightly positive net charge of +0.05 (Figure 38). Due to the high negative charge on the nitrogen atom, the anion coordinates to cations via nitrogen (Sheldrick et al., 1979).
Experimental and computational data of [[18]-crown-6-Na][P(CN)2] (bond lengths in Å; angles in °; wave numbers in cm−1).
| Exp./calc. | rP−C (Sheldrick et al., 1979) | rC−N (Sheldrick et al., 1979) | < (C–P–C) (Sheldrick et al., 1979) | < (P–C–N) (Sheldrick et al., 1979) | νsym.CN (Schmidpeter et al., 1985a) | νas.CN (Schmidpeter et al., 1985a) |
|---|---|---|---|---|---|---|
| Exp. – XRD, IR | 1.76(2)/1.72(2) | 1.155 | 95.0(10) | 169(2)/175(2) | 2120 | 2097 |
| STO-3G | 1.77 | 1.16 | 94.7 | 175.3 | – | – |
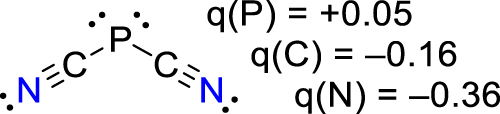
Calculated net charges of dicyanophosphide (Sheldrick et al., 1979).
In 31P NMR spectra the shift of the phosphorus in dicyanophosphides (δ = −193 ppm) is nearly independent of the solvent and the cation (Schmidpeter and Zwaschka, 1977b). With regard to 13C NMR spectra the chemical shift of the carbon is at about δ = 132.1 ppm with a coupling constant of 1J(31P–13C) = 106.2 Hz, which is similar to organic nitriles (Schmidpeter et al., 1985a).
Only recently it was found that salts containing the phosphaethynolate anion, [OCP]–, react with HCN, leading to the formation of the dicyanophosphide anion in a previously unknown decomposition process. In the case of [PPN][OCP], for example, the reaction with pure HCN as reaction medium and solvent led to the decomposition of the phosphaethynolate anion and finally to the formation of [PPN][P(CN⋅HCN)2] (Figure 39). This is an exergonic process according to calculations (ΔrG° = −35.8 kcal mol−1). It was assumed that formaldehyde is formed as a by-product according to the following reaction equation: [PCO]– + 4 HCN → [P(CN⋅HCN)2]– + H2CO. Its [PPN]+-salt crystallized in the form of an HCN solvate with the formation of a bent [P(CN⋅HCN)2]– anion (Figure 39) (Bläsing et al., 2020).
![Figure 39:
Structure of the [P(CN·HCN)2]– ion in [PPN][P(CN·HCN)2] (color code: P = orange, N = blue, C = grey, H = white) (Bläsing et al., 2020).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_039.jpg)
Structure of the [P(CN·HCN)2]– ion in [PPN][P(CN·HCN)2] (color code: P = orange, N = blue, C = grey, H = white) (Bläsing et al., 2020).
Dicyanophosphide can be regarded as a pseudohalide; accordingly, P(CN)3 could be described as an interpseudohalogen compound. This is consistent with the electrophilic transfer of the cyano radical to the dialkyl phosphite in the course of the above-mentioned synthesis.
Phosphorus tricyanide P(CN)3
The phosphorus tricyanide, P(CN)3, which was first mentioned by M. Cenedella in 1835 (Cenedella, 1835), is widely characterized. However, the start of the chemistry of phosphorus tricyanides dates back to 1863, when H. Hübner and G. Wehrhane described the isolation of “long, snow-white needles or hexagonal plates of cyanophosphorus” (German: Cyanphosphor), P(CN)3, in the reaction of PCl3 with three equivalents of AgCN at 393–413 K (Scheme 74) (Hübner and Wehrhane, 1863; Wehrhane and Hübner, 1864). In 1958, T. A. Bither et al. described the isolation of the entire series of E(CN)3 (E = P, As, Sb and Bi) obtained by treating pnictogen(III) chlorides ECl3 with three equivalents of TMS–CN in xylene under reflux conditions. All these species were characterized from elemental analyses only (Bither et al., 1958). In 1964, K. Emerson et al. carried out crystallographic studies on monocrystals (Emerson and Britton, 1964), which were synthesized by addition of phosphorus trichloride in excess to silver cyanide in carbon tetrachloride under reflux (Scheme 74) (Davies et al., 1976; Kirk and Smith, 1968).

Synthesis of P(CN)3 (Davies et al., 1976; Emerson and Britton, 1964; Kirk and Smith, 1968).
This synthesis was significantly improved in 1971 by C. E. Jones and K. J. Coskran (Jones and Coskran, 1971). When reacting PCl3 with AgCN in acetonitrile (MeCN) under a nitrogen atmosphere, the reaction already proceeds at room temperature within 2 h with good yield. Alternatively, PBr3 can also be reacted with AgCN in MeCN (Maier, 1963) or diethyl ether (Et2O) (Dillon and Platt, 1982). Another route is the reaction of phosphorus trichloride and methoxydimethylsilyl cyanide (Krolevets et al., 1991) achieving a 96.7% yield of P(CN)3 in 10–25 min at 303–323 K.
The phosphorus tricyanide sublimes at 323–333 K at normal pressure (Coskran and Jones, 1971). P(CN)3 is moisture-sensitive and hydrolyzes to H3PO3 and HCN (Gall and Schüppen, 1930). Work must therefore be carried out under protective gas and in dry solvents. Suitable solvents for P(CN)3 include MeCN, nitromethane (MeNO2) and 2-nitropropane (Wilkie and Parry, 1980) as well as dioxane and benzene (Kirk and Smith, 1968). In solution, P(CN)3 exists as a dimer or tetramer (Kirk and Smith, 1968). C. A. Wilkie and R. W. Parry found that the phosphorus atom in P(CN)3 can act as a Lewis acidic center (Wilkie and Parry, 1980). Due to the Lewis acidic character of the P atom, there are a number of adducts of the form [P(CN)3X]– (X = Cl−, Br−, I−) (Dillon et al., 1982; Sheldrick et al., 1981).
The crystals of P(CN)3 have the shape of flat colorless needles. The unit cell of the crystals is tetragonal (space group
Experimental average and computational data using different methods of phosphorus tricyanide (bond lengths in Å; angles in °; wave numbers in cm−1).
| Publication | Exp./calc. | r P−C | r C−N | < (C–P–C) | < (P–C–N) | ν sym. CN |
|---|---|---|---|---|---|---|
| Miller et al. (1965) | Exp. – Raman | – | – | – | – | 2206 |
| Emerson and D. Britton (1964) | Exp. – XRD | 1.803(29)/1.786(27)/1.774(27) | 1.124(47)/1.158(44)/1.165(44) | 93.7(2)/93.2(2)/93.6(2) | 170.7(3)/172.2(3)/171.9(3) | – |
| J. O. Jensen (2004a) | HF/6-311G** | 1.791 | 1.130 | 98.3 | 174.1 | 2567 |
| B3LYP/6-311G** | 1.790 | 1.155 | 98.6 | 172.4 | 2308 | |
| MP2/6-311G** | 1.786 | 1.179 | 97.6 | 173.1 | 2110 |
The standard formation enthalpy of gaseous P(CN)3 was reported to be ΔfH° = (494.6 ± 25.1) kJ mol−1 (Davies et al., 1976). The chemical shifts of P(CN)3 are summarized in Table 52 and the vibrational data in Table 53.
NMR data of phosphorus tricyanide (in acetonitrile, chemical shifts in ppm).
| Compound | Exp./calc. | δ (31P) | δ (13C) |
|---|---|---|---|
| P(CN)3 | Exp. | −138.3 (Coskran and Jones, 1971) | 110.54 (Tattershall, 1990) |
| −137.83 (Tattershall, 1990) |
Selected vibrational data (wave numbers in cm−1) of E(CN)3 and E(CN)3·2 THF (Arlt et al., 2016a).
| Compound | IR[a] | Raman[b] |
|---|---|---|
| P(CN)3[c] | 2205(m) | 2207(10) |
| As(CN)3[c] | 2209(m), 2199(s) | 2208(7), 2198(8) |
| Sb(CN)3 | 2293(w), 2251(m), 2188(m, br) | 2248(0.5), 2187(8) |
| Bi(CN)3 | 2291(m), 2254(s), 2174(m, br) | 2256(1), 2171(7) |
| Sb(CN)3·2 THF | 2170(m) | 2185(7,br) |
| Bi(CN)3·2 THF | 2154(m) | 2158(10,br) |
-
[a]Nicolet 380 FT-IR spectrometer with a Smart Orbit ATR at 298 K. [b]LabRAM HR 800 Horiba Jobin YVON Raman spectrometer (red laser 633 nm, 17 mW) at 298 K. [c]E(CN)3 (E = P, As) was prepared for comparison only.
Tetracyanidophosphate(III) anion [P(CN)4]– and tetracyanido phosphonium cation [P(CN)4]+
R. W. Parry et al. described the formation of the [P(CN)4]– ion in the reaction of potassium cyanide with phosphorus tricyanide in acetonitrile (Scheme 75). Only NMR data as well as vibrational data were given (Wilkie and Parry, 1980). C. A. Wilkie and R. W. Parry could not observe a signal in the 13C NMR spectrum, which clearly relates to the carbon atom of the cyanide groups. But they assumed, that the signal lies under the signal of the acetonitrile due to an expected downfield shift, which would mean a chemical shift of about δ (CN) ≈ 118 ppm. The added cyanide also causes a shift of the signal in the 31P NMR spectrum of about 53.6 ppm to more negative values (δ (31P) = −192.7 ppm). Compared to PCl3 and four times coordinated equivalent [PCl4]+ the direction of the chemical shift is opposite. C. A. Wilkie and R. W. Parry discussed the different charges of the ions and the different changes of symmetry as possible reasons, because PCl3 changes from C3v to Td by addition of a formal Cl+, but P(CN)3 changes from C3v to Cs by addition of the cyanide. The infrared spectrum of K[P(CN)4] displayed bands at 2195, 2160, 2097, and 2070 cm−1, suggesting a distorted trigonal-bipyramidal structure in the solid state (Table 54) (Wilkie and Parry, 1980).
![Scheme 75:
Top: Synthesis of the phosphorus tetracyanide anion by R. W. Parry et al. (Wilkie and Parry, 1980). Bottom: Decomposition of [P(CN)4]– (Schmidpeter et al., 1985b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_075.jpg)
Top: Synthesis of the phosphorus tetracyanide anion by R. W. Parry et al. (Wilkie and Parry, 1980). Bottom: Decomposition of [P(CN)4]– (Schmidpeter et al., 1985b).
IR data of [P(CN)4]– and related assignments (wave numbers in cm−1) (Wilkie and Parry, 1980).
| ν CN | Symmetry |
|---|---|
| 2070 | B1, sym., eq. |
| 2097 | B2, sym., ax. |
| 2160 | A1, sym., eq. |
| 2195 | A1, sym., ax. |
Investigations by A. Schmidpeter et al. have shown, however, that the [P(CN)4]– ion is indeed formed in solution by the addition of [CN]– to P(CN)3, but then rapidly decays in the sense of an intermolecular redox reaction. This produces dicyanophosphide, [P(CN)2]–, as well as the “unusual” anion [C10N10P2]2–, whose structure was published in the same paper (Schmidpeter et al., 1985b).
Furthermore, if one reacts [P(CN)2]– with X2 (X = Br, I), one obtains the [P(CN)2X2]– anion in the course of an oxidative addition. Similarly, X–CN adds to the [P(CN)3X]– ion. With Cl2, on the other hand, only P(CN)2Cl is formed (Schmidpeter and Zwaschka, 1979).
Tetracyanido phosphonium cation [P(CN) 4 ] + : In addition to the anion [P(CN)3X]– mentioned above, the cation [P(CN)4]+ was also found, but only in solution together with other tetra(pseudo)halogen phosphonium cations (Dillon and Platt, 1983). Different [PBr4–n(CN) n ]+ (n = 0–4) species were generated in the reaction of PBr5 with Zn(CN)2 in liquid bromine, which led to four new resonances in the 31P NMR spectrum. 31P NMR data of [PBr4–n(CN) n ]+ (n = 0–4; δ (31P) = −76.5, −49.3, −40.3, −33.3 and −41.9 ppm) were reported by K. B. Dillon et al. The [P(CN)4]+ assignment at −41.9 ppm was confirmed by measurements on a P(CN)3/(CN)2 system, where in addition to these resonances the signals of P(CN)3 (−130.6 ppm) and P(CN)5 (−98.4 ppm) are assigned (Dillon and Platt, 1983). Isolation of solid [P(CN)4]+ salts was not achieved.
Phosphorus pentacyanide P(CN)5
H. Gall and J. Schüppen carried out experiments dealing with penta-coordinated phosphates in 1930 (Gall and Schüppen, 1930). They tried to completely substitute the chloride atoms of phosphorus pentachloride PCl5 by cyanide and thiocyanate groups. When they heated a mixture of PCl5 and an excess of silver cyanide to a temperature of 403 K, they noticed the formation of large quantities of cyanogen (Scheme 76). But in this setup only phosphorus tricyanide was formed due to the high reaction temperature.

Release of cyanogen during the reaction of PCl5 and silver cyanide (Gall and Schüppen, 1930).
Based on this observation, they also heated a mixture of phosphorus tricyanide and cyanogen in a closed tube and realized a degradiation of the P(CN)3 crystals concluding that phosphorus pentacyanide was formed reversibly, which was confirmed by vapor pressure measurements (Scheme 77) (Gall and Schüppen, 1930). Due to the absence of analytical data of the product, the isolation must be questioned critically. Hence, so far, the in situ generation of P(CN)5 was only proposed and it should be noted that P(CN)5 should decompose rapidly at standard conditions.

Phosphorus pentacyanide formed in an equilibrium reaction (Gall and Schüppen, 1930).
Hexacyanidophosphate(V) anion [P(CN)6]‒
Despite many attempts, the hexacyanidophosphate(V) anion [P(CN)6]– has not been synthesized until now and has just been a hypothetical counterion in computational study e.g. by J. E. Bara and co-workers in 2012 or A. Schulz and co-workers in 2016 (Bläsing et al., 2016b; Shannon et al., 2012). However, as early as 1967, H. W. Roesky reported the synthesis of [ n Bu4N][PCl4(CN)2] starting from PCl5 and an excess of KCN (Scheme 78) (Roesky, 1967). Furthermore, H. W. Roesky was able to show that under the same reaction conditions he could isolate [Ph4P][P(N3)6], but not a salt containing the [P(CN)6]– ion.
![Scheme 78:
Attempted synthesis of [P(CN)6]– starting from PCl5 (Roesky, 1967).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_078.jpg)
Attempted synthesis of [P(CN)6]– starting from PCl5 (Roesky, 1967).
In 1982, K. B. Dillon and A. W. G. Platt reported the formation of [Et4N][PCl6–n(CN) n ] (n = 1–3) when they treated [PCl6]– salts with AgCN (Scheme 79). However, none of these salts were isolated and fully characterized (Dillon and Platt, 1982). P. J. Chevrier et al. published 19F NMR data of the [PF5(CN)]–-ion (Chevrier and Brownstein, 1980), and somewhat later K. B. Dillon et al. 31P NMR data of reaction mixtures containing [PF6–n(CN) n ]–- (1 ≤ n ≤ 4) and [PF3Cl3–n(CN) n ]–-ions (1 ≤ n ≤ 3) (Dillon and Platt, 1982). These reaction mixtures were obtained when PF5 was treated with [Et4N]CN or [PCl4(CN)2]– salts with AgF.

Synthesis of cyanido-chlorido and cyanido-fluorido phosphates (El-Yazal et al., 1997; Jensen, 2004b; Jung et al., 2010; Küppers et al., 2007; Leibovici, 1973; Sheldrick et al., 1981).
The first cyanido(fluorido)phosphate, [BMIm][PF3(CN)3] ([BMIm]+ = 1-butyl-3-methylimidazolium), was isolated in the reaction of [BMIm][PCl3(CN)3] and Ag[BF4] after four days of reaction (Tatsumi et al., 2012). Furthermore, [ n Bu4N][PF2(CN)4] was isolated in the reaction of [ n Bu4N][PF6] with an excess of TMS–CN under autogenous pressure in a steel autoclave at high temperatures (453–483 K, Scheme 79) (Bresien et al., 2015a, 2015b; Ellinger, Franke, et al., 2014; Ellinger, Harloff, et al., 2014; Sievert et al., 2015). By salt metathesis starting from [ n Bu4N][PF2(CN)4], a series of different M[PF2(CN)4] (M = Ag+, K+, Li+, [H5O2]+, [EMIm]+; [EMIm]+ = 1-ethyl-3-methylimidazolium) was successfully isolated and fully characterized.
The reaction of [PF6]–-salts with TMS–CN under Lewis acid catalyzed conditions was studied in detail by A. Schulz and co-workers (Bläsing et al., 2016b; Bresien et al., 2015b). This group was able to show that the best results, i.e. certain cyanido(fluorido)phosphate anions of the general formula [PF6–n(CN) n ]– (n = 1–4) can be generated by a very mild Lewis acid catalyzed synthesis protocol. Salts containing the [PF6–n(CN) n ]– ion (n = 1–4) can thus be isolated on a preparative scale (Scheme 80). It was also possible to detect the anion [PF(CN)5]– but not the anion [P(CN)6]–. The formation of the [P(CN)6]–-anion is thermodynamically not allowed as DFT calculations showed. All steps up to [PF(CN)5]– ion are exergonic only the 6th substitution step [PF(CN)5]– + CN− → [P(CN)6]– is endergonic (Table 55) in accord with the experimental observations (Bläsing et al., 2016b; Bresien et al., 2015b). NMR data of all fluorido-cyanido phosphates are listed in Table 56.
![Scheme 80:
Lewis acid catalyzed F−/[CN]–-exchange reactions leading to the formation of salts bearing the [PF6–n(CN)
n
]– (n = 1– 4) ion (Bläsing et al., 2016b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_080.jpg)
Lewis acid catalyzed F−/[CN]–-exchange reactions leading to the formation of salts bearing the [PF6–n(CN) n ]– (n = 1– 4) ion (Bläsing et al., 2016b).
Calculated gas phase Gibbs energies for the consecutive substitution reactions (ΔG298 in kcal mol−1, M06-2X/aug-cc-pVDZ level of theory) (Bläsing et al., 2016b; Bresien et al., 2015b).
| Reaction step | ΔG298 | ||
|---|---|---|---|
| [PF6]– + TMS–CN | → | [PF5CN]– + TMS–F | −9.12 |
| [PF5CN]– + TMS–CN | → | [cis-PF4(CN)2]– + TMS–F | −5.59 |
| [PF5CN]– + TMS–CN | → | [trans-PF4(CN)2]– + TMS–F | −6.83 |
| [trans-PF4(CN)2]– + TMS–CN | → | [fac-PF3(CN)3]– + TMS–F | −2.48 |
| [trans-PF4(CN)2]– + TMS–CN | → | [mer-PF3(CN)3]– + TMS–F | −3.07 |
| [cis-PF4(CN)2]– + TMS–CN | → | [fac-PF3(CN)3]– + TMS–F | −3.72 |
| [cis-PF4(CN)2]– + TMS–CN | → | [mer-PF3(CN)3]– + TMS–F | −4.31 |
| [mer-PF3(CN)3]– + TMS–CN | → | [trans-PF2(CN)4]– + TMS–F | + 0.24 |
| [fac-PF3(CN)3]– + TMS–CN | → | [trans-PF2(CN)4]– + TMS–F | −0.35 |
| [mer-PF3(CN)3]– + TMS–CN | → | [cis-PF2(CN)4]– + TMS–F | −2.47 |
| [fac-PF3(CN)3]– + TMS–CN | → | [cis-PF2(CN)4]– + TMS–F | −3.06 |
| [cis-PF2(CN)4]– + TMS–CN | → | [PF(CN)5]– + TMS–F | −0.09 |
| [trans-PF2(CN)4]– + TMS–CN | → | [PF(CN)5]– + TMS–F | −2.80 |
| [PF(CN)5]– + TMS–CN | → | [P(CN)6]– + TMS–F | + 2.48 |
| [PF6]– + 6 TMS–CN | → | [P(CN)6]– + 6 TMS–F | −19.10 |
13C, 19F and 31P NMR shifts (298 K in D2O) of all [PF6–n(CN) n ]− ions along with coupling constants (J) and multiplicity (M) (Bläsing et al., 2016b).
| Reaction step | δ31P | M | δ19F | M | δ13C | M | 1 J PF | 2 J FF | 1 J PC | 2 J FC |
|---|---|---|---|---|---|---|---|---|---|---|
| [PF6]– | −144.6 | sept | −73.0 | d | – | – | 707 | – | – | – |
| [PF5CN]– | −158.4 | dquin | −47.1 −75.1 |
dd dquin |
126.0 | dquin | 740 762 |
56 56 |
331 | 77 |
| [trans-PF4(CN)2]– | −170 | quin | −31.5 | d | 124.3 | dquin | 739 | – | 320 | 71 |
| [cis-PF4(CN)2]– | −183.1 | tt | −41.3 −54.3 |
dt dt |
127.7[a] | dddt | 710 764 |
43 43 |
247 | 37/41/53 |
| [mer-PF3(CN)3]– | −211.3 | dt | −8.7 −39.9 |
dt dd |
124.6 127.7 |
ddt ddt |
780 681 |
35 35 |
262 183 |
80/54/35/19 |
| [fac-PF3(CN)3]– | −219.3 | q | −40.5 | d | [b] | dq | 742 | – | [b] | 42[c] |
| [trans-PF2(CN)4]– | [b] | t | −48.8 | d | [b] | dt | 632 | – | [b] | [b] |
| [cis-PF2(CN)4]– | −269.1 | t | −6.1 | d | 125.7 128.1 |
dt dt |
730 | – | 218 143 |
65 38 |
| [PF(CN)5]– | −316.5 | d | 20.1 | d | [b] | dd | 708 | – | [b] | [b] |
| [P(CN)6]– | [b] | s | – | – | [b] | d | [b] | – | [b] | [b] |
-
[a]13C NMR data of the Li salt in d6-DMSO. [b]Not observed. [c]2JFC taken from 19F NMR spectrum.
Arsenic
Although As(CN)3 was first described in 1892 (Guenez, 1892), there are very few publications on the chemistry of arsenic cyanides. All of them essentially concern As(CN)3, i.e. with arsenic in the oxidation state + III. The experimental and theoretical IR and Raman data of As(CN)3 were also studied in detail (Cyvin et al., 1973; Edwards and Fawcett, 1987; Jensen, 2004b; Largo and Barrientos, 1991; Leibovici, 1973; Magnusson, 1986; Nagarajan, 1966).
Arsenic cyanide AsCN
The arsenic cyanide radical is rarely analyzed, but in 1968 it was part of a study by N. Basco and K. K. Yee, which deals with the absorption spectrum of flash photolysis of cyanogen and arsenic trichloride or arsane in an inert nitrogen atmosphere (Scheme 81).

Formation of arsenic cyanide by flash photolysis of cyanogen and arsane (Basco and Yee, 1968b).
AsCN was detectable during a period of τ = 150 ms, reaches its maximum of absorption intensity after τ = 3 ms and showed the largest triplet splitting of about 550 cm−1 of the 5th main group monocyanides (NCN: 40 cm−1, PCN: 104 cm−1) due to enhanced spin–orbit interaction (Basco and Yee, 1968b).
Ab initio calculations, using the G2 and G2(QCI) procedures, have been performed by S. Petrie on the various isomers of AsCN, which are summerized in Table 57 (Petrie, 1999).
Computational structural data of AsCN and AsNC (bond lengths in Å; angles in °) (Petrie, 1999).
| Compound | Method + basis set | r As–E | r C–N | < (As–A–B) |
|---|---|---|---|---|
| AsCN | MP2(full)/6-31G* | 1.853 | 1.191 | 85.58 |
| AsNC | QCISD(fc)/6-31G* | 1.816 | 1.200 | 81.01 |
In relation to both isomers, the triplet state is more stable than the singlet state associated with a triplet-singlet gap of about ΔH0K° = 90 kJ mol−1. Additionally, the cyanide isomer is 58.3 kJ mol−1 on G2 level and about 57 kJ mol−1 on G2(QCI) lower in energy than the isocyanide isomer, which implies the arsenic cyanide in triplet state is the most stable species (Table 58) (Petrie, 1999).
Computational data of the arsenic cyanide and isocyanide on different levels of theory (ZPE, ΔfH0K° and bond strength DAs–E in kJ mol−1; E0 in Eh including ZPE; AsNC in 1Σ at QCISD/6-31G*) (Petrie, 1999).
| Compound | State | HF/6-31G* | G2 | G2(QCI) | |||
|---|---|---|---|---|---|---|---|
| ZPE | E 0 | ΔfH0 K° | E 0 | ΔfH0 K° | D As–E, 0 K | ||
| AsCN | 1Σ | 19.6 | −2326.939 | 470.0 | −2326.940 | 473.0 | 276.5 |
| 3Σ | 16.6 | −2326.973 | 380.2 | −2326.974 | 383.2 | 366.2 | |
| AsNC | 1Σ[a] | 18.4 | −2326.917 | 528.3 | −2326.918 | 530.3 | 219.1 |
| 3Σ | 13.8 | −2326.951 | 438.5 | −2326.953 | 439.8 | 309.6 | |
-
[a]AsNC in 1Σ calculated at QCISD/6-31G*.
Dicyanoarsenide [As(CN)2]–
Similar to the synthesis of the analog [P(CN)2]– L. B. Macdonald and co-workers published a new approach to the [As(CN)2]–. The starting material [dppeAs][BPh4] (dppe = bis(diphenylphosphino)ethane) was obtained by a one-pot reaction of dppe, Na[BPh4] and arsenic trichloride AsCl3 as a pale-yellow solid based on M. Driess’ approach. Then the phosphino ligand dppe in [dppeAs][BPh4] was displaced by cyanide ligands using tetraphenylphosphonium cyanide [PPh4]CN as cyanide source similar to an approach reported by A. Schmidpeter (Binder et al., 2018).
Arsenic tricyanide As(CN)3
Arsenic tricyanide was synthesized for the first time in 1892 by M. E. Guenez by mixing cyanogen iodide and arsenic in carbon disulfide (Scheme 82) (Guenez, 1892). In 1958, T. A. Bither et al. described the isolation of As(CN)3, which were obtained by treating pnictogen chlorides ECl3 with three equivalents of TMS–CN in xylene under reflux conditions (Bither et al., 1958). As(CN)3 was only characterized on the basis of elemental analysis (Bither et al., 1958; Jones and Coskran, 1971).

Synthesis of arsenic tricyanide using arsenic and cyanogen iodide (Guenez, 1892).
The crystal structure was solved by K. Emerson and D. Britton in 1963. Their synthesis was an anhydrous reaction of silver cyanide and arsenic trichloride in acetonitrile by heating to reflux or using a glass tube under vacuum at 373 K (Scheme 83) (Emerson and Britton, 1963). The group of O. Sala reported a formation of arsenic trioxide in spite of careful exclusion of moisture during heating to reflux. Although, the use of the glass tube leads to smaller amounts of the oxide, the group carried out a sublimation to remove the oxide, but the product is still contaminated with arsenic trioxide (Miller et al., 1965). As(CN)3 can also be synthesized directly from AsCl3 with TMS–CN without the need for another solvent. This reaction already proceeds at room temperature and after the removal of all volatile products, a white substance is obtained which can be washed with n-hexane (decomposition point at 492 K) (Arlt et al., 2017). P. Deokar et al. carried out the reaction utilizing arsenic trifluorides and an excess trimethylsilyl cyanide in acetonitrile (Deokar et al., 2016).

Synthesis of arsenic tricyanide by K. Emerson and D. Britton (1963) S. Arlt et al. (Arlt et al., 2017) and P. Deokar et al. (Deokar et al., 2016).
As(CN)3 is highly moisture-sensitive and it should be noted that purification by vacuum sublimation increases decomposition (Edwards et al., 1976). IR and Raman data of all E(CN)3 (E = P, As, Sb, Bi) spezies are summarized in Table 53.
Arsenic tricyanide forms monoclinic crystals, space group C2 with four molecules in a unit cell. The molecules approximately exhibit C3v symmetry including one bent cyanide group showing external interaction to one arsenic of the neighboring layer. The molecules are orientated roughly vertical to the c-axis and to the chains in neighboring layers (Emerson and Britton, 1963). R. Haiges and co-workers have redetermined the As(CN)3 structure. The newly determined structure also shows an As(CN)3 molecule with the approximate C3v symmetry expected for an isolated trivalent As atom, containing three ligands and a sterically active free valence electron pair. The As–C bond distances are 1.956(3), 1.964(3) and 1.964(2) Å and the C–N distances of 1.133(3) and 1.134(3) Å, respectively, are very similar (Table 59). The arsenic atom is additionally coordinated by three N atoms from neighbouring cyano groups, resulting in a coordination number of six for the arsenic atom [3 + 3] (3 covalent + 3 van der Waals bonds). It can be seen that the As–C–N groups deviate significantly from linearity with an average angle of 174.8(4)°. The same is true for the non-linear geometry of P(CN)3. The three As⋯N bridge bond distances of 2.704(3), 2.823(2), and 2.837(3) Å are much shorter than the van der Waals value of 3.40 Å, indicating a strong association in the solid state (Deokar et al., 2016).
Furthermore, H. W. Roesky described the insertion of P(CN)3 and As(CN)3 into the dimer of hexafluorothioacetone (Scheme 84) (Roesky and Dhathathreyan, 1984; Roesky et al., 1985). R. Haiges and co-workers reported also on the adduct formation of As(CN)3 with 2,2′-bipyridine (2,2′-bipy) leading to the formation of As(CN)3⋅(2,2′-bipy) (Deokar et al., 2016).

Reaction of E(CN)3 species with hexafluorothioacetone (Roesky and Dhathathreyan, 1984; Roesky et al., 1985).
Experimental and computational data of As(CN)3 (bond lengths in Å; angles in °; wave numbers in cm−1).
| Publication | Exp./calc. | r As–C | r C–N | < (C–As–C) | < (As–C–N) | ν sym. CN |
|---|---|---|---|---|---|---|
| Emerson and Britton (1963) | Exp. – XRD | 1.82(5)/1.85(5)/1.96(5) | 1.13(7)/1.19(7)/1.14(7) | 87.6(3.5)/91.5(3.5)/91.5(3.5) | 175(8)/171(8)/168(8) | – |
| Edwards et al. (1976) | Exp. – Raman | – | – | – | – | 2205 |
| Deokar et al. (2016) | Exp. – XRD, IR | 1.964(3)/1.964(2)/1.956(3) | 1.134(3)/1.133(3)/1.138(3) | 89.94(10)/90.56(10)/90.47(10) | 174.5(3)/175.7(2)/174.5(2) | 2209 |
| B3LYP/aug-cc-pVDZ//-PP | 1.93518 | 1.16280 | 95.49 | 170.86 | 2289 |
Cyanidoarsenates and the unusual [AsC4N4]– ion
In principle, there are two ways to generate cyanidoarsenates (Scheme 85) in analogy to the cyanido phosphates or heavy pnictogens Sb and Bi. A. Schulz and co-workers reported two routes: (i) the reaction of E(CN)3 with weakly coordinating cyanide salts of the type [Cat]CN ([Cat]+ = e.g. [PPh4]+, [PPN]+ = [Ph3P−N−PPh3]+ and (ii) the treatment of EX3 with [Cat]CN in the presence of molar amounts of TMS–CN (X = halogen) (Schmidpeter et al., 1985b; Arlt et al., 2016b). Interestingly, the formation of [P(CN)4]– was described as a very labile intermediate in the reaction of P(CN)3 with CN− sources, which rapidly decomposes to [NC–P–CN]– and the unusual [C10N10P2]2– (Schmidpeter et al., 1985b), while the formation and isolation of the heavy cyanidopnictogen anions of the type [E(CN)3+n]n– (E = Sb, Bi) was only achieved when EX3 was treated with cyanide salts and TMS–CN (Arlt et al., 2016b), since E(CN)3 (E = Sb, Bi) are high polymer species that are almost insoluble in common organic solvents, so that no reaction was observed even after several days of reaction time and under reflux. It was shown that As(CN)3 does not form stable [E(CN)3+n]n– salts like phosphorus and unlike the heavier antimony and bismuth, which do form stable salts with [E(CN)3+n]n– ions.
![Scheme 85:
Synthesis of salts containing [As(CN)3+n]n– ions (X = F−, Cl−; n = 1–3; [Cat]+ = [PPh4]+, [PPN]+ = [Ph3P–N–PPh3]+; solvent = excess TMS–CN, CH3CN, ionic liquids) (Arlt et al., 2017).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_085.jpg)
Synthesis of salts containing [As(CN)3+n]n– ions (X = F−, Cl−; n = 1–3; [Cat]+ = [PPh4]+, [PPN]+ = [Ph3P–N–PPh3]+; solvent = excess TMS–CN, CH3CN, ionic liquids) (Arlt et al., 2017).
The chemistry of arsenic cyanides was found to be completely different from the chemistry of the heavier analogues antimony and bismuth as well as the lighter congener phosphorus. The reaction of As(CN)3 with cyanide salts led to the formation of an unknown cyanido arsazolide heterocycle, which is a structural isomer of the desired [As(CN)4]– (Arlt et al., 2017). The formation of this unusual heterocycle is discussed on the basis of computation featuring an arsenic-mediated C–C coupling of cyanides. [As(CN)4]– salts with various counterions such as [PPh4]+, [PPN]+, Ag+ and [BMIm]+ are reported, where [BMIm][AsC4N4] is a low temperature ionic liquid (TM = 211 K).
First the authors treated AsCl3 with MCN in TMS–CN which always led to the formation of [PPh4]2[As(CN)3Cl2] as shown in Figure 40. To avoid the Cl−/[CN]–-substitution problem, As(CN)3 was used in a second series of experiments (Scheme 85). Regardless of the stoichiometry, cyanide source and solvent (e.g. CH3CN, TMS–CN) or even ionic liquids such as [BMIm][OTf], [OTf]– = [F3CSO2O]–, always the structural isomer of [As(CN)4]–, the 4,5-dicyano-1,3,2-diazarsolide, [AsC4N4]– (Figure 40, Scheme 85), was isolated in quite good yields (up to 70%). As calculations show, heterocyclic [AsC4N4]– is formed in an unusual isomerization process starting from its structural isomer [As(CN)4]–, which is first formed as a transient species when As(CN)3 is treated with [CN]–. Then two arsenic-mediated C–C coupling steps occur, which finally lead to the formation of the aromatic ring system [AsC4N4]–, which can be regarded as a resonance-stabilized pseudohalide (Arlt et al., 2017).
![Figure 40:
Left: ORTEP representation of [As(CN)3Cl2]2– and Right: Heterocyclic [AsC4N4]– in the crystal (Arlt et al., 2017).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_040.jpg)
Left: ORTEP representation of [As(CN)3Cl2]2– and Right: Heterocyclic [AsC4N4]– in the crystal (Arlt et al., 2017).
[AsIII(CN) 6 ] 3– : There is only one report on this ion by L. Kahlenberg and J. V. Steinle (1923). They studied the redox potential of many salts and amongst these they also treated a solution of silver cyanide in aqueous potassium cyanide with arsenic. They write: “The reaction probably takes place as indicated by this equation: 3K[Ag(CN)2] + As = K3[As(CN)6] + 3Ag.” No analytical data are presented. Furthermore, [As V (CN) 6 ] – was mentioned in a Japanese patent on tetracyanidoborates, in which the authors calculated the highest occupied molecular orbital energy level of a great number of cyanide containing anions. Though the [As(CN)6]– has the smallest energy of −6.744 eV, which is also smaller than −6.561 eV of [P(CN)6]–, −5.961 eV of [Si(CN)5]– or −5.809 eV of [B(CN)4]– (Hagiwara et al., 2011).
Antimony
There are only very few publications on antimony cyanides, most of them are dedicated to the oxidation state(III). It was only in 2016 that the chemistry of Sb(CN)3 was taken up again by two research groups (Arlt et al., 2016a, 2016b; Deokar et al., 2016).
Antimony tricyanide Sb(CN)3
Sb(CN)3 was synthesized for the first time in 1958 by heating a mixture of trimethylsilyl cyanide TMS–CN dissolved in xylene and freshly sublimated antimony trichloride to reflux (Scheme 86). The white powder antimony tricyanide, which decomposes at temperatures higher than 473 K, was identified by elementary analysis (Bither et al., 1958; Jones and Coskran, 1971).

Synthesis of antimony tricyanide using antimony trichloride and TMS–CN (Bither et al., 1958).
Another synthetic route is the electrochemical preparation of Sb(CN)3, which was published by H. Schmidt and H. Meinert in 1958. The dissolved product is obtained at the antimony anode due to electrolysis of silver cyanide, which is also dissolved in pyridine, and can be isolated as colorless, hygroscopic crystals by evaporation of the solution (Schmidt and Meinert, 1958). The reaction of the antimony trifluorides with TMS–CN in acetonitrile in the presence of stoichiometric amounts of 2,2′-bipyridine (2,2′-bipy) led to the formation of the corresponding 2,2′-bipyridine adduct, Sb(CN)3⋅(2,2′-bipy) (Deokar et al., 2016).
A. Schulz and co-workers reacted pnictogen trifluoride EF3 with three equivalents of TMS–CN in various organic solvents such as acetonitrile or THF (Arlt et al., 2016a, 2016b). They also carried out the reaction in a large excess of TMS–CN. All these experiments resulted in the quantitative conversion of EF3 to E(CN)3 (E = Sb, Bi), which were obtained as highly amorphous white powders (Scheme 87). Both species are almost insoluble in standard organic solvents. Hence, it was not possible to produce single crystals of both E(CN)3 species by this method. Therefore, both species could only be characterized by elemental analysis and vibrational spectroscopy (Table 53). Mass spectrometric investigations showed no specific molecular peak. Moreover, both Sb(CN)3 and Bi(CN)3 hydrolyse slowly to E2O3, as shown by powder X-ray diffraction studies, and decompose without melting above 472 K (Sb(CN)3) and 439 K (Bi(CN)3), respectively. All these results indicate the presence of high polymer structured E(CN)3 species that exhibit strong intermolecular interactions, as predicted by the group of R. Dronskowski (George et al., 2014).

Synthesis of polymeric E(CN)3 species (E = Sb, Bi; solvent = MeCN, TMS–CN, THF) (Roesky, 1967).
However, it was shown that molecular Sb(CN)3 and Bi(CN)3 species, once formed in the synthesis process, can be stabilized in ionic liquids. To avoid oligomerization of the molecular E(CN)3 species, different ionic liquids were used as reaction media instead of (polar) organic solvents (Ahmed and Ruck, 2011; Freudenmann et al., 2011). By this approach it was possible to prevent the in situ formed E(CN)3 molecules from oligomerization by anion formation with the counterion of the ionic liquid (Scheme 3, Scheme 88). For example, in the reaction of SbF3 with TMS–CN in presence of [BMIm][OTf] ([BMIm]+ = 1-butyl-3-methylimidazolium, [OTf]– = triflate = [CF3SO3]–), it was possible to isolate and fully characterize [BMIm][Sb(CN)3(OTf)] in the first reaction step of the reaction sequence depicted in Scheme 88.
![Scheme 88:
Synthesis of E(CN)3⋅2 THF in ionic liquids ([BMIm]+ = 1-butyl-3-methylimidazolium, [EMIm]+ = 1-ethyl-3-methylimidazolium; [OTf]– = [CF3SO3]–) (Roesky, 1967).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_088.jpg)
Synthesis of E(CN)3⋅2 THF in ionic liquids ([BMIm]+ = 1-butyl-3-methylimidazolium, [EMIm]+ = 1-ethyl-3-methylimidazolium; [OTf]– = [CF3SO3]–) (Roesky, 1967).
The structure of the Sb(CN)3 adducts is characterized by a trigonal-pyramidal arrangement of the three cyanide groups as illustrated in Figure 41. In addition, there are three further van der Waals interactions with two solvent molecules and an adjacent CN group, so that here, too, one can speak of a formal [3 + 3] coordination or a distorted octahedral coordination sphere, quite in analogy to the lighter species E(CN)3 (E = P, As).

Molecular structure of Sb(CN)3⋅ 2 THF in the crystal.
Cyanidoantimonates [SbIII(CN)3+n]n– and [SbV(CN)6]–
The quest for cyanidoantimonates began with U. Müller who observed the formation of the [SbCl5(CN)]– anion upon addition of MCN (M = Na+, K+) to a solution of SbCl5 in SO2 (Müller, 1973). M. K. Rastogi studied the reaction of SbCl3 with KCN which afforded the formation of K2[SbCl3(CN)2] (Rastogi, 1987). In 2009 F. Renz and co-worker reported the isolation of an iron salt containing the [Sb(CN)6]– ion from the reaction of [SbCl6]– with KCN in acetone under reflux for six days (Renz et al., 2009b). In the light of the rather poor analytical data (ESI-MS and IR data, Mössbauer spectra) the formation of [Sb(CN)6]– ion seems to be rather doubtful. Actually, neither the mentioned ESI-MS and IR data, nor any elemental analysis data at all could be found. A retry clearly showed that it is impossible to get a complete Cl−/[CN]–-exchange by this procedure starting from [SbCl6]–.
When SbF3 is reacted with TMS–CN, only the insoluble polymer Sb(CN)3 is obtained, regardless of the solvent used. No molecular Sb(CN)3 solvent adducts could be isolated (Arlt et al., 2016b). However, when SbF3 is treated with three equivalents of TMS–CN in the presence of n equivalents of [WCC]CN (n = 1–3) in acetonitrile, [WCC]2[E(CN)5] salts are obtained (Scheme 89). In addition to [WCC]2[Sb(CN)5] (Arlt et al., 2016b), quantities of Sb(CN)3 were always detected, depending on the stoichiometry of [WCC]CN used, which is why the yields were rather low (between 20 and 30%). Thus, when only one equivalent of [WCC]CN was used, mixtures of Sb(CN)3 and [Sb(CN)5]2– were always observed. It was impossible to isolate or even observe the [WCC][Sb(CN)4] salts. When three equivalents of [WCC]CN were added, only [WCC]2[Sb(CN)5] salts could be isolated in addition to an excess of [WCC]CN. No formation of [WCC]3[Sb(CN)6] was observed (Arlt et al., 2016b). The structures of [PPN]2[Sb(CN)5] and [Ph4P]2[Sb(CN)5] have been determined (Figure 42). Both structures consist of well-separated cations and [Sb(CN)5]2– anions without significant cation⋯anion contacts. [Sb(CN)5]2– adopts a highly distorted square pyramidal geometry. There are no significant interanion interactions. The basal Sb–C distances range between 2.305(4) and 2.479(5) Å, while the apical Sb–C distances are much shorter at 2.142(4) Å (Arlt et al., 2016b).

Synthesis of cyanido antimonates (Arlt et al., 2016b).
![Figure 42:
Molecular structure of [Sb(CN)5]2– in the crystal (color code: blue = nitrogen, grey = carbon, green = antimony) (Arlt et al., 2016b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_042.jpg)
Molecular structure of [Sb(CN)5]2– in the crystal (color code: blue = nitrogen, grey = carbon, green = antimony) (Arlt et al., 2016b).
Bismuth
Bi(CN)3 is very similar to Sb(CN)3 in terms of synthesis and structure, as it also occurs as a polymer. The cyanido bismuthates, on the other hand, show a much greater variety.
Bismuth dicyanide Ph3Bi(CN)2
Until now, no homoleptic bismuth dicyanide has been published in literature, but several multivalent bismuth derivatives consisting of two cyanide groups and some organic substituents, for example phenyl groups. The first synthesis of triphenylbismuth dicyanide using triphenylbismuth dichloride and potassium cyanide was published by R. G. Goel and H. S. Prasad (Scheme 90).

Synthesis of triphenylbismuth dicyanide by R. G. Goel and H. S. Prasad (1973).
Furthermore, IR and Raman spectra of triphenylbismuth dicyanide, which can be obtained as colorless crystals, were published (Table 60) (Goel and Prasad, 1973). However, cyanide is also a common ligand of many different bismuth complexes, e.g. tetraethylamine-dicyano-diphenyl bismuthate. However, these organo-bismuth complexes are not part of this article and the reader is referred to the relevant literature (Allman et al., 1979).
Experimental spectroscopic data of the CN group of Ph3Bi(CN)2 (wave numbers in cm−1) (Goel and Prasad, 1973).
| spectrum | ν CN |
|---|---|
| IR | 2128, 2135, 2140 |
| Raman | 2136 |
Bismuth tricyanide Bi(CN)3
One of the first synthesis of bismuth tricyanide was published by H. Schmidt and H. Meinert in 1958. The synthetic route is similar to the electrochemical preparation of antimony tricyanide as the bismuth compound is obtained at the bismuth anode due to electrolysis of silver cyanide dissolved in pyridine (Scheme 91). As described for E(CN)3 (E = P, As, Sb, see above) also Bi(CN)3 can be prepared by treating BiCl3 with three equivalents of TMS–CN in xylene under reflux conditions (Bither et al., 1958; Jones and Coskran, 1971). The reaction of BiCl3 with TMS–CN was reported to be incomplete leading only to mixed chloride/cyanide bismuth species. However, Bi(CN)3 can be prepared utilizing the same procedure as discussed for Sb(CN)3 (see 7.4.1 Antimony tricyanide Sb(CN)3, Scheme 87, Scheme 88) (Arlt et al., 2016a). IR and Raman data of all E(CN)3 (E = P, As, Sb, Bi) spezies are summarized in Table 53. The bismuth tricyanide is also a colorless solid (Schmidt and Meinert, 1958).

Electrochemical preparation of bismuth tricyanide by H. Schmidt and H. Meinert (Schmidt and Meinert, 1958).
Crystals of Bi(CN)3⋅2 THF were studied exhibiting also a [3 + 3] coordination around the bismuth atom as found for the analogous antimony compound. Both THF adducts E(CN)3⋅2 THF (E = Sb, Bi) are highly moisture-sensitive and decompose above 485 and 409 K, respectively, under release of THF as shown by DSC/TGA experiments.
Cyanidobismuthates [BiIII(CN)3+n]n– and [BiV(CN)6]–
The reaction of in situ generated Bi(CN)3 with different amounts of [Ph4P]CN and [PPN]CN yielded salts with the ternary ions [Bi(CN)5]2–, [Bi2(CN)11]5– and [Bi(CN)6]3– (synthesis analogous to the reaction with Sb(CN)3; Scheme 86 - 88, Figure 43) (Arlt et al., 2016b). It was shown that the bismuth reaction mixture is highly dynamic with respect to the CN ligand sphere around the formal Bi3+ cation and that, depending on solubility, concentration and temperature, different products can be isolated as illustrated in Figure 43. All structures display an active valence electron pair at the central atom of the bismuthIII ions and Bi–C bonds that can be either asymmetrically (three shorter bonds + x longer bonds) or symmetrically (with rather long averaged bonds). In the presence of weakly coordinating cations (e.g. [Ph4P]+ and [PPN]+), the solid state structures of salts containing [Bi(CN)5]2– anions contain well-separated cations and monomeric anions that have a sterically active lone pair and a monomeric square-based pyramidal (pseudo-octahedral) structure. The [Bi(CN)5·MeCN]2– acetonitrile adduct ion shows a strongly distorted octahedral structure that is better understood as a [5 + 1] coordination. The [Ph4P]6[Bi2(CN)11]CN salt consists of separate cations and anions, as well as well-separated [Bi2(CN)11]5– and [CN]– ions. The structure of [Bi2(CN)11]5– ion can be described as two square-based pyramidal [Bi(CN)5]2– fragments connected by a disordered bridging [CN]– ion, resulting in a distorted octahedral environment around the two Bi centers. Here, the steric effect of the lone pair is much less pronounced, but still present.

Molecular structures of different cyanido bismuthates (Arlt et al., 2016b).
6th Main group
When an oxygen atom is formally linked to a [CN]– ion with its electron sextet, the cyanate ion, [OCN]–, is formed, which is a pseudohalogen just like the cyanide ion itself. The cyanate ion is by far the most important chalcogen-cyanide compound, followed by the thiocyanate, [SCN]–. The heavy chalcogen congeners are also pseudohalide ions (see 1.2 Cyanide, a classic pseudohalide). The cyanate is the eponym of all monocyanides of this main group. As typical pseudohalides they easily generate salts in presence of different metal cations. Cyanates are of great industrial (e.g. synthesis of polymers) as well as biological importance (Arnold et al., 1957; Barton et al., 1991, 1999; Bauer and Gnauck, 1987; Bauer et al., 1986, 1998; Bernsdorf and Köckerling, 2012; Britton and Dunitz, 1965a; Decius et al., 1965; Eyster and Gillette, 1940; Fang and Shimp, 1995; Grenier-Loustalot et al., 1996; Hamerton and Hay, 1998; Hendricks and Pauling, 1925; Kalmutzki et al., 2013; Kertes, 1979; Kotch et al., 1995; Maki and Decius, 1958, 1959; Marcos-Fernández et al., 1999; Moreno et al., 2013; Okubo and Ise, 1972; Osei Owusu et al., 1996; Rabalais et al., 1969a, 1969b; Reghunadhan Nair et al., 2001; Schalke, 2006; Shorter, 1978). Especially the latter also applies to the thiocyanates (Aguirre et al., 2010; Akcil, 2003; Anderson, 1980; Anderson et al., 1990; Azizitorghabeh et al., 2021; Bahta et al., 1997; Banerjee, 1996; Betts et al., 1979; Bezsudnova et al., 2007; Bhunia et al., 2000; Castanheiro et al., 2016; Dash et al., 2009; Du Plessis et al., 2001; Ebbs, 2004; Happold et al., 1958; Jensen and Tuan, 1993; Kelly and Baker, 1990; Kim and Katayama, 2000; Kwon et al., 2002; McDonald et al., 1969; Mudder et al., 1991; Palatinszky et al., 2015; Ryu et al., 2015; Stafford and Callely, 1969; Staib and Lant, 2007; Stott et al., 2001; Stratford et al., 1994; Sorokin et al., 2001; Wald et al., 1939; Watts and Moreau, 2016; Wilson and Harris, 1961; Wijeyasinghe and Anthopoulos, 2015; Willemin and Lumen, 2017; Wilson et al., 1960; Youatt, 1954). However, these aspects are not examined in this review, nor are the many inorganic salts of cyanates and thiocyanates (Bahta et al., 1997; Bernsdorf and Köckerling, 2012; Britton and Dunitz, 1965a; Schalke, 2006; McDonald et al., 1969; Norbury et al., 1973; Schultz, 1996; Wilson and Harris, 1961; Wijeyasinghe and Anthopoulos, 2015).
Formally, all chalcogenates, [ECN]– (E = O, S, Se, Te, and Po), can also be considered as chalcogen monocyanides, where the interaction between the chalcogen and the cyanide group decreases along the group. Accordingly, the charge between the oxygen and nitrogen atoms of the cyanate anion is approximately equally distributed, but the negative charge on the nitrogen atom increases the heavier the chalcogen is due to a decrease in electronegativity towards the tellurocyanate. Another aspect is the reduced influence of resonance structures, which reduce the π-interaction of the heavier elements.
The formal dicyanides of the type E(CN)2 are also known. The neutral chalcogen tetracyanide, E(CN)4, compounds are only known from selenium and tellurium, which are not stable in the long term. Their structure can be discussed as a pseudo-trigonal-bipyramidal arrangement and the unpaired electrons are in axial position.
Oxygen
Cyanate radical [OCN]⋅ and cyanate anion [OCN]–
Modern cyanate chemistry began with a dispute between Friedrich Wöhler and Justus v. Liebig (Liebig and Gay-Lussac, 1824a, 1824b; Liebig and Wöhler, 1830; Wöhler, 1824, 1825b). The latter was of the opinion that the elemental analysis of silver cyanate (AgNCO) published by F. Wöhler must be wrong, as it was the same as the one he had found for silver fulminate (AgCNO). The dispute was settled when the isomerism of these compounds was discovered (Berzelius, 1832). Incidentally, it is remarkable that the two congenial minds had been working with isomeric acids since their early youth: F. Wöhler with (iso)cyanic acid and J. v. Liebig with fulminic acid and their salts; already at the age of 15 J. v. Liebig had copied the preparation of silver fulminate from a market trader. Last but not least, F. Wöhler’s urea synthesis from ammonium cyanate was a milestone in the history of chemistry ([NH4]OCN → H2NC(O)NH2) (Wöhler, 1828).
Cyanate radical [OCN]⋅
Large sources of natural free cyanate radicals are not known, but smaller amounts of cyanates are reported to be part of carbon-star atmospheres in regard to CNO processing and intermediates of the cyanate radical are still existing in exhaust gases of modern combustion engines during reduction processes of nitric oxides (Du and Zhang, 2013; Morris and Wyller, 1967).
The cyanate radical has a linear structure and exhibits C∞v symmetry. Cyanate radicals can be generated by photolysis of isocyanic acid using UV light in a noble gas matrix at low pressure and low temperature. According to H. Okabe, HNCO is prepared by heating powdery cyanuric acid in an evacuated vessel, whereas D. E. Milligan and M. E. Jacox heated up the cyclic trimer. But also the reaction of carbon atoms of cyanogen azide with NO or the reaction of oxygen atoms with CN can result in the formation of [OCN]⋅. Using UV light the isocyanic acid can directly dissociate in hydrogen and the cyanate radical, but photolytic isomerization to HNCO and its photodissociation forming the cyanate radical is also possible (Scheme 92) (Milligan and Jacox, 1967a, 1967b; Okabe, 1970).

Formation of the cyanate radical by vacuum-ultraviolet photolysis (Milligan and Jacox, 1967a, 1967b; Okabe, 1970).
In 1985, D. D. Bell and R. D. Coombe published a study on the photolysis of chlorine isocyanate to form cyanate radicals and discussing several standard enthalpies of formation (Scheme 93) (Bell and Coombe, 1985). As early as 1974, C. Thomson and B. J. Wishart could confirm the linear structure of the cyanate radical by computations (Table 61). Based on population analysis data, the dipole moment of the radical was estimated to be 0.504 D (LCAO-MO-SCF/BA + P, Table 62) (Thomson and Wishart, 1974).

Photolysis of chlorine isocyanates to form the cyanate radical (Bell and Coombe, 1985).
Structural data of the cyanate radical (bond lengths in Å; angles in °; wave numbers in cm−1).
| Publication | Exp./calc. | r C–N | r C–O | < (O–C–N) | ν CO | ν CN |
|---|---|---|---|---|---|---|
| Chase et al. (1974) | Exp. | 1.23 | 1.18 | 180 | 1275 | 1922 |
| Thomson and Wishart (1974) | LCAO-MO-SCF/DZ + P | 1.259 | 1.191 | 180 | – | – |
| LCAO-MO-SCF/BA + P | 1.226 | 1.134 | 180 | – | – | |
| Yu et al. (1992) | HF/6-31G* | 1.231 | 1.160 | 180 | 1407 | 1959 |
| MP2/6-31G* | 1.253 | 1.166 | 180 | – | – | |
| Léonard et al. (2007) | MRCI/cc-pVQZ | 1.230 | 1.178 | 180 | 1270.8 | 1987.6 |
Population analysis of the valence orbitals within the cyanate radical as reported by B. Thomson and B. J. Wishart (1974).
| MO | N | C | O |
|---|---|---|---|
| 4s | 0.0228 | 0.5369 | 1.4403 |
| 5s | 1.2054 | 0.7653 | 0.0294 |
| 6s | 0.0537 | 0.2106 | 1.7357 |
| 7s | 1.7375 | 0.2118 | 0.0507 |
| 1p | 0.1064 | 1.2493 | 2.6434 |
| 2p | 1.9431 | 0.7588 | 0.2982 |
| Total | 7.0679 | 5.7339 | 8.1983 |
Cyanate anion [OCN]‒
The cyanate anion [OCN]– is much more stable than the radical and can be isolated as anion in salts with metal or WCCs as counter cation (Bernsdorf and Köckerling, 2012; Britton and Dunitz, 1965a; Decius et al., 1965; Galliart and Brown, 1972; Hendricks and Pauling, 1925; Hennings et al., 2011; Kertes, 1979; Maki and Decius, 1958; Schalke, 2006). Furthermore, it is an ambidentate nucleophile and able to bind protons or metal centers at both ends. If the proton is bound at the nitrogen atom, the product is called cyanic acid, if it is bound to the oxygen atom, the isomer is called isocyanic acid. At room temperature, isocyanic acid clearly predominates. However, esters of both isomers exist, e.g. ethyl cyanate, Et–OCN, and ethyl isocyanate, Et–NCO. The cyanate ion should be represented by at least three mesomeric structures (Scheme 94). The constitutional isomer of the cyanate is called fulminate [CNO]– and its corresponding acid is called fulminic acid. In comparison to the cyanates, the fulminates are more unstable, which also corresponds to the name fulminic acid derived from fulmination, which is a synonym of detonation or bang. But on the other hand, fulminate salts are stable enough to carry out X-ray structure analyses (Britton and Dunitz, 1965b).
![Scheme 94:
Lewis representation of the [OCN]– ion.](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_094.jpg)
Lewis representation of the [OCN]– ion.
Both KOCN and NaOCN are commercially available. Alkali metal cyanate is obtained by melting urea and alkali metal carbonate (Scheme 95) (Müller et al., 2014). Metal hydroxide is only useful for laboratory synthesis (Schalke, 2006). Urea is the preferred source for the cyanate part of the molecule, with ammonium carbonate being formed as a by-product in reversal of F. Wöhler’s urea synthesis. The endothermic reaction requires strong heating above 473 K for a prolonged period of time to achieve a reasonable conversion. This process does not achieve a complete conversion, but only a product with a purity of approx. 89–92%. The reaction takes place in a urea melt with a high content of dispersed solids. The main impurity is metal carbonate with small amounts of metal allophanate (H2NC(O)NHCO2M), cyanuric acid, biuret and urea. Higher purities > 95% are achieved by directly using technical-grade MOCN and urea to decrease the amount of by-products in the technical-grade MOCN. In this case, temperatures up to 873 K are required. Metathesis reactions can produce cyanate salts other than NaOCN and KOCN. The synthesis of [NH4]OCN can be generated by reversing F. Wöhler’s urea synthesis (H2NC(O)NH2 → [NH4]OCN) (Boatright, 1954). The synthesis of [NH4]OCN can also be achieved by a catalytic reaction (with the catalysts Pt, Cu/Ni, Ru or Os). In this process, NO is reduced with CO and H2 to [NH4]OCN (Cant et al., 2003, 2004, 2005a, 2005b; Chambers et al., 2001; Dalla Betta and Shelef, 1976; Unland, 1973; Voorhoeve and Thimble, 1978). It was the C. C. Cummins group that studied nitrogen fixation with transition metal complex to form the N2-derived terminal nitrido ligand in NMo–(N[ t Bu]Ar)3 (Ar = 3,5-Me2C6H3). The latter complex was shown to form OCN–Mo–(N[ t Bu]Ar)3 in the reaction with CO (Cozzolino et al., 2014). An elegent way to synthesize [WCC]OCN salts represents the nucleophilic desilylation of trimethylsilyl cyanate by methylcarbonate-containing ILs (Harloff et al., 2019a).

Synthesis of alkali metal and WCC cyanates (Müller et al., 2014).
The cyanate ion has a linear structure, exhibits C∞v symmetry (Cozzolino et al., 2014; Galliart and Brown, 1972; Hennings et al., 2011; MacLean et al., 2003; Schalke, 2006). Its silver salt forms monoclinic crystals, space group P21/m (Britton and Dunitz, 1965a). One could expect a coordination of the cation to both ends of the cyanate, so coordination to the nitrogen and oxygen, but in fact long chains are reported because of two silver cations linearly coordinating to one nitrogen atom. Though, the silver nitrogen distance of rAg–N = 2.115(8) Å is in the range of a typical silver cyanide bond. In contrast, the distance between the silver cation and the harder oxygen of rAg–O = 2.996(8) Å is much longer causing a weaker interaction (Britton and Dunitz, 1965a). Using neutron powder diffraction (MacLean et al., 2003), it was shown that in the structure of ammonium cyanate, the NH4+ cation forms N–H–N hydrogen bonds to four cyanate N atoms at alternate corners of a distorted cube (Figure 44), rather than the alternative arrangement with N–H⋅⋅⋅O hydrogen bonds to cyanate O atoms at the other four corners. With two formula units of [NH4]NCO in the tetragonal unit cell in the space group P4/nmm the linear cyanate anions lie along the fourfold axes, and the N atom of the ammonium cation is on a site with
![Figure 44:
Schematic illustration of the N–H⋅⋅⋅N hydrogen bonding in [NX4]OCN (X = H, D).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_044.jpg)
Schematic illustration of the N–H⋅⋅⋅N hydrogen bonding in [NX4]OCN (X = H, D).
Computational studies confirm the linear structure of the anion in the gas phase and an NBO analysis show a well-balanced charge distribution on nitrogen and oxygen (Table 63). In comparison to carbon both terminal elements are more electronegative having an electron-withdrawing effect on carbon and causing a positive partial charge (Lu et al., 2014). Selected experimental and computational data are listed in Table 64.
Natural Bond Orbital (NBO) atomic charges (in a.u.) of the cyanate ion at BP86/cc-pVQZ (Lu et al., 2014).
| NBO | O | C | N |
|---|---|---|---|
| q E | −0.72 | 0.50 | −0.78 |
Experimental and computational data of the cyanate anion and different cyanate salts (bond lengths in Å; angles in °; wave numbers in cm−1).
| Compound | Exp./calc. | r C–N | r C–O | < (O–C–N) | ν as. | ν sym. |
|---|---|---|---|---|---|---|
| [OCN]– | Exp. – UV (Jacox, 1990) | – | – | – | 2338 | 1289 |
| B3LYP/aug-cc-pVTZ (Moreno et al., 2013) | 1.184 | 1.221 | 180 | – | – | |
| CCSD(T)/cc-pVQZ (Lu et al., 2014) | 1.192 | 1.230 | 180 | 2188 | 1232 | |
| CCSD(T)/aug-cc-pV5Z (Léonard et al., 2010) | 1.193 | 180 | 2166.1 | 1228.0 | ||
| AgNCO | Exp. – XRD (Britton and Dunitz, 1965a) | 1.195(11) | 1.180(11) | – | – | – |
| NaOCN | Exp. – XRD (170 K)/Raman (Reckeweg et al., 2010) | 1.227(16) | 1.192(17) | 180 | 2178 | 1215 |
| KOCN | Exp. – XRD (Nambu, 2003)/Raman (Brooker and Wen, 1993) | – | 1.1988(4) | 180 | 2181 | 1207 |
| [ND4]OCN | Exp. – XRD (MacLean et al., 2003) (14 K) | 1.191(5) | 1.215(5) | 180 | – | – |
| Exp. – XRD (MacLean et al., 2003) (288 K) | 1.192(7) | 1.174(8) | 180 | – | – |
In 2018, T. M. Klapötke and co-workers reported the synthesis of isocyanic acid using potassium cyanate and stearic acid, which is similar to the synthesis of hydrogen cyanide by A. Schulz and co-workers in 2016 (Labbow et al., 2016). Based on the X-ray diffraction experiment similarities of carbon dioxide and the isocyanic acid, which can also be called the imide of CO2, are discussed. In the crystal the acid also forms zigzag chains with N⋯HN hydrogen bonds of 2.14 Å (Evers et al., 2018).
The trimerization and polymerization of cyanic acid are well known (Scheme 96). Trimerization reactions of isocyanates (R–NCO) have been investigated in many ways, e.g. for the preparation of cyclic cyanurate esters, which show great performance as adhesives, composites, resins etc. (Arnold et al., 1957; Barton et al., 1991, 1999; Bauer and Gnauck, 1987; Bauer et al., 1986, 1998; Fang and Shimp, 1995; Grenier-Loustalot et al., 1996; Hamerton and Hay, 1998; Kotch et al., 1995; Marcos-Fernández et al., 1999; Osei Owusu et al., 1996; Reghunadhan Nair et al., 2001; Seifer, 2002; Williams, 1948).

Oligomers and polymers of (iso)cyanic acid (Huthmacher and Most, 2000).
Although a large number of derivatives of cyanuric acid are known in organic chemistry, there are only a few structurally characterized metal cyanurates. Salts of cyanuric acid have been mentioned since the mid-eighteenth century (Kalmutzki et al., 2013; Seifer, 2002; Williams, 1948). The first reported compounds containing anions of cyanuric acid were transition metal cyanurate complexes, mainly of nickel, copper and cobalt. Structures with trivalent cyanurate anions were isolated and fully characterized by H. J. Meyer and co-workers (Scheme 97) (Kalmutzki et al., 2013). The preparations of cyanurate compounds LiSr(O3C3N3) and Li3Sr2F(O3C3N3)2 were possible due to using the concept of solid-state metathesis reactions.

Synthesis of salts bearing the trimeric cyanate ion (Kalmutzki et al., 2013).
Dicyanoether O(CN)2
Experimentally, dicyanoether O(CN)2 has been investigated by C. W. Hand and R. M. Hexter in 1970 (Hand and Hexter, 1970). Flash photolysis of a mixture of cyanogen and ozone and followed by a rapid scanning using an IR spectrometer led to the detection of the O(CN)2 molecule. Besides O(CN)2, a further product was observed. The asymmetrical product evolved from the terminal addition of oxygen in its ground state to the cyanogen. On the other hand, the symmetrical product O(CN)2 is the result of an insertion of singlet oxygen into the C–C bond of the cyanogen (Scheme 98).

Flash photolysis of cyanogen and ozone to form dicyanoether (Hand and Hexter, 1970).
One of the new absorption band at (2205 ± 10) cm−1, which appeared within 20 μs was assigned to the formation of O(CN)2. After 70 μs the band reaches the maximum of intensity and fell off gradually during several minutes (Hand and Hexter, 1970). In 1977, D. Poppinger and L. Radom published the first computations on the geometrical and energetical properties of R–OCN fragments (Poppinger and Radom, 1978). Due to limited computing capacity, the cyanate group was supposed to be linear, so the geometry optimization was restricted to the bond length and bond angle of the substituent R of R–OCN (Table 65). With the presumption of linear cyanate groups, the dicyanoether was found to exhibit C2v symmetry. Interestingly, dicyanoether does not represent the lowest lying isomer as it is 157.0 kJ mol−1 above the cyanoisocyanate, (NC)NCO.
Computational data of cyanogen isocyanate, cyanogen cyanate and cyanoformonitrile oxide using HF/STO-3G (bond lengths in Å; angles in °; Erel. in kJ mol−1) (Poppinger and Radom, 1978).
| Compound | Symmetry | r NC–X | < (C–X–Y) | E rel. |
|---|---|---|---|---|
| (NC)NCO | C s | 1.381 | 123.5 | 0.0 |
| (NC)O(CN) | C 2v | 1.369 | 111.4 | 157.0 |
| (NC)CNO | C ∞v | 1.392 | 180.0 | 261.3 |
Furthermore, the product of terminal addition of oxygen in its ground state is the most unstable compound. After all, these compounds with R = CN show the same order of stability as R = CH3 and the authors noted that the isocyanate structure is the most stable one, because the lone pair of nitrogen is a better p donor than the lone pair of oxygen. The change to a bigger basis set 4-31G including polarization functions leads to the bending of the angle < (O–C–N) = 175.6° and an decrease of symmetry from C2v to Cs (Poppinger and Radom, 1978). J. B. Moffat could confirm the bond lengths and bond angles reported by D. Poppinger and L. Radom. In regard to the compounds of Table 65 the C–N distances of the cyanide groups (rN–CX = 1.160/1.159/1.163 Å) are in the typical range of a triple bond and the C–O bonds of dicyanoether are slightly shortened due to a back-bonding of unpaired electrons of oxygen into antibonding orbitals of the cyanide groups (Moffat, 1979; Poppinger and Radom, 1978).
Oxocyanogen (OCN)2 – the cyanate dimer
In 1977, the formation of an (OCN)2– species, which was part of an equilibrium of the cyanate anion [OCN]– and a complex HNCOOH formed by irradiation of OH radical/[OCN]– mixture, was reported by J. G. Leopold and M. Faraggi (Scheme 99). By means of pulse radiolysis of cyanate anion solutions, kinetic studies led to the assumption of the in situ formation of an [(OCN)2]– species (Leopold and Faraggi, 1977).
![Scheme 99:
Pulse radiolysis to form the [(OCN)2]– dimer (Leopold and Faraggi, 1977).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_099.jpg)
Pulse radiolysis to form the [(OCN)2]– dimer (Leopold and Faraggi, 1977).
Experimental evidence of the existence of an (CNO)2 species, isomer of (OCN)2, was published in 1984. Time-resolved mass spectrometry of the decomposition products after thermal explosion (at 430 K) of mercury fulminate showed mass peak at m/z = 84 that could allocated to the (CNO)2 dimer (Scheme 100) which further breaks down into more stable species such as ONC–CN, O, N2O and (CN)2 (Tang et al., 1984).

Thermal explosion of mercury fulminate forming (OCN)2 as an intermediate of decomposition (Tang et al., 1984).
In 1981, M. S. Delgado and V. Fernandez published the reaction of silver cyanate and bromine and a stabilization of (OCN)2 using titanium tetrachloride to isolate the dimer, but polymerization was assumed, which hindered the unambiguous identification of the dimer (Delgado and Fernandez, 1981). In 1996, A. Schulz and T. M. Klapötke reported the formation of dioxocyan as a reactive intermediate in the reaction of silver cyanate and bromine, which dissociated under release of nitrogen (Scheme 101).

The reaction of silver cyanate and bromine leads to dioxocyan as an intermediate (Schulz and Klapötke, 1996).
Due to fast decomposition of the dioxocyan, its isolation and spectroscopic analyses were not possible. Computational studies at MP2(full)/6-31G(d,p) were also carried out to distinguish between isomers of O–O and N–N linked dimers showing that the diisooxocyan is the thermodynamically more stable isomer lying 343.7 kJ mol−1 lower in energy than the oxygen linked isomer. In addition, the N–N bond is significantly shortened and the bond length of rN–N = 1.385 Å is in between a typical single and double bond. In contrast to this shortened bond, the O–O bond of the dioxocyan is much weaker (rO–O = 1.622 Å, Table 66) (Schulz and Klapötke, 1996). In the same year, G. Maier et al. generated and identified (OCN)2 by matrix IR spectroscopy (Maier et al., 1996a). It was generated by UV irradiation of oxalic acid diazide and azidoformyl isocyanate in argon at 10 K and by high vacuum flash pyrolysis of the same precursors at 1173 K. Although (OCN)2 proved stable at 80 K, it polymerized and was not in pure form at room temperature. In 2008, T. Pasinszki also published detailed studies on cyanate dimers of different configurations and could also confirm, that the diisooxocyan is the thermodynamically most stable isomer (Table 67) (Pasinszki, 2008).
Computational data of (OCN)2 dimers (bond lengths in Å; angles in °).
| Compound ABC–DEF | Method + basis set | r A–B | r B–C | r C–D | r D–E | r E–F | < (A–B–C) | < (B–C–D) | < (C–D–E) | < (D–E–F) |
|---|---|---|---|---|---|---|---|---|---|---|
| OCN–NCO (Schulz and Klapötke, 1996) | MP2(FU)/6-31G(d,p) | 1.181 | 1.238 | 1.385 | 1.238 | 1.181 | 168.5 | 123.3 | 123.3 | 168.5 |
| NCO–OCN (Schulz and Klapötke, 1996) | MP2(FU)/6-31G(d,p) | 1.190 | 1.296 | 1.622 | 1.296 | 1.190 | 176.5 | 104.6 | 104.6 | 176.5 |
| ONC–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.201 | 1.165 | 1.340 | 1.165 | 1.201 | 180.0 | 180.0 | 180.0 | 180.0 |
| OCN–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.212 | 1.162 | 1.319 | 1.210 | 1.162 | 173.5 | 159.0 | 141.7 | 172.5 |
| NCO–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.196 | 1.180 | 1.351 | 1.307 | 1.151 | 168.5 | 135.9 | 117.0 | 175.1 |
| CNO–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.173 | 1.341 | 1.349 | 1.179 | 1.198 | 166.8 | 112.9 | 135.6 | 169.6 |
| OCN–NCO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.163 | 1.221 | 1.376 | 1.221 | 1.163 | 170.5 | 125.0 | 125.0 | 170.5 |
| NCO–NCO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.154 | 1.297 | 1.458 | 1.239 | 1.156 | 175.1 | 110.1 | 112.9 | 171.1 |
| CNO–NCO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | 1.173 | 1.321 | 1.465 | 1.241 | 1.155 | 171.6 | 108.2 | 111.9 | 171.5 |
Computational data of (OCN)2 dimers (total energy in a.u.; relative energy and dissociation energy at 298 K in kJ mol−1; wave numbers in cm−1).
| Compound ABC–DEF | Method + basis set | E tot. | E rel. | D ABC–DEF | ν CD | ν DEF |
|---|---|---|---|---|---|---|
| OCN–NCO (Schulz and Klapötke, 1996) | MP2(full)/6-31G(d,p) | −335.23422 | 0.0 | – | – | – |
| NCO–OCN (Schulz and Klapötke, 1996) | MP2(full)/6-31G(d,p) | −335.103437 | 82.9 | – | – | – |
| ONC–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −336.076636 | 347 | 332 | 634 | 1588 |
| OCN–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −336.152341 | 148 | 288 | 737 | 1596/1296[a] |
| NCO–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −336.072284 | 356 | 117 | – | 1429 |
| CNO–CNO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −335.980380 | 592 | 144 | – | 1410 |
| OCN–NCO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −336.209270 | 0 | 188 | 837 | 1514 |
| NCO–NCO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −336.134348 | 193 | 21 | 805 | 1324 |
| CNO–NCO (Pasinszki, 2008) | B3LYP/6-311 + G(2d) | −336.046988 | 418 | 56 | 797 | 1301 |
-
[a]CNO and NCO stretching vibration.
Sulfur
As for oxygen, the monocyanide [SCN]⋅ radical, the thiocyanide anion, [SCN]–, and the dicyanide, S(CN)2 are also known for sulfur. However, while the number of CN groups attached to the oxygen atom is (so far) limited to two, three and four CN groups have also been found for sulfur. Since thiocyanate plays a major role in both inorganic and bioorganic chemistry (see introduction), only the essentials such as synthesis and structure are discussed here, otherwise reference is made to the many review articles (Anderson, 1980; Azizitorghabeh et al., 2021; Bahta et al., 1997; Bezsudnova et al., 2007; Castanheiro et al., 2016; Dash et al., 2009; Ebbs, 2004; Happold et al., 1958; Jensen and Tuan, 1993; McDonald et al., 1969; Sorokin et al., 2001; Stott et al., 2001; Wald et al., 1939; Watts and Moreau, 2016; Willemin and Lumen, 2017; Wilson et al., 1960).
Thiocyanate radical [SCN]∙ and [SCN]– ion
Thiocyanate radical [SCN]∙
The thiocyanate radical, [SCN]⋅, can be generated by photolytic dissociation of methyl isothiocyanate, using KrF or ArF excimer laser. Prior to the dissociation, methyl isothiocyanate was very diluted by helium. [SCN]⋅ has also been detected after radio frequency discharge of methyl thiocyanate and the reaction between CN and OCS (Scheme 102). To avoid higher thermal excitation, conditions of supersonic free jet expansion are used to obtain fluorescence excitation spectra (Northrup and Sears, 1989).

Photolytic dissociation of methyl isothiocyanate to obtain the thiocyanate radical (Northrup and Sears, 1989).
Additional experimental studies focusing on the Renner-Teller effect of the linear thiocyanate radical, which exhibits C∞v symmetry, confirmed the 2Π state to be the electronic ground state (Northrup and Sears, 1990). Its fluorescent lifetime is reported to be t 0 = 165 ns (Ohtoshi et al., 1984). Furthermore, the bond lengths were calculated based on observed rotational constants (Table 68) (Maeda et al., 2007).
Experimental and calculated structural data of the thiocyanate radical (bond lengths in Å).
| Publication | Exp./calc. | r C–N | r S–N |
|---|---|---|---|
| Maeda et al. (2007) | Exp. – SWS | 1.1831(18) | 1.6301(14) |
| Li et al. (2012) | MP2/aug-cc-pVDZ | 1.1563 | 1.6879 |
| Chen et al. (2008) | MP2/6–311 + G(3df,2p) | 1.15 | 1.66 |
| B3LYP/6–311 + G(3df,2p) | 1.17 | 1.63 |
DFT calculations could confirm experimental values and the electron affinity of EA = 3.51 eV (339 kJ mol−1), which was calculated by J. G. Dillard and J. L. Franklin, was re-adjusted to be EA = 3.52 eV by H.-L. Chen et al. using CCSD(T)/aug-cc-PVQZ//B3LYP/6–311 + G(3df,2p) (Chen et al., 2008; Dillard and Franklin, 1968). Photoionization mass spectrometry led to an adiabatic ionization potential of IP = 10.689(5) eV and the standard formation enthalpy was reported to be ΔfH° = (304.4 ± 3.3) kJ mol−1 (Ruscic and Berkowitz, 1994).
Thiocyanate anion [SCN]‒
Thiocyanates are the salts of thiocyanic acid, HSCN (initially known as Schwefelblausäure = German for sulfur blue acid). Thiocyanic acid was observed as early as 1790 by Winterl, 1799 by C. Buchholz (Buchholz, 1799), 1804 by Rink and produced in 1808 by R. Porret (Köhler, 1914). Its composition was determined by J. J. Berzelius in 1820 and he also introduced the old name rhodanic acid (from Greek for red), since ferric thiocyanate is a deep red complex. Thiocyanate salts can be prepared from cyanides and sulfur in the melt (Scheme 103) (Köhler, 1914). In addition to thiocyanic acid (HSCN), an isomer is also known, namely isothiocyanic acid (HNCS also thiocarbimide). Both types of acid are labile with respect to decomposition. The thiocyanate ion, like the azide, cyanide or cyanate, is called a linear pseudohalide, which easily forms salts with alkali metals such as potassium thiocyanate (KSCN, also potassium rhodanide) (Birckenbach and Kellermann, 1925a; Brand et al., 2006, 2007; Kauffman et al., 1968; Stopenko et al., 1986). But also non-metal main group thionates are known (Bahta et al., 1997; McDonald et al., 1969; Wijeyasinghe and Anthopoulos, 2015). As early as 1873, L. Lössner et al. converted phosphorus trichloride with KSCN into P(SCN)3 in an alcoholic solution (Lössner et al., 1873). Shortly thereafter, the reaction of PCl3, AsCl3 and SbCl3 with metal thiocyanates (MSCN, M = alkali metal) was studied by different groups (Dixon, 1901). The formation of the thiocyanates P(SCN)3, As(SCN)3 and Sb(SCN)3 was assumed since upon contact of these E(SCN)3 (E = P, As, and Sb) species with water, thiocyanic acid was observed. In 1902, A. E. Dixon studied the reaction of PCl3 with KSCN in detail in order to investigate the behavior of P(SCN)3 towards alcohol (Dixon, 1902). It was E. Söderbäck who advanced the thiocyanate-chemistry and dealt in particular with the synthesis of free HSCN and its salts. In the course of these investigations, he examined the effect of HSCN-ether solutions on pure arsenic and antimony, whereby he could isolate a yellow hygroscopic oily solid (Söderbäck, 1919). To date, various synthetic routes to P(SCN)3 have been published, most of them starting from PCl3 and metal thiocyanates such as AgSCN, Hg(SCN)2, or [NH4]SCN (Fluck et al., 1965; Gall and Schüppen, 1930). Also, pure heavy E(NCS)3 (E = Sb, Bi) species were obtained from the reaction of EF3 and an excess of TMS–SCN (Arlt et al., 2019). Furthermore, the cluster ion [SCN(HCN)3]− was isolated when WCC[SCN] is recrystallized from liquid HCN (Bläsing et al., 2020)

Synthesis of potassium thiocyanate (Köhler, 1914).
Thiocyanates fulfill several functions in organisms, for example degradation product of the cyanide detoxication or oxidation of thiocyanate by enzymes like eosinophil peroxidase to form hypothiocyanous acid HOSCN, which is present in the oral cavity, airway or alimentary tract and has got antimicrobial properties (Chandler and Day, 2015). Also the thiocyanate anion itself is analyzed due to biological activities, for example the affinity to bind to hemoglobin (Kumar Sau et al., 2003). Furthermore, the formation of bonds to organic substituents leading to isothiocyanates is possible, which is interesting in respect to bonding of nitric monoxide radicals or the prevention of inhibition of the immune system by reduced macrophage migration (Noshita et al., 2009; Spencer et al., 2015).
Structurally, many thiocyanate salts have already been characterized. For example, in 1971 Z. Igbal published structural data of sodium thiocyanate, which were rectified by P. H. van Rooyen and J. C. A. Boeyens in 1975 using single crystal XRD and observed characteristic vibrational bands (Table 69). The sodium thiocyanate forms orthorhombic crystals, space group Pnma, the sodium is octahedral coordinated by three sulfur and three nitrogen atoms in fac arrangement and the thiocyanate anion can be regarded to be linear (Iqbal, 1971; van Rooyen and Boeyens, 1975). The crystal structures of thiocyanate salts (M = alkali, [NH4]+, Tl+), their crystal growth, properties and phase transitions in comparison to cyanates, azides and bifluorides ([HF2]–) have been reviewed in detail by A. Fuith (1997).
Experimental and computational data of sodium and potassium thiocyanate (bond lengths in Å; angles in °; wave numbers in cm−1).
| Compound | Exp./calc. | r C–N | r S–C | < (S–C–N) | ν CN | ν SC |
|---|---|---|---|---|---|---|
| NaSCN | Exp. – XRD, IR (Iqbal, 1971; van Rooyen and Boeyens, 1975) | 1.168(18) | 1.663(9) | 179.0(8) | 2070/2090 | 750/760 |
| KSCN | Exp. – XRD, Raman (Akers et al., 1968; Dao and Wilkinson, 1973) | 1.149(14) | 1.689(13) | 178.3(1) | 2050(2) | 749(2) |
| KSCN | Rigid ion model (Kanamori et al., 1981) | 1.15 | 1.79 | 180 | 2050 | 749 |
The calculations on KSCN by K. Urabe and co-workers were carried out in the framework of the rigid ion model. To analyze the mesomeric stability of this compound they also calculated the distribution of negative charge (q(K) = 0.747, q(S) = −0.194, q(C) = −0.161, q(N) = −0.392) identifying a main focus of negative charge on the nitrogen (Kanamori et al., 1981). In contrast to sodium thiocyanate the space group of potassium thiocyanate is Pbcm and it exhibits longer S–C bonds and shorter C–N bonds. Although, the sulfur-carbon bond is 0.12 Å shorter than a typical single bond, the Lewis picture should include at least mesomeric structures as depicted in Figure 45 (Akers et al., 1968). The chemical shift of 15N and 13C were measured using Li[SCN] dissolved in DMF (δ[15N] = −160 ppm, δ[13C] = 150–152 ppm) (Vaes et al., 1978).

Mesomeric structures of the thiocyanate anion.
Dirhodan, NCS–SCN (Allenstein and Lattewitz, 1964; Fieser et al., 2006; Nolan et al., 1975; Kuhn and Mecke, 1960, 1961b; Kuhnhen, 1977; Schoneshofer et al., 1970; Seel and Wesemann, 1953; Wizemann et al., 1969). As classical pseudohalide thiosulfate can be oxidized to form dirhodan, the dimer of the [SCN]⋅ radical. As early as 1919, E. Söderbäck tried to oxidize the [SCN]– ion to dirhodan using nitrous acid (Scheme 104) (Söderbäck, 1919). However, when an aqueous solution of potassium rhodanide and potassium nitrite was acidified, the result was not dirhodan but the intensely red nitrosyl rhodanide, which had already been observed in 1852 by J. Besnou and later also by E. W. Davy (Scheme 104) (Seel and Wesemann, 1953).

Different routes for the synthesis of (SCN)2 (Emerson, 1966; Fitzgerald and Bowie, 2004; Rogers and Gross, 1952; van Rooyen and Boeyens, 1975).
All the authors mentioned report that nitrosyl rhodanide can easily be decomposed with the release of nitrogen oxide. Since dirhodan is hydrolysed by water (Schoneshofer et al., 1970), nitrosyl rhodanide must be prepared in a non-aqueous solvent (Seel and Wesemann, 1953). The preparation of a solution of nitrosyl rhodanide in liquid sulfur dioxide, which is obtained by reacting nitrosyl chloride with a solution of potassium rhodanide, proved to be particularly advantageous. In this quantitative reaction, potassium chloride precipitates. After careful evaporation of the intensely red solution in vacuo and at low temperatures, the solid nitrosyl rhodanide is obtained, which rapidly changes into completely colorless dirhodan with the elimination of nitrogen oxide. The decomposition of the dirhodan (TM = 288–289 K) leads to an orange-red solid substance. Another method for the synthesis of dirhodan is based on Pb(SCN)2 and bromine in ether at temperatures between 273 and 283 K (Scheme 104) (Kuhn and Mecke, 1960).
K(SCN) 3 . If three equivalent KSCN are treated with two equivalent [NO]SCN, the interesting yellow potassium tri-rhodanide, K(SCN)3, which is analogous to the better-known potassium triiodide, KI3, is obtained after NO cleavage.
Thiocyanate cation [SCN]+
Equally to the [SCN]⋅ radical only a few experimental studies of the thiocyanate cation [SCN]+ are published in literature. The first evidence of the cation was published by B. Ruscic and J. Berkowitz in 1994 as a result of hydrogen abstraction of HNCS by atomic fluorine, but the spectra showed several autoionizing features. A second route was the formation of the dirhodane (dithiocyane, see above) using silver thiocyanate and chlorine and in the second step it was photolytically ionized in a mass spectrometer (Scheme 105). The correlated standard formation enthalpy was reported to be ΔfH° = (1336.4 ± 3.8) kJ mol−1 (Ruscic and Berkowitz, 1994).

Formation of the thiocyanate cation (Ruscic and Berkowitz, 1994).
In 2004, mass spectrometric studies on methylene-isothiocyanate, which was formed by dissociative electron ionization of 2-mercaptoimidazole, were reported and confirmed by tandem mass spectrometry. The neutralization-reionization spectrum showed a peak at m/z = 58, which was allocated to the thiocyanate cation due to removal of the methylene group from [H2C–NCS]+ (Reddy et al., 2004). M. Fitzgerald and J. H. Bowie could also generate the thiocyanate cation by charge reversal of the thiocyanate anion directly in the collision cell of a mass spectrometer by single collisions with oxygen. In their computational studies they could additionally show that the isomerization from thiocyanate cations in the triplet state to thiofulminate cations is endothermic by 86.2 kJ mol−1 and correlated to an activation barrier of 188.8 kJ mol−1, whereas the change of the isomers in the singlet state is less endothermic by 76.2 kJ mol−1. The calculated parameters of the triplet ground state and the singlet state are shown in Table 70 and all isomers exhibit C∞v symmetry (Fitzgerald and Bowie, 2004).
Computational data of [SCN]+ in different electronic states (Etot. in Hartree; bond lengths in Å; dipole moment μ in D) (Fitzgerald and Bowie, 2004).
| Compound | State | E tot. [a] | r A–B [b] | r B–C [b] | μ |
|---|---|---|---|---|---|
| [SCN]+ | 1Π | −489.83203 | 1.593 | 1.211 | 1.98 |
| [SNC]+ | 1Π | −489.80298 | 1.547 | 1.245 | 1.63 |
| [SCN]+ | 3Σ | −489.87300 | 1.594 | 1.213 | 2.25 |
| [SNC]+ | 3Σ | −489.84011 | 1.561 | 1.232 | 2.07 |
-
[a]Etot. calculated at CCSD(T)/aug-cc-pVDZ and zpe included. [b]Bond length calculated at B3LYP/6-31G + (d).
Evidence for the formation of [SCN]+ ion was also found in the fragmentation reaction of methylenethiocyanate investigated by E. Cortés et al. in 2009. Based on photoelectron photoion coincidence (PEPICO) and photoelectron photoion photoion coincidence (PEPIPICO) spectra it was shown that the fragmentation processes leading to formation of [CH3]+ and [SCN]+ ions dominate the dissociation of CH3SCN excited at the S 2p levels (Cortés et al., 2009).
Sulfur dicyanide S(CN)2
In 1919, E. Söderbäck published a detailed article dealing with the free thiocyanate radical, starting with J. v. Liebig’s experiments (Ag[SCN] + Cl2) to obtain the radical, presenting several attempts to synthesize the free radical and collecting characteristic chemical properties (Söderbäck, 1919). In this context, he also described F. Linnemann’s synthesis (Linnemann, 1861) of sulfur dicyanide S(CN)2 due to a reaction of cyanogen iodide and silver thiocyanate and R. Schneider’s attempt (Schneider, 1885) to synthesize dirhodane, (SCN)2, but due to several analyses R. Schneider assumed a rapid decomposition of the dimer to form the dicyansulfide and dicyantrisulfide S3(CN)2 (Scheme 106) (Söderbäck, 1919).

Reactions of F. Linnemann and R. Schneider obtaining dicyansulfide (Söderbäck, 1919).
At that time the homologous series of cyanosulfanes was extended quickly and also contained the dithiocyanate (SCN)2, sulfur dithiocyanate S(SCN)2 and disulfur dithiocyanate S2(SCN)2 based on the same synthetic route of mercury thiocyanate and sulfur–halogen compounds (Scheme 107).

Synthetic route of homologous cyanosulfanes (Feher and Weber, 1958).
Isolated colorless crystals of S(CN)2 sublime easily at normal pressure, melt at 335 K and in addition to its low sublimation temperature the compound tends to polymerize easily (Feher and Weber, 1958). The dipole moment of SCN was determined to be μ = 2.28 D using electron diffraction experiments and under assumption of an carbon-sulfur-carbon angle of < (C–S–C) = 105° identical to dimethyl sulfide (Rogers and Gross, 1952). Furthermore, several results of structure determination, for example using single crystal XRD experiments, were published during the 1960s and during the last 15 years (Table 72). K. Emerson as well as K.-H. Linke and F. Lemmer reported different parameters for each CN group, which led to an asymmetric molecule, but this fact could not be confirmed by later studies (Emerson, 1966; Linke and Lemmer, 1966a). The small distances of neighboring molecules in the crystal, which are smaller than the van der Waals radii, are striking and could be interpreted using the σ-hole concept established some years ago (Donald and Tawfik, 2013; Murray et al., 2007b; Politzer et al., 2007), which implies, that the half-filled p bonding orbital of group V to VII atoms showing electron deficiency interacts with a nucleophile, whereas the intensity depends on its electronegativity, polarizability and the electron-withdrawing character of the group to which it is bonded. So, the outer surface of the chalcogen atoms has regions of positive electrostatic potential, which noncovalently interact with the lone pairs of the nitrogen atom of a neighboring cyanide group causing a denser package in the crystal (Murray et al., 2007b; Politzer et al., 2007).
Several spectroscopic studies specified the structure determination (Table 71), for example, D. A. Long and D. Steele could confirm C 2v symmetry and discussed various C–S–C angles due to different assumed bond dipole moments using IR and Raman spectra (Long and Steele, 1963).
IR and Raman data of S(CN)2 in different matrices (wave numbers in cm−1).
| Publication | Method | Matrix | ν sym. SC | ν sym. CN |
|---|---|---|---|---|
| Long and Steele (1963) | Exp. – IR | Solid | 752 | 2190 |
| Exp. – Raman | Solid | – | 2191 | |
| Fehér and Weber (1958) | Exp. – Raman | In bromoethane | 680 | 2186 |
| Burchell et al. (2006) | Exp. – IR | In KBr | 697 | – |
| Exp. – Raman | Solid | 693 | 2196 |
Experimental and computational data of S(CN)2 reported in different studies (bond lengths in Å; angles in °).
| Publication | Exp./calc. | r S–C | r C–N | < (S–C–N) | < (C–S–C) | Space group |
|---|---|---|---|---|---|---|
| Emerson (1966) | Exp. – XRD | 1.736(15)/1.718(18) | 1.118(21)/1.134(23) | 177.49(1.34)/176.43(1.70) | 95.60(78) | Pbca |
| Linke and Lemmer (1966a) | Exp. – XRD | 1.87/2.07 | 1.02/1.20 | 177/144 | 96 | Pbca |
| Burchell et al. (2006) | Exp. – XRD | 1.74(2)/1.72(2) | 1.12(2)/1.13(2) | 177(1)/176(2) | 95.6(8) | – |
| B3LYP/6-311 + G* | 1.71 | 1.16 | 175 | 99.6 | – | |
| Kisiel et al. (2013) | FIR spectra, CCSD(T)/aug-cc-pVTZ | 1.6972(4) | 1.1602(4) | 175.14(5) | 98.36(3) | – |
| CCSD(T)/aug-cc-pVTZ | 1.7059 | 1.1617 | 175.37 | 97.77 | – |
Experimental and computational data of chemical shifts fit well, especially regarding the 13C species the difference between theory and experiment is about Δδ ≈ 5 ppm (δ (13C, exp., CD2Cl2) = 100.1 ppm, δ (13C, calc.) = 105.0 ppm), regarding 14N the difference is large (δ (14N, exp., CD2Cl2) = 293.4 ppm, δ (14N, calc.) = 338.4 ppm). All calculations were carried out at B3LYP/6-311 + G* (Burchell et al., 2006).
Furthermore, dicyansulfide is an alkaline compound due to its lone pairs at the sulfur atom and both nitrogen atoms, which can also interact with electrophiles. In accordance to the HSAB principle of Pearson, pairs of acids and bases of the same strength are most stable, which implies, that the nitrogen atoms can interact with two Lewis acids, for example antimony or arsenic pentafluoride, forming strong diadducts (Minkwitz et al., 1991; Pearson, 1968).
Thiocyanogen (SCN)2
The syntheses of thiocyanogen published by F. Linnemann and R. Schneider based on the reaction of silver cyanide and disulfur dicyanide and subsequently formation of sulfur dicyanide S(CN)2 and sulfur dithiocyanate S(SCN)2 (Scheme 106) (Söderbäck, 1919) as well as the reaction of mercury thiocyanate and sulfur–halogen compounds (Scheme 107) (Feher and Weber, 1958) reported by F. Fehér and H. Weber have already been named. In the 2000s, J. D. Woollins and co-workers also dealed with sulfur-cyanogen compounds and reported a synthesis of thiocyanogen based on the reaction of silver thiocyanate and molecular bromine at −20 °C. They reported carbon and nitrogen NMR data and also noticed the polymerization of thiocyanogen forming polycyanogen at ambient temperature (Burchell et al., 2004, 2006).
Tricyanosulfonium cation [S(CN)3]+
There is only one article in literature published by R. Minkwitz and V. Gerhard dealing with the [S(CN)3]+ cation. The synthesis is a halogen-pseudohalogen exchange at tribromosulfonium hexafluoroarsenate in liquid sulfur dioxide at lower temperatures (Scheme 108). The reaction takes 24 h under exclusion of light, when silver bromide and an excess of silver cyanide are removed from the dissolved product by filtration.
![Scheme 108:
Synthesis of the [S(CN)3]+ cation (Minkwitz and Gerhard, 1991).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_108.jpg)
Synthesis of the [S(CN)3]+ cation (Minkwitz and Gerhard, 1991).
The product is a colorless solid, is sensitive to hydrolysis and decomposes at 364 K. The results of their spectroscopic measurements are listed in Table 73. The 13C NMR spectrum shows one signal at δ = 85.4 ppm, which implies a significant high field shift in comparison to S(CN)2 (δ = 100–102 ppm) (Minkwitz and Gerhard, 1991).
Experimental data of IR and Raman spectra of [S(CN)3]+ (wave numbers in cm−1) (Minkwitz and Gerhard, 1991).
| Method | Matrix | ν sym. SC | ν as. SC | ν sym. CN |
|---|---|---|---|---|
| IR | Solid | – | 564 | 2230 |
| Raman | Solid | 638 | 568 | 2242 |
Tetracyanosulfonium cation [S(CN)4]2+
The tetracyanosulfonium dication [S(CN)4]2+ was described theoretically by I. A. Koppel et al. (B3LYP/6-311 + G**) (Koppel et al., 2002). This publication deals with the generalized principle of superstrong Brønsted acids, in which sulfur tetracyanide is a fragment of a major compound and substitutes the oxygen atom of nitrous acid, acetalaldehyde or nitroxyl (Figure 46). The calculated distances of the four sulfur–cyanide bonds are not equal and listed in Table 74 as well as the calculated deprotonation enthalpy ΔH°acid and acidity ΔG°acid to estimate the Brønsted acid behavior.

The oxygen atom of nitrous acid, acetalaldehyde and nitroxyl is substituted by R = S(CN)4 (Koppel et al., 2002).
Computational data of R = S(CN)4 containing compounds (bond lengths in Å; gas phase acidity and proton affinity in kJ mol−1) (Koppel et al., 2002).
| Compound | r S–C | ΔH°acid | ΔG°acid |
|---|---|---|---|
| HON=S(CN)4[a] | 1.78/1.91/1.84/1.83 | – | – |
| CH3CHC=S(CN)4 | 1.92/1.92/2.22/2.22 | 1022.0 | 983.5 |
| [CH2HC=S(CN)4]– | 1.92/1.92/2.23/2.23 | – | – |
| HN=S(CN)4 | 1.79/1.79/1.90/1.83 | 1277.0 | 1240.1 |
-
[a]This structure forms fragments during structure optimization.
Selenium
Selenium cyanide hydrogen compounds are unstable acids, but some of their salts are storable for longer periods of time. For example, they can be found in wastewater of oil and gas industries, especially coal-fired power plants using flue gas desulfurization, and are toxic (Petrov et al., 2012; Vadhanavikit and Ganther, 1988). In regard to chemical properties and resulting biological activity selenium and sulfur are very similar to each other. For example, in some cases the sulfur atom of the amino acid cysteine is replaced by selenium forming selenocysteine. Accordingly, selenium cyanide compounds possess several biological activities similar to the sulfur cyanide compounds discussed before (Badiello, 1992; Huber and Criddle, 1967; Shrift and Virupaksha, 1963). In some studies the application of organo selenium compounds as cancerostatic drugs are tested, which are synthesized using selenocyanates (Bouchet et al., 2011; El-Bayoumy and Sinha, 2004).
Selenocyanate [SeCN]− and the triselenocyanate [(SeCN)3]– ion
Selenocyanate, [SeCN]–, is known for a long time and its stability enables commercial trade. The contact to air leads to decomposition of these hydroscopic compounds forming potassium cyanide and red selenium, which necessitates its handling under inert gas atmosphere. The synthesis runs in reverse direction in terms of an insertion of elementary, powdered selenium into potassium cyanide (Scheme 109) (Kaufmann and Kögler, 1926).

Insertion of red selenium into potassium cyanide to form potassium selenocyanate (Kaufmann and Kögler, 1926).
The first crystal structure determination of the selenocyanate anion (with K+ as counter ion) was published by D. D. Swank and R. D. Willett in 1965 reporting a nearly linear structure, which is slightly perturbed due to packing effects (Swank and Willett, 1965). The space group of potassium selenocyanate crystals is P21/c. Within the standard deviation, the CN bond length is in the range of a triple bond, but the Se–C bond length of rSe–C(exp.) = 1.829 Å is significantly shorter than the expected single bond length of rSe–C(calc.) = 1.94 Å, which indicates a partial double bond character, but does not affect the CN bond due to the higher difference in electronegativities (Table 75) (Swank and Willett, 1965).
Experimental and computational structural data of the selenocyanate anion (bond lengths in Å; angle in °).
| Publication | Exp./calc. | r Se–C | r C–N | < (Se–C–N) |
|---|---|---|---|---|
| Swank and Willett (1965) | Exp. – XRD | 1.829(25) | 1.117(26) | 178.8(2.5) |
| Petrie (1999a) | MP2(full)/6-31G* | 1.832 | 1.147 | – |
Detailed information about all alkali metal selenocyanates MSeCN with M = Li to Cs are summarized by F. Tambornino and co-workers including syntheses, spectroscopic data as well as crystal structures and computations (Shlyaykher et al., 2021).
Silver selenocyanate complexes dissolved in acetonitrile were studied by means of IR spectroscopy (νSeC = 530–560 cm−1, νCN = 2070–2110 cm−1, νbent = 380–410 cm−1) (Singh, 1980).
Selenocyanogen (SeCN)2 and selenium dicyanide Se(CN)2
Selenium dicyanide Se(CN)2 is significantly more stable than the oxygen species O(CN)2 and can be obtained as a pure substance. The crucial step of its synthesis is the disproportionation of selenocyanogen in selenium dicyanide and selenium diselenocyanate, which can be separated by sublimation at 323 K. The selenocyanogen can be synthesized on two different routes. The first one is a reaction of potassium selenocyanate and iodine pentafluoride and the second one is a salt metathesis of potassium selenocyanate in presence of silver nitrate and following oxidation by iodine (Scheme 110) (Aynsley et al., 1964; Burchell et al., 2004, 2006; Linke and Lemmer, 1966b).

Synthesis of selenocyanogen and its disproportionation to selenium dicyanide (Linke and Lemmer, 1966b).
The first structure determination by single crystal XRD was published by A. C. Hazell in 1963. He assumed that selenium dicyanide would form orthorhombic crystals, space group Cmca (Hazell, 1963). Detailed structural data are shown in Table 76. In 1966, K.-H. Linke and F. Lemmer also published a study on the structure of selenium dicyanide correcting the space group to Pbca and lowering the symmetry to C s because of two non-equivalent cyanide groups, which differ due to the carbon–nitrogen bond and the bond angle < (Se–C–N). After all, they suggest to call it selenocyanocyanide instead of selenium dicyanide (Linke and Lemmer, 1966b). Publications of T. M. Klapötke et al. and J. D. Woollins and co-workers confirmed the results of K.-H. Linke and F. Lemmer in principle, but they all differ due to the C–Se–C angle. On the other hand, they all report very small intermolecular distances of nitrogen and selenium atoms in the crystal of 2.74–2.82 Å, which is significantly smaller than the sum of the van der Waals radii (3.45 Å) and is explained by the σ-hole concept mentioned in chapter 8.2.2 (Burchell et al., 2006; Klapötke et al., 2008). The first IR and Raman spectroscopic measurements were carried out in 1964 and were confirmed experimentally and computationally (Table 77). NMR spectroscopic data of Se(CN)2 in deuterated dichloromethane were reported by J. D. Woollins and co-workers in 2006 (Table 78) (Burchell et al., 2006).
Experimental and computational data of Se(CN)2 (bond lengths in Å; angles in °).
| Publication | Exp./calc. | r Se–C | r C–N | < (C–Se–C) | < (Se–C–N) |
|---|---|---|---|---|---|
| Hazell (1963) | Exp. – XRD | 1.86(10) | 1.42(15) | 119(6) | 177(6) |
| Linke and Lemmer (1966b) | Exp. – XRD | 2.08/2.01 | 1.07/1.27 | 99 | 168/155 |
| Klapötke et al. (2008) | Exp. – XRD | 1.862(7)/1.870(7) | 1.138(8)/1.131(8) | 91.3(3) | 179.2(7)/176.3(6) |
| Burchell et al. (2006) | B3LYP/6-311 + G* | 1.86 | 1.16 | 97 | 175 |
Some spectroscopic data of Se(CN)2 (wave numbers in cm−1).
| Publication | Exp./calc. | ν sym. SeC | ν as. SeC | ν sym. CN | ν as. CN |
|---|---|---|---|---|---|
| Aynsley et al. (1964) | IR (KBr) | 516 | (608) | 2183 | 2175 |
| Raman (in CH3CN) | 514 | 598 | – | 2178 | |
| Burchell et al. (2006) | IR (KBr) | – | 509 | – | 2179 |
| Raman (solid) | 512 | – | 2188 | – | |
| B3LYP/6-311 + G* | 540 | 524 | 2282 | 2269 |
Experimental and computational NMR data of Se(CN)2 in CD2Cl2 (chemical shifts in ppm) (Burchell et al., 2006).
| Compound | δ (13C{1H}, 67.9 MHz) | δ (14N, 21.7 MHz) | δ (77Se, 67.9 MHz) |
|---|---|---|---|
| Se(CN)2 in CD2Cl2 | 91.5 | 299.8 | 0.29 |
| Se(CN)2 at B3LYP/6-311 + G* | 106.9 | 355.2 | 574.87 |
The mesomerism of π-bond electrons and lone pairs causes an interaction of the selenium and carbon atom, which is stronger than a typical single bond and correlates with calculated bond orders of 1.27 regarding Se–C and 2.92 with regard to C–N (Burchell et al., 2006). In 1981, G. Jonkers et al. also analyzed the ionization energy using ultraviolet photoelectron spectroscopy and asserted that the ionization energy EI = 10.96 eV of Se(CN)2 is in the range of elementary carbon (Table 79) (Jonkers et al., 1981).
Experimental and computational (Hartree-Fock-Slater/double zeta) ionization energies (in eV) (Jonkers et al., 1981).
| Method | 1st EI | 2nd EI |
|---|---|---|
| Exp. – UVPE | 10.96(5) | 13.82(5) |
| HFS/DZ | 10.9 | 12.2 |
Selenium diselenocyanate Se(SeCN)2, triselenocyanate [(SeCN)3]–, and selenium triselenocyanate [Se(SeCN)3]–
Selenium diselenocyanate, Se(SeCN) 2 , synthesized from KSeCN in an oxidation process, was first described by A. Verneuil (Verneuil, 1886). Se(SeCN)2 is formed along with Se(CN)2, as rearrangement product of selenocyanogen, (SeCN)2 (Scheme 111). The same products are obtained when Se2Br2 is treated with AgCN or Pb[(SeCN)2] + Br2. W. Muthmann and E. Schröder observed the formation of Se(SeCN)2 in the reaction of KSeCN with nitrogen dioxide (Aksnes et al., 1954; Challenger et al., 1926; Kaufmann and Kögler, 1926; Muthmann and Schröder, 1900; Rogers and Gross, 1952). The molecule is C2 symmetric with Se–C–N angles of 164° and Se–Se bond lengths of 2.33 Å and Se–C bond lengths of 1.83 Å (Figure 47).

Synthesis of Se(SeCN)2 (X = Br−, I−) (Verneuil, 1886).
![Figure 47:
Schematic representation of the molecular structure of Se(SeCN)2, [(SeCN)3]– and [Se(SeCN)3]–.](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_047.jpg)
Schematic representation of the molecular structure of Se(SeCN)2, [(SeCN)3]– and [Se(SeCN)3]–.
Triselenocyanate ion [(SeCN) 3 ] – . Probably the first triselenocyanate salt with potassium as counterion was produced by A. Verneuil in 1886 (Verneuil, 1886). The cesium salt bearing the triselenocyanate ion was first isolated by L. Birckenbach and K. Kellermann in 1925 (Scheme 112) (Birckenbach and Kellermann, 1925b). Half a century later in a series of papers S. Hauge et al. isolated and fully characterized the alkali salts M[(SeCN)3] (M = K+, Rb+, Cs+). The potassium and rubidium salts were isolated as hemihydrates (Hauge et al., 1971a, 1971b, 1975). The alkali metal salts are prepared by oxidation of the corresponding selenocyanates dissolved in water with bromine dissolved in benzene (Scheme 112). Furthermore, it was found that at ambient temperature a mixture of [(SeCN)3]– and [Se(SeCN)3]– crystallizes indicating an equilibrium (Scheme 112). An alternative is the reaction of Se(SeCN)2 with [CN]– (Hauge et al., 1971a). Cs[(SeCN)3] crystallized in monoclinic space group C2/c (Hauge et al., 1975). The three selenium chains are almost linear (178.3(1)°) with Se–Se bond lengths of 2.650(3) Å, which is significantly longer than the Se–Se single bond (cf. Σrcov(Se) = 2.32 Å, Figure 47) (Pyykkö and Atsumi, 2009).
![Scheme 112:
Synthesis of [(SeCN)3]– (Birckenbach and Kellermann, 1925b).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_112.jpg)
Synthesis of [(SeCN)3]– (Birckenbach and Kellermann, 1925b).
Selenium triselenocyanate, [Se(SeCN) 3 ] – , was synthesized by the reaction of Se(SeCN)2 and MCN in water (M = K+, Rb+, Cs+) in a rather complex reaction (Hauge et al., 1971a).
Selenium tetracyanide Se(CN)4
The selenium tetracyanide Se(CN)4 could not be synthesized until now. A reaction of selenium tetrafluoride and trimethylsilyl cyanide TMS–CN at 223 K, which was carried out by T. M. Klapötke et al. in 2008, did not lead to Se(CN)4, but its decomposition products Se(CN)2 and (CN)2 could be detected (Scheme 113) which indicates the in situ generation.

Reaction to synthesize Se(CN)4, which could not be isolated but its decomposition products (Klapötke et al., 2008).
Due to computational studies the selenium dicyanide is significantly more stable than its oxidized species and in detail the reductive decomposition represented in Scheme 113 implies a gain of energy of ΔE = −300.4 kJ mol−1 calculated at MP2/cc-pVTZ (Klapötke et al., 2008). Additional computational data of Se(CN)2 and Se(CN)4 are represented in Table 80.
Calculated data of Se(CN)2 and Se(CN)4 (E in a.u.; zpe in kJ mol−1; bond lengths in Å; angles in °) (Klapötke et al., 2008).
| Se(CN)2 | Se(CN)4 | |
|---|---|---|
| Point group | C 2v | C 2v |
| State | 1 A 1 | 1A′ |
| –E | 194.610124 | 379.814761 |
| zpe | 40.82 | 78.92 |
| r Se–C | 1.842 | 1.854 (eq.) 2.066 (ax.) |
| r C–N | 1.176 | 1.178 (eq.) 1.180 (ax.) |
| < (C–Se–C) | 94.9 | 102.5 (eq.–eq.) 167.1 (ax.–ax.) 86.0 (ax.–eq.) |
The calculated Se(CN)4 exhibits pseudo trigonal-bipyramidal structure and the free valance electron pair is located at an equatorial position. The calculated bond lengths also indicate a significant elongation of the Se–C bond in axial positions and a slightly elongated bond of the axial cyanide groups (Klapötke et al., 2008).
Tellurium
Tellurocyanate [TeCN]−
Salts of tellurocyanate, [TeCN]–, with alkali metal cations like sodium or potassium could not be isolated, so weakly coordinating cations (WCC) are necessary to stabilize the anion. The first synthesis was published by A. W. Downs in 1968 based on a new signal in the Raman spectrum at 2080 cm−1. After a reaction of tellurium and tetraethylammonium cyanide in DMF slightly yellow crystals were obtained, which included a stoichiometric amount of DMF (Scheme 114) (Downs, 1968).
Synthesis of the tellurium monocyanide anion (Downs, 1968).
To obtain crystals, which include no solvent molecules, are more stable and less sensitive to hydrolysis, the reaction was carried out in acetonitrile using tetramethylammonium cyanide or tetraphenylarsonium cyanide (Austad et al., 1971). Due to the increased stability, XRD experiments of the solvent free tetraphenylarsonium tellurocyanate were possible and the stretching mode of the cyanide group was observed at 2081 cm−1 in the IR spectrum.
Nine years later in 1977, M. P. Cava and co-workers published the first synthesis of an alkali metal tellurocyanate salt obtained in a reaction of tellurium and alkaline cyanide dissolved in dry dimethyl sulfoxide at 373 K under inert atmosphere leading to a pale-yellow solution (Scheme 115). But the salt could not be isolated and tellurium was formed after addition of water, acetone or diethylether (Spencer et al., 1977).

First synthesis of potassium tellurocyanate (Spencer et al., 1977).
Only a few spectroscopic studies of the tellurocyanate were reported due to its sensitivity and instability. Some of these spectroscopic data are shown in Table 81. The calculated data were obtained by extrapolation of force constants k and interaction force constants kEC of experimental values of the preceding cyanates [OCN]–, [SCN]– and [SeCN]– (Greenwood et al., 1964).
Selected experimental spectroscopic data of K[TeCN] and computational data of the tellurocyanate anion (bond lengths in Å; wave numbers in cm−1).
| Method | r Te–C | r C–N | ν TeC | ν CN |
|---|---|---|---|---|
| Exp. (Loewenschuss and Marcus, 1996) | 1.904 | 1.153 | 459 | 2086 |
| Extrapolation (Greenwood et al., 1964) | – | – | 455–457 | 2083–2086 |
Tellurium dicyanide Te(CN)2
The first synthesis and elementary analysis of tellurium dicyanide Te(CN)2 was published by H. E. Cocksedge in 1908. The synthesis was based on a reaction of tellurium tetrabromide and silver cyanide (Scheme 116). The product was sublimed to obtain crystals of 70.9% tellurium (calc. 71.0%) and 28.8% CN (calc. 29.0%), which are sensitive to hydrolysis and decomposed into tellurium and cyanogen at 373 K (Cocksedge, 1908).

First synthesis of tellurium dicyanide (Cocksedge, 1908).
More than 50 years later H. P. Fritz and H. Keller repeated the synthesis published by H. E. Cocksedge, the product was purified by high vacuum sublimation and an IR measurement was carried out using the received pale-pink crystals. The bands at νCN = 2181 cm−1 and νCN = 2179 cm−1 were assigned to the C–N stretching mode and the asymmetrical tellurium–carbon stretching mode was observed at νTeC = 403 cm−1. Due to two bands of the cyanide group and in comparison to other chalcogene halide compounds they assumed a bent structure of the Te(CN)2, although only one fundamental of the Te–C bond was found. They also reported the thermal decomposition of Te(CN)2 at 353 K (Fritz and Keller, 1961).
A complete structure characterization including XRD measurements were reported by T. M. Klapötke et al. in 2008. In addition to the synthetic route of H. E. Cocksedge using tellurium tetrabromide, T. M. Klapötke et al. could report a synthesis starting with tellurium tetraiodide. In regard to the IR and Raman spectrum they also observed two bands of the cyanide group at νCN = 2176 cm−1 and νCN = 2168 cm−1, but additionally they could identify two fundamentals at νTeC = 410 cm−1 and νTeC = 398 cm−1 originating from the Te–C bond and finally confirming the bent structure. The two signals in the mass spectrum (EI) were related to the parent ion [Te(CN)2]+ (m/z = 182) and to the fragment [TeCN]+ (m/z = 156). To obtain NMR spectra, the product was dissolved in deuterated THF. The chemical shift of the C atom is at δ (13C) = 86.2 ppm, the chemical shift of the N atom at δ (14N) = −70 ppm and the chemical shift of Te at δ (125Te) = 567 ppm. The coupling constant of C and Te is reported to be 1J(125Te-13C) = 330.2 Hz. In the 14N NMR spectrum extra signals of dinitrogen (δ (14N) = −71.5 ppm) and cyanogen (δ (14N) = −123 ppm) were found. A solution of Te(CN)2 in diethyl ether was layered by n-heptane and cooled to 248 K receiving colorless needles suitable for X-ray structure elucidation. Decomposition being faster than crystallization was problematic. The space group is
The tellurium dicyanide exhibits C 2v symmetry in gas phase, but in regard to the experimental crystal structure the symmetry is decreased due to electrostatic interaction of lone pairs of neighboring cyanide groups, which is explained by the σ-hole concept mentioned in chapter 8.2.2. This causes a nearly square-planar coordination of Te(CN)2 due to solvent molecules, which are included in the crystal. Both effects cause a significant variation of experimental and computational data (George et al., 2015).
Tellurium tricyanide Te(CN)3
Homoleptic threefold cyanide substituted tellurides Te(CN)3 are not stable and require one more halogen to gain a certain level of stability (Fritz et al., 2008).
Tellurium tetracyanide Te(CN)4
Tellurium tetracyanide Te(CN)4 is less investigated due to its high sensitivity and its tendency to explosively decompose at air or temperatures higher than 253 K. In 2004, T. M. Klapötke et al. published a synthesis of Te(CN)4 based on a reaction of tellurium tetrafluoride and four equivalents of trimethylsilyl cyanide, TMS–CN, dissolved in dichloromethane, which is similar to the reaction of Se(CN)4 (Scheme 117) (Table 82).
Synthesis of Te(CN)4 accordingly the synthesis of Se(CN)4 (Klapötke et al., 2004; Klapötke et al., 2008).
Selected computational data of Te(CN)2 (zpe in kJ mol−1; bond lengths in Å; angles in °; wave numbers in cm−1) (Klapötke et al., 2008).
| Method + basis set | zpe | r Te–C | r C–N | < (C–Te–C) | ν CN |
|---|---|---|---|---|---|
| B3LYP/cc-pVQZ | 40.57 | 2.054 | 1.153 | 94.46 | 2274/2266 |
| MP2(fc)/cc-pVQZ | 39.05 | 2.021 | 1.177 | 93.19 | 2101/2100 |
The product was isolated in form of a colorless powder and was analyzed by Raman spectroscopy due to its low solubility in common solvents (Table 83). The identification of four different carbon–nitrogen stretching modes implies the presence of two axial and two equatorial cyanide groups. Furthermore, computational studies of Te(CN)4 were carried out using different methods and basis sets (Table 84).
C–N stretching modes of Te(CN)4 in the Raman spectrum (wave numbers in cm−1) (Klapötke et al., 2004, 2008).
| Compound | ν CN, 1 | ν CN, 2 | ν CN, 3 | ν CN, 4 |
|---|---|---|---|---|
| Te(CN)4 | 2204 | 2191 | 2149 | 2129 |
Selected computational data of Te(CN)4 (total energies in a.u.; zpe in kJ mol−1; bond lengths in Å; angles in °; wave numbers in cm−1) (Klapötke et al., 2004, 2008).
| Method + basis set | E tot. | zpe | r Te–C | r C–N | < (C–Te–C) | ν CN |
|---|---|---|---|---|---|---|
| B3LYP/cc-pVTZ | −639.4585 | 88.91 | 2.074/2.227 | 1.152/1.155 | 84.35/104.06 | 2300/2295/2265/2263 |
| MP2(FC)/cc-pVTZ | −637.7936 | 77.00 | 2.042/2.180 | 1.176/1.180 | 83.53/105.60 | 2089/2086/2057/2053 |
The computational results imply two pairs of cyanide groups and C 2v symmetry leading to a trigonal-bipyramidal structure. The reductive decomposition of tellurium tetracyanide forming tellurium dicyanide and cyanogen is exothermic (ΔE = −357.8 kJ mol−1, MP2(fc)/cc-pVTZ) illustrating the high instability of Te(CN)4 (Scheme 118) (Klapötke et al., 2004, 2008).

Decomposition of tellurium tetracyanide (Klapötke et al., 2004, 2008).
Tellurium hexacyanide Te(CN)6
Tellurium hexacyanide Te(CN)6 could not be synthesized until now and computational studies of this compound are rare. T. M. Klapötke et al. published results of calculations at B3LYP/cc-pVTZ and MP2(fc)/cc-pVTZ in 2004 (Table 85).
Selected computational data of Te(CN)6 (zpe in kJ mol−1; bond lengths in Å; angles in °; wave numbers in cm−1) (Klapötke et al., 2004).
| Method + basis set | zpe | r Te–C | r C–N | < (C–Te–C) | ν CN |
|---|---|---|---|---|---|
| B3LYP/cc-pVTZ | 125.49 | 2.118 | 1.153 | 90 | 2292–2297 |
| MP2(fc)/cc-pVTZ | 118.69 | 2.078 | 1.179 | 90 | 2062–2070 |
The centered tellurium atom is octahedral coordinated by cyanide groups (point group O h ). The reductive decomposition of tellurium hexacyanide forming tellurium tetracyanide and cyanogen is exothermic (ΔE = −343.32 kJ mol−1, MP2(fc)/cc-pVTZ) illustrating the high instability of Te(CN)6 (Scheme 119). However, the received tellurium tetracyanide itself also decomposes yielding tellurium dicyanide and cyanogen (Klapötke et al., 2004).

Reductive decomposition of tellurium hexacyanide (Klapötke et al., 2004).
Polonium
Since polonium is extremely rare on earth, e.g. traces in uranium pitchblende (uraninite), and is also highly radioactive, it is not surprising that nothing is known about polonium cyanide.
7th Main group
Formally, all halogen cyanides (also cyanogen halides), X–CN (X = F−–I−) can be regarded as halogen-pseudohalogen compounds. Their aggregate state ranges from gaseous to solid at ambient temperatures and normal pressure and the melting and boiling points increase in accordance to the atomic number. Their reaction behavior is manifold and they tend to oligomerize forming the corresponding cyanuric halides, e.g. (XCN)3. The solid structure of cyanogen fluoride is not experimentally confirmed, but there are many indications, that all cyanogen halides are crystallizing in the cyanogen bromide or iodide type. But in general, it can be assumed that all XCN molecules show a chain-like orientation of the molecules in the crystal.
The application of cyanogen halides of the smaller homologues fluorine, chlorine and bromine has not been intensively studied and is limited due to the instability of these compounds, quite in contrast to ICN, which has a quite diverse chemistry.
Fluorine
Cyanogen fluoride FCN, isocyanic fluoride FNC and their ions
Cyanogen fluoride FCN and isocyanic fluoride FNC
The first description of cyanogen fluoride FCN was published by V. E. Cosslett in 1931 being the latest cyanogen halide that was found. The colorless solid sublimes at 201 K and forms a colorless, sharp smelling gas. Synthesis was achieved in the reaction of silver fluoride and cyanogen iodide or bromide carried out in an evacuated glass ampulla at 493 K (Scheme 120) (Cosslett, 1931). It should be noted that this synthesis could not be repeated by E. E. Aynsley et al. in 1959 (Aynsley et al., 1959).

Synthesis of cyanogen fluoride published by Cosslett (X = Br−, I−) (Cosslett, 1931).
Moreover, it was reported that the pyrolysis of cyanuric fluoride in a stream of nitrogen at 1573 K and 67 mbar led to the formation of cyanogen fluoride (Scheme 121).

Synthesis of cyanogen fluoride by thermolysis of cyanuric fluoride (Fawcett and Lipscomb, 1960; Panas, 1994).
In this context, F. S. Fawcett and R. D. Lipscomb reported a boiling point of TB = 229 K, a melting point at TM = 191 K and characteristic bands at νCN = 2290 cm−1 and νCF = 1078 cm−1 were allocated to the cyanogen fluoride in good accordance to earlier assignments (Fawcett and Lipscomb, 1960; Panas, 1994). But they also studied alternative routes of pyrolysis to obtain FCN and at the end of several tests of different precursors they patent their formation of FCN by pyrolysis (Fawcett and Lipscomb, 1964; Lipscomb and Smith, 1961).
A synthetic route to obtain FCN, but requiring smaller instrumental effort was published by E. Mayer in 1969 and is based on the reaction of cesium fluoride and tetracyanomethane, although this method was already used to obtain the heavier compounds cyanogen chloride and bromide (Scheme 122) (Mayer, 1969c).

Synthesis of FCN reported by E. Mayer (Mayer, 1969c).
And in the early 1970s, Du Pont submitted another patent on the formation of FCN which describes fluorination of a solution of sodium cyanide using elemental fluorine (Scheme 123) and analyzing the escaping gas stream after 10 mins of reaction (Grakauskas, 1970).

Fluorination of sodium cyanide in solution patented by Du Pont (Grakauskas, 1970).
The MW spectra of cyanogen fluoride suggest a C–F bond length of about 1.262 Å and a carbon–nitrogen bond length of about 1.159 Å, which is in good accordance to computations (rC–F = 1.26–1.29 Å, rC–N = 1.15–1.18 Å) (Bhattacharyya et al., 2007a; Degli Esposti et al., 1982; Lee and Racine, 1995; Lee et al., 1995; Ruoff, 1970; Schmiedekamp et al., 1980; Tyler and Sheridan, 1963). Based on MW spectra the dipole moment was determined being μ = 2.17 D, which is also in good accordance to computational studies (μ = 2.13–2.31 D) (Bhattacharyya et al., 2007a; Lee and Racine, 1995; Tyler and Sheridan, 1963; Sheridan et al., 1960; Zúñiga et al., 2012).
The crystal structures of the homologous cyanogen halides are known for several years, but until now the structure determination of cyanogen fluoride by single crystal XRD has not been possible. Based on computational studies two possible crystal structures were proposed, which are analogous to the non-polar, crystalline cyanogen chloride or polar hydrogen cyanide depending on the chosen structure modell. For each basic type two unit cells, an orthorhombic and a tetragonal one, can be derived (Table 88) (Panas, 1994).
The extremely unstable isocyanic fluoride was synthesized by D. E. Milligan and M. E. Jacox by ultraviolet photolysis of cyanogen fluoride and characterized using IR spectroscopy (Milligan and Jacox, 1967a, 1967b). The patent of Du Pont also reports the formation of isocyanic fluoride using potassium isocyanide instead of sodium cyanide (Scheme 123) (Grakauskas, 1970). Additionally, some theoretical studies are dealing with the constitution isomers of cyanogen fluoride (Tables 86 and 87). In regard to computations the N–F bond length is in the range of 1.31–1.33 Å and the C–N bond length in the range of 1.15–1.20 Å (Lee et al., 1995; Schmiedekamp et al., 1980). Furthermore, bands of streching modes of νCN = 2214 cm−1 and νNF = 964 cm−1 are supposed and the mechanism of photochemical isomerization of cyanogen fluoride in contrast to dissociation is discussed (Zhang et al., 2007).
Some experimental and computational data of cyanogen fluoride (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r F–C | r C–N | ν FC | ν CN |
|---|---|---|---|---|---|
| Aynsley et al. (1959) | Exp. – IR | 1.26 | 1.16 | 1077 | 2290 |
| Fawcett and Lipscomb (1960) | Exp. – IR | – | – | 1078 | 2290 |
| Tyler and Sheridan (1963) | Exp. – MW | 1.262 | 1.159 | – | – |
| Esposti et al. (1982) | Exp. – MW | 1.26240 | 1.15928 | – | – |
| Lee et al. (1995) | CCSD(T)/cc-pVDZ | 1.2826 | 1.1779 | – | – |
| CCSD(T)/cc-pVTZ | 1.2701 | 1.1632 | 1077 | 2312 | |
| CCSD(T)/cc-pVQZ | 1.2670 | 1.1596 | – | – | |
| Lee and Racine (1995) | CCSD/TZ2P | 1.270 | 1.153 | 1087 | 2404 |
| CCSD(T)/TZ2P | 1.275 | 1.162 | 1060 | 2331 | |
| Wang et al. (1998) | CASSCF(6,6)/6-31G* | 1.2630 | 1.1698 | 1132.3 | 2409.8 |
| MP2/6-31G* | 1.2828 | 1.1825 | 1060.6 | 2267.4 | |
| MP2/6-311G(2df) | 1.2640 | 1.1689 | 1082.2 | 2291.5 | |
| CCSD(T)/6-311G* | 1.2717 | 1.1665 | 1068.2 | 2350.6 | |
| CCSD(T)/6-311G(2d) | 1.2733 | 1.1625 | 1055.8 | 2322.2 | |
| Bhattacharyya et al. (2007a) | MP2/cc-pVTZ | 1.27 | 1.17 | 1074.16 | 2277.52 |
| CCSD/cc-pVTZ | 1.27 | 1.15 | 1104.23 | 2422.87 | |
| CCSD(T)/cc-pVTZ | 1.27 | 1.16 | 1078.47 | 2350.36 | |
| Zhang et al. (2007) | B3LYP/6-311G** | – | – | 1088.0 | 2419.6 |
Selected experimental and computational data of isocyanic fluoride (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r F–N | r C–N | ν FN | ν CN |
|---|---|---|---|---|---|
| Milligan and Jacox (1967a), (1967b) | Exp. – IR | – | – | 928 | 2123 |
| Lee et al. (1995) | CCSD(T)/cc-pVDZ | 1.3199 | 1.2011 | – | – |
| CCSD(T)/cc-pVTZ | 1.3097 | 1.1842 | 935 | 2109 | |
| Lee and Racine (1995) | CCSD/TZ2P | 1.310 | 1.173 | 967 | 2220 |
| CCSD(T)/TZ2P | 1.321 | 1.184 | 912 | 2126 | |
| Bhattacharyya et al. (2007a) | MP2/cc-pVTZ | 1.30 | 1.19 | 987.20 | 2127.65 |
| CCSD/cc-pVTZ | 1.30 | 1.17 | 997.44 | 2248.01 | |
| CCSD(T)/cc-pVTZ | 1.31 | 1.18 | 948.01 | 2157.39 | |
| Zhang et al. (2007) | B3LYP/6-311G** | – | – | 963.5 | 2214.0 |
Possible crystal structures of cyanogen fluoride (dimensions of unit cell in Å; volumes in Å3) (Panas, 1994).
| Parameter | ClCN type tetragonal | ClCN type orthorhombic | HCN type tetragonal | HCN type orthorhombic |
|---|---|---|---|---|
| Space group | P4/nmm | Pmmn | I4mm | Imm |
| a | 4.76 | 4.50 | 4.29 | – |
| b | 4.76 | 4.87 | 4.29 | – |
| c | 5.40 | 5.50 | 5.45 | – |
| V | 122.35 | 120.53 | 100.30 | – |
| Z | 2 | 2 | 2 | 2 |
Cyanuric fluoride, (FCN) 3 (also known as trifluorotriazine, cyanuric trifluoride, trifluoro-s-triazine, 2,4,6-Trifluorotriazine, 2,4,6-trifluro-s-triazin, trifluoro-1,3,5-triazine), which is the trimer of FCN, is commercially available as it is widely used as a fluorinating agent in the conversion of carboxylic acids into acyl fluorides. It is used as a precursor for fibre-reactive dyes and a specific reagent for tyrosine residues in enzymes (Anderson and Frick, 1972; Chambers and Tamura, 1985; Fawcett and Lipscomb, 1964; Grisley et al., 1958; Groβ et al., 2000; Kober and Grundmann, 1959; Kober et al., 1962; Maxwell et al., 1958; Nenajdenko, 2014; Rusinov et al., 2014; Sheridan et al., 1960; Shmel’kova et al., 1989; Zúñiga et al., 2012). Most synthesis processes for the preparation of cyanuric fluorides are based on cyanuric chloride, which can be converted to cyanuric fluoride by Cl−/F−-exchange using various fluoride sources (Scheme 124). When using alkali fluorides, it is necessary to use COF2 as a catalyst to allow lower temperatures (Anderson and Frick, 1972).

Synthesis of cyanuric fluoride starting from cyanuric chloride (FS = fluoride source, MF, SbF3; M = alkali metal with COF2 as catalyst) (Anderson and Frick, 1972).
Cyanogen fluoride anion [FCN]− and isocyanic fluoride anion [FNC]−
Neither the cyanogen fluoride anion, which is an odd electron species, nor the isocyanic fluoride anion could be synthesized or isolated until now. The isomeric structures and other properties were analyzed in several computational studies. Some of these data, bond angles, bond lengths and spectroscopic data for example, reported by different working groups are represented in Tables 89 and 90.
Selected computational data of the cyanogen fluoride anion (bond lengths in Å; angles in °; wave numbers in cm−1).
| Publication | Exp./calc. | r F–C | r C–N | < (F–C–N) | ν FC | ν CN |
|---|---|---|---|---|---|---|
| Gauld et al. (1997) | MP2/6-31G(d) | 1.607 | 1.201 | 130.6 | – | – |
| QCISD/6-31G(d) | 1.616 | 1.205 | 129.1 | – | – | |
| QCISD/6-311 + G(2d,f,p) | 1.531 | 1.197 | 128.1 | – | – | |
| PW-P86/IGLO-III | 1.718 | 1.192 | 131.3 | – | – | |
| Guerra (1999) | UQCISD/6-311G(d,p) | 1.638 | 1.195 | 128.9 | – | – |
| UQCISD/6-311 + G(d,p) | 1.603 | 1.199 | 128.1 | – | – | |
| Sommerfeld (2001) | MP2/TZV(d) + p | 1.6538 | 1.1887 | 130.4 | – | – |
| SDCI/TZV(d) + p | 1.6007 | 1.1799 | 128.5 | – | – | |
| CCD/TZV(d) + p | 1.6423 | 1.1826 | 128.7 | – | – | |
| CCSD/TZV(d) + p | 1.7056 | 1.1848 | 131.1 | 534.8 | 2013.1 | |
| CCSD/TZV(2df) + sp | 1.5314 | 1.1928 | 128.6 | – | – | |
| CCSD(T)/TZV(d) + p | 1.7215 | 1.1920 | 131.6 | – | – | |
| CCSD(T)/TZV(2df) + sp | 1.5661 | 1.1989 | 128.8 | – | – | |
| Bhattacharyya et al. (2007b) | B3LYP/cc-pVTZ | 1.67 | 1.18 | 129.9 | 534.86 | 1972.17 |
| MP2/cc-pVTZ | 1.58 | 1.19 | 131.0 | 816.17 | 2056.13 | |
| CCSD/cc-pVTZ | 1.57 | 1.19 | 127.7 | 605.22 | 1887.52 | |
| CCSD(T)/cc-pVTZ | 1.59 | 1.20 | 129.0 | 612.11 | 1891.68 |
Selected computational data of the isocyanic fluoride anion (bond lengths in Å; angles in °; wave numbers in cm−1).
| Publication | exp./calc. | r F–N | r C–N | < (F–N–C) | ν FN | ν CN |
|---|---|---|---|---|---|---|
| Sommerfeld (2001) | MP2/TZV(d) + p | 1.8592 | 1.1937 | 141.6 | – | – |
| SDCI/TZV(d) + p | 2.1002 | 1.1682 | 180.0 | – | – | |
| CCD/TZV(d) + p | 2.0173 | 1.1769 | 150.3 | – | – | |
| CCSD/TZV(d) + p | 2.0128 | 1.1803 | 180.0 | 324.0 | 2117.1 | |
| CCSD/TZV(2df) + sp | 1.9874 | 1.1747 | 180.0 | – | – | |
| CCSD(T)/TZV(d) + p | 2.0000 | 1.1880 | 165.3 | – | – | |
| CCSD(T)/TZV(2df) + sp | 1.9625 | 1.1837 | 161.9 | – | – | |
| Bhattacharyya et al. (2007b) | B3LYP/cc-pVTZ | 2.06 | 1.17 | 150.1 | 307.39 | 2105.08 |
| MP2/cc-pVTZ | 1.86 | 1.19 | 144.1 | 368.36 | 1949.63 | |
| CCSD/cc-pVTZ | 2.00 | 1.18 | 180.0 | 335.20 | 2114.95 | |
| CCSD(T)/6–311 + G(2d,2p) | 1.98 | 1.18 | 180.0 | 372.66 | 2052.87 |
The calculated bond lengths significantly vary depending on the chosen method and basis set. The F–C bond length of the cyanogen fluoride anion is in the range of 1.53–1.72 Å, whereas the F–N bond length of the isocyanic fluoride anion is in the range of 1.86–2.10 Å. The variation of the C–N bond length is smaller, but also depends on method, basis set and isomer ([FCN]–: 1.18–1.21 Å; [FNC]–: 1.17–1.19 Å).
Cyanogen fluoride cation [FCN]+
Similar to the cyanogen fluoride anion the cation species could also not be synthesized until now. But several computational studies have been published during the last decades, for example by A. K. Das and co-workers (Bhattacharyya et al., 2007a) and D.-C. Wang et al. (1998). Selected structural and spectroscopic data are listed in Table 91.
Some computational data of [FCN]+ (bond lengths in Å; wave numbers in cm−1).
| Author | Exp./calc. | r F–C | r C–N | ν FC | ν CN |
|---|---|---|---|---|---|
| D.-C. Wang et al. (1998) | CASSCF(5,6)/6-31G* | 1.1979 | 1.2362 | 1170.1 | 2219.1 |
| MP2/6-31G* | 1.2139 | 1.2128 | 1350.5 | 3021.0 | |
| MP2/6-311G(2df) | 1.1962 | 1.2106 | 1339.7 | 2763.0 | |
| CCSD(T)/6-311G* | 1.2050 | 1.2315 | 1145.8 | 2195.1 | |
| CCSD(T)/6-311G(2d) | 1.2062 | 1.2299 | 1133.9 | 2159.8 | |
| IFCA | 1.205 | 1.223 | 1134 | 2160 | |
| Bhattacharyya et al. (2007a) | MP2/cc-pVTZ | 1.20 | 1.21 | 1342.26 | 2795.49 |
| CCSD/cc-pVTZ | 1.20 | 1.22 | 1170.21 | 2218.79 | |
| CCSD(T)cc-pVTZ | 1.21 | 1.23 | 1151.81 | 2187.14 |
Difluorocyan cation [F2(CN)]+
Calculations of the difluorocyan cation [F2(CN)]+ and its structure were published by P. Pyykkö and N. Runeberg in 1991 and T. M. Klapötke and co-workers considered the linear cation to be an adduct of a fluorine atom and the cyanogen fluoride cation (Pyykkö and Runeberg, 1991). Their synthesis is a two-step-reaction. After the reaction of hydrogen cyanide and arsenic pentafluoride in liquid hydrogen fluoride the received cation [HCNH]+ is fluorinated using an excess of elementary fluorine (Scheme 125).

Two-step-synthesis of the difluorocyan cation by Tornieporth-Oetting et al. (1991).
T. M. Klapötke and co-workers could characterize the cation using IR and Raman spectroscopy, but could not isolate the salt due to decomposition at ambient temperatures. The experimental data reported by T. M. Klapötke and co-workers, their computational results and the data of P. Pyykkö and N. Runeberg are summarized in Table 92 (Pyykkö and Runeberg, 1991; Tornieporth-Oetting et al., 1991).
Experimental Raman data and computational data of [F2(CN)][AsF6] (bond lengths in Å; wave numbers in cm−1).
| Exp./calc. | r F–CNF | r FC–NF | r FCN–F | ν (σg) | ν (σu) | ν (σu) | ν (πg) | ν (πg) |
|---|---|---|---|---|---|---|---|---|
| Exp. – Raman (Tornieporth-Oetting et al., 1991) | – | – | – | 2587 | 1378* | 729 | 456 | 243 |
| HF/6-31G[a] (Tornieporth-Oetting et al., 1991) | – | – | – | 2565 | 1312 | 774 | 457 | 230 |
| HF/6-31G[a] (Pyykkö and Runeberg, 1991) | 1.215 | 1.108 | 1.261 | 3017 | 1544 | 911 | 537 | 270 |
| MP2/6-31G[a] (Pyykkö and Runeberg, 1991) | 1.241 | 1.147 | 1.282 | – | – | – | – | – |
-
[a]IR data.
Chlorine
Cyanogen chloride ClCN, isocyanic chloride ClNC and their ions
Cyanogen chloride (also known as chlorine cyanide, chlorocyanogen, chlorcyan) is both an industrial chemical needed to build a variety of organic/inorganic products (e.g. cyan amide, H2N–CN, in the reaction with ammonia), but it is also a chemical warfare agent (1.5 HCN sources, toxicology, treatment, and mode of action (Newhouse and Chiu, 2010)) (Romano et al., 2007). It hydrolyzes in water to form HCl and HOCN.
Cyanogen chloride ClCN and isocyanic chloride ClNC
The first synthesis of cyanogen chloride ClCN was carried out by C.-L. Berthollet in 1787 and several studies were published since that time (Jennings and Scott, 1919). During the World War I it was used as a warfare agent due to its toxicity. Many different synthetic routes have been reported in the earlier years, especially wet chemical methods based on the reaction of cyanides and elementary chlorine in aqueous solution. One example is the reaction of potassium cyanide and chlorine water carried out by A. Hantzsch and L. Mai (Scheme 126). To avoid side reaction, an excess of chlorine water is necessary (Hantzsch and Mai, 1895).

Synthesis of cyanogen chloride by A. Hantzsch and L. Mai (Hantzsch and Mai, 1895).
Therefore, several working groups tried to find synthetic routes with less experimental effort resulting in the use of in situ formed potassium tetracyanozincate instead of potassium cyanide (Scheme 127) (Klemenc and Wagner, 1938; Schröder, 1958).

Synthesis of cyanogen chloride using potassium tetracyanozincate (Schröder, 1958).
An anhydrous synthesis of cyanogen chloride is the reaction of chlorine and a mixture of potassium cyanide and sand to distribute heat (Jennings and Scott, 1919). R. Varma and A. J. Signorelli reported a similar route using dichlorine monoxide and silver cyanide (Scheme 128) (Varma and Signorelli, 1969).

Anhydrous synthesis of cyanogen chloride by R. Varma and and A. J. Signorelli (Varma and Signorelli, 1969).
W. Sundermeyer also discussed molten salts and their use as reaction media. As an example of halogen-pseudohalogen exchange, he studied the reaction of potassium cyanide and chlorine to form cyanogen and potassium chloride. Discussing the cyanogen chloride as a possible intermediate, he reported that the reaction of potassium cyanide and chlorine may lead to cyanogen chloride in nearly quantitative yields in the first step (Sundermeyer, 1965). Additionally, several industrial synthetic routes were developed. For example, a patent of Degussa includes the reaction of chlorine and hydrogen cyanide, which is formed in situ using hydrogen peroxide and hydrogen chloride (Scheme 129).

Industrial formation of cyanogen chloride protected by patent of Degussa (Heilos et al., 1971; Heimberger and Schreyer, 1975).
Catalysts are Lewis acids, copper dichloride or aluminum trichloride for example (Heilos et al., 1971; Heimberger and Schreyer, 1975). The immediate reaction of chlorine and hydrogen cyanide in liquid phase at low temperatures and in presence of a catalyst is protected by a patent owned by the Bayer AG (Scheme 130). Catalysts are Lewis acids like phosphorus trichloride and side products of this reaction are cyanuric chloride and hydrogen chloride (Enders, 1972). In 2007, Evonik published a patent for a continuous process for ClCN that is particularly suitable for industrial scale. The process is based on sodium cyanide, which is reacted with chlorine in aqueous solution to form cyanogen chloride (Tautz and Blumberg, 2007).

Industrial formation of cyanogen chloride protected by patent of Bayer AG (Enders, 1972).
Under standard conditions cyanogen chloride is a colorless gas, which liquefies under normal pressure at 286 K and changes to solid state at 266 K (Chadwick and Edwards 1973). Especially MW spectroscopy was used to extensively investigate cyanogen chloride in gaseous state. The carbon–nitrogen bond length is reported to be in the range of 1.14–1.17 Å and the chlorine–carbon bond length in the range of 1.62–1.64 Å (Townes et al., 1947, 1948; Tyler and Sheridan, 1963; Smith et al., 1948). In regard to MW spectroscopy the experimental dipole moment of cyanogen chloride is μ = 2.80 D, which is in accordance to the calculated value of μ = 2.869 D (Kellö and Sadlej, 1992; Tyler and Sheridan, 1963). Based on IR spectra the band at 2219 cm−1 was allocated to the C–N stretching mode and the band at 714 cm−1 was assigned to the C–Cl stretching mode by W. O. Freitag and E. R. Nixon, which is in good accordance to the Raman spectra of M. Pézolet and R. Savoie (Freitag and Nixon, 1956; Pézolet and Savoie, 1971).
In 1956 the first structure determination using single crystal XRD of solid cyanogen chloride was published by R. B. Heiart and G. B. Carpenter. It forms orthorhombic crystals, space group Pmmn, and each unit cell contains two molecules. Similar to the later homologs the molecules exhibit a substructure of chains along the c-axis. They argued that the bond lengths are decreased to rCl–C = 1.57(1) Å and rC–N = 1.16(2) Å and due to these intermolecular interactions along the chains, respectively (Heiart and Carpenter, 1956).
Next to experimental results, several computational studies have been published in literature (Bhattacharyya et al., 2007a; Destro et al., 1988; Lee and Racine, 1995; Lee et al., 1995; Schmiedekamp et al., 1980; Støgard, 1976; Zhang et al., 2007). A summary of these data in comparison to the experimental values are represented in Tables 93 and 94. Computational data of the isocyanic chloride are also included, although the isomerization product could not be isolated, but D. E. Milligan and M. E. Jacox reported that photolysis experiments similar to the formation of isocyanic fluoride and spectroscopic analysis may indicate the formation of ClNC (Milligan and Jacox, 1967a, 1967b).
Some experimental and computational data of cyanogen chloride (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r Cl–C | r C–N | ν ClC | ν CN |
|---|---|---|---|---|---|
| Townes et al. (1947) | Exp. – MW | 1.64 | 1.15 | – | – |
| Smith et al. (1948) | Exp. – MW | 1.630 | 1.163 | – | – |
| Townes et al. (1948) | Exp. – MW | 1.629 | 1.163 | – | – |
| Freitag and Nixon (1956) | Exp. – IR | – | – | 714 | 2219 |
| Heiart and Carpenter (1956) | Exp. – XRD | 1.57(1) | 1.16(2) | – | – |
| Tyler and Sheridan (1963) | Exp. – MW | 1.631 | 1.159 | – | – |
| Pézolet and Savoie (1971) | Exp. – Raman | – | – | 728 | 2205 |
| Støgård (1976) | HF (no 3d of Cl) | 1.684 | 1.150 | – | – |
| HF (with 3d of Cl) | 1.657 | 1.151 | – | – | |
| Lee and Racine (1995) | CCSD/TZ2P | 1.652 | 1.155 | 729 | 2313 |
| CCSD(T)/TZ2P | 1.655 | 1.164 | 716 | 2236 | |
| Lee et al. (1995) | CCSD(T)/cc-pVDZ | 1.6604 | 1.1800 | – | – |
| CCSD(T)/cc-pVTZ | 1.6437 | 1.1656 | 707 | 2209 | |
| CCSD(T)/cc-pVQZ | 1.6384 | 1.1619 | – | – | |
| Bhattacharyya et al. (2007a) | MP2/cc-pVTZ | 1.63 | 1.17 | 754.55 | 2139.38 |
| CCSD/cc-pVTZ | 1.64 | 1.16 | 749.54 | 2318.78 | |
| CCSD(T)/cc-pVTZ | 1.64 | 1.17 | 737.80 | 2241.87 | |
| Zhang et al. (2007) | B3LYP/6-311G** | – | – | 737.4 | 2316.4 |
Some experimental and computational data of isocyanic chloride (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r Cl–N | r C–N | ν ClN | ν CN |
|---|---|---|---|---|---|
| Milligan and Jacox (1967a, 1967b) | Exp. – IR | – | – | 615 | 2074 |
| Destro et al. (1988) | SCF/STO-3G | 1.693 | 1.179 | – | – |
| SCF/3-21G | 1.690 | 1.165 | – | – | |
| SCF/6-31G* | 1.619 | 1.157 | – | – | |
| Lee et al. (1995) | CCSD(T)/cc-pVDZ | 1.6612 | 1.2009 | – | – |
| CCSD(T)/cc-pVTZ | 1.6371 | 1.1845 | 702 | 2069 | |
| Bhattacharyya et al. (2007a) | MP2/cc-pVTZ | 1.62 | 1.19 | 734.87 | 2074.88 |
| CCSD/cc-pVTZ | 1.63 | 1.18 | 733.94 | 2185.78 | |
| CCSD(T)/cc-pVTZ | 1.64 | 1.18 | 706.99 | 2105.81 | |
| Zhang et al. (2007) | B3LYP/6-311G** | – | – | 685.2 | 2149.8 |
Cyanuric chloride (ClCN) 3 , the trimer of cyanogen chloride, is commonly produced on a large scale worldwide by trimerization of cyanogen chloride (Burger and Hornbaker, 1953; Grundmann and Kober, 1956; Grundmann and Kreutzberger, 1954, 1955; Goubeau et al., 1954; Huthmacher and Most, 2000; Ruoff, 1970; Kreutzberger, 2006; Romano et al., 2007; Tautz and Blumberg, 2007). The synthesis of (ClCN)3 usually proceeds in two steps. The hydrocyanic acid is converted into cyanogen chloride, which is directly trimerized to cyanuric chloride. The dry cyanogen chloride is trimerized at 573 K on activated carbon. The ClCN yield in this process exceeds 95%; the (ClCN)3 yield exceeds 90% (Scheme 131) (Huthmacher and Most, 2000).

Two-step synthesis of cyanuric chloride (Huthmacher and Most, 2000).
Catalysts for the cyanogen chloride trimerization as activated carbon are, among others, molten cyanuric acid chloride, molten aluminum chloride, eutectic mixtures of tetrachloroaluminates, aluminum silicates, and Zeolites doped with metal oxides or sulphides. In some of these processes, ’’tetrameric cyanogen chloride’’ (2,4-dichloro-6-isocyanodichloro-5-triazine) is a by-product; its conversion to cyanuric chloride is catalyzed by iron oxide. In the laboratory, ClCN trimerizes in benzene or chloroform solution in the presence of hydrogen chloride (Diels, 1899). For further details on the chemistry of cyanuric chloride, we refer to the review article by K. Huthmacher (Huthmacher and Most, 2000).
Cyanogen chloride anion [ClCN] − and isocyanic chloride anion [ClNC] −
Similar to the fluorine species, the cyanogen chloride anion [ClCN]– and the isocyanic chloride anion [ClNC]–could not be isolated until now. The data of a theoretical study are represented in Table 95 (Bhattacharyya et al., 2007b).
Computational data of the isomers [ClCN]– and [ClNC]– reported by I. Bhattacharyya et al. (bond lengths in Å; angles in °; wave numbers in cm−1) (Bhattacharyya et al., 2007b).
| Compound | Method + basis set | r Cl–E | r C–N | < (F–A–B) | ν ClE | ν CN |
|---|---|---|---|---|---|---|
| [ClCN]– | B3LYP/cc-pVTZ | 2.44 | 1.16 | 167.9 | 251.74 | 2169.11 |
| MP2/cc-pVTZ | 2.22 | 1.18 | 131.2 | 1072.59 | 2086.36 | |
| CCSD/cc-pVTZ | 2.37 | 1.17 | 179.7 | 293.05 | 2181.28 | |
| CCSD(T)/cc-pVTZ | 2.33 | 1.18 | 158.0 | 338.83 | 2123.1 | |
| [ClNC]– | B3LYP/cc-pVTZ | 2.46 | 1.17 | 178.6 | 204.58 | 2126.79 |
| MP2/cc-pVTZ | 2.27 | 1.19 | 144.0 | 352.30 | 2053.93 | |
| CCSD/cc-pVTZ | 2.34 | 1.18 | 180.0 | 274.90 | 2126.68 | |
| CCSD(T)/6–311 + G(2d,2p) | 2.37 | 1.18 | 179.9 | 269.61 | 2067.89 |
Some experimental and computational data of the cyanogen chloride cation (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r Cl–C | r C–N | ν ClC | ν CN |
|---|---|---|---|---|---|
| Fulara et al. (1985) | Exp – PES | – | – | 827 | 1914 |
| Jacox and Thompson (2007) | Exp. – IR | – | – | 832.0 | 1919.4 |
| Wang et al. (1998) | CASSCF(5,6)/6-31G* | 1.5717 | 1.2326 | 819.8 | 2005.4 |
| MP2/6-31G* | 1.5592 | 1.1921 | 928.6 | 2975.7 | |
| MP2/6-311G(2df) | 1.5526 | 1.1885 | 913.2 | 2878.4 | |
| CCSD(T)/6-311G* | 1.5732 | 1.2152 | 826.5 | 1981.2 | |
| CCSD(T)/6-311G(2d) | 1.5800 | 1.2127 | 800.2 | 1959.8 | |
| IFCA | 1.556 | 1.214 | 800 | 1960 | |
| Bhattacharyya et al. (2007a) | MP2/cc-pVTZ | 1.56 | 1.19 | 912.29 | 2897.20 |
| CCSD/cc-pVTZ | 1.57 | 1.21 | 837.73 | 2040.04 | |
| CCSD(T)/cc-pVTZ | 1.57 | 1.21 | 833.96 | 2062.42 |
Cyanogen chloride cation [ClCN]+ and isocyanic chloride cation [ClNC]+
Besides some computational studies of the cyanogen chloride cation [ClCN]+ and its isomer there are only two experimental investigations (Fulara et al., 1985; Jacox and Thompson, 2007). M. E. Jacox and W. E. Thompson reported vibrational data of the cations. After formation of the cations using cyanogen chloride, which was exposed to a beam of neon atoms excited in a microwave discharge, the reactive species were included in a neon matrix and analyzed IR spectroscopically (Jacox and Thompson, 2007). J. P. Maier and co-workers used a combination of present emission spectra and photoelectron spectroscopy (PES) to identify the cation and to determine its structural parameters (Table 96) (Fulara et al., 1985).
On excitation of ClCN at energies between 16.6 and 16.85 eV, followed by rapid quenching of the products in solid neon at 4.3 K (see above), prominent infrared absorptions result from both photoisomerization and photoionization of the ClCN. In regard to the isocyanic chloride cation, a band at 1952.8 cm−1 was allocated to the C–N stretching mode and the band at 734.8 cm−1 was assigned to the Cl–N stretching mode by M. E. Jacox and W. E. Thompson (2007).
Bromine
Cyanogen bromide BrCN, isocyanic bromide BrNC and their ions
Just like ClCN, BrCN is a commercially available substance that is frequently used in both inorganic and organic chemistry because, unlike ClCN, it is a solid (Bhattacharyya et al., 2010; Chau et al., 1993; Geller and Schawlow, 1955; Hartman and Dreger, 1931; Holland et al., 2004; Hollas and Sutherley, 1971; Jacox and Thompson, 2007; Kellö and Sadlej, 1992; Lee, 1995; Lord and Woolf, 1954; Mishra et al., 2006; Nolan et al., 1975; Oberhauser, 1927; Pasternack and Dagdigian, 1976; Pézolet and Savoie, 1971; Rösslein et al., 1989; Ruoff, 1970; Salud et al., 1993; Smith et al., 1948; Slotta, 1934; Tyler and Sheridan, 1963; Zhang et al., 2007).
Cyanogen bromide BrCN and isocyanic bromide BrNC
At standard conditions cyanogen bromide BrCN is a colorless, transparent solid. In spite of its toxicity, cyanogen bromide is a frequently used reagent in biochemical and biological studies. To synthesize this compound in laboratory scale, a reaction of dissolved sodium or potassium cyanide and elementary bromine at low temperature is used (Scheme 132). Purification of the product by distillation is difficult due to the boiling point of TB = 335 K which is quite close to its melting point of TM = 325 K (Baum, 1908; Scholl, 1896; Slotta, 1934; Steinkopf, 1925).
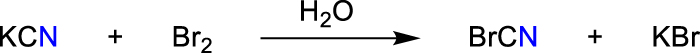
Synthesis of cyanogen bromide published by R. Scholl (1896).
Another synthetic route of cyanogen bromide is the reaction of bromine and potassium thiocyanate leading to cyanogen bromide and some side products, potassium bromide and hydrogen bromide for example (Scheme 133) (König, 1911).

Synthesis of cyanogen bromide published by W. König (1911).
Cyanogen bromide can also be isolated after a reaction of bromine monochloride and sodium cyanide. To avoid the use of bromine monochloride, this method has been modified to form this compound in situ by addition of chlorine to a solution of sodium cyanide and sodium bromide (Scheme 134) (Dodonow, 1926).

Synthesis of BrCN using bromine monochloride (Dodonow, 1926).
Additionally, several industrial synthetic routes were developed. The synthetic route of cyanogen chloride, which is protected by a patent of Degussa (Scheme 129), was modified and can also be used to isolate the bromine species (Scheme 135). Similar to the formation of cyanogen chloride catalysts are Lewis acids, copper dichloride or aluminum trichloride for example (Heimberger and Schreyer, 1975).

Industrial formation of cyanogen bromide protected by patent of Degussa (Heimberger and Schreyer, 1975).
The anhydrous formation of cyanogen bromide based on a reaction of trimethylsilyl cyanide TMS–CN and homo- or heteronuclear halogens, bromine monochloride for example, is another industrial process and protected by a patent of Bayer AG (Scheme 136).

Industrial generation of cyanogen bromide (patent of Bayer AG) (Fauß et al., 1984).
The synthesis of cyanogen bromide using sodium cyanide and sodium hypobromite, which is formed in situ, was protected by a patent of Syngenta Limited in 2001 (Scheme 137). A small temperature range is preferred to avoid hydrolysis of the product and buffer, especially borates, maintain the preferred pH range (Jackson et al., 2001).

Industrial formation of cyanogen bromide protected by patent of Syngenta Limited (Jackson et al., 2001).
The cyanogen bromide has been structurally investigated several times. Studies of the gas phase using MW spectroscopy led to bond lengths of rBr–C = 1.79 Å and rC–N = 1.16 Å, respectively (Le Guennec et al., 1992; Smith et al., 1948; Tyler and Sheridan, 1963; Townes et al., 1947, 1948). J. K. Tyler and J. Sheridan also reported an experimental dipole moment μ = 2.94 D, while the calculated dipole moment is μ = 3.187 D (Kellö and Sadlej, 1992; Tyler and Sheridan, 1963). Several working groups also analyzed IR and Raman spectra in respect of the BrC and CN stretching mode. Depending on the publication, bands at about νBrC = 575 cm−1 were assigned to the BrC and bands at νCN = 2187–2200 cm−1 to the CN stretching mode (Bandy et al., 1970; Freitag and Nixon, 1956; Klapötke and Schulz, 1996; Pézolet and Savoie, 1971; Thomas and Orville-Thomas, 1969).
The first study of cyanogen bromide determining bond lengths and molecular constitution using electron diffraction experiments was published by J. Y. Beach and A. Turkevich in 1939 (Beach and Turkevich, 1939). Cyanogen bromide forms orthorhombic crystals, space group Pnmm, and each unit cell contains two molecules. The reported bond lengths are rBr–C = 1.79(2) Å and rC–N = 1.13(4) Å being nearly identical to the bond lengths in gas phase (vide supra). Similar to the cyanogen chloride discussed above the molecules exhibit a substructure of chains along the a-axis corresponding to intermolecular interactions along these chains and shortening intermolecular distances (Geller and Schawlow, 1955). Additionally, several computational studies have been published in literature and selected data are listed in Table 97 along with reported experimental data (Bhattacharyya et al., 2010; Holland et al., 2004; Klapötke and Schulz, 1996; Lee, 1995; Mishra et al., 2006; Zhang et al., 2007).
Some experimental and computational data of the cyanogen bromide (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r Br–C | r C–N | ν BrC | ν CN |
|---|---|---|---|---|---|
| Beach and Turkevich (1939) | Exp. – ED | 1.79(2) | 1.13(4) | – | – |
| Townes et al. (1947) | Exp. – MW | 1.79 | 1.15 | – | – |
| Smith et al. (1948) | Exp. – MW | 1.789 | 1.160 | – | – |
| Townes et al. (1948) | Exp. – MW | 1.790 | 1.158 | – | – |
| Geller and Schawlow (1955) | Exp. – XRD | 1.79 | 1.15 | – | – |
| Freitag and Nixon (1956) | Exp. – IR | – | – | 575 | 2200 |
| Tyler and Sheridan (1963) | Exp. – MW | 1.789 | 1.158 | – | – |
| Thomas and Orville-Thomas (1969) | Exp. – IR | – | – | 577 | 2188 |
| Bandy et al. (1970) | Exp. – IR | – | – | 573 | 2193 |
| Pézolet and Savoie (1971) | Exp. – Raman | – | – | 577 | 2187 |
| Le Guennec et al. (1992) | Exp. – MW | 1.7891 | 1.1586 | – | – |
| Klapötke and Schulz (1996) | Exp. – IR | – | – | 574 | 2200 |
| MP2/6-31G* | 1.791 | 1.185 | – | – | |
| Lee (1995) | CCSD(T)/TZ2P | 1.810 | 1.165 | 577 | 2218 |
| Holland et al. (2004) | MP2/6-311G** | 1.790 | 1.178 | – | – |
| Mishra et al. (2006) | B3LYP/aug-cc-pVTZ(RECP) | 1.7899 | 1.1531 | 585 | 2289 |
| CCSD(T)/aug-cc-pVTZ(RECP) | 1.7966 | 1.1662 | – | – | |
| Zhang et al. (2007) | B3LYP/6-311G** | – | – | 581.1 | 2294.0 |
| Bhattacharyya et al. (2010) | MP2/(SDB-aug-)cc-pVTZ | 1.78 | 1.18 | 601.81 | 2111.70 |
| CCSD/(SDB-aug-) cc-pVTZ | 1.79 | 1.16 | 600.44 | 2281.64 | |
| CCSD(T)/(SDB-aug-) cc-pVTZ | 1.80 | 1.17 | 594.00 | 2197.07 |
Similar to the lighter homologs, isocyanic bromide has also been investigated computationally, whereas its isolation has failed until now and only one experimental evidence of its formation was published in literature (Table 98) (Bhattacharyya et al., 2010; Lee, 1995; Milligan and Jacox, 1967a, 1967b; Zhang et al., 2007).
Some experimental and computational data of the isocyanic bromide (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r Br–N | r C–N | ν BrN | ν CN |
|---|---|---|---|---|---|
| Milligan and Jacox (1967a, 1967b) | Exp. – IR | – | – | – | 2067 |
| Lee (1995) | CCSD(T)/TZ2P | 1.800 | 1.183 | 561 | 2089 |
| Zhang et al. (2007) | B3LYP/6-311G** | – | – | 543.8 | 2134.4 |
| Bhattacharyya et al. (2010) | MP2/(SDB-aug-)cc-pVTZ | 1.77 | 1.19 | 593.36 | 2061.01 |
| CCSD/(SDB-aug-)cc-pVTZ | 1.78 | 1.18 | 587.90 | 2142.73 | |
| CCSD(T)/(SDB-aug-)cc-pVTZ | 1.79 | 1.19 | 568.88 | 2090.18 |
Cyanuric bromide (BrCN) 3 , can be obtained by the polymerization of BrCN similar to the methods described for the trimerization of cyanuric chloride, (ClCN)3 (see Scheme 131) (Smolin and Rapoport, 1959). For example, by allowing a BrCN solution in ether to stand, to add HBr at ambient temperatures or to add AlBr3 in nitromethane. Also the reaction of K3[Fe(CN)6] with bromine yields (BrCN)3 at 473 K in good yields. (BrCN)3 also features the same s-triazine ring structure as discussed for (XCN)3 (X = F−, Cl−; vide supra).
Cyanogen bromide cation [BrCN]+
The cyanogen bromide cation [BrCN]+ was formed by exposure of cyanogen bromide to a beam of excited helium atoms and studied by laser excitation spectroscopy (LES). Based on the analysis of recorded spectra M. A. Hanratty and co-workers could report Br–C and C–N bond lengths of the cation as listed in Table 99 (Rösslein et al., 1989). IR spectroscopy was used by M. E. Jacox and W. E. Thompson to identify the cation in a similar way to the formation of the cyanogen chloride cation described before (Jacox and Thompson, 2007). The bond lengths, which were determined using photoelectron spectroscopy (PES), are similar to those of LES and in the range of the calculated values (Chau et al., 1993).
Some experimental and computational data of the cyanogen bromide cation (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r Br–C | r C–N | ν BrC | ν CN |
|---|---|---|---|---|---|
| Rösslein et al. (1989) | Exp – LES | 1.745(14) | 1.195(16) | – | – |
| Salud et al. (1993) | Exp – IR | – | – | – | 1905.93 |
| Chau et al. (1993) | Exp. – PES | 1.745(14) | 1.195(16) | – | – |
| Jacox and Thompson (2007) | Exp. – IR | – | – | – | 1907.7 |
| Mishra et al. (2006) | B3LYP/aug-cc-pVTZ(RECP) | 1.7304 | 1.1868 | 640 | 1989 |
| CCSD(T)/aug-cc-pVTZ(RECP) | 1.7218 | 1.2039 | 640 | 1925 |
Bromine bromocyanate [Br(BrCN)]– and bromine tribromocyanate [Br(BrCN)3]–
The polyhalogen pseudohalide [Br(BrCN)]– was synthesized by adding one equivalent of BrCN to [PNP]Br ([PNP] = [Ph3PNPPh3]) in acetonitrile (Schmidt et al., 2019). The compound crystallizes in the triclinic space group
![Scheme 138:
Important Lewis representations of [Br(BrCN)]– and [Br(BrCN)3]–.](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_138.jpg)
Important Lewis representations of [Br(BrCN)]– and [Br(BrCN)3]–.
The polypseudohalogen anion is comparable to the known trihalides such as [Br3]– or [Cl3]–. Quantum chemical calculations show that the energy required for the cleavage of [Br]– in [Br3]– is 37 kJ mol−1 higher than that for [Br(BrCN)]–.
From a mixture of three equivalents of BrCN and [PNP]Br in acetonitrile, the pseudohalogen salt [PNP][Br(BrCN)3] (monoclinic space group P21/n; Scheme 138) crystallizes. In the anion the central bromide ion is coordinated by three BrCN molecules in a distorted pyramidal structure. The Br⋯BrCN bond lengths are significantly elongated (3.089(1) Å) compared to a Br–Br single bond (cf. Σrcov(Br) = 2.28 Å). All Br–Br–C angles are nearly linear (169°–176°). Both ions, [Br(BrCN)]– and [Br(BrCN)3]–, can be referred to as BrCN adducts of a bromide anion of the type [Br]–⋅n (BrCN) with n = 1 and 3.
Iodine
In contrast to the cyanide compounds of chlorine and bromine, the cyanogen iodide compounds show a quite manifold chemistry. Some polyiodine-pseudohalide anions are synthesized and purified in form of their alkali metal salts enabling structural characterizations. Their structural properties are quite similar to the well-known poly-iodide anions.
Cyanogen iodide ICN and derived ions
Cyanogen iodide ICN
Studies on cyanogen iodide ICN go back to the nineteenth century when F. Wöhler and other chemists investigated these compounds systematically. The cyanogen iodide is a very poisonous, colorless solid forming crystalline needles, melts at about 420 K and decomposes at higher temperatures leading to cyanogen and iodine (Chadwick and Edwards 1973). One of the first synthetic routes was published by H. Davy and F. Wöhler. They observed that the reaction of iodine and mercury cyanide or silver cyanide led to the formation of cyanogen iodide (Scheme 139). F. Wöhler preferred silver cyanide as starting material due to an easier purification by sublimation of the cyanogen iodide (Wöhler, 1821).

Synthesis of cyanogen iodide published by F. Wöhler (1821).
During his studies on nitrogen triiodide in the 1880s, F. Raschig observed that the reaction of this compound and an aqueous solution of potassium cyanide or potassium thiocyanate led to the formation of cyanogen iodide, which was easily isolated by extraction (Scheme 140) (Raschig, 1885).

Reaction of nitrogen triiodide and potassium cyanide or potassium thiocyanate to form ICN (Raschig, 1885).
Cyanogen iodide can also be obtained by partial transformation of hydrogen cyanide with iodine into ICN and HI which is described as an equilibrium reaction (Scheme 141) (von Meyer, 1887).

Equilibrium of hydrogen cyanide and iodine with ICN and hydrogen iodide (von Meyer, 1887).
In regard to the smaller homologs chlorine and bromine, the formation of cyanogen halides using the elemental halogen has already been discussed. In this context the formation of cyanogen iodide using iodine and a metal cyanide as cyanide source is also possible (Scheme 142) (Bak and Hillebert, 1952). Additionally, the reaction of potassium cyanide and heteronuclear instead of homonuclear halogens, for example I–Cl, also yields cyanogen iodide (Scheme 142) (Birckenbach and Huttner, 1929; Cornog et al., 1938).

Cyanogen iodide formed by alkaline cyanide and homonuclear, elemental halogen (M = Na+, K+; top) or potassium cyanide and I–Cl (bottom) (Bak and Hillebert, 1952; Birckenbach and Huttner, 1929; Cornog et al., 1938).
Studies of the gas phase of cyanogen iodide using MW spectroscopy led to the determination of the bond lengths (rI–C = 2.00 Å and rC–N = 1.16 Å) (Smith et al., 1948; Townes et al., 1948; Tyler and Sheridan, 1963). J. K. Tyler and J. Sheridan also reported an experimental dipole moment μ = 3.71 D, while the calculated dipole moment is μ = 3.628 D (Kellö and Sadlej, 1992; Tyler and Sheridan, 1963). The vibrational modes of cyanogen iodide were analyzed using IR, Raman and photoelectron spectroscopy. In general the IC stretching band is in the range of 452–487 cm−1 and the CN stretching band at about 2171–2188 cm−1 (Freitag and Nixon, 1956; Hemple and Nixon, 1967; Hollas and Sutherley, 1971).
Based on a crystal structure determination of cyanogen iodide, published by J. A. A. Ketelaar and J. W. Zwartsenberg in the 1930s, this compound forms rhombohedral crystals with the space group
The metastabile isocyanic iodide, INC, was formed from its constitution isomer ICN by photoisomerization and was analyzed using spectroscopic measurements combined with computations (Larsen et al., 2002; Samuni et al., 1994). Computational data compared to experimental data are listed in Tables 100 and 101.
Some experimental and computational data of the cyanogen iodide (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r I–C | r C–N | ν IC | ν CN |
|---|---|---|---|---|---|
| Smith et al. (1948) | Exp. – MW | 1.995 | 1.159 | – | – |
| Townes et al. (1948) | Exp. – MW | 1.995 | 1.158 | – | – |
| Freitag and Nixon (1956) | Exp. – IR | – | – | 451.5 | 2176 |
| Tyler and Sheridan (1963) | Exp. – MW | 1.994 | 1.159 | – | – |
| Hemple and Nixon (1967) | Exp. – IR | – | – | 485.8 | 2188 |
| Hollas and Sutherley (1971) | Exp. – PES | 1.994 | 1.159 | 452 | 2176 |
| Simpson et al. (1972) | Exp. – MW | 1.995(1) | 1.158(1) | – | – |
| Samuni et al. (1994) | Exp. – IR | 1.992 | 1.160 | 487 | 2171 |
| HF/D95v(RECP) | 1.998 | 1.140 | 530.1 | 2543.5 | |
| QCISD(T)/D95v(RECP) | 2.013 | 1.185 | 486.6 | 2178.6 | |
| HF/(SBK)RECP | 2.005 | 1.153 | 531.7 | 2547.5 | |
| QCISD(T)/(SBK)RECP | 2.026 | 1.197 | 480.3 | 2159.5 | |
| Holland et al. (2004) | MP2/6-311G** | 2.003 | 1.178 | – | – |
| Bhattacharyya et al. (2010) | MP2/(SDB-aug-)cc-pVTZ | 1.99 | 1.18 | 516.49 | 2086.44 |
| CCSD/(SDB-aug-)cc-pVTZ | 2.00 | 1.16 | 507.99 | 2278.54 | |
| CCSD(T)/(SDB-aug-) cc-pVTZ | 2.01 | 1.17 | 499.73 | 2200.32 |
Some experimental and computational data of the isocyanic iodide (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r I–N | r C–N | ν IN | ν CN |
|---|---|---|---|---|---|
| Samuni et al. (1994) | Exp. – IR | – | – | 494 | 2057.5 |
| HF/D95v(RECP) | 1.950 | 1.163 | 551.0 | 2364.6 | |
| QCISD(T)/D95v(RECP) | 1.991 | 1.203 | 479.2 | 2073.2 | |
| HF/(SBK)RECP | 1.966 | 1.173 | 550.4 | 2348.2 | |
| QCISD(T)/(SBK)RECP | 2.014 | 1.214 | 469.9 | 2026.5 | |
| Larsen et al. (2002) | Exp. – IR | 2.004 | 1.203 | 459 | 2073 |
| Bhattacharyya et al. (2010) | MP2/(SDB-aug-)cc-pVTZ | 1.97 | 1.19 | 516.52 | 2040.96 |
| CCSD/(SDB-aug-)cc-pVTZ | 1.97 | 1.18 | 516.03 | 2157.65 | |
| CCSD(T)/(SDB-aug-) cc-pVTZ | 1.98 | 1.19 | 500.71 | 2052.40 |
Cyanur iodide (ICN) 3 , was first prepared by P. Klason. It can either be obtained by a Cl−/I−-exchange starting from cyanuric chloride or by trimerization of ICN (Scheme 143) (Klason, 1886; Smolin and Rapoport, 1959).

Synthesis of cyanuric iodide (M = H, alkaline metal) (Klason, 1886; Smolin and Rapoport, 1959).
ICN adducts. Normally, in the ICN molecule, the nitrogen atom is a strong Lewis basic center, while the iodine can already act as a weak Lewis acid. To give one example: The reaction behavior of trimethylsilylphosphanimine, TMS–N–PPh3, towards the pseudohalogen species X–CN (X = Cl−, Br− and I−), in particular the intermediate formation of [TMS–N(PPh3)–XCN] adduct complexes, was investigated in solution (Kempe et al., 2001). It was shown that only the ICN adduct is metastable in solution with respect to further reaction to Ph3PNCN and TMS–X and can be trapped. Raman and X-ray data of the ICN adduct showed a very labile donor-acceptor complex in which the iodine atom of the ICN moiety is loosely bound to the nitrogen atom of TMS–NPPh3. There are two different, rather long N⋯I bonds with bond lengths of 2.634(1) and 2.739(14) Å (Kempe et al., 2001).
Cyanogen iodide anion [ICN]−
In 2012, E. M. Miller et al. described photoelectron spectroscopy experiments on the [ICN]– anion, which was previously formed in a gas matrix using an electron beam. In order to generate [ICN]– or [ICN]– Ar, CO2 or Ar gas was passed over a solid sample of ICN. The gas was expanded into the source chamber through a heated, pulsed general valve nozzle. The gas pulse interacts with a counterpropagating 1-keV electron beam, producing anions through secondary electron attachment. The reported results are presented in Table 102 in comparison to a computational study of A. B. McCoy (McCoy, 2013; Miller et al., 2012).
Selected experimental and computational data of the cyanogen iodide anion (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r I–C | r C–N | ν IC |
|---|---|---|---|---|
| Miller et al. (2012) | Exp. – PES | 2.65 | – | 235.2 |
| McCoy (2013) | MR-SO-CISD/aug-cc-pVTZ(-PP) | 2.65 | 1.16 | 235 |
Cyanogen iodide cation [ICN]+
This transient cyanogen iodide cation [ICN]+ was formed by collision ionization in a special apparatus and was characterized by photoelectron spectroscopy (Fulara et al., 1985; Hollas and Sutherley, 1971). The structural and spectroscopic data are given in Table 103.
Some experimental data of [ICN]+ (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r I–C | r C–N | ν IC | ν CN |
|---|---|---|---|---|---|
| Hollas and Sutherley (1971) | Exp. – PES | 1.934 | 1.180 | 500 | 2050 |
| Fulara et al. (1985) | Exp. – PES | – | – | 535 | 2082 |
Cyanoiodine cation [ICN]2+
In 1997, R. Minkwitz and T. Hertel reported the synthesis of the cyanoiodine cation [ICN]2+ in form of its nitrate, which is just stable at temperatures below 228 K, carrying out a reaction of cyanogen iodide and excess of chlorine nitrate at 203 K (Scheme 144). The received yellow solid was characterized using IR and Raman spectroscopy. The band at 2200–2212 cm−1 was assigned to the CN stretching mode and the band at 469 cm−1 in the IR spectrum and 473 cm−1 in the Raman spectrum was allocated to the IC stretching mode (Minkwitz and Hertel, 1997). Cyanoiodine(III) dinitrate is a yellow, hydrolysis-sensitive solid that decomposes above 228 K to release ICN and N2O5. At 203 K it can be stored undecomposed for several weeks.

Synthesis of the cyanoiodine cation by Minkwitz and Hertel (1997).
Dicyanoiodate anion [I(CN)2]−
The dicyanoiodate anion [I(CN)2]– is formed in an aqueous solution of cyanogen iodide and an excess of cyanide anions (Scheme 145). Furthermore, in 1933, D. M. Yost and W. E. Stone determined the stability of the anions in the chemical equilibrium as illustrated in Scheme 145 and estimated the equilibrium constant (Yost and Stone, 1933).

Cyanogen iodide and cyanide forming the dicyanoiodate anion in equilibrium (Yost and Stone, 1933).
The first systematic study on salts containing the dicyanoiodate anion was carried out by B. M. Chadwick et al. Counterions of these salts were cesium, tetraphenylphosphonium and tetraphenylarsonium cations.
In case of the cesium species, the cesium chloride was added to a mixture of saturated aqueous solution of potassium cyanide and a saturated solution of cyanogen iodide in ethanol. The salts of these two bulky cations ([Ph4P]+, [Ph4As]+) were obtained using a similar route after a reaction of potassium cyanide and saturated aqueous solution of cyanogen iodide and following addition of the chloride salt dissolved in water (Scheme 146). All salts were finally characterized by IR and Raman spectroscopy (Chadwick et al., 1980).
![Scheme 146:
Synthesis of [I(CN)2]– containing salts by B. M. Chadwick et al. (E = P, As) (Chadwick et al., 1980).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_146.jpg)
Synthesis of [I(CN)2]– containing salts by B. M. Chadwick et al. (E = P, As) (Chadwick et al., 1980).
In the 1980s, K.-F. Tebbe and co-workers published several syntheses and characterizations of different salts containing the dicyanoiodate anion. The synthesis of the pure potassium salt, based on a reaction of potassium cyanide and elemental iodine, was reported in 1988 (Scheme 147). After the addition of solid iodine to a saturated aqueous solution of potassium cyanide at ambient temperature, a colorless solid is formed and slow cooling of the solution leads to crystals suitable for single crystal XRD.

Synthesis of the pure potassium salt published by Tebbe and Krauß (1988).
The use of cyanogen iodide instead of pure iodine also led to the formation of the dicyanoiodate anion (Scheme 148). Therefore, a saturated aqueous solution of potassium cyanide was added to a solution of cyanogen iodide using different solvents. The crystallisation started at 255 K after six weeks (Tebbe and Krauß, 1988).

Synthesis of the potassium salt using cyanogen iodide instead of iodine (Tebbe and Krauß, 1988).
The addition of rubidium bromide to this reaction mixture in a solvent mixture of water and ethanol also led to a salt of dicyanoiodate. But this salt contained two different types of cations, the potassium and rubidium cation (Scheme 149, x = 0.6) (Tebbe and Fröhlich, 1983a).

Synthesis of a dicyanoiodide salt of two different cations ((EtOCNH)2 = (diimino)oxalic acid diethyl ether; x = 0.6) (Tebbe and Fröhlich, 1983a).
Due to variation of the reaction conditions they used different solvents and they observed that the use of ethanol caused the formation of an additional product, which coordinated to the expected salt forming adducts (Scheme 150).

Formation of (diimino)oxalic acid diethyl ether by solvolysis of cyanogen iodide in presence of ethanol (Tebbe and Fröhlich, 1983b; Tebbe and Krauß, 1988).
They proposed that the formation of the (diamino)oxalic acid diethyl ether was caused by partial solvolysis of cyanogen iodide to iodine and cyanogen, which already reacted with two solvent molecules (Tebbe and Fröhlich, 1983b; Tebbe and Krauß, 1988).
The [I(CN)2]– anion is nearly linear in the solid state as predicted by the VSEPR model. This is true for both the C–I–C unit and the two ICN units. The I–C bond can be understood as a 3-center-4-electron bond (Scheme 151). Experimental data of salts containing the [I(CN)2]– ion are summarized in Tables 104 and 105 (Chadwick et al., 1980).
![Scheme 151:
3-Center-4-electron bond in [I(CN)2]– (Chadwick et al., 1980).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_151.jpg)
3-Center-4-electron bond in [I(CN)2]– (Chadwick et al., 1980).
Experimental data of salts containing [I(CN)2]– (bond lengths in Å; wave numbers in cm−1).
| Compound | r I–C [a] | r C–N [a] | ν sym. IC [b] | ν as. IC [c] | ν sym. CN [b] | ν as. CN [c] |
|---|---|---|---|---|---|---|
| Cs[I(CN)2] (Chadwick et al., 1980) | – | – | 434 | 431 | 2118 | 2109 |
| [Ph4P][I(CN)2] (Chadwick et al., 1980) | – | – | 440 | – | 2124 | 2110 |
| [Ph4As][I(CN)2] (Chadwick et al., 1980) | – | – | – | – | 2121.5 | 2110 |
| K[I(CN)2] (Tebbe and Krauβ, 1988) | 2.298(6) | 1.129(7) | – | – | – | 2115 |
| K[I(CN)2]·(EtOHNC)2 (Tebbe and Fröhlich, 1983b) | 2.302(3) | 1.121(5) | 422 | 425 | 2126 | 2108 |
| K0.6Rb0.4[I(CN)2]·0.5 (EtOCNH)2 (Tebbe and Fröhlich, 1983a) | 2.265(8)/2.351(10) | 1.117(12)/1.096(12) | – | 415–440 | – | 2155/2100/2060/22016 |
-
[a]Bond lengths obtained by XRD. [b]Wave numbers of symmetric vibrations obtained by Raman spectroscopy. [c] Wave numbers of asymmetric vibrations obtained by IR spectroscopy.
Crystallographic data of salts synthesized by K.-F. Tebbe et al. (bond lengths in Å; angles in °; volumes in Å3).
| parameter | K[I(CN)2] (Tebbe and Krauβ, 1988) monoclinic | K[I(CN)2]·(EtOHNC)2 (Tebbe and Fröhlich, 1983b) triclinic | K0.6Rb0.4[I(CN)2]·0.5 (EtOCNH)2 (Tebbe and Fröhlich, 1983a) monoclinic |
|---|---|---|---|
| Space group | C2/m |
|
P21/c |
| a | 7.3637(9) | 4.285(1) | 4.3634(8) |
| b | 4.5144(4) | 9.259(2) | 19.335(3) |
| c | 9.0800(8) | 10.350(2) | 12.405(2) |
| α | 90.00 | 63.41(1) | 90.00 |
| β | 92.56(1) | 76.33(2) | 92.30(2) |
| γ | 90.00 | 78.58(2) | 90.00 |
| V | 301.54(5) | 354.8 | 1046 |
| Z | 2 | 1 | 4 |
(Iodocyano)iodine ions [I2(CN)]− and [I2(CN)]+
(Iodocyano)iodide anion [I2(CN)]−
Similar to the measurement of the equilibrium constant of the dicyanoiodate anion, D. M. Yost and W. E. Stone also determined the equilibrium constant of the formation of the (iodocyano)iodide anion [I2(CN)]– (Scheme 152), which is the only report on this anion (Yost and Stone, 1933). As discussed before for the [I(CN)2]– ion, the [I2(CN)]– anion should be linear and the bonding within the I–I–C moiety can also be understood as a 3-center-4-electron bond.

Equilibrium of cyanogen iodide, iodide and the (iodocyano)iodide anion (Yost and Stone, 1933).
(Iodocyano)iodonium cation [I2(CN)]+
In contrast to BrCN and ClCN, which do not form stable salts of the type [XCNX]+[AsF6]– (X = Cl− or Br−) due to the thermodynamically favourable adduct formation XCN⋅AsF5 and elimination of Cl–F and Br–F, ICN does react with [I]+ donors to yield compounds containing the [ICNI]+ = [I2(CN)]+ ion as demonstrated by I. Tornieporth-Oetting and T. M. Klapötke. They investigated the reactions of triiodonium hexafluoroarsenate, [I3][AsF6], and different cyanide containing compounds. For example, the reaction of [I3][AsF6] and cyanogen iodide led to the formation of the (iodocyano)iodonium hexafluoroarsenate, [I2(CN)][AsF6], which was characterized by IR and Raman spectroscopy. To obtain [I2(CN)][AsF6], a suspension of [I3][AsF6] in trichlorofluoromethane was added to cyanogen iodide dissolved in carbon disulfide at 283 K or it can be directly generated from ICN, I2 and AsF5 (Scheme 153).
![Scheme 153:
Synthesis of [I2(CN)][AsF6] (Binnewies et al., 2016; Pettersson et al., 1998; Werner et al., 1988).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_153.jpg)
Synthesis of [I2(CN)][AsF6] (Binnewies et al., 2016; Pettersson et al., 1998; Werner et al., 1988).
Due to its high instability, crystals of the [I2(CN)][AsF6] could not be obtained (Tornieporth-Oetting and Klapötke, 1990, 1991), but structural data were computed (Klapötke, 1997). The [ICNI]+ cation adopts a linear structure with a I–C–N–I connectivity (Scheme 154). Selected experimental and theoretical data are summarized in Table 106. Strong hyperconjugative effects within the π-electronic system along the I–C–N–I are discussed [p-LP(I) → π*(C–N)].
![Scheme 154:
Important Lewis representations of the [ICNI]+ cation (Klapötke, 1997).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_154.jpg)
Important Lewis representations of the [ICNI]+ cation (Klapötke, 1997).
Some experimental and computational data of the (iodocyano)iodonium cation (bond lengths in Å; wave numbers in cm−1).
| Publication | Exp./calc. | r I–C | r I–N | r C–N | ν IC, NI | ν IC, NI | ν CN |
|---|---|---|---|---|---|---|---|
| Tornieporth-Oetting and Klapötke (1990) | Exp. – IR | – | – | – | 470 | 700 | 2330 |
| Klapötke (1997) | Exp. – IR, Raman | – | – | – | 179 | 590 | 2200 |
| AM1/6-31G(d) (LANL2DZ) | 1.939 | 1.916 | 1.163 | 223 | 760 | 2494 | |
| PM3/6-31G(d) (LANL2DZ) | 1.896 | 1.892 | 1.165 | 287 | 1032 | 2438 | |
| RHF/6-31G(d) (LANL2DZ) | 2.023 | 2.030 | 1.132 | 181 | 615 | 2585 | |
| RMP2/6-31G(d) (LANL2DZ) | 1.986 | 0.020 | 1.176 | 179 | 624 | 2205 | |
| RMP4(SDQ1)/6-31G(D) (LANL2DZ) | 2.001 | 0.021 | 1.167 | 178 | 615 | 2285 |
(Diiododicyano)iodide anion [I3(CN)2]−
The first synthesis of the dicyanotriiodide or (diiododicyano)iodide anion, [I(ICN)2]– = [I3(CN)2]– (Figure 48), was reported by K.-F. Tebbe and co-workers. The potassium, rubidium and cesium salts were obtained by addition of two equivalents of cyanogen iodide to the potassium, rubidium or cesium iodide in aqueous solution (Scheme 155) (Frölich and Tebbe, 1982; Krauβ and Tebbe, 1986; Tebbe et al., 1983c).
![Figure 48:
V-shaped structure of [I3(CN)2]− (Tebbe and Grafe-Kavoosian, 1996).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_fig_048.jpg)
V-shaped structure of [I3(CN)2]− (Tebbe and Grafe-Kavoosian, 1996).
![Scheme 155:
Synthesis of [I3(CN)2]− salts by K.-F. Tebbe and co-workers (M = K+, Rb+, Cs+) (Frölich and Tebbe, 1982; Krauβ and Tebbe, 1986; Tebbe et al., 1983c).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_155.jpg)
Synthesis of [I3(CN)2]− salts by K.-F. Tebbe and co-workers (M = K+, Rb+, Cs+) (Frölich and Tebbe, 1982; Krauβ and Tebbe, 1986; Tebbe et al., 1983c).
Additionally, a salt of the [I3(CN)2]− anion having a potassium-2.2.2-cryptand cation ([(Crypt)K]+) was synthesized adding an excess of cyanogen iodide to the KI crown ether complex dissolved in ethanol (Scheme 156) (Tebbe and Grafe-Kavoosian, 1996).
![Scheme 156:
Synthesis of a complex containing the [I3(CN)2]− anion (Tebbe and Grafe-Kavoosian, 1996).](/document/doi/10.1515/revic-2021-0044/asset/graphic/j_revic-2021-0044_scheme_156.jpg)
Synthesis of a complex containing the [I3(CN)2]− anion (Tebbe and Grafe-Kavoosian, 1996).
All mentioned salts were characterized using single crystal XRD and IR spectroscopy (Tables 107 and 108). Similar to the pentaiodide anion, the anions of these salts also show a V-shaped structure in solid state (Figure 48) (Tebbe and Grafe-Kavoosian, 1996). The [I3(CN)2]– ion is best described as [I(ICN)2]–, that is as an iodide, I−, briding two ICN molecules.
Crystallographic data of [I3(CN)2]− salts synthesized by K.-F. Tebbe and co-workers (lengths in Å; angles in °; volumes in Å3; [CryptK] = [potassium-2.2.2-cryptand]).
| Parameter | K[I3(CN)2] (Krauß and Tebbe, 1986) monoclinic | Rb[I3(CN)2] (Tebbe et al., 1983c) orthorhombic | Cs[I3(CN)2] (Frölich and Tebbe, 1982) orthorhombic | [CryptK][I3(CN)2] (Tebbe and Grafe-Kavoosian, 1996) triclinic |
|---|---|---|---|---|
| Space group | C2/m | Pmmn | Pmmn |
|
| a | 7.364 | 16.246(7) | 16.494(4) | 11.923(2) |
| b | 4.514 | 6.795(4) | 6.726(1) | 11.972(2) |
| c | 9.080 | 4.397(2) | 4.592(1) | 12.781(3) |
| α | 90.00 | 90.00 | 90.00 | 69.79(2) |
| β | 92.56 | 90.00 | 90.00 | 72.67(2) |
| γ | 90.00 | 90.00 | 90.00 | 75.79(1) |
| V | 301.5 | 485.4 | 509.4 | 1613.5(8) |
| Z | 2 | 2 | 2 | 2 |
Experimental data of salts containing the [I3(CN)2]– anion (bond lengths in Å; angles in °; wave numbers in cm−1).
| Compound | r I–I [a] | r I–C [a] | r C–N [a] | < (I–I–I)[a] | < (I–C–N)[a] | ν IC [b] | ν CN [b] |
|---|---|---|---|---|---|---|---|
| Rb[I3(CN)2] (Tebbe et al., 1983c) | 3.271(1) | 2.11(2) | 1.13(3) | 128.69(5) | 177(1) | 415 | 2154 |
| Cs[I3(CN)2] (Tebbe et al., 1983c; Frölich and Tebbe, 1982) | 3.306(1) | 2.08(2) | 1.07(2) | 123.12(5) | 179(2) | – | 2168 |
| [CryptK][I3(CN)2][c] (Tebbe and Grafe-Kavoosian, 1996) | 3.2025(7) | 2.075(7)/2.100(5) | 1.108(10)/1.070(7) | 88.64(1) | 178.(6)/174.5(6) | – | – |
-
[a]Bond lengths and angles obtained by XRD. [b]Wave numbers obtained by IR spectroscopy. [c][CryptK] = [(Cryptand-2.2.2)K].
8th Main group
Noble gas cyanides
The chemistry of the nobel gases mainly focuses on the later homologs krypton and xenon due to the increasing strength of Rg–E bonds the heavier the rare gas (Rg). Especially xenon is able to form manifold compounds with electronegative elements, several xenon fluorides for example (Binnewies et al., 2016).
As a consequence of the group electronegativity of the cyanide group, which is significantly smaller in comparison to the strongly electronegative fluorine, noble gas cyanides could not be received on common synthetic routes and could not be formed in industrial scales. Nevertheless, some working groups reported indications of the existence of short-lived nobel gas cyanide species in excited states. And additionally, several computational studies were published to maintain experimental data or predict compounds, which have not been synthesized until now (Arppe et al., 2012; Fiedler et al., 2002; Han et al., 2008a, 2008b; Jana et al., 2018; Lin and Heaven, 1991; Lin et al., 1994; Pan et al., 2015; Pettersson et al., 1998; Thoma et al., 1994; van de Burgt et al., 1984; Werner et al., 1988; Zhu et al., 2015).
For example, H.-J. Werner et al. studied excited helium cyanide radical complexes. Assuming very weak intermolecular interaction, they carried out several calculations of their structures, although there is no experimental evidence of their existence (Werner et al., 1988).
Studies on the chemistry of (cyanide) radicals in nobel gas matrices by M. Heaven led to the proof of different complexes formed by excited radicals and nobel gas atoms (Heaven, 1992). Using a special experimental setup they could obtain short-lived neon cyanide radical complexes by laser induced photochemical excitation of cyanogen iodide anions in a beam of neon and helium gas (70%: 30%) and verified their existence by LIF spectroscopy (Lin and Heaven, 1991; van de Burgt et al., 1984). They could also form argon cyanide radical cluster and exciplexes of cyanide radicals and argon atoms on a similar way using a beam of argon and helium (14%: 86%) (Lin et al., 1994; Han et al., 2008a, 2008b).
The exciplex of cyanide radicals of krypton and xenon atoms was reported by V. A. Apkarian et al. in 2002. A thin layer of a mixture of the nobel gas and deuterated hydrogen cyanide in the guest-to-host ratio of 1:1000 was deposited on magnesium difluoride at 20–30 K. The exciplex is formed by photolysis of the hydrogen cyanide leading to the cyanide radicals. The existence of this exciplex in noble gas matrix was proven by UV/vis and fluorescence spectroscopy. The visible absorption and the ground state vibrational infrared emission of the CN–Xe complex were studied in an argon matrix by A. Thoma et al. in 1994. In this early study cyanide radicals being precursor of those exciplexes were received from dicyanoacetylene in a self-igniting pulsed discharge source (Fiedler et al., 2002; Thoma et al., 1994).
Another working group carried out computations and predicted the formation of organo xenon and radon compounds (Scheme 157). The formation of xenon or radon CN compounds is an exergonic process of ΔE(Xe) = −68 kJ mol−1 and ΔE(Rn) = −71 kJ mol−1, respectively. They also published structural parameters of Rg(CN)2 (bond lengths of rXe–C = 2.192 Å and rRn–C = 2.268 Å) (Fitzsimmons and Klobukowski, 2013). Earlier computational studies on xenon complexes by C. C. Lovallo et al., dealing with the same reaction, revealed bond lengths of rRg–C = 2.202–2.246 Å. The predicted C–N bond lengths are in the range of rC–N = 1.133–1.180 Å depending on the basis set (Lovallo and Klobukowski, 2002).

Simulated reaction to synthesize nobel gas dicyanide compounds (Rg = Xe, Rn) (Fitzsimmons and Klobukowski, 2013).
Besides binary noble gas cyanide species, E–Rg–CN (E = H, Au, halogens etc., Rg = rare gase), were also investigated (Arppe et al., 2012; Pan et al., 2015; Pettersson et al., 1998; Zhu et al., 2015). For example, the synthesis of the new noble gas compounds HXeCN, HXeNC and HKrCN was reported. HKrCN was the first stable compound with a Kr–C bond. These molecules were formed in solid Xe and Kr by first photolysing monomeric HCN with a 193 nm ArF laser at 7.5 K. The photolysis generates isolated hydrogen atoms and CN radicals, as detected by IR spectroscopy and laser-induced fluorescence (Pettersson et al., 1998). Another publication reports on the preparation and characterization of noble gas molecules of the type ClXeCN, ClXeNC and BrXeCN. These molecules were synthesized by 193 nm photolysis and thermal annealing of ClCN and BrCN in a xenon matrix (Arppe et al., 2012). A similar report deals with FRgCN (Rg = Kr, Xe), which were also prepared in low-temperature Kr and Xe matrices. These F–Rg–CN molecules were produced by UV photolysis of FCN in the matrices and subsequent thermal annealing. The FCN precursor is produced by depositing the (FCN)3 containing matrix gas by a microwave discharge (Zhu et al., 2015).
Final remarks
More than 315 years have passed since the beginning of cyanide chemistry with the discovery of Berlin Blue in 1706. Starting from the pseudohalogen concept, according to which the cyanide ion is to be regarded as such, cyanide chemistry of the main group elements developed enormously at the end of the nineteenth and in the twentieth century. Since then, both organic cyanide chemistry, which has not been covered here, and inorganic cyanide chemistry have become important fields with industrial, biological and fundamental significance. Although cyanide compounds are mostly toxic, they have found their way into modern industrial society. Despite all this, we are still far from fully understanding the cyanide ion in its compounds. For example, the oligomerization reactions, cyclisation reactions or redox reactions are often still not fully understood, as are such unusual reactions as the isomerization of [As(CN)4]– to an unusual cyclic cyanido-arsenate ion of the type [AsC4N4]–.
We are confident that further experimental research also in basic research as well as improved computational methods will further highlight the unique properties of this very versatile two-atomic CN ligand.
About the authors

Axel Schulz studied chemistry at the Humboldt-University and Technical University of Berlin (1987–1992), with a diploma thesis (1993) in inorganic chemistry and a doctoral thesis (1993–1994) in inorganic chemistry (Thomas M. Klapötke, nitrogen chemistry). Thereafter he was a research associate at the university of Glasgow (1995–1997) and a postdoctoral fellow at the ANU in Canberra with Leo Radom (computational chemistry). In 1997, he went to the LMU Munich where he received his second PhD (Dr. Habil.) in 2001 (elementorganic and theoretical chemistry). In 2006 he was appointed full professor for inorganic chemistry at the University of Rostock. Research in the Schulz group largely focuses on fundamental main group element chemistry and the quantum mechanical description of chemical bonding. The group’s more applied interests include the synthesis of new hydrogen acids of pseudohalogens, their stabilization using Lewis acids, the study of their chemical properties and their application in noval reaction pathways.

In 2019, Jonas Surkau joined the group of Axel Schulz at the University of Rostock to work on his BSc thesis on the topic of pseudohalogen chemistry. For his MSc (2021) and PhD he set his focus on the stabilization of hydrogen acids by Lewis acids, the influence of adduct formation on the tautomeric equilibrium and their reaction behavior towards hydrocarbons.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: None declared.
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
Abelson, P. H. Chemical events on the primitive earth. Proc. Natl. Acad. Sci. U.S.A. 1966, 55, 1365.10.1073/pnas.55.6.1365Search in Google Scholar PubMed PubMed Central
Adams, W. S. Some results with the COUDÉ Spectrograph of the mount Wilson Observatory. Astrophys. J. 1941, 93, 11.10.1086/144237Search in Google Scholar
Adrian, F. J.; Cochran, E. L.; Bowers, V. A.; Weatherley, B. C. ESR spectrum and structure of HCN− in KCl at 4°K. Phys. Rev. 1969, 177 (1), 129–135.10.1103/PhysRev.177.129Search in Google Scholar
Aguirre, N. V.; Vivas, B. P.; Montes-Morán, M. A.; Ania, C. O. Adsorption of thiocyanate anions from aqueous solution onto adsorbents of various origin. Adsorpt. Sci. Technol. 2010, 28 (8–9), 705–716.10.1260/0263-6174.28.8-9.705Search in Google Scholar
Ahmed, E.; Ruck, M. Homo- and heteroatomic polycations of groups 15 and 16. Recent advances in synthesis and isolation using Room temperature ionic liquids. Coord. Chem. Rev. 2011, 255 (23–24), 2892–2903.10.1016/j.ccr.2011.06.011Search in Google Scholar
Akcil, A. Destruction of cyanide in gold mill effluents: biological versus chemical treatments. Biotechnol. Adv. 2003, 21 (6), 501–511.10.1016/S0734-9750(03)00099-5Search in Google Scholar
Akers, C.; Peterson, S. W.; Willett, R. D. A refinement of the crystal structure of KSCN. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1968, 24 (8), 1125–1126.10.1107/S0567740868003808Search in Google Scholar
Aksnes, O.; Foss, O.; Schultz, G.; Sörensen, N. A. The crystal structure of selenium diselenocyanate. Acta Chem. Scand. 1954, 8, 1787–1795.10.3891/acta.chem.scand.08-1787Search in Google Scholar
Al-Azmi, A.; Elassar, A. Z. A.; Booth, B. L. The chemistry of diaminomaleonitrile and its utility in heterocyclic synthesis. Tetrahedron 2003, 59 (16), 2749–2763.10.1016/S0040-4020(03)00153-4Search in Google Scholar
Alkorta, I.; Elguero, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998, 27 (2), 163.10.1039/a827163zSearch in Google Scholar
Allen, T. L.; Goddard, J. D.; Schaefer, H. F. A possible role for triplet H2CN+ Isomers in the formation of HCN and HNC in Interstellar Clouds. J. Chem. Phys. 1980, 73 (7), 3255–3263.10.1063/1.440520Search in Google Scholar
Allenstein, E.; Lattewitz, E. Versuche Zur Darstellung von N-Rhodanaminen. Z. Anorg. Allg. Chem. 1964, 333 (1–3), 1–9.10.1002/zaac.19643330102Search in Google Scholar
Allman, T.; Goel, R. G.; Prasad, H. S. Organobismuth compounds. J. Organomet. Chem. 1979, 166 (3), 365–371.10.1016/S0022-328X(00)82555-3Search in Google Scholar
Altman, R. S.; Crofton, M. W.; Oka, T. High resolution infrared spectroscopy of the v1 (NH stretch) and v2 (CH stretch) bands of HCNH+. J. Chem. Phys. 1984a, 81 (10), 4255–4258.10.1063/1.447433Search in Google Scholar
Altman, R. S.; Crofton, M. W.; Oka, T. Observation of the infrared v2 band (CH stretch) of protonated hydrogen cyanide HCNH+. J. Chem. Phys. 1984b, 80 (8), 3911–3912.10.1063/1.447173Search in Google Scholar
Amano, T.; Tanaka, K. Difference frequency laser spectroscopy of HCNH+: observation of the isotopic species and the hot bands. J. Mol. Spectrosc. 1986, 116 (1), 112–119.10.1016/0022-2852(86)90257-2Search in Google Scholar
Ambient Water Quality Criteria: Report from US Environmental Protection Agency (EPA), the Office of Water Regulations and Standards [Criteria and Standards Division], the Office of Research and Development [Environmental Criteria and Assessment Office], Carcinogen Assessment Group and Environmental Research Laboratories; Washington D.C., 1980.Search in Google Scholar
Anastassiou, A. G.; Simmons, H. E. Cyanonitrene. Reaction with saturated hydrocarbons. J. Am. Chem. Soc. 1967, 89 (13), 3177–3184.10.1021/ja00989a018Search in Google Scholar
Andersen, P.; Klewe, B. X-ray investigation of potassium tricyano-methanide (KC(CN)3). Nature 1963, 200, 464.10.1038/200464a0Search in Google Scholar
Anderson, P. M. Purification and properties of the inducible enzyme cyanase. Biochemistry 1980, 19 (13), 2882–2888.10.1021/bi00554a010Search in Google Scholar PubMed
Anderson, R. W.; Frick, H. R. Synthesis of cyanuric fluoride 1972, 3641020.Search in Google Scholar
Anderson, P. M.; Sung, Y.; Fuchs, J. A. The cyanase operon and cyanate metabolism. FEMS Microbiol. Lett. 1990, 87 (3–4), 247–252.10.1111/j.1574-6968.1990.tb04920.xSearch in Google Scholar PubMed
Anderson, M. A.; Steimle, T. C.; Ziurys, L. M. The millimeter and submillimeter rotational spectrum of the MgCN radical (X2Σ+). Astrophys. J. 1994, 429, L41.10.1086/187408Search in Google Scholar
Andreas, H. “Cyan-industrie” – Anfänge Einer Chemischen Industrie in Deutschland Im 18. Jahrhundert. Mitt. - Ges. Dtsch. Chem., Fachgruppe Gesch. Chem. 2017, 25, 47–56.Search in Google Scholar
Andrussow, L. Über die Katalytische Oxidation von Cyanwasserstoffsäure und Ammoniak (Ein Vergleich). Ber. Dtsch. Chem. Ges. 1926, 59 (3), 458–461.10.1002/cber.19260590320Search in Google Scholar
Andrussow, L. Über die Katalytische Oxidation von Ammoniak-Methan-Gemischen Zu Blausäure. Angew. Chem. 1935, 48 (37), 593–595.10.1002/ange.19350483702Search in Google Scholar
Andrussow, L. Blausäuresynthese und die Schnell Verlaufenden Katalytischen Prozesse in Strömenden Gasen. Chem. Ing. Tech. 1955, 27 (8–9), 469–472.10.1002/cite.330270804Search in Google Scholar
Anex, D. S.; Davidson, E. R.; Douketis, C.; Ewing, G. E. Vibrational spectroscopy of hydrogen cyanide clusters. J. Phys. Chem. 1988, 92 (10), 2913–2925.10.1021/j100321a041Search in Google Scholar
Anseeuw, K.; Delvau, N.; Burillo-Putze, G.; De Iaco, F.; Geldner, G.; Holmström, P.; Lambert, Y.; Sabbe, M. Cyanide poisoning by fire smoke inhalation. Eur. J. Emerg. Med. 2013, 20 (1), 2–9.10.1097/MEJ.0b013e328357170bSearch in Google Scholar PubMed
Apponi, A. J.; McCarthy, M. C.; Gottlieb, C. A.; Thaddeus, P. The radio spectra of SiCCH, SiCN, and SiNC. Astrophys. J. 2000, 536 (1), 55–58.10.1086/312719Search in Google Scholar
Arlt, S.; Harloff, J.; Schulz, A.; Stoffers, A.; Villinger, A. Molecular E(CN)3 (E = Sb, Bi) species synthesized from ionic liquids as solvates. Chem. Eur J. 2016a, 22 (45), 16012–16016.10.1002/chem.201603849Search in Google Scholar PubMed
Arlt, S.; Harloff, J.; Schulz, A.; Stoffers, A.; Villinger, A. Cyanido antimonate(III) and bismuthate(III) anions. Inorg. Chem. 2016b, 55 (23), 12321–12328.10.1021/acs.inorgchem.6b02174Search in Google Scholar PubMed
Arlt, S.; Harloff, J.; Schulz, A.; Stoffers, A.; Villinger, A. Arsenic-mediated C−C coupling of cyanides leading to cyanido arsazolide [AsC4N4]−. Chem. Eur J. 2017, 23 (52), 12735–12738.10.1002/chem.201703317Search in Google Scholar PubMed
Arlt, S.; Harloff, J.; Schulz, A.; Stoffers, A.; Villinger, A. Heavy neutral and anionic pnictogen thiocyanates. Inorg. Chem. 2019, 58 (8), 5305–5313.10.1021/acs.inorgchem.9b00378Search in Google Scholar PubMed
Arlt, S.; Bläsing, K.; Harloff, J.; Laatz, K. C.; Michalik, D.; Nier, S.; Schulz, A.; Stoer, P.; Stoffers, A.; Villinger, A. Pseudohalogen chemistry in ionic liquids with non‐innocent cations and Anions. ChemistryOpen 2021, 10 (2), 62–71.10.1002/open.202000252Search in Google Scholar PubMed PubMed Central
Armstrong, J.; Degoricija, L.; Hildebrand, A.; Koehne, J.; Fleming, P. E. The ionization energies of the isomers of CN2. Chem. Phys. Lett. 2000, 332 (5–6), 591–596.10.1016/S0009-2614(00)01299-9Search in Google Scholar
Arnold, R. G.; Nelson, J. A.; Verbanc, J. J. Recent advances in isocyanate chemistry. Chem. Rev. 1957, 57 (1), 47–76.10.1021/cr50013a002Search in Google Scholar
Arp, H. P. H.; Decken, A.; Passmore, J.; Wood, D. J. Preparation, characterization, X-ray crystal structure, and energetics of cesium 5-cyano-1,2,3,4-tetrazolate: Cs[NCCNNNN]. Inorg. Chem. 2000, 39 (9), 1840–1848.10.1021/ic990828qSearch in Google Scholar PubMed
Arppe, T.; Khriachtchev, L.; Lignell, A.; Domanskaya, A. V.; Räsänen, M. Halogenated xenon cyanides ClXeCN, ClXeNC, and BrXeCN. Inorg. Chem. 2012, 51 (7), 4398–4402.10.1021/ic3002543Search in Google Scholar PubMed
Arulsamy, N.; Bohle, D. S.; Doletski, B. G. Synthesis and thermal decomposition studies of new nitroso- and nitrodicyanomethanide salts. Inorg. Chem. 1999, 38 (11), 2709–2715.10.1021/ic9814064Search in Google Scholar
Atoji, M. Neutron structure analysis of cubic CaC2 and KCN. J. Chem. Phys. 1971, 54 (8), 3514–3516.10.1063/1.1675375Search in Google Scholar
Austad, T.; Songstad, J.; Åse, K.; Omfeldt, M.; Lagerlund, I.; Ehrenberg, L. Stable salts of the tellurocyanate ion. Acta Chem. Scand. 1971, 25, 331–333.10.3891/acta.chem.scand.25-0331Search in Google Scholar
Aynsley, E. E.; Dodd, R. E.; Little, R. Proc. Chem. Soc., London 1959, 241–284.Search in Google Scholar
Aynsley, E. E.; Greenwood, N. N.; Sprague, M. J. The oxidation of potassium selenocyanate by iodine pentafluoride. J. Chem. Soc. 1964, 704–708.10.1039/jr9640000704Search in Google Scholar
Azizitorghabeh, A.; Wang, J.; Ramsay, J. A.; Ghahreman, A. A review of thiocyanate gold leaching – chemistry, thermodynamics, kinetics and processing. Miner. Eng. 2021, p. 106689.10.1016/j.mineng.2020.106689Search in Google Scholar
Badiello, R. Physical-chemical properties of selenium-containing inorganic radicals and their reactivity with amino acids and enzymes. Phosphorus, Sulfur Silicon Relat. Elem. 1992, 67 (1–4), 7–10.10.1080/10426509208045813Search in Google Scholar
Baglin, F. G.; Coulter, G. L.; Durig, J. R. Low frequency vibrations of molecular crystals, VIII. The far infrared spectrum of polycrystalline hydrogen and deuterium cyanide. Mol. Cryst. Liq. Cryst. 1970, 10 (1–2), 47–60.10.1080/15421407008083486Search in Google Scholar
Bahta, A.; Parker, G. A.; Tuck, D. G. Critical survey of stability constants of complexes of thiocyanate ion (technical report). Pure Appl. Chem. 1997, 69 (7), 1489–1548.10.1351/pac199769071489Search in Google Scholar
Bak, B.; Hillebert, A. Cyanogen iodide. Org. Synth. 1952, 32, 29.10.1002/0471264180.os032.12Search in Google Scholar
Bak, B.; Svanholt, H. The existence of gaseous cyanoform as observed by microwave spectra. J. Mol. Struct. 1977, 37 (1), 153–156.10.1016/0022-2860(77)87015-4Search in Google Scholar
Bak, B.; Clementi, E.; Kortzeborn, R. N. Structure, vibrational spectra, dipole moment, and stability of gaseous LiCN and LiNC. J. Chem. Phys. 1970, 52 (2), 764–772.10.1063/1.1673052Search in Google Scholar
Bandy, A. R.; Friedrich, H. B.; Person, W. B. Absolute infrared intensities of the fundamental absorption bands in solid cyanogen halides. J. Chem. Phys. 1970, 53 (2), 674–681.10.1063/1.1674043Search in Google Scholar
Banerjee, G. Phenol‐ and thiocyanate‐based wastewater treatment in RBC reactor. J. Environ. Eng. 1996, 122 (10), 941–948.10.1061/(ASCE)0733-9372(1996)122:10(941)Search in Google Scholar
Banert, K.; Chityala, M.; Hagedorn, M.; Beckers, H.; Stüker, T.; Riedel, S.; Rüffer, T.; Lang, H. Tricyanomethane and its ketenimine tautomer: generation from different precursors and analysis in solution, argon matrix, and as a single crystal. Angew. Chem. Int. Ed. 2017, 56 (32), 9582–9586.10.1002/anie.201704561Search in Google Scholar PubMed
Bányai, I.; Glaser, J.; Losonczi, J. Equilibrium dynamics in the thallium(III)-cyanide system in aqueous solution. Inorg. Chem. 1997, 36 (25), 5900–5908.10.1021/ic970366lSearch in Google Scholar PubMed
Bányai, I.; Glaser, J.; Tóth, I. Cyanide exchange on Tl(CN)4− in aqueous solution studied by 205Tl and 13C NMR spectroscopy. Eur. J. Inorg. Chem. 2001, 2001 (7), 1709–1717.10.1002/1099-0682(200107)2001:7<1709::AID-EJIC1709>3.0.CO;2-SSearch in Google Scholar
Barrientos, C.; Largo, A. Ionization and protonation of (MgCN): an ab initio study of some gas-phase properties of the first magnesium compound in space. J. Mol. Struct.: Theochem. 1995, 336 (1), 29–37.10.1016/0166-1280(94)04099-ESearch in Google Scholar
Barton, J. M.; Greenfield, D. C. L.; Hamerton, I.; Jones, J. R. A study of the cyclotrimerisation and polymerisation of aryl cyanates using 13C and 15N nuclear magnetic resonance spectroscopy, Fourier transform infra-red spectroscopy and differential scanning calorimetry. Polym. Bull. 1991, 25 (4), 475–482.10.1007/BF00310239Search in Google Scholar
Barton, J. M.; Chaplin, A.; Hamerton, I.; Howlin, B. J. A new synthetic route for the preparation of alkenyl functionalized aryl cyanate ester monomers. Polymer. 1999, 40 (19), 5421–5427.10.1016/S0032-3861(98)00739-3Search in Google Scholar
Basco, N.; Yee, K. K. Spectra attributed to the PCN and HPCN free radicals. Chem. Commun. (London) 1968a, 152–153.10.1039/c19680000152Search in Google Scholar
Basco, N.; Yee, K. K. Spectrum attributed to the AsCN free radical. Chem. Commun. (London) 1968b, 153–154.10.1039/c19680000153Search in Google Scholar
Baskin, S. I.; Horowitz, A. M.; Nealley, E. W. The antidotal action of sodium nitrite and sodium thiosulfate against cyanide poisoning. J. Clin. Pharmacol. 1992, 32 (4), 368–375.10.1002/j.1552-4604.1992.tb03849.xSearch in Google Scholar
Bassett, H.; Corbet, A. S. Ccxvii. A phase rule study of the cupro-, argento-, auro- and thallo-cyanides of potassium. J. Chem. Soc. Trans. 1924, 125 (1660), 1660–1675.10.1039/CT9242501660Search in Google Scholar
Batta, G.; Banyai, I.; Glaser, J. 2D NMR exchange spectroscopy with spy nuclei for thallium(III) cyano complexes in aqueous solution. J. Am. Chem. Soc. 1993, 115 (15), 6782–6785.10.1021/ja00068a040Search in Google Scholar
Batten, S. R.; Murray, K. S. Structure and magnetism of coordination polymers containing dicyanamide and tricyanomethanide. Coord. Chem. Rev. 2003, 246 (1–2), 103–130.10.1016/S0010-8545(03)00119-XSearch in Google Scholar
Bauer, M.; Gnauck, R. Thermischer und Thermisch-Oxidativer Abbau von Polycyanuraten und deren Kombinationen Mit Polyglycidethern. Acta Polym. 1987, 38 (12), 658–661.10.1002/actp.1987.010381205Search in Google Scholar
Bauer, M.; Bauer, J.; Kühn, G. Analytik von Prepolymeren und Polymeren Aromatischer Cyansäureester. 1. IR‐spektroskopie. Acta Polym. 1986, 37 (4), 218–220.10.1002/actp.1986.010370408Search in Google Scholar
Bauer, J.; Höper, L.; Bauer, M. Cyclotrimerization reactivities of mono- and difunctional cyanates. Macromol. Chem. Phys. 1998, 199 (11), 2417–2423.10.1002/(SICI)1521-3935(19981101)199:11<2417::AID-MACP2417>3.0.CO;2-KSearch in Google Scholar
Baum, F. Zur Darstellung von Bromcyan und Cyanamid-Derivaten. Ber. Dtsch. Chem. Ges. 1908, 41 (1), 523–524.10.1002/cber.19080410198Search in Google Scholar
Bauschlicher, C. W.; Partridge, H. Mg+-ligand binding energies. Chem. Phys. Lett. 1991, 181 (2–3), 129–133.10.1016/0009-2614(91)90344-9Search in Google Scholar
Bauschlicher, C. W.; Langhoff, S. R.; Partridge, H. Ab initio study of BeCN, MgCN, CaCN and BaCN. Chem. Phys. Lett. 1985, 115 (2), 124–129.10.1016/0009-2614(85)80663-1Search in Google Scholar
Beach, J. Y.; Turkevich, A. The electron diffraction investigation of the molecular structures of cyanogen chloride and cyanogen bromide. J. Am. Chem. Soc. 1939, 61 (2), 299–303.10.1021/ja01871a022Search in Google Scholar
Beaton, S. A.; Ito, Y.; Brown, J. M. Laser excitation spectroscopy of the Ã3Πu–X3Σ−g transition of the NCN radical. J. Mol. Spectrosc. 1996, 178 (1), 99–107.10.1006/jmsp.1996.0162Search in Google Scholar
Beaumont, R. C.; Aspin, K. B.; Demas, T. J.; Hoggatt, J. H.; Potter, G. E. Oxidation of the tricyanomethanide ion: the tricyanocarbonium ion. Inorg. Chim. Acta. 1984, 84 (2), 141–147.10.1016/S0020-1693(00)82399-3Search in Google Scholar
Bebarta, V. S.; Pitotti, R. L.; Dixon, P.; Lairet, J. R.; Bush, A.; Tanen, D. A. Hydroxocobalamin versus sodium thiosulfate for the treatment of acute cyanide toxicity in a Swine (Sus Scrofa) model. Ann. Emerg. Med. 2012, 59 (6), 532–539.10.1016/j.annemergmed.2012.01.022Search in Google Scholar
Bebarta, V. S.; Brittain, M.; Chan, A.; Garrett, N.; Yoon, D.; Burney, T.; Mukai, D.; Babin, M.; Pilz, R. B.; Mahon, S. B.; Brenner, M.; Boss, G. R. Sodium nitrite and sodium thiosulfate are effective against acute cyanide poisoning when administered by intramuscular injection. Ann. Emerg. Med. 2017, 69 (6), 718–725.10.1016/j.annemergmed.2016.09.034Search in Google Scholar PubMed PubMed Central
Beelitz, J.; Kill, C.; Feldmann, C.; Wulf, H.; Vogt, N.; Veit, F.; Dersch, W. Resuscitation after smoke inhalation with cyanide intoxication: influence of hydroxycobalamin on the change of blood cyanide level during resuscitation. Resuscitation 2017, 118, e94.10.1016/j.resuscitation.2017.11.009Search in Google Scholar
Behera, S.; Jena, P. Stability and spectroscopic properties of singly and doubly charged anions. J. Phys. Chem. 2012, 116 (23), 5604–5617.10.1021/jp210095qSearch in Google Scholar PubMed
Beichert, P.; Pfeiler, D.; Knözinger, E. HCN clusters in molecular beams and cryogenic matrices. Ber. Bunsenges. Phys. Chem. 1995, 99 (12), 1469–1478.10.1002/bbpc.199500111Search in Google Scholar
Belbruno, J. J.; Tang, Z.-C.; Smith, R.; Hobday, S. The structure and energetics of carbon-nitrogen clusters. Mol. Phys. 2001, 99 (11), 957–967.10.1080/00268970110040219Search in Google Scholar
Bell, D. D.; Coombe, R. D. Photodissociation of chlorine isocyanate. J. Chem. Phys. 1985, 82 (3), 1317–1322.10.1063/1.448962Search in Google Scholar
Berger, B. Toxikologie. Eine Einführung Für Naturwissenschaftler und Mediziner. Angew. Chem. 1997, 109 (3), 307.10.1002/ange.19971090336Search in Google Scholar
Bergstrom, F. W. The reaction between mercuric cyanide and certain metals in liquid ammonia. J. Am. Chem. Soc. 1924, 46 (7), 1559–1568.10.1021/ja01672a002Search in Google Scholar
Berkei, M.; Bernhardt, E.; Schürmann, M.; Mehring, M.; Willner, H. Darstellung, Spektroskopische Charakterisierung und Kristallstrukturen von Quecksilber(II)-Bis(Tetracyanoborat) Hg[B(CN)4]2 und Diquecksilber(I)-Bis(Tetracyanoborat) Hg2[B(CN)4]2. Z. Anorg. Allg. Chem. 2002, 628 (8), 1734.10.1002/1521-3749(200208)628:8<1734::AID-ZAAC1734>3.0.CO;2-QSearch in Google Scholar
Berlinerblau, J. Über die Einwirkung von Chlorcyan Auf Ortho- und Auf Para-Amidophenetol. J. Prakt. Chem. 1884, 30 (1), 97–115.10.1002/prac.18840300110Search in Google Scholar
Bernhardt, E.; Henkel, G.; Willner, H. Die Tetracyanoborate M[B(CN)4], M = [Bu4N]+, Ag+, K+. Z. Anorg. Allg. Chem. 2000, 626 (2), 560–568.10.1002/(SICI)1521-3749(200002)626:2<560::AID-ZAAC560>3.0.CO;2-ESearch in Google Scholar
Bernhardt, E.; Henkel, G.; Willner, H.; Pawelke, G.; Bürger, H. Synthesis and properties of the tetrakis(trifluoromethyl)borate anion, [B(CF3)4]−: structure determination of Cs[B(CF3)4] by single-crystal X-ray diffraction. Chem. Eur J. 2001, 7 (21), 4696–4705.10.1002/1521-3765(20011105)7:21<4696::AID-CHEM4696>3.0.CO;2-5Search in Google Scholar
Bernhardt, E.; Berkei, M.; Willner, H.; Schürmann, M. Die Reaktionen von M[BF4] (M = Li, K) Und (C2H5)2O·BF3 Mit (CH3)3SiCN. Bildung von M[BFx(CN)4-x] (M = Li, K; x = 1, 2) Und (CH3)3SiNCBFx(CN)3-x, (x = 0, 1). Z. Anorg. Allg. Chem. 2003a, 629 (4), 677–685.10.1002/zaac.200390115Search in Google Scholar
Bernhardt, E.; Finze, M.; Willner, H. Eine Effiziente synthese von Tetracyanoboraten Durch Sinterprozesse. Z. Anorg. Allg. Chem. 2003b, 629 (78), 1229–1234.10.1002/zaac.200300047Search in Google Scholar
Bernhardt, E.; Bernhardt-Pitchougina, V.; Willner, H.; Ignatiev, N. “Umpolung” at boron by reduction of [B(CN)4]− and formation of the dianion [B(CN)3]2−. Angew. Chem. Int. Ed. 2011a, 50 (50), 12085–12088.10.1002/anie.201105848Search in Google Scholar
Bernhardt, E.; Finze, M.; Willner, H. Mechanistic study on the fluorination of K[B(CN)4] with ClF enabling the high yield and large scale synthesis of K[B(CF3)4] and K[(CF3)3BCN]. Inorg. Chem. 2011b, 50 (20), 10268–10273.10.1021/ic201319hSearch in Google Scholar
Bernsdorf, A.; Köckerling, M. Chains of alkaline metal cations and tetracyanidoborato anions - syntheses, structures and properties of the new [AI(18-Crown-6)][B(CN)4)] (AI = Cs, Rb). Eur. J. Inorg. Chem. 2009, 2009 (29–30), 4547–4553.10.1002/ejic.200900285Search in Google Scholar
Bernsdorf, A.; Köckerling, M. Crystal structure of tetraphenylphosphonium cyanate, [P(C6H5)4]OCN. Z. Kristallogr. - New Cryst. Struct. 2012, 227 (1), 85–86.10.1524/ncrs.2012.0038Search in Google Scholar
Bernsdorf, A.; Brand, H.; Hellmann, R.; Köckerling, M.; Schulz, A.; Villinger, A.; Voss, K. Synthesis, structure, and bonding of weakly coordinating anions based on CN adducts. J. Am. Chem. Soc. 2009a, 131 (25), 8958–8970.10.1021/ja902450bSearch in Google Scholar
Bernsdorf, A.; Brand, H.; Hellmann, R.; Köckerling, M.; Schulz, A.; Villinger, A.; Voss, K. Synthesis, structure, and bonding of weakly coordinating anions based on CN adducts. J. Am. Chem. Soc. 2009b, 131 (25), 8958–8970.10.1021/ja902450bSearch in Google Scholar
Berthollet, C.-L. Méthode de Nomenclature Chimique. Mem. Acad. r. Sci. Paris 1787, 148.Search in Google Scholar
Berzelius, J. J. Unterscheidung der Isomerie von Analogen Zuständen. Ann. Phys. 1832, 102 (10), 320–322.10.1002/andp.18321021012Search in Google Scholar
Bessler, E.; Goubeau, J. Darstellung Einiger Cyan-Borverbindungen. Z. Anorg. Allg. Chem. 1967, 352 (1–2), 67–76.10.1002/zaac.19673520111Search in Google Scholar
Bessler, E. Darstellung und Eigenschaften von AgB(CN)4 und CuB(CN)4. Z. Anorg. Allg. Chem. 1977, 430 (1), 38–42.10.1002/zaac.19774300103Search in Google Scholar
Betts, P. M.; Rinder, D. F.; Fleeker, J. R. Thiocyanate utilization by an arthrobacter. Can. J. Microbiol. 1979, 25 (11), 1277–1282.10.1139/m79-201Search in Google Scholar PubMed
Beuermann, G.; Hausmann, A. Elektronenspinresonanz von U2-Folgezentren in Alkalihalogenid-Kristallen. Z. Phys. 1967, 204 (5), 425–442.10.1007/BF01325734Search in Google Scholar
Beyer, H.; Walter, W. Organische Chemie; Hirzel Verlag: Stuttgart, 1991.Search in Google Scholar
Bezsudnova, E. Y.; Sorokin, D. Y.; Tikhonova, T. V.; Popov, V. O. Thiocyanate hydrolase, the primary enzyme initiating thiocyanate degradation in the novel obligately chemolithoautotrophic halophilic sulfur-oxidizing bacterium thiohalophilus thiocyanoxidans. Biochim. Biophys. Acta, Proteins Proteomics 2007, 1774 (12), 1563–1570.10.1016/j.bbapap.2007.09.003Search in Google Scholar PubMed
Bhattacharyya, I.; Bera, N. C.; Das, A. K. Theoretical study of structural properties and dissociation pathways of FCN and ClCN. Eur. Phys. J. D 2007a, 42 (2), 221–226.10.1140/epjd/e2007-00027-7Search in Google Scholar
Bhattacharyya, I.; Bera, N. C.; Das, A. K. Theoretical study of XCN− (X=F,Cl) anions. Int. J. Quantum Chem. 2007b, 107 (3), 680–684.10.1002/qua.21138Search in Google Scholar
Bhattacharyya, I.; Mondal, B.; Bera, N. C.; Das, A. K. Structure and dissociation of cyanogen halides BrCN and ICN. Int. J. Quantum Chem. 2010, 110, 1165–1171.10.1002/qua.22163Search in Google Scholar
Bhunia, F.; Saha, N. C.; Kaviraj, A. Toxicity of Thiocyanate to fish, Plankton, Worm and Aquatic Ecosystem. Bull. Environ. Contam. Toxicol. 2000, 64 (2), 197–204.10.1007/s001289910030Search in Google Scholar PubMed
Bijvoet, J. M.; Lely, J. A. Eine Rhombische Modifikation des Kaliumcyanids. Über die lage des Umwandlungspunkts des Natriumcyanids in Abhängigkeit von Beimischungen. Recl. Trav. Chim. Pays-Bas 2010, 59 (9), 908–912.10.1002/recl.19400590911Search in Google Scholar
Biltz, W. Raumchemie der Festen Stoffe. Ber. Dtsch. Chem. Ges. 1935, 68 (7), A91–A108.10.1002/cber.19350680738Search in Google Scholar
Binder, J. F.; Kosnik, S. C.; St Onge, P. B. J.; Macdonald, C. L. B. Synthesis of heavy dicyanamide homologues from air-stable precursors. Chem. Eur J. 2018, 24 (55), 14644–14648.10.1002/chem.201803941Search in Google Scholar PubMed
Binnewies, M.; Finze, M.; Jäckel, M.; Schmidt, P.; Willner, H.; Rayner-Canham, G. Allgemeine Und Anorganische Chemie; Springer Berlin Heidelberg: Berlin, Heidelberg, Vol. 58; 2016.10.1007/978-3-662-45067-3Search in Google Scholar
Binsch, G.; Roberts, J. D. Nitrogen-15 magnetic resonance spectroscopy. Coupling constants in hydrogen cyanide. J. Phys. Chem. 1968, 72 (12), 4310–4311.10.1021/j100858a064Search in Google Scholar
Birckenbach, L.; Huttner, K. Über Pseudohalogene, III.: Über Das Pseudohalogen Tricyanmethyl Und Das Mischhalogen Brom-Tricyanmethyl. Ber. Dtsch. Chem. Ges. 1929, 62 (1), 153–163.10.1002/cber.19290620122Search in Google Scholar
Birckenbach, L.; Kellermann, K. Über Pseudohalogene (I). Ber. Dtsch. Chem. Ges. 1925a, 58 (4), 786–794.10.1002/cber.19250580429Search in Google Scholar
Birckenbach, L.; Kellermann, K. Über Pseudohalogene (II.): 1. Der Knallsäure-Rest. 2. Das Gleichgewicht Zwischen Jod, Selenocyan und den Entsprechenden Silbersalzen. 3. Polypseudohalogenide. Ber. Dtsch. Chem. Ges. 1925b, 58 (10), 2377–2386.10.1002/cber.19250581022Search in Google Scholar
Birckenbach, L.; Kellermann, K. Verein Deutscher Chemiker. Angew. Chem. 1930, 43 (31), 700–702.10.1002/ange.19300433112Search in Google Scholar
Birckenbach, L.; Kolb, H. Über Reaktionen und die Tautomerie von Cyanaten (XXIII. Mitteil. Zur Kenntnis der Pseudohalogene). Ber. Dtsch. Chem. Ges. 1933, 66 (10), 1571–1577.10.1002/cber.19330661025Search in Google Scholar
Birckenbach, L.; Linhard, M. Über Pseudohalogene, V.: Über Das Mischhalogen Brom-Oxycyan. Ber. Dtsch. Chem. Ges. 1929, 62 (8), 2261–2277.10.1002/cber.19290620864Search in Google Scholar
Birckenbach, L.; Linhard, M. Über Pseudohalogene, X.: Über Dichlor-Dioxycyan, Nω-Mono- und Nω-Dichlor-Allophansäure-Äthylester und Nω-Chlor-Allophansäure-Chlorid. Ber. Dtsch. Chem. Ges. 1930, 63 (9), 2528–2544.10.1002/cber.19300630929Search in Google Scholar
Birckenbach, L.; Huttner, K.; Stein, W. Über Pseudohalogene, IV.: Die Hydrolysen-Konstanten Des Brom-Tricyanmethyls Und Des Chlor-, Brom- Und Jod-Trinitromethyls. Ber. Dtsch. Chem. Ges. 1929a, 62 (8), 2065–2075.10.1002/cber.19290620826Search in Google Scholar
Birckenbach, L.; Huttner, K.; Stein, W. Über Pseudohalogene, IV.: Die Hydrolysen-Konstanten des Brom-Tricyanmethyls und des Chlor-, Brom- und Jod-Trinitromethyls. Ber. Dtsch. Chem. Ges. 1929b, 62 (8), 2065–2075.10.1002/cber.19290620826Search in Google Scholar
Bither, T. A.; Knoth, W. H.; Lindsey, R. V.; Sharkey, W. H. Trialkyl- and Triaryl(Iso)Cyanosilanes. J. Am. Chem. Soc. 1958, 80 (16), 4151–4153.10.1021/ja01549a010Search in Google Scholar
Black, J. H.; van Dishoeck, E. F. Electron densities and the excitation of CN in molecular clouds. Astrophys. J. 1991, 369, 9.10.1086/185947Search in Google Scholar
Blanksby, S. J.; Dua, S.; Bowie, J. H.; Schröder, D.; Schwarz, H. Syntheses of NCN and NC3N from ionic precursors in the gas phase and an unusual rearrangement of neutral NC3N: a joint experimental and theoretical study. J. Phys. Chem. 2000, 104 (47), 11248–11256.10.1021/jp001590qSearch in Google Scholar
Bläsing, K.; Ellinger, S.; Harloff, J.; Schulz, A.; Sievert, K.; Täschler, C.; Villinger, A.; Zur Täschler, C. Lewis acid catalyzed synthesis of cyanidoborates. Eur. J. Inorg. Chem. 2016a, 2016 (8), 1175–1183.10.1002/ejic.201501485Search in Google Scholar
Bläsing, K.; Ellinger, S.; Harloff, J.; Schulz, A.; Sievert, K.; Täschler, C.; Villinger, A.; Zur Täschler, C. Lewis acid catalyzed synthesis of cyanidophosphates. Chem. Eur J. 2016b, 22 (12), 4175–4188.10.1002/chem.201504523Search in Google Scholar PubMed
Bläsing, K.; Bresien, J.; Labbow, R.; Schulz, A.; Villinger, A. A dimer of hydrogen cyanide stabilized by a Lewis acid. Angew. Chem. Int. Ed. 2018, 57 (29), 9170–9175.10.1002/anie.201804193Search in Google Scholar PubMed
Bläsing, K.; Harloff, J.; Schulz, A.; Stoffers, A.; Stoer, P.; Villinger, A. Salts of HCN‐cyanide aggregates: [CN(HCN)2]− and [CN(HCN)3]−. Angew. Chem. Int. Ed. 2020, 59 (26), 10508–10513.10.1002/anie.201915206Search in Google Scholar PubMed PubMed Central
Blixt, J.; Gyori, B.; Glaser, J. Determination of stability constants for thallium(III) cyanide complexes in aqueous solution by means of carbon-13 and thallium-205 NMR. J. Am. Chem. Soc. 1989, 111 (20), 7784–7791.10.1021/ja00202a019Search in Google Scholar
Blixt, J.; Glaser, J.; Mink, J.; Persson, I.; Persson, P.; Sandstroem, M. Structure of thallium(III) chloride, bromide, and cyanide complexes in aqueous solution. J. Am. Chem. Soc. 1995, 117 (18), 5089–5104.10.1021/ja00123a011Search in Google Scholar
Boatright, L. G. Preparation of ammonium cyanate from urea. US2712491, 1954.Search in Google Scholar
Böhland, H.; Golub, A. M.; Köhler, H.; Lisko, T. P.; Samoilenko, V. M.; Skopenko, V. V.; Cincadze, G. V. Chemie Der Pseudohalogenide; DVW: Berlin, 1978.Search in Google Scholar
Booth, M. R.; Frankiss, S. G. Trimethylsilyl isocyanide. Chem. Commun. (London) 1968, 1347–1348.10.1039/c19680001347Search in Google Scholar
Borron, S. W.; Baud, F. J.; Barriot, P.; Imbert, M.; Bismuth, C. Prospective study of hydroxocobalamin for acute cyanide poisoning in smoke inhalation. Ann. Emerg. Med. 2007, 49 (6), 794–801.10.1016/j.annemergmed.2007.01.026Search in Google Scholar PubMed
Bouchet, L. M.; Peñéñory, A. B.; Argüello, J. E. Synthesis of arylselenide ethers by photoinduced reactions of selenobenzamide, selenourea and selenocyanate anions with aryl halides. Tetrahedron Lett. 2011, 52 (9), 969–972.10.1016/j.tetlet.2010.11.115Search in Google Scholar
Bourson, P.; Ndtoungou, A.; Bouillot, J.; Soubeyroux, J. L.; Durand, D. Phase transition of RbCN. Phys. B (Amsterdam, Neth.) 1992, 180–181, 351–353.10.1016/0921-4526(92)90756-ISearch in Google Scholar
Bozorth, R. M. The crystal sructure of potassium cyanide. J. Am. Chem. Soc. 1922, 44 (2), 317–323.10.1021/ja01423a010Search in Google Scholar
Bradforth, S. E.; Kim, E. H.; Arnold, D. W.; Neumark, D. M. Photoelectron spectroscopy of CN−, NCO−, and NCS−. J. Chem. Phys. 1993, 98 (2), 800–810.10.1063/1.464244Search in Google Scholar
Brand, H.; Mayer, P.; Schulz, A.; Weigand, J. J. Nitro(Nitroso)Cyanmethanide. Angew. Chem. 2005a, 117 (25), 3998–4001.10.1002/ange.200500495Search in Google Scholar
Brand, H.; Mayer, P.; Schulz, A.; Weigand, J. J. Nitro(Nitroso)Cyanomethanides. Angew. Chem. Int. Ed. 2005b, 44 (25), 3929–3932.10.1002/anie.200500495Search in Google Scholar PubMed
Brand, H.; Liebman, J. F.; Schulz, A.; Mayer, P.; Villinger, A. Nonlinear, resonance-stabilized pseudohalides: from alkali methanides to ionic liquids of methanides. Eur. J. Inorg. Chem. 2006, 2006 (21), 4294–4308.10.1002/ejic.200600668Search in Google Scholar
Brand, H.; Schulz, A.; Villinger, A. Modern aspects of pseudohalogen chemistry: news from CN- and PN-chemistry. Z. Anorg. Allg. Chem. 2007, 633 (1), 22–35.10.1002/zaac.200600239Search in Google Scholar
Brand, H.; Liebman, J. F.; Schulz, A. Cyano-, nitro- and nitrosomethane derivatives: structures and gas-phase acidities. Eur. J. Org Chem. 2008, 2008 (27), 4665–4675.10.1002/ejoc.200800583Search in Google Scholar
Brand, H.; Martens, J.; Mayer, P.; Schulz, A.; Seibald, M.; Soller, T. Salts and ionic liquids of resonance stabilized amides. Chem. Asian J. 2009, 4 (10), 1588–1603.10.1002/asia.200900181Search in Google Scholar PubMed
Brazier, C. R.; O’Brien, L. C.; Bernath, P. F. Fourier transform detection of laser‐induced fluorescence from the CCN free radical. J. Chem. Phys. 1987, 86 (6), 3078–3081.10.1063/1.452018Search in Google Scholar
Bresien, J.; Ellinger, S.; Harloff, J.; Schulz, A.; Sievert, K.; Stoffers, A.; Täschler, C.; Villinger, A.; Zur Täschler, C. Tetracyanido(difluorido)phosphates M+[PF2(CN)4]−. Angew. Chem. Int. Ed. 2015a, 54 (15), 4474–4477.10.1002/anie.201412209Search in Google Scholar PubMed
Bresien, J.; Ellinger, S.; Harloff, J.; Schulz, A.; Sievert, K.; Stoffers, A.; Täschler, C.; Villinger, A.; Zur Täschler, C. Tetracyanido(difluorido)phosphate M+[PF2(CN)4]−. Angew. Chem. 2015b, 127 (15), 4556–4559.10.1002/ange.201412209Search in Google Scholar
Britton, D.; Dunitz, J. D. The crystal structure of silver cyanate. Acta Crystallogr. 1965a, 18 (3), 424–428.10.1107/S0365110X65000944Search in Google Scholar
Britton, D.; Dunitz, J. D. The crystal structure of silver fulminate. Acta Crystallogr. 1965b, 19 (4), 662–668.10.1107/S0365110X6500405XSearch in Google Scholar
Britton, D. The crystal structure of tetracyanomethane, C(CN)4. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30 (7), 1818–1821.10.1107/S0567740874005863Search in Google Scholar
Brooker, M. H.; Wen, N. Raman studies of cyanate: fermi resonance, hydration and hydrolysis to urea. Can. J. Chem. 1993, 71 (10), 1764–1773.10.1139/v93-219Search in Google Scholar
Brotherton, T. K.; Lynn, J. W. The synthesis and chemistry of cyanogen. Chem. Rev. 1959, 59 (5), 841–883.10.1021/cr50029a003Search in Google Scholar
Brousseau, L. C.; Williams, D.; Kouvetakis, J.; O’Keeffe, M. Synthetic routes to Ga(CN)3 and MGa(CN)4 (M = Li, Cu) framework structures. J. Am. Chem. Soc. 1997, 119 (27), 6292–6296.10.1021/ja9702024Search in Google Scholar
Brown, R. D.; Godfrey, P. D.; Winkler, D. A. The microwave spectrum of HCN dimer. J. Mol. Spectrosc. 1981, 89 (2), 352–355.10.1016/0022-2852(81)90029-1Search in Google Scholar
Brupbacher, T.; Bohn, R. K.; Jäger, W.; Gerry, M. C. L.; Pasinszki, T.; Westwood, N. P. C. Microwave spectrum and geometry of cyanogen N-oxide, NCCNO. J. Mol. Spectrosc. 1997, 181 (2), 316–322.10.1006/jmsp.1996.7180Search in Google Scholar
Buchholz, C. F. Bemerkungen, Gemacht Bei Gelegenheit Einiger Versuche Über die Reinigung der Blausauren Salze Vom Eisen. In Beiträge zur Erweiterung und Berichtigung der Chemie; Erfurt, 1799; pp. 81–92.Search in Google Scholar
Buljan, A.; Alemany, P.; Ruiz, E. Electronic structure and dynamic properties of solid alkali cyanides. J. Phys. Chem. 1997, 101 (7), 1393–1399.10.1021/jp962875zSearch in Google Scholar
Bundhun, A.; Abdallah, H. H.; Ramasami, P.; Gaspar, P. P.; Schaefer, H. F. Dicyanogermylenes: a tale of isomers and interconversions. Inorg. Chem. 2012, 51 (22), 12152–12164.10.1021/ic301225wSearch in Google Scholar PubMed
Burchell, C. J.; Aucott, S. M.; Robertson, S. D.; Slawin, A. M. Z.; Woollins, J. D. The chemistry of (ECN)2 (E = S, Se) and related compounds. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179 (4–5), 865–868.10.1080/10426500490427619Search in Google Scholar
Burchell, C. J.; Kilian, P.; Slawin, A. M. Z.; Woollins, J. D.; Tersago, K.; Van Alsenoy, C.; Blockhuys, F. E2(CN)2 (E = S, Se) and related compounds. Inorg. Chem. 2006, 45 (2), 710–716.10.1021/ic0515103Search in Google Scholar PubMed
Burger, A.; Hornbaker, E. D. Some reactions in the 1,3,5-triazine series. J. Am. Chem. Soc. 1953, 75 (18), 4579–4580.10.1021/ja01114a502Search in Google Scholar
Burkholder, J. B.; Sinha, A.; Hammer, P. D.; Howard, C. J. High-resolution Fourier transform infrared spectroscopy of the fundamental bands of HNC. J. Mol. Spectrosc. 1987, 126 (1), 72–77.10.1016/0022-2852(87)90077-4Search in Google Scholar
Burns, W. A.; Leopold, K. R. Unusually large gas–solid structure differences: a crystallographic study of HCN–BF3 [hydrogen cyanide-boron trifluoride]. J. Am. Chem. Soc. 1993, 115 (24), 11622–11623.10.1021/ja00077a081Search in Google Scholar
Buser, H. J.; Schwarzenbach, D.; Petter, W.; Ludi, A. The crystal structure of prussian blue: Fe4[Fe(CN)6]3 xH2O. Inorg. Chem. 1977, 16 (11), 2704–2710.10.1021/ic50177a008Search in Google Scholar
Buxton, L. W.; Campbell, E. J.; Flygare, W. H. The vibrational ground state rotational spectroscopic constants and structure of the HCN dimer. Chem. Phys. 1981, 56 (3), 399–406.10.1016/0301-0104(81)80161-9Search in Google Scholar
Camargo, S. S.; von der Weid, J. P. Optical absorption of triplet molecular excitons in alkali cyanides. Solid State Commun. 1982, 44 (8), 1227–1229.10.1016/0038-1098(82)91091-2Search in Google Scholar
Campbell, R.; Davis, M. F.; Fazakerley, M.; Portius, P. Taming Tin(IV) polyazides. Chem. Eur J. 2015, 21 (51), 18690–18698.10.1002/chem.201503478Search in Google Scholar PubMed
Cant, N. W.; Chambers, D. C.; Liu, I. O. Y. The reduction of NO by CO in the presence of water vapour on supported platinum catalysts: formation of isocyanic acid (HNCO) and ammonia. Appl. Catal. B 2003, 46 (3), 551–559.10.1016/S0926-3373(03)00318-7Search in Google Scholar
Cant, N. W.; Chambers, D. C.; Liu, I. O. Y. The formation of isocyanic acid and ammonia during the reduction of NO over supported platinum group metals. Catal. Today 2004, 93–95, 761–768.10.1016/j.cattod.2004.06.068Search in Google Scholar
Cant, N. W.; Chambers, D. C.; Liu, I. O. Y. The amplification of ammonia by reaction with NO and CO over dual function platinum and palladium catalyst systems with isocyanic acid as an intermediate. Appl. Catal. B 2005a, 60 (1–2), 57–63.10.1016/j.apcatb.2005.02.021Search in Google Scholar
Cant, N. W.; Chambers, D. C.; Liu, I. O. Y. The formation of isocyanic acid during the reaction of NH3 with NO and excess CO over silica-supported platinum, palladium, and rhodium. J. Catal. 2005b, 231 (1), 201–212.10.1016/j.jcat.2005.01.022Search in Google Scholar
Carrasco, N.; Schmitz-Afonso, I.; Bonnet, J.-Y.; Quirico, E.; Thissen, R.; Dutuit, O.; Bagag, A.; Laprévote, O.; Buch, A.; Giulani, A.; Adandé, G.; Ouni, E.; Hadamcik, E.; Szopa, C.; Cernogora, G.Chemical characterization of Titan’s tholins: solubility, morphology and molecular structure revisited. J. Phys. Chem. 2009, 113 (42), 11195–11203.10.1021/jp904735qSearch in Google Scholar PubMed
Castanheiro, T.; Suffert, J.; Donnard, M.; Gulea, M. Recent advances in the chemistry of organic thiocyanates. Chem. Soc. Rev. 2016, pp. 494–505.10.1039/C5CS00532ASearch in Google Scholar
Cenedella, M. Cyanure de Phosphore. J. Pharm. (Lahore) 1835, 11, 683.Search in Google Scholar
Cerny, D.; Bacis, R.; Guelachvili, G.; Roux, F. Extensive analysis of the red system of the CN molecule with a high resolution Fourier spectrometer. J. Mol. Spectrosc. 1978, 73 (1), 154–167.10.1016/0022-2852(78)90201-1Search in Google Scholar
Chacko, S. A.; Krouse, I. H.; Hammad, L. A.; Wenthold, P. G. In situ generation of HCN for mass spectrometric studies. J. Am. Soc. Mass Spectrom. 2006, 17 (1), 51–55.10.1016/j.jasms.2005.08.018Search in Google Scholar PubMed
Chadwick, B. M.; Long, D. A.; Qureshi, S. U. The Raman and infrared spectra of the dicyanoiodate (I) ion. J. Raman Spectrosc. 1980, 9 (1), 1–4.10.1002/jrs.1250090103Search in Google Scholar
Challenger, F.; Peters, A. T.; Halévy, J. CCXV.–The introduction of the selenocyano-group into aromatic compounds. J. Chem. Soc. 1926, 129, 1648–1655.10.1039/JR9262901648Search in Google Scholar
Chambers, R. D.; Tamura, M. Fluorinated 1,2,3-triazines. J. Fluor. Chem. 1985, 29 (1–2), 127.10.1016/S0022-1139(00)83363-1Search in Google Scholar
Chambers, D. C.; Angove, D. E.; Cant, N. W. The formation and hydrolysis of isocyanic acid during the reaction of NO, CO, and H2 mixtures on supported platinum, palladium, and rhodium. J. Catal. 2001, 204 (1), 11–22.10.1006/jcat.2001.3359Search in Google Scholar
Chan, W.-T.; Hamilton, I. P. Geometries and vibrational frequencies for calcium and strontium radical salts of H, F, Cl, Br, I, OH, SH, CN, NC, CCH, NNN, NCO, NCS, NH2, CH3, OCH3, SCH3, HCOO, CH3COO, HCONH and HCONCH3. Chem. Phys. Lett. 1998, 297 (3–4), 217–224.10.1016/S0009-2614(98)01106-3Search in Google Scholar
Chandler, J. D.; Day, B. J. Biochemical mechanisms and therapeutic potential of pseudohalide thiocyanate in human health. Free Radical Res. 2015, 49 (6), 695–710.10.3109/10715762.2014.1003372Search in Google Scholar PubMed PubMed Central
Chase, M. W.; Curnutt, J. L.; Hu, A. T.; Prophet, H.; Syverud, A. N.; Walker, L. C. JANAF thermochemical tables, 1974 supplement. J. Phys. Chem. Ref. Data 1974, 3 (2), 311–480.10.1063/1.3253143Search in Google Scholar
Chau, F. T.; McDowell, C. A.; Tang, Y. W. Determination of geometries and molecular properties of XCN+ ions (where X is Cl, Br, I) through Franck–Condon analyses on the corresponding photoelectron spectra. J. Electron Spectrosc. 1993, 61 (2), 217–229.10.1016/0368-2048(93)80052-NSearch in Google Scholar
Chadwick, B. M.; Edwards, H. G. M. Vibrational and vibrational-rotational spectroscopy of the cyanide ion, the cyano-radical, the cyanogen molecules, and the triatomic cyanides XCN (X=H, F, Cl, Br, and I). In Molecular spectroscopy; Barrow, R. F., Long, D. A., Millen, D. J., Eds.; The Chemical Society: London, 1973; pp. 516–522.10.1039/9781847556684-00446Search in Google Scholar
Chaudhuri, R. K.; Krishnamachari, S. L. N. G. Theoretical Study on the ground and excited states of dicyanocarbene (c3n2) and its isomers: a low-temperature matrix emission spectrum attributable to 3-cyano-2h-azirenylidene. J. Phys. Chem. 2007, 111 (22), 4849–4854.10.1021/jp066358kSearch in Google Scholar PubMed
Chavant, C.; Constant, G.; Jeannin, Y.; Morancho, R. Structure Cristalline Du Pentachloroformonitrileniobium(V). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1975, 31 (7), 1823–1827.10.1107/S0567740875006309Search in Google Scholar
Chen, Y.; Bai, X. A review on quantum dots modified g-C3N4-based photocatalysts with improved photocatalytic activity. Catalysts 2020, 10 (1), 142.10.3390/catal10010142Search in Google Scholar
Chen, H.-L.; Zhu, R.; Chen, H.-T.; Li, H.-J.; Ju, S.-P. Ab initio study on mechanisms and kinetics for reaction of NCS with NO. J. Phys. Chem. 2008, 112 (24), 5495–5501.10.1021/jp8013805Search in Google Scholar PubMed
Chevrier, P. J.; Brownstein, S. Complex fluoroanions in solution – X: complexes of phosphorus and arsenic fluorides with simple anions. J. Inorg. Nucl. Chem. 1980, 42 (10), 1397–1405.10.1016/0022-1902(80)80103-5Search in Google Scholar
Chizmeshya, A. V. G.; Ritter, C. J.; Groy, T. L.; Tice, J. B.; Kouvetakis, J. Synthesis of molecular adducts of beryllium, boron, and gallium cyanides: theoretical and experimental correlations between solid-state and molecular analogues. Chem. Mater. 2007, 19 (24), 5890–5901.10.1021/cm071275hSearch in Google Scholar
Chow, Y. M.; Britton, D. Silver dicyanonitrosomethanide, AgC(CN)2NO. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30 (4), 1117–1118.10.1107/S0567740874004377Search in Google Scholar
Christie, A. Sparkling Cyanide; Dodd Mead and Company: New York, 1945.Search in Google Scholar
Cioslowski, J.; Mixon, S. T.; Fleischmann, E. D. Electronic structures of trifluoro-, tricyano-, and trinitromethane and their conjugate bases. J. Am. Chem. Soc. 1991, 113 (13), 4751–4755.10.1021/ja00013a007Search in Google Scholar
Clementi, E.; Kistenmacher, H.; Popkie, H. Study of the electronic structure of molecules. XVIII. Interaction between a lithium atom and a cyano group as an example of a polytopic bond. J. Chem. Phys. 1973, 58 (6), 2460–2466.10.1063/1.1679525Search in Google Scholar
Clifford, E. P.; Wenthold, P. G.; Lineberger, W. C.; Petersson, G. A.; Ellison, G. B. Photoelectron spectroscopy of the NCN– and HNCN– ions. J. Phys. Chem. 1997, 101 (24), 4338–4345.10.1021/jp964067dSearch in Google Scholar
Coates, G. E.; Mukherjee, R. N. Dimethylaluminium cyanide and its gallium, indium, and thallium analogues; beryllium and methylberyllium cyanide. J. Chem. Soc. 1963, 35, 229–233.10.1039/jr9630000229Search in Google Scholar
Cochran, E. L.; Adrian, F. J.; Bowers, V. A. ESR detection of the cyanogen and methylene imino free radicals. J. Chem. Phys. 1962, 36 (7), 1938–1942.10.1063/1.1701294Search in Google Scholar
Cocksedge, H. E. CCXVII.–Tellurium dicyanide. J. Chem. Soc. Trans. 1908, 93, 2175–2177.10.1039/CT9089302175Search in Google Scholar
Coleman, G. H.; Leeper, R. W.; Schulze, C. C.; Scortt, L. D.; Fernelius, W. C. Cyanogen chloride. In Inorganic synthesis; McGraw-Hill Book Company, 2, 2007; pp. 90–94.10.1002/9780470132333.ch25Search in Google Scholar
Coll, P.; Coscia, D.; Gazeau, M. C.; Guez, L.; Raulin, F. Review and latest results of laboratory investigations of Titan’s aerosols. Origins Life Evol. Biospheres 1998, 28 (2), 195–213.10.1023/A:1006521601133Search in Google Scholar PubMed
Conrad, M. P.; Schaefer, H. F. Role of different isomers of the H2CN+ ion in the formation of interstellar HCN and HNC. Nature 1978, 274 (5670), 456–457.10.1038/274456a0Search in Google Scholar
Coogan, C. K.; Gutowsky, H. S. NMR studies of phase transitions. II. NaCN. J. Chem. Phys. 1964, 40 (11), 3419–3425.10.1063/1.1725016Search in Google Scholar
Cook, T. J.; Levy, D. H. Optically detected gas phase magnetic resonance spectrum of the CN radical. J. Chem. Phys. 1973, 58 (9), 3547–3557.10.1063/1.1679698Search in Google Scholar
Cook, R. P.; Robinson, P. L. Certain physical properties of cyanogen and its halides. J. Chem. Soc. 1935, 1001–1005.10.1039/jr9350001001Search in Google Scholar
Corain, B. The coordination chemistry of hydrogen cyanide, cyanogen and cyanogen halides. Coord. Chem. Rev. 1982, 47 (1–2), 165–200.10.1016/0010-8545(82)85013-3Search in Google Scholar
Cornog, J.; Horrabin, H. W.; Karges, R. A. Iodine monochloride. II. Reactions with salts. J. Am. Chem. Soc. 1938, 60 (2), 429–432.10.1021/ja01269a057Search in Google Scholar
Cortés, E.; Erben, M. F.; Geronés, M.; Romano, R. M.; Della Védova, C. O. Dissociative photoionization of methyl thiocyanate, CH3SCN, in the proximity of the sulfur 2p edge. J. Phys. Chem. 2009, 113 (3), 564–572.10.1021/jp807230sSearch in Google Scholar PubMed
Coskran, K. J.; Jones, C. E. Synthesis and spectroscopic properties of some cyanophosphines. Inorg. Chem. 1971, 10 (7), 1536–1537.10.1021/ic50101a049Search in Google Scholar
Cosslett, V. E. Darstellung und Eigenschaften von Fluorcyan. Z. Anorg. Allg. Chem. 1931, 201 (1), 75–80.10.1002/zaac.19312010108Search in Google Scholar
Costain, C. C. Determination of molecular structures from ground state rotational constants. J. Chem. Phys. 1958, 29 (4), 864–874.10.1063/1.1744602Search in Google Scholar
Cotton, F. A.; Wilkinson, G.; Fritz, H. P. Anorganische Chemie, XIII; DVW: Berlin, 1967.Search in Google Scholar
Cozzolino, A. F.; Silvia, J. S.; Lopez, N.; Cummins, C. C. Experimental and computational studies on the formation of cyanate from early metal terminal nitrido ligands and carbon monoxide. Dalton Trans. 2014, 43 (12), 4639–4652.10.1039/C3DT52738GSearch in Google Scholar PubMed
Cravens, T. E.; Robertson, I. P.; Waite, J. H.; Yelle, R. V.; Kasprzak, W. T.; Keller, C. N.; Ledvina, S. A.; Niemann, H. B.; Luhmann, J. G.; McNutt, R. L.; Ip, W.-H.; De La Haye, V.; Mueller-Wodarg, I.; Wahlung, J.-E.; Anicich, V. G.; Vuitton, V. Composition of Titan’s ionosphere. Geophys. Res. Lett. 2006, 33 (7), L07105.10.1029/2005GL025575Search in Google Scholar
Cummins, P. L.; Bacskay, G. B.; Hush, N. S.; Jönsson, B. On the structure, lattice energy and 14N nuclear quadrupole coupling constant of solid HCN. Chem. Phys. Lett. 1988, 145 (5), 399–406.10.1016/0009-2614(88)80198-2Search in Google Scholar
Cyvin, S. J.; Ratkje, S. K.; Devarajan, V.; Nayar, V. U.; Aruldhas, G. Spectroscopic calculations on phosphorus and arsenic tricyanides: harmonic force fields, mean amplitudes of vibration and vibrational assignments. J. Mol. Struct. 1973, 17 (2), 371–375.10.1016/0022-2860(73)85178-6Search in Google Scholar
Dagg, I. R.; Anderson, A.; Yan, S.; Smith, W.; Joslin, C. G.; Read, L. A. A. The Quadrupole moment of cyanogen: a comparative study of collision-induced absorption in gaseous C2N2, CO2, and mixtures with argon. Can. J. Phys. 1986, 64 (11), 1475–1481.10.1139/p86-261Search in Google Scholar
Dalla Betta, R. A.; Shelef, M. Isocyanates from the reaction of NO and CO on supported noble-metal catalysts. J. Mol. Catal. 1976, 1 (6), 431–434.10.1016/0304-5102(76)85004-3Search in Google Scholar
Dammeier, J.; Friedrichs, G. Thermal decomposition of NCN3 as a high-temperature NCN radical source: singlet−triplet relaxation and absorption cross section of NCN(3Σ). J. Phys. Chem. 2010, 114 (50), 12963–12971.10.1021/jp1043046Search in Google Scholar PubMed
Dao, N. Q.; Wilkinson, G. R. Far‐infrared and Raman spectra of the crystal KNCS. J. Chem. Phys. 1973, 59 (3), 1319–1324.10.1063/1.1680186Search in Google Scholar
Darkwah, W. K.; Ao, Y. Mini review on the structure and properties (photocatalysis), and preparation techniques of graphitic carbon nitride nano-based particle, and its applications. Nanoscale Res. Lett. 2018, 13 (1), 388.10.1186/s11671-018-2702-3Search in Google Scholar PubMed PubMed Central
Dash, R. R.; Gaur, A.; Balomajumder, C. Cyanide in industrial wastewaters and its removal: a review on biotreatment. J. Hazard Mater. 2009, 163 (1), 1–11.10.1016/j.jhazmat.2008.06.051Search in Google Scholar PubMed
Daubert, J.; Knorr, K.; Dultz, W.; Jex, H.; Currat, R. Lattice dynamics of KCN. J. Phys. C Solid State Phys. 1976, 9 (15), L389–L391.10.1088/0022-3719/9/15/001Search in Google Scholar
Davies, R. H.; Finch, A.; Gardner, P. J.; Hameed, A.; Stephens, M. The standard enthalpies of hydrolysis and formation of tricyanophosphine. J. Chem. Soc., Dalton Trans. 1976, 6, 556–558.10.1039/dt9760000556Search in Google Scholar
Davydova, E. I.; Sevastianova, T. N.; Suvorov, A. V.; Timoshkin, A. Y. Molecular complexes formed by halides of group 4, 5, 13 – 15 elements and the thermodynamic characteristics of their vaporization and dissociation found by the static tensimetric method. Coord. Chem. Rev. 2010, 254 (17–18), 2031–2077.10.1016/j.ccr.2010.04.001Search in Google Scholar
Decius, J. C.; Jacobson, J. L.; Sherman, W. F.; Wilkinson, G. R. Cyanate ion in alkali halides: new vibrational levels and interpretation of localized lattice modes. J. Chem. Phys. 1965, 43 (7), 2180–2186.10.1063/1.1697108Search in Google Scholar
Decker, D. L.; Beyerlein, R. A.; Roult, G.; Worlton, T. G. Neutron diffraction study of KCN III and KCN IV at high pressure. Phys. Rev. B 1974, 10 (8), 3584–3593.10.1103/PhysRevB.10.3584Search in Google Scholar
Deflon, V. M.; Lopes, C. C. de S.; Bessler, K. E.; Romualdo, L. L.; Niquet, E. Preparation, characterization and crystal structure of lead(II) tricyanomethanide. Z. Naturforsch., B: J. Chem. Sci. 2006, 61 (1), 33–36.10.1515/znb-2006-0107Search in Google Scholar
DeFrees, D. J.; McLean, A. D. Molecular orbital predictions of the vibrational frequencies of some molecular ions. J. Chem. Phys. 1985, 82 (1), 333–341.10.1063/1.448805Search in Google Scholar PubMed
Degli Esposti, C.; Favero, P. G.; Serenellini, S.; Cazzoli, G. Rotational spectra of excited vibrational states of F12C15N. Equilibrium structure of cyanogen fluoride. J. Mol. Struct. 1982, 82 (3–4), 221–236.10.1016/0022-2860(82)80037-9Search in Google Scholar
Delbrück, H. Über Das cyan und Paracyan. J. Prakt. Chem. 1847, 41 (1), 161–180.10.1002/prac.18470410123Search in Google Scholar
Delgado, M. S.; Fernandez, V. Die Komplexe von Dioxocyan (OCN)2 Mit TiCl4 und ZrCl4. Z. Anorg. Allg. Chem. 1981, 476 (5), 149–152.10.1002/zaac.19814760517Search in Google Scholar
DeLong, M. C.; Rosenberger, F. High purity cyanides: a dying technology revived. J. Cryst. Growth 1986, 75 (1), 164–172.10.1016/0022-0248(86)90237-XSearch in Google Scholar
Deokar, P.; Leitz, D.; Stein, T. H.; Vasiliu, M.; Dixon, D. A.; Christe, K. O.; Haiges, R. Preparation and characterization of antimony and arsenic tricyanide and their 2,2′‐bipyridine adducts. Chem. Eur J. 2016, 22 (37), 13251–13257.10.1002/chem.201602436Search in Google Scholar PubMed
Derry, T. K.; Williams, T. I. A short history of technology: from the earliest times to A.D. 1900; Dover Publications: New York, 1993.Search in Google Scholar
Desiderato, R.; Sass, R. L. The crystal structure of ammonium tricyanomethide, NH4C(CN)3. Acta Crystallogr. 1965, 18 (1), 1–4.10.1107/S0365110X65000014Search in Google Scholar
Destro, R.; Merati, F.; Ortoleva, E. An ab initio study of the N–Cl bond. Chem. Phys. Lett. 1988, 145 (3), 193–199.10.1016/0009-2614(88)80177-5Search in Google Scholar
Dibeler, V. H.; Liston, S. K. Mass‐spectrometric study of photoionization. IX. Hydrogen cyanide and acetonitrile. J. Chem. Phys. 1968, 48 (10), 4765–4768.10.1063/1.1668059Search in Google Scholar
Diels, O. Zur Kenntnis der Cyanurverbindungen. Ber. Dtsch. Chem. Ges. 1899, 32 (1), 691–702.10.1002/cber.189903201103Search in Google Scholar
Dillard, J. G.; Franklin, J. L. Reactions of negative ions in the gas phase. II. NH2−. J. Chem. Phys. 1968, 48 (5), 2353–2358.10.1063/1.1669436Search in Google Scholar
Dillon, K. B.; Marshall, A. Tin-119 nuclear magnetic resonance studies of some pseudohalogeno-derivatives of [SnX6]2– (X = Cl or Br). J. Chem. Soc., Dalton Trans. 1987, No. 2, 315–317.10.1039/DT9870000315Search in Google Scholar
Dillon, K. B.; Platt, A. W. G. Cyano- and thiocyanato-derivatives of the hexachlorophosphate ion (PCl6–). J. Chem. Soc. Dalton Trans. 1982, No. 7, 1199–1204.10.1039/DT9820001199Search in Google Scholar
Dillon, K. B.; Platt, A. W. G. Cyano- and thiocyanato-derivatives of tetrahalophosphonium ions. Polyhedron 1983, 2 (7), 641–643.10.1016/S0277-5387(00)81526-0Search in Google Scholar
Dillon, K. B.; Platt, A. W. G.; Schmidpeter, A.; Zwaschka, F.; Sheldrick, W. S. Tetrachloro- und Tricyanochlorophosphat(III) Strukturbilder Einer Auf Halbem Wege Stehengebliebenen Addition. Z. Anorg. Allg. Chem. 1982, 488 (1), 7–26.10.1002/zaac.19824880102Search in Google Scholar
Dixon, D. A.; Calabrese, J. C.; Miller, J. S. Crystal and molecular structure of the charge-transfer salt of decamethylferrocenium and tricyanomethanide, [Fe(C5Me5)2]+[C(CN)3]–. The electronic structure and spectra of the tricyanomethanide ion. J. Am. Chem. Soc. 1986, 108 (10), 2582–2588.10.1021/ja00270a015Search in Google Scholar
Dixon, A. ELIX.–A form of tautomerism occurring amongst the thiocyanates of electronegative radicles. J. Chem. Soc. Trans. 1901, 79, 541–552.10.1039/CT9017900541Search in Google Scholar
Dixon, A. E. XVII.–The action of phosphorus trithiocyanate on alcohol. J. Chem. Soc. Trans. 1902, 81 (168), 168–171.10.1039/CT9028100168Search in Google Scholar
do Carmo, L. C. S.; Lüty, F.; Holstein, T.; Orbach, R. Cooperative stretching vibration absorption in alkali cyanides. Phys. Rev. B 1981, 23 (7), 3186–3196.10.1103/PhysRevB.23.3186Search in Google Scholar
Dodonow, J. Bemerkungen Zur Arbeit von E. Zmaczyński: »Über Ein Neues Verfahren Zum Bromieren Und Jodieren von Organischen Verbindungen«. Ber. Dtsch. Chem. Ges. 1926, 59 (9), 2208–2209.10.1002/cber.19260590905Search in Google Scholar
Donald, K. J.; Tawfik, M. The weak helps the strong: sigma-holes and the stability of MF4·base complexes. J. Phys. Chem. 2013, 117 (51), 14176–14183.10.1021/jp407657dSearch in Google Scholar PubMed
Douay, M.; Bernath, P. F. Laser spectroscopy of CaNC and SrNC. Chem. Phys. Lett. 1990, 174 (3–4), 230–234.10.1016/0009-2614(90)85337-CSearch in Google Scholar
Douglas, A. E.; Sharma, D. Rotation‐vibration spectra of diatomic and simple polyatomic molecules with long absorbing paths. IX. The spectra of the HCN and DCN molecules from 2.5 μ to 0.5 μ. J. Chem. Phys. 1953, 21 (3), 448–458.10.1063/1.1698927Search in Google Scholar
Downs, A. W. The isolation of a salt of the tellurocyanate anion. Chem. Commun. (London) 1968, 1290–1291.10.1039/c19680001290Search in Google Scholar
Du, B.; Zhang, W. Theoretical study on the water-assisted reaction of NCO with HCHO. J. Phys. Chem. 2013, 117 (31), 6883–6892.10.1021/jp405687cSearch in Google Scholar PubMed
Du Plessis, C. A.; Barnard, P.; Muhlbauer, R. M.; Naldrett, K. Empirical model for the autotrophic biodegradation of thiocyanate in an activated sludge reactor. Lett. Appl. Microbiol. 2001, 32 (2), 103–107.10.1046/j.1472-765x.2001.00859.xSearch in Google Scholar PubMed
Dua, S.; Blanksby, S. J.; Bowie, J. H. Electron capture of tetracyanoethylene oxide in the gas phase. Rearrangement of the parent radical anion to form [(NC)3C]−. A joint experimental and ab initio study. Int. J. Mass Spectrom. 2000, 194 (2–3), 165–170.10.1016/S1387-3806(99)00124-4Search in Google Scholar
Duboudin, F.; Cazeau, P.; Babot, O.; Moulines, F. Utilisation en synthese organique de MeSi(CN)3 prepare. Tetrahedron Lett. 1983, 24 (40), 4335–4336.10.1016/S0040-4039(00)88334-3Search in Google Scholar
Dulmage, W. J.; Lipscomb, W. N. The crystal structures of hydrogen cyanide, HCN. Acta Crystallogr. 1951, 4 (4), 330–334.10.1107/S0365110X51001070Search in Google Scholar
Dultz, W.; Krause, H. Light-scattering studies of the polymorphism of potassium cyanide under hydrostatic pressure. Phys. Rev. B 1978, 18 (1), 394–400.10.1103/PhysRevB.18.394Search in Google Scholar
Dultz, W.; Otto, H. H.; Krause, H.; Buevoz, J. L. Elastic neutron scattering investigations of new high-pressure phases of KCN. Phys. Rev. B 1981, 24 (3), 1287–1291.10.1103/PhysRevB.24.1287Search in Google Scholar
Durand, D.; do Carmo, L. C. S.; Anderson, A.; Lüty, F. Raman and infrared studies of rotational-translational modes in stress-aligned ferroelastic KCN and NaCN. Phys. Rev. B 1980, 22 (8), 4005–4021.10.1103/PhysRevB.22.4005Search in Google Scholar
Earnshawb, J. C.; Ireland, N. Infrared spectra of (HCN)n clusters. J. Mol. Struct. 1995, 348, 273–276.Search in Google Scholar
Easley, W. C.; Weltner, W. ESR of the CN radical in inert matrices. J. Chem. Phys. 1970, 52 (1), 197–205.10.1063/1.1672665Search in Google Scholar
Ebbs, S. Biological degradation of cyanide compounds. Curr. Opin. Biotechnol. 2004, 15 (3), 231–236.10.1016/j.copbio.2004.03.006Search in Google Scholar PubMed
Edwards, H. G. M.; Fawcett, V. Force constants of phosphorus(III) cyanide and arsenic(III) cyanide. Spectrochim. Acta, Part A 1987, 43 (11), 1345–1348.10.1016/S0584-8539(87)80008-9Search in Google Scholar
Edwards, H. G. M.; Ingman, J. S.; Long, D. A. The Raman spectra of phosphorus and arsenic tricyanides, P(CN)3 and as(CN)3. Spectrochim. Acta, Part A 1976, 32 (4), 731–738.10.1016/0584-8539(76)80141-9Search in Google Scholar
Ehrhardt, K. D.; Press, W.; Lefebvre, J.; Haussühl, S. Lattice dynamics in RbCN. Solid State Commun. 1980, 34 (7), 591–593.10.1016/0038-1098(80)90157-XSearch in Google Scholar
El-Bayoumy, K.; Sinha, R. Mechanisms of mammary cancer chemoprevention by organoselenium compounds. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2004, 551 (1–2), 181–197.10.1016/j.mrfmmm.2004.02.023Search in Google Scholar PubMed
Ellinger, S.; Franke, P.; Harloff, J.; Rijksen, C.; Täschler, C.; Ott, L.; Schulz, A.; Sievert, K.; Stoffers, A.; Zur Täschler, C. Fluoro and chloro cyano compounds of the 15th group, 2014, WO 2014167034 A2, 20141016.Search in Google Scholar
Ellinger, S.; Harloff, J.; Ott, L.; Rijksen, C.; Schulz, A.; Sievert, K. Method for preparation of tricyanidofluoroborates in 2 steps. EP 2772495 A1 20140903: Visp, 2014.Search in Google Scholar
Elliott, N.; Hastings, J. Neutron diffraction investigation of KCN. Acta Crystallogr. 1961, 14 (10), 1018.10.1107/S0365110X61002977Search in Google Scholar
El-Yazal, J.; Martin, J. M. L.; François, J.-P. Structure and vibrations of the C2P and CNP radicals and their cations using density functional and coupled cluster theories. J. Phys. Chem. 1997, 101 (44), 8319–8326.10.1021/jp970614zSearch in Google Scholar
Emerson, K.; Britton, D. The crystal and molecular structure of arsenic tricyanide. Acta Crystallogr. 1963, 16 (2), 113–118.10.1107/S0365110X63000268Search in Google Scholar
Emerson, K.; Britton, D. The crystal and molecular structure of phosphorus tricyanide. Acta Crystallogr. 1964, 17 (9), 1134–1139.10.1107/S0365110X64002948Search in Google Scholar
Emerson, K. The crystal and molecular structure of sulfur dicyanide. Acta Crystallogr. 1966, 21 (6), 970–974.10.1107/S0365110X66004262Search in Google Scholar
Emri, J.; Györi, B. On the existence of isocyanotrihydroborate. Polyhedron 1994, 13 (15–16), 2353–2357.10.1016/S0277-5387(00)88147-4Search in Google Scholar
Enders, E. Process for the production of cyanogen chloride together with cyanuric chloride and tetrameric cyanogen chloride, 1972, 3666427.Search in Google Scholar
Epps, G. D. van.; Reid, E. E. Studies in the preparation of nitriles. II. The preparation of aliphatic nitriles. J. Am. Chem. Soc. 1916, 38 (10), 2120–2128.10.1021/ja02267a022Search in Google Scholar
Ergöçmen, D.; Goicoechea, J. M. Synthesis, structure and reactivity of a cyapho‐cyanamide salt. Angew. Chem. Int. Ed. 2021, 60 (48), 25286–25289.10.1002/anie.202111619Search in Google Scholar PubMed
Essers, R.; Tennyson, J.; Wormer, P. E. S. An SCF potential energy surface for lithium cyanide. Chem. Phys. Lett. 1982, 89 (3), 223–227.10.1016/0009-2614(82)80046-8Search in Google Scholar
Evers, J.; Krumm, B.; Axthammer, Q. J.; Martens, J.; Blaha, P.; Steemann, F. X.; Reith, T.; Mayer, P.; Klapötke, T. M. Molecular structure of isocyanic acid, HNCO, the imide of carbon dioxide. J. Phys. Chem. 2018, 122 (12), 3287–3292.10.1021/acs.jpca.8b00557Search in Google Scholar PubMed
Eyster, E. H.; Gillette, R. H. The vibration spectra of hydrazoic acid, methyl azide, and methyl isocyanate: the thermodynamic functions of hydrazoic acid. J. Chem. Phys. 1940, 8 (5), 369–377.10.1063/1.1750669Search in Google Scholar
Fang, T.; Shimp, D. A. Polycyanate esters: science and applications. Prog. Polym. Sci. 1995, 61–118.10.1016/0079-6700(94)E0006-MSearch in Google Scholar
Fauß, R.; Linker, K.-H.; Findeisen, K. Verfahren Zur Herstellung von Halogencyanen. 1984, 3229416.Search in Google Scholar
Fawcett, F. S.; Lipscomb, R. D. Cyanogen fluoride. J. Am. Chem. Soc. 1960, 82 (6), 1509–1510.10.1021/ja01491a064Search in Google Scholar
Fawcett, F. S.; Lipscomb, R. D. Cyanogen fluoride: synthesis and properties. J. Am. Chem. Soc. 1964, 86 (13), 2576–2579.10.1021/ja01067a011Search in Google Scholar
Feher, F.; Weber, H. Beiträge Zur Chemie des Schwefels, 49. Über die Homologe Reihe der Cyansulfane Sn(CN)2. Chem. Ber. 1958, 91 (3), 642–650.10.1002/cber.19580910326Search in Google Scholar
Feller, D.; Peterson, K. A.; Dixon, D. A. A survey of factors contributing to accurate theoretical predictions of atomization energies and molecular structures. J. Chem. Phys. 2008, 129 (20), 204105.10.1063/1.3008061Search in Google Scholar PubMed
Felsing, W. A.; Drake, G. W. The determination of the heat capacities and the heat capacity ratios of gaseous hydrogen cyanide and of hydrogen sulfide. J. Am. Chem. Soc. 1936, 58 (9), 1714–1717.10.1021/ja01300a063Search in Google Scholar
Ferrario, M.; McDonald, I. R.; Klein, M. L. Anion ordering in alkali cyanide crystals. J. Chem. Phys. 1986, 84 (7), 3975–3985.10.1063/1.450107Search in Google Scholar
Ferus, M.; Kubelík, P.; Kawaguchi, K.; Dryahina, K.; Španěl, P.; Civiš, S. HNC/HCN ratio in acetonitrile, formamide, and BrCN discharge. J. Phys. Chem. 2011, 115 (10), 1885–1899.10.1021/jp1107872Search in Google Scholar PubMed
Fiacco, D. L.; Hunt, S. W.; Leopold, K. R. Structural change at the onset of microsolvation: rotational spectroscopy of HCN···HCN−SO3. J. Phys. Chem. 2000, 104 (36), 8323–8327.10.1021/jp001983pSearch in Google Scholar
Fichter, F.; Richard, S. Über Magnesiumcyanid. Helv. Chim. Acta 1922, 5 (3), 396–400.10.1002/hlca.19220050311Search in Google Scholar
Fichter, F.; Schölly, C. Das Verhalten von Magnesiumnitrid Gegenüber Kohlenoxid und Kohlendioxid. Helv. Chim. Acta 1920, 3 (1), 298–304.10.1002/hlca.19200030128Search in Google Scholar
Fiedler, S. L.; Vaskonen, K.; Ahokas, J.; Kunttu, H.; Eloranta, J.; Apkarian, V. A. Host–guest charge transfer states: CN doped Kr and Xe. J. Chem. Phys. 2002, 117 (19), 8867–8878.10.1063/1.1513463Search in Google Scholar
Fieser, L. F.; Fieser, M.; Ho, T. Fieser and Fieser’s reagents for organic synthesis; Wiley: Hoboken, 2006.10.1002/9780471264194Search in Google Scholar
Finger, L. H.; Sundermeyer, J. Halide-free synthesis of hydrochalcogenide ionic liquids of the type [cation][HE] (E=S, Se, Te). Chem. Eur J. 2016, 22 (12), 4218–4230.10.1002/chem.201504577Search in Google Scholar PubMed
Fitzgerald, M.; Bowie, J. H. The generation of the thiocyanate radical and cation from the thiocyanate anion [SCN]– in the gas phase. The rearrangements of neutral and cationic SCN. A joint experimental and theoretical study. J. Phys. Chem. 2004, 108 (16), 3668–3674.10.1021/jp0311308Search in Google Scholar
Fitzsimmons, A.; Klobukowski, M. Structure and stability of organic molecules containing heavy rare gas atoms. Theor. Chem. Acc. 2013, 132 (2), 1314.10.1007/s00214-012-1314-8Search in Google Scholar
Flemming, A.; Hoffmann, M.; Köckerling, M. Tetracyanidoborates with sterically demanding phosphonium cations – thermally resistant lonic liquids. Z. Anorg. Allg. Chem. 2010, 636 (3–4), 562–568.10.1002/zaac.200900469Search in Google Scholar
Flores, J. R. A. Theoretical study of the excited electronic states of the SiCN system. Chem. Phys. 2005, 310 (1–3), 303–310.10.1016/j.chemphys.2004.11.008Search in Google Scholar
Fluck, E.; Goldmann, F. L.; Rümpler, K.-D. Pseudohalogenide nichtmetallischer elemente. II. Phosphor(III)-isothiocyanat und phosphor(III)-isocyanat die systeme PCl3/P(NCS)3, PBr3/P(NCS)3 und PCl3/P(NCO)3. Z. Anorg. Allg. Chem. 1965, 338 (1–2), 52–57.10.1002/zaac.19653380109Search in Google Scholar
Fontaine, D. Structure Du cyanure de Sodium en Phase Ordonnee. C. R. Seances Acad. Sci., Ser. B 1975, 281, 443–444.Search in Google Scholar
Forney, D.; Thompson, W. E.; Jacox, M. E. The vibrational spectra of molecular ions isolated in solid neon. IX. HCN+, HNC+, and CN−. J. Chem. Phys. 1992, 97 (3), 1664–1674.10.1063/1.463963Search in Google Scholar
Forthomme, D.; McRaven, C. P.; Sears, T. J.; Hall, G. E. Collinear two-color saturation spectroscopy in CN A–X (1–0) and (2–0) bands. J. Mol. Spectrosc. 2014, 296, 36–42.10.15278/isms.2014.TJ15Search in Google Scholar
Fowler, P. W.; Legon, A. C.; Peebles, S. A. Rotational spectrum of HCN···HI and a comparison of properties in the series HCN···HX (X = F, Cl, Br, I and CN). Chem. Phys. Lett. 1994, 226 (5–6), 501–508.10.1016/0009-2614(94)00760-8Search in Google Scholar
Fox, J. L.; Yelle, R. V. Hydrocarbon ions in the ionosphere of Titan. Geophys. Res. Lett. 1997, 24 (17), 2179–2182.10.1029/97GL02051Search in Google Scholar
Franck, H. H.; Freitag, C. Die Darstellung von Calciumcyanid. Angew. Chem. 1926, 39 (47), 1430–1432.10.1002/ange.19260394706Search in Google Scholar
Franklin, E. C. The nitrogen system of compounds; Reinhold Publishing Cooperation: ACS Monograph Series: New York, 1935.10.1021/j150366a034Search in Google Scholar
Freitag, W. O.; Nixon, E. R. Infrared spectra of gaseous and crystalline cyanogen halides. J. Chem. Phys. 1956, 24 (1), 109–114.10.1063/1.1700809Search in Google Scholar
Frenking, G.; Schwarz, H. HCN/HNC versus HCN+/HNC+ - Zur Ursache der Stabilitätsumkehr Beim Übergang von Den Neutralmolekülen Zu den Radikalkationen. Sci. Nat. 1982, 69 (9), 446–447.10.1007/BF00404767Search in Google Scholar
Freudenmann, D.; Wolf, S.; Wolff, M.; Feldmann, C. Ionic liquids: new perspectives for inorganic synthesis? Angew. Chem. Int. Ed. 2011, 50 (47), 11050–11060.10.1002/anie.201100904Search in Google Scholar PubMed
Fridh, C.; Åsbrink, L. Photoelectron and electron impact spectrum of HCN. J. Electron Spectrosc. 1975, 7 (2), 119–138.10.1016/0368-2048(75)80045-4Search in Google Scholar
Fritz, H. P.; Keller, H. Spektroskopische Untersuchungen an Organometallischen Verbindungen, V. Infrarot-spektren von Organotellurverbindungen. Chem. Ber. 1961, 94 (6), 1524–1533.10.1002/cber.19610940616Search in Google Scholar
Fritz, S.; Lentz, D.; Szwak, M. Synthesis and structure determination of selenium(IV) cyanides. Eur. J. Inorg. Chem. 2008, 2008 (30), 4683–4686.10.1002/ejic.200800712Search in Google Scholar
Frölich, R.; Tebbe, K.-F. Untersuchungen an Polyhalogeniden. 2. Cesiumdicyanotriiodid. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982, 38 (1), 71–75.10.1107/S0567740882002076Search in Google Scholar
Fuith, A. The KSCN family: structural properties and phase transitions of crystals with three-atomic linear anions. Phase Transitions 1997, 62 (1–2), 1–93.10.1080/01411599708220061Search in Google Scholar
Fukushima, M.; Ishiwata, T. Vibrationally hot bands of the SiCN Ã2Δ – X̃2Π system. J. Phys. Chem. 2013, 117 (39), 9435–9443.10.1021/jp311684eSearch in Google Scholar PubMed
Fukushima, M. Laser induced fluorescence spectroscopy of AlNC/AlCN in supersonic free expansions. Chem. Phys. Lett. 1998, 283 (5–6), 337–344.10.1016/S0009-2614(97)01408-5Search in Google Scholar
Fulara, J.; Klapstein, D.; Kuhn, R.; Maier, J. P. Emission spectra of supersonically cooled halocyanide cations, XCN+ (X = Cl, Br, I): Ã2Σ+ → X̃2Π and B2Π → X̃2Π band systems. J. Phys. Chem. 1985, 89 (20), 4213–4219.10.1021/j100266a014Search in Google Scholar
Gail, E.; Gos, S.; Kulzer, R.; Lorösch, J.; Rubo, A.; Sauer, M. Cyano compounds, inorganic. In Ullmann’s encyclopedia of industrial chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004.10.1002/14356007.a08_159.pub2Search in Google Scholar
Gall, H.; Schüppen, J. Über die Valenz-Grenze Bei Phosphorcyaniden und Phosphorrhodaniden. Ber. Dtsch. Chem. Ges. 1930, 63 (2), 482–487.10.1002/cber.19300630237Search in Google Scholar
Galliart, A.; Brown, T. M. Ammonium and tetraalkylammonium cyanates. Synth. React. Inorg. Met.-Org. Chem. 1972, 2 (4), 273–275.10.1080/00945717208069612Search in Google Scholar
Gao, H.; Joo, Y.-H.; Twamley, B.; Zhou, Z.; Shreeve, J. M. Hypergolic ionic liquids with the 2,2-dialkyltriazanium cation. Angew. Chem. Int. Ed. 2009, 48 (15), 2792–2795.10.1002/anie.200900094Search in Google Scholar PubMed
Garand, E.; Yacovitch, T. I.; Neumark, D. M. Slow photoelectron velocity-map imaging spectroscopy of C2N−, C4N−, and C6N−. J. Chem. Phys. 2009, 130 (6), 064304–064310.10.1063/1.3076320Search in Google Scholar PubMed
Gardner, P. J.; Preston, S. R.; Siertsema, R.; Steele, D. Ab initio molecular orbital studies for compounds of magnesium. J. Comput. Chem. 1993, 14 (12), 1523–1533.10.1002/jcc.540141215Search in Google Scholar
Gauld, J. W.; Eriksson, L. A.; Radom, L. Assessment of procedures for calculating radical hyperfine structures. J. Phys. Chem. 1997, 101 (7), 1352–1359.10.1021/jp961328aSearch in Google Scholar
Gay-Lussac, L. J. Recherches Sur l’acide Prussique. Ann. Chim. Phys. 1815, 95, 136–231.Search in Google Scholar
Geier, J.; Willner, H. Tris(trifluoromethyl)borane adducts of cyanogen halides. Z. Anorg. Allg. Chem. 2008, 634 (11), 1863–1866.10.1002/zaac.200800227Search in Google Scholar
Geller, S.; Schawlow, A. L. Crystal structure and quadrupole coupling of cyanogen bromide, BrCN. J. Chem. Phys. 1955, 23 (5), 779–783.10.1063/1.1742121Search in Google Scholar
George, J.; Deringer, V. L.; Dronskowski, R. Cooperativity of halogen, chalcogen, and pnictogen bonds in infinite molecular chains by electronic structure theory. J. Phys. Chem. 2014, 118 (17), 3193–3200.10.1021/jp5015302Search in Google Scholar PubMed
George, J.; Deringer, V. L.; Dronskowski, R. Dimensionality of intermolecular interactions in layered crystals by electronic-structure theory and geometric analysis. Inorg. Chem. 2015, 54 (3), 956–962.10.1021/ic5023328Search in Google Scholar PubMed
Georgieva, M. K.; Binev, I. G. The computational study on the structural and force field changes, caused by the conversion of dicyanamide into the azanion. J. Mol. Struct. 2005, 752 (1–3), 14–19.10.1016/j.molstruc.2005.05.014Search in Google Scholar
Georgieva, M. K.; Velcheva, E. A. Computational and experimental studies on the IR spectra and structure of the simplest nitriles (C1 and C2), their anions, and radicals. Int. J. Quantum Chem. 2006, 106 (6), 1316–1322.10.1002/qua.20887Search in Google Scholar
Gerasimov, I.; Yang, X.; Dagdigian, P. J. Laser fluorescence excitation spectra of the AlNC and AlCN isomers. J. Chem. Phys. 1999, 110 (1), 220–228.10.1063/1.478097Search in Google Scholar
Gettler, A. O.; Baine, J. O. The toxicology of cyanide. Am. J. Med. Sci. 1938, 195 (2), 182–197.10.1097/00000441-193802000-00007Search in Google Scholar
Giauque, W. F.; Ruehrwein, R. A. The entropy of hydrogen cyanide. Heat capacity, heat of vaporization and vapor pressure. hydrogen bond polymerization of the gas in chains of indefinite length. J. Am. Chem. Soc. 1939, 61 (10), 2626–2633.10.1021/ja01265a017Search in Google Scholar
Glasnov, T. N.; Holbrey, J. D.; Kappe, C. O.; Seddon, K. R.; Yan, T. Methylation using dimethylcarbonate catalysed by ionic liquids under continuous flow conditions. Green Chem. 2012, 14 (11), 3071–3076.10.1039/c2gc36226kSearch in Google Scholar
Gleiter, R.; Hyla-Kryspin, I.; Pfeifer, K.-H. On the stability of the tetramers of carbon monoxide, hydrogen isocyanide, and vinylidene. a molecular orbital theoretical rationalization. J. Org. Chem. 1995, 60 (18), 5878–5883.10.1021/jo00123a025Search in Google Scholar
Glidewell, C.; Thomson, C. Ab initio calculations on the effect of different basis sets and electron correlation on the transition state for the reactions HNC ⇌ HCN and BCN ⇌ BNC. J. Comput. Chem. 1984, 5 (1), 1–10.10.1002/jcc.540050102Search in Google Scholar
Glidewell, C. Jahn-Teller effects in the MNDO approximation: structures of the molecular cations of some simple organosilanes. J. Organomet. Chem. 1981, 217 (1), 11–18.10.1016/S0022-328X(00)86018-0Search in Google Scholar
Glockler, G.; Baker, H. T. Raman line of cyanide ion. J. Chem. Phys. 1942, 10 (5), 305–306.10.1063/1.1723727Search in Google Scholar
Glöcklhofer, F.; Lunzer, M.; Fröhlich, J. Facile synthesis of cyanoarenes from quinones by reductive aromatization of cyanohydrin intermediates. Synlett 2015, 26 (07), 950–952.10.1055/s-0034-1380150Search in Google Scholar
Goel, R. G.; Prasad, H. S. Organobismuth compounds. V. Preparation, characterization, and properties of triphenylbismuth diazide and dicyanide. J. Organomet. Chem. 1973, 50 (1), 129–134.10.1016/S0022-328X(00)95098-8Search in Google Scholar
Goggin, P. L.; McColm, I. J.; Shore, R. The oxyhalides of indium. J. Chem. Soc. A 1966a, 1004–1008.10.1039/j19660001004Search in Google Scholar
Goggin, P. L.; McColm, I. J.; Shore, R. Indium tricyanide and indium trithiocyanate. J. Chem. Soc. A 1966b, 1314–1317.10.1039/j19660001314Search in Google Scholar
Goubeau, J.; Jahn, E. L.; Kreutzberger, A.; Grundmann, C., Triazines, X. The infrared and Raman spectra of 1,3,5-triazine. J. Phys. Chem. 1954, 58 (12), 1078–1081.10.1021/j150522a008Search in Google Scholar
Gracia, R.; Shepherd, G. Cyanide poisoning and its treatment. Pharmacotherapy 2004, 24 (10), 1358–1365.10.1592/phco.24.14.1358.43149Search in Google Scholar
Grakauskas, V. Method for preparation of cyanogen fluoride. 3532475, 1970.Search in Google Scholar
Graninger, D. M.; Herbst, E.; Öberg, K. I.; Vasyunin, A. I. The HNC/HCN ratio in star-forming regions. Astrophys. J. 2014, 787 (1), 74.10.1088/0004-637X/787/1/74Search in Google Scholar
Grant Hill, J.; Mitrushchenkov, A.; Yousaf, K. E.; Peterson, K. A. Accurate ab initio ro-vibronic spectroscopy of the X̃2Π CCN radical using explicitly correlated methods. J. Chem. Phys. 2011, 135 (14), 144309–144320.10.1063/1.3647311Search in Google Scholar
Greenwood, N. N.; Little, R.; Sprague, M. J. The tellurocyanate ion, TeCN–. J. Chem. Soc. 1964, 1292–1295.10.1039/jr9640001292Search in Google Scholar
Greenwood, N. N.; Earnshaw, A.; Hückmann, K. Chemie Der Elemente, 1st ed.; Wiley VCH: Weinheim, 1988.Search in Google Scholar
Greetham, G. M.; Ellis, A. M. Ultraviolet laser spectroscopy of jet-cooled CaNC and SrNC free radicals: observation of bent excited electronic states. J. Chem. Phys. 2000, 113 (20), 8945–8952.10.1063/1.1319344Search in Google Scholar
Grenier-Loustalot, M. F.; Lartigau, C.; Metras, F.; Grenier, P. Mechanism of thermal polymerization of cyanate ester systems: chromatographic and spectroscopic studies. J. Polym. Sci., Part A: Polym. Chem. 1996, 34 (14), 2955–2966.10.1002/(SICI)1099-0518(199610)34:14<2955::AID-POLA13>3.0.CO;2-JSearch in Google Scholar
GrisleyJrD.; Gluesenkamp, E.; Heininger, S. Notes – reactions of nucleophilic reagents with cyanuric fluoride and cyanuric chloride. J. Org. Chem. 1958, 23 (11), 1802–1804.10.1021/jo01105a620Search in Google Scholar
Gröger, H.; Asano, Y. Cyanide-free enantioselective catalytic strategies for the synthesis of chiral nitriles. J. Org. Chem. 2020, 85 (10), 6243–6251.10.1021/acs.joc.9b02773Search in Google Scholar PubMed
Groß, S.; Laabs, S.; Scherrmann, A.; Sudau, A.; Zhang, N.; Nubbemeyer, U. Improved syntheses of cyanuric fluoride and carboxylic acid fluorides. J. Prakt. Chem. 2000, 342 (7), 711–714.10.1002/1521-3897(200009)342:7<711::AID-PRAC711>3.0.CO;2-MSearch in Google Scholar
Groutas, W. C. T-butyldimethylsilyl cyanide. e-EROS Encycl. Reagents Org. Synth. 2001, 1 (4), 1–6.10.1002/047084289X.rb374Search in Google Scholar
Grundmann, C.; Beyer, E. Triazines. VII. The reaction of cyanuric chloride with lithium aluminum hydride. J. Am. Chem. Soc. 1954, 76 (7), 1948–1949.10.1021/ja01636a065Search in Google Scholar
Grundmann, C.; Fulton, M. B. Zur Kenntnis des Cyanformaldehyds. Chem. Ber. 1964, 97 (2), 566–574.10.1002/cber.19640970237Search in Google Scholar
Grundmann, C.; Kreutzberger, A. Triazines. IX. 1,3,5-triazine and its formation from hydrocyanic acid. J. Am. Chem. Soc. 1954, 76 (22), 5646–5650.10.1021/ja01651a013Search in Google Scholar
Grundmann, C.; Kreutzberger, A. Triazines. XIII. The ring cleavage of s-triazine by primary amines. A new method for the synthesis of heterocycles. J. Am. Chem. Soc. 1955, 77 (24), 6559–6562.10.1021/ja01629a041Search in Google Scholar
Grundmann, C.; Kober, E. Triazines. XVII. s-Triazine from s-triazine-2,4,6-tricarbonic acid. J. Org. Chem. 1956, 21 (12), 1392–1394.10.1021/jo01118a017Search in Google Scholar
Guélin, M.; Cernicharo, J.; Kahane, C.; Gomez-Gonzalez, J. A new free radical in IRC + 10216. Astron. Astrophys. 1986, 157 (2), 17–20.Search in Google Scholar
Guélin, M.; Muller, S.; Cernicharo, J.; McCarthy, M. C.; Thaddeus, P. Detection of the SiNC radical in IRC+10216. Astron. Astrophys. 2004, 426 (2), 49-L52.10.1051/0004-6361:200400074Search in Google Scholar
Guenez, M. E. Sur Le Preparation et Les Proprietes Du Cyanure d’arsenic. C. R. Hebd. Seances Acad. Sci. 1892, 114, 1186–1189.Search in Google Scholar
Guennoun, Z.; Couturier-Tamburelli, I.; Piétri, N.; Aycard, J. P. UV photoisomerisation of cyano and dicyanoacetylene: the first identification of CCNCH and CCCNCN isomers – matrix isolation, infrared and ab initio study. Chem. Phys. Lett. 2003, 368 (5–6), 574–583.10.1016/S0009-2614(02)01898-5Search in Google Scholar
Guernsey, E. W.; Sherman, M. S. The thermal dissociation of sodium cyanide. J. Am. Chem. Soc. 1926, 48 (3), 695–704.10.1021/ja01414a022Search in Google Scholar
Guerra, M. Role of standard diffuse functions for computing hyperfine splitting constants in radical anions. J. Phys. Chem. 1999, 103 (30), 5983–5988.10.1021/jp990012aSearch in Google Scholar
Günther, P.; Meyer, R.; Müller-Skjold, F. Zur Thermochemie der Stickstoffwasserstoffsäure. Z. Phys. Chem. 1935, 175 (1), 154–169.10.1515/zpch-1935-17510Search in Google Scholar
Guo, B.; Pasinszki, T.; Westwood, N. P. C.; Zhang, K.; Bernath, P. F. High resolution infrared spectroscopy of cyanogen N‐oxide, NCCNO. J. Chem. Phys. 1996, 105 (11), 4457–4460.10.1063/1.472297Search in Google Scholar
Gutowsky, H. S.; Germann, T. C.; Augspurger, J. D.; Dykstra, C. E. Structure and dynamics of the H2O–HCN dimer. J. Chem. Phys. 1992, 96 (8), 5808–5816.10.1063/1.462680Search in Google Scholar
Gutsev, G. L.; Boldyrev, A. I. DVM-xα calculations on the ionization potentials of MXk+1− complex anions and the electron affinities of MXk+1 “Superhalogens”. Chem. Phys. 1981, 56 (3), 277–283.10.1016/0301-0104(81)80150-4Search in Google Scholar
Haas, A. Kovalente Kohlenstoffverbindungen. In Handbuch der präparativen anorganischen Chemie in drei Bänden; Ferdinand Enke Verlag: Stuttgart, 1978; pp. 629–630.Search in Google Scholar
Hackspill, L.; Grandadam, R. Contribution to the study of some potassium and sodium salts. Ann. Chim. 1925, 5, 218–250.Search in Google Scholar
Hagiwara, Y.; Ochi, T.; Ohata, K.; Kasahara, T.; Toba, T.; Mizuta, K.; Katsuyama, H.; Ishida, S.; Nishida, T. Ionic compound, method for producing the same, and ion-conductive material comprising the same. US2011150736A1, 2011.Search in Google Scholar
Haiges, R.; Schneider, S.; Schroer, T.; Christe, K. O. High-energy-density materials: synthesis and characterization of N5+[P(N3)6]−, N5+[B(N3)4]−, N5+[HF2]−⋅n HF, N5+[BF4]−, N5+ [PF6]−, and N5+[SO3F]−. Angew. Chem. Int. Ed. 2004, 43 (37), 4919–4924.10.1002/anie.200454242Search in Google Scholar PubMed
Hajgató, B.; Flammang, R.; Veszprémi, T.; Nguyen, M. T. Experimental and theoretical study of dicyanocarbene C(CN)2. Mol. Phys. 2002, 100 (11), 1693–1702.10.1080/00268970110101356Search in Google Scholar
Hakuta, K.; Uehara, H. Laser‐induced fluorescence spectrum of the CCN radical with an Ar+ laser. J. Chem. Phys. 1983, 78 (11), 6484–6489.10.1063/1.444687Search in Google Scholar
Halfen, D. T.; Sun, M.; Clouthier, D. J.; Ziurys, L. M. The microwave and millimeter rotational spectra of the PCN radical (X̃3Σ−). J. Chem. Phys. 2012, 136 (14), 144312.10.1063/1.3696893Search in Google Scholar
Hall, A. H.; Dart, R.; Bogdan, G. Sodium thiosulfate or hydroxocobalamin for the empiric treatment of cyanide poisoning? Ann. Emerg. Med. 2007, 49 (6), 806–813.10.1016/j.annemergmed.2006.09.021Search in Google Scholar
Hamerton, I.; Hay, J. N. Recent developments in the chemistry of cyanate esters. Polym. Int. 1998, 47 (4), 465–473.10.1002/(SICI)1097-0126(199812)47:4<465::AID-PI88>3.0.CO;2-SSearch in Google Scholar
Han, J.; Heaven, M. C.; Schnupf, U.; Alexander, M. H. Experimental and theoretical studies of the CN–Ar van Der Waals complex. J. Chem. Phys. 2008a, 128 (10), 104308.10.1063/1.2834932Search in Google Scholar
Han, J.; Heaven, M. C.; Schnupf, U. Spectroscopy, dissociation dynamics, and potential energy surfaces for CN(A)−Ar. J. Chem. Phys. 2008b, 128 (22), 224309.10.1063/1.2936123Search in Google Scholar
Hanawalt, J. D.; Rinn, H. W.; Frevel, L. K. Chemical analysis by X-ray diffraction. Ind. Eng. Chem., Anal. Ed. 1938, 10 (9), 457–512.10.1021/ac50125a001Search in Google Scholar
Hand, C. W.; Hexter, R. M. Flash Photolysis of ozone-cyanogen mixtures. J. Am. Chem. Soc. 1970, 92 (7), 1828–1831.10.1021/ja00710a005Search in Google Scholar
Hantzsch, A.; Mai, L. Über Imidokohlensäureether und die sogen. normalen Cyansäureether. Ber. Dtsch. Chem. Ges. 1895, 28 (3), 2466–2472.10.1002/cber.18950280328Search in Google Scholar
Hantzsch, A.; Osswald, G. Über cyanoform. Ber. Dtsch. Chem. Ges. 1899, 32 (1), 641–650.10.1002/cber.18990320193Search in Google Scholar
Happold, F. C.; Jones, G. L.; Pratt, D. B. Utilization of thiocyanate by thiobacillus thioparus and t. thiocyanoxidans. Nature 1958, 182 (4630), 266–267.10.1038/182266a0Search in Google Scholar
Harloff, J.; Schulz, A.; Stoer, P.; Villinger, A. Pseudo halide chemistry in ionic liquids with decomposable anions. Z. Anorg. Allg. Chem. 2019a, 645 (12), 835–839.10.1002/zaac.201900055Search in Google Scholar
Harloff, J.; Michalik, D.; Nier, S.; Schulz, A.; Stoer, P.; Villinger, A. Cyanidosilicates – synthesis and structure. Angew. Chem. Int. Ed. 2019b, 58 (16), 5452–5456.10.1002/anie.201901173Search in Google Scholar PubMed
Harloff, J.; Laatz, K. C.; Lerch, S.; Schulz, A.; Stoer, P.; Strassner, T.; Villinger, A. Hexacyanidosilicates with functionalized imidazolium counterions. Eur. J. Inorg. Chem. 2020a, 2020 (25), 2457–2464.10.1002/ejic.202000281Search in Google Scholar
Harloff, J.; Schulz, A.; Stoer, P.; Villinger, A. Pseudohalide HCN aggregate ions: [N3(HCN)3]−, [OCN(HCN)3]−, [SCN(HCN)2]− and [P(CN·HCN)2]−. Dalton Trans. 2020b, 49 (38), 13345–13351.10.1039/D0DT02973DSearch in Google Scholar PubMed
Harrison, P. G.; Stobart, S. R. Derivatives of divalent germanium, tin, and lead. Part I. The protolysis of cyclopentadienyltin(II) compounds by hydroxy-derivatives. Tin(II) Oximes and hydroxylpmines. J. Chem. Soc., Dalton Trans. 1973, 9, 940–943.10.1039/dt9730000940Search in Google Scholar
Harrison, P. G. Inorganic tin(II) derivatives. J. Chem. Soc., Chem. Commun. 1972, No. 9, 544.10.1039/c39720000544Search in Google Scholar
Hartman, W. W.; Dreger, E. E. Cyanogen bromide. Org. Synth. 1931, 11, 30.10.1002/0471264180.os011.09Search in Google Scholar
Hartwig, A. Gesundheitsschädliche Arbeitsstoffe - Toxikologisch-Arbeitsmedizinische Begründungen von MAK-Werten; Wiley VCH: Weinheim, 1972.Search in Google Scholar
Hauge, S.; Kimmel, E. C.; Holm, B.; Widmark, G.; Koskikallio, J.; Kachi, S. Syntheses and crystal data on salts of the triselenocyanate and seleniumtriselenocyanate ions. Acta Chem. Scand. 1971a, 25, 3081–3093.10.3891/acta.chem.scand.25-3081Search in Google Scholar
Hauge, S.; Sletten, J.; Rasmussen, S. E.; Svensson, S.; Koskikallio, J.; Kachi, S. The crystal structure of potassium triselenocyanate hemihydrate. Acta Chem. Scand. 1971b, 25, 3094–3102.10.3891/acta.chem.scand.25-3094Search in Google Scholar
Hauge, S.; Hordvik, A.; Sæthre, L. J.; Andresen, A. F.; Southern, J. T.; Edlund, K.; Eliasen, M.; Herskind, C.; Laursen, T.; Pedersen, P. M. The crystal structure of cesium triselenocyanate. Acta Chem. Scand. 1975, 29a, 163–169.10.3891/acta.chem.scand.29a-0163Search in Google Scholar
Hausmann, A. Elektronenspin-resonanz in Alkalihalogenid-Kristallen Mit Schwefel- und Selenzusätzen. Z. Phys. 1966, 192 (3), 313–328.10.1007/BF01325806Search in Google Scholar
Hazell, A. C. The crystal structures of selenium dicyanide and sulphur dicyanide. Acta Crystallogr. 1963, 16 (8), 843–844.10.1107/S0365110X63002206Search in Google Scholar
Heaven, M. Spectroscopy and dynamics of open-shell van der Waals molecules. Annu. Rev. Phys. Chem. 1992, 43 (1), 283–310.10.1146/annurev.pc.43.100192.001435Search in Google Scholar
Heckathorn, J. W.; Kruger, M. B.; Gerlich, D.; Jeanloz, R. High-pressure behavior of the alkali cyanides KCN and NaCN. Phys. Rev. B 1999, 60 (2), 979–983.10.1103/PhysRevB.60.979Search in Google Scholar
Heiart, R. B.; Carpenter, G. B. The crystal structure of cyanogen chloride. Acta Crystallogr. 1956, 9 (11), 889–895.10.21236/AD0081100Search in Google Scholar
Heilos, J.; Heimberger, W.; Lüssling, T.; Weigert, W. Verfahren Zur Herstellung von Chlorcyan. 2027957, 1971.Search in Google Scholar
Heimberger, W.; Schreyer, G. Process for the production of cyanogen chloride or cyanogen bromide. 1525343, 1975.Search in Google Scholar
Hemple, S.; Nixon, E. R. Infrared spectrum of cyanogen iodide vapor. J. Chem. Phys. 1967, 47 (10), 4273–4274.10.1063/1.1701619Search in Google Scholar
Henderson, T. J.; Cullinan, D. B. Purity analysis of hydrogen cyanide, cyanogen chloride and phosgene by quantitative 13C NMR spectroscopy. Magn. Reson. Chem. 2007, 45 (11), 954–961.10.1002/mrc.2081Search in Google Scholar PubMed
Hendricks, S. B.; Pauling, L. The crystal structures of sodium and potassium trinitrides and potassium cyanate and the nature of the trinitride group. J. Am. Chem. Soc. 1925, 47 (12), 2904–2920.10.1021/ja01689a008Search in Google Scholar
Henninger, A. A. Henninger, Aus Paris, 7. 1876. Ber. Dtsch. Chem. Ges. 1876, 9 (2), 1430–1435.10.1002/cber.187600902112Search in Google Scholar
Hennings, E.; Schmidt, H.; Voigt, W. Structure and thermal properties of lithium cyanate. Z. Anorg. Allg. Chem. 2011, 637 (9), 1199–1202.10.1002/zaac.201100081Search in Google Scholar
Henretig, F. M.; Kirk, M. A.; McKay, C. A. Hazardous chemical emergencies and poisonings. N. Engl. J. Med. 2019, 380 (17), 1638–1655.10.1056/NEJMra1504690Search in Google Scholar PubMed
Herbst, E.; Klemperer, W. The formation and depletion of molecules in dense interstellar clouds. Astrophys. J. 1973, 185 (9), 505–533.10.1086/152436Search in Google Scholar
Henriques, A. B.; von der Weid, J. P. Decay kinetics of triplet molecular excitons in alkali cyanides. Phys. Status Solidi B 1985, 132(2), 627–633.10.1515/9783112495445-036Search in Google Scholar
Herbst, E. Chemistry in the interstellar medium. Annu. Rev. Phys. Chem. 1995, 46 (1), 27–54.10.1146/annurev.pc.46.100195.000331Search in Google Scholar
Hering, C.; von Langermann, J.; Schulz, A. The elusive cyanoformate: an unusual cyanide shuttle. Angew. Chem. Int. Ed. 2014, 53 (32), 8282–8284.10.1002/anie.201405339Search in Google Scholar PubMed
Herz, W.; Neukirch, E. Die Quantitative Bestimmung des Bleis Als Cyanid. Z. Anorg. Allg. Chem. 1923, 130 (1), 343–344.10.1002/zaac.19231300130Search in Google Scholar
Hester, R. E.; Lee, K. M.; Mayer, E. Tetracyanomethane as a pseudo-(carbon tetrahalide). J. Phys. Chem. 1970, 74 (18), 3373–3376.10.1021/j100712a011Search in Google Scholar
Hinks, D. G.; Price, D. L.; Rowe, J. M.; Susman, S. Single crystal growth of sodium cyanide and potassium cyanide. J. Cryst. Growth 1972, 15 (3), 227–230.10.1016/0022-0248(72)90122-4Search in Google Scholar
Hipps, K. W.; Aplin, A. T. The tricyanomethanide ion: an infrared, raman, and tunneling spectroscopy study including isotopic substitution. J. Phys. Chem. 1985, 89 (25), 5459–5464.10.1021/j100271a029Search in Google Scholar
Ho, W.-C.; Blom, C. E.; Liu, D.-J.; Oka, T. The infrared v5 band (HNC bend) of protonated hydrogen cyanide, HCNH+. J. Mol. Spectrosc. 1987, 123 (1), 251–253.10.1016/0022-2852(87)90275-XSearch in Google Scholar
Ho, T.-L.; Fieser, M.; Fieser, L.; Danheiser, R.; Roush, W. Fieser and Fieser’s reagents for organic synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2006.10.1002/9780471264194Search in Google Scholar
Hoffman, R. E.; Hornig, D. F. The infra‐red spectrum of solid hydrogen cyanide. J. Chem. Phys. 1949, 17 (11), 1163.10.1063/1.1747132Search in Google Scholar
Hoffmann, R.; Zeiss, G. D.; Van Dine, G. W. The electronic structure of methylenes. J. Am. Chem. Soc. 1968, 90 (6), 1485–1499.10.1021/ja01008a017Search in Google Scholar
Holbrey, J. D.; Rogers, R. D.; Shukla, S. S.; Wilfred, C. D. Optimised microwave-assisted synthesis of methylcarbonate salts: a convenient methodology to prepare intermediates for ionic liquid libraries. Green Chem. 2010, 12 (3), 407–413.10.1039/B918713HSearch in Google Scholar
Holland, D. M.; Powis, I.; Karlsson, L.; Trofimov, A.; Schirmer, J.; von Niessen, W. A study of the photoionisation dynamics of the cyanogen halides. Chem. Phys. 2004, 297 (1–3), 55–73.10.1016/j.chemphys.2003.09.027Search in Google Scholar
Hollas, J. M.; Sutherley, T. A. Geometry of cyanogen halide positive ions from photoelectron spectroscopy. Mol. Phys. 1971, 22 (2), 213–223.10.1080/00268977100102491Search in Google Scholar
Hörst, S. M.; Yelle, R. V.; Buch, A.; Carrasco, N.; Cernogora, G.; Dutuit, O.; Quirico, E.; Sciamma-O’Brien, E.; Smith, M. A.; Somogyi, A.; Szopa, C.; Thissen, R.; Vuitton, V. Formation of amino acids and nucleotide bases in a Titan atmosphere simulation experiment. Astrobiology 2012, 12 (9), 809–817.10.1089/ast.2011.0623Search in Google Scholar PubMed PubMed Central
Huber, R. E.; Criddle, R. S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122 (1), 164–173.10.1016/0003-9861(67)90136-1Search in Google Scholar PubMed
Hübner, H.; Wehrhane, G. Über eine Verbindung des Cyans Mit Phosphor. Justus Liebigs Ann. Chem. 1863, 128 (2), 254–256.10.1002/jlac.18631280218Search in Google Scholar
Huthmacher, K.; Most, D. Cyanuric acid and cyanuric chloride. In Ullmann’s encyclopedia of industrial chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000.10.1002/14356007.a08_191Search in Google Scholar
Hvastijová, M.; Kohout, J.; Kožíšek, J.; Svoboda, I. X-ray crystallographic evidence for addition of water to nitrosodicyanomethanide in the 3d-metal coordination sphere. J. Coord. Chem. 1999, 47 (4), 573–579.10.1080/00958979908024356Search in Google Scholar
Ingold, C. K. CVI. The form of the vapour pressure curve at high temperatures. Part II. The curve for sodium cyanide. J. Chem. Soc. Trans. 1923, 123, 885–891.10.1039/CT9232300885Search in Google Scholar
Iqbal, Z. Vibrational spectrum of single crystal sodium thiocyanate. J. Mol. Struct. 1971, 7 (1–2), 137–146.10.1016/0022-2860(71)90014-7Search in Google Scholar
Irran, E.; Jürgens, B.; Schnick, W. Trimerization of alkali dicyanamides M[N(CN)2] and formation of tricyanomelaminates M3[C6N9] (M = K, Rb) in the melt: crystal structure determination of three polymorphs of K[N(CN)2], two of Rb[N(CN)2], and one of K3[C6N9] and Rb3[C6N9] from X-ray powder. Chem. Eur J. 2001, 7 (24), 5372–5381.10.1002/1521-3765(20011217)7:24<5372::AID-CHEM5372>3.0.CO;2-#Search in Google Scholar
Irran, E.; Jürgens, B.; Schnick, W. Synthesis., Crystal structure determination from X-ray powder diffractometry and vibrational spectroscopy of the tricyanomelaminate monohydrates M3[C6N9]·H2O (M=K, Rb). Solid State Sci. 2002, 4 (10), 1305–1311.10.1016/S1293-2558(02)00012-2Search in Google Scholar
Irvine, W. M.; Schloerb, F. P. Cyanide and isocyanide abundances in the cold, dark cloud TMC-1. Astrophys. J. 1984, 282 (9), 516–521.10.1086/162229Search in Google Scholar
Isetti, G.; Neubert, T. J. Light-scattering and color centers in sodium cyanide. J. Chem. Phys. 1957, 26 (2), 337–344.10.1063/1.1743295Search in Google Scholar
Ishii, K.; Hirano, T.; Nagashima, U.; Weis, B.; Yamashita, K. An ab initio prediction of the spectroscopic constants of MgNC – the first Mg-bearing molecule in space. Astrophys. J. 1993, 410, L43–L44.10.1086/186875Search in Google Scholar
Ishii, K.; Hirano, T.; Nagashima, U.; Weis, B.; Yamashita, K. Ab initio study on low-lying vibrational states and spectroscopic constants of MgNC (X2Σ+). J. Mol. Struct.: Theochem. 1994, 305 (C), 117–125.10.1016/0166-1280(94)80147-9Search in Google Scholar
Ishii, K.; Taketsugu, T.; Hirano, T. Theoretical study on the potential energy surfaces of CaNC and CaCN. Chem. Phys. Lett. 2003, 374 (5–6), 506–512.10.1016/S0009-2614(03)00738-3Search in Google Scholar
Ismail, Z. K.; Hauge, R. H.; Margrave, J. L. Infrared study of matrix‐isolated lithium isocyanide. J. Chem. Phys. 1972, 57 (12), 5137–5142.10.1063/1.1678202Search in Google Scholar
Israël, G.; Szopa, C.; Raulin, F.; Cabane, M.; Niemann, H. B.; Atreya, S. K.; Bauer, S. J.; Brun, J. F.; Chassefière, E.; Coll, P.; Condé, E.; Cascia, D.; Hauchecorne, A.; Millian, P.; Nguyen, M. J.; Owen, T.; Riedler, W.; Samuelson, R. E.; Siguier, J. M.; Steller, M.; Sternberg, R.; Vidal-Madjar, C. Complex organic matter in Titan’s atmospheric aerosols from in situ pyrolysis and analysis. Nature 2005, 438 (7069), 796–799.10.1038/nature04349Search in Google Scholar
Jackson, D. A.; Atherton, J. H.; Lennon, M.; Cox, L. C. Process for the preparation of cyanogen bromide. WO0142138, 2001.Search in Google Scholar
Jacobs, K.; Siche, D.; Klimm, D.; Rost, H.-J.; Gogova, D. Pseudohalide vapour growth of thick GaN layers. J. Cryst. Growth 2010, 312 (6), 750–755.10.1016/j.jcrysgro.2009.12.055Search in Google Scholar
Jacox, M. E.; Thompson, W. E. Infrared spectra of ClCN+, ClNC+, and BrCN+ trapped in solid neon. J. Chem. Phys. 2007, 126 (24), 244311.10.1063/1.2746868Search in Google Scholar PubMed
Jacox, M. E. Vibrational and electronic energy levels of polyatomic transient molecules: supplement 1. J. Phys. Chem. Ref. Data 1990, 19 (6), 1387–1546.10.1063/1.555848Search in Google Scholar
Jäger, L.; Kretschmann, M.; Köhler, H. Pseudohalogenverbindungen. Darstellung und Reaktionsverhalten von Alkyl- und Arylammoniumdicyanamiden, -Tricyammethaniden und -Dicyanmethanidoacetaten [RNH3]X (X = N(CN)2, C(CN)3, C(CN)2C(O)CH3). Z. Anorg. Allg. Chem. 1992, 611 (5), 68–72.10.1002/zaac.19926110511Search in Google Scholar
Jana, G.; Pan, S.; Osorio, E.; Zhao, L.; Merino, G.; Chattaraj, P. K. Cyanide–isocyanide isomerization: stability and bonding in noble gas inserted metal cyanides (metal = Cu, Ag, Au). Phys. Chem. Chem. Phys. 2018, 20 (27), 18491–18502.10.1039/C8CP02837KSearch in Google Scholar PubMed
Jennings, W. L.; Scott, W. B. The preparation of cyanogen chloride. J. Am. Chem. Soc. 1919, 41 (8), 1241–1248.10.1021/ja02229a011Search in Google Scholar
Jensen, J. N.; Tuan, Y. J. Chemical oxidation of thiocyanate ion by ozone. Ozone Sci. Eng. 1993, 15 (4), 343–360.10.1080/01919519308552494Search in Google Scholar
Jensen, J. O. Vibrational frequencies and structural determination of phosphorous tricyanide. Spectrochim. Acta, Part A 2004a, 60 (11), 2537–2540.10.1016/j.saa.2003.12.032Search in Google Scholar PubMed
Jensen, J. O. Vibrational frequencies and structural determination of arsenous tricyanide. J. Mol. Struct.: Theochem. 2004b, 678 (1–3), 195–199.10.1016/j.theochem.2004.02.038Search in Google Scholar
Jiang, N.; Xu, S.; Ostrikov, K. N.; Tsakadze, E. L.; Long, J. D.; Chai, J. W.; Tsakadze, Z. L. Synthesis and characterization of ternary Al-C-N compound. Int. J. Mod. Phys. B 2002, 16 (06n07), 1132–1137.10.1142/S0217979202010993Search in Google Scholar
Jiang, Z.-Y.; Xu, X.-H.; Wu, H.-S.; Jin, Z.-H. Theoretical study of structures and stabilities of CmN2 (M=1–14) Ions. Int. J. Mass Spectrom. 2003, 230 (1), 33–39.10.1016/j.ijms.2003.08.002Search in Google Scholar
Jin-chai, L.; Xian-feng, L.; Zi-hong, Z.; Huai-xi, G.; Ming-sheng, Y. The preparation and characterization of amorphous SiCN thin films. Wuhan Univ. J. Nat. Sci. 2001, 6 (3), 665–669.10.1007/BF02830280Search in Google Scholar
Joannis, A. Cyanure de Sodium et Hydrates. In Annales de chimie et de physique; V. Masson: Paris, 1882a; pp. 484–486.Search in Google Scholar
Joannis, A. Cyanure de Sodium et Hydrates. In Annales de chimie et de physique; V. Masson: Paris, 1882b; pp. 494–496.Search in Google Scholar
Johansson, A.; Kollman, P.; Rothenberg, S. The electronic structure of the HCN dimer and trimer. Theor. Chim. Acta 1972, 26 (1), 97–100.10.1007/BF00527658Search in Google Scholar
Johns, I. B.; DiPietro, H. R. Synthesis and reactions of anhydrous lithium cyanide. J. Org. Chem. 1964, 29 (7), 1970–1971.10.1021/jo01030a076Search in Google Scholar
Jones, C. E.; Coskran, K. J. The synthesis and spectroscopic properties of some cyanophosphines. Inorg. Chem. 1971, 10 (7), 1536–1537.10.1021/ic50101a049Search in Google Scholar
Jonkers, G.; Mooyman, R.; de Lange, C. A. Ultraviolet photoelectron spectroscopy of the unstable molecules ClSeCN and BrSeCN and the stable Se(CN)2. Mol. Phys. 1981, 43 (3), 655–667.10.1080/00268978100101581Search in Google Scholar
Jost, M.; Finger, L. H.; Sundermeyer, J.; von Hänisch, C. Simple access to ionic liquids and organic salts containing the phosphaethynolate (PCO−) and zintl (Sb113−) Anions. Chem. Commun. 2016, 52 (78), 11646–11648.10.1039/C6CC06620HSearch in Google Scholar
Jucks, K. W.; Miller, R. E. Near infrared spectroscopic observation of the linear and cyclic isomers of the hydrogen cyanide trimer. J. Chem. Phys. 1988, 88 (4), 2196–2204.10.1063/1.454052Search in Google Scholar
Jung, S.; Renz, F.; Klein, M.; Menzel, M.; Boča, R.; Stößer, R. Molecular switching in iron complexes bridged via tin-cyanides observed by Mössbauer and ESR spectroscopy. J. Phys. Conf. Ser. 2010, 217 (1), 012027–012032.10.1088/1742-6596/217/1/012027Search in Google Scholar
Jürgens, B.; Milius, W.; Morys, P.; Schnick, W. Trimerisierung von Dicyanamid-Ionen C2N3− Im Festkörper – Synthesen, Kristallstrukturen und Eigenschaften von NaCs2(C2N3)3 und Na3C6N9 · 3 H2O. Z. Anorg. Allg. Chem. 1998, 624 (1), 91–97.10.1002/(SICI)1521-3749(199801)624:1<91::AID-ZAAC91>3.0.CO;2-ISearch in Google Scholar
Jürgens, B.; Irran, E.; Schneider, J.; Schnick, W. Trimerization of NaC2N3 to Na3C6N9 in the solid: ab initio crystal structure determination of two polymorphs of NaC2N3 and of Na3C6N9 from X-ray powder diffractometry. Inorg. Chem. 2000, 39 (4), 665–670.10.1021/ic991044fSearch in Google Scholar PubMed
Jürgens, B.; Irran, E.; Schnick, W. Syntheses, vibrational spectroscopy, and crystal structure determination from x-ray powder diffraction data of alkaline earth dicyanamides M[N(CN)2]2 with M = Mg, Ca, Sr, and Ba. J. Solid State Chem. 2001, 157 (2), 241–249.10.1006/jssc.2000.9036Search in Google Scholar
Jürgens, B.; Höppe, H. A.; Schnick, W. Synthesis., Crystal structure, vibrational spectroscopy, and thermal behaviour of lead dicyanamide Pb[N(CN)2]2. Solid State Sci. 2002, 4 (6), 821–825.10.1016/S1293-2558(02)01325-0Search in Google Scholar
Jürgens, B.; Irran, E.; Schnick, W. Synthesis and characterization of the rare-earth dicyanamides Ln[N(CN)2]3 with Ln=La, Ce, Pr, Nd, Sm, and Eu. J. Solid State Chem. 2005, 178 (1), 72–78.10.1016/j.jssc.2004.10.030Search in Google Scholar
Jursic, B. S. Ab initio and density functional theory studies of the potential energy surface of the LiNC → LiCN isomerization. J. Mol. Struct.: Theochem. 1998, 428 (1–3), 41–47.10.1016/S0166-1280(97)00259-5Search in Google Scholar
Jursic, B. S. Complete basis set ab initio study of potential energy surfaces of the dissociation recombination reaction HCNH+ + E−. J. Mol. Struct.: Theochem. 1999, 487 (3), 211–220.10.1016/S0166-1280(98)00591-0Search in Google Scholar
Kahlenberg, L.; Steinle, J. V. On the single potential of arsenic and its power to replace other metals in solutions. Trans. Electrochem. Soc. 1923, 44, 493.Search in Google Scholar
Kaiser, R. I.; Mebel, A. M. On the formation of polyacetylenes and cyanopolyacetylenes in Titan’s atmosphere and their role in astrobiology. Chem. Soc. Rev. 2012, 41 (16), 5490–5501.10.1039/c2cs35068hSearch in Google Scholar PubMed
Kakimoto, M.; Kasuya, T. Doppler-limited dye laser excitation spectroscopy of the CCN radical. J. Mol. Spectrosc. 1982, 94 (2), 380–392.10.1016/0022-2852(82)90014-5Search in Google Scholar
Kalescky, R.; Kraka, E.; Cremer, D. Identification of the strongest bonds in chemistry. J. Phys. Chem. 2013, 117 (36), 8981–8995.10.1021/jp406200wSearch in Google Scholar PubMed
Kalikhman, I.; Gostevskii, B.; Kertsnus, E.; Botoshansky, M.; Tessier, C. A.; Youngs, W. J.; Deuerlein, S.; Stalke, D.; Kost, D. Competitive molecular rearrangements in hexacoordinate cyano-silicon dichelates. Organometallics 2007, 26 (10), 2652–2658.10.1021/om061176ySearch in Google Scholar
Kalmutzki, M.; Ströbele, M.; Meyer, H. J. From cyanate to cyanurate: cyclotrimerization reactions towards the novel family of metal cyanurates. Dalton Trans. 2013, 42 (36), 12934–12939.10.1039/c3dt51002fSearch in Google Scholar PubMed
Kanamori, H.; Okamoto, H.; Urabe, K. Lattice vibrational modes in potassium thiocyanate. J. Phys. Chem. Solids 1981, 42 (3), 197–202.10.1016/0022-3697(81)90081-0Search in Google Scholar
Kapp, J.; Schleyer, P. v. R. M(CN)2 species (M = Be, Mg, Ca, Sr, Ba): cyanides, nitriles, or neither? Inorg. Chem. 1996, 35 (8), 2247–2252.10.1021/ic9511837Search in Google Scholar PubMed
Karpfen, A. Linear and cyclic clusters of hydrogen cyanide and cyanoacetylene: a comparative ab initio and density functional study on cooperative hydrogen bonding. J. Phys. Chem. 1996, 100 (32), 13474–13486.10.1021/jp960599iSearch in Google Scholar
Kassaee, M. Z.; Musavi, S. M.; Buazar, F.; Ghambarian, M. Novel triplet ground state silylenes: H–N=C=Si, CN–N=C=Si, and MeO–N=C=Si at DFT Levels. Monatsh. Chem. 2006, 137 (11), 1385–1400.10.1007/s00706-006-0539-4Search in Google Scholar
Kauffman, G. B.; Foust, G. E.; Tun, P. Pseudohalogens: a general chemistry laboratory experiment. J. Chem. Educ. 1968, 45 (2), 141–146.10.1021/ed045p141Search in Google Scholar
Kaufmann, H. P.; Kögler, F. Blei(4)-Salze der Pseudohalogene. Ber. Dtsch. Chem. Ges. 1926, 59 (2), 178–186.10.1002/cber.19260590212Search in Google Scholar
Kawaguchi, K.; Suzuki, T.; Saito, S.; Hirota, E.; Kasuya, T. Dye Laser Excitation spectroscopy of the CCN radical: the A ̃2Δi-X ̃2Πr (0,1,0)-(0,1,0) and (0,2,0)-(0,2,0) Bands. J. Mol. Spectrosc. 1984, 106 (2), 320–329.10.1016/0022-2852(84)90162-0Search in Google Scholar
Kawaguchi, K.; Kagi, E.; Hirano, T.; Takano, S.; Saito, S. Laboratory spectroscopy of MgNC – the first radioastronomical identification of Mg-bearing molecule. Astrophys. J. 1993, 406, L39–L42.10.1086/186781Search in Google Scholar
Keller, C. N.; Anicich, V. G.; Cravens, T. E. Model of Titans ionosphere with detailed hydrocarbon ion chemistry. Planet. Space Sci. 1998, 46 (9–10), 1157–1174.10.1016/S0032-0633(98)00053-1Search in Google Scholar
Kellö, V.; Sadlej, A. J. Electron correlation and relativistic contributions to molecular electric properties: dipole and quadrupole moments of cyanogen halides. Mol. Phys. 1992, 75 (1), 209–220.10.1080/00268979200100161Search in Google Scholar
Kelly, D. P.; Baker, S. C. The organosulphur cycle: aerobic and anaerobic processes leading to turnover of C1‐sulphur compounds. FEMS Microbiol. Lett. 1990, 87 (3–4), 241–246.10.1111/j.1574-6968.1990.tb04919.xSearch in Google Scholar
Kempe, R.; Kessenich, E.; Schulz, A. [Me3SiN(PPh3)·ICN]: a new labile donor–acceptor complex. Inorg. Chem. 2001, 40 (20), 5182–5187.10.1021/ic0102687Search in Google Scholar PubMed
Kertes, A. S., Ed. Silver azide, cyanide, cyanamides, cyanate, selenocyanate and thiocyanate; Elsevier, 1979.Search in Google Scholar
Ketelaar, J. A. A.; Zwartsenberg, J. W. The crystal structure of the cyanogen halides. I. The structure of cyanogen iodide. Recl. Trav. Chim. Pays-Bas 1939, 58 (5), 448–452.10.1002/recl.19390580508Search in Google Scholar
Khalouf-Rivera, J.; Carvajal, M.; Santos, L. F.; Pérez-Bernal, F. Calculation of transition state energies in the HCN–HNC isomerization with an algebraic model. J. Phys. Chem. 2019, 123 (44), 9544–9551.10.1021/acs.jpca.9b07338Search in Google Scholar PubMed
Khazaei, M.; Liang, Y.; Bahramy, M. S.; Pichierri, F.; Esfarjani, K.; Kawazoe, Y. High-pressure phases of hydrogen cyanide: formation of hydrogenated carbon nitride polymers and layers and their electronic properties. J. Phys. Condens. Matter 2011, 23 (40), 405403–405414.10.1088/0953-8984/23/40/405403Search in Google Scholar PubMed
Kichigina, G. A.; Mozhaev, P. S.; Kiryukhin, D. P.; Barkalov, I. M. Low-temperature radiation polymerization of cyanogen bromide. Mendeleev Commun. 1998, 8 (4), 159–160.10.1070/MC1998v008n04ABEH000939Search in Google Scholar
Kim, S. J.; Katayama, Y. Effect of growth conditions on thiocyanate degradation and emission of carbonyl sulfide by thiobacillus thioparus THI115. Wat. Res. 2000, 34 (11), 2887–2894.10.1016/S0043-1354(00)00046-4Search in Google Scholar
King, B. F.; Weinhold, F. Structure and spectroscopy of (HCN)n clusters: cooperative and electronic delocalization effects in C–H⋅⋅⋅N hydrogen bonding. J. Chem. Phys. 1995, 103 (1), 333–347.10.1063/1.469645Search in Google Scholar
Kirk, P. G.; Smith, T. D. The molecular complexity of phosphorus tricyanide in organic solvents. J. Inorg. Nucl. Chem. 1968, 30 (3), 892–893.10.1016/0022-1902(68)80454-3Search in Google Scholar
Kisiel, Z.; Winnewisser, M.; Winnewisser, B. P.; De Lucia, F. C.; Tokaryk, D. W.; Billinghurst, B. E. Far-infrared spectrum of S(CN)2 measured with synchrotron radiation: global analysis of the available high-resolution spectroscopic data. J. Phys. Chem. 2013, 117 (50), 13815–13824.10.1021/jp408255mSearch in Google Scholar PubMed
Klapötke, T. M.; Schulz, A. Reaction of AgOCN with NO, NO2, ClNO2, ClNO, and BrNO: evidence of the formation of OCN−NO2 and OCN−NO. Inorg. Chem. 1996, 35 (26), 7897–7904.10.1021/ic960707fSearch in Google Scholar
Klapötke, T. M.; McIntyre, G.; Schulz, A. Nitration of hydrogen cyanide with nitryl tetrafluoroborate. J. Chem. Soc., Dalton Trans. 1996, No. 15, 3237–3241.10.1039/DT9960003237Search in Google Scholar
Klapötke, T. M.; Krumm, B.; Gálvez-Ruiz, J. C.; Nöth, H.; Schwab, I. Experimental and theoretical studies of homoleptic tellurium cyanides Te(CN)X: crystal structure of Te(CN)2. Eur. J. Inorg. Chem. 2004, 2004 (24), 4764–4769.10.1002/ejic.200400763Search in Google Scholar
Klapötke, T. M.; Krumm, B.; Scherr, M. Homoleptic selenium cyanides: attempted preparation of Se(CN)4 and redetermination of the crystal structure of Se(CN)2. Inorg. Chem. 2008, 47 (15), 7025–7028.10.1021/ic801011gSearch in Google Scholar PubMed
Klapötke, T. M. The [ICNI]+ cation: a combined experimental and theoretical study. Reaction of [ICNI]+[AsF6]− with CsN3. J. Chem. Soc. Dalton Trans. 1997, 3 (4), 553–558.10.1039/a605349aSearch in Google Scholar
Klason, P. Über Das Radical Cyanur und Seine Verbindungen Mit Halogenen. J. Prakt. Chem. 1886, 34 (1), 152–160.10.1002/prac.18860340119Search in Google Scholar
Klein, M. L.; Goddard, J. D.; Bounds, D. G. An ab initio molecular orbital study of NaCN and KCN. J. Chem. Phys. 1981, 75 (8), 3909–3915.10.1063/1.442547Search in Google Scholar
Klemenc, A.; Wagner, G. Bemerkungen Über Das Cyanchlorid. Z. Anorg. Allg. Chem. 1938, 235 (4), 427–430.10.1002/zaac.19382350412Search in Google Scholar
Knight, J. S.; Petrie, S. A. H.; Freeman, C. G.; McEwan, M. J.; McLean, A. D.; DeFrees, D. J. Structural Isomers of C2N+: a selected-ion flow tube study. J. Am. Chem. Soc. 1988, 110 (16), 5286–5290.10.1021/ja00224a008Search in Google Scholar PubMed
Knopp, G.; Knorr, K.; Loidl, A.; Haussühl, S. On the phase transition in CsCN. Z. Phys. B: Condens. Matter 1983, 51 (3), 259–263.10.1007/BF01307681Search in Google Scholar
Knözinger, E. Cluster formation of hydrogen cyanide in solid and liquid CCl4 matrices. J. Chem. Phys. 1986, 85 (9), 4881.10.1063/1.451723Search in Google Scholar
Kober, E.; Grundmann, C. Triazines. XXII. Fluoro-s-Triazines. J. Am. Chem. Soc. 1959, 81 (14), 3769–3770.10.1021/ja01523a069Search in Google Scholar
Kober, E.; Schroeder, H.; Ratz, R. F. W.; Ülrich, H.; Gruxdmann, C. Synthesis of polyfluorinated heterocycles by indirect fluorination with silver fluorides. I. Fluoro-s-triazines and reactions of cyanuric fluoride. J. Org. Chem. 1962, 27 (7), 2577–2580.10.1021/jo01054a065Search in Google Scholar
Kohguchi, H.; Ohshima, Y.; Endo, Y. Laser-induced fluorescence spectroscopy of the C̃2Σ+−X̃2Π1/2 band system of jet-cooled CCN radical. J. Chem. Phys. 1997, 106 (13), 5429–5438.10.1063/1.473568Search in Google Scholar
Köhler, H.; Lux, G. Zur Kenntnis des Nitrosodicyanmethanid-Ions [ONC(CN)2]−. Inorg. Nucl. Chem. Lett. 1968, 4 (3), 133–136.10.1016/0020-1650(68)80153-9Search in Google Scholar
Köhler, H.; Seifert, B. Beiträge Zur Chemie des Dicyanamid- und des Tricyanmethanid-Ions XI. Diorganozinn-pseudohalogenide. J. Organomet. Chem. 1968, 12 (1), 253–255.10.1016/S0022-328X(00)90924-0Search in Google Scholar
Köhler, H.; Kolbe, A.; Lux, G. Metall-pseudohalogenide. 27. Zur Struktur der Dicyanamide Zweiwertiger 3d-Metalle M(N(CN)2)2. Z. Anorg. Allg. Chem. 1977, 428 (1), 103–112.10.1002/zaac.19774280113Search in Google Scholar
Köhler, H. Die Fabrikation von Cyanverbindungen aus Tierischen Abfällen und Produkten der Trockenen Destillation. In Die Industrie der Cyanverbindungen; Vieweg: Wiesbaden, 1914; pp. 60–101.10.1007/978-3-322-99005-1_3Search in Google Scholar
Köhler, H. Beitrage Zur Chemie des Dicyanamid- und des Tricyanmethanidions. I. Die Bildung von Übergangsmetall-Pyridin-Komplexen. Z. Anorg. Allg. Chem. 1964, 331 (5–6), 237–248.10.1002/zaac.19643310502Search in Google Scholar
Kohout, J.; Jäger, L.; Hvastijová, M.; Kožíšek, J. Tricyanomethanide and dicyanamide complexes of Cu(II), Ni(II), Co(II), their structures and properties. J. Coord. Chem. 2000, 51 (2), 169–218.10.1080/00958970008055127Search in Google Scholar
Komornicki, A.; Ishida, K.; Morokuma, K.; Ditchfield, R.; Conrad, M. Efficient determination and characterization of transition states using ab-initio methods. Chem. Phys. Lett. 1977, 45 (3), 595–602.10.1016/0009-2614(77)80099-7Search in Google Scholar
Kondo, Y.; Schoemaker, D.; Lüty, F. Molecular motion and ordering in rubidium cyanide, studied with dielectric and raman techniques. Phys. Rev. B 1979, 19 (8), 4210–4216.10.1103/PhysRevB.19.4210Search in Google Scholar
König, W. Untersuchungen aus Dem Organ.-Chem. Laboratorium der Technischen Hochschule Zu Dresden. XCVII. Über die Umsetzung von Rhodaniden Mit Brom in Wäßriger Lösung. J. Prakt. Chem. 1911, 84 (1), 558–560.10.1002/prac.19110840139Search in Google Scholar
Konnert, J.; Britton, D. The crystal structure of AgC(CN)3. Inorg. Chem. 1966, 5 (7), 1193–1196.10.1021/ic50041a026Search in Google Scholar
Koppe, K.; Bilir, V.; Frohn, H.-J.; Mercier, H. P. A.; Schrobilgen, G. J. Syntheses., Solution multi-NMR characterization, and reactivities of [C6F5Xe]+ salts of weakly coordinating borate anions, [BY4]– (Y = CF3, C6F5, CN, or OTeF5). Inorg. Chem. 2007, 46 (22), 9425–9437.10.1021/ic7010138Search in Google Scholar PubMed
Koppel, I. A.; Burk, P.; Koppel, I.; Leito, I. Generalized principle of designing neutral superstrong brønsted acids. J. Am. Chem. Soc. 2002, 124 (19), 5594–5600.10.1021/ja0255958Search in Google Scholar PubMed
Koschel, D.; Gmelin, L.; Meyer, R. J.; Pietsch, E. H. E.; Fluck, E. Carbon nitrogen compounds. In Gmelin handbook of inorganic and organometallic chemistry; Verl. Chemie: Berlin, 1971; p. 475.Search in Google Scholar
Kotch, T. G.; Lees, A. J.; Fuerniss, S. J.; Papathomas, K. I. Photocatalyzed polymerization of aromatic dicyanate esters by iron-arene complexes. Chem. Mater. 1995, 7 (4), 801–805.10.1021/cm00052a028Search in Google Scholar
Kozirovski, Y.; Folman, M. Infra-red spectrum and spectral shifts of HCN adsorbed on evaporated alkali halides. Trans. Faraday Soc. 1966, 62, 808–820.10.1039/tf9666200808Search in Google Scholar
Kraemer, W. P.; Bunker, P. R.; Yoshimine, M. A theoretical study of the rotation-vibration energy levels and dipole moment functions of CCN+, CNC+, and C3. J. Mol. Spectrosc. 1984, 107 (1), 191–207.10.1016/0022-2852(84)90276-5Search in Google Scholar
Kraft, A. “Notitia Caerulei Berolinensis Nuper Inventi” on the 300th anniversary of the first publication on Prussian blue. Bull. Hist. Chem. 2011, 36 (1), 3–9.Search in Google Scholar
Krause, A. Zum Mechanismus der Blausäuresynthese aus Ammoniak, Methan und Luftsauerstoff Am Platinkontakt. Monatsh. Chem. 1965, 96 (4), 1132–1133.10.1007/BF00904256Search in Google Scholar
Krauß, N.; Tebbe, K.-F. Polypseudohalogenide des Kaliums. Z. für Kristallogr. – Cryst. Mater. 1986, 174 (1–4), 124–125.Search in Google Scholar
Kreutzberger, A. Die Chemie des S-Triazins. In Fortschritte der Chemischen Forschung; Springer-Verlag: Berlin, 2006; pp. 273–300.10.1007/BFb0051921Search in Google Scholar
Kroeker, R. M.; Pacansky, J. Observation of electron beam induced chemistry by tunneling spectroscopy. J. Chem. Phys. 1982, 77 (10), 4955–4956.10.1063/1.443713Search in Google Scholar
Krolevets, A. A.; Antipova, V. V.; Martynov, I. V. Organosilicon and organophosphorus compounds with pseudohalide groups. 2. Reactions of alkoxydimethylsilyl cyanides with chlorides of tricoordinated phosphorus. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1991, 40 (12), 2483–2485.10.1007/BF00959728Search in Google Scholar
Krüss, G. Über eine Neue darstellungsmethode Für Nitrile. Ber. Dtsch. Chem. Ges. 1884, 17 (2), 1766–1768.10.1002/cber.18840170245Search in Google Scholar
Kuhn, M.; Mecke, R. Die Reaktion von Dirhodan Mit Aliphatischen Aminen. Chem. Ber. 1960, 93 (3), 618–621.10.1002/cber.19600930312Search in Google Scholar
Kuhn, M.; Mecke, R. IR-Spektroskopische Untersuchungen Am Dicyan-amid-Anion, [N(CN)2]. Chem. Ber. 1961a, 94 (11), 3010–3015.10.1002/cber.19610941128Search in Google Scholar
Kuhn, M.; Mecke, R. IR-spektroskopische Untersuchungen Am Dicyan-amid-Anion, [N(CN)2]–. Chem. Ber. 1961b, 94 (11), 3010–3015.10.1002/cber.19610941128Search in Google Scholar
Kuhnhen, H. B. Notizen: Reaktion von Dirhodan Mit Substituierten Carbonyl(η-Cyclopentadienyl)Iodoeisen-Komplexen/reaction of Thiocyanogen with Substituted Carbonyl(η-Cyclopentadienyl)Iodoiron Complexes. Z. Naturforsch. 1977, 32 (6), 718–720.10.1515/znb-1977-0628Search in Google Scholar
Kulsha, A. V.; Sharapa, D. I. Superhalogen and superacid. J. Comput. Chem. 2019, 40 (26), 2293–2300.10.1002/jcc.26007Search in Google Scholar PubMed
Kumar Sau, A.; Currell, D.; Mazumdar, S.; Mitra, S. An NMR and circular dichroism study of the interaction of thiocyanate with human and cross-linked hemoglobin: identification of Lys-α-99 as a possible dissociation linked binding site. Biophys. Chem. 2003, 106 (3), 233–240.10.1016/S0301-4622(03)00154-6Search in Google Scholar
Küppers, T.; Bernhardt, E.; Willner, H.; Rohm, H. W.; Köckerling, M. Tetracyanoborate salts M[B(CN)4] with M = singly charged cations: properties and structures. Inorg. Chem. 2005, 44 (4), 1015–1022.10.1021/ic048780qSearch in Google Scholar PubMed
Küppers, T.; Bernhardt, E.; Lehmann, C. W.; Willner, H. Die Tetracyanoborsäuren H[B(CN)4]n H2O, n = 0, 1, 2. Z. Anorg. Allg. Chem. 2007, 633 (10), 1666–1672.10.1002/zaac.200700097Search in Google Scholar
Kurnig, I. J.; Lischka, H.; Karpfen, A. Linear versus cyclic (HCN)3: an ab initio study on structure, vibrational spectra, and infrared intensities. J. Chem. Phys. 1990, 92 (4), 2469–2477.10.1063/1.457990Search in Google Scholar
Kwon, H. K.; Woo, S. H.; Park, J. M. Thiocyanate degradation by acremonium strictum and inhibition by secondary toxicants. Biotechnol. Lett. 2002, 24 (16), 1347–1351.10.1023/A:1019825404825Search in Google Scholar
Labbow, R.; Michalik, D.; Reiß, F.; Schulz, A.; Villinger, A. Isolation of labile pseudohalogen NSO species. Angew. Chem. Int. Ed. 2016, 55 (27), 7680–7684.10.1002/anie.201601878Search in Google Scholar PubMed
Ladenburg, A. Vorträge Über Die Entwicklungsgeschichte Der Chemie Von Lavoirsier Bis Zur Gegenwart; Friedrich Vieweg & Sohn: Braunschweig, 1907.10.1007/978-3-663-20167-0Search in Google Scholar
Landmann, J.; Keppner, F.; Hofmann, D. B.; Sprenger, J. A. P.; Häring, M.; Zottnick, S. H.; Müller-Buschbaum, K.; Ignat’ev, N. V.; Finze, M. Deprotonation of a hydridoborate anion. Angew. Chem. Int. Ed. 2017, 56 (10), 2795–2799.10.1002/anie.201611899Search in Google Scholar PubMed
Langel, W.; Kollhoff, H.; Knözinger, E. From isolated HCN molecules to the crystal: inelastic neutron scattering, far infrared spectroscopy, and normal coordinate analysis. J. Chem. Phys. 1989, 90 (7), 3430–3442.10.1063/1.455852Search in Google Scholar
Lanzisera, D. V.; Andrews, L. Reactions of laser-ablated beryllium atoms with hydrogen cyanide in excess argon. FTIR spectra and quantum chemical calculations on BeCN, BeNC, HBeCN, and HBeNC. J. Am. Chem. Soc. 1997a, 119 (27), 6392–6398.10.1021/ja970740gSearch in Google Scholar
Lanzisera, D. V.; Andrews, L. Reactions of laser-ablated Al, Ga, in, and Tl atoms with hydrogen cyanide in excess argon. Matrix infrared spectra and density functional theory calculations on new cyanide and isocyanide products. J. Phys. Chem. 1997b, 101 (50), 9660–9665.10.1021/jp972333xSearch in Google Scholar
Lanzisera, D. V.; Andrews, L.; Taylor, P. R. Reactions of laser-ablated boron atoms with HCN during condensation in argon. A comparison of matrix infrared and DFT, CCSD(T), and CASSCF frequencies of BNC, BCN, HBNC, and HBCN. J. Phys. Chem. 1997, 101 (38), 7134–7140.10.1021/jp971116tSearch in Google Scholar
Largo-Cabrerizo, A.; Barrientos, C. A comparative theoretical study of the C2N+ and SiCN+ ions and their formation processes. Chem. Phys. Lett. 1988, 148 (1), 79–85.10.1016/0009-2614(88)87264-6Search in Google Scholar
Largo, A.; Barrientos, C. Theoretical studies of possible processes for the interstellar production of phosphorus compounds: reaction of phosphorus(1+) with hydrogen cyanide and protonation of cyanogen phosphide compounds. J. Phys. Chem. 1991, 95 (24), 9864–9868.10.1021/j100177a046Search in Google Scholar
Largo-Cabrerizo, A. A theoretical study of the SiCN radical. Chem. Phys. Lett. 1988, 147 (1), 95–98.10.1016/0009-2614(88)80230-6Search in Google Scholar
Larsen, J.; Madsen, D.; Poulsen, J.-A.; Poulsen, T. D.; Keiding, S. R.; Thøgersen, J. The photoisomerization of aqueous ICN studied by subpicosecond transient absorption spectroscopy. J. Chem. Phys. 2002, 116 (18), 7997–8005.10.1063/1.1467897Search in Google Scholar
Larson, J. W.; McMahon, T. B. Gas-phase bihalide and pseudobihalide ions. An ion cyclotron resonance determination of hydrogen bond energies in XHY- species (X, Y = F, Cl, Br, CN). Inorg. Chem. 1984, 23 (14), 2029–2033.10.1021/ic00182a010Search in Google Scholar
Le Bihan, M. Structure Du cyanure de Sodium Hydraté. Acta Crystallogr. 1958, 11 (11), 770–773.10.1107/S0365110X58002188Search in Google Scholar
Le Guennec, M.; Wlodarczak, G.; Chen, W. D.; Bocquet, R.; Demaison, J. Rotational spectrum and equilibrium structure of cyanogen bromide. J. Mol. Spectrosc. 1992, 153 (1–2), 117–132.10.1016/0022-2852(92)90463-XSearch in Google Scholar
Lee, T. J.; Racine, S. C. A coupled-cluster study of the molecular structure, vibrational spectrum and relative energies of the XCN and XNC (X = F, Cl) isomers. Mol. Phys. 1995, 84 (4), 717–725.10.1080/00268979500100491Search in Google Scholar
Lee, T. J.; Schaefer, H. F. Vibrational frequencies and infrared intensities for H2CN+, protonated HCN. J. Chem. Phys. 1984, 80 (6), 2977–2978.10.1063/1.447009Search in Google Scholar
Lee, T. J.; Martin, J. M. L.; Dateo, C. E.; Taylor, P. R. Accurate ab initio quartic force fields, vibrational frequencies, and heats of formation for FCN, FNC, ClCN, and ClNC. J. Phys. Chem. 1995, 99 (43), 15858–15863.10.1021/j100043a025Search in Google Scholar
Lee, D.; Lee, Y. S.; Hagebaum-Reignier, D.; Jeung, G.-H. First-order correction for bond energy applied to polar molecules: alkali halides, alkali cyanides, LiCH3, and CH3F. Chem. Phys. 2006, 327 (2–3), 406–414.10.1016/j.chemphys.2006.05.018Search in Google Scholar
Lee, D.; Lim, I. S.; Lee, Y. S.; Hagebaum-Reignier, D.; Jeung, G.-H. Molecular properties and potential energy surfaces of the cyanides of the groups 1 and 11 metal atoms. J. Chem. Phys. 2007, 126 (24), 244313–244321.10.1063/1.2749504Search in Google Scholar PubMed
Lee, S. Time‐dependent hartree approximation applied to the photodissociation of ICN. J. Chem. Phys. 1992, 97 (1), 227–235.10.1063/1.463620Search in Google Scholar
Lee, T. J. Ab initio characterization of triatomic bromine molecules of potential interest in stratospheric chemistry. J. Phys. Chem. 1995, 99 (41), 15074–15080.10.1021/j100041a024Search in Google Scholar
Legon, A. C.; Millen, D. J.; Mjöberg, P. J. The hydrogen cyanide dimer: identification and structure from microwave spectroscopy. Chem. Phys. Lett. 1977, 47 (3), 589–591.10.1016/0009-2614(77)85048-3Search in Google Scholar
Leibovici, C. A. Quantum-mechanical evaluation of the tilt of the cyano group in difluorocyanamide, cyanodifluoro-phosphine and phosphorus tricyanide. J. Mol. Struct. 1973, 18 (2), 343–345.10.1016/0022-2860(73)85239-1Search in Google Scholar
Lely, J. A.; Bijvoet, J. M. The crystal structure of lithium cyanide. Recl. Trav. Chim. Pays-Bas 1942, 61 (4), 244–252.10.1002/recl.19420610402Search in Google Scholar
Léonard, C.; Gritli, H.; Chambaud, G. Ab initio study of the spectroscopy of the X2Π electronic ground states of CNO and NCO. J. Mol. Spectrosc. 2007, 243 (1), 90–98.10.1016/j.jms.2007.04.005Search in Google Scholar
Léonard, C.; Gritli, H.; Chambaud, G. New study of the stability and of the spectroscopy of the molecular anions NCO− and CNO−. J. Chem. Phys. 2010, 133 (12), 124318–124327.10.1063/1.3483463Search in Google Scholar PubMed
Leopold, J. G.; Faraggi, M. Pulse radiolysis of the cyanate anion in aqueous solution. J. Phys. Chem. 1977, 81 (8), 803–806.10.1021/j100523a024Search in Google Scholar
LeSar, R.; Gordon, R. G. Density‐functional theory for the solid alkali cyanides. J. Chem. Phys. 1982, 77 (7), 3682–3692.10.1063/1.444271Search in Google Scholar
Letts, E. A. Neue Bildungsweisen der Amide und Nitrile. Ber. Dtsch. Chem. Ges. 1872, 5 (2), 669–674.10.1002/cber.18720050228Search in Google Scholar
Li, X.; Sayah, N.; Jackson, W. M. Laser measurements of the effects of vibrational energy on the reactions of CN. J. Chem. Phys. 1984, 81 (2), 833–840.10.1063/1.447717Search in Google Scholar
Li, X.; Zeng, Y.; Zhang, X.; Zheng, S.; Meng, L. Computational studies of σ-type weak interactions between NCO/NCS radicals and XY (X = H, Cl; Y = F, Cl, and Br). Sci. China Chem. 2012, 55 (7), 1395–1404.10.1007/s11426-012-4611-0Search in Google Scholar
Liebig, J.; Gay-Lussac, J. L. Zerlegung des Knallsauren Silberoxids. Ann. Phys. Chem. 1824a, 77, 117–124.10.1002/andp.18240770505Search in Google Scholar
Liebig, J.; Gay-Lussac, J. L. Analyse Du Fulminate. Ann. Chim. Phys. 1824b, 25, 285–311.Search in Google Scholar
Liebig, J.; Wöhler, F. Untersuchungen Über die Cyansäure. Ann. Phys. 1830, 96 (11), 369–400.10.1002/andp.18300961102Search in Google Scholar
Lin, Y.; Heaven, M. C. Observation and analysis of the CN–Ne B–X transition. J. Chem. Phys. 1991, 94 (8), 5765–5768.10.1063/1.460459Search in Google Scholar
Lin, H.-S.; Erickson, M. G.; Lin, Y.; Basinger, W. H.; Lawrence, W. G.; Heaven, M. C. Vibronic relaxation dynamics of CN in Ar matrices and clusters. Chem. Phys. 1994, 189 (2), 235–243.10.1016/0301-0104(94)00206-1Search in Google Scholar
Linke, K.-H.; Lemmer, F. Röntgenographische Kristallstrukturanalyse von Schwefeldicyanid. Z. Anorg. Allg. Chem. 1966a, 345 (3–4), 203–210.10.1002/zaac.19663450310Search in Google Scholar
Linke, K.-H.; Lemmer, F. Röntgenographische Kristallstrukturanalyse von Selendicyanid. Z. Anorg. Allg. Chem. 1966b, 345 (3–4), 211–216.10.1002/zaac.19663450311Search in Google Scholar
Linnemann, F. Untersuchung Über Das Cyansulfid. Justus Liebigs Ann. Chem. 1861, 120 (1), 36–47.10.1002/jlac.18611200103Search in Google Scholar
Lipscomb, R. D.; Smith, W. C. Preparation of cyanogen fluoride. 3008, 1961.Search in Google Scholar
Liszt, H.; Lucas, R. Comparative chemistry of diffuse clouds. Astron. Astrophys. 2001, 370 (2), 576–585.10.1051/0004-6361:20010260Search in Google Scholar
Liu, R.; Chen, Z.; Yao, Y.; Li, Y.; Cheema, W. A.; Wang, D.; Zhu, S. Recent advancements in g-C3N4-based photocatalysts for photocatalytic CO2 reduction: a mini review. RSC Adv. 2020, 10 (49), 29408–29418.10.1039/D0RA05779GSearch in Google Scholar
Livinghouse, T. Trimethylsilyl cyanide: cyanosilation of p-benzoquinone. Org. Synth. 1981, 60, 126.10.15227/orgsyn.060.0126Search in Google Scholar
Löber, M.; Dolabdjian, K.; Ströbele, M.; Romao, C. P.; Meyer, H.-J. Synthesis, structure, and electronic properties of Sn(CN2) and Sn4Cl2(CN2)3. Inorg. Chem. 2019, 58 (12), 7845–7851.10.1021/acs.inorgchem.9b00527Search in Google Scholar PubMed
Loew, G. H.; Chang, S. Quantum chemical Study of HCN dimer and its role in chemical evolution. Tetrahedron 1971, 27 (13), 2989–3001.10.1016/S0040-4020(01)98091-3Search in Google Scholar
Loew, G. H. Conformation of hydrogen cyanide dimer and its role in chemical evolution. J. Theor. Biol. 1971, 33 (1), 121–130.10.1016/0022-5193(71)90220-7Search in Google Scholar PubMed
Loewenschuss, A.; Marcus, Y. Standard thermodynamic functions of some isolated ions at 100–1000 K. J. Phys. Chem. Ref. Data 1996, 25 (6), 1495–1507.10.1063/1.555990Search in Google Scholar
Loidl, A.; Knorr, K.; Daubert, J.; Dultz, W.; Fitzgerald, W. J. Inelastic neutron scattering by coupled rotational and translational modes in KCN. Z. Phys. B: Condens. Matter 1980a, 38 (2), 153–163.10.1007/BF01598756Search in Google Scholar
Loidl, A.; Knorr, K.; Kjems, J. I.; Haussuhl, S. Inelastic neutron scattering by coupled rotational and translational modes in CsCN. J. Phys. C Solid State Phys. 1980b, 13 (14), L349–L355.10.1088/0022-3719/13/14/002Search in Google Scholar
Loidl, A.; Haussühl, S.; Kjems, J. K. Elastic properties of CsCN. Z. Phys. B: Condens. Matter 1983, 50 (3), 187–192.10.1007/BF01304126Search in Google Scholar
Long, D. A.; Steele, D. Spectroscopic studies of compounds containing the —CN group—II: vibrational spectra, assignments and configuration of sulphur dicyanide S(CN2). Spectrochim. Acta 1963, 19 (11), 1731–1737.10.1016/0371-1951(63)80192-7Search in Google Scholar
Long, D. A.; Carrington, R. A. G.; Gravenor, R. B. Vibrational spectra and structure of the tricyanomethanide ion C(CN)−3. Nature 1962, 196, 371–372.10.1038/196371a0Search in Google Scholar
Lord, G.; Woolf, A. A. The cyanogen halides. Part III. Their heats of formation and free energies. J. Chem. Soc. 1954, 2546.10.1039/jr9540002546Search in Google Scholar
Lössner, L.; Kretzschmar, A.; Peitzsch, B.; Salomon, F. Einwirkung von Phosphorchlorid und von Benzoylchlorid Auf Rhodankalium. J. Prakt. Chem. 1873, 7 (1), 474–480.10.1002/prac.18730070151Search in Google Scholar
Lotsch, B. V.; Senker, J.; Schnick, W. Characterization of the thermally induced topochemical solid-state transformation of NH4[N(CN)2] into NCN[double bond]C(NH2)2 by means of X-ray and neutron diffraction as well as Raman and solid-state NMR spectroscopy. Inorg. Chem. 2004, 43 (3), 895–904.10.1021/ic034984fSearch in Google Scholar PubMed
Loupy, A.; Corset, J. Infrared spectral studies of ion association in nonaqueous solutions of cyanide salts with consequences for their nucleophilic reactivity. J. Solution Chem. 1976, 5 (12), 817–831.10.1007/BF01167237Search in Google Scholar
Lovallo, C. C.; Klobukowski, M. Improved model core potentials: application to the thermochemistry of organoxenon complexes. Int. J. Quantum Chem. 2002, 90 (3), 1099–1107.10.1002/qua.10256Search in Google Scholar
Lu, Y.; Wang, H.; Xie, Y.; Liu, H.; Schaefer, H. F. The cyanate and 2-phosphaethynolate anion congeners ECO– (E = N, P, as, Sb, Bi): prelude to experimental characterization. Inorg. Chem. 2014, 53 (12), 6252–6256.10.1021/ic500780hSearch in Google Scholar PubMed
Lucchese, R. R.; Schaefer, H. F. Dicyanocarbene. Triplet and singlet structures and energetics. J. Am. Chem. Soc. 1977, 99 (1), 13–14.10.1021/ja00443a003Search in Google Scholar
Ma, B.; Yamaguchi, Y.; Schaefer, H. F. Spectroscopic constants and potential energy surfaces for the possible interstellar molecules AlNC and AlCN. Mol. Phys. 1995, 86 (6), 1331–1337.10.1080/00268979500102771Search in Google Scholar
MacLean, E. J.; Harris, K. D. M.; Kariuki, B. M.; Kitchin, S. J.; Tykwinski, R. R.; Swainson, I. P.; Dunitz, J. D. Ammonium cyanate shows N−H···N hydrogen bonding, not N−H···O. J. Am. Chem. Soc. 2003, 125 (47), 14449–14451.10.1021/ja021156xSearch in Google Scholar PubMed
Madelung, W.; Kern, E. Über Dicyanamid. Justus Liebigs Ann. Chem. 1922a, 427 (1), 1–26.10.1002/jlac.19224270102Search in Google Scholar
Madelung, W.; Kern, E. Über Tricyanmelamin. Justus Liebigs Ann. Chem. 1922b, 427 (1), 26–34.10.1002/jlac.19224270103Search in Google Scholar
Maeda, A.; Habara, H.; Amano, T. Submillimetre-wave spectrum of NCS. Mol. Phys. 2007, 105 (5–7), 477–495.10.1080/00268970701233364Search in Google Scholar
Magnusson, E. The electronic structures of P(III) compounds with P–C bonds. Phosphorous Sulfur Relat. Elem. 1986, 28 (3), 379–394.10.1080/03086648608072830Search in Google Scholar
Mai, K.; Patil, G. Alkylsilyl cyanides as silylating agents. J. Org. Chem. 1986, 51 (18), 3545–3548.10.1021/jo00368a030Search in Google Scholar
Maier, G.; Reisenauer, H. P. Reactions of silicon atoms with cyanogen: generation and matrix-spectroscopic identification of five C2N2Si isomers. Eur. J. Org Chem. 2005, 2005 (10), 2015–2021.10.1002/ejoc.200400767Search in Google Scholar
Maier, G.; Teles, J. H. Isolierung und Photoisomerisierung von Einfach Substituierten Nitriloxiden. Angew. Chem. 1987a, 99 (2), 152–153.10.1002/ange.19870990221Search in Google Scholar
Maier, G.; Teles, J. H. Isolation and photoisomerization of simply substituted nitrile oxides. Angew. Chem. Int. Ed. 1987b, 26 (2), 155–156.10.1002/anie.198701551Search in Google Scholar
Maier, G.; Naumann, M.; Reisenauer, H. P.; Eckwert, J. Diisocyanate. Angew. Chem. Int. Ed. 1996a, 35 (15), 1696–1697.10.1002/anie.199616961Search in Google Scholar
Maier, G.; Naumann, M.; Reisenauer, H. P.; Eckwert, J. Diisocyanat. Angew. Chem. 1996b, 108 (15), 1800–1801.10.1002/ange.19961081518Search in Google Scholar
Maier, G.; Reisenauer, H. P.; Eckwert, J.; Naumann, M.; De Marco, M. Isomers of the elemental composition CN2O. Angew. Chem. Int. Ed. 1997a, 36 (16), 1707–1709.10.1002/anie.199717071Search in Google Scholar
Maier, G.; Reisenauer, H.; Eckwert, J.; Naumann, M.; Marco, M. Isomere der Elementarzusammensetzung CN2O. Angew. Chem. 1997b, 109 (16), 1785–1787.10.1002/ange.19971091609Search in Google Scholar
Maier, G.; Reisenauer, H. P.; Egenolf, H.; Glatthaar, J. Reaction of silicon atoms with hydrogen cyanide: generation and matrix-spectroscopic identification of CHNSi and CNSi isomers. Eur. J. Org Chem. 1998, 1998 (7), 1307–1311.10.1002/(SICI)1099-0690(199807)1998:7<1307::AID-EJOC1307>3.0.CO;2-ZSearch in Google Scholar
Maier, G.; Reisenauer, H. P.; Ruppel, R. Dicyanocarbene and its isomers: a matrix spectroscopic study. Eur. J. Org Chem. 2003, 2003 (14), 2695–2701.10.1002/ejoc.200300204Search in Google Scholar
Maier, L. Organische Phosphorverbindungen. IX. Über Einige Reaktionen des Methyldibromphosphins [CH3PBr2]. Helv. Chim. Acta 1963, 46 (7), 2667–2676.10.1002/hlca.19630460725Search in Google Scholar
Maillard, J.; Schmitz-Afonso, I.; Gautier, T.; Afonso, C.; Carrasco, N. Suggested plausible structures for Titan’s haze analogs using tandem mass spectrometry. Icarus 2021, 358, 114181.10.1016/j.icarus.2020.114181Search in Google Scholar
Makarewicz, J.; Ha, T.-K. Vibrational states of LiCN calculated from an ab initio potential energy surface. Chem. Phys. Lett. 1995, 232 (5–6), 497–502.10.1016/0009-2614(94)01387-BSearch in Google Scholar
Maki, A.; Decius, J. C. Infrared spectrum of cyanate ion as a solid solution in a potassium iodide lattice. J. Chem. Phys. 1958, 28 (5), 1003–1004.10.1063/1.1744260Search in Google Scholar
Maki, A.; Decius, J. C. Vibrational spectrum of cyanate ion in various alkali halide lattices. J. Chem. Phys. 1959, 31 (3), 772–782.10.1063/1.1730461Search in Google Scholar
Maki, A. G.; Sams, R. L. High temperature, high resolution infrared spectral measurements on the HNC–HCN equilibrium system. J. Chem. Phys. 1981, 75 (9), 4178–4182.10.1063/1.442645Search in Google Scholar
Mamajanov, I.; Herzfeld, J. HCN polymers characterized by solid state NMR: chains and sheets formed in the neat liquid. J. Chem. Phys. 2009, 130 (13), 1–6.10.1063/1.3092908Search in Google Scholar
Mandal, S. K.; Roesky, H. W. Interstellar molecules: guides for new chemistry. Chem. Commun. (London) 2010, 46 (33), 6016–6041.10.1039/c0cc01003kSearch in Google Scholar
Marcos-Fernández, A.; Posadas, P.; Rodríguez, A.; González, L. Synthesis and characterization of new dicyanate monomers. A way to obtain fully aromatic crosslinked poly(ether ketone)S. J. Polym. Sci. Part A: Polym. Chem. 1999, 37 (16), 3155–3168.10.1002/(SICI)1099-0518(19990815)37:16<3155::AID-POLA13>3.0.CO;2-0Search in Google Scholar
Marín-Yaseli, M. R.; Cid, C.; Yagüe, A. I.; Ruiz-Bermejo, M. Detection of macromolecular fractions in HCN polymers using electrophoretic and ultrafiltration techniques. Chem. Biodiversity 2017, 14 (2), e1600241.10.1002/cbdv.201600241Search in Google Scholar
Markley, T. J.; Toby, B. H.; Pearlstein, R. M.; Ramprasad, D. New synthesis routes to lithium and cesium cyanide salts. Inorg. Chem. 1997, 36 (15), 3376–3378.10.1021/ic961395jSearch in Google Scholar
Marszalek, M.; Fei, Z.; Zhu, D.-R.; Scopelliti, R.; Dyson, P. J.; Zakeeruddin, S. M.; Grätzel, M. Application of ionic liquids containing tricyanomethanide [C(CN)3]− or tetracyanoborate [B(CN)4]− anions in dye-sensitized solar cells. Inorg. Chem. 2011, 50 (22), 11561–11567.10.1021/ic201513mSearch in Google Scholar
Martin, G.; Barbour, W. The cyanide and Prussiate Industry; Crosby Lockwood and Son: London, 1915; Vol. Chapter VI.Search in Google Scholar
Martin, J. M. L.; Taylor, P. R. Ab initio study of the isoelectronic molecules BCN, BNC, and C3 including anharmonicity. J. Phys. Chem. 1994, 98 (24), 6105–6109.10.1021/j100075a010Search in Google Scholar
Martin, J. M. L.; Taylor, P. R.; François, J. P.; Gijbels, R. Ab initio study of the spectroscopy and thermochemistry of the C2N and CN2 molecules. Chem. Phys. Lett. 1994, 226 (5–6), 475–483.10.1016/0009-2614(94)00758-6Search in Google Scholar
Matsuo, T.; Suga, H.; Seki, S. Thermodynamic properties and phase transitions of sodium cyanide crystal. Bull. Chem. Soc. Jpn. 1968, 41 (3), 583–593.10.1246/bcsj.41.583Search in Google Scholar
Maxwell, A. F.; Fry, J. S.; Bigelow, L. A. The indirect fluorination of cyanuric chloride. J. Am. Chem. Soc. 1958, 80 (3), 548–549.10.1021/ja01536a010Search in Google Scholar
Maya, L. Paracyanogen reexamined. J. Polym. Sci. Part A: Polym. Chem. 1993, 31 (10), 2595–2600.10.1002/pola.1993.080311020Search in Google Scholar
Mayer, E. Einfache Darstellung von Cyanfluorid. Angew. Chem. 1969a, 81 (16), 627.10.1002/ange.19690811614Search in Google Scholar
Mayer, E. Darstellung und Eigenschaften von Tetracyanmethan. Monatsh. Chem. 1969b, 100 (2), 462–468.10.1007/BF00904089Search in Google Scholar
Mayer, E. Simple preparation of cyanogen fluoride. Angew. Chem. Int. Ed. 1969c, 8 (8), 601.10.1002/anie.196906011Search in Google Scholar
McCarthy, M. C.; Apponi, A. J.; Gottlieb, C. A.; Thaddeus, P. Rotational spectra of SiCN, SiNC, and the SiCnH (N = 2, 4–6) radicals. J. Chem. Phys. 2001, 115 (2), 870–877.10.1063/1.1370068Search in Google Scholar
McCoy, A. B. Potential energy surfaces and properties of ICN− and ICN. Int. J. Quantum Chem. 2013, 113 (3), 366–374.10.1002/qua.24011Search in Google Scholar
McDonald, J. R.; Scherr, V. M.; McGlynn, S. P. Lower‐energy electronic states of HNCS, NCS−, and thiocyanate salts. J. Chem. Phys. 1969, 51 (5), 1723–1731.10.1063/1.1672256Search in Google Scholar
McEwan, M. J.; Anicich, V. G.; Huntress, W. T.; Kemper, P. R.; Bowers, M. T. Reactions of CN+ and C2N+ ions. Int. J. Mass Spectrom. Ion Phys. 1983, 50 (1–2), 179–187.10.1016/0020-7381(83)80008-4Search in Google Scholar
McGibbon, G. A.; Kingsmill, C. A.; Terlouw, J. K.; Burgers, P. C. The isomeric C2NO+ ions NCCO+, CNCO+, CCNO+ and their neutral counterparts are stable species in the gas phase. Int. J. Mass Spectrom. Ion Processes 1992, 121 (1–2), R11–R18.10.1016/0168-1176(92)80079-GSearch in Google Scholar
Meloni, G.; Gingerich, K. A. Thermodynamic investigation of the AlNC and AlCN Isomers by Knudsen cell mass spectrometry. J. Chem. Phys. 1999, 111 (3), 969–972.10.1063/1.479379Search in Google Scholar
Menzer, W. Zur Kenntnis des Germaniumtetracyanids. Angew. Chem. 1958, 70 (21), 656.10.1002/ange.19580702107Search in Google Scholar
Meot-Ner, M.; Speller, C. V. Multicomponent cluster ions. 2. Comparative stabilities of cationic and anionic hydrogen-bonded networks. Mixed clusters of water and hydrogen cyanide. J. Phys. Chem. 1989, 93 (9), 3663–3666.10.1021/j100346a058Search in Google Scholar
Meot-Ner, M.; Cybulski, S. M.; Scheiner, S.; Liebman, J. F. Is cyanide significantly anisotropic? Comparison of cyanide vs chloride: clustering with hydrogen cyanide and condensed-phase thermochemistry. J. Phys. Chem. 1988, 92 (10), 2738–2745.10.1021/j100321a009Search in Google Scholar
Meot-Ner, M. Solvation of the proton by hydrogen cyanide and acetonitrile. Condensation of hydrogen cyanide with ions in the gas phase. J. Am. Chem. Soc. 1978, 100 (15), 4694–4699.10.1021/ja00483a012Search in Google Scholar
Meredith, T. J.; Jacobsen, D.; Haines, J. A.; Berger, J. C.; Heijst, A. N. P. Antidotes for poisoning by cyanide, 2nd ed.; Cambridge University Press: Cambridge, 1993.Search in Google Scholar
Merer, A. J.; Travis, D. N. Absorption spectrum of the CCN radical. Can. J. Phys. 1965, 43 (10), 1795–1830.10.1139/p65-173Search in Google Scholar
Merer, A. J.; Travis, D. N. The absorption spectrum of CNC. Can. J. Phys. 1966, 44 (2), 353–372.10.1139/p66-029Search in Google Scholar
Messer, C. E.; Ziegler, W. T.III. Rotation of groups in ionic lattices. The heat capacities of sodium and potassium cyanides. J. Am. Chem. Soc. 1941, 63 (10), 2703–2708.10.1021/ja01855a058Search in Google Scholar
Metzger, F. J. Calcium cyanide – “powdered hydrocyanic acid”. Ind. Eng. Chem. 1926, 18 (2), 161–163.10.1021/ie50194a017Search in Google Scholar
Meyer, R. E. Zur Kenntnis des Indiums. Justus Liebigs Ann. Chem. 1869, 150 (2), 137–160.10.1002/jlac.18691500202Search in Google Scholar
Meyer, J. Zur Kenntnis Der Alkalicyanide. Z. Anorg. Allg. Chem. 1921, 115 (1), 203–217.10.1002/zaac.19211150114Search in Google Scholar
Middleton, W. J.; Little, E. L.; Coffman, D. D.; Engelhardt, V. A. Cyanocarbon chemistry. V. Cyanocarbon acids and their salts. J. Am. Chem. Soc. 1958, 80 (11), 2795–2806.10.1021/ja01544a055Search in Google Scholar
Mihrin, D.; Jakobsen, P. W.; Voute, A.; Manceron, L.; Wugt Larsen, R. High-resolution synchrotron terahertz investigation of the large-amplitude hydrogen bond librational band of (HCN)2. Phys. Chem. Chem. Phys. 2018, 20 (12), 8241–8246.10.1039/C7CP08412ASearch in Google Scholar
Mikhailov, V.; Wheeler, M. D.; Ellis, A. M. Laser-induced fluorescence spectroscopy of the BaNC free radical in a supersonic jet. J. Phys. Chem. 2003, 107 (22), 4367–4372.10.1021/jp027617iSearch in Google Scholar
Miller, F. A.; Wilkins, C. H. Infrared spectra and characteristic frequencies of inorganic ions. Anal. Chem. 1952, 24 (8), 1253–1294.10.1021/ac60068a007Search in Google Scholar
Miller, F. A.; Carlson, G. L.; Bentley, F. F.; Jones, W. H. Infrared spectra of inorganic ions in the cesium bromide region (700–300 cm−1). Spectrochim. Acta 1960, 16 (1–2), 135–235.10.1016/0371-1951(60)80077-XSearch in Google Scholar
Miller, F. A.; Frankiss, S. G.; Sala, O. Infrared and Raman spectra of P(CN)3 and as(CN)3. Spectrochim. Acta 1965, 21 (4), 775–781.10.1016/0371-1951(65)80035-2Search in Google Scholar
Miller, E. M.; Sheps, L.; Lu, Y.-J.; Case, A. S.; McCoy, A. B.; Carl Lineberger, W. New view of the ICN A continuum using photoelectron spectroscopy of ICN−. J. Chem. Phys. 2012, 136 (4), 044313.10.1063/1.3679170Search in Google Scholar PubMed
Milligan, D. E.; Jacox, M. E. Matrix‐isolation study of the photolysis of cyanogen azide. II. The symmetric stretching fundamental of the free radical NCN. J. Chem. Phys. 1966, 45 (5), 1387–1391.10.1063/1.1727771Search in Google Scholar
Milligan, D. E.; Jacox, M. E. Matrix‐isolation study of the infrared and ultraviolet spectra of the free radical NCO. J. Chem. Phys. 1967a, 47 (12), 5157–5168.10.1063/1.1701773Search in Google Scholar
Milligan, D. E.; Jacox, M. E. Spectroscopic study of the vacuum-ultraviolet photolysis of matrix-isolated HCN and halogen cyanides. Infrared spectra of the species CN and XNC. J. Chem. Phys. 1967b, 47 (1), 278–285.10.1063/1.1711859Search in Google Scholar
Milligan, D. E.; Jacox, M. E.; Bass, A. M. Matrix isolation study of the photolysis of cyanogen azide. The infrared and ultraviolet spectra of the free radical NCN. J. Chem. Phys. 1965, 43 (9), 3149–3160.10.1063/1.1697289Search in Google Scholar
Minkwitz, R.; Gerhard, V. Preparation and spectroscopic characterization of cyansulfonium salts. S(CN)3+AsF6– and (CH3)2SCN+MF6–. Z. Naturforsch. 1991, 46b (3), 265–269.10.1515/znb-1991-0301Search in Google Scholar
Minkwitz, R.; Hertel, T. Formation of cyanoiodine dinitrate NCI(ONO2)2. Z. Naturforsch. B: J. Chem. Sci. 1997, 52 (10), 1191–1193.10.1515/znb-1997-1007Search in Google Scholar
Minkwitz, R.; Nowicki, J.; Jahnkow, B.; Koch, M. Darstellung von Cyanhalogenmethyl- Und Dicyanhalogensulfoniumsalzen CH3(CN)SHal+MF6– und (CN)2SHal+MF6– (Hal = F, Cl, Br, I; M = As, Sb) sowie von Methylthionitrillium- und (μ-Thio)bisnitriliumhexafluorometallaten CH3SCNX+MF6– und S(CNH)22+ (MF6–)2 (X = H, D). Z. Anorg. Allg. Chem. 1991, 596 (1), 77–88.10.1002/zaac.19915960111Search in Google Scholar
Mishra, S.; Vallet, V.; Poluyanov, L. V.; Domcke, W. Calculation of the vibronic structure of the X̃2Π photoelectron spectra of XCN, X = F, Cl, and Br. J. Chem. Phys. 2006, 124 (4), 044317.10.1063/1.2161186Search in Google Scholar PubMed
Moffat, J. B.; Tang, K. F. A theoretical study of the reactive dimerization of HCN. J. Theor. Biol. 1976, 58 (1), 83–95.10.1016/0022-5193(76)90140-5Search in Google Scholar PubMed
Moffat, J. B. A quantum-chemical Study of BCN. J. Mol. Struct. 1971, 7 (3–4), 474–477.10.1016/0022-2860(71)87017-5Search in Google Scholar
Moffat, J. B. Cyanide-isocyanide isomerization in the structural isomers of cyanogen isocyanate. Int. J. Quantum Chem. 1979, 15 (5), 547–557.10.1002/qua.560150510Search in Google Scholar
Mokross, B. J.; Pirc, R. Elastic interaction of tunneling impurities in solids. J. Chem. Phys. 1978, 68 (11), 4823–4831.10.1063/1.435662Search in Google Scholar
Moreno, M. A.; Maté, B.; Rodríguez-Lazcano, Y.; Gálvez, O.; Gómez, P. C.; Herrero, V. J.; Escribano, R. The structure and spectroscopy of cyanate and bicarbonate ions. Astrophysical implications. J. Phys. Chem. 2013, 117 (39), 9564–9573.10.1021/jp3122616Search in Google Scholar PubMed
Morris, S.; Wyller, A. A. Molecular dissociative equilibria in carbon stars. Astrophys. J. 1967, 150, 877–907.10.1086/149390Search in Google Scholar
Morris, D. F. C. Crystal radius of the cyanide ion. Acta Crystallogr. 1961, 14 (5), 547–548.10.1107/S0365110X6100173XSearch in Google Scholar
Mowry, D. T. The preparation of nitriles. Chem. Rev. 1948, 42 (2), 189–283.10.1021/cr60132a001Search in Google Scholar PubMed
Mudder, T.; Botz, M.; Smith, A. Chemistry and treatment of cyanidation wastes, 2nd ed.; Mining Journal Books Ltd: London, 1991.Search in Google Scholar
Müller, P.; Müller-Dolezal, H.; Stoltz, R.; Söll, H. Peroxides, carbonic acid derivatives, carboxylic acids, carboxylic acid derivatives. In Methods of organic chemistry; Thieme - Houben Weyl: Stuttgart, 2014; p. 775.Search in Google Scholar
Müller, U. Darstellung, Eigenschaften Und Schwingungsspektren von K[SbCl5N3], K[SbCl5NCO] Und K[SbCl5CN]. Z. Anorg. Allg. Chem. 1973, 396 (2), 187–198.10.1002/zaac.19733960210Search in Google Scholar
Murray, J. S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007a, 107 (12), 2286–2292.10.1002/qua.21352Search in Google Scholar
Murray, J. S.; Lane, P.; Clark, T.; Politzer, P. σ-Hole bonding: molecules containing group VI atoms. J. Mol. Model. 2007b, 13 (10), 1033–1038.10.1007/s00894-007-0225-4Search in Google Scholar PubMed
Murrell, J. N.; Derzi, A. A. Calculations on the ground states of HCN+ and HNC+. J. Chem. Soc., Faraday Trans. 2 1980, 76 (3), 319–323.10.1039/f29807600319Search in Google Scholar
Muthmann, W.; Schröder, E. Einige Beobachtungen Über Cyanselenverbindungen. Ber. Dtsch. Chem. Ges. 1900, 33 (2), 1765–1769.10.1002/cber.19000330256Search in Google Scholar
Nagarajan, G. Potential field and force constants of phosphorus and arsenic tricyanides. Proc. Int. Conf. Cosmic Rays, 11th 1966, 20 (4), 323–329.10.1007/BF03156708Search in Google Scholar
Nagy, P.; Fischer, A.; Glaser, J.; Ilyukhin, A.; Maliarik, M.; Tóth, I. Solubility, complex formation, and redox reactions in the Tl2O3−HCN/CN−−H2O system. Crystal structures of the cyano compounds Tl(CN)3·H2O, Na[Tl(CN)4]·3H2O, K[Tl(CN)4], and TlI[TlIII(CN)4] and of TlI2C2O4. Inorg. Chem. 2005, 44 (7), 2347–2357.10.1021/ic0485918Search in Google Scholar PubMed
Nambu, H. X-ray diffraction study of KOCN at room temperature. J. Phys. Chem. Solids 2003, 64 (11), 2269–2272.10.1016/S0022-3697(03)00258-0Search in Google Scholar
Nauta, K.; Miller, R. E.; Series, N.; Mar, N. Nonequilibrium self-assembly of long chains of polar molecules in superfluid helium. Science 1999, 283 (5409), 1895–1897.10.1126/science.283.5409.1895Search in Google Scholar PubMed
Nauth, A. M.; Opatz, T. Non-toxic cyanide sources and cyanating agents. Org. Biomol. Chem. 2019, 17 (1), 11–23.10.1039/C8OB02140FSearch in Google Scholar
Nenajdenko, V. Fluorine in heterocyclic chemistry, Vol. 2; Nenajdenko, V., Ed.; Springer International Publishing: Cham, 2014.10.1007/978-3-319-04435-4Search in Google Scholar
Neubert, T. J.; Susman, S. Purification and single‐crystal growth of potassium cyanide. J. Chem. Phys. 1964, 41 (3), 722–729.10.1063/1.1725951Search in Google Scholar
Neubner, R. Die Umwandlungsgleichung Ba(CN)2 → BaCN2 + C Im Temperaturgebiet von 500 Bis 1000 °C. Z. Elektrochem. Angew. Phys. Chem. 1934, 40 (10), 693–698.10.1002/bbpc.19340401005Search in Google Scholar
Neukirch, M.; Tragl, S.; Meyer, H.-J.; Küppers, T.; Willner, H. M[B(CN)4]2: Zwei Neue Tetracyanoborate Mit Zweiwertigen Kationen (M = Zn, Cu). Z. Anorg. Allg. Chem. 2006, 632 (6), 939–944.10.1002/zaac.200500421Search in Google Scholar
Newhouse, K.; Chiu, N. Toxicological review of hydrogen cyanide and cyanide salts; EPA - United States Environmental Protection Agency: Washington D.C., 2010.Search in Google Scholar
Ng, V. M.; Xu, M.; Huang, S. Y.; Long, J. D.; Xu, S. Assembly and photoluminescence of SiCN nanoparticles. Thin Solid Films 2006, 506–507, 283–287.10.1016/j.tsf.2005.08.075Search in Google Scholar
Ngassam, V.; Orel, A. E.; Suzor-Weiner, A. Ab initio study of the dissociative recombination of HCNH+. J. Phys. Conf. Ser. 2005, 4, 224–228.10.1088/1742-6596/4/1/032Search in Google Scholar
Nitschke, C.; Köckerling, M. A new transition metal tetracyanidoborate: synthesis, structure and properties of Co[B(CN)4]2·2H2O. Z. Anorg. Allg. Chem. 2009, 635 (3), 503–507.10.1002/zaac.200801234Search in Google Scholar
Nitschke, C.; Köckerling, M. Iron salts with the tetracyanidoborate anion: [FeIII(H2O)6][B(CN)4]3, coordination polymer [FeII(H2O)2{κ2N[B(CN)4]}2], and [FeII(DMF)6][B(CN)4]2. Inorg. Chem. 2011, 50 (10), 4313–4321.10.1021/ic102278zSearch in Google Scholar PubMed
Nitschke, C.; Köckerling, M.; Bernhardt, E.; Küppers, T.; Willner, H. Structural diversity and spectral and thermal properties of the first alkaline earth metal tetracyanidoborates: [Mg(H2O)6][B(CN)4]2, [Mg(H2O)2][B(CN)4]2, [Mg(DMF)6][B(CN)4]2, [Ca(H2O)3][B(CN)4]2, and [Ca(H2O)2(CH3CN)][B(CN)4]2. Dalton Trans. 2014, 43 (19), 7128–7138.10.1039/C3DT51945GSearch in Google Scholar
Nolan, M. F.; Pendlebury, J. N.; Smith, R. H. Kinetics of the reactions Br2 + HCN, BrCN + I−, S(CN)2 + I− in aqueous acid solution. Int. J. Chem. Kinet. 1975, 7 (2), 205–214.10.1002/kin.550070205Search in Google Scholar
Norbury, A. H.; Thompson, M.; Songstad, J. X-ray photoelectron spectroscopy of the cynate, thiocyanate, selenocyanate and tellurocyanate ions. Inorg. Nucl. Chem. Lett. 1973, 9 (3), 347–350.10.1016/0020-1650(73)80243-0Search in Google Scholar
North, M.; Omedes-Pujol, M.; Young, C. Kinetics and mechanism of the racemic addition of trimethylsilyl cyanide to aldehydes catalysed by Lewis bases. Org. Biomol. Chem. 2012, 10 (21), 4289–4298.10.1039/c2ob25188dSearch in Google Scholar PubMed
Northrup, F. J.; Sears, T. J. Laser‐induced fluorescence spectroscopy of NCS in a free jet expansion. J. Chem. Phys. 1989, 91 (2), 762–774.10.1063/1.457128Search in Google Scholar
Northrup, F. J.; Sears, T. J. Renner–Teller Spin-orbit and Fermi-resonance interactions in X2Π NCS Investigated by LIF spectroscopy. Mol. Phys. 1990, 71 (1), 45–64.10.1080/00268979000101631Search in Google Scholar
Noshita, T.; Kidachi, Y.; Funayama, H.; Kiyota, H.; Yamaguchi, H.; Ryoyama, K. Anti-nitric oxide production activity of isothiocyanates correlates with their polar surface area rather than their lipophilicity. Eur. J. Med. Chem. 2009, 44 (12), 4931–4936.10.1016/j.ejmech.2009.08.005Search in Google Scholar PubMed
Oberhammer, H. Die Molekülstruktur des Tetracyanmethans. Z. Naturforsch., A: Phys. Sci. 1971, 26a (12), 2043–2046.10.1515/zna-1971-1213Search in Google Scholar
Oberhauser, F. Das Verhalten des Bromcyans Gegenüber Metallsalzen. Ber. Dtsch. Chem. Ges. 1927, 60 (6), 1434–1439.10.1002/cber.19270600628Search in Google Scholar
Ohshima, Y.; Endo, Y. Fourier-transform microwave spectroscopy of CCN (X 2Π1/2). J. Mol. Spectrosc. 1995, 172 (1), 225–232.10.1006/jmsp.1995.1170Search in Google Scholar
Ohtoshi, H.; Tsukiyama, K.; Yanagibori, A.; Shibuya, K.; Obi, K.; Tanaka, K. Laser induced fluorescence of NCS in the gas phase. Chem. Phys. Lett. 1984, 111 (1–2), 136–140.10.1016/0009-2614(84)80451-0Search in Google Scholar
Okabayashi, T.; Tanimoto, M. Millimeter and submillimeter wave spectroscopy of HNC and DNC in the vibrationally excited states. J. Chem. Phys. 1993, 99 (5), 3268–3271.10.1063/1.465135Search in Google Scholar
Okabe, H.; Mele, A. Photodissociation of NCN3 in the vacuum‐ultraviolet production of CN B2Σ and NCN A3Π. J. Chem. Phys. 1969, 51 (5), 2100–2106.10.1063/1.1672305Search in Google Scholar
Okabe, H. Photodissociation of HNCO in the vacuum ultraviolet; production of NCO A2Σ and NH(A3π, πc1). J. Chem. Phys. 1970, 53 (9), 3507–3515.10.1063/1.1674525Search in Google Scholar
Okubo, T.; Ise, N. Catalysis of the ammonium cyanate-urea conversion by polyelectrolytes. Proc. R. Soc. London, Ser. A 1972, 327 (1570), 413–424.10.1098/rspa.1972.0054Search in Google Scholar
Olah, G. A.; Kiovsky, T. E. Stable carbonium ions. LXV. Protonation of hydrogen cyanide and alkyl nitriles in FSO3H-SbF5-SO2 solution. Comparative study of Meerwein’s N-alkylnitrilium ions. J. Am. Chem. Soc. 1968, 90 (17), 4666–4672.10.1021/ja01019a028Search in Google Scholar
Olah, G. A.; White, A. M. Stable carbonium ions. XCI. Carbon-13 nuclear magnetic resonance spectroscopic study of carbonium ions. J. Am. Chem. Soc. 1969, 91 (21), 5801–5810.10.1021/ja01049a017Search in Google Scholar
Oliphant, N.; Lee, A.; Bernath, P. F.; Brazier, C. R. Fourier transform emission spectroscopy of the jet‐cooled CCN free radical. J. Chem. Phys. 1990, 92 (4), 2244–2247.10.1063/1.458016Search in Google Scholar
Olsson, L.; Ottosson, C.-H.; Cremer, D. Properties of R3SiX compounds and R3Si+ ions: do silylium ions exist in solution? J. Am. Chem. Soc. 1995, 117 (28), 7460–7479.10.1021/ja00133a019Search in Google Scholar
Ong, W.-J.; Tan, L.-L.; Ng, Y. H.; Yong, S.-T.; Chai, S.-P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 2016, 116 (12), 7159–7329.10.1021/acs.chemrev.6b00075Search in Google Scholar PubMed
Onyszchuk, M.; Castel, A.; Riviere, P.; Satge, J. Germanium(II) pseudohalides: Ge(CN)2, Ge(NCO)2 and Ge(NCS)2; syntheses and reactivities. J. Organomet. Chem. 1986, 317 (3), C35–C37.10.1016/0022-328X(86)80548-4Search in Google Scholar
Ortiz-Lopez, J.; Luty, F. Optical studies of thermal cycling and hysteresis effects in elastic order-disorder phase transformations. I. Pure alkali-metal cyanide crystals. Phys. Rev. B 1988, 37 (10), 5452–5460.10.1103/PhysRevB.37.5452Search in Google Scholar
Osei Owusu, A.; Martin, G. C.; Gotro, J. T. Triazine formation in cyanate-based resin systems at room temperature conditions. Polymer. 1996, 37 (21), 4869–4872.10.1016/S0032-3861(96)00407-7Search in Google Scholar
Ott, L.; Rijksen, C.; Ellinger, S.; Sievert, K.; Harloff, J.; Schulz, A. Method for the preparation of tetraalkylammonium tetracyanidoborates. WO 2014029834 A1 20140227, 2014a.Search in Google Scholar
Ott, L.; Rijksen, C.; Ellinger, S.; Sievert, K.; Harloff, J.; Schulz, A. Method for the preparation of tetraalkylammonium or tetraalkylphosphonium tricyanidofluoroborates. WO 2014029833 A1 20140227, 2014b.Search in Google Scholar
Pacansky, J.; Dalal, N. S.; Bagus, P. S. SCF ab-initio ground state potential energy surfaces for HCN and HCN−. Chem. Phys. 1978, 32 (2), 183–187.10.1016/0301-0104(78)87050-5Search in Google Scholar
Pacansky, J. The infrared spectrum of a molecular aggregate. The hydrocyanic acid dimer isolated in an argon matrix. J. Phys. Chem. 1977, 81 (24), 2240–2243.10.1021/j100539a007Search in Google Scholar
Palatinszky, M.; Herbold, C.; Jehmlich, N.; Pogoda, M.; Han, P.; Von Bergen, M.; Lagkouvardos, I.; Karst, S. M.; Galushko, A.; Koch, H.; Berry, D.; Daims, H.; Wagner, M. Cyanate as an energy source for nitrifiers. Nature 2015, 524 (7563), 105–108.10.1038/nature14856Search in Google Scholar PubMed PubMed Central
Pan, S.; Gupta, A.; Saha, R.; Merino, G.; Chattaraj, P. K. A coupled-cluster study on the noble gas binding ability of metal cyanides versus metal halides (metal = Cu, Ag, Au). J. Comput. Chem. 2015, 36 (29), 2168–2176.10.1002/jcc.24190Search in Google Scholar PubMed
Panas, I. A self-consistent crystal field approach to the structures of molecular crystals applied to solid HCN. Chem. Phys. Lett. 1992, 194 (3), 239–246.10.1016/0009-2614(92)85541-HSearch in Google Scholar
Panas, I. Two hypothetical structures of solid FCN. Theor. Chim. Acta 1994, 87 (4–5), 335–342.10.1007/BF01113389Search in Google Scholar
Parent, D. C. Reactions of the carbene ions CnN+ with labeled methane: mechanistic interpretation. J. Am. Chem. Soc. 1990, 112 (16), 5966–5973.10.1021/ja00172a009Search in Google Scholar
Park, D. J.; Stern, A. G.; Wilier, R. L. A convenient laboratory preparation of cyanogen. Synth. Commun. 1990, 20 (18), 2901–2906.10.1080/00397919008051503Search in Google Scholar
Park, J. The cyanide process of gold extraction; Nabu Press: London, 2014.Search in Google Scholar
Parry, G. S. Studies in domain crystallography. II. A new low-temperature crystal structure of potassium cyanide. Acta Crystallogr. 1962, 15 (6), 601–607.10.1107/S0365110X62001516Search in Google Scholar
Pascoli, G.; Lavendy, H. Are CnN− clusters really bent? Chem. Phys. Lett. 1999, 312 (2–4), 333–340.10.1016/S0009-2614(99)00963-XSearch in Google Scholar
Pasinszki, T. Quantum-chemical study of the structure and stability of pseudohalogens: OCN–NCO and its isomers. Phys. Chem. Chem. Phys. 2008, 10 (10), 1411.10.1039/b713832fSearch in Google Scholar PubMed
Pasternack, L.; Dagdigian, P. J. Laser‐fluorescence study of the reactions of alkaline earth atoms with BrCN: spectroscopic observation of the alkaline earth monocyanides. J. Chem. Phys. 1976, 65 (4), 1320–1326.10.1063/1.433228Search in Google Scholar
Pau, C. F.; Hehre, W. J. Heat of formation of hydrogen isocyanide by ion cyclotron double resonance spectroscopy. J. Phys. Chem. 1982, 86 (3), 321–322.10.1021/j100392a006Search in Google Scholar
Paul, D. E.; Dalby, F. W. Kinetics of disappearance of the CN radical formed from C2N2. J. Chem. Phys. 1962, 37 (3), 592–598.10.1063/1.1701380Search in Google Scholar
Pauling, L.; Springall, H. D.; Palmer, K. J. The electron diffraction investigation of methylacetylene, dimethylacetylene, dimethyldiacetylene, methyl cyanide, diacetylene, and cyanogen. J. Am. Chem. Soc. 1939, 61 (4), 927–937.10.1021/ja01873a047Search in Google Scholar
Pauling, L. The rotational motion of molecules in crystals. Phys. Rev. 1930, 36 (3), 430–443.10.1103/PhysRev.36.430Search in Google Scholar
Pauling, L. Die Natur Der Chemischen Bindung, 3. Aufl.; Verl. Chemie: Weinheim/Bergstr., 1968.Search in Google Scholar
Pearson, P. K.; Schaefer, H. F. Some properties of H2CN+: a potentially important interstellar species. Astron. J. 1974, 192 (2), 33–36.10.1086/153031Search in Google Scholar
Pearson, P. K.; Schaefer, H. F.; Wahlgren, U. Potential energy surface for the model unimolecular reaction HNC → HCN. J. Chem. Phys. 1975, 62 (2), 350–354.10.1063/1.430492Search in Google Scholar
Pearson, R. G. Hard and soft acids and bases, HSAB, Part 1: fundamental principles. J. Chem. Educ. 1968, 45 (9), 581–587.10.1021/ed045p581Search in Google Scholar
Pedersen, L. The structure of the triatomic free radicals HCO, HCN–, and FCO. J. Mol. Struct. 1970, 5 (1–2), 21–26.10.1016/0022-2860(70)87018-1Search in Google Scholar
Penna-Franca, E.; Dodson, R. W. The effect of cyanide on the rate of the Thallous-Thallic exchange reaction. J. Am. Chem. Soc. 1955, 77 (9), 2651–2653.10.1021/ja01614a098Search in Google Scholar
Penneman, R. A.; Staritzky, E. Infra-red and crystallographic investigation of the TlCN–KCN–H2O system; the lack of Tl(I)-cyanide complexes. J. Inorg. Nucl. Chem. 1958, 6 (2), 112–118.10.1016/0022-1902(58)80055-XSearch in Google Scholar
Penzias, A. A.; Wilson, R. W.; Jefferts, K. B. Hyperfine structure of the CN radical determined from astronomical observations. Phys. Rev. Lett. 1974, 32 (13), 701–703.10.1103/PhysRevLett.32.701Search in Google Scholar
Perry, J. H.; Bardwell, D. C. The vapor pressures of solid and liquid cyanogen. J. Am. Chem. Soc. 1925, 47 (11), 2629–2632.10.1021/ja01688a002Search in Google Scholar
Petersen, G.; Franck, H. H. Beiträge Zur Kenntnis des Calciumcyanides, Insbesondere Zur Umwandlung Calciumcyanid ⇄ Calciumcyanamid. Z. Anorg. Allg. Chem. 1938, 237 (1), 1–37.10.1002/zaac.19382370102Search in Google Scholar
Peterson, P. E.; Abu-Omar, M.; Johnson, T. W.; Parham, R.; Goldin, D.; Henry, C.; Cook, A.; Dunn, K. M. Ab initio predictions of vibrational frequencies for cationic species. J. Phys. Chem. 1995, 99 (16), 5927–5933.10.1021/j100016a030Search in Google Scholar
Petrie, S.; Freeman, C. G.; Meot-Ner, M.; McEwan, M. J.; Ferguson, E. E. Experimental study of HCN+ and HNC+ ion chemistry. J. Am. Chem. Soc. 1990, 112 (20), 7121–7126.10.1021/ja00176a006Search in Google Scholar
Petrie, S. Structural trends in the monocyanides of the second-row metal ions Na+, Mgm+ (m = 1, 2), and Aln+ (n = 1−3). J. Phys. Chem. 1996, 100 (28), 11581–11588.10.1021/jp960285vSearch in Google Scholar
Petrie, S. Trends in M(CN) isomerism: a computational study of monocyanides of the main-group third row atoms. Phys. Chem. Chem. Phys. 1999a, 1 (12), 2897–2905.10.1039/a901143iSearch in Google Scholar
Petrie, S. Magnesium dicyanide: three isomers or seven? J. Phys. Chem. 1999b, 103 (13), 2107–2116.10.1021/jp983401ySearch in Google Scholar
Petrov, P. K.; Charters, J. W.; Wallschläger, D. Identification and determination of selenosulfate and selenocyanate in flue gas desulfurization waters. Environ. Sci. Technol. 2012, 46 (3), 1716–1723.10.1021/es202529wSearch in Google Scholar PubMed
Pettersson, M.; Lundell, J.; Khriachtchev, L.; Räsänen, M. Neutral rare-gas containing charge-transfer molecules in solid matrices. III. HXeCN, HXeNC, and HKrCN in Kr and Xe. J. Chem. Phys. 1998, 109 (2), 618–625.10.1063/1.476599Search in Google Scholar
Pézolet, M.; Savoie, R. Raman spectra of liquid and crystalline HCN and DCN. Can. J. Chem. 1969, 47 (16), 3041–3048.10.1139/v69-502Search in Google Scholar
Pézolet, M.; Savoie, R. Raman spectra of liquid and crystalline ClCN and BrCN. J. Chem. Phys. 1971, 54 (12), 5266–5270.10.1063/1.1674824Search in Google Scholar
Pham, T. A.; Kim, D.-P.; Lim, T.-W.; Park, S.-H.; Yang, D.-Y.; Lee, K.-S. Three-dimensional SiCN ceramic microstructures via nano-stereolithography of inorganic polymer photoresists. Adv. Funct. Mater. 2006, 16 (9), 1235–1241.10.1002/adfm.200600009Search in Google Scholar
Pham-Tran, N.-N.; Hou, X. J.; Nguyen, M. T. On the nature of the CP group adjacent to a valence-deficient atom: phosphaethynyl substituent vs. phosphorus center. J. Phys. Org. Chem. 2006, 19 (3), 167–172.10.1002/poc.1021Search in Google Scholar
Plumet, J.; Roscales, S. Mini-review: organic catalysts in the 1,3-dipolar cycloaddition reactions of nitrile oxides. Heterocycles 2019, 99 (2), 725.10.3987/REV-18-SR(F)4Search in Google Scholar
Poletto, G.; Rigutti, M. The A2Π and X2Σ states of the CN molecule from the Berkeley analysis of the CN red system. Nuovo Cimento 1965, 39 (2), 519–530.10.1007/BF02735821Search in Google Scholar
Politzer, P.; Murray, J. S.; Lane, P. σ-Hole bonding and hydrogen bonding: competitive interactions. Int. J. Quantum Chem. 2007, 107 (15), 3046–3052.10.1002/qua.21419Search in Google Scholar
Pople, J. A.; Krishnan, R.; Schlegel, H. B.; Binkley, J. S. Electron correlation theories and their application to the study of simple reaction potential surfaces. Int. J. Quantum Chem. 1978, 14 (5), 545–560.10.1002/qua.560140503Search in Google Scholar
Poppinger, D.; Radom, L. A. Theoretical study of substituted CHNO isomers. J. Am. Chem. Soc. 1978, 100 (12), 3674–3685.10.1021/ja00480a004Search in Google Scholar
Price, C. J.; Chen, H.-Y.; Launer, L. M.; Miller, S. A. Weakly coordinating cations as alternatives to weakly coordinating anions. Angew. Chem. Int. Ed. 2009, 48 (5), 956–959.10.1002/anie.200802605Search in Google Scholar PubMed
Provasi, P. F.; Aucar, G. A.; Sanchez, M.; Alkorta, I.; Elguero, J.; Sauer, S. P. A. Interaction energies and NMR indirect nuclear spin-spin coupling constants in linear NCH and CNH complexes. J. Phys. Chem. 2005, 109, 6555–6564.10.1021/jp051600tSearch in Google Scholar PubMed
Purdy, A. P.; Houser, E.; George, C. F. Lithium dicyanamide, its reactions with cyanuric chloride, and the crystal structures of LiN(CN)2(MeCN)2 and LiCN(C5H5N)2. Polyhedron 1997, 16 (20), 3671–3679.10.1016/S0277-5387(97)00097-1Search in Google Scholar
Pyykkö, P.; Atsumi, M. Molecular double-bond covalent radii for elements Li-E112. Chem. Eur J. 2009, 15 (46), 12770–12779.10.1002/chem.200901472Search in Google Scholar PubMed
Pyykkö, P.; Runeberg, N. Ab initio studies of bonding trends. J. Mol. Struct.: Theochem. 1991, 234 (C), 269–277.10.1016/0166-1280(91)89017-USearch in Google Scholar
Pyykko, P.; Zhao, Y. Ab initio study of bonding trends. 4. The 22-electron A=B=C series: possible new anions down to NCB4- and possible new cations up to FNF3+. J. Phys. Chem. 1990, 94 (20), 7753–7759.10.1021/j100383a002Search in Google Scholar
Qin, L.; Yu, J.; Kuang, S.; Xiao, C.; Bai, X. Few-atomic-layered boron carbonitride nanosheets prepared by chemical vapor deposition. Nanoscale 2012, 4 (1), 120–123.10.1039/C1NR11387ASearch in Google Scholar
Rabalais, J. W.; McDonald, J. R.; McGlynn, S. P. Electronic states of HNCO, cyanate salts, and organic isocyanates. II. Absorption studies. J. Chem. Phys. 1969a, 51 (11), 5112–5117.10.1063/1.1671908Search in Google Scholar
Rabalais, J. W.; McDonald, J. R.; McGlynn, S. P. Electronic states of HNCO, cyanate salts, and organic isocyanates. I. Luminescence studies. J. Chem. Phys. 1969b, 51 (11), 5095–5102.10.1063/1.1671907Search in Google Scholar
Radford, H. E.; Broida, H. P. Rotational perturbations in CN. Zero-field theory, optical zeeman effect, and microwave transition probabilities. Phys. Rev. 1962, 128 (1), 231–242.10.1103/PhysRev.128.231Search in Google Scholar
Raksit, A. B.; Bohme, D. K. Flow tube studies of reactions of cyanogen with ions selected from cyanogen. Int. J. Mass Spectrom. Ion Processes 1985, 63 (2–3), 217–229.10.1016/0168-1176(85)80026-4Search in Google Scholar
Ramos, M. N.; Taft, C. A.; Tostes, J. G. R.; Lester, W. A. An spectroscopic study of the HCN…HCN and HCN…HNC linear complexes. J. Mol. Struct. 1988, 175, 303–306.10.1016/S0022-2860(98)80092-6Search in Google Scholar
Rao, V. S.; Vijay, A.; Chandra, A. K. A comparative study of the energetics, structures, and mechanisms of the HCN ↔ HNC and LiCN ↔ LiNC isomerizations. Can. J. Chem. 1996, 74 (6), 1072–1077.10.1139/v96-120Search in Google Scholar
Raschig, F. Zur Kenntniss des Jodstickstoffs. Justus Liebigs Ann. Chem. 1885, 230 (2), 212–221.10.1002/jlac.18852300203Search in Google Scholar
Rastogi, M. K. Synthesis and characterization of some cyclopentadienylantimony(III) pseudohalides. Synth. React. Inorg. Met.-Org. Chem. 1987, 17 (5), 525–537.10.1080/00945718708070216Search in Google Scholar
Reckeweg, O.; Schulz, A.; Leonard, B.; DiSalvo, F. J. Single-crystal X-ray diffraction study of Na[OCN] at 170 K and its vibrational spectra. Z. Naturforsch. B 2010, 65 (4), 528–532.10.1515/znb-2010-0416Search in Google Scholar
Reddy, P. N.; Srikanth, R.; Bhanuprakash, K.; Srinivas, R. Generation and characterization of ionic and neutral methylene isothiocyanate by a combined tandem mass spectrometry and computational study. Rapid Commun. Mass Spectrom. 2004, 18 (17), 1939–1946.10.1002/rcm.1574Search in Google Scholar
Redmon, L. T.; Purvis, G. D.; Bartlett, R. J. Correlation effects in the isomeric cyanides: HNC ↔ HCN, LiNC ↔ gLiCN, and BNC ↔ gBCN. J. Chem. Phys. 1980, 72 (2), 986–991.10.1063/1.439218Search in Google Scholar
Reeve, S. W.; Burns, W. A.; Lovas, F. J.; Suenram, R. D.; Leopold, K. R. Microwave spectra and structure of hydrogen cyanide-boron trifluoride: an almost weakly bound complex. J. Phys. Chem. 1993, 97 (41), 10630–10637.10.1021/j100143a018Search in Google Scholar
Reghunadhan Nair, C. P.; Mathew, D.; Ninan, K. N. Cyanate ester resins, recent developments. Advances in polymer science. Springer, Berlin, Heidelberg 2001, pp. 1–99.Search in Google Scholar
Rehwald, W.; Sandercock, J. R.; Rossinelli, M. Elastic properties of KCN and K(CN)1−xClx. Phys. Status Solidi 1977, 42 (2), 699–705.10.1002/pssa.2210420235Search in Google Scholar
Reifsig, T. Über Einige Rubidiumverbindungen. Justus Liebigs Ann. Chem. 1863, 127 (1), 33–37.10.1002/jlac.18631270104Search in Google Scholar
Renz, F.; Jung, S.; Klein, M.; Menzel, M.; Thünemann, A. F. Molecular switching complexes with iron and tin as central atom. Polyhedron 2009a, 28 (9–10), 1818–1821.10.1016/j.poly.2008.12.028Search in Google Scholar
Renz, F.; Zaba, C.; Roßberg, L.; Jung, S.; Klein, M.; Klingelhöfer, G.; Wünsche, A.; Reinhardt, S.; Menzel, M. Spin transition in heptanuclear star-shaped iron(III)–antimony(V) NCS- and CN-bridged compounds. Polyhedron 2009b, 28 (9–10), 2036–2038.10.1016/j.poly.2008.12.027Search in Google Scholar
Rhimi, B.; Wang, C.; Bahnemann, D. W. Latest progress in g-C3N4 based heterojunctions for hydrogen production via photocatalytic water splitting: a mini review. J. Phys. Energy 2020, 2 (4), 042003.10.1088/2515-7655/abb782Search in Google Scholar
Richardson, N. A.; Yamaguchi, Y.; Schaefer, H. F. Isomerization of the interstellar molecule silicon cyanide to silicon isocyanide through two transition states. J. Chem. Phys. 2003, 119 (24), 12946–12955.10.1063/1.1627290Search in Google Scholar
Riemenschneider, W. Neue Einstufen-Synthese von Dicyan Oder Oxamid aus Blausäure. Chem. Ing. Tech. 1978, 50 (1), 55.10.1002/cite.330500116Search in Google Scholar
Riggs, N.; Radom, L. Ab initio studies on amides: cyanamide, dicyanamide and tricyanamide. Aust. J. Chem. 1985, 38 (6), 835.10.1071/CH9850835Search in Google Scholar
Robles, N. L.; Flores Antognini, A.; Romano, R. M. Formation of XNCO species (X=F, Cl) through matrix-isolation photochemistry of XSO2NCO molecules. J. Photochem. Photobiol., A 2011, 223 (2–3), 194–201.10.1016/j.jphotochem.2011.08.018Search in Google Scholar
Roesky, H. W.; Dhathathreyan, K. S. Insertion of P(CN)3 and as(CN)3 as their isonitrile forms into the dimer of hexafluorothioacetone. J. Chem. Soc., Chem. Commun. 1984, 1053–1054.10.1039/c39840001053Search in Google Scholar
Roesky, H. W.; Dhathathreyan, K. S.; Noltemeyer, M.; Sheldrick, G. M. Reactions of the hexafluorothioacetone dimer with cyanides of phosphorus, arsenic and germanium. Z. Naturforsch., B: J. Chem. Sci. 1985, 40 (2), 240–246.10.1515/znb-1985-0216Search in Google Scholar
Roesky, H. W. Preparation of tetrachlorodicyanophosphates and the existence of tetrachlorodifluorophosphates. Angew. Chem. Int. Ed. 1967, 6 (4), 363.10.1002/anie.196703631Search in Google Scholar
Rogers, M. T.; Gross, K. J. The electric moments of some sulfur and selenium compounds. J. Am. Chem. Soc. 1952, 74 (21), 5294–5296.10.1021/ja01141a018Search in Google Scholar
Romano, J. A.; Lukey, B. J.; Salem, H. Chemical warfare agents chemistry, pharmacology, toxicology, and therapeutics; CRC Press: Boca Raton, 2007.Search in Google Scholar
Root, K. D. J.; Symons, M. C. R.; Weatherley, B. C. Electron spin resonance spectrum of HCN− in alkali halide matrices. Mol. Phys. 1966, 11 (2), 161–164.10.1080/00268976600100991Search in Google Scholar
Rösslein, M.; Hanratty, M. A.; Maier, J. P. The rs-structure of bromocyanogen cation: BrCN+ [Xtilde]2Π. Mol. Phys. 1989, 68 (4), 823–833.10.1080/00268978900102571Search in Google Scholar
Rossmanith, K. Neue Wege Zur Herstellung von Reinem Lithiumcyanid und Lithiumcyanoargentat. Monatsh. Chem. 1965, 96 (6), 1690–1694.10.1007/BF01185887Search in Google Scholar
Roth, K. Berliner Blau: Alte Farbe in Neuem Glanz. Chem. Unserer Zeit 2003, 37 (2), 150–151.10.1002/ciuz.200390024Search in Google Scholar
Rowe, J. M.; Rush, J. J.; Prince, E.; Chesser, N. J. Neutron scattering studies of crystal dynamics and order-disorder phase transitions in alkali cyanides. Ferroelectrics 1977a, 16 (1), 107–109.10.1080/00150197708237131Search in Google Scholar
Rowe, J. M.; Rush, J. J.; Prince, E. Neutron diffraction study of the structure and phase transitions of alkali cyanide crystals. J. Chem. Phys. 1977b, 66 (11), 5147–5149.10.1063/1.433776Search in Google Scholar
Rowe, J. M.; Rush, J. J.; Susman, S. Neutron powder diffraction study of phase transitions and structures of (KCN)x(KBr)1−x mixed crystals. Phys. Rev. B 1983, 28 (6), 3506–3511.10.1103/PhysRevB.28.3506Search in Google Scholar
Rowe, J. M.; Rush, J. J.; Lüty, F. Crystal structure of rubidium cyanide at 4 K determined by neutron powder diffraction. Phys. Rev. B 1984, 29 (4), 2168–2170.10.1103/PhysRevB.29.2168Search in Google Scholar
Rubo, A.; Kellens, R.; Reddy, J.; Steier, N.; Hasenpusch, W. Alkali metal cyanides. In Ullmann’s encyclopedia of industrial chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 1–16.10.1002/14356007.i01_i01Search in Google Scholar
Rüger, C. P.; Maillard, J.; Le Maître, J.; Ridgeway, M.; Thompson, C. J.; Schmitz-Afonso, I.; Gautier, T.; Carrasco, N.; Park, M. A.; Giusti, P.; Afonso, C. Structural study of analogues of Titan’s haze by trapped ion mobility coupled with a fourier transform ion cyclotron mass spectrometer. J. Am. Soc. Mass Spectrom. 2019, 30 (7), 1169–1173.10.1007/s13361-019-02205-7Search in Google Scholar PubMed
Ruiz-Bermejo, M.; Zorzano, M.-P.; Osuna-Esteban, S. Simple organics and biomonomers identified in HCN polymers: an overview. Life 2013, 3 (3), 421–448.10.3390/life3030421Search in Google Scholar PubMed PubMed Central
Ruoff, R. S.; Emilsson, T.; Klots, T. D.; Chuang, C.; Gutowsky, H. S. Rotational spectrum and structure of the linear HCN trimer. J. Chem. Phys. 1988, 89 (1), 138–148.10.1063/1.455515Search in Google Scholar
Ruoff, A. Die Kraftkonstanten der Moleküle FCN, ClCN, BrCN und ICN. Spectrochim. Acta, Part A 1970, 26 (3), 545–556.10.1016/0584-8539(70)80101-5Search in Google Scholar
Ruscic, B.; Berkowitz, J. The H–NCS bond energy, ΔH○f (HNCS), ΔH○f (NCS), and IP(NCS) from photoionization mass spectrometric studies of HNCS, NCS, and (NCS)2. J. Chem. Phys. 1994, 101 (9), 7975–7989.10.1063/1.468495Search in Google Scholar
Rusinov, V. L.; Nosova, E. V.; Charushin, V. N. Fluorinated triazines. In Fluorine in heterocyclic chemistry, Vol. 2; Springer International Publishing: Cham, 2014; 673–716.10.1007/978-3-319-04435-4_8Search in Google Scholar
Ryu, I.; Murai, S.; Shinonaga, A.; Horiike, T.; Sonoda, N. Dimethyldicyanosilane: a reagent for concurrent silylation and cyanosilylation of β-diketones. J. Org. Chem. 1978, 43 (4), 780–782.10.1021/jo00398a075Search in Google Scholar
Ryu, B. G.; Kim, W.; Nam, K.; Kim, S.; Lee, B.; Park, M. S.; Yang, J. W. A comprehensive study on algal-bacterial communities shift during thiocyanate degradation in a microalga-mediated process. Bioresour. Technol. 2015, 191, 496–504.10.1016/j.biortech.2015.03.136Search in Google Scholar PubMed
Ryu, I. Dicyanodimethylsilane. e-EROS Encycl. Reagents Org. Synth. 2001, 2 (8), 5–6.10.1002/047084289X.rd143Search in Google Scholar
Saal, T.; Christe, K. O.; Haiges, R. Lewis adduct formation of hydrogen cyanide and nitriles with arsenic and antimony pentafluoride. Dalton Trans. 2019, 48 (1), 99–106.10.1039/C8DT03970DSearch in Google Scholar PubMed
Sagan, C.; Khare, B. N. Tholins: organic chemistry of interstellar grains and gas. Nature 1979, 277 (5692), 102–107.10.1038/277102a0Search in Google Scholar
Sakai, S.; Inagaki, S. Abnormally narrow carbon-metal-carbon bond angle in M(L1)L2 complexes (M = silicon, aluminum, magnesium, sodium; L1 and L2 = CO, CN). J. Am. Chem. Soc. 1990, 112 (22), 7961–7964.10.1021/ja00178a017Search in Google Scholar
Salud, C.; Feher, M.; Amano, T. Infrared diode laser detection of the v1 fundamental band of BrCN+. J. Mol. Spectrosc. 1993, 162 (1), 172–177.10.1006/jmsp.1993.1277Search in Google Scholar
Salzner, U.; von Raguè Schleyer, P. CH4−nXn: a comparison between the stabilized X=F series and the destabilized X=CN series. Chem. Phys. Lett. 1992, 190 (5), 401–406.10.1016/0009-2614(92)85163-5Search in Google Scholar
Samuni, U.; Kahana, S.; Fraenkel, R.; Haas, Y.; Danovich, D.; Shaik, S. The ICN-INC system: experiment and quantum chemical calculations. Chem. Phys. Lett. 1994, 225 (4–6), 391–397.10.1016/0009-2614(94)87100-0Search in Google Scholar
Sato, T.; Narazaki, A.; Kawaguchi, Y.; Niino, H.; Bucher, G. Dicyanocarbodiimide and trinitreno-s-triazine generated by consecutive photolysis of triazido-s-triazine in a low-temperature nitrogen matrix. Angew. Chem. Int. Ed. 2003, 42 (42), 5206–5209.10.1002/anie.200351879Search in Google Scholar PubMed
Sato, T.; Narazaki, A.; Kawaguchi, Y.; Niino, H.; Bucher, G.; Grote, D.; Wolff, J. J.; Wenk, H. H.; Sander, W. Generation and photoreactions of 2,4,6-trinitreno-1,3,5-triazine, a septet trinitrene. J. Am. Chem. Soc. 2004, 126 (25), 7846–7852.10.1021/ja031794vSearch in Google Scholar PubMed
Schalke, P. M. Cyanates, inorganic salts. In Ullmann’s encyclopedia of industrial chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006.10.1002/14356007.a08_157.pub2Search in Google Scholar
Schalley, C. A.; Hornung, G.; Schrdder, D.; Schwarz, H. Mass spectrometry as a tool to probe the gas-phase reactivity of neutral molecules. Int. J. Mass Spectrom. Ion Processes 1998, 172 (3), 181–208.10.1016/S0168-1176(97)00115-8Search in Google Scholar
Scheers, J.; Lim, D.-H.; Kim, J.-K.; Paillard, E.; Henderson, W. A.; Johansson, P.; Ahn, J.-H.; Jacobsson, P. All fluorine-free lithium battery electrolytes. J. Power Sources 2014, 251, 451–458.10.1016/j.jpowsour.2013.11.042Search in Google Scholar
Schiavon, R. P.; Caldwell, N.; Morrison, H.; Harding, P.; Courteau, S.; MacArthur, L. A.; Graves, G. J. Star clusters in M31. IV. A comparative analysis of absorption line indices in old M31 and milky way clusters. Astron. J. 2012, 143 (1), 14–32.10.1088/0004-6256/143/1/14Search in Google Scholar
Schilke, P.; Walmsley, C. M.; Pineau Des Forets, G.; Roueff, E.; Flower, D. R.; Guilloteau, S. A study of HCN, HNC and their isotopometers in OMC-1. I. Abundances and chemistry. Astron. Astrophys. 1992, 256, 595–612.Search in Google Scholar
Schmidpeter, A.; Zwaschka, F. Dicyanphosphid. Angew. Chem. 1977a, 89 (10), 747.10.1002/ange.19770891020Search in Google Scholar
Schmidpeter, A.; Zwaschka, F. Dicyanophosphide. Angew. Chem. Int. Ed. 1977b, 16 (10), 704–705.10.1002/anie.197707042Search in Google Scholar
Schmidpeter, A.; Zwaschka, F. Cyanohalophosphates(III) – hypervalent anions containing lamdba5-phosphorus(III). Angew. Chem. Int. Ed. 1979, 18 (5), 411–412.10.1002/anie.197904112Search in Google Scholar
Schmidpeter, A.; Burget, G.; Zwaschka, F.; Sheldrick, W. S. Cyanphosphorverbindungen. IX. Cyanidabbau von Weißem Phosphor Zu Dicyanphosphiden und die Dicyanphosphid-Struktur. Z. Anorg. Allg. Chem. 1985a, 527 (8), 17–32.10.1002/zaac.19855270803Search in Google Scholar
Schmidpeter, A.; Zwaschka, F.; Sheldrick, W. S. Cyanphosphorverbindungen, 8. Das Ungewöhnliche anion P2C10N102–. Chem. Ber. 1985b, 118 (3), 1078–1085.10.1002/cber.19851180325Search in Google Scholar
Schmidt, H.; Meinert, H. Elektrolysen von Cyaniden. III. Elektrolysen von Cyaniden in Wasserfreiem Pyridin. Z. Anorg. Allg. Chem. 1958, 295 (3–4), 173–184.10.1002/zaac.19582950304Search in Google Scholar
Schmidt, F.; Gruber, L.; Knorr, K. Effects of pressure and of thermal history on the structural phase transition of KCN. Z. Phys. B: Condens. Matter 1992, 87 (1), 127–131.10.1007/BF01308267Search in Google Scholar
Schmidt, B.; Schröder, B.; Sonnenberg, K.; Steinhauer, S.; Riedel, S. From polyhalides to polypseudohalides: chemistry based on cyanogen bromide. Angew. Chem. Int. Ed. 2019, 58 (30), 10340–10344.10.1002/anie.201903539Search in Google Scholar PubMed
Schmidtmann, H. Über Einige derivate des Malonitrils. Ber. Dtsch. Chem. Ges. 1896, 29 (2), 1168–1175.10.1002/cber.18960290204Search in Google Scholar
Schmiedekamp, A.; Bock, C. W.; George, P. An ab initio study of the structures and the harmonic and anharmonic stretching force constants of the cyanides HCN, LiCN, FCN, ClCN and isocyanides HNC, LiNC, FNC, ClNC. J. Mol. Struct. 1980, 67, 107–119.10.1016/0022-2860(80)80355-3Search in Google Scholar
Schneider, R. Über Das Verhalten des Cyansilbers Zum Schwefelchlorür. J. Prakt. Chem. 1885, 32 (1), 187–210.10.1002/prac.18850320119Search in Google Scholar
Schulz, A.; Klapötke, T. M. Zum Koordinationsverhalten Der Lewis-Basen HCN Und DCN Gegenüber Cp2Ti(AsF6)2. J. Organomet. Chem. 1992, 436 (2), 179–183.10.1016/0022-328X(92)85045-XSearch in Google Scholar
Scholl, R. Zur Darstellung des Bromcyans sowie des Mono- und Dibromnitromethans. Ber. Dtsch. Chem. Ges. 1896, 29 (2), 1822–1825.10.1002/cber.189602902129Search in Google Scholar
Schoneshofer, M.; Beck, G.; Henglein, A. Pulsradiolytische Untersuchung der Hydrolyse des Dirhodans. Chem. Ber. 1970, 74, 1011–1015.Search in Google Scholar
Schrems, O.; Huth, M.; Kollhoff, H.; Wittenbeck, R.; Knözinger, E. FTIR-spectroscopy of molecular clusters in liquid solutions and cryogenic solids. Ber. Bunsenges. Phys. Chem. 1987, 91 (11), 1261–1266.10.1002/bbpc.19870911136Search in Google Scholar
Schröder, H. Zur Darstellung von Cyanchlorid. Z. Anorg. Allg. Chem. 1958, 297 (5–6), 296–299.10.1002/zaac.19582970504Search in Google Scholar
Schultz, P. W. Ab initio calculations of ionic and hydrogen bonding interactions with the OCN–, SCN– and SeCN– anions. Mol. Phys. 1996, 88 (1), 217–246.10.1080/00268979650026712Search in Google Scholar
Schulz, A.; Klapötke, T. M. Does Diisooxocyan (OCN−NCO) exist? Inorg. Chem. 1996, 35 (16), 4791–4793.10.1021/ic960049zSearch in Google Scholar
Schulz, A.; Villinger, A. Pseudohalonium ions: [Me3Si-X-SiMe3]+ (X=CN, OCN, SCN, and NNN). Chem. Eur J. 2010, 16 (24), 7276–7281.10.1002/chem.201000289Search in Google Scholar PubMed
Schulz, A.; Surkau, J.; Zander, E. Synthesis of Pb(CN)2. Priv. Commun. 2021.Search in Google Scholar
Schulz, O. No title, Göttingen, 1856a.Search in Google Scholar
Schulz, C. Verbindungen des Cyans Mit den Metallen der Alkalischen Erden. J. Prakt. Chem. 1856b, 68 (1), 257–279.10.1002/prac.18560680145Search in Google Scholar
Schulz, A. Computed spectroscopic data of some basic CN species. Priv. Commun. 2021.Search in Google Scholar
Seel, F.; Wesemann, D. Eine Neue Darstellungsweise Für Dirhodan. Chem. Ber. 1953, 86 (9), 1107–1110.10.1002/cber.19530860912Search in Google Scholar
Seifer, G. B. Cyanuric acid and cyanurates. Koord. Khim. 2002, 28 (5), 301–324.10.1023/A:1015531315785Search in Google Scholar
Selliez, L.; Maillard, J.; Cherville, B.; Gautier, T.; Thirkell, L.; Gaubicher, B.; Schmitz‐Afonso, I.; Afonso, C.; Briois, C.; Carrasco, N. High‐resolution mass spectrometry for future space missions: comparative analysis of complex organic matter with LAb‐CosmOrbitrap and laser desorption/ionization Fourier transform ion cyclotron resonance. Rapid Commun. Mass Spectrom. 2020, 34 (10), 8645.10.1002/rcm.8645Search in Google Scholar PubMed
Sequeira, A. On the detection of cyanide-ion rotation in potassium cyanide by neutron diffraction. Acta Crystallogr. 1965, 18 (2), 291–292.10.1107/S0365110X65000622Search in Google Scholar
Shannon, M. S.; Tedstone, J. M.; Danielsen, S. P. O.; Hindman, M. S.; Irvin, A. C.; Bara, J. E. Free volume as the basis of gas solubility and selectivity in imidazolium-based ionic liquids. Ind. Eng. Chem. Res. 2012, 51 (15), 5565–5576.10.1021/ie202916eSearch in Google Scholar
Shapiro, H.; Hnizda, V. F.; Calingaert, G. Manufacture of lead cyanide. US2674519, 1954.Search in Google Scholar
Sheldrick, W. S.; Kroner, J.; Zwaschka, F.; Schmidpeter, A. Struktur des Dicyanphosphid-Ions Im Kronenether-Natriumsalz. Angew. Chem. 1979, 91 (12), 998–1000.10.1002/ange.19790911208Search in Google Scholar
Sheldrick, W. S.; Schmidpeter, A.; Zwaschka, F.; Dillon, K. B.; Platt, A. W. G.; Waddington, T. C. The structures of hypervalent phosphorus(III) anions P(CN)4–nBrn–. Transition from ψ-trigonal-bipyramidal to ψ-octahedral co-ordination and deviation from valence shell electron pair repulsion theory. J. Chem. Soc., Dalton Trans. 1981, No. 2, 413–418.10.1039/DT9810000413Search in Google Scholar
Sheridan, J.; Tyler, J. K.; Aynsley, E. E.; Dodd, R. E.; Little, R. Microwave spectrum of fluorine cyanide. Nature 1960, 185 (4706), 96.10.1038/185096a0Search in Google Scholar
Shi, D.; Li, W.; Sun, J.; Zhu, Z. MRCI study on spectroscopic and molecular properties of several low-lying electronic states of the CN radical. J. Quant. Spectrosc. Radiat. Transfer 2011, 112 (14), 2335–2346.10.1016/j.jqsrt.2011.06.002Search in Google Scholar
Shimada, T.; Matsuo, T.; Suga, H.; Luty, F. Phase transition and glass transition in rubidium cyanide. J. Chem. Phys. 1986, 85 (6), 3530–3536.10.1063/1.450924Search in Google Scholar
Shimada, Y.; Chikamatsu, K.; Kimura, C.; Aoki, H.; Sugino, T. Effect of plasma treatment on interface property of BCN/GaN structure. Appl. Surf. Sci. 2006, 253 (3), 1459–1463.10.1016/j.apsusc.2006.02.016Search in Google Scholar
Shlyaykher, A.; Ehmann, M.; Karttunen, A. J.; Tambornino, F. A comprehensive study on the full series of alkali‐metal selenocyanates AI[SeCN] (AI = Li−Cs). Chem. Eur J. 2021, 27 (54), 13552–13557.10.1002/chem.202102058Search in Google Scholar PubMed PubMed Central
Shmel’kova, T. K.; Ignatenko, A. V.; Krukovskii, S. P.; Ponomarenko, V. A. Synthesis of fluoro-containing substituted 1,3,5-triazines. Bull. Acad. Sci. USSR Div. Chem. Sci. 1989, 38 (4), 836–840.10.1007/BF00953302Search in Google Scholar
Shorter, J. The conversion of ammonium cyanate into urea – a saga in reaction mechanisms. Chem. Soc. Rev. 1978, 1–14.10.1039/CS9780700001Search in Google Scholar
Shrift, A.; Virupaksha, T. K. Biosynthesis of Se-methylselenocysteine from selenite in selenium-accumulating plants. Biochim. Biophys. Acta 1963, 71, 483–485.10.1016/0006-3002(63)91113-2Search in Google Scholar PubMed
Sidorov, L. N. Mass spectrometric investigation into inorganic complex molecules and stability of BeF3, AlF4, FeF4. Koord. Khim. 1977, 3 (8), 1128–1139.Search in Google Scholar
Sievert, K.; Schulz, A.; Harloff, J.; Ellinger, St.; Täschler, C. Method for preparation of fluoro cyano compounds of the 15th group with a Lewis acid. WO 2015067404, 2015.Search in Google Scholar
Simmons, J. W.; Anderson, W. E.; Gordy, W. Microwave spectrum and molecular constants of hydrogen cyanide. Phys. Rev. 1950, 77 (1), 77–79.10.1103/PhysRev.77.77Search in Google Scholar
Simpson, J. B.; Smith, J. G.; Whiffen, D. H. Microwave spectrum of ICN including IC15N. J. Mol. Spectrosc. 1972, 44 (3), 558–570.10.1016/0022-2852(72)90264-0Search in Google Scholar
Singh, P. Bimetallic tetrathiocyanates and selenocyanates as Lewis acids. Coord. Chem. Rev. 1980, 32 (1), 33–65.10.1016/S0010-8545(00)80369-0Search in Google Scholar
Sinosaki, H.; Hara, R. Unknown. Technol. Rep. Tohoku Univ. 1929, 8 (297).Search in Google Scholar
Šišak, D.; McCusker, L. B.; Buckl, A.; Wuitschik, G.; Wu, Y.-L.; Schweizer, W. B.; Dunitz, J. D. The search for tricyanomethane (cyanoform). Chem. Eur J. 2010, 16 (24), 7224–7230.10.1002/chem.201000559Search in Google Scholar PubMed
Skatrud, D. D.; De Lucia, F. C.; Blake, G. A.; Sastry, K. V. L. N. The millimeter and submillimeter spectrum of CN in its first four vibrational states. J. Mol. Spectrosc. 1983, 99 (1), 35–46.10.1016/0022-2852(83)90290-4Search in Google Scholar
Slotta, K. H. Bromcyan Und Wasserfreie Blausäure. Ber. Dtsch. Chem. Ges. 1934, 67 (6), 1028–1030.10.1002/cber.19340670622Search in Google Scholar
Smallwood, Z. M.; Davis, M. F.; Hill, J. G.; James, L. J. R.; Portius, P. Syntheses, structures, and infrared spectra of the hexa(cyanido) complexes of silicon, germanium, and tin. Inorg. Chem. 2019, 58 (7), 4583–4591.10.1021/acs.inorgchem.9b00150Search in Google Scholar PubMed
Smith, A. G.; Ring, H.; Smith, W. V.; Gordy, W. Interatomic distances and nuclear quadrupole couplings in ClCN, BrCN, and ICN. Phys. Rev. 1948, 74 (4), 370–372.10.1103/PhysRev.74.370Search in Google Scholar
Smith-Gicklhorn, A. M.; Frankowski, M.; Bondybey, V. E.Tetracyanoethylene, its ions and ionic fragments. Phys. Chem. Chem. Phys. 2002, 4 (8), 1425–1431.10.1039/b110032gSearch in Google Scholar
Smolin, E. M.; Rapoport, L. S-Triazines and derivatives; Interscience Publishers INC.: New York, 1959.Search in Google Scholar
Smuczyńska, S.; Skurski, P. Halogenoids as ligands in superhalogen anions. Inorg. Chem. 2009, 48 (21), 10231–10238.10.1021/ic901253rSearch in Google Scholar PubMed
Söderbäck, E. Studien Über Das Freie Rhodan. Justus Liebigs Ann. Chem. 1919, 419 (3), 217–322.10.1002/jlac.19194190302Search in Google Scholar
Sommerfeld, T.; Bhattarai, B. Al(CN)3−6 and Al(NC)3−6 trianions. Phys. Chem. Chem. Phys. 2011, 13 (41), 18393.10.1039/c1cp21570aSearch in Google Scholar PubMed
Sommerfeld, T. FCN– and FNC– radical anions. Phys. Chem. Chem. Phys. 2001, 3 (12), 2394–2399.10.1039/b102112pSearch in Google Scholar
Somogyi, Á.; Thissen, R.; Orthous-Daunay, F. R.; Vuitton, V. The role of ultrahigh resolution Fourier transform mass spectrometry (FT-MS) in astrobiology-related research: analysis of meteorites and tholins. Int. J. Mol. Sci. 2016, 17(4), 439–453.10.3390/ijms17040439Search in Google Scholar PubMed PubMed Central
Sorensen, T. E.; England, W. B. Valence states of the cyano radical Feynman’s way. Int. J. Quantum Chem. 2002, 90 (2), 516–533.10.1002/qua.928Search in Google Scholar
Sorokin, D. Y.; Tourova, T. P.; Lysenko, A. M.; Kuenen, J. G. Microbial thiocyanate utilization under highly alkaline conditions. Appl. Environ. Microbiol. 2001, 67 (2), 528–538.10.1128/AEM.67.2.528-538.2001Search in Google Scholar PubMed PubMed Central
Soto-Blanco, B. Cyanide: occurrence, characteristics and applications; Nova Science Publishers: New York, 2013.Search in Google Scholar
Spencer, H. K.; Lakshmikantham, M. V.; Cava, M. P. Organotellurium chemistry. 1. Benzyl tellurocyanate: a stable alkyl tellurocyanate. J. Am. Chem. Soc. 1977, 99 (5), 1470–1473.10.1021/ja00447a032Search in Google Scholar
Spencer, E. S.; Dale, E. J.; Gommans, A. L.; Rutledge, M. T.; Vo, C. T.; Nakatani, Y.; Gamble, A. B.; Smith, R. A. J.; Wilbanks, S. M.; Hampton, M. B.; Tyndall, J. D. A. Multiple binding modes of isothiocyanates that inhibit macrophage migration inhibitory factor. Eur. J. Med. Chem. 2015, 93, 501–510.10.1016/j.ejmech.2015.02.012Search in Google Scholar PubMed
Sprenger, J. A. P.; Landmann, J.; Drisch, M.; Ignat’ev, N.; Finze, M. Syntheses of tricyanofluoroborates M[BF(CN)3] (M = Na, K): (CH3)3SiCl catalysis, countercation effect, and reaction intermediates. Inorg. Chem. 2015, 54 (7), 3403–3412.10.1021/ic503077cSearch in Google Scholar PubMed
Stafford, D. A.; Callely, A. G. The utilization of thiocyanate by a heterotrophic bacterium. J. Gen. Microbiol. 1969, 55 (2), 285–289.10.1099/00221287-55-2-285Search in Google Scholar PubMed
Staib, C.; Lant, P. Thiocyanate degradation during activated sludge treatment of Coke–Ovens wastewater. Biochem. Eng. J. 2007, 34 (2), 122–130.10.1016/j.bej.2006.11.029Search in Google Scholar
Starynowicz, P. Structure of caesium dicyanamide. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1991, 47 (10), 2198–2199.10.1107/S0108270191003700Search in Google Scholar
Steimle, T. C.; Fletcher, D. A.; Jung, K. Y.; Scurlock, C. T. Molecular beam optical stark spectroscopy of calcium monocyanide. J. Chem. Phys. 1992, 97 (5), 2909–2919.10.1063/1.463032Search in Google Scholar
Steimle, T. C.; Fletcher, D. A.; Jung, K. Y.; Scurlock, C. T. Erratum: Molecular beam optical stark spectroscopy of calcium monocyanide [J. Chem. Phys. 97, 2909 (1992)]. J. Chem. Phys. 1994, 100 (5), 4025–4026.10.1063/1.467281Search in Google Scholar
Steinkopf, W. Apparatives aus Dem Laboratorium. J. Prakt. Chem. 1925, 109 (1), 347–351.10.1002/prac.19251090117Search in Google Scholar
Støgard, Å. Ab initio calculations on CICN and ONCI. Chem. Phys. Lett. 1976, 40 (3), 429–432.10.1016/0009-2614(76)85112-3Search in Google Scholar
Stokes, H. T.; Decker, D. L.; Nelson, H. M.; Jorgensen, J. D. Structure of potassium cyanide at low temperature and high pressure determined by neutron diffraction. Phys. Rev. B 1993, 47 (17), 11082–11092.10.1103/PhysRevB.47.11082Search in Google Scholar PubMed
Stopenko, V. V.; Golub, A. M.; Köhler, H. Chemistry of pseudohalides; Elsevier: Amsterdam, 1986.Search in Google Scholar
Stott, M. B.; Franzmann, P. D.; Zappia, L. R.; Watling, H. R.; Quan, L. P.; Clark, B. J.; Houchin, M. R.; Miller, P. C.; Williams, T. L. Thiocyanate removal from saline CIP process water by a rotating biological contactor, with reuse of the water for bioleaching. Hydrometallurgy 2001, 62 (2), 93–105.10.1016/S0304-386X(01)00185-2Search in Google Scholar
Stratford, J.; Dias, A. E. X. O.; Knowles, C. J. The utilization of thiocyanate as a nitrogen source by a heterotrophic bacterium: the degradative pathway involves formation of ammonia and tetrathionate. Microbiology 1994, 140 (10), 2657–2662.10.1099/00221287-140-10-2657Search in Google Scholar PubMed
Strössner, K.; Hochheimer, H. D.; Hönle, W.; Werner, A. High‐pressure Raman and X‐ray studies of the alkali cyanides up to 27 GPa. J. Chem. Phys. 1985, 83 (5), 2435–2440.10.1063/1.449289Search in Google Scholar
Suga, H.; Matsuo, T.; Seki, S. Thermodynamic properties and order-disorder phase transitions of the potassium cyanide crystal. Bull. Chem. Soc. Jpn. 1965, 38 (7), 1115–1124.10.1246/bcsj.38.1115Search in Google Scholar
Sugino, T.; Etou, Y.; Tai, T.; Mori, H. Dielectric constant of boron carbon nitride films synthesized by plasma-assisted chemical-vapor deposition. Appl. Phys. Lett. 2002, 80 (4), 649–651.10.1063/1.1436522Search in Google Scholar
Sugisaki, M.; Matsuo, T.; Suga, H.; Seki, S. Phase transitions and thermodynamic properties of rubidium and cesium cyanides. Bull. Chem. Soc. Jpn. 1968, 41 (8), 1747–1756.10.1246/bcsj.41.1747Search in Google Scholar
Sulzer, H. Verfahren Zur Darstellung von Ammoniak und Ameisensäure aus Kalkstickstoff. Angew. Chem. 1912, 25 (25), 1268–1273.10.1002/ange.19120252503Search in Google Scholar
Sun, Y.; Metz, M. V.; Stern, C. L.; Marks, T. J. Al-, Nb-, and Ta-based perfluoroaryloxide anions as cocatalysts for metallocene-mediated Ziegler–Natta olefin polymerization. Organometallics 2000, 19 (11), 1625–1627.10.1021/om990946lSearch in Google Scholar
Sundermeyer, W. Salzschmelzen und Ihre Verwendung Als Reaktionsmedien. Angew. Chem. 1965, 77 (6), 241–258.10.1002/ange.19650770603Search in Google Scholar
Swank, D. D.; Willett, R. D. The crystal structure of potassium selenocyanate. Inorg. Chem. 1965, 4 (4), 499–501.10.1021/ic50026a013Search in Google Scholar
Swanson, H.; Tatge, E. Standard X-ray diffraction powder patterns; Washington D.C.: Washington D.C., I, 1953.Search in Google Scholar
Swenson, J. S.; Renaud, D. J. Dicyanocarbene. J. Am. Chem. Soc. 1965, 87 (6), 1394.10.1021/ja01084a050Search in Google Scholar
Szopa, C.; Cernogora, G.; Boufendi, L.; Correia, J. J.; Coll, P. PAMPRE: a dusty plasma experiment for Titan’s tholins production and study. Planet. Space Sci. 2006, 54 (4), 394–404.10.1016/j.pss.2005.12.012Search in Google Scholar
Talbi, D.; Ellinger, Y. Potential energy surfaces for the electronic dissociative recombination of HCNH+: astrophysical implications on the HCN/HNC abundance ratio. Chem. Phys. Lett. 1998, 288 (1), 155–164.10.1016/S0009-2614(98)00213-9Search in Google Scholar
Talbi, D.; Herbst, E. An extensive ab initio study of the C+ + NH3 reaction and its relation to the HNC/HCN abundance ratio in interstellar clouds. Astron. Astrophys. 1998, 333 (3), 1007–1015.Search in Google Scholar
Talbi, D. An extensive ab initio study of a process of astrophysical interest: the N+(N) + CH3(CH3+) reaction. Chem. Phys. Lett. 1999, 312 (2–4), 291–298.10.1016/S0009-2614(99)00907-0Search in Google Scholar
Tang, T. B.; Swallowe, G. M.; Mohan, V. K. Application of continuously recorded time-resolved mass spectrometry to the study of thermal explosions. J. Solid State Chem. 1984, 55 (2), 239–242.10.1016/0022-4596(84)90271-8Search in Google Scholar
Tao, Y.; Ding, Y.; Liu, J.; Li, Z.; Huang, X.; Sun, C.-C. Theoretical mechanistic study on the ion–molecule reactions of CCN+/CNC+ with H2O and HCO+/HOC+ with HCN/HNC. J. Chem. Phys. 2002a, 116 (5), 1892–1910.10.1063/1.1431272Search in Google Scholar
Tao, Y.; Ding, Y.; Liu, J.; Li, Z.; Huang, X.; Sun, C.-C. Theoretical mechanistic study on the ion−molecule reactions of CCN+/NC+ with H2S. J. Phys. Chem. 2002b, 106 (12), 2949–2962.10.1021/jp013892dSearch in Google Scholar
Tatsumi, R.; Aoki, Y.; Maeda, S.; Hori, M.; Hayakawa, S. Ionic liquid, electrolyte, lithium secondary battery comprising same, and process for producing ionic liquid. EP 2410601 A1: Osaka-shi, 2012.Search in Google Scholar
Tattershall, B. W. Phosphorus-carbon nuclear magnetic coupling constants in phosphorus(III) cyanides. Polyhedron 1990, 9 (4), 553–555.10.1016/S0277-5387(00)86233-6Search in Google Scholar
Tautz, H.; Blumberg, W. Verfahren Zur Herstellung von Chlorcyan; DE102007052538A1: Essen, 2007.Search in Google Scholar
Taylor, E. C.; Andrade, J. G.; John, K. C.; McKillop, A. Thallium in organic synthesis. 50. A convenient synthesis of thallium(I) cyanide, a useful reagent in organic synthesis. J. Org. Chem. 1978, 43 (11), 2280–2282.10.1021/jo00405a042Search in Google Scholar
Tebbe, K.-F.; Fröhlich, R. Untersuchungen an Polypseudohalogeniden. 4. Darstellung und Kristallstrukur von Kalium(0,6)Rubidium(0,4) Dicyanoiodat(I)-Diiminooxalsäurediethylester (2/1) KxRb1-x[I(CN)2] x 1/2 C6H12N2O2 (x = 0,6). Z. Anorg. Allg. Chem. 1983a, 505 (10), 19–31.10.1002/zaac.19835051003Search in Google Scholar
Tebbe, K.-F.; Fröhlich, R. Untersuchungen an Polypseudohalogeniden. 3. Darstellung und Kristallstruktur von Kaliumdicyanoiodat(I)-Diiminooxalsäurediethylester (1/1), K[I(CN)2] x C6H12N2O2. Z. Anorg. Allg. Chem. 1983b, 505 (10), 7–18.10.1002/zaac.19835051002Search in Google Scholar
Tebbe, K.; Fröhlich, R. Studies on the polypseudohalides, II. Preparation and crystal structure of Rb[I(ICN)2]. Z. Naturforsch. B: J. Chem. Sci. 1983c, 38 (5), 549–553.10.1515/znb-1983-0504Search in Google Scholar
Tebbe, K.; Gräfe-Kavoosian, A. Studies on polypseudohalides, VII preparation and crystal structure of [K(Crypt-2.2.2)] I(ICN)2. Z. Naturforsch. B: J. Chem. Sci. 1996, 51 (7), 1007–1010.10.1515/znb-1996-0716Search in Google Scholar
Tebbe, K.; Krauß, N. Studies on the polypseudohalides, V preparation and crystal structure of K[I(CN)2]. Z. Naturforsch. B 1988, 43 (2), 149–152.10.1515/znb-1988-0203Search in Google Scholar
Thalhammer, F.; Trautz, H. Verfahren Zur Herstellung von Natrium-Dicyanamid; DE19904877A1: Trostberg, 2000.Search in Google Scholar
Thaulow, H. Über die Einwirkung der Schwefelsäure Auf Ferrocyankalium und Über die Medizinische Blausäure. J. Prakt. Chem. 1844, 31 (1), 234–256.10.1002/prac.18440310134Search in Google Scholar
Thielemann, G.; Spange, S. Polarity of tetraalkylammonium-based ionic liquids and related low temperature molten salts. New J. Chem. 2017, 41 (16), 8561–8567.10.1039/C7NJ00443ESearch in Google Scholar
Thoma, A.; Schallmoser, G.; Smith, A. M.; Wurfel, B. E.; Bondybey, V. E. Visible absorption and infrared emission of CN–Xe in an argon Matrix. J. Chem. Phys. 1994, 100 (7), 5387–5389.10.1063/1.467155Search in Google Scholar
Thomas, B. H.; Orville-Thomas, W. J. Molecular parameters and bond structure. J. Mol. Struct. 1969, 3 (3), 191–206.10.1016/0022-2860(69)87003-1Search in Google Scholar
Thompson, J. P.; Marrs, T. C. Hydroxocobalamin in cyanide poisoning. Clin. Toxicol. 2012, 50 (10), 875–885.10.3109/15563650.2012.742197Search in Google Scholar PubMed
Thompson, W. R.; McDonald, G. D.; Sagan, C. The Titan haze revisited: magnetospheric energy sources and quantitative tholin yields. Icarus 1994, 112 (2), 376–381.10.1006/icar.1994.1191Search in Google Scholar PubMed
Thompson, M. R. Purification and analysis of alkali cyanides. Bur. Stand. J. Res. 1931, 6 (6), 1051–1059.10.6028/jres.006.065Search in Google Scholar
Thomson, C.; Wishart, B. J. Electronic structure of unstable intermediates. Theor. Chim. Acta 1974, 35 (3), 261–265.10.1007/BF00546911Search in Google Scholar
Thomson, C. An INDO investigation of the electronic structure and hyperfine coupling constants of the radicals HBO–, HCO and HCN–. Theor. Chim. Acta 1970, 17 (4), 320–322.10.1007/BF00527459Search in Google Scholar
Timoshkin, A. Y.; Schaefer, H. F. Ab initio and DFT investigations of Al, Ga, and in tricyanides and triisocyanides. J. Struct. Chem. 2000, 41 (1), 35–40.10.1007/BF02684725Search in Google Scholar
Tokue, I.; Nanbu, S. Theoretical transition probabilities for the Ã1Π-X̃1Σ+ system of AlNC and AlCN isomers based on global potential energy surfaces. J. Chem. Phys. 2006, 124 (22), 224301.10.1063/1.2207611Search in Google Scholar PubMed
Tokue, I.; Nanbu, S. Isomerization reaction between linear AlNC and AlCN including the X̃1Σ+ and Ã1Π states studied by three-dimensional wave packet propagation. J. Chem. Phys. 2011, 135 (2), 024305.10.1063/1.3608915Search in Google Scholar PubMed
Tornieporth-Oetting, I.; Klapötke, T. Synthese Und Charakterisierung von (Iodcyan)Iodonium-Hexafluoroarsenat, [ICNI]+[AsF6]-. Chem. Ber. 1990, 123 (6), 1343–1344.10.1002/cber.19901230622Search in Google Scholar
Tornieporth-Oetting, I.; Klapötke, T. Untersuchung Zum Reaktionsverhalten von Nitrilen RCN Gegenüber [I3]+[AsF6]− (R = CH3, CF3, Br, I, H). Chem. Ber. 1991, 124 (7), 1571–1573.10.1002/cber.19911240715Search in Google Scholar
Tornieporth-Oetting, I. C.; Gowik, P.; Klapötke, T. M. [FCNF]+[AsF6]−, Ein Salz des Linearen 22-Valenzelektronen-Kations [FCNF]+. Angew. Chem. 1991, 103 (11), 1490–1492.10.1002/ange.19911031111Search in Google Scholar
Tornieporth-Oetting, I. C.; Klapötke, T. M.; Cameron, T. S.; Valkonen, J.; Rademacher, P.; Kowski, K. Reactivity of Lewis acids towards nitriles; crystal structure and electron deformation density of C2N2·SbF5 and photoelectron spectrum of AsF5. J. Chem. Soc. Dalton Trans. 1992, 2 (4), 537–543.10.1039/DT9920000537Search in Google Scholar
Torrie, B. H.; Powell, B. M. Cyanogen: structure, dynamics and inter-molecular potentials. J. Phys. Condens. Matter 1989, 1 (34), 5827–5835.10.1088/0953-8984/1/34/001Search in Google Scholar
Townes, C. H.; Holden, A. N.; Merritt, F. R. Rotational spectra of some linear molecules near 1-cm wave-length. Phys. Rev. 1947, 71 (1), 64.10.1103/PhysRev.71.64Search in Google Scholar
Townes, C. H.; Holden, A. N.; Merritt, F. R. Microwave spectra of some linear XYZ molecules. Phys. Rev. 1948, 74 (9), 1113–1133.10.1103/PhysRev.74.1113Search in Google Scholar
Trofimenko, S.; Little, E. L.; Mower, H. F. Tricyanomethane (cyanoform), carbamyldicyanomethane, and their derivatives. J. Org. Chem. 1962, 27 (2), 433–438.10.1021/jo01049a021Search in Google Scholar
Tsuda, S.; Yokohata, A.; Umaba, T. Measurement of negative ions formed by electron impact. VIII. Ionization efficiency curves of negative ions from methyl and ethyl cyanides. Bull. Chem. Soc. Jpn. 1971, 44 (6), 1486–1491.10.1246/bcsj.44.1486Search in Google Scholar
Tyler, J. K.; Sheridan, J. Structural studies of linear molecules by microwave spectroscopy. Trans. Faraday Soc. 1963, 59, 2661.10.21236/AD0409439Search in Google Scholar
Tyuzyo, K. Stoichiometric investigations of the liquid state IV. Viscosity of associated liquid. Bull. Chem. Soc. Jpn. 1957, 30 (7), 782–789.10.1246/bcsj.30.782Search in Google Scholar
Tzialla, O.; Veziri, C.; Papatryfon, X.; Beltsios, K. G.; Labropoulos, A.; Iliev, B.; Adamova, G.; Schubert, T. J. S.; Kroon, M. C.; Francisco, M.; Zubeir, L. F.; Romanos, G. E.; Karanikolos, G. N. Zeolite imidazolate framework–ionic liquid hybrid membranes for highly selective CO2 separation. J. Phys. Chem. C 2013, 117 (36), 18434–18440.10.1021/jp4051287Search in Google Scholar
Ugarov, M.; Ageev, V.; Karabutov, A.; Loubnin, E.; Pimenov, S.; Konov, V.; Bensaoula, A. UV laser induced interfacial synthesis of CN–BCN layers on diamond films in borazine and ammonia. Appl. Surf. Sci. 1999, 138–139 (1–4), 359–363.10.1016/S0169-4332(98)00427-9Search in Google Scholar
Unknown. Notitia Caerulei Berolinensis Nuper Inventi. Miscnea. Berol. Soc. Sci. 1710, 1, 377–378.Search in Google Scholar
Unland, M. L. Isocyanate intermediates in the reaction of NO and CO over noble metal catalysts. J. Catal. 1973, 31 (3), 459–465.10.1016/0021-9517(73)90318-7Search in Google Scholar
Vadhanavikit, S.; Ganther, H. E. Nutritional availability and chronic toxicity of selenocyanate in the rat. J. Nutr. 1988, 118 (6), 718–722.10.1093/jn/118.6.718Search in Google Scholar PubMed
Vaes, J.; Chabanel, M.; Martin, M. L. Ionic interactions in lithium thiocyanate solutions. Nitrogen-15 and lithium-7 nuclear magnetic resonance studies. J. Phys. Chem. 1978, 82 (22), 2420–2423.10.1021/j100511a015Search in Google Scholar
van de Burgt, L. J.; Nicolai, J.; Heaven, M. Laser induced fluorescence Study of the HeBr2 van Der Waals Complex. J. Chem. Phys. 1984, 81 (12), 5514–5520.10.1063/1.447652Search in Google Scholar
van Rooyen, P. H.; Boeyens, J. C. A. Sodium thiocyanate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1975, 31 (12), 2933–2934.10.1107/S0567740875009326Search in Google Scholar
van Vaals, J. J.; Meerts, W. L.; Dymanus, A. Rotational spectrum, hyperfine spectrum and structure of lithium isocyanide. Chem. Phys. 1983, 82 (3), 385–393.10.1016/0301-0104(83)85243-4Search in Google Scholar
van Vaals, J. J.; Meerts, W. L.; Dymanus, A. High-resolution molecular-beam spectroscopy of NaCN and Na13CN. Chem. Phys. 1984a, 86 (1–2), 147–159.10.1016/0301-0104(84)85163-0Search in Google Scholar
van Vaals, J. J.; Leo Meerts, W.; Dymanus, A. Molecular beam electric resonance study of KCN, K13CN and KC15N. J. Mol. Spectrosc. 1984b, 106 (2), 280–298.10.1016/0022-2852(84)90159-0Search in Google Scholar
Varma, R.; Signorelli, A. J. A new synthesis of cyanogen chloride. Inorg. Nucl. Chem. Lett. 1969, 5 (12), 1017–1019.10.1016/0020-1650(69)80114-5Search in Google Scholar
Vazquez, G. J.; Gouyet, J.-F. SCF CI potential energy surfaces for the HCN α HCN isomerisation reaction. Chem. Phys. Lett. 1981, 77 (2), 233–238.10.1016/0009-2614(81)80136-4Search in Google Scholar
Verneuil, A. Recherches Sur Quelques Combinaisons Azotees Du Selenium. Ann. Chim. Phys. 1886, 9, 289.Search in Google Scholar
Verweel, Η. J.; Bijvoet, J. M. Die Kristallstruktur von NaCN. Z. für Kristallogr. Cryst. Mater. 1939, 100 (1–6), 201–207.10.1524/zkri.1939.100.1.201Search in Google Scholar
Vichietti, R. M.; Haiduke, R. L. A. Kinetic study of the isomerization reaction HCnN → HCn-1NC (n = 1, 3 and 5). Mon. Not. R. Astron. Soc. 2014, 437 (3), 2351–2360.10.1093/mnras/stt2053Search in Google Scholar
Völker, T. Polymere Blausäure. Angew. Chem. 1960, 72 (11), 379–384.10.1002/ange.19600721104Search in Google Scholar
von der Weid, J. P.; do Carmo, L. C. S.; Ribeiro, S. C. Substitutional HCN− molecular ions in KCN crystals: a paramagnetic probe in a ferroelastic material. J. Phys. C: Solid State Phys. 1979, 12 (22), 4927–4938.10.1088/0022-3719/12/22/032Search in Google Scholar
von Ittner, F. Beiträge Zur Geschichte Der Blausäure Mit Versuchen Über Ihre Verbindungen Und Wirkungen Auf Den Tierischen Organismus; Universität Freiburg: Freiburg, 1809.Search in Google Scholar
von Meyer, E. Zur Kenntniss der Blausäure und des Jodcyans. J. Prakt. Chem. 1887, 36 (1), 292–299.10.1002/prac.18870360126Search in Google Scholar
von Ragué Schleyer, P.; Sawaryn, A.; Reed, A. E.; Hobza, P. The remarkable structure of lithium cyanide/isocyanide. J. Comput. Chem. 1986, 7(5), 666–672.10.1002/jcc.540070509Search in Google Scholar
Voorhoeve, R. J. H.; Thimble, L. E. Synthesis of ammonium cyanate and urea from NO over Pt, Os, Ru, and CuNi catalysts. J. Catal. 1978, 53 (2), 251–259.10.1016/0021-9517(78)90073-8Search in Google Scholar
Vörös, T.; Bazsó, G.; Tarczay, G.; Pasinszki, T. Matrix-isolation spectroscopic and computational study of [2C, 2N, 2S] isomers: photochemical generation of SCNNCS and NCSNCS from NCSSCN. J. Mol. Struct. 2012, 1025, 117–123.10.1016/j.molstruc.2012.04.021Search in Google Scholar
Vörös, T.; Pacsai, B.; Magyarfalvi, G.; Tarczay, G. Generation and spectroscopic identification of NCXNC and NCNCX (X = S, Se) in low-temperature inert matrices. J. Mol. Spectrosc. 2015, 316, 95–104.10.1016/j.jms.2015.07.003Search in Google Scholar
Voss, K.; Becker, M.; Villinger, A.; Emel’yanenko, V. N.; Hellmann, R.; Kirchner, B.; Uhlig, F.; Verevkin, S. P.; Schulz, A. Ionic liquids containing the triply negatively charged tricyanomelaminate anion and a B(C6F5)3 adduct anion. Chem. Eur J. 2011, 17 (48), 13526–13537.10.1002/chem.201101946Search in Google Scholar PubMed
Vuitton, V.; Bonnet, J. Y.; Frisari, M.; Thissen, R.; Quirico, E.; Dutuit, O.; Schmitt, B.; Le Roy, L.; Fray, N.; Cottin, H.; Sciamma-O’Brien, E.; Carrasco, N.; Szopa, C. Very high resolution mass spectrometry of HCN polymers and tholins. Faraday Discuss. 2010, 147, 495–508.10.1039/c003758cSearch in Google Scholar PubMed
Wald, M. H.; Lindberg, H. A.; Barker, M. H. The toxic manifestations of the thiocyanates. JAMA J. Am. Med. Assoc. 1939, 112 (12), 1120.10.1001/jama.1939.02800120006002Search in Google Scholar
Walden, P. C. W. S. Ein Gedenkblatt Zu Seinem 200. Geburtstage. Z. Anorg. Allg. Chem. 1943, 250 (3–4), 230–235.10.1002/zaac.19432500302Search in Google Scholar
Walker, K. A.; Evans, C. J.; Suh, S.-H. K.; Gerry, M. C. L.; Watson, J. K. G. Fourier transform microwave spectroscopy of cyanides and isocyanides of Al, Ga, and in. J. Mol. Spectrosc. 2001, 209 (2), 178–191.10.1006/jmsp.2001.8421Search in Google Scholar
Walsh, B.; Barnes, A. J.; Suzuki, S.; Orville-Thomas, W. J. Studies of intermolecular interactions by matrix isolation vibrational spectroscopy and normal coordinate analysis. J. Mol. Spectrosc. 1978, 72 (1), 44–56.10.1016/0022-2852(78)90042-5Search in Google Scholar
Wang, C.-R.; Huang, R.-B.; Liu, Z.-Y.; Zheng, L.-S. Laser generation and ab initio studies of CnN− clusters. Chem. Phys. Lett. 1995, 237 (5–6), 463–467.10.1016/0009-2614(95)00355-8Search in Google Scholar
Wang, D.-C.; Chau, F.-T.; Lee, E. P. F.; Leung, A. K.-M.; Dyke, J. M. The X2Π and A2Σ states of FCN+ and ClCN+: ab initio calculations and simulation of the HeI photoelectron spectra of FCN and ClCN. Mol. Phys. 1998, 93 (6), 995–1005.10.1080/00268979809482286Search in Google Scholar
Wang, Q.; Ding, Y.; Sun, C. Theoretical study on germanium cyanide radical GeCN and its ions. J. Chem. Phys. 2005, 122 (20), 204305–204312.10.1063/1.1900083Search in Google Scholar PubMed
Wang, J.; Ding, Y.; Sun, C. Cyanomethylidyne: a reactive carbyne radical. ChemPhysChem 2006, 7 (3), 710–722.10.1002/cphc.200500548Search in Google Scholar PubMed
Wang, Z.; Zhang, J.; Wu, J.; Cao, W. Theoretical investigation on intermolecular interactions between HCN and HNC: the nature and thermodynamic properties. J. Mol. Struct.: Theochem. 2007, 806 (1–3), 239–246.10.1016/j.theochem.2006.11.030Search in Google Scholar
Wasserman, E.; Barash, L.; Yager, W. A. The electron paramagnetic resonance of triplet CNN, NCN, and NCCCN. J. Am. Chem. Soc. 1965, 87 (9), 2075–2076.10.1021/ja01087a058Search in Google Scholar
Wasylishen, R. E.; Jeffrey, K. R. Nitrogen‐14 NMR study of molecular motion in rubidium and cesium cyanide. J. Chem. Phys. 1983, 78 (2), 1000–1002.10.1063/1.444813Search in Google Scholar
Watts, M. P.; Moreau, J. W. New insights into the genetic and metabolic diversity of thiocyanate-degrading microbial consortia. Applied microbiology and biotechnology, 3rd ed.; Springer-Verlag, 100, 2016; pp. 1101–1108.10.1007/s00253-015-7161-5Search in Google Scholar PubMed
Wehrhane, G.; Hübner, H. Über Den Cyanphosphor. Justus Liebigs Ann. Chem. 1864, 132 (3), 277–289.10.1002/jlac.18641320305Search in Google Scholar
Weidinger, D.; Houchins, C.; Owrutsky, J. C. Vibrational dynamics of tricyanomethanide. Chem. Phys. Lett. 2012, 525–526, 60–63.10.1016/j.cplett.2011.12.060Search in Google Scholar
Weinberg, J. M.; Fishburne, E. S.; Narahari Rao, K. Infrared bands of the CN red system (A2Π-X2Σ). J. Mol. Spectrosc. 1967, 22 (1–4), 406–418.10.1016/0022-2852(67)90187-7Search in Google Scholar
Wen, J.; Xie, J.; Chen, X.; Li, X. A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 2017, 391, 72–123.10.1016/j.apsusc.2016.07.030Search in Google Scholar
Werner, H.; Follmeg, B.; Alexander, M. H. Adiabatic and diabatic potential energy surfaces for collisions of CN(X2Σ+, A2Π) with He. J. Chem. Phys. 1988, 89 (5), 3139–3151.10.1063/1.454971Search in Google Scholar
Whitham, C. J.; Soep, B.; Visticot, J.; Keller, A. Observation and spectroscopy of metallic free radicals produced by reactive collisions during a supersonic expansion. J. Chem. Phys. 1990, 93 (2), 991–1000.10.1063/1.459126Search in Google Scholar
Wiberg, K. B.; Rablen, P. R. Origin of the stability of carbon tetrafluoride: negative hyperconjugation reexamined. J. Am. Chem. Soc. 1993, 115 (2), 614–625.10.1021/ja00055a034Search in Google Scholar
Wijeyasinghe, N.; Anthopoulos, T. D. Copper(I) thiocyanate (CuSCN) as a hole-transport material for large-area opto/electronics. Semicond. Sci. Technol. 2015, 30 (10), 104002.10.1088/0268-1242/30/10/104002Search in Google Scholar
Wilcox, D. E.; Bromley, L. A. Computer estimation of heat and free energy of formation for simple inorganic compounds. Ind. Eng. Chem. 1963, 55 (7), 32–39.10.1021/ie50643a006Search in Google Scholar
Wilkie, C. A.; Parry, R. W. Halocyanophosphines, P(CN)YX3-y, and related cyanophosphine derivatives. Inorg. Chem. 1980, 19 (6), 1499–1502.10.1021/ic50208a015Search in Google Scholar
Willemin, M.-E.; Lumen, A. Thiocyanate: a review and evaluation of the kinetics and the modes of action for thyroid hormone perturbations. Crit. Rev. Toxicol. 2017, 47 (7), 543–569.10.1080/10408444.2017.1281590Search in Google Scholar PubMed
Williams, D. R.; Damrauer, R. Molecular orbital calculations on substituted carbodiimides. Theor. Chim. Acta 1971, 23 (2), 195–202.10.1007/BF00526432Search in Google Scholar
Williams, D.; Kouvetakis, J.; O’Keeffe, M. Synthesis of nanoporous cubic in(CN)3 and In1−xGax(CN)3 and corresponding inclusion compounds. Inorg. Chem. 1998, 37 (18), 4617–4620.10.1021/ic971638nSearch in Google Scholar PubMed
Williams, D.; Pleune, B.; Kouvetakis, J.; Williams, M. D.; Andersen, R. A. Synthesis of LiBC4N4, BC3N3, and related C−N compounds of boron: new precursors to light element ceramics. J. Am. Chem. Soc. 2000, 122 (32), 7735–7741.10.1021/ja0006752Search in Google Scholar
Williams, D.; Pleune, B.; Leinenweber, K.; Kouvetakis, J. Synthesis and structural properties of the binary framework C–N compounds of Be, Mg, Al, and Tl. J. Solid State Chem. 2001, 159 (1), 244–250.10.1006/jssc.2001.9192Search in Google Scholar
Williams, H. E. The chemistry of cynanogen compounds; J. & A. Churchill: London, 1915.Search in Google Scholar
Williams, H. E. Cyanogen compounds: their chemistry, detection and estimation, 2nd ed.; Edward Arnold: London, 1948.Search in Google Scholar
Wilson, I. R.; Harris, G. M. The oxidation of thiocyanate ion by hydrogen peroxide. II. The acid-catalyzed reaction. J. Am. Chem. Soc. 1961, 83 (2), 286–289.10.1021/ja01463a007Search in Google Scholar
Wilson, I. R.; Harris, G. M.; Wilson, I. R.; Harris, G. M. The oxidation of thiocyanate ion by hydrogen peroxide. I. The PH-independent reaction. J. Am. Chem. Soc. 1960, 82 (17), 4515–4517.10.1021/ja01502a017Search in Google Scholar
Wittig, G.; Bille, H. Notizen: Über aluminium-tricyanid und lithium-aluminium-tetracyanid. Z. Naturforsch. B: J. Chem. Sci. 1951, 6 (4), 226.10.1515/znb-1951-0412Search in Google Scholar
Wittig, G.; Raff, P. Notizen: Darstellung von Lithium-Monocyano-Borhydrid. Z. Naturforsch. B: J. Chem. Sci. 1951, 6 (4), 225.10.1515/znb-1951-0411Search in Google Scholar
Wizemann, T.; Müller, H.; Seybold, D.; Dehnicke, K. Reaktionen von Rirhodan Mit Einigen Organometall-Verbindungen. J. Organomet. Chem. 1969, 20 (1), 211–217.10.1016/S0022-328X(00)80108-4Search in Google Scholar
Wofford, B. A.; Bevan, J. W.; Olson, W. B.; Lafferty, W. J. Rotational analysis and vibrational predissociation in the Ν2 band of HCN dimer. J. Chem. Phys. 1986, 85 (1), 105–108.10.1063/1.451646Search in Google Scholar
Wöhler, F. Über Einige Verbindungen des Cyans (Blaustoffs). Ann. Phys. 1821, 69 (11), 271–282.10.1002/andp.18210691109Search in Google Scholar
Wöhler, F. Analytische Versuche Über die Cyansäure. Ann. Phys. 1824, 77 (5), 117–124.10.1002/andp.18240770506Search in Google Scholar
Wöhler, F. Über Cyan Verbindungen. Ann. Phys. 1825a, 79 (2), 177–182.10.1002/andp.18250790208Search in Google Scholar
Wöhler, F. Über die Zusammensetzung der Cyansäure. Ann. Phys. 1825b, 81 (11), 386–388.10.1002/andp.18250811110Search in Google Scholar
Wöhler, F. Ueber Künstliche Bildung des Harnstoffs. Ann. Phys. 1828, 88 (2), 253–256.10.5962/bhl.title.62545Search in Google Scholar
Wootten, A.; Lichten, S. M.; Sahai, R.; Wannier, P. G. CN abundance variations in the shell of IRC + 10216. Astrophys. J. 1982, 257, 151.10.1086/159973Search in Google Scholar
Wormer, P. E. S.; Tennyson, J. Ab initio SCF calculations on the potential energy surface of potassium cyanide (KCN). J. Chem. Phys. 1981, 75 (3), 1245–1252.10.1063/1.442174Search in Google Scholar
Wu, W. W.; Chadik, P. A.; Schmidt, C. J. An in situ synthesis of cyanogen chloride as a safe and economical aqueous standard. Wat. Res. 1998, 32 (9), 2865–2869.10.1016/S0043-1354(98)00029-3Search in Google Scholar
Wu, C.; Xiong, Y.; Gao, Z.; Kong, F.; Lu, H.; Yang, X.; Xu, Z. Ionization and dissociation of acetonitrile by intense femtosecond laser pulse. Chin. Sci. Bull. 2000, 45 (21), 1953–1955.10.1007/BF02909685Search in Google Scholar
Yamamoto, J.; Okabe, Y. Ab initio molecular dynamics simulation on SiN + CH and SiC + NH reactions. Comput. Theor. Chem. 2011, 963 (1), 24–33.10.1016/j.comptc.2010.09.004Search in Google Scholar
Yan, G.; Zhang, Y.; Wang, J. Recent advances in the synthesis of aryl nitrile compounds. Adv. Synth. Catal. 2017, 359 (23), 4068–4105.10.1002/adsc.201700875Search in Google Scholar
Yang, Z.; Luty, F. Theoretical investigation of ferroelastic phase transition of pure and mixed alkali cyanide systems by the elastic dipole model. II. The cubic-monoclinic phase transition of pure rubidium cyanide. Phys. Status Solidi B 1989, 154 (1), 181–194.10.1002/pssb.2221540116Search in Google Scholar
Yang, D. L.; Yu, T.; Lin, M. C.; Melius, C. F. CN radical reactions with hydrogen cyanide and cyanogen: comparison of theory and experiment. J. Chem. Phys. 1992, 97 (1), 222–226.10.1063/1.463945Search in Google Scholar
Yang, Z.; Cole, C. A.; MartinezJr.O.; Carpenter, M. Y.; Snow, T. P.; Bierbaum, V. M. Experimental and theoretical studies of reactions between h atoms and nitrogen-containing carbanions. Astrophys. J. 2011, 739 (1), 19–28.10.1088/0004-637X/739/1/19Search in Google Scholar
Yoshimura, Y.; Shinohara, S.; Tsuda, N.; Iwasaki, H. X-ray diffraction study of the low temperature phase of rubidium cyanide. J. Phys. Soc. Jpn. 1996, 65 (7), 2099–2105.10.1143/JPSJ.65.2099Search in Google Scholar
Yoshimura, Y. Stucture determination of the monoclinic form of KCN. J. Phys. Soc. Jpn. 1989, 58 (11), 3993–3998.10.1143/JPSJ.58.3993Search in Google Scholar
Yost, D. M.; Stone, W. E. The complex ions formed by iodine cyanide with cyanide and iodide ions. The vapor pressure, free energy and dissociation of iodine cyanide. J. Am. Chem. Soc. 1933, 55 (5), 1889–1895.10.1021/ja01332a015Search in Google Scholar
Youatt, J. B. Studies on the metabolism of thiobacillus thiocyanoxidans. J. Gen. Microbiol. 1954, 11 (2), 139–149.10.1099/00221287-11-2-139Search in Google Scholar PubMed
Yu, D.; Rauk, A.; Armstrong, D. A. Electron affinities and thermodynamic properties of some triatomic species. J. Phys. Chem. 1992, 96 (14), 6031–6038.10.1021/j100193a071Search in Google Scholar
Yu, J.-T.; Teng, F.; Cheng, J. The construction of X-CN (X = N, S, O) bonds. Adv. Synth. Catal. 2017, 359 (1), 26–38.10.1002/adsc.201600741Search in Google Scholar
Zarges, W.; Marsch, M.; Harms, K.; Boche, G. [{Li2(Me3SiCCN)}12(Et2O)6(C6H14)], Kristallstruktur Mit Dem Trimethylsilylacetonitril-Dianion. Chem. Ber. 1989, 122 (7), 1307–1311.10.1002/cber.19891220714Search in Google Scholar
Zhan, C.; Iwata, S. Ab initio studies on the structures, vertical electron detachment energies, and fragmentation energies of CnN− clusters. J. Chem. Phys. 1996, 104 (22), 9058–9064.10.1063/1.471638Search in Google Scholar
Zhang, L.-Y.; Shi, L.-X.; Chen, Z.-N. Syntheses, structures, and electronic interactions of dicyanamide/tricyanomethanide-bridged binuclear organometallic complexes. Inorg. Chem. 2003, 42 (2), 633–640.10.1021/ic025879tSearch in Google Scholar PubMed
Zhang, M.; Gong, J.; Ma, A.; Rice, S. A. Infrared multiphoton induced isomerization and dissociation of FCN, ClCN, and BrCN in liquid Ar: a classical simulation study. J. Chem. Phys. 2007, 127 (14), 144501.10.1063/1.2772620Search in Google Scholar PubMed
Zhang, C.; Li, Y.; Shuai, D.; Shen, Y.; Xiong, W.; Wang, L. Graphitic carbon nitride (g-C3N4)-based photocatalysts for water disinfection and microbial control: a review. Chemosphere 2019, 214, 462–479.10.1016/j.chemosphere.2018.09.137Search in Google Scholar PubMed
Zhao, W.; Pan, J.; Huang, F. Nonaqueous synthesis of metal cyanamide semiconductor nanocrystals for photocatalytic water oxidation. Chem. Commun. (London) 2018, 54 (13), 1575–1578.10.1039/C7CC09699BSearch in Google Scholar PubMed
Zheng, Z. Q.; Wang, J.; Wu, T. H.; Zhou, X. P. Alkylation of ammonium salts catalyzed by imidazolium-based ionic liquid catalysts. Adv. Synth. Catal. 2007, 349 (7), 1095–1101.10.1002/adsc.200600451Search in Google Scholar
Zhu, Z.; Zhang, Z.; Huang, C.; Pei, L.; Chen, C.; Chen, Y. Kinetics of CCN radical reactions with a series of normal alkanes. J. Phys. Chem. 2003, 107 (48), 10288–10291.10.1021/jp030763jSearch in Google Scholar
Zhu, C.; Räsänen, M.; Khriachtchev, L. Fluorinated noble-gas cyanides FKrCN, FXeCN, and FXeNC. J. Chem. Phys. 2015, 143 (7), 074306.10.1063/1.4928649Search in Google Scholar PubMed
Ziegler, K. Über die Darstellung Wasserfreier Blausäure. Ber. Dtsch. Chem. Ges. 1921, 54 (1), 110–112.10.1002/cber.19210540111Search in Google Scholar
Zúñiga, J.; Picón, J. A. G.; Bastida, A.; Requena, A. A spectroscopic potential energy surface for FCN. J. Quant. Spectrosc. Radiat. Transfer 2012, 113 (11), 1155–1169.10.1016/j.jqsrt.2012.01.023Search in Google Scholar
© 2022 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Research progress of catalytic oxidation of volatile organic compounds over Mn-based catalysts – a review
- Recent progress on fabrication methods of graphene-based membranes for water purification, gas separation, and energy sustainability
- In-vitro anticancer profile of recent ruthenium complexes against liver cancer
- Main group cyanides: from hydrogen cyanide to cyanido-complexes
- Quantitative phase analysis of anhydrous clinker Portland using Rietveld method
Articles in the same Issue
- Frontmatter
- Research progress of catalytic oxidation of volatile organic compounds over Mn-based catalysts – a review
- Recent progress on fabrication methods of graphene-based membranes for water purification, gas separation, and energy sustainability
- In-vitro anticancer profile of recent ruthenium complexes against liver cancer
- Main group cyanides: from hydrogen cyanide to cyanido-complexes
- Quantitative phase analysis of anhydrous clinker Portland using Rietveld method